 John R. Klein
John R. Klein- Department of Diagnostic and Biomedical Sciences, School of Dentistry, The University of Texas Health Science Center at Houston, Houston, TX, United States
Thyroid stimulating hormone (TSH), a glycoprotein hormone produced by the anterior pituitary, controls the production of thyroxine (T4) and triiodothyronine (T3) in the thyroid. TSH is also known to be produced by the cells of the immune system; however, the physiological importance of that to the organism is unclear. We identified an alternatively-spliced form of TSHβ that is present in both humans and mice. The TSHβ splice variant (TSHβv), although produced at low levels by the pituitary, is the primary form made by hematopoietic cells in the bone marrow, and by peripheral leukocytes. Recent studies have linked TSHβv functionally to a number of health-related conditions, including enhanced host responses to infection and protection against osteoporosis. However, TSHβv also has been associated with autoimmune thyroiditis in humans. Yet to be identified is the process by which the TSHβv isoform is produced. Here, a set of genetic steps is laid out through which human TSHβv is generated using splicing events that result in a novel transcript in which exon 2 is deleted, exon 3 is retained, and the 3′ end of intron 2 codes for a signal peptide of the TSHβv polypeptide.
Introduction
Structure and Function of TSH
TSH consists of an α and a β subunit. The β subunit determines hormone specificity. Binding of TSH to the TSH receptor (TSHR) in the thyroid induces the release of thyroxine (T4) and a limited amount of triiodothyronine (T3). In tissue cells, most T4 is converted by deiodination to T3, the more biologically-active form of thyroid hormone. Regulation of TSH release from the pituitary is governed by levels of circulating TSH, T4, and T3. Considerable homology exists at both the gene and protein levels between human and mouse TSHβ (1, 2). Within the hypothalamus-pituitary-thyroid (HPT) axis, TSH regulates a broad spectrum of cellular and organismic activities, including cell physiology, development, and cellular and organismic metabolism. The TSHβ polypeptide is coded for by exons 4 and 5 of the mouse TSHβ gene, and by exons 2 and 3 of the human TSHβ gene.
Immune System TSH
The pioneering work of Blalock and colleagues was the first to demonstrate that TSH is made by the immune system. The authors reported that leukocytes cultured with known forms of immune stimuli, such as Staphylococcus enterotoxin A or TRH, secreted immunoreactive TSH (3–5). Other studies demonstrated that splenic dendritic cells from mice produced TSH endogenously, as well as following stimulation with Staphylococcus enterotoxin B (6, 7). Bone marrow (BM) hematopoietic cells, in particular CD11b+ monocyte/macrophage precursors and to a lesser extent granulocyte and lymphocyte precursors, were shown to be a source of TSH (8). Additionally, intraepithelial lymphocytes and lamina propria lymphocytes in the intestinal mucosa of mice produced TSH locally following experimental virus infection (9–11). In studies aimed at understanding the integration of TSH immune-endocrine interactions, we observed that BM-derived CD11b+ cells traffic to the thyroid where they are deposited around thyroid follicles (7), and produce TSHβ intrathyroidally (6). Those studies collectively provided evidence that immune system-derived TSH may co-regulate the synthesis and release of thyroid hormones under normal homeostatic conditions (7).
There is longstanding evidence linking TSH to immune system function. TSH has been shown to directly influence the cellular responses of leukocytes in both positive and negative ways. This includes enhancement of antibody responses to T cell-dependent antigens (12), and iodine uptake inhibition (13, 14). T3 stimulates thymocyte proliferation, which may be the consequence of TSH-mediated increase in thyroid hormones (15). In that light, recent studies have documented a protective effect of TSH on thymocyte apoptosis (16). These and other directional interactions of HPT hormones on immunity have been detailed in several review articles (17, 18).
The TSHβ Splice Variant (TSHβV)
Studies in our laboratory revealed that at both the transcript and protein levels, TSHβ produced in the thyroid as well as by leukocytes of mice and humans was significantly smaller in size than pituitary-derived TSHβ (19, 20), though it retained functional activity as determined by cAMP cell signaling (19). Further analysis of leukocyte-derived TSHβ identified a novel TSHβ splice variant (TSHβv) transcript coded for by exon 5 and the 3′ end of intron 4 in mice (19), and exon 3 and the 3′ end of intron 2 in humans (20, 21). Those splicing patterns for human native TSHβ and TSHβv are shown in Figure 1A.

Figure 1. Structural organization and splicing patterns of the native and splice variant forms of human tshβ transcripts. (A) The TSHβv transcript is produced by removal of the region coded for by exon 2, and splicing into the 3′end of intron 2 (arrow), yielding a truncated TSHβ transcript coded for by exon 3. Gray boxes are transcript regions from non-coding exons. Blue boxes are transcript regions from coding exons. Hatched regions code for signal peptides, which in native TSHβ is at the beginning of exon 2, and in TSHβv is at the 3′ end of intron 2. (B) Unspliced TSHβ. Gray underlined nucleotides, TATAAA hexamer box of transcriptional start site. Upper yellow nucleotides, exon 1. Blue nucleotides, portion of intron 1. Middle yellow nucleotides, exon 2. Green nucleotides, intron 2. Bottom yellow nucleotides, exon 3. Red nucleotides, potential splice donor sites. Purple nucleotides, potential splice acceptor sites. Underlined TAA, stop codon at end of exon 3. White nucleotides, untranslated portion of exon 3. Potential donor sites in intron 1: TATTTGTAAGAT, TCAAGGTGATCA; both with donor site scores ≥83.4. Potential acceptor sites in intron 2: TTTTGTGTCCCAGCT, AATTCTTTCCCAGTT; both with acceptor site scores ≥88.0. (C) Spliced human TSHβ resulting in TSHβv transcript. Blue, intron 1-coded nucleotides. Green, intron 2-coded nucleotides. Yellow, exon 3-coded nucleotides. Combined red/purple, splice site of intron 1 with intron 2 resulting in deletion of exon 2-coded nucleotides but leaving exon 3-coded nucleotides. Bolded 69 nucleotide sequence in intron 2 codes for a putative 23 amino acid signal peptide of the TSHβv protein molecule with translation being initiated at the AUG codon (underlined). A second smaller signal peptide could be coded for beginning at the AUG (underlined) that is located 27 nucleotides upstream from the beginning of exon 3. White nucleotides, untranslated portion of exon 3.
Experiments using mouse BM cells confirmed that TSHβv is the principal form of TSHβ made by hematopoietic cells, particularly by CD11B+ myeloid cells (19, 21, 22). Similar findings were obtained using mouse splenic leukocytes (22). Zymosan, lipopolysaccharide, and CD14 and TLR2 triggering induced the release of TSHβv from the mouse AM macrophage cell line (22). Similarly, the THP-1 human macrophage cell line expressed the TSHβv gene, but not the native TSHβ gene (23). Interestingly, the TSHα gene was expressed at low levels in mouse BM cells (19) and human THP-1 cells (23), suggesting that TSHβv may operate either as a monomeric molecule or as a dimer. Further evidence of the latter is supported by studies demonstrating that TSHβv is dimerized with TSHα in the human circulation (24), a finding that is consistent with the presence of an eighteen amino acid “seatbelt” region coded for by human exon 3 that is used for attachment of TSHβ to TSHα (2). Thus, the TSHβv protein may under certain circumstances be coupled to TSHα.
Following polarization into M1 and M2 macrophages, TSHβv was primarily produced by M2 macrophages in mice (21), and by both M1 and M2 cells generated from the human THP-1 cell line (23). In vivo treatment of mice with T4 for 21 days resulted in differential effects on TSHβv expression in the BM and the pituitary, such that it was significantly increased in BM cells and suppressed in pituitary tissue (23). T3-treated F4/80+ RAW 264.7 mouse macrophages had dose-dependent increases in TSHβv expression (23).
A Putative Basis of TSHβv Splicing
The findings described above demonstrate that the TSHβv isoform is selectively expressed in hematopoietic cells. An as yet unresolved question pertains to the process by which the isoform is generated. Given that the immune system makes TSHβv and little if any native TSHβ, there must be well-regulated process for doing this. Understanding the underlying mechanism of that will likely provide valuable insights into how TSHβv is functionally linked to the homeostatic balance of thyroid hormone synthesis, to disease, and/or to the control of host metabolic activity.
Alternative splicing is a complex process by which cells generate variable protein constructs from a common gene source (25, 26). Numerous mechanisms have been described through which alternative isoforms can be made. These include exon skipping, alternative use of 3′ or 5′ splice sites, and intron retention (27, 28). In hormone biology, the presence of receptor isoforms produced by alternative splicing is common, as is seen in secretin and gastrin receptors, for example (29). However, alternatively-spliced isoforms may produce receptors associated with neoplastic cell growth, as occurs in HER-2/neu (30).
In classical splicing situations involving intron removal, a spliceosome directs the splicing process by recognizing splicing signals in the transcript and catalyzing intron removal and exon joining (31). However, the process of alternative splicing for TSHβv differs notably from traditional approaches, including those in which exon skipping occurs, because a portion of the 3′ end of the intron preceding the last coding exon (exon 3 in humans and exon 5 in mice) is incorporated into the new isoform. Although utilization of introns in alternatively-spliced genes is known to occur (32), the majority of those retain the entire intron, as is the case with the tissue factor gene in which a new exon is generated from an intronal sequence. (33). Proteins in malignant tissues also have been shown to utilize intron inclusion (34). The reasons why this occurs are unclear, although they may be tied to mechanisms of dysregulated gene expression that leads to neoplasia (35). While there is no evidence of this for immune system TSHβv, it is possible that under some circumstances TSHβv may be expressed autonomously in the absence of known stimulatory signals (21).
The structure of the human unspliced TSHβ gene is shown in Figure 1B. Initiation of transcription is believed to occur at the TATAAA hexamer 22 nucleotides upstream from exon 1 (36). To gain insights into how TSHβv might be generated, the spliceport on-line prediction tool http://spliceport.cbcb.umd.edu/SplicingAnalyser.html was applied to the human unspliced TSHβ gene (36), from which a number of possible splice sites were identified. These consisted of donor sites in intron 1 and acceptor sites in intron 2 (Figure 1B). All four of those have predictive splicing scores ≥80.0. Note that splicing in using those would result in the complete deletion of exon 2, the portion of the TSHβ transcript that codes for the native TSHβ but not the TSHβv polypeptide. The final rearranged TSHβv transcript after splicing is shown in Figure 1C. This results in the retention of a piece of the 3′ end of intron 2 that may code for two possible signal peptides, one beginning with an AUG translational start site located at position −69 from the 3′ end of intron 2, the second located −27 nucleotides from the 3′ end of intron 2 (Figure 1C, underlined AUGs). These would code for 23 and 9 amino acid peptide sequences, respectively, as shown in Figure 2A.
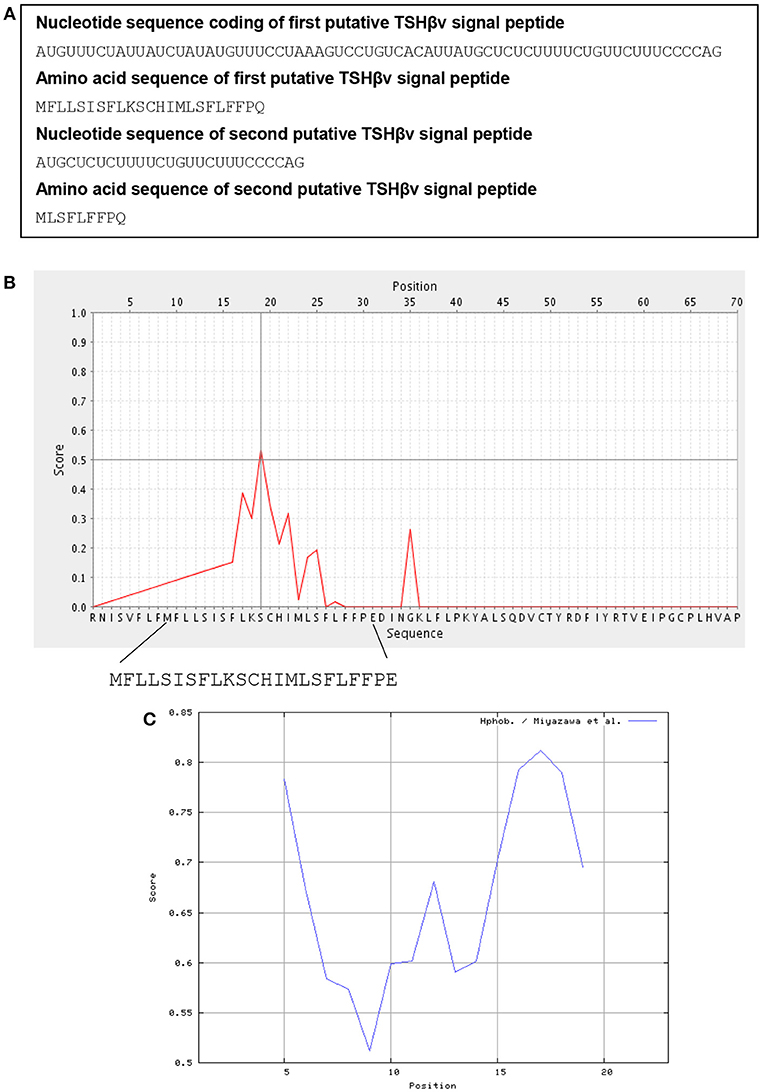
Figure 2. Signal peptide analysis of TSHβv in intron 2. (A) The nucleotide and amino acid sequences at the 3′ end of intron 2 (see Figure 1C, bold sequences) that code for a putative 23 and 9 amino acid signal peptides of TSHβv. (B) Biophysical properties of the putative 23 amino acid TSHβv signal peptide as determined using a PrediSi signal prediction program, the 23 amino acid peptide with the serine reside at position 11 likely representing the cleavage site. (C) The 23 amino acid signal peptide has hydrophobicity scores between 0.5 and 0.85 in a range from 0.0 (lowest hydrophobicity) to 1.0 (highest hydrophobicity).
Signal Peptide Analysis of TSHβv
Using on-line signal peptide prediction tools http://www.predisi.de/, the 23 amino acid peptide shown in Figure 2A, prior to and contiguous with exon 3, bears physiochemical properties that are typical of signal peptides as shown by the large number of hydrophobic and polar non-charged residues (Figures 2B,C). Peptide sequences such as those are commonly used to feed mature proteins through the membrane translocon for extracellular secretion. Although the nine amino acid signal peptide would be substantially shorter, it too retains most of the elements needed for a signal peptide. Which of those two is preferentially used may eventually be predicted from sequence analyses studies of TSHβv. While neither of the translational start sites contain classic Kozak features, a number of new mechanisms for initiating translation has been identified in eukaryotic cells (37), thus making it conceivable that either one of the two could serve as signal peptides for human TSHβv.
Transcriptional Regulation of TSHβv
The predictions presented here provide a potential process whereby the human TSHβv isoform may be generated in cells of the immune system. Still to be learned is the process that regulates when TSHβv is made, given the selective use of that form of TSHβ by leukocytes. Although transcription could be initiated in intron 2 at the standard transcriptional start site used for the native TSHβ gene, it also could begin with the TATAA box in intron 3, (see Figure 1B). Early studies into TSHβ gene regulation identified Pit-1 and GATA-2 as potential transcription factors acting on the TSHβ promoter in both rat and human TSHβ (38–42). However, it has since been learned that Pit-1 is probably minimally involved, and that GATA-2 may be the primary activator of TRH-driven transcription of the TSHβ gene (43–46). Other studies have shown that thyroid hormone negatively regulates TSHβ by interfering with the binding of transcription factors near the proximal promoter region (47, 48). In the case of immune system TSHβv, no transcription factors have, as yet, been linked to gene activation.
Biological Significance of TSHβv
Although it is now clear that TSHβv is made by cells of the immune system, many unanswered question remain regarding the functional role of the TSHβv isoform (49), and why the production of it is primarily relegated to the cells of the immune system rather than as an isoform made in the pituitary through the conventional HPT axis. In that vein, the immune system is by nature involved in sensing and responding to a wide range of external stimuli, in particular to a variety of threats to the safety and well-being of the host. An example of this is the situation of acute or chronic immune stress due to infection. Although some TSHβv-producing spleen cells routinely migrate to the thyroid in healthy animals (7), our studies demonstrated a marked increase in TSHβv gene expression in the thyroid of mice following virus infection with reovirus (19) or bacterial infection with Listeria monocytogenes (22). In the case of the latter, trafficking of splenic CD14+, Ly6C+, Ly6G+ cells to the thyroid was accompanied by robust intrathyroidal TSHβv synthesis (22). Importantly, splenic leukocytes from L. monocytogenes-infected mice homed to the thyroid when injected into non-infected mice, thus pointing to an active and integrated immune-endocrine host response during infection (22). We have speculated that this constitutes an immune system-driven host response used to manipulate, specifically to curtail, metabolic activity at a time when energy conservation is crucial (50).
Additional work is needed to understand the significance of TSHβv in terms of thyroid hormone synthesis and metabolic regulation, as well as to elucidate its action within the thyroid. This could occur if the TSHβv polypeptide, either as a monomer or as a TSHα/TSHβv dimer, interfered with the binding of native TSHβ to the TSHR. It is known, for example, that naturally-occurring minor biophysical changes in the human TSHβ subunit can have dramatic effects on the ability of TSH to bind to the TSHR (51–53). Given that the human TSHβv polypeptide retains the seat-belt region coded for by exon 3 that is used to couple to TSHα, a TSHα/TSHβv complex would almost certainly exhibit altered physiochemical properties that could substantially affect the binding of the conventional TSHα/TSHβ complex to the TSHR. Additional studies will be needed to understand the effects of competitive binding between TSHβv vs. native TSHβ in driving the release of thyroid hormone. Moreover, because thyroid hormones directly influence peripheral leukocyte function, as has been shown for neutrophils (54, 55), monocytes and macrophages (56), and dendritic cells (57), a loop may exist between TSHβv produced by the immune system, and the regulatory effects of TSHβv-mediated thyroid hormone release on the immune system itself.
Immune system TSHβv has been tied to the host response in other physiological scenarios. Various studies have implicated low TSH levels, such as occurs in hyperthyroidism, with an increased risk of osteoporosis (58, 59). This is reinforced by studies demonstrating a reduction in bone loss concomitant with TSH binding to TSHRs on bone osteoblasts (60). Perhaps most striking, however, was the finding that the TSHβv made by bone marrow cells has an osteoprotective effect by acting directly on bone TSHRs. Thus, TSHβv may play a key role in bone health (21, 61–63).
In addition to the use of immune system TSHβv in response to infection and for bone remodeling, a number of human conditions tied to thyroid dysregulation, many of which are associated with inflammation or autoimmunity, have yet to be fully understood. These include Graves' disease and Hashimoto's thyroiditis (64–66), Graves' Ophthalmopathy (67, 68), Pendred's Syndrome (69), Lyme disease (70), inflammatory bowel disease (71), rheumatoid arthritis (72), systemic lupus erythematosus (73, 74), psoriasis (75), asthma (76), and sepsis (77, 78). Of particular interest, patients with Hashimoto's thyroiditis (HT) were found to have elevated transcript levels of TSHβv in PB compared to normal controls (24). Treatment of HT patients with prednisone reduced TSHβv transcript levels in patients having disease of < 9 months compared to patients with a disease >18 months (24). Also in that study, dexamethasone treatment suppressed in a dose-dependent fashion the expression of TSHβv in PBLs of HT patients (24). A second study implicated plasma cell-derived TSHβv in the pathogenesis of HT (79).
Summary
A model is presented in which the TSHβv transcript is produced in cells of the immune system using a mechanism of alternative splicing that results in deletion of exon 2 and retention of exon 3 coded portions of the transcript. A unique feature of this splicing process is that the transcript codes for a signal peptide derived from the 3′ end of intron 2 prior to exon 3. While it is anticipated that the model presented here will add to our understanding of the biological and inherent functional role of immune system TSHβv in human health, it should be noted that caution is warranted when ascribing splice site locations using predictive information (34, 80, 81). Even sequences that have a high potential as a splice site may be pseudosplice sites with limited functional significance. Sequence analyses may help to resolve this. Additionally, it may be possible to directly assess the functional role of TSHβv in a number of ways. In mice, for example, it may be possible using Crispr/Cas9 gene editing to selectively disrupt the expression of intron 4 and exon 5 in order to eliminate the TSHβv splice variant without affecting expression of a native TSHβ. An approach such as this, if successful, would permit experimental analyses into the extent to which immune system TSHβv contributes to thyroid biology and overall host metabolism. Although parallel studies cannot be done in humans, a version of that could be addressed in vitro using human cell lines. In summary, it is hoped that the system laid out here will provide a point from which to launch additional experiments into the role of TSHβv in health and disease.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by grants AI-135293 and AI133313 from the National Institutes of Health.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gordon DF, Wood WM, Ridgway EC. Organization and nucleotide sequence of the gene encoding the beta-subunit of murine thyrotropin. DNA (1988)7:17–26. doi: 10.1089/dna.1988.7.17
2. Szkudlinski MW, Fremont V, Ronin C, Weintraub BD. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev. (2002) 82:473–502. doi: 10.1152/physrev.00031.2001
3. Smith EM, Phan M, Kruger TE, Coppenhaver DH, Blalock JE. Human lymphocyte production of immunoreactive thyrotropin. Proc Natl Acad Sci USA. (1983) 80:6010–3. doi: 10.1073/pnas.80.19.6010
4. Kruger TE, Smith LR, Harbour DV, Blalock JE. Thyrotropin: an endogenous regulator of the in vitro immune response. J Immunol. (1989) 142:744–7.
5. Harbour DV, Kruger TE, Coppenhaver D, Smith EM, Meyer WJ. Differential expression and regulation of thyrotropin (TSH) in T cell lines. Mol Cell Endocrinol. (1989) 64:229–41. doi: 10.1016/0303-7207(89)90150-0
6. Bagriacik EU, Zhou Q, Wang HC, Klein JR. Rapid and transient reduction in circulating thyroid hormones following systemic antigen priming: implications for functional collaboration between dendritic cells and thyroid. Cell Immunol. (2001) 212:92–100. doi: 10.1006/cimm.2001.1846
7. Klein JR, Wang HC. Characterization of a novel set of resident intrathyroidal bone marrow-derived hematopoietic cells: potential for immune-endocrine interactions in thyroid homeostasis. J Exp Biol. (2004) 207 (Pt. 1):55–65. doi: 10.1242/jeb.00710
8. Wang HC, Dragoo J, Zhou Q, Klein JR. An intrinsic thyrotropin-mediated pathway of TNF-alpha production by bone marrow cells. Blood (2003) 101:119–23. doi: 10.1182/blood-2002-02-0544
9. Wang J, Klein JR. Thymus-neuroendocrine interactions in extrathymic T cell development. Science (1994) 265:1860–2. doi: 10.1126/science.8091211
10. Scofield VL, Montufar-Solis D, Cheng E, Estes MK, Klein JR. Intestinal TSH production is localized in crypt enterocytes and in villus 'hotblocks' and is coupled to IL-7 production: evidence for involvement of TSH during acute enteric virus infection. Immunol Lett. (2005) 99:36–44. doi: 10.1016/j.imlet.2004.12.010
11. Wang J, Whetsell M, Klein JR. Local hormone networks and intestinal T cell homeostasis. Science (1997) 275:1937–9. doi: 10.1126/science.275.5308.1937
12. Blalock JE, Johnson HM, Smith EM, Torres BA. Enhancement of the in vitro antibody response by thyrotropin. Biochem Biophys Res Commun. (1984) 125:30–4. doi: 10.1016/S0006-291X(84)80329-0
13. Stolc V. Inhibitory effect of pituitary factor on phagocytosis and iodine metabolism in human leukocytes. Endocrinology (1972) 91:835–9. doi: 10.1210/endo-91-4-835
14. Stolc V. Regulation of iodine metabolism in human leukocytes by adenosine 3',5' -monophosphate. Biochim Biophys Acta (1972) 264:285–8. doi: 10.1016/0304-4165(72)90292-9
15. Fabris N, Mocchegiani E, Provinciali M. Pituitary-thyroid axis and immune system: a reciprocal neuroendocrine-immune interaction. Hormone Res. (1995) 43:29–38. doi: 10.1159/000184234
16. Wu K, Zhao M, Ma C, Zhang H, Liu X, Zhou L, et al. Thyrotropin alters T cell development in the thymus in subclinical hypothyroidism mouse model. Scandinavian J Immunol. (2017) 85:35–42. doi: 10.1111/sji.12507
17. Jara EL, Munoz-Durango N, Llanos C, Fardella C, Gonzalez PA, Bueno SM, et al. Modulating the function of the immune system by thyroid hormones and thyrotropin. Immunol Lett. (2017) 184:76–83. doi: 10.1016/j.imlet.2017.02.010
18. Kelley KW, Weigent DA, Kooijman R. Protein hormones and immunity. Brain Behav Immun. (2007) 21:384–92. doi: 10.1016/j.bbi.2006.11.010
19. Vincent BH, Montufar-Solis D, Teng BB, Amendt BA, Schaefer J, Klein JR. Bone marrow cells produce a novel TSHb splice variant that is upregulated in the thyroid following systemic virus infection. Genes Immun. (2009) 10:18–26. doi: 10.1038/gene.2008.69
20. Schaefer JS, Klein JR. A novel thyroid stimulating hormone b-subunit isoform in human pituitary, peripheral blood leukocytes, and thyroid. Gen Comp Endocrinol. (2009) 162:241–4. doi: 10.1016/j.ygcen.2009.04.006
21. Baliram R, Chow A, Huber AK, Collier L, Ali MR, Morshed SA, et al. Thyroid and bone: macrophage-derived tsh-beta splice variant increases murine osteoblastogenesis. Endocrinology (2013) 154:4919–26. doi: 10.1210/en.2012-2234
22. Montufar-Solis D, Klein JR. Splenic leukocytes traffic to the thyroid and produce a novel TSHbeta isoform during acute listeria monocytogenes infection in mice. PLoS ONE (2016) 11:e0146111. doi: 10.1371/journal.pone.0146111
23. Baliram R, Latif R, Morshed SA, Zaidi M, Davies TF. T3 regulates a human macrophage-derived TSH-beta splice variant: implications for human bone biology. Endocrinology (2016) 157:3658–67. doi: 10.1210/en.2015-1974
24. Liu C, Li L, Ying F, Xu C, Zang X, Gao Z. A newly identified TSHbeta splice variant is involved in the pathology of Hashimoto's thyroiditis. Mol Biol Rep. (2012) 39:10019–30. doi: 10.1007/s11033-012-1871-x
25. Ding WQ, Miller LJ. Signaling from novel splice variants of hormone receptors in cancer. Int J Gastrointest Cancer (2002) 31:31–9. doi: 10.1385/IJGC:31:1-3:31
26. Ding WQ, Kuntz SM, Miller LJ. A misspliced form of the cholecystokinin-B/gastrin receptor in pancreatic carcinoma: role of reduced sellular U2AF35 and a suboptimal 3'-splicing site leading to retention of the fourth intron. Cancer Res. (2002) 62:947–52.
27. Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. (2000) 25:381–8. doi: 10.1016/S0968-0004(00)01604-2
28. Galante PA, Sakabe NJ, Kirschbaum-Slager N, de Souza SJ. Detection and evaluation of intron retention events in the human transcriptome. RNA (2004) 10:757–65. doi: 10.1261/rna.5123504
29. Ding WQ, Kuntz S, Bohmig M, Wiedenmann B, Miller LJ. Dominant negative action of an abnormal secretin receptor arising from mRNA missplicing in a gastrinoma. Gastroenterology (2002) 122:500–11. doi: 10.1053/gast.2002.31039
30. Scott GK, Robles R, Park JW, Montgomery PA, Daniel J, Holmes WE, et al. A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol Cell Biol. (1993) 13:2247–57. doi: 10.1128/MCB.13.4.2247
31. Dauksaite V, Akusjarvi G. The second RNA-binding domain of the human splicing factor ASF/SF2 is the critical domain controlling adenovirus E1A alternative 5'-splice site selection. Biochem J. (2004) 381 (Pt. 2):343–50. doi: 10.1042/BJ20040408
32. Sironen A, Thomsen B, Andersson M, Ahola V, Vilkki J. An intronic insertion in KPL2 results in aberrant splicing and causes the immotile short-tail sperm defect in the pig. Proc Natl Acad Sci USA. (2006) 103:5006–11. doi: 10.1073/pnas.0506318103
33. Chand HS, Ness SA, Kisiel W. Identification of a novel human tissue factor splice variant that is upregulated in tumor cells. Int J Cancer (2006) 118:1713–20. doi: 10.1002/ijc.21550
34. Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. (2005) 42:737–48. doi: 10.1136/jmg.2004.029538
35. Mauger O, Lemoine F, Scheiffele P. Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron (2016) 92:1266–78. doi: 10.1016/j.neuron.2016.11.032
36. Tatsumi K, Hayashizaki Y, Hiraoka Y, Miyai K, Matsubara K. The structure of the human thyrotropin beta-subunit gene. Gene (1988) 73:489–97. doi: 10.1016/0378-1119(88)90513-6
37. Kochetov AV. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays (2008) 30:683–91. doi: 10.1002/bies.20771
38. Shupnik MA, Rosenzweig BA, Showers MO. Interactions of thyrotropin-releasing hormone, phorbol ester, and forskolin-sensitive regions of the rat thyrotropin-beta gene. Mol Endocrinol. (1990) 4:829–36. doi: 10.1210/mend-4-6-829
39. Mason ME, Friend KE, Copper J, Shupnik MA. Pit-1/GHF-1 binds to TRH-sensitive regions of the rat thyrotropin beta gene. Biochemistry (1993) 32:8932–8. doi: 10.1021/bi00085a026
40. Steinfelder HJ, Radovick S, Mroczynski MA, Hauser P, McClaskey JH, Weintraub BD, et al. Role of a pituitary-specific transcription factor (pit-1/GHF-1) or a closely related protein in cAMP regulation of human thyrotropin-beta subunit gene expression. J Clin Invest. (1992) 89:409–19. doi: 10.1172/JCI115600
41. Haugen BR, Gordon DF, Nelson AR, Wood WM, Ridgway EC. The combination of Pit-1 and Pit-1T have a synergistic stimulatory effect on the thyrotropin beta-subunit promoter but not the growth hormone or prolactin promoters. Mol Endocrinol. (1994) 8:1574–82.
42. Gordon DF, Lewis SR, Haugen BR, James RA, McDermott MT, Wood WM, et al. Pit-1 and GATA-2 interact and functionally cooperate to activate the thyrotropin beta-subunit promoter. J Biol Chem. (1997) 272:24339–47. doi: 10.1074/jbc.272.39.24339
43. Kapiloff MS, Farkash Y, Wegner M, Rosenfeld MG. Variable effects of phosphorylation of Pit-1 dictated by the DNA response elements. Science (1991) 253:786–9. doi: 10.1126/science.1652153
44. Gordon DF, Haugen BR, Sarapura VD, Nelson AR, Wood WM, Ridgway EC. Analysis of Pit-1 in regulating mouse TSH beta promoter activity in thyrotropes. Mol Cell Endocrinol. (1993) 96:75–84. doi: 10.1016/0303-7207(93)90097-4
45. Kashiwabara Y, Sasaki S, Matsushita A, Nagayama K, Ohba K, Iwaki H, et al. Functions of PIT1 in GATA2-dependent transactivation of the thyrotropin beta promoter. J Mol Endocrinol. (2009) 42:225–37. doi: 10.1677/JME-08-0099
46. Ohba K, Sasaki S, Matsushita A, Iwaki H, Matsunaga H, Suzuki S, et al. GATA2 mediates thyrotropin-releasing hormone-induced transcriptional activation of the thyrotropin beta gene. PloS ONE (2011) 6:e18667. doi: 10.1371/journal.pone.0018667
47. Wood WM, Kao MY, Gordon DF, Ridgway EC. Thyroid hormone regulates the mouse thyrotropin beta-subunit gene promoter in transfected primary thyrotropes. J Biol Chem. (1989) 264:14840–7.
48. Yusta B, Alarid ET, Gordon DF, Ridgway EC, Mellon PL. The thyrotropin beta-subunit gene is repressed by thyroid hormone in a novel thyrotrope cell line, mouse T alphaT1 cells. Endocrinology (1998) 139:4476–82. doi: 10.1210/endo.139.11.6283
49. Wang SH, Koenig RJ. A locally secreted thyrotropin variant may regulate thyroid function in thyroid inflammatory disorders. Thyroid (2009) 19:5–6. doi: 10.1089/thy.2008.1564
50. Schaefer JS, Klein JR. Immunological regulation of metabolism–a novel quintessential role for the immune system in health and disease. FASEB J. (2011) 25:29–34. doi: 10.1096/fj.10-168203
51. Dacou-Voutetakis C, Feltquate DM, Drakopoulou M, Kourides IA, Dracopoli NC. Familial hypothyroidism caused by a nonsense mutation in the thyroid-stimulating hormone beta-subunit gene. Am J Human Genet. (1990) 46:988–93.
52. Medeiros-Neto G, Herodotou DT, Rajan S, Kommareddi S, de Lacerda L, Sandrini R, et al. A circulating, biologically inactive thyrotropin caused by a mutation in the beta subunit gene. J Clin Invest. (1996) 97:1250–6. doi: 10.1172/JCI118540
53. Biebermann H, Liesenkotter KP, Emeis M, Oblanden M, Gruters A. Severe congenital hypothyroidism due to a homozygous mutation of the betaTSH gene. Pediatr Res. (1999) 46:170–3. doi: 10.1203/00006450-199908000-00007
54. van der Spek AH, Bloise FF, Tigchelaar W, Dentice M, Salvatore D, van der Wel NN, et al. The thyroid hormone inactivating enzyme type 3 deiodinase is present in bactericidal granules and the cytoplasm of human neutrophils. Endocrinology (2016) 157:3293–305. doi: 10.1210/en.2016-1103
55. Mezosi E, Szabo J, Nagy EV, Borbely A, Varga E, Paragh G, et al. Nongenomic effect of thyroid hormone on free-radical production in human polymorphonuclear leukocytes. J Endocrinol. (2005) 185:121–9. doi: 10.1677/joe.1.05968
56. Perrotta C, Buldorini M, Assi E, Cazzato D, De Palma C, Clementi E, et al. The thyroid hormone triiodothyronine controls macrophage maturation and functions: protective role during inflammation. Am J Pathol. (2014) 184:230–47. doi: 10.1016/j.ajpath.2013.10.006
57. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity (2013) 39:38–48. doi: 10.1016/j.immuni.2013.07.004
58. Bassett JH, Williams GR. The skeletal phenotypes of TRalpha and TRbeta mutant mice. J Mol Endocrinol. (2009) 42:269–82. doi: 10.1677/JME-08-0142
59. Sun L, Vukicevic S, Baliram R, Yang G, Sendak R, McPherson J, et al. Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci USA. (2008) 105:4289–94. doi: 10.1073/pnas.0712395105
60. Abe E, Marians RC, Yu W, Wu XB, Ando T, Li Y, et al. TSH is a negative regulator of skeletal remodeling. Cell (2003) 115:151–62. doi: 10.1016/S0092-8674(03)00771-2
61. Williams GR, Bassett JHD. Thyroid diseases and bone health. J Endocrinol Invest. (2018) 41:99–109. doi: 10.1007/s40618-017-0753-4
62. Bassett JH, Williams GR. Critical role of the hypothalamic-pituitary-thyroid axis in bone. Bone (2008) 43:418–26. doi: 10.1016/j.bone.2008.05.007
63. Baliram R, Latif R, Zaidi M, Davies TF. Expanding the role of thyroid-stimulating hormone in skeletal physiology. Front Endocrinol. (2017) 8:252. doi: 10.3389/fendo.2017.00252
64. Salmaso C, Bagnasco M, Pesce G, Montagna P, Brizzolara R, Altrinetti V, et al. Regulation of apoptosis in endocrine autoimmunity: insights from Hashimoto's thyroiditis and Graves' disease. Ann NY Acad Sci. (2002) 966:496–501. doi: 10.1111/j.1749-6632.2002.tb04253.x
65. Salmaso C, Olive D, Pesce G, Bagnasco M. Costimulatory molecules and autoimmune thyroid diseases. Autoimmunity (2002) 35:159–67. doi: 10.1080/08916930290013441
66. Iwatani Y, Hidaka Y, Matsuzuka F, Kuma K, Amino N. Intrathyroidal lymphocyte subsets, including unusual CD4+ CD8+ cells and CD3loTCR alpha beta lo/-CD4-CD8- cells, in autoimmune thyroid disease. Clini Exp Immu. (1993) 93:430–36. doi: 10.1111/j.1365-2249.1993.tb08196.x
67. Mooij P, Drexhage HA. Interactions between the immune system and the thyroid. Regulatory networks in health and disease. Thyroidology (1992) 4:45–8.
68. Douglas RS, Afifiyan NF, Hwang CJ, Chong K, Haider U, Richards P, et al. Increased generation of fibrocytes in thyroid-associated ophthalmopathy. J Clini Endocrinol Metabol. (2010) 95:430–8. doi: 10.1210/jc.2009-1614
69. Camargo R, Limbert E, Gillam M, Henriques MM, Fernandes C, Catarino AL, et al. Aggressive metastatic follicular thyroid carcinoma with anaplastic transformation arising from a long-standing goiter in a patient with Pendred's syndrome. Thyroid (2001) 11:981–8. doi: 10.1089/105072501753211073
70. Vaccaro M, Guarneri F, Borgia F, Cannavo SP, Benvenga S. Association of lichen sclerosus and autoimmune thyroiditis: possible role of Borrelia burgdorferi? Thyroid (2002) 12:1147–8. doi: 10.1089/105072502321085261
71. Bianchi GP, Marchesini G, Gueli C, Zoli M. Thyroid involvement in patients with active inflammatory bowel diseases. Ital J Gastroenterol. (1995) 27:291–5.
72. Maksymowych WP, Brown MA. Genetics of ankylosing spondylitis and rheumatoid arthritis: where are we at currently, and how do they compare? Clini Exp Rheumatol. (2009) 27 (4 Suppl. 55):S20–25.
73. Parente Costa L, Bonfa E, Martinago CD, de Oliveira RM, Carvalho JF, Pereira RM. Juvenile onset systemic lupus erythematosus thyroid dysfunction: a subgroup with mild disease? J Autoimmun. (2009) 33:121–4. doi: 10.1016/j.jaut.2009.04.001
74. Mader R, Mishail S, Adawi M, Lavi I, Luboshitzky R. Thyroid dysfunction in patients with systemic lupus erythematosus (SLE): relation to disease activity. Clini Rheumatol. (2007) 26:1891–4. doi: 10.1007/s10067-007-0602-5
75. Gul U, Gonul M, Kaya I, Aslan E. Autoimmune thyroid disorders in patients with psoriasis. Eur J Dermatol. (2009) 19:221–3. doi: 10.1684/ejd.2009.0632
76. Manzolli S, Macedo-Soares MF, Vianna EO, Sannomiya P. Allergic airway inflammation in hypothyroid rats. J Allergy Clini Immunol. (1999) 104(3 Pt. 1):595–600. doi: 10.1016/S0091-6749(99)70329-5
77. Collins J, Gough S. Autoimmunity in thyroid disease. Eur J Nuclear Med Mol Imaging. (2002) 29 (Suppl. 2):S417–24. doi: 10.1007/s00259-002-0848-8
78. Lodha R, Vivekanandhan S, Sarthi M, Arun S, Kabra SK. Thyroid function in children with sepsis and septic shock. Acta paediatr. (2007) 96:406–9. doi: 10.1111/j.1651-2227.2007.00135.x
79. Liu CR, Miao J, Zhao ZK, Li LY, Liu YM, Zhang YL, et al. Functional human TSHbeta splice variant produced by plasma cell may be involved in the immunologic injury of thyroid in the patient with Hashimoto's thyroiditis. Mol Cell Endocrinol. (2015) 414:132–142. doi: 10.1016/j.mce.2015.06.009
80. Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Mol Cell Biol. (2000) 20:6414–25. doi: 10.1128/MCB.20.17.6414-6425.2000
Keywords: alternative splicing, hormone, immune-endocrine, leukocyte, thyroid
Citation: Klein JR (2019) Novel Splicing of Immune System Thyroid Stimulating Hormone β-Subunit—Genetic Regulation and Biological Importance. Front. Endocrinol. 10:44. doi: 10.3389/fendo.2019.00044
Received: 29 November 2018; Accepted: 18 January 2019;
Published: 05 February 2019.
Edited by:
Louise Metherell, Queen Mary University of London, United KingdomReviewed by:
Eusebio Chiefari, University Magna Graecia of Catanzaro, ItalyLaura Ellestad, University of Georgia, United States
Copyright © 2019 Klein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John R. Klein, am9obi5yLmtsZWluQHV0aC50bWMuZWR1