 María Carolina Barbosa1
María Carolina Barbosa1 Claudio Marcelo Fader
Claudio Marcelo Fader- 1Laboratorio de Biología Celular y Molecular, Instituto de Histología y Embriología (IHEM), Universidad Nacional de Cuyo, CONICET, Mendoza, Argentina
- 2Facultad de Odontología, Universidad Nacional de Cuyo, Mendoza, Argentina
Autophagy is a major protein turnover pathway by which cellular components are delivered into the lysosomes for degradation and recycling. This intracellular process is able to maintain cellular homeostasis under stress conditions, and its dysregulation could lead to the development of physiological alterations. The autophagic activity has been found to decrease with age, likely contributing to the accumulation of damaged macromolecules and organelles during aging. Interestingly, failure of the autophagic process has been reported to worsen aging-associated diseases, such as neurodegeneration or cancer, among others. Likewise, it has been proposed in different organisms that maintenance of a proper autophagic activity contributes to extending longevity. In this review, we discuss recent papers showing the impact of autophagy on cell activity and age-associated diseases, highlighting the relevance of this process to the hallmarks of aging. Thus, understanding how autophagy plays an important role in aging opens new avenues for the discovery of biochemical and pharmacological targets and the development of novel anti-aging therapeutic approaches.
The Autophagic Process
Autophagy, literally meaning “self-eating,” is an evolutionarily conserved catabolic process in eukaryotic cells by means of which intracellular components and extracellular incorporated material are delivered into lysosomes, where their degradation occurs (1). Since its discovery, autophagy has been associated with the maintenance of cellular homeostasis, as well as the cytoplasmic quality control process (1, 2). Its dysregulation has being related to a diversity of pathological or physiological processes such as neurodegenerative, infectious, and metabolic disorders, as well as cancer and aging, among others (3–5). Several studies have demonstrated that autophagy can be very selective in targeting its cargo for degradation. Three major types of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy (hereafter referred to as autophagy) begins with the extension of a specialized membrane, known as the phagophore, derived from the endoplasmic reticulum (ER), the mitochondria, and the Golgi cisternae (6, 7). The phagophore engulfs the molecules and organelles to be eliminated, forming a double membrane vesicle called autophagosome (7, 8). Finally, autophagosomes are targeted to lysosomes and fusion occurs, the sequestered material is degraded and released back into the cytosol (8). In microautophagy, the lysosome picks up cytosolic components directly via invagination of the lysosomal membrane (9). On the other hand, CMA is a process involving the direct transport of cytosolic components across the lysosomal membrane via chaperone proteins. Several studies have demonstrated that CMA is a highly regulated and degradative event, involving HSC70 (heat shock protein 70 complex) and multimerization of the LAMP2A receptor (lysosome-associated membrane protein type 2A). Interestingly, not all proteins are able to be CMA substrates. To undergo CMA degradation, proteins must contain a KFERQ motif in their amino acid sequences, which is necessary to bind the chaperone HSC70 (10, 11). Substrate and the HSC70 complex can bind a 12-amino-acid cytosolic tail of LAMP2A for lysosomal docking. In addition, LAMP2A multimerization is necessary for substrate translocation into the lysosomal lumen. Cytosolic HSC70 is released from the multimeric complex, and then a chaperone HSP90 (located at the lumen of the lysosomal membrane) interacts with LAMP2A, stabilizing it during the substrate translocation. Finally, a luminal chaperone HSC70 is required to end the translocation process, and once inside the targeted protein is degraded by the lysosomal enzymes (12) (Figure 1).
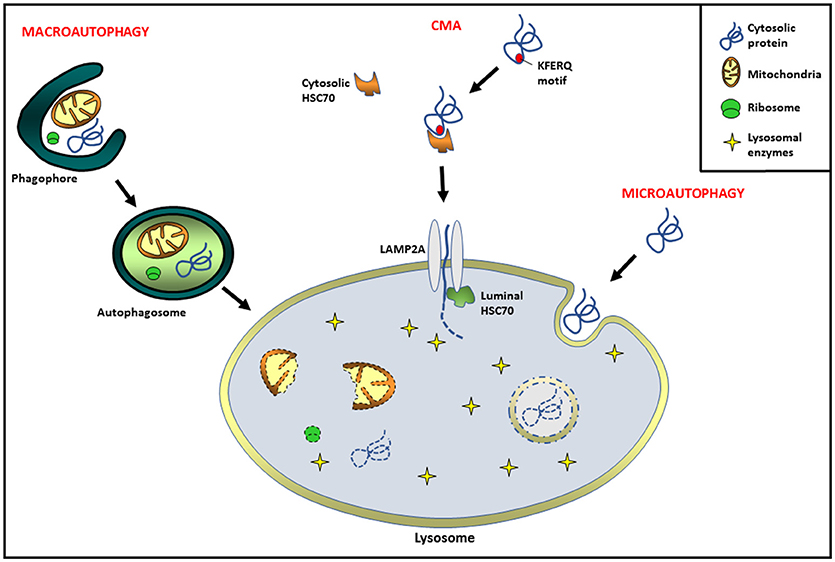
Figure 1. Different types of autophagy pathways in mammals. Macroautophagy: extension of a specialized membrane (phagophore) surrounds molecules and organelles, forming a double membrane vesicle called autophagosome. Finally, the fusion of autophagosomes with lysosomes leads to cargo degradation. Chaperone-mediated autophagy (CMA): proteins containing a KFERQ motif are delivered to lysosome via cytosolic HSC70 chaperone complex. The receptor lysosome-associated membrane protein type 2A (LAMP2A) is necessary for substrate translocation into the lysosomal lumen, where the degradation occurs. Microautophagy: invagination of the lysosomal membrane engulfs cytosolic cargo in small vesicles for its degradation inside.
Autophagy can be induced by a variety of stressors, and nutrient restriction is one of the major stimuli, capable of rapidly activating the autophagic process with the concomitant inhibition of protein synthesis (1). Studies in both yeasts and mammals have characterized at least 40 autophagy-related genes (Atg), which encode proteins that participate in autophagy (13). In addition, the canonical autophagy pathway includes the inactivation of mammalian target of rapamycin complex 1 (mTORC1), allowing the phosphorylation and activation of the Unc-51-like kinase complex (Ulk1/2), with the subsequent cascade activation of the other ULK complex members such as FIP200 and ATG13 (14, 15). Another important complex that is activated is the BECLIN1, in which VPS34, one of its members, is translocated into the ER membranes and it produces high levels of phosphatidylinositol-3-phosphate, which is necessary for the recruitment of other effectors such as WIPI2b (16). Next, this effector interacts and recruits ATG16L, which binds ATG5-ATG12 conjugated to generate the ATG12-ATG5-ATG16L complex. This complex is required for the lipidation of LC3 (Microtubule-associated protein light chain 3), because it determines the site where LC3 will be conjugated and activated to LC3-II (17). Moreover, ATG3 (an E2-like protein) is associated with LC3-I and it binds to the complex through ATG12, allowing the conjugation of LC3-I with phosphatidylethanolamine to generate LC3-II. LC3-II, which is present in both inner and outer membranes of autophagosomal structures and is necessary for phagophore extension, cargo engulfment, and vesicle closure to form the autophagosome. Additionally, the targeted cargo is able to bind receptor/adaptor molecules like p62, NDP52, and NIX. These proteins contain a LC3 interacting region (LIR), which allows the recognition of elements to be engulfed by the phagophore and eliminated in an autophagic manner (18, 19).
In addition to degradation, autophagy, or part of its machinery, can mediate a regulated cell death, named autophagy-dependent cell death (ADCD). Moreover, autophagy can participate in other cell death types [reviewed in reference (20)]. Interestingly, despite the fact that regulated cell death of malignant cells is a pro-survival mechanism at the whole organism level, it can also lead to tissue degeneration and function loss, and this can reduce the fitness of the aged individual (21).
Aging
Aging, the natural event occurring in all living organisms, can be defined as a deterioration of the cell functioning due to damage accumulation over time (22–25). This is an important biological, demographic and socio-economic issue all over the world. Dr. Barja points out that all living organisms have different longevity, indicating that evolution has played an important role in regulation and flexibilization of aging between species, in a relatively fast process (26). The understanding of the molecular basis of aging and longevity could let us manipulate it somehow in the future. In this regard, in the last 50 years numerous investigations related to aging have emerged, trying to explain this unstoppable process.
Despite the general accepted concept that aging is a multifactorial process, several theories have emerged in an attempt to explain it as a single predominant age-related change. A popular aging theory is the “Stochastic Theory,” which suggests that aging results from random damage accumulation. This can be due to external and internal sources over time, in addition to a failure of the repairing capacity. On the other hand, other theories support the idea that aging is a regulated process, mainly by the genetic code, such as the telomere length, the number of divisions that a somatic cell can go through (the “Hayflick limit”) and spatio-temporal regulation of gene expression (27, 28). Nevertheless, one of the most popular theories is the Free Radicals (or Oxidative Stress) Theory of Aging, which hypothesizes that an accumulation of Reactive Oxygen Species (hereafter ROS) falls into an oxidative damage of biomolecules, with the consequent cell functioning decline (27–29). A considerable body of evidence supports this theory, because it points to an increase in ROS cellular levels as we age, due to a higher production of them as well as a failure in the anti-oxidant systems (30, 31).
Autophagy in Aging
Several animal models have contributed to our understanding of how the impairment of autophagy and redox homeostasis can result in age-related diseases. In the same way, numerous studies involving genetic ablation or induction of autophagic genes have revealed the importance of this process in aging of yeast, nematodes, flies, and mammals (32). The most important work that links an overexpression of a single Atg gene with an increment in mammals' lifespan was conducted by Pyo and collaborators. The authors overexpressed Atg5 in mice and found an enhancement of the autophagy process and anti-aging features, compared with the wild type mice. The mean lifespan was also incremented, suggesting the importance of autophagy in the longevity of mice (33). Another approach that demonstrates the importance of autophagy in aging has been done in Ana María Cuervo's laboratory. In aged mice, they generated a double transgenic mouse model, in which it was possible to modulate the expression of the lysosomal receptor for CMA. The results revealed that the enhancement of this receptor can prevent features of aging at cellular and organ levels (34). In addition, mice overexpressing Atg5 showed a better resistance to age-related obesity and enhanced insulin sensitivity, exhibiting an improved metabolism in aged individuals (33). Despite the mentioned studies, several others failed to demonstrate that upregulation of a single autophagic component can extend lifespan (32). Moreover, several KO mouse models have been shown to have extended lifespan, although the molecular mechanisms behind it and the connection with aging are not yet clear (35).
Notably, another relationship between autophagy augmentation and extended lifespan has been reported in exceptionally healthy centenarian humans, who have increased levels of BECLIN1, compared to young people (36). We hope that in the next years these preliminary studies in humans will be more advanced, providing insights into our species longevity mechanisms from clinical case studies.
The knockout for essential Atg genes is lethal in mice, and tissue-specific ablation has a less-dramatic phenotype, manifesting premature signs of aging (37). Specific-Atg5 or Atg7 KO leads to neurodegeneration or tissue abnormalities in most of the cases available in the literature [for a more detailed summary see reference (37)].
Finally, as we age, the incidence of cancer rises, probably because of the decline of homeostatic processes and the increase in the accumulation of potentially harmful molecules such as ROS and protein aggregates. Autophagy has been proposed to have a dual role in tumorigenesis, being important both in suppression as well as in tumor progression and surveillance (38, 39).
ROS Generation and Aging
The ROS are considered metabolites of molecular oxygen during cellular respiration, being very reactive due to an unpaired electron (40). Mitochondria are the major ROS producers and perhaps the organelle most affected by them. In order to avoid detrimental effects of ROS, two important processes arise: Mitophagy and antioxidant system. Mitochondrial ROS can activate the autophagic pathway upon starvation by the activation of ATG4 (41), and this in turn leads to autophagic degradation of mitochondria (mitophagy) in order to reduce the ROS levels by limiting the number of mitochondria per cell (42). In addition, hypoxia and exercise can also trigger redox-dependent autophagy, suggesting that ROS might regulate the autophagic response to several stresses (43). Regarding the second process, the antioxidant system consists of several enzymes and molecules that react with ROS and neutralize them somehow, but the connection of antioxidants with lifespan is controversial (44). Notably, overexpression of a mitochondrial-targeted catalase in mice extends lifespan and reduces overall ROS, reinforcing the Free Radicals Theory of Aging in such model (45). Moreover, these mice showed a reduction in age-related pathologies (46). Additionally, Mn-superoxide dismutase (SOD2) heterozygous mice showed a life-long reduction, but surprisingly they did not have an accelerated aging phenotype. Nevertheless, this Sod2+/− mice showed a higher oxidative damage to DNA and had higher cancer incidence compared with wild type individuals (47). By contrast, knockout of 17 genes involved in the antioxidant system exhibited no effect in lifespan: Only the knockout for Cu/Zn-superoxide dismutase (Sod1) resulted in a decrease in longevity and premature aging as well (48). This mouse model showed an increase in senescent markers, suggesting that the oxidative stress that Sod1−/− mice suffer leads to DNA damage, promoting an irreversible state of quiescence (49). In addition, these Sod1−/− mice showed an accelerated sarcopenia, manifesting muscular mass loss and altered neuromuscular junctions (50). Despite these controversial and unexpected results in mice, the relationship between antioxidants and their role in healthy or pathologic aging needs to be deeply studied in the future.
Finally, it is important to highlight that ROS have been proposed to be implicated in proliferation and survival signaling in certain conditions (42). A new concept has emerged recently in the aging field, termed “hormesis,” according to which low doses of a stressor can improve the cell response for a more detrimental condition in the future (32, 51). This could increase lifespan and cellular fitness (52). In this context, low levels of ROS can be beneficial due to the trigger of homeostatic responses, but its disproportional augmentation can lead to damage or aging (42, 53). From an autophagic perspective, an augmentation in ROS levels and a decline in mitophagy occur simultaneously, leading to aging (43, 52, 54).
Hallmarks of Aging: An Autophagic View
In the last years, aging has begun to be seen as an active and highly regulated process (55). Age-related changes at cellular level include an increase in ROS, loss of proteostasis, genome instability, and telomere exhaustion, among others (23, 56, 57). These characteristic features of aging were termed “hallmarks of aging” by López-Otín et al. (52). In the following sections, we discuss how autophagy plays an important role in some of these hallmarks of aging, in both health and disease.
Loss of Proteostasis
Proteostasis is one of the major functions of autophagy in normal tissues. Imbalance of proteostasis due to aging leads to protein aggregation, accumulation of misfolded proteins and in the end to cellular dysfunction, among others (23, 56, 57). Notably, carbonylation due to oxidative stress is one of the changes that leads to loss of proteostasis (44). To avoid cell death or dysfunction, numerous homeostatic mechanisms turn on, mainly autophagy (58) and the Ubiquitin-Proteasome-System (UPS). Because autophagy is considered one of the most important intracellular homeostatic processes, an alteration or deterioration of this pathway could modify the normal cell functioning, including a variety of diseases and normal cell physiology declination. Autophagosomes and lysosomes decline in an age-dependent manner in muscles (59), heart (43), and several other tissues. Moreover, CMA has also been implicated in removing oxidized and potentially dangerous proteins by direct lysosomal degradation (60).
The UPS is mostly implicated in the degradation of misfolded proteins, as well as short- and long-lived proteins by their ubiquitination. This process is achieved thanks to three major proteins that sequentially activate the ubiquitin tag (E1), transfer it to a second enzyme (E2), and finally ligate the ubiquitin tag to the target molecule (by E3 ligase), which eventually reaches the proteasome for degradation (61, 62). It is important to note that almost all regulatory proteins are substrates for this system (61, 63), and UPS declines with age [reviewed in (64)]. Interestingly, mTORC1 was found to regulate not only lysosomal protein degradation, but also proteasomal proteolysis of long-lived proteins, independently of protein synthesis, suggesting a common regulation of both proteolytic systems by nutrient-sensing (63). In addition, overexpression of a sole subunit of the proteasome enhanced its activity and the survival against several oxidants in two cell lines as well as primary culture of human fibroblasts (65). Moreover, proteasome activity decreased in an age-dependent manner (66). Overexpression of proteasome subunits in aged dermal human fibroblasts ameliorated the aged phenotype and restored the oxidized and ubiquitinated proteins to young levels (66). In the same way, transgenic mice with reduced proteasomal activity accumulated oxidized and ubiquitinated proteins, accelerating the aging phenotype and the age-related metabolic diseases (67). Besides, inhibition of proteasome activity impaired cell proliferation and shortened lifespan (68), reinforcing the importance of a correct proteostasis in healthy aging and longevity.
Several studies have been done on neurodegenerative diseases related to aging and autophagy, including those most relevant for their high impact on human population. Most of them share the accumulation of ROS, misfolded proteins, and damaged organelles, aging being the main risk factor (69, 70). This accumulation interferes with proper axonal traffic, enhancing neurotoxicity. Both autophagy and CMA impairment hamper the correct protein-aggregates degradation and the remodeling of dendrites and axons, thus diminishing the nervous plasticity (71, 72). In Parkinson's disease (PD), the cytoplasmic aggregates are formed by α-synuclein and ubiquitin (or Lewy bodies) in dopaminergic neurons of substantia nigra, leading to their death (32). Alterations in UPS (71) and also in CMA can develop the disease too. It is worth noting that overexpression of Lamp2A improved CMA performance and decreased α-synuclein cytoplasmic levels (73). In the same way, Alzheimer's disease (AD) is characterized by intracellular accumulation of tau protein as well as β-amyloids (Aβ), derived from the amyloid precursor protein (APP). This aggregate formation impairs normal cell function, finally leading to cell death (32). Also, extracellular Aβ-plaques secreted by autophagosomes can interrupt intercellular communication (72), another hallmark of aging (52). Besides, Apolipoprotein E4 (ApoE4) is the main genetic risk for sporadic AD and was found to promote the disruption of the lysosomal membrane together with Aβ, leading to neuronal degeneration (72, 74). Mutations in Presenilin1 or 2 (PS1 and PS2, two transmembrane subunits of gamma-secretase), as well as in tau protein or in APP are common causes of the familial AD (72). Other neurodegenerative diseases implicate alterations in the autophagic process, i.e., SENDA, Huntington, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia disorders [reviewed in (70, 75, 76)]. In all these cases, the lack of proper degradation by autophagy promotes the aggregation of several proteins and the consequent malfunctioning of axonal transport.
Regarding metabolic diseases and autophagy, it is well known that islet amyloid deposition leads to type 2 diabetes in humans due to the amyloidogenic property of human islet amyloid polypeptide (hIAPP). It is important to note that mice do not develop such aggregation. In order to bypass this model difference, Kim et al. developed transgenic mice expressing hIAPP specifically in β-cells and bred them with Atg7Δβ−cell mice. Male mice had premature diabetes, while females had hyperglycemia but never developed the disease, suggesting a synergism between autophagy deficiency and human amyloid overexpression. Moreover, primary culture of monkey islet cells overexpressing precursors of hIAPP showed that autophagic inhibition by 3-methyladenine (3-MA) increased pro-hIAPP dimer or trimer accumulation, blocking the autophagic activity in these pancreatic cells (77). It is also important to highlight that diabetes or glucose handling deficiencies are risk factors for the AD, as the amyloids properties of proteins implicated in metabolic diseases and AD are similar and probably interconnected (78). More studies regarding the connection between metabolic and neurodegenerative diseases are required for a better understanding of the molecular basis of such relationships at systemic level.
Finally, sarcopenia is characterized by a progressive loss of muscle mass and strength thanks to an imbalance between production and degradation of proteins (79). Aged-related declination of autophagy (both mitophagy as well as CMA) promotes sarcopenia by protein accumulation interference with normal myofibers functioning, but an exacerbation of autophagy can also result in cellular stress and finally death (79). Thus, an age-related imbalance of proteostasis could drive a variety of diseases involving both protein accumulation and degradation.
Mitochondrial Dysfunction
Mitophagy is a basal process involved in the autophagic degradation of mitochondria (76, 80, 81). It is necessary in normal differentiation of certain cell types such as red blood cells (82), in embryogenesis, immune response, cell programming, and cell death (80). Mitophagy is required not only to remove damaged mitochondria, but also to promote the biosynthesis of new ones, supporting the mitochondrial quality control (76, 80). Given that mitochondria are implicated in bioenergetics and ROS production, the mitophagy plays an important role in cell homeostasis. Additionally, a decrease in mitophagy is observed in aged animals and this contributes to aging phenotype (81).
Canonically, mitophagy is triggered by the cytosolic exposition of mitochondrial outer membrane (MOM) proteins, which have a LIR domain. The mitophagy is tightly regulated by several molecules, NIX and BNIP being two of the most widely characterized mitochondrial adaptors for autophagic machinery (83). NIX activation is associated with an increment in mitochondrial degradation in HeLa cells, protecting them against cellular stress (84). Interestingly, NIX has a LIR domain which binds LC3 once it is activated by phosphorylation (84). Additionally, PINK1 and PARKIN have been involved in the regulation of mitophagy when the mitochondrion loses its membrane potential (80). These proteins have been considered as key components in controlling the activation of mitophagy (85) and also as participants of mitophagy-associated cancer resistance. PINK1 and PARKIN are activated in response to an increment of intracellular ROS levels, which stimulate the MAPK and ERK1/2 signaling cascades, triggering parapoptosis in non-malignant cells, which bypass the caspases activation and, thus, the apoptosis (86) (Figure 2).
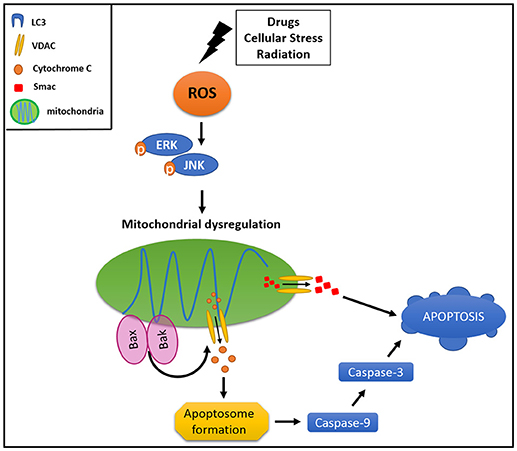
Figure 2. Mitophagy protects cancer cell from apoptosis. Different stimuli could drive an activation of mitochondrial dysregulation, triggering signaling pathways involved in activation of pro-apoptotic proteins (BAK and BAX). This results in MOM damage and the consequent cytochrome c and SMAC release to the cytoplasm, activating intrinsic apoptotic pathway.
Additionally, Mitofusin 2 (MFN2) is a mitochondrial membrane fusing protein involved in several processes, including mitochondria fusion and mitophagy. Its expression declines with age, and its deficiency provokes precocious sarcopenia, accumulation of damaged mitochondria, and metabolic disorders in young mice (87). In addition, Humanin, an antiapoptotic mitochondrial protein, is capable of activating the CMA machinery, thus protecting several cell types from oxidative stress (88). Interestingly, both CMA and Humanin decline with age (89, 90), contributing to the age-related deterioration of proteostasis and mitochondrial functionality. As can be seen, several proteins regulate the mitophagy and contribute to the mitochondria homeostasis. As almost all of them decline with age, the modulation of mitophagy regulatory proteins could be a novel anti-aging therapeutic approach in the future. Despite this, more studies are needed in order to understand the complex regulation of mitophagy and the relationships between the players.
Different compounds, intracellular changes or stimuli could drive an activation of mitochondrial dysfunction. Normally, ROS oxidative stress, loss of membrane potential, MOM permeability, and aging are able to cause mitochondrial dysregulation. This imbalance triggers signaling pathways involving activation of pro-apoptotic proteins of the BCL-2 family such as BAK and BAX, resulting in an MOM damage and the consequent release of cytochrome c and SMAC (second mitochondrial-derived activator of caspase) to the cytoplasm (91), activating the intrinsic apoptotic pathway through caspase 9 (92, 93) (Figure 2). Furthermore, hypoxia inducible factor 1α (HIF1α) is able to trigger mitophagy by stabilization and activation of NIX protein, and is also responsible for autophagic activation through VMP1 (vacuole membrane protein 1) promoter, causing colon cancer resistance to photodynamic therapy (94).
Disruption in mitophagy, and thus in redox homeostasis, can produce different cardiovascular pathologies (43). PINK1-KO mice developed a decline in cardiac function due to dysfunctional mitochondria and an increase in oxidative stress (95). Notably, the same features were observed in human end-stage heart failure samples, where diminished PINK1 levels were also found (95). Despite the ubiquitous KO model, the authors were capable of demonstrating the importance of PINK1 in heart functioning. Additionally, altered mitophagy due to elevated ROS production has been linked to Alzheimer Disease (AD), but there is controversy as to whether this disturbance in the autophagic pathway is a cause or a consequence of AD (72). Remarkably, PARKIN and PINK1 have been found mutated in Parkinson Disease (PD) patients, suggesting the importance of this pathway in dysfunctional mitochondria clearance by autophagy (76, 96). In 2015, Sun et al. published a new approach for measuring mitophagy in vivo, using a transgenic mouse model consisting in the mitochondria-targeted overexpression of a fluorescence reporter named Keima (81). We think that this tool could be very interesting for the in vivo study of mitophagy and its regulation under a wide variety of conditions. Deeper studies are then required to fully understand this process and its role in healthy and pathologic aging.
Deregulated Nutrient Sensing
Cellular response to nutrient privation implies some kind of intracellular sensor, capable of triggering the corresponding survival mechanisms. It has been proved that nutrient sensing is a highly conserved process across eukaryotes (97). Several nutrient-related signaling pathways converge on mTOR [mammalian Target Of Rapamycin; (97)], which triggers the response to growth factors, energy, glucose or amino acid changes (38, 97–100). Interestingly, nutrient sensors can also be activated under oxidative stress conditions, suggesting a common regulatory mechanism linking redox homeostasis and nutrients availability [reviewed in (101)].
The kinase mTOR is capable of linking environmental conditions with reproduction and somatic maintenance, thus influencing the individual lifespan (102). In addition, mTOR and mslt8 (a positive regulator of mTORC1) haploinsufficient female mice showed an increment in lifespan, compared to wild type mice (103). Mice carrying hypomorphic alleles of mTOR also had an increase in lifespan, a reduction in aging biomarkers, and a normal metabolism (104), showing the importance of nutrient sensing in aging. Moreover, Ribosomal S6 kinase 1 (S6K1) knockout mice showed an extended lifespan, compared with wild type counterparts, demonstrating again the importance of the mTOR pathway inhibition in longevity (105).
Besides, mTOR forms two distinct complexes, the Complex 1 (mTORC1) being capable of integrating different responses depending on nutrients availability (99, 106, 107). Upstream mTORC1, there are multiple regulating complexes, responding to each amino acid, glucose or growth factor input (99, 106, 107). Notably, p62 is one of the mTOR interactors upon amino acid stimulation, and it has been proposed to specifically stabilize the activated mTORC1 at the lysosome surface (108). It is well known that p62 is an autophagic adaptor protein whose role in nutrient sensing pathway might be another connection between mTOR and the regulation of autophagy.
Because autophagy is a catabolic mechanism, it can be assumed to be implicated in cellular and systemic metabolism. Metabolic stress responses could be compromised due to a decline in autophagic activity (109). As an important process regulating the general cellular status, autophagy can also crosslink metabolic pathways to maintain the homeostasis under a variety of conditions (43). In this sense, it has been demonstrated that, after nutrient or growth factor deprivation, ULK1 and ULK2 are activated, and these kinases phosphorylate and activate several glycolytic enzymes as well as autophagic proteins. This makes it possible to obtain metabolites thanks to glucose uptake, gluconeogenic pathway blockage, and autophagic degradation of cytosolic components (110). Supporting this, mTOR hyperactivation was found in several diseases such as obesity, metabolic syndrome, and type 2 diabetes (100), which highlights the importance of a tight regulation of autophagy as well as the nutrient sensing pathway.
Given that mTOR is capable of sensing the nutritional state of the cell, it was proposed to play an important role in Caloric Restriction (CR) therapy. Indeed, mTOR signaling network was shown to mediate lifespan extension by CR. Notably, the sole amino acid-restriction is enough to promote CR-response (111). Sirtuins 1 and 3 (NAD+ deacetylases) are activated in response to CR, as well as SOD1, in order to change the metabolism accordingly and prevent oxidative damage, respectively. Such ROS regulation rules out the Free Radicals Theory of Aging, at least partially (101, 112, 113). It is important to highlight the fact that inhibition of mTOR (specifically mTORC1) in embryogenesis is lethal, while the ablation of these pathways in adulthood can extend life [reviewed in (111)]. In fact, weight loss is suggested as prevention as well as therapy for a variety of age-related diseases (98).
Genomic Instability
In the last decade, several studies have demonstrated that autophagy or autophagic-related molecules act as a “safeguard” of genome stability both directly (DNA repair modulation) and indirectly (by acting as a homeostatic response) (114). Several mouse models have provided substantial information regarding genomic instability and its connection with healthy and pathological aging (55).
Regarding oxidative stress and DNA damage, ROS increase is thought to be mainly harmful for the mitochondrial DNA (mtDNA), generating the mutagenic 8-hydroxy-20-deoxyguanosine (8-OHdG), as well as mutations and deletions in mtDNA that result in a dysfunctional mitochondrion (69). Moreover, mitochondrial dysfunction promotes telomere attrition, telomere loss, and chromosome alterations, culminating in apoptosis in mouse embryos (115). Besides, Donati and collaborators demonstrated that, upon autophagic stimulation with an anti-lipolytic agent in 16-month-old rats, 8-OHdG accumulation in liver was successfully supressed, reaching the values obtained from young animals in only 6 h. When they measured the cytochrome c oxidase activity, they found that this decrease was not associated with lower mitochondrial enzyme activity, demonstrating the selective mitophagy of a small population of 8-OHdG-positive mitochondria and the importance of this proteostatic process in anti-aging mechanisms (54, 116). In the same way, dietary restriction reduced 8-OHdG levels in mitochondrial DNA (mtDNA) of aged rats and mice compared with those fed ad libitum (117), supporting the importance of dietary restriction in prevention of mtDNA damage by ROS in aged animals. Notably, Sod2−/− mice accumulated high levels of 8-OHdG both in nuclear and mitochondrial DNA, compared with wild type mice. Nevertheless, they showed no changes in lifespan or age biomarkers (47). On the other hand, Atg7−/− mouse keratinocytes presented premature aging after oxidative stress induction, supporting the importance of autophagy in healthy aging (118). In addition, Bender et al. found high levels of mtDNA deletions in dopaminergic neurons of PD patients, compared to controls (119). As we have already mentioned, PARKIN and PINK1 are mutated in PD, thus altered mitophagy can explain, in part, the accumulation of mtDNA damage in PD patients.
Autophagy has emerged as an important process in genome maintenance. After treatment with several cell cycle blockers, human osteosarcoma cells (U2OS) increased the micronuclei frequency as well as autophagosomes. Importantly, the authors observed a small but significant colocalization between them. Knockdown of Atg5 or Atg7 abolished this colocalization. P62/SQSTM1 also colocalized with micronuclei, indicating that micronuclei can be degraded by autophagy and this may contribute to genome stability (120). Moreover, NDP52 and p62-dependent autophagy can degrade retrotransposon RNA, preventing new insertions into the genome of long and short interspersed elements (121). Additionally, autophagy deficiency leads to an accumulation of RHOA with p62. This phenotype drives cytokinesis failure, aneuploidy, and multinucleation due to inappropriate formation of contractile ring (122). Furthermore, allelic loss of Beclin1 promotes tumorigenesis and activation of DNA-damage response in neoplasic cells. In this context, autophagy deficiency leads to genome instability under metabolic stress in these mouse mammary epithelial cells (123). Artificially aneuploid mouse cells showed increased autophagy to protect cells from genome instability (124).
Autophagic adaptor p62 has been found to be implicated in genome instability in several studies. Accumulation of p62 led to the activation of DNA-damage response (125). By contrast, overexpression of p62 (or autophagy deficiency) suppressed DNA-damage response by its direct inhibitory interaction with RNF168, an important E3 ligase for histone H2A ubiquitination and DNA-damage response (126). In this regard, p62 downregulates the protein levels of several molecules involved in homologous recombination (HR) of damaged DNA, inducing at the same time non-homologous end-joining (NHEJ), stressing the importance of p62 nuclear accumulation upon several stresses (127). More studies are needed to completely elucidate the role of p62 and other autophagic components in genome maintenance throughout life.
Epigenetic Alterations
Epigenetic changes due to external or internal factors drive several processes, including development and aging (128). In muscular and hematopoietic stem cells, different histone modifications help to establish the quiescence state with age (129). Besides, epigenetic alterations lead to different responses in aging and longevity in several mouse models [reviewed in (55)]. Notably, macrophages of old mice had hypermethylated LC3 and Atg5 promoter regions, compared with those of young mice, downregulating the protein levels and promoting aging decline of autophagy (130).
The natural polyphenol resveratrol, an autophagic inducer, has been proved to exert its action by inositol 1,4,5-triphosphate receptor (IP3R) and by protein acetylation decrease (131, 132). In this sense, a balance between different degrees of protein acetylation is presumed to be important for autophagy (32). High levels of acetyl-CoA, which serves as an acetyl group donor, were found to promote histone hyperacetylation, downregulating the expression of Atg genes, thus inhibiting autophagy and shortening lifespan in aged yeast (133). In addition, these authors found that the knockout of a certain acetyl-CoA in Drosophila melanogaster brain enhances autophagy and prolongs lifespan. Morselli et al. used specific siRNA knockdown of Sirtuin1, a NAD+ deacetylase in HCT116 cell line, which resulted in a suppression of the autophagic response to resveratrol. Importantly, resveratrol induced autophagy by AMP-dependent kinase/mTOR-independent pathway and changed the acetylation of 375 proteins, half of them involved in autophagy. On the other hand, deacetylations were often observed in metabolism-related proteins, thus activating autophagy (131). Another autophagic inducer, spermidine, is a well-known pro-longevity polyamine (56), largely studied as an anti-aging agent. Like resveratrol, spermidine can induce both acetylation and deacetylation changes that promote the autophagic pathways via AMP-dependent kinase signaling. In this regard, low doses of this polyamine together with low doses of resveratrol synergistically caused the same autophagic response as higher doses of each inducer separately (131).
Taken together, organismal models as well as in vitro studies highlight the importance of epigenetics throughout life. The relationship between epigenetic changes and autophagy needs to be deeply studied in order to understand the regulatory loop that seems to be involved in development and aging.
Telomere Attrition
Telomeres are specific repetitive sequences at the end of chromosomes, the telomerase is the special polymerase involved in the elongation of this protective zone. It is important to note that somatic cells of mammalian species lack the expression of telomerase. Thus, telomerase is highly regulated in a spatio-temporal manner (134). In each cell cycle, the telomere becomes shorter and shorter, leading to a vulnerability of the chromosomal end. This process is called telomere attrition and is considered another hallmark of aging (52).
Telomere shortening can cause genomic instability, and thus contributes to cancer or age-related diseases (115). This progressive diminishment in telomere length can influence gene expression and provoke several pathologies without damaging DNA (134). Moreover, ROS can trigger telomere attrition or loss and genomic instability, which can be prevented by using an antioxidant (115). In addition, telomerase activity can support cell cycle progression by preventing the arrest due to short telomeres, leading to a putative malignancy. Remarkably, overexpression of Beclin1 in HeLa cells revealed that telomerase activity is reduced after autophagy induction (39). This approach argues in favor of the hypothesis that autophagy plays an important tumor suppressor role by the modulation of telomerase activity in somatic cells. Similarly, Guanosine-rich zones, like telomeres, can suffer the formation of G-quadruplexes, interfering with gene expression and cell growth (135). In this regard, melanoma cells treated with an anthracene-based ligand (Ant 1,5, capable of stabilizing G-quadruplexes) showed an increase in genome instability and telomere dysfunction. Furthermore, Ant 1,5 induces a p21-dependent augmentation of autophagy levels. This autophagic response arises in order to avoid genome instability and telomeric dysfunction, thus promoting cell survival (135).
Other Hallmarks of Aging
Cellular senescence is considered another hallmark of aging (52). Senescence can be induced by DNA damage, telomere attrition or other stress signals, with the consequent cell cycle arrest. Several mouse models have contributed to the current knowledge of senescence and its characteristics (55). For example, Apolipoprotein E (ApoE) knockout mice present atherosclerotic plaques, which can be prevented by the addition of rapamycin. Furthermore, autophagy regulates the senescence of vascular smooth muscle cells (VSMCs) of ApoE−/− mice, involving the mTORC1/ULK1/ATG13 pathway in atherosclerosis progression (101). In addition, senescent MEF cells accumulate copper due to higher import and lower export, enhancing antioxidant defense mechanisms. In addition, rapamycin treatment can prevent and reverse copper accumulation, suggesting that autophagy mediates the copper homeostasis (136). Along the same lines, primary cultures from human fibroblasts depleted for Atg7, Atg12, or Lamp2 showed cell cycle arrest and high levels of SA-β-Gal staining, a characteristic feature of replicative senescence cells (137). Intriguingly, autophagy can mediate the transition to a senescent phenotype in IMR90 human diploid oncogene-induced senescence fibroblasts, making possible the protein remodeling needed to establish the senescent phenotype under oncogene activation (138). An interesting review conducted by Kwon et al. put forward a “toolkit” of differential diagnosis to resolve the apparent contradiction of autophagy in the cellular senescence. They proposed that type of autophagy, the exact moment when it acts, and the place where it occurs can define the pro or anti-senescence role of autophagy (139).
Another hallmark of aging, according to López-Otín et al., is stem cell (SC) exhaustion, with the consequent decline in tissue regenerative potential (52). Self-renewal is important to maintain the population of tissue-specific stem cells throughout life. Importantly, as we age, stem-cell activity decreases (140). In addition, SCs function is highly regulated in response to external stimuli (129). Ho et al. have shown that autophagy is necessary for preservation and quiescence of hematopoietic stem cells (HSCs). The authors demonstrated, in an Atg12-KO mice model, that autophagy-defective HSCs resemble old HSCs, in terms of accumulation of mitochondria, augmentation of myeloid-to-lymphoid ratio, diminishment of the regenerative potential, and decrease in self-renewal (141). This study supports the importance of autophagy in blood system homeostasis (142). Autophagy is also important to maintain stemness in bone marrow-derived mesenchymal stem cells (BMMSCs), and induction with rapamycin restores the biological properties of BMMSCs (143). In addition, Atg7 loss in aged muscle stem cells (satellite cells) of transgenic mice caused altered mitophagy and an accumulation of ROS, all features of senescence that diminish the regenerative potential of aged satellite cells (144). It is important to note that autophagic modulation could be an interesting therapeutic approach to prevent stem cell senescence and decline. Due to SC complexity, more studies are required to fully elucidate the role of autophagy in maintenance of stem cells.
Current and Future Autophagic Treatments in Aging and Age-Related Diseases
It can be presumed that induction or restoration of autophagy and antioxidant cellular systems could alleviate aging symptoms. Three major anti-aging therapies were evaluated over the last 30 years: Autophagic inducers, antioxidant (polyamine-rich) consumption, and caloric restriction.
Rapamycin, an immunosuppressive macrolide, is a well-known autophagic activator via mTORC1 inhibition of the mTOR complex. This pharmacological treatment has proved to increase lifespan in flies, nematodes, yeast, and mice [reviewed in (113)]. Moderate doses of this drug can alleviate atherosclerosis, achieving the same effects as higher concentrations (101). Furthermore, rapamycin added during reperfusion after heart infarction in a C57 myocardial ischemia mouse model improved the survival and cardiac functioning, reducing the infarcted zone size as well as apoptosis post-ischemia/reperfusion. In addition, AKT phosphorylation increased after treatment, suggesting that AKT-ERK pathways were selectively activated by rapamycin (145). Finally, rapamycin improves whole metabolism in several ways, strengthening the importance of regulating autophagy activity by external compounds in order to ameliorate metabolic diseases at cellular and whole organism levels (100, 109).
In addition, exogenously administered spermidine extends lifespan in mice treated throughout life or at pre-aged adulthood, and in hypertensive rats as well. In addition, spermidine dietary intake was inversely correlated with cardiovascular pathologies in a human population-based cohort (146). Spermidine, thus, has been proposed to be a neuro and cardioprotector in aging models and humans, highlighting the importance of polyamine-rich diets (56).
Caloric restriction (CR) is the reduction of total calorie intake by 30-40% without malnutrition. There is strong evidence supporting this therapy as one of the most effective in reducing oxidative stress in rats and mice, prolonging lifespan (31, 113). CR has been found to provoke a decrease in ROS generation (69) and a diminishment of DNA damage (31). Regarding CR and autophagy, it was demonstrated that 8%-CR in combination with exercise or 8%-CR alone were capable of upregulating Atg7, LC3, and LAMP2 in type II skeletal muscle in rats (147). The authors also found that aging augmented BECLIN1 protein expression and oxidative stress, but CR alone or with exercise diminished this modification as well as the apoptotic index, both correlating negatively with LAMP2 gene expression (147). Sod−/− mice treated with CR attenuated the age-related-like phenotype of this knockout in terms of DNA damage, cellular senescence, and inflammation (49). CR could be beneficial to human health, according to epidemiological studies (37). As CR regulates several pathways, more integrative studies are required in order to fully understand its anti-aging effect.
Finally, several reviews and research studies highlight the importance of autophagic modulation as an anti-aging therapy for the future (32, 37, 43). Rapamycin, resveratrol, polyamines, and CR are possible candidates to be tested more carefully in order to improve the putative treatments for human age-related diseases.
Concluding Remarks
Aging involves several features that can promote the development of a variety of disorders in aged individuals, such as neurodegenerative, heart, and metabolic diseases, as well as cancer. These age-related characteristics involve a gradual increase in ROS production and genome instability, and a progressive decrease in antioxidant, DNA repairing, and proteostatic systems, among others (Figure 3). Autophagy, as a homeostatic process, plays an important role in the maintenance of cell physiology and avoidance of any internal or external damage that could eventually appear. Several attempts were made to improve age-related features, such as caloric restriction as well as antioxidants and autophagy inducers. Rapamycin, resveratrol, and polyamines are autophagic inductors clinically available that could improve aging and some age-related disorders. It is important to highlight the fact that chemotherapies combined with autophagic inhibitors (i.e., chloroquine and derivatives) could be more effective in cancer treatment. Further studies are required to make autophagy modulation a more promising anti-aging and anti-tumoral therapy in the next decades.
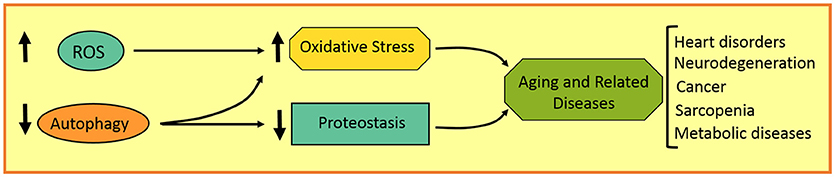
Figure 3. Schematic representation of aging-related disorders in autophagy and redox imbalance.
Author Contributions
MB analyzed the data, wrote the manuscript, and critically read the manuscript. RG wrote the manuscript. CF designed the research, analyzed the data, wrote the manuscript, produced the figures and critically read the manuscript.
Funding
This work was partly supported by grants from Agencia Nacional de Promoción Científica y Tecnológica PICT 2013-2335, PICTO 2016-0055, SeCTyP K013 (Universidad Nacional de Cuyo) to CF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. (2013) 15:713–20. doi: 10.1038/ncb2788
2. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. (2017) 36:1811–36. doi: 10.15252/embj.201796697
3. Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol. (2010) 298:C776–85. doi: 10.1152/ajpcell.00507.2009
4. White E. The role for autophagy in cancer. J Clin Invest. (2015) 125:42–46. doi: 10.1172/JCI73941
5. Yang Z, Goronzy JJ, Weyand CM. Autophagy in autoimmune disease. J Mol Med. (2015) 93:707–17. doi: 10.1007/s00109-015-1297-8
6. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. (2008) 182:685–701. doi: 10.1083/jcb.200803137
7. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. (2013) 14:759–74. doi: 10.1038/nrm3696
8. Militello RD, Colombo MI. A membrane is born: origin of the autophagosomal compartment. Curr Mol Med. (2011) 11:197–203. doi: 10.2174/156652411795243441
9. Jiang ZF, Shao LJ, Wang WM, Yan XB, Liu RY. Decreased expression of Beclin-1 and LC3 in human lung cancer. Mol Biol Rep. (2012) 39:259–267. doi: 10.1007/s11033-011-0734-1
10. Dice JF,. Altered Degradation of Proteins Microinjected into Senescent Human Fibroblasts. (1982). Available online at: at: http://www.jbc.org/content/257/24/14624.full.pdf (Accessed July 27, 2018).
11. Dice JF. Peptide sequences that target proteins for lysosomal degradation. J Control Release (1990) 11:291–6. doi: 10.1016/0168-3659(90)90141-F
12. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
13. Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. (2015) 16:1014–24. doi: 10.1038/ni.3273
14. Wirth M, Joachim J, Tooze SA. Autophagosome formation—the role of ULK1 and Beclin1–PI3KC3 complexes in setting the stage. Semin Cancer Biol. (2013) 23:301–9. doi: 10.1016/J.SEMCANCER.2013.05.007
15. Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci. (2013) 126:5224–38. doi: 10.1242/jcs.132415
16. Fader CM, Aguilera MO, Colombo MI. Autophagy response: manipulating the mTOR-controlled machinery by amino acids and pathogens. Amino Acids (2015) 47:2101–12. doi: 10.1007/s00726-014-1835-7
17. Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. (2013) 14:284–91. doi: 10.1038/embor.2013.6
18. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. (2010) 11:45–51. doi: 10.1038/embor.2009.256
19. Zhu Y, Wang C, Yu M, Cui J, Liu L, Xu Z. ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression. Protein Cell (2013) 4:711–721. doi: 10.1007/s13238-013-3910-3
20. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
21. Zinger A, Cho WC, Ben-Yehuda A. Cancer and aging - the inflammatory connection. Aging Dis. (2017) 8:611–27. doi: 10.14336/AD.2016.1230
22. Vellai T, Takács-Vellai K, Sass M, Klionsky DJ. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol. (2009) 19:487–94. doi: 10.1016/J.TCB.2009.07.007
23. Chuang SY, Lin CH, Fang JY. Natural compounds and aging: between autophagy and inflammasome. Biomed Res Int. (2014) 2014:297293. doi: 10.1155/2014/297293
24. Nakamura S, Yoshimori T. Autophagy and longevity. Mol Cells (2018) 41:65–72. doi: 10.14348/molcells.2018.2333
25. Ageing and Health. Available online at: http://www.who.int/news-room/fact-sheets/detail/ageing-and-health (Accessed July 27, 2018)
26. Barja G. The Cell Aging Regulation System (CARS). Barja G React Oxyg Species (2017) 148:148–83. doi: 10.20455/ros.2017.829
27. Kamel HK, Mooradian AD, Mir T. Biological theories of aging. Disease-a-Month (2015) 61:460–6. doi: 10.1016/J.DISAMONTH.2015.09.005
28. Wahab Pathath A. Theories of aging. Artic Int J Indian Psychol. (2017) 4:15–22. doi: 10.25215/0403.142
29. Harman D. The free radical theory of aging. Antioxid Redox Signal. (2003) 5:557–61. doi: 10.1089/152308603770310202
30. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature (2000) 408:239–47. doi: 10.1038/35041687
31. Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech Ageing Dev. (2004) 125:811–26. doi: 10.1016/J.MAD.2004.07.009
32. Markaki M, Metaxakis A, Tavernarakis N. The role of autophagy in aging: molecular mechanisms. Autoph Cancer Other Pathol Inflamm Immunity Infect Aging (2017) 123–38. doi: 10.1016/B978-0-12-812146-7.00002-0
33. Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. (2013) 4:2300. doi: 10.1038/ncomms3300
34. Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. (2008) 14:959–65. doi: 10.1038/nm.1851
35. Liang H, Masoro EJ, Nelson JF, Strong R, McMahan CA, Richardson A. Genetic mouse models of extended lifespan. Exp Gerontol. (2003) 38:1353–64. doi: 10.1016/J.EXGER.2003.10.019
36. Emanuele E, Minoretti P, Sanchis-Gomar F, Pareja-Galeano H, Yilmaz Y, Garatachea N, et al. Can enhanced autophagy be associated with human longevity? Serum levels of the autophagy biomarker Beclin-1 are increased in healthy centenarians. Rejuvenation Res. (2014) 17:518–24. doi: 10.1089/rej.2014.1607
37. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell (2011) 146:682–95. doi: 10.1016/J.CELL.2011.07.030
38. Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest. (2015) 125:47–54. doi: 10.1172/JCI73942
39. Taji F, Kouchesfahani HM, Sheikholeslami F, Romani B, Baesi K, Vahabpour R, et al. Autophagy induction reduces telomerase activity in HeLa cells. Mech Ageing Dev. (2017) 163:40–5. doi: 10.1016/J.MAD.2016.12.011
40. Abbas G, Salman A, Rahman SU, Ateeq MK, Usman M, Sajid S, et al. Aging Mechanisms: Linking oxidative stress, obesity and inflammation. Matrix Sci Med. (2017) 1:30–3. doi: 10.26480/msm.01.2017.30.33
41. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. (2007) 26:1749–60. doi: 10.1038/sj.emboj.7601623
42. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell (2012) 48:158–67. doi: 10.1016/J.MOLCEL.2012.09.025
43. Yan Y, Finkel T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic Biol Med. (2017) 109:108–13. doi: 10.1016/J.FREERADBIOMED.2016.12.003
44. Santos AL, Sinha S, Lindner AB. The good, the bad, and the ugly of ROS: new insights on aging and aging-related diseases from eukaryotic and prokaryotic model organisms. Oxid Med Cell Longev. (2018) 2018:1–23. doi: 10.1155/2018/1941285
45. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Q2: extension of murine life span by overexpression of catalase targeted to mitochondria. Science (2003) 299:1342–6. doi: 10.1126/science.1077991
46. Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF, Martin GM, et al. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol Ser A Biol Sci Med Sci. (2008) 63:813–22. doi: 10.1093/gerona/63.8.813
47. Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics (2003) 16:29–37. doi: 10.1152/physiolgenomics.00122.2003
48. Pérez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, et al. Is the oxidative stress theory of aging dead? Biochim Biophys Acta (2009) 1790:1005–14. doi: 10.1016/J.BBAGEN.2009.06.003
49. Zhang Y, Unnikrishnan A, Deepa SS, Liu Y, Li Y, Ikeno Y, et al. A new role for oxidative stress in aging: the accelerated aging phenotype in Sod1−/− mice is correlated to increased cellular senescence. Redox Biol. (2017) 11:30–7. doi: 10.1016/J.REDOX.2016.10.014
50. Deepa SS, Van Remmen H, Brooks SV, Faulkner JA, Larkin L, McArdle A, et al. Accelerated sarcopenia in Cu/Zn superoxide dismutase knockout mice. Free Radic Biol Med. (in press) doi: 10.1016/J.FREERADBIOMED.2018.06.032
51. Calabrese EJ, Baldwin LA. Defining hormesis. Hum Exp Toxicol. (2002) 21:91–7. doi: 10.1191/0960327102ht217oa
52. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi: 10.1016/J.CELL.2013.05.039
53. Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. (2011) 21:569–76. doi: 10.1016/J.TCB.2011.06.008
54. Cavallini G, Donati A, Taddei M, Bergamini E. Evidence for selective mitochondrial autophagy and failure in aging. Autophagy (2007) 3:26–7. doi: 10.4161/auto.3268
55. Folgueras AR, Freitas-Rodríguez S, Velasco G, López-Otín C. Mouse models to disentangle the hallmarks of human aging. Circ Res. (2018) 123:905–24. doi: 10.1161/CIRCRESAHA.118.312204
56. Bhukel A, Madeo F, Sigrist SJ. Spermidine boosts autophagy to protect from synapse aging. Autophagy (2017) 13:444–5. doi: 10.1080/15548627.2016.1265193
57. Gong Z, Tasset I. Humanin enhances the cellular response to stress by activation of chaperone-mediated autophagy. Oncotarget (2018) 9:10832–3. doi: 10.18632/oncotarget.24396
58. Amm I. Protein quality control and elimination of protein waste: the role of the ubiquitin–proteasome system. Biochim Biophys Acta Mol Cell Res. (2014) 1843:182–96. doi: 10.1016/J.BBAMCR.2013.06.031
59. Jiao J, Demontis F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr Opin Pharmacol. (2017) 34:1–6. doi: 10.1016/J.COPH.2017.03.009
60. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell (2004) 15:4829–40. doi: 10.1091/mbc.e04-06-0477
61. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. (2002) 82:373–428. doi: 10.1152/physrev.00027.2001
62. Callis J. The ubiquitination machinery of the ubiquitin system. Arab B (2014) 12:e0174. doi: 10.1199/tab.0174
63. Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci USA. (2015) 112:15790–7. doi: 10.1073/pnas.1521919112
64. Höhn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B, et al. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. (2017) 11:482–501. doi: 10.1016/J.REDOX.2016.12.001
65. Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES. Overexpression of Proteasome β 5 Subunit Increases Amount of Assembled Proteasome and Confers Ameliorated Response to Oxidative Stress and Higher Survival Rates. (2005). Available online at: http://www.jbc.org/ (Accessed November 15, 2018).
66. Hwang JS, Hwang JS, Chang I, Kim S. Age-associated decrease in proteasome content and activities in human dermal fibroblasts: restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. J Gerontol Ser A Biol Sci Med Sci. (2007) 62:490–9. doi: 10.1093/gerona/62.5.490
67. Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol. (2012) 180:963–72. doi: 10.1016/J.AJPATH.2011.11.012
68. Torres C, Lewis L, Cristofalo VJ. Proteasome inhibitors shorten replicative life span and induce a senescent-like phenotype of human fibroblasts. J Cell Physiol. (2006) 207:845–853. doi: 10.1002/jcp.20630
69. Barja G. Free radicals and aging. Trends Neurosci. (2004) 27:595–600. doi: 10.1016/J.TINS.2004.07.005
70. Nassif M, Woehlbier U, Manque PA. The delicate balance of autophagy in neurodegeneration. Autoph Cancer, Other Pathol Inflamm Immunity Infect Aging (2017)387–99. doi: 10.1016/B978-0-12-812146-7.00019-6
71. Rezzani R, Stacchiotti A, Rodella LF. Morphological and biochemical studies on aging and autophagy. Ageing Res Rev. (2012) 11:10–31. doi: 10.1016/J.ARR.2011.09.001
72. Li Q, Liu Y, Sun M. Autophagy and Alzheimer's disease. Cell Mol Neurobiol. (2017) 37:377–88. doi: 10.1007/s10571-016-0386-8
73. Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, et al. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain (2013) 136:2130–46. doi: 10.1093/brain/awt131
74. Ji ZS, Müllendorff K, Cheng IH, Dennis Miranda R, Huang Y, Mahley RW. Reactivity of apolipoprotein E4 And Amyloid β peptide: lysosomal stability and neurodegeneration. J Biol Chem. (2005) 281:2683–92 doi: 10.1074/jbc.M506646200
75. Fîlfan M, Elena Sandu R, Zăvăleanu A, Greşită A, Glăvan D, Olaru D, et al. Autophagy in aging and disease. Rom J Morphol Embryol. (2017):27–31.
76. Shi R, Guberman M, Kirshenbaum LA. Mitochondrial quality control: the role of mitophagy in aging. Trends Cardiovasc Med. (2018) 28:246–60. doi: 10.1016/J.TCM.2017.11.008
77. Kim J, Cheon H, Jeong YT, Quan W, Kim KH, Cho JM, et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J Clin Invest. (2014) 124:3311–24. doi: 10.1172/JCI69625
78. Loos B, Klionsky DJ, Wong E. Augmenting brain metabolism to increase macro- and chaperone-mediated autophagy for decreasing neuronal proteotoxicity and aging. Prog Neurobiol. (2017) 156:90–106. doi: 10.1016/J.PNEUROBIO.2017.05.001
79. Fan J, Kou X, Jia S, Yang X, Yang Y, Chen N. Autophagy as a potential target for sarcopenia. J Cell Physiol. (2016) 231:1450–9. doi: 10.1002/jcp.25260
80. Um JH, Yun J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. (2017) 50:299–307. doi: 10.5483/BMBREP.2017.50.6.056
81. Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, et al. Measuring in vivo mitophagy. Mol Cell (2015) 60:685–96. doi: 10.1016/J.MOLCEL.2015.10.009
82. Grosso R, Fader CM, Colombo MI. Autophagy: a necessary event during erythropoiesis. Blood Rev. (2017) 31:300–5. doi: 10.1016/J.BLRE.2017.04.001
83. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature (2008) 454:232–5. doi: 10.1038/nature07006
84. Rogov VV, Suzuki H, Marinković M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. (2017) 7:1131. doi: 10.1038/s41598-017-01258-6
85. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. (2010) 8:e1000298. doi: 10.1371/journal.pbio.1000298
86. Han H, Chou CC, Li R, Liu J, Zhang L, Zhu W, et al. Chalcomoracin is a potent anticancer agent acting through triggering oxidative stress via a mitophagy- and paraptosis-dependent mechanism. Sci Rep. (2018) 8:9566. doi: 10.1038/s41598-018-27724-3
87. Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz-Bonilla V, Sala D, et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. (2016) 35:1677–93. doi: 10.15252/embj.201593084
88. Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J Cell Biol. (2018) 217:635–47. doi: 10.1083/jcb.201606095
89. Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. (2000) 275:31505–13. doi: 10.1074/jbc.M002102200
90. Muzumdar RH, Huffman DM, Atzmon G, Buettner C, Cobb LJ, Fishman S, et al. Humanin: a novel central regulator of peripheral insulin action. PLoS ONE (2009) 4:e6334. doi: 10.1371/journal.pone.0006334
91. Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. (2010) 11:621–32. doi: 10.1038/nrm2952
92. Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene (2004) 23:2861–74. doi: 10.1038/sj.onc.1207523
93. Danish L, Imig D, Allgöwer F, Scheurich P, Pollak N. Bcl-2-mediated control of TRAIL-induced apoptotic response in the non-small lung cancer cell line NCI-H460 is effective at late caspase processing steps. PLoS ONE (2018) 13:e0198203. doi: 10.1371/journal.pone.0198203
94. Rodríguez ME, Catrinacio C, Ropolo A, Rivarola VA, Vaccaro MI. A novel HIF-1α/VMP1-autophagic pathway induces resistance to photodynamic therapy in colon cancer cells. Photochem Photobiol Sci. (2017) 16:1631–42. doi: 10.1039/C7PP00161D
95. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. (2011) 108:9572–7. doi: 10.1073/pnas.1106291108
96. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci (2015) 40:200–10. doi: 10.1016/J.TIBS.2015.02.003
97. Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes (2012) 61:23–9. doi: 10.2337/db11-0555
98. Blagosklonny M V. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle (2010) 9:683–8. doi: 10.4161/cc.9.4.10766
99. González A, Hall MN. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. (2017) 36:397–408. doi: 10.15252/embj.201696010
100. Das A, Reis F, Maejima Y. Cai Z, Ren J. mTOR Signaling in Cardiometabolic Disease, Cancer, and Aging. (2017). Available online at: http://downloads.hindawi.com/journals/omcl/2017/6018675.pdf (Accessed July 19, 2018).
101. Luo Z, Xu W, Ma S, Qiao H, Gao L, Zhang R, et al. Moderate autophagy inhibits vascular smooth muscle cell senescence to stabilize progressed atherosclerotic plaque via the mTORC1/ULK1/ATG13 signal pathway. Oxid Med Cell Longev (2017) 2017:1–9. doi: 10.1155/2017/3018190
102. Templeman NM, Murphy CT. Regulation of reproduction and longevity by nutrient-sensing pathways. J Cell Biol. (2018) 217:93–106. doi: 10.1083/jcb.201707168
103. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance. Science (2012) 335 1638–43. doi: 10.1126/science.1215135
104. Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mtor expression. Cell Rep. (2013) 4:913–20. doi: 10.1016/J.CELREP.2013.07.030
105. Selman C, Tullet JMA, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science (2009) 326:140–4. doi: 10.1126/science.1177221
106. Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science (2017) 358:813–8. doi: 10.1126/science.aao3265
107. Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature (2015) 517:302–10. doi: 10.1038/nature14190
108. Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell (2011) 44:134–46. doi: 10.1016/J.MOLCEL.2011.06.038
109. Kim J, Lim YM, Lee MS. The role of autophagy in systemic metabolism and human-type diabetes. Mol Cells (2018) 41:11–7. doi: 10.14348/molcells.2018.2228
110. Li TY, Sun Y, Liang Y, Liu Q, Shi Y, Zhang CS, et al. ULK1/2 constitute a bifurcate node controlling glucose metabolic fluxes in addition to autophagy. Mol Cell (2016) 62:359–70. doi: 10.1016/J.MOLCEL.2016.04.009
111. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PWL, Thomas EL, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. (2010) 11:453–65. doi: 10.1016/J.CMET.2010.05.001
112. Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. (2010) 31:212–20. doi: 10.1016/J.TIPS.2010.02.003
113. Tsang CK, Chen M, Cheng X, Qi Y, Chen Y, Das I, et al. SOD1 phosphorylation by mTORC1 couples nutrient sensing and redox regulation. Mol Cell (2018) 70:502–15.e8. doi: 10.1016/J.MOLCEL.2018.03.029
114. Hewitt G, Korolchuk VI. Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol. (2017) 27:340–51. doi: 10.1016/J.TCB.2016.11.011
115. Liu L, Trimarchi JR, Smith PJS, Keefe DL. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell (2002) 1:40–6. doi: 10.1046/j.1474-9728.2002.00004.x
116. Donati A, Taddei M, Cavallini G, Bergamini E. Stimulation of macroautophagy can rescue older cells from 8-OHdG mtDNA accumulation: a safe and easy way to meet goals in the SENS agenda. Rejuvenation Res. (2006) 9:408–12. doi: 10.1089/rej.2006.9.408
117. Van Remmen H, Hamilton ML, Richardson A. Oxidative damage to DNA and aging. Exerc Sport Sci Rev. (2003) 31:149–53. doi: 10.1097/00003677-200307000-00009
118. Song X, Narzt MS, Nagelreiter IM, Hohensinner P, Terlecki-Zaniewicz L, Tschachler E, et al. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biol. (2017) 11:219–30. doi: 10.1016/J.REDOX.2016.12.015
119. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. (2006) 38:515–7. doi: 10.1038/ng1769
120. Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Mariño G, et al. Autophagic removal of micronuclei. Cell Cycle (2012) 11:170–6. doi: 10.4161/cc.11.1.18564
121. Guo H, Chitiprolu M, Gagnon D, Meng L, Perez-Iratxeta C, Lagace D, et al. Autophagy supports genomic stability by degrading retrotransposon RNA. Nat Commun. (2014) 5:5276. doi: 10.1038/ncomms6276
122. Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. (2013) 73:4311–22. doi: 10.1158/0008-5472.CAN-12-4142
123. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. (2007) 21:1621–35. doi: 10.1101/gad.1565707
124. Ariyoshi K, Miura T, Kasai K, Fujishima Y, Oshimura M, Yoshida MA. Induction of genomic instability and activation of autophagy in artificial human aneuploid cells. Mutat Res Mol Mech Mutagen. (2016) 790:19–30. doi: 10.1016/J.MRFMMM.2016.06.001
125. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell (2009) 137:1062–75. doi: 10.1016/J.CELL.2009.03.048
126. Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K, et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol Cell (2016) 63:34–48. doi: 10.1016/J.MOLCEL.2016.05.027
127. Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, Nelson G, et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy (2016) 12:1917–30. doi: 10.1080/15548627.2016.1210368
128. Gabbianelli R, Malavolta M. Epigenetics in ageing and development. Mech Ageing Dev. (2018) 174:1–2. doi: 10.1016/j.mad.2018.05.005
129. Brunet A, Rando TA. Interaction between epigenetic and metabolism in aging stem cells. Curr Opin Cell Biol. (2017) 45:1–7. doi: 10.1016/J.CEB.2016.12.009
130. Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, et al. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics (2016) 11:381–8. doi: 10.1080/15592294.2016.1144007
131. Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol. (2011) 192:615–29. doi: 10.1083/jcb.201008167
132. Luyten T, Welkenhuyzen K, Roest G, Kania E, Wang L, Bittremieux M, et al. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2+. Biochim Biophys Acta Mol Cell Res. (2017) 1864:947–56. doi: 10.1016/J.BBAMCR.2017.02.013
133. Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Küttner V, Bhukel A, et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. (2014) 19:431–44. doi: 10.1016/J.CMET.2014.02.010
134. Shay JW. Telomeres and aging. Curr Opin Cell Biol. (2018) 52:1–7. doi: 10.1016/J.CEB.2017.12.001
135. Orlotti NI, Cimino-Reale G, Borghini E, Pennati M, Sissi C, Perrone F, et al. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy (2012) 8:1185–96. doi: 10.4161/auto.20519
136. Masaldan S, Clatworthy SAS, Gamell C, Smith ZM, Francis PS, Denoyer D, et al. Copper accumulation in senescent cells: interplay between copper transporters and impaired autophagy. Redox Biol. (2018) 16:322–31. doi: 10.1016/J.REDOX.2018.03.007
137. Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE (2011) 6:e23367. doi: 10.1371/journal.pone.0023367
138. Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. (2009) 23:798–803. doi: 10.1101/gad.519709
139. Kwon Y, Kim JW, Jeoung JA, Kim MS, Kang C. Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol Cells (2017) 40:607–12. doi: 10.14348/molcells.2017.0151
140. Rossi DJ, Jamieson CHM, Weissman IL. Stems cells and the pathways to aging and cancer. Cell (2008) 132:681–96. doi: 10.1016/J.CELL.2008.01.036
141. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature (2017) 543:205–10. doi: 10.1038/nature21388
142. Doulatov S, Daley GQ. Autophagy: it's in your blood. Dev Cell (2017) 40:518–20. doi: 10.1016/J.DEVCEL.2017.03.011
143. Ma Y, Qi M, An Y, Zhang L, Yang R, Doro DH, et al. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell (2018) 17:e12709. doi: 10.1111/acel.12709
144. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature (2016) 529:37–42. doi: 10.1038/nature16187
145. Filippone SM, Samidurai A, Roh SK, Cain CK, He J, Salloum FN, et al. Reperfusion therapy with rapamycin attenuates myocardial infarction through activation of AKT and ERK. Oxid Med Cell Longev. (2017) 2017:1–16. doi: 10.1155/2017/4619720
146. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22:1428–38. doi: 10.1038/nm.4222
Keywords: autophagy, ROS, aging, hallmarks of aging, mitophagy
Citation: Barbosa MC, Grosso RA and Fader CM (2019) Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 9:790. doi: 10.3389/fendo.2018.00790
Received: 30 July 2018; Accepted: 17 December 2018;
Published: 09 January 2019.
Edited by:
Maria Ines Vaccaro, Universidad de Buenos Aires, ArgentinaReviewed by:
Rong Yuan, Southern Illinois University Carbondale, United StatesClaudio Acuña-Castillo, Universidad de Santiago de Chile, Chile
Copyright © 2019 Barbosa, Grosso and Fader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Marcelo Fader, Y2ZhZGVyQGZjbS51bmN1LmVkdS5hcg==