 Giovanni Lombardi
Giovanni Lombardi Ewa Ziemann
Ewa Ziemann Giuseppe Banfi1,3
Giuseppe Banfi1,3- 1Laboratory of Experimental Biochemistry & Molecular Biology, IRCCS Istituto Ortopedico Galeazzi, Milan, Italy
- 2Department of Physiology and Pharmacology, Faculty of Rehabilitation and Kinesiology, Gdansk University of Physical Education and Sport, Gdansk, Poland
- 3Vita-Salute San Raffaele University, Milan, Italy
Bone tissue can be seen as a physiological hub of several stimuli of different origin (e.g., dietary, endocrine, nervous, immune, skeletal muscle traction, biomechanical load). Their integration, at the bone level, results in: (i) changes in mineral and protein composition and microarchitecture and, consequently, in shape and strength; (ii) modulation of calcium and phosphorous release into the bloodstream, (iii) expression and release of hormones and mediators able to communicate the current bone status to the rest of the body. Different stimuli are able to act on either one or, as usual, more levels. Physical activity is the key stimulus for bone metabolism acting in two ways: through the biomechanical load which resolves into a direct stimulation of the segment(s) involved and through an indirect load mediated by muscle traction onto the bone, which is the main physiological stimulus for bone formation, and the endocrine stimulation which causes homeostatic adaptation. The third way, in which physical activity is able to modify bone functions, passes through the immune system. It is known that immune function is modulated by physical activity; however, two recent insights have shed new light on this modulation. The first relies on the discovery of inflammasomes, receptors/sensors of the innate immunity that regulate caspase-1 activation and are, hence, the tissue triggers of inflammation in response to infections and/or stressors. The second relies on the ability of certain tissues, and particularly skeletal muscle and adipose tissue, to synthesize and secrete mediators (namely, myokines and adipokines) able to affect, profoundly, the immune function. Physical activity is known to act on both these mechanisms and, hence, its effects on bone are also mediated by the immune system activation. Indeed, that immune system and bone are tightly connected and inflammation is pivotal in determining the bone metabolic status is well-known. The aim of this narrative review is to give a complete view of the exercise-dependent immune system-mediated effects on bone metabolism and function.
Introduction
Exercise, particularly when energetically demanding and sustained, affects all human tissues and organs. Different kinds of exercise (e.g., endurance, high intensity, resistance) have different effects on tissues and organs homeostasis and, consequently, different kinds of training require different adaptive changes that might take place (1). Bone is importantly affected by exercise and bone cells metabolism forcefully adapts to training. This metabolic adaptation reflects, on long-term basis, in a micro-architectural, and possibly macro-architectural, redefinition of bone shape and structure. The biological meaning of this phenomena resides in the physical adaptation of bones (in terms of shape, mass, and strength) to the changed environmental conditions (applied forces) (2). However, the bony response to exercise does not only depend on the biomechanical stimulus but equally relies on other systemic mechanisms that make the bone a center for the integration of the signals generated during and following the exercise (3). A main signal is the metabolic one: bone metabolism is highly-demanding in terms of energy; parallel, during exercise the energy needs increase with the increasing duration and intensity. Being not a lifesaving function, bone metabolism is partially blocked in response to acute exercise, regardless the degree of biomechanical load. This block mainly pertains the anabolic function (osteoblastic function, i.e., bone formation) while leaves almost unaltered the catabolic side (osteoclastic function, i.e., bone resorption). The acute unbalance toward bone resorption makes the calcium, stored in the bone matrix, available to skeletal muscles (SKM) and cardiac muscle for contraction and to the brain to sustain the neuromuscular function (4). On the other hand, the chronically established bone metabolic response to training is driven by the degree of load: weight-bearing/high impact activities (e.g., plyometric) shift the balance toward bone formation and on average increase bone mass and bone mineral density (BMD). Equally demanding activities featured by a low/absent biomechanical load (e.g., swimming, cycling) shift the balance toward resorption and causes a decrease in the average bone mass and BMD (3). Another key signal, generated during and following exercise and affecting bone metabolism, is the immune/inflammatory one. Inflammation, acute and chronic, is a main determinant for bone metabolism and the plethora of inflammatory mediators, produced under either physiologic or pathologic conditions, affects bone cells. Noteworthy, in last 15 years an interdisciplinary branch of study embracing (but not limited to) endocrinology, immunology, orthopedics, and rheumatology, namely osteoimmunology, has developed quickly thus becoming a central subject in metabolic diseases of bone (5). A key role of the osteoimmune networking has been highlighted by the clinical success and safety, over the classical anti-resorptive drugs (6, 7), of the fully human monoclonal antibody denosumab used in the treatment of primary and secondary osteoporosis, in both males and females (8), which is an inhibitor of the prototypic osteoimmunological signaling pathway, the RANK/RANKL system, as described elsewhere (9). Acute exercise represents a powerful inflammatory stimulus itself while the inflammatory response to training is a handbook example of adaptation to a continuous stimulus. Exercise, indeed, initiates a series of inflammatory events, which ultimately, if chronically continued, positively affects health. The inflammatory response to exercise, however, takes place in the immune system (IS), involving mainly the innate compartment with fallouts on the humoral immunity, as well as in tissues other than the IS, e.g., SKM, white and brown adipose tissues (WAT, BAT), brain, liver (3, 10, 11). Notably, the acute response elicited by exercise in terms of inflammatory mediators is often very similar to that observed during pathological acute and chronic inflammatory conditions (e.g., sepsis, obesity, autoimmune diseases, tumors) but what determines the net result of this response (pro- vs. anti-inflammatory) is the milieu in which this response is generated, together with its temporal length and the site of production. As an example, interleukin (IL)-6 expression is strongly induced by time, intensity and type of muscle contractions-dependently as well as in sepsis (various origin) but, in the latter case, it is driven by the previous increase in another cytokine, the tumor necrosis factor (TNF)-α (12). Based on the interdependence between bone and IS, it is implicit that the acute inflammatory response induced by exercise- and the modulation of the inflammatory status induced by chronic training both affect bone cells differentiation and function and, in turn, the bone metabolism. It should be kept in mind that, other than a target of these endocrine and inflammatory stimuli, the bone is itself a source of these mediators and, hence, it actively enters in the regulation/modulation network of the homeostasis (3).
This review aims at summarizing the experimental evidences about the exercise- and training-dependent effects on bone mediated by the IS and the other inflammatory sources in order to depict additional, and less explored, link between bone and inflammation as further mechanisms by which the physically active status is a main determinant for health.
In order to go deep into the description, it is necessary to introduce some key concepts, relative to the exercise physiology, that will help the reader in fully understand the biological meaning underlying the homeostatic response. About the types of muscle actions, one can distinguish between static (isometric) and dynamic (isotonic) actions. During the static (isometric) action, muscles generate force without changing length due to an external resistance (weight of an object) greater than the force produced by the muscle; despite the energy expenditure, no work is done due to lack of movement. The isotonic action, instead, can be either (i) concentric, when muscles produce enough force to overcome the external resistance and the muscle contraction results in a work, or (ii) eccentric, when the muscles lengthens while generate a force due to the opposite movement of the external resistance and the sarcomere shortening (13). Another series of concepts regard the terms physical activity (PA), exercise, and training. PA is defined as any bodily movement produced by SKM resulting in an energy consumption. Everyone performs PA in order to sustain life but the amount is subject to personal. The term “exercise,” instead, although often erroneously used interchangeably with PA, is defined as a PA featured by planning, structure, and repetitiveness, which is aimed at maintaining or improving the physical fitness (14). The common and distinctive features of PA and exercise are summarized in Table 1.
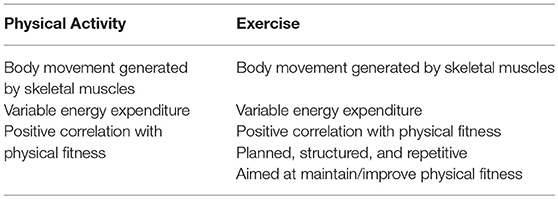
Table 1. Common and distinctive features of physical activity and exercise.
When a single bout of exercise (acute exercise) is continued over the time, in the same fashion, it is defined training (exercise training). Finally, the different types of exercise and training can be categorized as follows: (i) endurance, mainly based on the aerobic metabolism (e.g., distance running, road cycling, swimming, triathlon), (ii) resistance (also known as strength), mainly based on the anaerobic metabolism (e.g., weight lifting, discus, hammer, and javelin throw) (15).
How Do Exercise and Training Affect Bone Metabolism?
The responsiveness of bone to mechanical stimulation was first theorized by Frost who postulated, with the “mechanostat” hypothesis, bone mass and structure remain constant around a certain threshold of mechanical strains. Bone formation takes place when the strain increases above this threshold, and it results in an increased bone stiffness. When the strain experienced by the bone segment is lower than this threshold bone loss can take place (16). Later, it was shown that the threshold itself is modifiable by several factors, mainly endocrine [parathyroid hormone (PTH), sex hormones, etc.] (17). However, despite its importance, the mechanical strains induced by strenuous PA is very small degree attesting to up to 0.3% (3,000 microstrain) (18). Based on that, it is likely that bone cells are exposed to and integrate different PA-generated mechanical stimuli that altogether imply an amplification of the environmental stimulation. A further level of complexity is due to the fact that different types of bone cells are anatomically exposed to different combinations of stimuli. Bone marrow and endosteal osteoblasts experience the pressure forces generated within the marrow cavity. Osteocytes buried into the matrix with their interconnecting long cellular processes running within the fluid-filled canalicular network experience dynamic fluid flow pressure, shear stress forces, and dynamic electric fields (due to the transit of charged ions in the interstitial fluid). Mature osteoclasts and their precursors, residing in the bone marrow, may be exposed to mechanical stimulation due to dynamic pressure (19). Bone mechanosensitivity is mediated by several cellular components (e.g., membrane, membrane proteins, cytoskeleton, cilia, ion channels). Shear stress and pressure deform the plasma membrane and, consequently, to the cytoskeleton and, in turn, through integrins to the protein machinery mediating the cell-to-matrix adhesion and to the nucleus where it induces the expression of downstream genes (20). In osteoblast, the deformation of the plasma membrane is associated with the activation of ion channels (21), as in osteocytes, whose cilia, protruding out of the dendritic extensions, sense fluid flow and activate channel-mediated ion fluxes that modulate the Wnt signaling pathway (22).
The different nature of the mechanical stimuli together with the number of cell structures involved in mechanosensitivity imply the integration of the different signals generated (19). Indeed, the physical stimulus is translated into several chemical signals including calcium, mitogen-activated protein kinase (MAPK), Wnt, and RhoA/ROCK pathways. For instance, the Frizzled-LRP5/6-mediated activation of Wnt induces the expression of osteoblastic factors, as RUNX2 that promotes the commitment of mesenchymal stem cells (MSCs) toward the osteoblast lineage, induces proliferation and differentiation of pre-osteoblast, and stimulates mineralization. Hence, exercise shifts the adipogenic-to-osteogenic equilibrium, governing the MSC fate, toward the osteoblastic commitment (23, 24). By modulating the OPG and RANKL expression in osteoblasts, Wnt signaling also downregulates osteoclastogenesis and osteoclast activity (25). A key mechanism regulating the Wnt activity that underlies the exercise-associated effects on bone is mediated by sclerostin (Sost) (26). Osteocytes constitutively produce Sost that inhibits the Wnt pathway, thereby osteoblastogenesis and bone formation. Loading activates a molecular response that inhibits Sost expression and, then, allow the activation of Wnt signaling. Noteworthy, also prostaglandin E2 (PGE2), which is induced by strain sensing, by activating its receptors EP2 and EP4, facilitates the nuclear translocation of β-catenin and its transcriptional action (27). Finally, the intracellular signaling generated following mechanical stimulation can be propagated to neighbor cells as calcium (e.g., between osteoblasts and osteocytes) or adenosine triphosphate (ATP; e.g., osteoblast-to-osteoclast) transients through the gap junction network (28, 29).
Another way by which exercise and recovery beneficially affects bone metabolism, mineral content, and structure is by increasing the blood flow to the bone and the consequent improved supply of nutrients supporting its metabolic needs (30, 31). Indeed, also from this point of view, bone is a really active tissue as demonstrated by the fact that blood flow to the bone and glucose uptake within the bone tissue are increased in response to exercise (32). However, as exercise load increases blood flow offs due to the sympathetic response that shifts the flow to the active SKM. Moreover, perfusion in bone, which ranges over a window wider than that expected during exercise, is mainly ruled by chemoreceptors, and hence nutrients, rather than by hypoxia (33). The exercise-induced enhanced bone perfusion also causes an increased efflux of stem cells from bone marrow (e.g., endothelial precursors) (34), consequently to the endothelial production of nitric oxide (35) and sympathetic nervous system activation (36). Finally, exercise-induced muscular-derived adenosine could solve another key role in bone blood flow determination (37).
The Effect of Acute Exercise
Despite the key role of loading, the bone tissue response of the to a single bout of exercise is mainly driven by the exercise-dependent metabolic requests of noble organs and of those organs that are directly involved in the activity. In other terms, during PA, and mostly dependent upon its intensity and duration, the metabolic needs of non-life saving organs (e.g., the bone, skin, gut) are somehow “sacrificed” in order to have all the fuels available for being used by brain, SKM, and liver (3). After the conclusion of the PA, during recovery, and with the chronic and/or long-term repetition of the act, the loading/impact-induced anabolic response takes place. This means that during PA the bone metabolism is mostly unbalanced toward resorption. This is also a consequence of the key role of bone in calcium homeostasis: PA implies the usage of calcium in terms of functioning of the contractile machinery of the SKM, release, and recycling of neurotransmitters and hormones. Being resorbed, bone make the calcium promptly available. Indeed, two activities both characterized by high energy expenditures but differentiated by kind of biomechanical loading, one featured by high load and impact (mountain ultra-trail) while the other non-impact/loading (cycling), both display an anti-anabolic/pro-catabolic acute response of the bone to the activity (38, 39).
By considering the biomechanical aspects, only dynamic stimuli can generate an osteogenic response that, instead, is not induced by static loading (40) and strain amplitude, number of cycles and interval between the cycles are important, as well (41, 42). Indeed, the anabolic response can be desensitized by long-lasting activities while interval rest between the load cycles may have an osteogenic effect (43, 44). On the contrary, inter-cycles resting activates some mechanotransduction-sensitizing mechanisms that sustain the osteogenic response (28). However, too high magnitudes or number of cycles of load may fatigue the bone tissue and consequently can cause microdamage that, in turns, result in a series of catabolic events including local bone resorption (45).
The Effect of Chronic Exercise and Training
Life-long PA is associated with a better bone quality, thus potentially resulting in a stronger bone, e.g., improved cross-sectional area, BMD, and moments of inertia. These features, for example, and have been observed in gymnasts vs. non-gymnasts (46, 47), and dominant vs. non-dominant limbs of racquet sports players (48) or triple jumpers (49). By applying the same external force, the deformation experienced by “weak bones” is greater than that of “strong bones” and, consequently, it elicits in larger tissue strains. This results in a greater anabolic response in the weaker bone that attempts to become stronger (50). In experimental models, loads causing high strains induce bone formation in loaded areas, while areas with lower peak strains featured by reduced bone formation or even increased bone catabolism (51). Site-specific adaptation to loads takes also place in human: in women, after skeletal maturity, the adaptation to load is related to the energy equivalent strain, which means that regions undergone to high-level strain experience bone apposition than regions undergone to low-level strain (50).
However, the bone adaptation to chronic PA and training mainly depends upon the kind of activity. This was clearly demonstrated by Nikander et al. (52), who evaluate the bone quality of the narrowest section of the femoral neck [areal BMD (aBMD), hip structure analysis (HSA), cross-sectional area (CSA), subperiosteal width (W), and section modulus of strength (Z)] in 225 premenopausal women performing different sports. Women performing high-impact (volleyball, hurdling) and odd-impact (squash, soccer, speed skating, step-aerobic) loading sports displayed the highest aBMD (+23 and +29% vs. non-athletic women), CSA (+22 and +27%), and Z (+22 and +26%) even following adjustment for age-, weight-, and height. Contrarily, low-impact (orienteering, cross-country skiing) and non-impact (cycling, swimming) repetitive loading activities were associated with no gains in bone quality, compared with the inactive controls (52). A Cochrane review evaluating the preventive and therapeutic effects of training on postmenopausal osteoporosis established that exercise has a small (about 3%) but significant effect on bone mass and BMD. According to the meta-analysis, femoral bone mass mostly benefits from high-force non- weight-bearing exercises (e.g., resistance strength training of the lower limbs). Vertebral bone mass, instead, mostly benefits from the combination of exercises featured by different types of dynamic loading. Consequently, the risk of fracture across all exercise groups was not significantly different compared to the control groups (53). Exercise-based, either preventing or therapeutic, strategies aimed at affecting bone health might account for this finding. Indeed, based on our recent overview of systematic reviews and meta-analyses, lifelong age-specific exercise is effective in sustaining bone health in women. School-based short bouts of high-impact plyometric exercises positively affect peak bone mass in young girls, while combined-impact exercises represent the best exercise modality to preserve/improve BMD in both pre- and post-menopausal women (54).
Beside the quite well-depicted effects of chronic exercising on the physical feature of the bone, the effects on blood flow or metabolism of human bone are scarce (33). Although neglected, these studies are of key importance since they highlight a role for bone in regulating the whole-body metabolism (55–57) and also because the bone is central in the release of vascular precursor and immune cells. Thus, it emerges that by influencing bone status, exercise training could potentially affect vascular impairments in pathological conditions such as diabetes (33) and it may regulate the release of immune cells into the circulation and, hence, to directly control the inflammatory status.
As important as loading, or even more important, is detraining. Constant load (use of the skeleton) is essential for osteocyte survival and in case of bone immobilization osteocyte apoptosis occurs (58). Parallel, detraining causes bone loss and, hence, training might be continued to maintain bone mass (59) since unloading and disuse increase bone resorption rate. This situation is encountered, for instance, in astronauts under weightlessness conditions, spinal cord injured patients, and elderly people forced to either partial or total immobilization (19). Unloading affects both the cortical and trabecular portions of the bone: spinal cord injured subjects experience a 20–40% decrease in cortical thickness (60) and an average 14% (range 2–80%) in trabecular density (61); for astronauts the mean BMD loss of the trabecular compartment was 4% at the lumbar spine and 12% at the proximal femur (62).
How do Exercise and Training Affect the Inflammatory Response and the Immune Function?
Exercise profoundly affects the normal functioning of the IS with immune responses to single bouts being transient while an immune adaptation is likely to take place with training. Exercise dose is important in determining the entity of the immune response: prolonged intense training can have depressive effects (e.g., increased infection risk), while regular moderate-intensity exercise has more balanced effects that mainly results in the improvement of the baseline immune reactivity (63). Several evidences support this effect, chronic exercise has been demonstrated to improve immune, and hence health and behavioral outcomes, in several conditions of deregulated immune response, such as aging, obesity, cancer, and chronic viral infections (e.g., HIV) as well as in preventing their onset (63–66). Interestingly, acute exercise is effective in improving vaccines response (63). These effects are mediated, from one hand, by the cells of the IS belonging to both the innate and adaptive branches and, from the other hand, by all the cells of the body that are induced to express either a pro-inflammatory or anti-inflammatory phenotype. Exercise activates inflammatory cascades involving cells belonging to both the innate and adaptive immunity branches, cytokines, and mediators with active roles in inflammation (myokines, adipokines) that are in turn responsible for the generation of an environment in which recovery, regeneration, and adaptation take place. Exercise duration, mode, and intensity are the determinants for the exercise-induced inflammatory response. At the same time, training exerts anti-inflammatory actions through several distinct mechanisms involving metabolic, endocrine, and immune mediators of various tissues and organs (67). However, exercise exerts its anti-inflammatory effect only after the activation of pro-inflammatory cascades (67).
A special mention might be spent about cortisol. Although this adrenal hormone has anti-inflammatory immunosuppressant actions, and its occasionally increased circulating concentration (e.g., following PA) are beneficial in reducing inflammation, its chronically high levels (i.e., hypercortisolism) and deregulated rhythms are associated with aging and age-associated low-grade inflammation (68). Cortisol and the other endogenous glucocorticoids (GCs) are the final products of the neuroendocrine hypothalamus-pituitary-adrenal cortex (HPA) axis that is responsible for the regulation of both the energy balance and stress response. The physiological circadian fluctuations of GC levels allow the correct functioning of the intermediate metabolism and the development and the maintenance of the homeostasis of a wide all the body tissues, including the bone. Indeed, GCs are essential for bone modeling and remodeling as they promote osteoblastogenesis to maintain the bone architecture (69). Excessive energy intake (unbalanced energy intake-to-energy expenditure ratio) and adiposity are associated with chronic inflammation and stress which are in turn responsible for a deregulated of the HPA axis. Moreover, hypercortisolism is associated with a deregulated energy metabolism that in turn is responsible for the maintenance of the chronic inflammatory state (70). PA, by acting as a chronoenhancer, also impacting on the HPA axis, is able to improve the GC response in healthy subjects as well as to restore the circadian rhythm in age-associated low-grade inflammation dependent hypercortisolism and, hence, to improve the related comorbidities (68).
Effects of Exercise and Training on Inflammation
Effect of Acute Exercise
A single exercise bout starts a series of timely-defined inflammatory events, which mainly depend upon mode, intensity, duration, and training status (i.e., familiarity with the exercise). This cascade starts with a pro-inflammatory phase (1.5–24 h post-exercise) which is then followed by an anti-inflammatory phase that sustains SKM regeneration (24–72 h post-exercise). The exercise-induced inflammatory response is evidenced by the rise of the circulating levels of myokines (i.e., IL-6) and anti-inflammatory mediators (IL-10, IL-1ra). Moreover, exercise downregulates the expression, on the surface of antigen-presenting cells (APCs, e.g., monocytes) of those receptors involved in the recognition of danger signals, i.e., toll-like receptors (TLRs) (67). TLRs are highly evolutionarily conserved transmembrane proteins involved in the recognition of classes of molecules non-specifically associated with pathogens (pathogen-associated molecular patterns, PAMPs) and “danger signals” non-specifically induced/released following tissue damage due to physical, chemical, or biological agents (danger-associated molecular patterns, DAMPs) (71). TLRs activation leads to the expression of inflammatory cytokines. Acute exercise affects the expression of TLRs on monocytes and, hence, by desensitizing these cells to pro-inflammatory stimulation, this results in a push toward the anti-inflammatory phenotype. 2.5 h of cycling at 60% of VO2max induces a significant decrease in the expression of TLRs on CD14+ monocytes, compared to rest, immediately (TLR2, about −25%) and 1 h post-exercise (TLR1, −60%, TLR2 and TLR4, −50%, TLR3, −30%) (72).
Effect of Chronic Exercise and Training
There are several potential tissue-specific anti-inflammatory mechanisms associated with regular PA and these include reductions in body fat (particularly, visceral fat), enhanced expression and release of contracting muscle-derived anti-inflammatory mediators, downregulated expression of TLRs in monocytes and macrophages, increased expression of anti-oxidant species counteracting the exercise-associated rise in reactive oxygen species (ROS) generation (73).
Exercise causes transient elevations in IL-6 coming from exercising SKM (74). Contrarily, during inflammation, and especially chronic low-grade inflammation, IL-6 is produced in a slightly chronically elevated manner and, in this case, the source is represented by immune cells and hepatocytes (3). Muscle-derived IL-6 has anti-inflammatory effects by inducing other anti-inflammatory cytokines (e.g., IL-1ra and IL-10) that antagonize the pro-inflammatory IL-1β and TNF-α (12). IL-6, but also the exercise-related increased energy needs, stimulate the release of cortisol that has an immunosuppressant activity (74). The logic around the IL-6 release by the contracting muscle resides in its activity aimed at increasing the usage and delivery of energetic substrates to the myocytes, in concert with the stress hormones (e.g., cortisol, epinephrine). By acting in autocrine and paracrine fashions, indeed, IL-6 stimulates the cellular uptake and the oxidation of glucose and fatty acids by the SKM itself; contemporary, by acting in an endocrine fashion, it induces lipolysis at the AT level and glycogenolysis and gluconeogenesis. The insulin-mimetic effect of IL-6 on glucose uptake is of particular interest. The binding of IL-6 with the IL-6Rα/gp130Rβ receptor complex leads to the activation of AMPK that triggers the plasma membrane translocation of intracellular vesicles bearing the insulin-dependent glucose transporter (GLUT4) allowing glucose uptake regardless the insulin status. This accounts for the beneficial effects of PA on the metabolic function also in impaired glucose tolerance and insulin resistance (75). Moreover, other than stimulating the release of IL-6, exercise training generates an appropriate environment making the IL-6 effect distinctly anti-inflammatory: for instance, aerobic training reduces the expression of TNF-α and IL-1α by mononuclear cells and induces IL-4, IL-10, and TGF-β1 in subjects at high-risks of heart disease (76).
Training exerts its effects by also reducing the activation potential of the innate immune response activation in terms of TLRs in an age-independent manner. Young (18–35 years of age) and elderly (65–80 years of age) active subjects have a one third reduced expression of TLR4 on CD14+ monocytes' surface, compared to their inactive counterparts. Moreover, 12-week of either endurance or resistance training halved TLR4 expression in monocytes from these inactive (old and young) subjects to a level comparable to those found in active age-matched controls, while the intervention was ineffective in the already active subjects (72).
The anti-inflammatory effect of training might is also the result of the modulation of nitric oxide (NO) and ROS production and the consequent activation of their downstream pathways. Exercise induces the synthesis of NO and ROS which are important in inducing anti-inflammatory defensive mechanisms especially by targeting muscle gene expression (77). In the case of ROS, with training, the cyclic exercise-induced spiked production (contrarily to what happens in chronic inflammation) causes the activation of an adaptive response that, in turn, protects SKM from exposure to the exercise-dependent increase of ROS itself. This phenomenon accounts also for the decreased expression of TNFα that may further inflammation (77).
The AT is determinant in defining the inflammatory status. The association of physical inactivity and high caloric intake results in adipocyte and AT hypertrophy. As adipocytes grow, the oxygen supply becomes limiting and the consequent hypoxic stress leads to cell death and necrosis. Necrosis recalls macrophages and potently induces an inflammatory response (78). Notably, this process seems to involve mainly the visceral AT that has a higher inflammatory potential than the subcutaneous one. Exercise-induced caloric imbalance causes lipolysis, aimed at mobilizing fats to be used as fuel by the exercising muscle, with a reduction of the adipocytes' size and, thereby, hypoxic stress and inflammation (79).
Effects of Exercise and Training on Immune Functions
Exercise profoundly affects the IS functioning. An exercise bout causes an important redistribution of leukocytes as a consequence of the hemodynamic response and the increased blood levels of catecholamine and glucocorticoids, but the effects depend upon exercise intensity and duration. Prolonged periods of intensive training can impair immune functions, and particularly those of T-cells, natural killer (NK)-cells, and neutrophils; in elite athletes, during periods of heavy training and competition, mucosal immunity is also affected determining an increased risk of infections of the upper respiratory tract. Contrarily, regular moderate-intensity activities are beneficial having immune-enhancing effects and, as stated above, throughout the reductions of inflammation, increased immune cells turnover, enhanced immune surveillance, and improvement of psychological stress status (63).
Effect of Acute Exercise
An exercise bout increases both the absolute and relative leukocyte counts. Transient leucocytosis takes place already after brief (minutes) dynamic exercise and is more sustained in the case of prolonged endurance exercise (80) and returns to pre-exercise levels within 6–24 h (63). Neutrophils and lymphocytes are mainly involved in this response while a smaller contribute is given by monocytes. The response, however, differs for these cells: during the early phase of recovery (30–60 min after exercise), the neutrophilia is associated with lymphocytopenia that can be particularly pronounced until clinically relevant low levels (<1.0·109/L, which means −30 to −50% compared to pre-exercise values) and can last up to 6 h (80). Other features also characterize this response. Exercise tends to mobilize cytotoxic cells (e.g., NK-cells, and CD8+ and γδ T-cells) (81) and non-lymphocyte effector cells (e.g., CD16+ monocytes and CD16- neutrophils) (82, 83). The exercise-dependent mobilization mainly involves those cells with a higher migration potential, e.g., leukocytes expressing high levels of integrins and intracellular adhesion molecules (84) and a wide range of chemokine receptors (85). Finally, in these leukocytes the expression of adrenoreceptors (β2-ARs) and glucocorticoid receptors is upregulated, and are therefore they are highly responsive to catecholamines and cortisol (81, 86, 87), this indicates that leukocyte trafficking between the blood and tissues is strongly influenced by both the sympathetic branch of the nervous system and the HPA axis activation.
Other than being dislodged from liver, lung, and spleen endothelia due to the exercise-induced increased blood pressure- and cardiac output-mediated shear stress (80), leucocytes come from lymph nodes, intestines, bone marrow, thymus, and SKM that contain large numbers of white cells. The contribution of primary (i.e., bone marrow, thymus) and some secondary (i.e., lymph nodes), lymphoid organs to the initial exercise-induced leucocytosis is limited, since their content in mature/differentiated cells. The bone marrow likely sustains neutrophilia during recovery from prolonged exercise, while lymph nodes and thymus mainly sustain the restoration of the blood lymphocyte count following the transient lymphocytopenia (63).
About innate immunity, submaximal exercises enhance neutrophil chemotaxis (88), phagocytosis (89), and spontaneous degranulation (90). Also the neutrophil oxidative burst is affected by the exercise although in an intensity-and duration-dependent fashion: cycling at 50 and 80% of VO2max have enhancing and impairing effects, respectively; moreover, during recovery from moderate-intensity exercise the oxidative burst is enhanced while it is impaired following exhaustive and prolonged activities (91, 92). Also NK-cell cytotoxicity is quickly induced by exercise but this response is followed by a delayed suppression during recovery but this likely mirroring changes in their number (93).
Adaptive immunity appears to be both augmented and inhibited depending on intensity, duration, and modality of exercise (63). In trained triathletes, following a half-Ironman race, an intradermal inoculation containing several recall antigens caused a reduced 48 h-delayed-type compared to both resting triathletes and moderately trained healthy men (94).
Effect of Chronic Exercise and Training
In general, high-intensity and high-volume training is thought to cause short- or long-term immune depressive states that can increase infection risk. Repeated bouts of strenuous exercise, performed without adequate recovery, result in a chronic state of impaired immunity (80). The decline in the count of circulating immune cells is associated with the increased susceptibility to infections: although athletes and healthy age-matched controls have comparable absolute and relative leukocyte counts, endurance athletes may experience reduced resting lymphocyte (runners) and NK-cell (swimmers, cyclists) counts (95–97). Functional declines in adaptive immunity associated with prolonged intensive training are related to unbalanced expression of pro- and anti-inflammatory cytokines and increased plasma levels of stress hormone (e.g., cortisol) (98).
Contrary to the strenuous, exhaustive exercise typically practiced by athletes, moderate-intensity training has beneficial effects on immune function (99). Moreover, exercise mode (i.e., aerobic, resistance, or combined) as well as the condition on which the intervention is addressed are main determinants of the immune effects. Indeed, moderate-intensity exercise training associates with a life-long improvement/maintenance of several aspects of the immune function (99, 100), such as increased response to vaccine (101, 102), viral infections (103, 104), and tumors (105–107), enhanced neutrophil phagocytic activity (108), T-cell proliferation (93, 109), NK-cell cytotoxic activity (93, 110), basal level of cytokines (111) and IL-2 production (112) and decreased number of senescent T-cells (113) and inflammatory response to bacterial challenge (114).
The enhanced adaptive immune response is also sustained by the improvement in systemic low-grade inflammation, driven by the improved inflammatory status of the AT, and hence in the associated adipokine profile (described above). Indeed, the downregulation of TLRs on the surface of monocytes (115) together with the direct exercise-induced M1 (pro-inflammatory)-to-M2 (anti-inflammatory) shift in macrophage phenotype, reduces the infiltration of the AT and, hence, its inflammatory status (116). The stress-related hormones released during exercise that have anti-inflammatory properties are responsible of a further stimulus: cortisol acting as an immunomodulatory and immunosuppressant compound and adrenaline that downregulates the expression of the inflammatory mediators IL-1β and TNF (73, 117). Exercise also decreases the content of cholesterol of the cell membranes that may improve T-cell receptor signaling and the translocation of MHC molecules for antigen presentation (118).
Immuno-Mediated Effects of Exercise and Training on Bone
Inflammasome Activation and Bone Metabolism
The innate immune function depends upon the recognition, by germline-encoded pattern-recognition receptors (PRRs), of PAMPs, derived from invading pathogens, and DAMPs, induced by endogenous stresses. PAMPs-/DAMPs-dependent activation of PRRs triggers the downstream signaling cascades and induces the expression of type I interferons (IFN-α, IFN-β) and pro-inflammatory cytokines (119). Inflammasomes are multimeric protein complexes assembling within the cytosol after sensing PAMPs or DAMPs (120, 121). They serve as scaffolds to recruit the inactive zymogen pro-caspase-1 that oligomerizes allowing the auto-proteolytic cleavage into active caspase-1. Active caspase-1 cleaves the precursor cytokines pro-IL-1β and pro-IL-18 generating the biologically active forms (122–124). Furthermore, when activated, caspase-1 can activate a series of intracellular events that lead to a form of cell-death mediated by inflammation which is known with the term of pyroptosis (125, 126). Several PRRs families are involved in inflammasomes activation, in both mice and humans, including the nucleotide-binding domain, leucine-rich repeat containing proteins (NLRs, NOD-like receptors), and absent in melanoma 2-like receptors (ALRs, AIM2-like receptors) (127). Following stimulation, the relevant NLR or ALR oligomerizes and becomes a caspase-1-activating scaffold. Inflammasomes have been linked to several auto-inflammatory and autoimmune diseases, neurodegenerative diseases (e.g., multiple sclerosis, Alzheimer's disease, Parkinson's disease), and metabolic disorders [atherosclerosis, type-2 diabetes (T2DM), obesity] (126). Inflammasomes play either causative or contributing roles in inflammatory diseases onset, and also increase the severity of the condition in response to host-derived factors (119). Many PRRs can sense metabolic signals, such as free fatty acids (FFAs) and ceramides (CERs), whose blood concentrations increase during aging. These signals activate critical inflammatory signaling cascade pathways, such as IκBα kinase/nuclear factor-κB (IKK/NF-κB), endoplasmic reticulum (ER) stress-induced unfolded protein response (UPR), and NLRP3 inflammasome. Notably, other than in immune cells, PRRs are expressed in several metabolically active tissues (liver, SKM, AT) where they prime the inflammatory cascades (128).
Besides the established role of inflammation (and age-associated low-grade inflammation) in the pathogenesis of osteoporosis (129–131), very recent findings have linked it to inflammasomes activation. PA counteracts all the molecular mechanisms involved in inflammatory signaling cascades and inflammasome complexes activation (128). In post-menopausal osteoporosis, IL-18 blood levels are increased while those of its antagonist, IL-18BP, are decreased. According to Mansoori et al., IL-18BP enhanced murine osteoblast differentiation and inhibits the activation of NLRP3 inflammasome and caspase-1, in vitro, and improved the metabolic and bone statuses in ovariectomized rats (a rodent model of post-menopausal OP) (132). Further evidences derived from the association of NLRP3 mutations with arthropathy and OP (133) and the SIRT1-dependent inhibition of osteogenic differentiation and enhancement adipogenic differentiation, in mesenchymal stem cells (MSC), following lipopolysaccharide (LPS)-induced NLRP3 inflammasome activation (134). It is known that bone matrix organic and inorganic components, released during high-rate bone turnover (e.g., chronic low-grade inflammation, estrogen deficiency, primary hyperparathyroidism), promote osteoclastogenesis. This process, however, was importantly reduced in Nlrp3−/− cells and mice and pharmacologic inhibition of bone resorption (with bisphosphonates, e.g., zoledronic acid) attenuated inflammasome activation in vivo. These evidences suggest that the DAMPs-NLRP3 inflammasome axis may represent a novel mechanism supporting bone resorption (135).
PA effectively counteracts all the molecular mechanisms involved in the inflammatory signaling cascades (e.g., IKK/NF-κB, ER-UPR, inflammasomes) although the evidences about the effects of exercise on inflammasome are currently limited to NLRP3 activation and only in mouse models and in obesity. According to Ringseis, in obese mice both endurance (treadmill, 80% VO2max, 10 weeks) and resistance exercise (intermittent vertical holding, 10 weeks) decrease NLRP3 mRNA in AT and IL-18 in plasma (128). A number of human studies demonstrated that PA reduces plasma IL-18 levels providing the evidence for the exercise-dependent NLRP3 pathway inhibition: 12-week aerobic interval training in males and females with metabolic syndrome; 6-month aerobic training (50–85% VO2max) in overweight T2DM individuals; 8-week high-intensity training on a rowing ergometer (≥70% VO2max) in obese. In diet-induced obese rats, exercise strongly reverses TLR4 signaling and IKKβ phosphorylation in AT, SKM, and liver, suggesting a priming role for the exercise-induced inhibition of NLRP3 inflammasome. Key primers of NLRP3 activation are saturated FFA and CERs, whose circulating levels are increased in aging and metabolic dysfunctions while are decreased in response to exercise in obese animals and humans. Exercise may also reduce ER stress that primes NLRP3 activation via ROS production and NF-κB activation (128).
Extra-Immune Systemic Inflammation and Bone Metabolism
Beside the above described prototypic adipo-myokine IL-6 with its anti-inflammatory actions (induction of IL-10 and IL-1ra and inhibition of IL-1β and TNFα), SKM and AT secrete a plethora of active molecules that act in autocrine, paracrine, and hormone-like fashion the blood concentrations of many of which have been associated with several metabolic, immune-related, and age-related pathological conditions (3). It is well-known that the post-exercise rise of circulating IL-6 is supported by SKM (136) but contrarily, chronically slightly elevated blood IL-6 are found in metabolic conditions such as metabolic syndrome and insulin resistance, obesity, and T2DM (137–139). In these cases, the main source of IL-6 is represented by the visceral AT, the liver, and the activated immune cells upon NF-κB signaling (140). Indeed, in both overweight and lean males the contribution of the AT to the circulating amount of IL-6 is mainly in the post-exercise phase (141).
In bone, IL-6 stimulates bone resorption by enhancing osteoclastogenesis/osteoclast differentiation throughout the induction of RANKL expression (142, 143) and by inducing prostaglandin E2 (PGE2) expression in osteoblasts (144–146). Bones from IL-6 transgenic mice developed osteoporosis in association with an increased number of osteoclasts and decreased osteoblasts while, on the contrary, IL-6 knock out (KO) improved the arthritis phenotype, associated with a reduced osteoclast recruitment at the erosion sites, in a murine model of arthritis (147, 148). Ovariectomy in rats, model of postmenopausal osteoporosis, decreased trabecular bone volume (TBV) and impaired hormone and inflammatory profile (decreased estradiol and calcitonin and increased bone-derived IL-1β, IL-6, and cyclooxygenase-2). Contrarily, the treadmill-exercised counterparts displayed an overall improved phenotype (149). Parallel, postmenopausal women have BMD and muscle strength correlated with soluble IL-6 (150).
Low-impact high-intensity interval training (HIIT) acutely increased bone alkaline phosphatase activity (BAP) and the expression of OPG, RANKL, and pro-inflammatory cytokines (IL-1α, IL-1β, IL-6, TNFα) while decreased bone resorption [N-terminal cross-linked telopeptide of type I collagen (NTx)] (151). In obese subjects (n = 173), the degree of obesity and BMD were related to IL-6 levels (in males), osteocalcin (in females), C-reactive protein (CRP), and leptin indicating that adiposity and systemic inflammation are associated with low BMD (152). In obese, a 32-week combined loading training improved muscle strength and BMD at various sites along with an improved metabolic/inflammatory status (decreased CRP, interferon (IFN)-γ, IL-6) (153).
The leukemia inhibitory factor (LIF), a myokine belonging to the IL-6 superfamily, stimulates the proliferation of satellite cell which is essential in post-injury muscle regeneration (e.g., exercise-induced muscle damage, EIMD) and SKM hypertrophy (154). It was induced, at least in term of mRNA, by acute aerobic and resistance exercises (155). In bone, LIF stimulates bone turnover, osteoblast proliferation and bone matrix deposition, and prostaglandin-induced bone resorption depending on the differentiation stage of the target cells: enhancement of differentiation in progenitors, inhibition of function (e.g., mineralization) in mature osteoblasts (156). Exercise-induced LIF acts on periosteal osteoblast in order to modulate their activity (157).
IL-7 is essential for T-cell and B-cell development (136) but it is also expressed by the contracting muscles where it acts paracrinally to induce migration in satellite cells (158). As such, IL-7 mediates the oestrogens deficiency-induced bone loss: it induces RANKL and TNFα expression in T-cells (159) and, hence, it activates mature osteoclast and stimulates the progenitors differentiation (160) as also observed in vivo in mice following systemic administration (161). It also promotes survival and differentiation of dendritic cells, B220+ subset, into osteoclasts (162). However, despite their beneficial effects on bone, exercise and training seem to increase the SKM expression of IL-7. Indeed, plasma IL-7 has been found to be induced, in elite female soccer players, after a 90-min soccer games (163), 30-min post resistance exercise but not after 12 weeks of resistance training (164), while the mRNA expression level was induced in SKM by 11-week long strength training (158). Therefore, as IL-6, the biological significance of SKM- and IS-derived IL-7 resides into basal-to-peak ratio (3).
In addition, IL-15 is considered an exercise-related myokine although the effect of exercise on its expression and secretion by the SKM are not well-understood. Current evidences account for a role of IL-15 in the first phase of adaptation to exercise since it has been found upregulated (in muscle and in blood) in inactive/normally active subjects following an acute bout of exercise but not following the completion of a training program (165). Indeed, IL-15 mRNA expression in SKM was not affected by a 3-h treadmill run in trained subjects (marathon runners) (166) as well as in healthy physically active men after 3-h cycling (167). However, despite no changes in plasma levels after 12-week endurance training, the protein content in SKM was increased (167). Increased blood IL-15 was found after a 30-min run on a treadmill at 70% of maximum heart rate in untrained healthy young men (168), acute resistance exercise in young healthy inactive subjects but not after 10-week chronic training (169) as well as SKM mRNA, in healthy normally active men, after an heavy bout resistance exercise (170). IL-15 is a powerful inducer of TNF-α expression, and hence of RANKL, in osteoblasts and stromal cells, resulting in enhanced osteoclastogenesis. Furthermore, in rat bone marrow cultures, it stimulates pre-osteoclasts differentiation independently from TNF-α (171). IL-15 acts synergistically with RANKL in osteoclastogenesis by activating ERK (172). These data suggest that IL-15 positively regulate osteoclastogenesis (173).
Myostatin, is a member of the transforming growth factor beta (TGF-β) superfamily [also known as growth-differentiation factor (GDF)-8], negatively regulates SKM hypertrophy and hyperplasia (174) and it may cause SKM mass loss during aging (sarcopenia) and with metabolic and inflammatory conditions (175, 176). As such it is negatively regulated by PA (acute endurance (177), acute resistance (178–180), chronic (6-month) aerobic training in overweight and obese men (181). The recovery strategy seems to have in determining the net effect of exercise (182). Different factors act as antagonist of myostatin and among them follistatin (FST) (183), follistatin-like 1 (FSTL1) (184), and decorin (185). Acute resistance exercise did not affect follistatin mRNA expression in SKM from lean young and old men (180) while it had enhancing effects in postmenopausal women (179). Decorin mRNA expression in SKM was induced by chronic combined strength and endurance training (186) but also by acute endurance exercise (187) and, in terms of plasma levels, by acute resistance exercise (186). FSTL1 plasma levels were increased in young healthy men after acute endurance exercise (188) while chronic strength training induced mRNA expression in SKM (189). Myostatin has direct effects on osteoclastogenesis (190). Indeed, osteoclasts number on trabecular bone surfaces is increased in unloading conditions in both wild-type and myostatin KO mice (191), however myostatin deficiency suppresses subperiosteal resorption with unloading, suggesting that at least a part of the effects myostatin on osteoclasts are localized to the muscle-bone interface (192). Wnt-independent bone resorption consequent to strong endurance effort (e.g., ultramarathon) has been associated to increased myostatin and decreased FST (193). FST and its related factors (FSTL1, FSTL3, decorin) are induced by exercise and their importance in bone and muscle development is evidenced in the severe phenotypes consequent to their mutation. Fstl3−/− mice experienced frequent fractures together with the loss of mechanosensitivity which led to the loss of bone gain and Sost response to exercise. Importantly, a decreased FSTL3 expression is associated with aging (176). FST is induced by hyper-gravity and inhibited by microgravity (194).
Although its indisputable role as a myokine (195), brain-derived neurotrophic factor (BDNF) is mostly expressed in the brain, as is its receptor (196, 197), indeed 70–80% of the circulating protein origins from brain (9). BDNF plasma levels are raised by acute endurance and high-intensity, but not low-intensity, exercise in both males and females (195, 198–200) and also in response to chronic endurance training in young adult males (201). On the other hand, 10-week chronic resistance exercise did not affect BDNF serum levels compared to physically inactive (202). Interestingly, BDNF plasma concentrations were found to be lower in young compared to middle-aged women. After high-intensity resistance exercise BDNF follows a biphasic response featured by a decrease 1 h and an increase 24 h post exercise (203). On the contrary, 12-week moderate aerobic training (nordic walking) increased circulating BDNF in middle-aged women in association with improvement in cognitive functions (204). The BDNF receptor, TrkB, is expressed by active trabecular osteoblasts, growth plate hypertrophic chondrocytes during intramembranous ossification, and in osteoblasts and endothelial cells in fracture healing site (205, 206). Mice BDNF conditional KO in brain, beside the metabolic phenotype (hyperphagia, increased abdominal AT, obesity, leptin resistance), displayed increased femur length, high BMD and BMC (207).
The chemokine MCP-1 (also known as CCL2) is the primary ligand for the CCR2 receptor, which is normally expressed on monocyte/macrophages. As such it is a key regulator of osteoclastogenesis and has a pivotal role in inflammation and tumor-induced osteolysis (208). It is also an adipo-myokine acting as chemoattractant for monocytes and T lymphocytes (209). MCP-1 expression in SKM is strongly induced by acute and chronic resistance exercise, in terms of both mRNA and protein (164, 210), in healthy young and elderly (211). Also acute endurance activities (70% VO2max for 40 min, high-intensity treadmill running) increase mRNA expression in SKM in lean, obese, and T2DM (164), and circulating protein in well-trained male runners subjects (212). Therefore, expression of MCP-1 seems to be influenced by the intensity of the exercise rather than the kind of activity. Moreover, MCP-1 is affected by acute exercise while its response is not affected by training in both young and elderly healthy subjects, males and females, regardless the type of activity (211, 213).
The pro-inflammatory TNFα, the prototypic early mediator of local inflammation and initiator of the acute phase response, is expressed, other than from the immune cells, also by AT and SKM. In the AT, its expression is related to the fat mass (140) while in the SKM, the mRNA expression is inhibited following training (endurance and resistance) (214, 215). The muscle expression is neither affected by the metabolic status, being comparable between lean, overweight, and T2DM, nor by acute exercise (141, 216, 217). Circulating TNFα concentrations, however, are inversely related with the amount of PA (218) but, contrarily to moderate-intensity exercise, high-intensive training causes a temporary rise in systemic inflammation (e.g., TNFα) during recovery in response to muscle damage (10) as a propaedeutic step needed for the following regeneration (219). TNF-α is a powerful stimulus for bone resorption and is strikingly implicated in inflammatory bone diseases (220). By activating NF-κB signaling, it induces osteoclast differentiation form progenitors in the presence of M-CSF and in the absence of RANKL (221) and it also enhance RANKL sensitivity in osteoclast progenitors by inducing the expression of RANK (222). TNF-α can accelerate RANKL-dependent osteoclastogenesis by activating NF-κB and AP-1 throughout TRAF2/5 and MAPKs cascades (223) and RANKL enhances TNFα-induced osteoclastogenesis via TRAF6-independent signaling (224). TNF-α stimulates osteoclastogenesis also indirectly by inducing the expression of M-CSF and RANKL stromal cells, osteoblasts, and activated T cells (161, 225).
Visfatin/NAMP (alternatively known as pre-B cell colony-enhancing factor, PEBF) (226) is synthesized as both an intracellular form, acting as nicotinamide phosphoribosyltransferase (eNAMPT) in NAD biosynthesis, and extracellular one, visfatin, mainly secreted by the visceral AT, acting as an insulin-mimetic, pro-inflammatory/immuno-modulating adipokine (227). Its circulating levels are associated with obesity/fat mass, insulin resistance (228), and the energy-bone crosstalk (57). NAMPT expression was two-fold higher in SKM of athletes compared to that found in SKM from sedentary obese, non-obese, and T2DM subjects (229). However, in obese (>30 kg/m2) men no difference circulating visfatin concentrations were found between subjects with high and low cardiorespiratory fitness (230). NAMPT mRNA expression and protein content in the SKM of the exercising leg was doubled compared to the non-exercising limb, of non-obese sedentary individuals after 3 weeks of one-legged endurance exercise training endurance training (231). Acute exercise, instead, affects eNAMPT/visfatin in an intensity-dependent manner: 3-h cycling at 60% VO2max had no effect (232) while an acute bout of high-intensity running-based anaerobic sprint exercise had an inductive effect (233). In vitro visfatin stimulates osteoblast proliferation through the activation of insulin-receptor (234), induces osteoblastic differentiation in association with inhibition of OC expression (235), inhibits osteoclastogenesis throughout the suppression of RANK and NF-AT pathways (236), stimulates adipogenesis in mesenchymal stem cells (MSC) throughout the induction of PPAR-γ (237). Plasma visfatin did not differ between less than moderately trained subjects and experienced ultramarathon runners but, in this latter group it was two-fold induced after a mountain ultramarathon (39). Similarly, despite the worse metabolic profile, sedentary subjects had comparable serum visfatin concentration than professional rowers (238). Patients with metabolic syndrome had higher plasma visfatin than their age-matched counterparts which was correlated with lumbar spine BMD in men (239). Furthermore, in different women population (Chinese, Iranian) visfatin independently predicted BMD (240, 241) although it was not correlated with either BMD or BMC in a cohort of adolescent female athletes, participating in different sports (242).
Adiponectin is a prototypic adipokines that increases fatty acid oxidation and glucose uptake in SKM while inhibiting hepatic gluconeogenesis (243) and its circulating levels are inversely related to BMI and adiposity (244). It has anti-inflammatory effects since it inhibits expression and secretion of TNFα in macrophages and induces the expression of IL-10 (245, 246). Although it is considered a classical adipokine, adiponectin is also expressed by the SKM (247, 248). Plasma adiponectin levels are decreased in obesity and insulin resistance but the SKM expression of its receptors (AdipoR1 and R2) is increased (249). Acute exercise has no clear effects on circulating adiponectin with researches depicting inductive (250, 251), depressing (141) or no effects (252–256), regardless the metabolic state of the subjects. Contrarily, in healthy lean and overweight and obese subjects with impaired glucose tolerance, endurance training increased plasma adiponectin levels and induced the expression of AdipoR1/R2 in SKM (257). In highly-trained professional cyclists, plasma adiponectin was increased during a 3-week stage race (38). Fatouros and co-workers, instead, reported that high-intensity, but not moderate-intensity, either acute or chronic resistance exercise increased plasma adiponectin levels (258, 259), suggest that exercise intensity is a key determinant of the regulation of adiponectin release in blood. Among the adipokines, adiponectin is the most closely associated with BMD (negative) and fracture risk (positively) regardless gender and menopausal status (260). Hence, it exerts negative effects on bone mass although it is inversely associated with fat mass, promotes insulin sensitivity, and fat oxidation. However, bone osteocalcin induces adiponectin expression in adipocytes that, in turn, improves glucose tolerance (261, 262). AdipoRs and adiponectin are expressed by osteoblasts and osteoclasts (263, 264). Current evidences suggest that adiponectin acts autocrinally/paracrinally to simulate osteoblast function, while systemic adiponectin has inhibitory effects on osteoblasts activity while enhances osteoclastogenesis (265, 266).
Leptin is another adipokine involved in the regulation of energy homeostasis (267) it is an adiposity signal that suppresses appetite. As for adiponectin, also leptin is expressed in SKM (268) but the relative contribution of AT and SKM to circulating leptin has been not fully understood (165). Leptin and leptin receptor KO mice (ob−/− and db−/−) which are obese have also higher bone mass and intracerebroventricular infusions improved the metabolic status and reverted the high bone mass phenotype (269). Leptin effects on bone formation are mediated by the sympathetic nervous system (SNS), independently from AT, indeed by high bone mass phenotype obtained following the inhibition adrenergic signaling cannot corrected by intracerebroventricular infusion of leptin (270, 271). In strenuous exercise-induced hypogonadal women, leptin induced oestrogens that partially improved the bone phenotype (272). Circulating leptin was decreased in highly trained professional cyclists during a 3-week stage race (38) and in experienced ultramarathon runners after a mountain ultramarathon (39). However, in these runners resting levels of leptin were significantly lower than their less than moderately trained counterparts (39). Interestingly, in competing professional cyclists the decrease in leptin was associated with increased bone resorption and GluOC-to-GlaOC ratio (38). Similarly, 8-week aerobic training decreased fat mass and leptin, improved insulin sensitivity, and increased both total OC and GluOC in obese young males experienced (273). On the contrary, in competing ultramarathon runners, a comparable trend in leptin was associated with a reduced GluOC-to-GlaOC ratio (39). Finally, soluble leptin, insulin, and OC were increased by bed rest independently from resistive vibration exercises (274). These data indicate that load may regulate leptin release. Leptin mRNA expression in AT after acute endurance exercise were found either unaffected (275) or decreased in lean and overweight subjects (141). Several studies have shown a delayed (24–48 h post exercise) reduction of circulating leptin levels in healthy active men (276–279). Taken together, the current evidences suggest that exercise training decreases plasma leptin levels, while is ineffective on mRNA expression in AT (165). Weight loss in elderly obese accelerated bone turnover but PA can attenuates BMD decrease and stimulated a greater decrease in circulating leptin (280). The detrimental bony effects of leptin also depends upon its pro-inflammatory action: stimulation of neutrophil chemotaxis and phagocytic function, induction of pro-inflammatory cytokines in monocytes, and induction of T helper (Th)-1 cytokines (246). These data suggest that the exercise-dependent beneficial effects on bone may be also mediated by the exercise-dependent reduction in circulating leptin (3).
Resistin, an inflammatory marker, is positively associated with fat mass, waist circumference, and obesity-related diseases, and it causes oxidative stress and nitric oxide production downregulating, thus, determining endothelial dysfunction (281, 282). Both circulating levels and AT mRNA expression are not affected by acute endurance training in overweight and lean males (141, 253). However, the baseline training status of the subjects seems to affect the resistance exercise-dependent response to exercise with regularly training subjects experiencing a decrease over 6 months (281). Also osteoclasts, osteoblasts, and bone marrow-derived MSC express resistin, and in vitro it may stimulate both osteoclastogenesis and osteoblastogenesis (234, 235). Similarly to visfatin, resistin induces PPAR-γ expression in MSC and, thus, the adipogenic differentiation (237). It has been negatively associated with BMD (240, 283), although not definitively (284–286), and in postmenopause its circulating levels are doubled compared to premenopause (287). Resistin correlated positively with previous osteoporotic fractures and much more in the presence of diabetes (288).
Irisin is a newly discovered myokine released into the circulation following the cleavage, mediated by unknown proteases, of the transmembrane glycoprotein fibronectin type III domain containing 5 (FNDC5). In target cells, mainly white adipocytes, it induces the expression of the mitochondrial uncoupling protein 1 (Ucp1) that uncouples the respiratory chain from the oxidative phosphorylation: the energy derived from the oxidation of energy substrates (e.g., carbohydrates, fatty acids), and generated by the passage along electrochemical gradient of the electrons, is released as heat. This process normally occurs in the thermogenic BAT rather than in fat-storing WAT. Irisin induces a metabolic shift in the white adipocytes, namely browning, making them expressing an intermediate beige phenotype (289). Irisin is induced by exercise and its circulating levels are higher in trained, males and young subjects than in sedentary, females, and elderly, as a function of the muscle mass and the muscle activation level (290). Along with animal studies (291), researches in human have highlighted that high-intensity acute exercise (292), endurance training (293, 294), cold exposure (295), lifestyle changes as in obese children (296) and pregnant women (297) all increase blood irisin in association with an improved metabolic status. However, these results have not been always replicated and there are still doubts about its physiology (182, 291). Such discrepancies could be, at least partially, imputed to the methodological issues emerged about some commercially available immunoassays when compared to the gold standard mass spectrometry-based method (298). Irisin is also expressed by the AT and respond to PA as for its muscle counterpart (299, 300). Other than being directly induced by exercise, irisin expression is regulated by several exercise-modulated factors such as BDNF, myostatin, follistatin, TGFβ, FFAs, cytokines, betatrophin (291). Irisin could be a link between exercise and BDNF expression (289). Indeed, 30-day voluntary free running-wheel induced FNDC5 expression in mice hippocampus, which in turn into and increased expression of BDNF (301). This correlation might support the neuroprotective effects of exercise. Still, data directly evaluating the impact of exercise (especially in human), its time duration and intensity on irisin, BDNF, and cognitive function are unclear. By affecting adiposity, irisin can improve the inflammatory status and, recently, a direct relationship between irisin concentrations and inflammatory markers in metabolic syndrome has been described (302). Irisin is involved in the SKM-bone endocrine connection and it its involvement in bone mass gain in muscle disease-associated osteopenia has been proposed (303). Irisin levels have been, indeed, associated with osteoporotic fractures in postmenopausal women (304) with an inverse correlation with fat mass and PA status (305). In vitro, irisin promotes osteoblast differentiation while in vivo it induces osteoblast proliferation and differentiation, inhibits osteoclast activity, increases cortical BMD (306), and prevents muscle atrophy-induced bone loss (307).
Adaptive Immunity Activation and Bone Metabolism
Besides the role of innate immunity, several examples exist about the interplay between bone and IS. First of all, as already stated, osteoclasts are monocytic/macrophagic origin and M-CSF, a key cytokine for this lineage is also important in osteoclast differentiation (308) and antigen-presenting cells, such as dendritic cells, retain the capability to transdifferentiate into bone-resorbing osteoclasts (309, 310). Moreover, several soluble mediators regulate osteoblasts and osteoclasts differentiation and activity (311). Interestingly, a subset of osteogenic cells, called N-cadherin-positive spindle-shaped osteoblasts are an integral part of the hematopoietic stem cell (HSC) niche and solve a key role in the maintenance of the HSC pool that gives rise to all blood and immune cells (312).
When activate, under inflammatory conditions (e.g., autoimmune diseases, inflammatory bowel diseases, periodontal infections), T- and B-lymphocytes secrete RANKL and TNFα that stimulate osteoclast differentiation and function and, therefore, bone resorption (313). However, under physiological conditions, B-cells represent an important source of the osteoclast inhibitor OPG. For instance, human tonsil B-cells secrete OPG and the activation of the CD40 costimulatory pathway on these cells, in vitro, further induced OPG expression (314); parallel, bone marrow B-cells contribute up to 64% of the total OPG, in mice (315). Consequently, B-cells KO mice experienced increased bone resorption rate, reduced BMD and bone mass, in association with low circulating OPG; the restoration of the B-cells pool into young B-cell prevented the bone phenotype. The ligand of CD40, CD40L, is mainly expressed by activated T-cells and the deletion of either CD40 or CD40L on T-cells caused a powerful inhibition of OPG expression in B-cells and bone loss (315). It is, thus, suggested that under physiological conditions B-cells, regulated by the T-cells co-stimulatory action, protect the skeleton by secreting OPG, while under inflammatory conditions B- and T-cells negatively affect bone metabolism by secreting RANKL and inflammatory cytokines (316).
Postmenopause-related estrogen deficiency gives an explicative example. Indeed, oestrogens mediate powerful anti-inflammatory effects and loss of estrogen causes significant proliferation of T- and B-lymphocytes (129, 317). This was demonstrated by the fact that while ovariectomized wild-type rats experienced bone loss, T-cell-deficient null mice were protected by osteoclastic bone resorption (318). RANKL is secreted by activated T but not in conditions of estrogen deficiency, in mice. Contrary, in this condition, circulating and tissue TNFα is raised (318, 319) and TNFα and TNFRI (p55) KO in mice prevented ovariectomy-induced bone loss (318). In agreement to this model, estrogen loss causes the expansion of TNFα-secreting T-cells and TNFα sustains and amplifies the RANKL-induced osteoclast-mediated bone resorption (320). IL-7 expression in several tissues anticipates T-cells expansion (321); this cytokine increases the sensitivity of T-cells to otherwise tolerogenic antigens and, hence, decreases the antigen-dependent T-cell activation threshold (320). Consequently, the differentiation of T-cells into the different T helper subsets (i.e., Th1) leads to TNFα secretion along with IFNγ, that upregulates the expression of CIITA in macrophage, a transcription factor that in turn upregulates MHCII expression and, hence, antigen presentation to T-cells, further amplifying T-cells activation (317). Also the Th17 subset is induced in this process; these cells secrete IL-17A a pro-osteoclastogenic cytokine that induces expression RANKL in osteoblasts. IL-17 expression is raised by ovariectomy (322): treatment with anti-IL-17 antibodies (323) or the IL-17 gene deletion (324) improves bone loss in ovariectomized mice. The downregulation of TGFβ is another step in this process. TGFβ is expressed in response to oestrogens and has immunosuppressive effects by inducing regulatory T-cells (Tregs) that down-regulate T-cells activation (316).
Importantly, T-cell activation-dependent ovariectomy-induced bone loss depends upon antigen stimulation. Indeed, ovariectomy in mice with silenced antigen presentation due to a mutated T-cell receptor, only responsive to chicken ovalbumin: when no antigen is presented, mice were fully protected from ovariectomy-induced bone loss while, after exogenous administration of ovalbumin (i.e., the antigen) the bone response was retained (317). Evidence suggests that, in human, these antigens are derived from the gut microbiota (325, 326) since the gut permeability is regulated by oestrogens (326).
T-cells are critical in the mechanisms of action of parathyroid Hormone (PTH) in bone (327). Chronic elevated production of PTH (hyperparathyroidism, HPT) causes skeletal and extra-skeletal diseases: primary HPT (PHPT) is associated with increased bone turnover and osteopenia (328), while secondary HPT (SHPT) is involved in the pathogenesis of age-associated osteoporosis (329). Continuous PTH infusion mimics PHPT and SHPT, while intermittent administration has pro-anabolic effects on bone (330). T-cells express PTH-1R, the functional G protein coupled PTH receptor and they may contribute to the catabolic effect of PTH, in vivo (327, 331). Continuous PTH treatment at doses that mimic HPT failed to induce osteoclast formation, bone resorption, and cortical bone depletion in mice deficient for T-cells (331). On the contrary, intermittent PTH stimulates Wnt10b expression in bone marrow CD8+ T-cells and activate the canonical Wnt signaling in pre-osteoblasts (332).
Currently, there are no available study depicting the effects of the exercise on the relationship between adaptive IS and bone cell function. Hence, in order to improve the use of PA as a therapy for bone loss it is necessary to increase the knowledge in this field.
Practical Implications, Conclusions, and Perspectives
The complex net of physiological connections linking bone metabolism and IS branches in relationship with physical exercise is, now, only a little depicted. The great majority of the current knowledge concern the inflammation-mediated effects of PA while, only a few is known about the adaptive immunity-mediated effects.
Chronic PA is a powerful stimulus for bone and lifelong exercising is the most effective strategy to improve bone mass (in childhood and adolescence) and to keep bone health (in adulthood and older ages). However, there is no consensus on the best kind of PA to be prescribed at this purpose. There are evidences that sustain the effectiveness of load and impact and this is further improved when the activity is carried on in an intermittent way (54). Therefore, the role of loading is central in this discussion but, importantly, the direct effect of the applied forces onto the skeleton on the immune function are not known and this point must be developed in the next years.
Author Contributions
GL conception, drafting, and reviewing the article. EZ drafting and reviewing the article. GB drafting and reviewing.
Funding
This work has been funded by an unrestricted grant from the Italian Ministry of Health and grant from the National Science Centre (Poland) No. 2018/29/B/NZ7/02094.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McLaughlin M, Jacobs I. Exercise is medicine, but does it interfere with medicine? Exerc Sport Sci Rev. (2017) 45:127–35. doi: 10.1249/JES.0000000000000111
2. Banfi G, Lombardi G, Colombini A, Lippi G. Bone metabolism markers in sports medicine. Sports Med. (2010) 40:697–714. doi: 10.2165/11533090-000000000-00000
3. Lombardi G, Sanchis-Gomar F, Perego S, Sansoni V, Banfi G. Implications of exercise-induced adipo-myokines in bone metabolism. Endocrine (2016) 54:284–305. doi: 10.1007/s12020-015-0834-0
4. Daly RM, Duckham RL, Gianoudis J. Evidence for an interaction between exercise and nutrition for improving bone and muscle health. Curr Osteoporos Rep. (2014) 12:219–26. doi: 10.1007/s11914-014-0207-2
5. Terashima A, Takayanagi H. Overview of osteoimmunology. Calcif Tissue Int. (2018) 102:503–11. doi: 10.1007/s00223-018-0417-1
6. Miller PD, Bolognese MA, Lewiecki EM, McClung MR, Ding B, Austin M, et al. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone (2008) 43:222–9. doi: 10.1016/j.bone.2008.04.007
7. McClung MR, Lewiecki EM, Geller ML, Bolognese MA, Peacock M, Weinstein RL, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int. (2013) 24:227–35. doi: 10.1007/s00198-012-2052-4
8. Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al. Clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int. (2014) 25:2359–81. doi: 10.1007/s00198-014-2794-2
9. Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. (2004) 19:1059–66. doi: 10.1359/JBMR.040305
10. Pedersen BK, Akerstrom TC, Nielsen AR, Fischer CP. Role of myokines in exercise and metabolism. J Appl Physiol. (2007) 103:1093–8. doi: 10.1152/japplphysiol.00080.2007
11. Weigert C, Hoene M, Plomgaard P. Hepatokines-a novel group of exercise factors. Pflugers Arch. (2018). doi: 10.1007/s00424-018-2216-y. [Epub Ahead of Print].
12. Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. (2005) 98:1154–62. doi: 10.1152/japplphysiol.00164.2004
13. Rivera-Brown AM, Frontera WR. Principles of exercise physiology: responses to acute exercise and long-term adaptations to training. PM R (2012) 4:797–804. doi: 10.1016/j.pmrj.2012.10.007
14. Caspersen CJ, Powell KE, Christenson GM. Physical activity, exercise, and physical fitness: definitions and distinctions for health-related research. Public Health Rep. (1985) 100:126–31.
15. Hawley JA. Molecular responses to strength and endurance training: are they incompatible? Appl Physiol Nutr Metab. (2009) 34:355–61. doi: 10.1139/H09-023
16. Frost HM. Bone's mechanostat: a 2003 update. Anat Rec A Discov Mol Cell Evol Biol. (2003) 275:1081–101. doi: 10.1002/ar.a.10119
17. Price JS, Sugiyama T, Galea GL, Meakin LB, Sunters A, Lanyon LE. Role of endocrine and paracrine factors in the adaptation of bone to mechanical loading. Curr Osteoporos Rep. (2011) 9:76–82. doi: 10.1007/s11914-011-0050-7
18. Rubin CT. Skeletal strain and the functional significance of bone architecture. Calcif Tissue Int. (1984) 36(Suppl. 1):S11–8. doi: 10.1007/BF02406128
19. Kjaer M, Jorgensen NR, Heinemeier K, Magnusson SP. Exercise and regulation of bone and collagen tissue biology. Prog Mol Biol Transl Sci. (2015) 135:259–91. doi: 10.1016/bs.pmbts.2015.07.008
20. Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. (2004) 279:12001–4. doi: 10.1074/jbc.R300038200
21. Duncan RL, Hruska KA, Misler S. Parathyroid hormone activation of stretch-activated cation channels in osteosarcoma cells (UMR-106.01). FEBS Lett. (1992) 307:219–23. doi: 10.1016/0014-5793(92)80771-8
22. Coughlin TR, Voisin M, Schaffler MB, Niebur GL, McNamara LM. Primary cilia exist in a small fraction of cells in trabecular bone and marrow. Calcif Tissue Int. (2015) 96:65–72. doi: 10.1007/s00223-014-9928-6
23. Zayzafoon M, Gathings WE, McDonald JM. Modeled microgravity inhibits osteogenic differentiation of human mesenchymal stem cells and increases adipogenesis. Endocrinology (2004) 145:2421–32. doi: 10.1210/en.2003-1156
24. David V, Martin A, Lafage-Proust MH, Malaval L, Peyroche S, Jones DB, et al. Mechanical loading down-regulates peroxisome proliferator-activated receptor gamma in bone marrow stromal cells and favors osteoblastogenesis at the expense of adipogenesis. Endocrinology (2007) 148:2553–62. doi: 10.1210/en.2006-1704
25. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. (2011) 17:1235–41. doi: 10.1038/nm.2448
26. Lombardi G, Lanteri P, Colombini A, Mariotti M, Banfi G. Sclerostin concentrations in athletes: role of load and gender. J Biol Regul Homeost Agents (2012) 26:157–63.
27. Qin YX, Hu M. Mechanotransduction in musculoskeletal tissue regeneration: effects of fluid flow, loading, and cellular-molecular pathways. Biomed Res Int. (2014) 2014:863421. doi: 10.1155/2014/863421
28. Jorgensen NR, Henriksen Z, Brot C, Eriksen EF, Sorensen OH, Civitelli R, et al. Human osteoblastic cells propagate intercellular calcium signals by two different mechanisms. J Bone Miner Res. (2000) 15:1024–32. doi: 10.1359/jbmr.2000.15.6.1024
29. Jorgensen NR, Henriksen Z, Sorensen OH, Eriksen EF, Civitelli R, Steinberg TH. Intercellular calcium signaling occurs between human osteoblasts and osteoclasts and requires activation of osteoclast P2X7 receptors. J Biol Chem. (2002) 277:7574–80. doi: 10.1074/jbc.M104608200
30. McCarthy I. The physiology of bone blood flow: a review. J Bone Joint Surg Am. (2006) 88(Suppl. 3):4–9. doi: 10.2106/00004623-200611001-00002
31. Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. (2011) 6:121–45. doi: 10.1146/annurev-pathol-011110-130203
32. Heinonen I, Kemppainen J, Kaskinoro K, Langberg H, Knuuti J, Boushel R, et al. Bone blood flow and metabolism in humans: effect of muscular exercise and other physiological perturbations. J Bone Miner Res. (2013) 28:1068–74. doi: 10.1002/jbmr.1833
33. Heinonen I, Kalliokoski KK, Hannukainen JC, Duncker DJ, Nuutila P, Knuuti J. Organ-specific physiological responses to acute physical exercise and long-term training in humans. Physiology (Bethesda) (2014) 29:421–36. doi: 10.1152/physiol.00067.2013
34. Schuler G, Adams V, Goto Y. Role of exercise in the prevention of cardiovascular disease: results, mechanisms, and new perspectives. Eur Heart J. (2013) 34:1790–9. doi: 10.1093/eurheartj/eht111
35. Laufs U, Werner N, Link A, Endres M, Wassmann S, Jurgens K, et al. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation (2004) 109:220–6. doi: 10.1161/01.CIR.0000109141.48980.37
36. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell (2006) 124:407–21. doi: 10.1016/j.cell.2005.10.041
37. Ham J, Evans BA. An emerging role for adenosine and its receptors in bone homeostasis. Front Endocrinol. (2012) 3:113. doi: 10.3389/fendo.2012.00113
38. Lombardi G, Lanteri P, Graziani G, Colombini A, Banfi G, Corsetti R. Bone and energy metabolism parameters in professional cyclists during the Giro d'Italia 3-weeks stage race. PLoS ONE (2012) 7:e42077. doi: 10.1371/journal.pone.0042077
39. Sansoni V, Vernillo G, Perego S, Barbuti A, Merati G, Schena F, et al. Bone turnover response is linked to both acute and established metabolic changes in ultra-marathon runners. Endocrine (2017) 56:196–204. doi: 10.1007/s12020-016-1012-8
40. Burr DB, Robling AG, Turner CH. Effects of biomechanical stress on bones in animals. Bone (2002) 30:781–6. doi: 10.1016/S8756-3282(02)00707-X
41. Rubin CT, Lanyon LE. Regulation of bone formation by applied dynamic loads. J Bone Joint Surg Am. (1984) 66:397–402. doi: 10.2106/00004623-198466030-00012
42. Rubin CT, Lanyon LE. Regulation of bone mass by mechanical strain magnitude. Calcif Tissue Int. (1985) 37:411–7. doi: 10.1007/BF02553711
43. Robling AG, Burr DB, Turner CH. Partitioning a daily mechanical stimulus into discrete loading bouts improves the osteogenic response to loading. J Bone Miner Res. (2000) 15:1596–602. doi: 10.1359/jbmr.2000.15.8.1596
44. Srinivasan S, Weimer DA, Agans SC, Bain SD, Gross TS. Low-magnitude mechanical loading becomes osteogenic when rest is inserted between each load cycle. J Bone Miner Res. (2002) 17:1613–20. doi: 10.1359/jbmr.2002.17.9.1613
45. Kennedy OD, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone (2014) 64:132–7. doi: 10.1016/j.bone.2014.03.049
46. Bareither ML, Troy KL, Grabiner MD. Bone mineral density of the proximal femur is not related to dynamic joint loading during locomotion in young women. Bone (2006) 38:125–9. doi: 10.1016/j.bone.2005.07.003
47. Dowthwaite JN, Rosenbaum PF, Scerpella TA. Mechanical loading during growth is associated with plane-specific differences in vertebral geometry: a cross-sectional analysis comparing artistic gymnasts vs. non-gymnasts. Bone (2011) 49:1046–54. doi: 10.1016/j.bone.2011.07.033
48. Kontulainen S, Sievanen H, Kannus P, Pasanen M, Vuori I. Effect of long-term impact-loading on mass, size, and estimated strength of humerus and radius of female racquet-sports players: a peripheral quantitative computed tomography study between young and old starters and controls. J Bone Miner Res. (2003) 18:352–9. doi: 10.1359/jbmr.2003.18.2.352
49. Heinonen A, Sievanen H, Kyrolainen H, Perttunen J, Kannus P. Mineral mass, size, and estimated mechanical strength of triple jumpers' lower limb. Bone (2001) 29:279–85. doi: 10.1016/S8756-3282(01)00574-9
50. Bhatia VA, Edwards WB, Johnson JE, Troy KL. Short-term bone formation is greatest within high strain regions of the human distal radius: a prospective pilot study. J Biomech Eng. (2015) 137. doi: 10.1115/1.4028847
51. Lambers FM, Schulte FA, Kuhn G, Webster DJ, Muller R. Mouse tail vertebrae adapt to cyclic mechanical loading by increasing bone formation rate and decreasing bone resorption rate as shown by time-lapsed in vivo imaging of dynamic bone morphometry. Bone (2011) 49:1340–50. doi: 10.1016/j.bone.2011.08.035
52. Nikander R, Sievanen H, Heinonen A, Kannus P. Femoral neck structure in adult female athletes subjected to different loading modalities. J Bone Miner Res. (2005) 20:520–8. doi: 10.1359/JBMR.041119
53. Howe TE, Shea B, Dawson LJ, Downie F, Murray A, Ross C, et al. Exercise for preventing and treating osteoporosis in postmenopausal women. Cochrane Database Syst Rev. (2011) CD000333. doi: 10.1002/14651858.CD000333.pub2
54. Xu J, Lombardi G, Jiao W, Banfi G. Effects of exercise on bone status in female subjects, from young girls to postmenopausal women: an overview of systematic reviews and meta-analyses. Sports Med. (2016) 46:1165–82. doi: 10.1007/s40279-016-0494-0
55. Motyl KJ, McCabe LR, Schwartz AV. Bone and glucose metabolism: a two-way street. Arch Biochem Biophys. (2010) 503:2–10. doi: 10.1016/j.abb.2010.07.030
56. Karsenty G, Ferron M. The contribution of bone to whole-organism physiology. Nature (2012) 481:314–20. doi: 10.1038/nature10763
57. Lombardi G, Perego S, Luzi L, Banfi G. A four-season molecule: osteocalcin. Updates in its physiological roles. Endocrine (2015) 48:394–404. doi: 10.1007/s12020-014-0401-0
58. Basso N, Heersche JN. Effects of hind limb unloading and reloading on nitric oxide synthase expression and apoptosis of osteocytes and chondrocytes. Bone (2006) 39:807–14. doi: 10.1016/j.bone.2006.04.014
60. Lee TQ, Shapiro TA, Bell DM. Biomechanical properties of human tibias in long-term spinal cord injury. J Rehabil Res Dev. (1997) 34:295–302.
61. Frey-Rindova P, de Bruin ED, Stussi E, Dambacher MA, Dietz V. Bone mineral density in upper and lower extremities during 12 months after spinal cord injury measured by peripheral quantitative computed tomography. Spinal Cord (2000) 38:26–32. doi: 10.1038/sj.sc.3100905
62. Lang T, LeBlanc A, Evans H, Lu Y, Genant H, Yu A. Cortical and trabecular bone mineral loss from the spine and hip in long-duration spaceflight. J Bone Miner Res. (2004) 19:1006–12. doi: 10.1359/JBMR.040307
63. Simpson RJ, Kunz H, Agha N, Graff R. Exercise and the regulation of immune functions. Prog Mol Biol Transl Sci. (2015) 135:355–80. doi: 10.1016/bs.pmbts.2015.08.001
64. Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature (2008) 454:463–9. doi: 10.1038/nature07206
65. Beavers KM, Brinkley TE, Nicklas BJ. Effect of exercise training on chronic inflammation. Clin Chim Acta (2010) 411:785–93. doi: 10.1016/j.cca.2010.02.069
66. Lemanne D, Cassileth B, Gubili J. The role of physical activity in cancer prevention, treatment, recovery, and survivorship. Oncology (Williston Park) (2013) 27:580–5.
67. Allen J, Sun Y, Woods JA. Exercise and the regulation of inflammatory responses. Prog Mol Biol Transl Sci. (2015) 135:337–54. doi: 10.1016/bs.pmbts.2015.07.003
68. Vitale JA, Lombardi G, Weydahl A, Banfi G. Biological rhythms, chronodisruption and chrono-enhancement: the role of physical activity as synchronizer in correcting steroids circadian rhythm in metabolic dysfunctions and cancer. Chronobiol Int. (2018) 35:1185–97. doi: 10.1080/07420528.2018.1475395
69. Suarez-Bregua P, Guerreiro PM, Rotllant J. Stress, glucocorticoids and bone: a review from mammals and fish. Front Endocrinol. (2018) 9:526. doi: 10.3389/fendo.2018.00526
70. Paredes S, Ribeiro L. Cortisol: the villain in metabolic syndrome? Rev Assoc Med Bras. (2014) 60:84–92. doi: 10.1590/1806-9282.60.01.017
71. Mifsud EJ, Tan AC, Jackson DC. TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front Immunol. (2014) 5:79. doi: 10.3389/fimmu.2014.00079
72. Gleeson M, McFarlin B, Flynn M. Exercise and Toll-like receptors. Exerc Immunol Rev. (2006) 12:34–53.
73. Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol. (2011) 11:607–15. doi: 10.1038/nri3041
74. Steensberg A, Fischer CP, Keller C, Moller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. (2003) 285:E433–7. doi: 10.1152/ajpendo.00074.2003
75. Pedersen BK. The diseasome of physical inactivity–and the role of myokines in muscle–fat cross talk. J Physiol. (2009) 587(Pt 23):5559–68. doi: 10.1113/jphysiol.2009.179515
76. Smith JK, Dykes R, Douglas JE, Krishnaswamy G, Berk S. Long-term exercise and atherogenic activity of blood mononuclear cells in persons at risk of developing ischemic heart disease. JAMA (1999) 281:1722–7. doi: 10.1001/jama.281.18.1722
77. Scheele C, Nielsen S, Pedersen BK. ROS and myokines promote muscle adaptation to exercise. Trends Endocrinol Metab. (2009) 20:95–9. doi: 10.1016/j.tem.2008.12.002
78. Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. (2013) 93:1–21. doi: 10.1152/physrev.00017.2012
79. Vieira VJ, Valentine RJ, Wilund KR, Antao N, Baynard T, Woods JA. Effects of exercise and low-fat diet on adipose tissue inflammation and metabolic complications in obese mice. Am J Physiol Endocrinol Metab. (2009) 296:E1164–71. doi: 10.1152/ajpendo.00054.2009
80. Walsh NP, Gleeson M, Shephard RJ, Gleeson M, Woods JA, Bishop NC, et al. Position statement. Part one: Immune function and exercise. Exerc Immunol Rev. (2011) 17:6–63.
81. Anane LH, Edwards KM, Burns VE, Drayson MT, Riddell NE, van Zanten JJ, et al. Mobilization of gammadelta T lymphocytes in response to psychological stress, exercise, and beta-agonist infusion. Brain Behav Immun. (2009) 23:823–9. doi: 10.1016/j.bbi.2009.03.003
82. Steppich B, Dayyani F, Gruber R, Lorenz R, Mack M, Ziegler-Heitbrock HW. Selective mobilization of CD14(+)CD16(+) monocytes by exercise. Am J Physiol Cell Physiol. (2000) 279:C578–86. doi: 10.1152/ajpcell.2000.279.3.C578
83. Peake J, Wilson G, Hordern M, Suzuki K, Yamaya K, Nosaka K, et al. Changes in neutrophil surface receptor expression, degranulation, and respiratory burst activity after moderate- and high-intensity exercise. J Appl Physiol. (2004) 97:612–8. doi: 10.1152/japplphysiol.01331.2003
84. Simpson RJ, Florida-James GD, Whyte GP, Guy K. The effects of intensive, moderate and downhill treadmill running on human blood lymphocytes expressing the adhesion/activation molecules CD54 (ICAM-1), CD18 (beta2 integrin) and CD53. Eur J Appl Physiol. (2006) 97:109–21. doi: 10.1007/s00421-006-0146-4
85. Bosch JA, Berntson GG, Cacioppo JT, Dhabhar FS, Marucha PT. Acute stress evokes selective mobilization of T cells that differ in chemokine receptor expression: a potential pathway linking immunologic reactivity to cardiovascular disease. Brain Behav Immun. (2003) 17:251–9. doi: 10.1016/S0889-1591(03)00054-0
86. Schedlowski M, Hosch W, Oberbeck R, Benschop RJ, Jacobs R, Raab HR, et al. Catecholamines modulate human NK cell circulation and function via spleen-independent beta 2-adrenergic mechanisms. J Immunol. (1996) 156:93–9.
87. Dimitrov S, Lange T, Born J. Selective mobilization of cytotoxic leukocytes by epinephrine. J Immunol. (2010) 184:503–11. doi: 10.4049/jimmunol.0902189
88. Ortega E. Neuroendocrine mediators in the modulation of phagocytosis by exercise: physiological implications. Exerc Immunol Rev. (2003) 9:70–93.
89. Nieman DC, Nehlsen-Cannarella SL, Fagoaga OR, Henson DA, Utter A, Davis JM, et al. Effects of mode and carbohydrate on the granulocyte and monocyte response to intensive, prolonged exercise. J Appl Physiol. (1998) 84:1252–9. doi: 10.1152/jappl.1998.84.4.1252
90. Bishop NC, Gleeson M, Nicholas CW, Ali A. Influence of carbohydrate supplementation on plasma cytokine and neutrophil degranulation responses to high intensity intermittent exercise. Int J Sport Nutr Exerc Metab. (2002) 12:145–56. doi: 10.1123/ijsnem.12.2.145
91. Pyne DB. Regulation of neutrophil function during exercise. Sports Med. (1994) 17:245–58. doi: 10.2165/00007256-199417040-00005
92. Suzuki K, Nakaji S, Yamada M, Liu Q, Kurakake S, Okamura N, et al. Impact of a competitive marathon race on systemic cytokine and neutrophil responses. Med Sci Sports Exerc. (2003) 35:348–55. doi: 10.1249/01.MSS.0000048861.57899.04
93. Nieman DC, Miller AR, Henson DA, Warren BJ, Gusewitch G, Johnson RL, et al. Effects of high- vs. moderate-intensity exercise on natural killer cell activity. Med Sci Sports Exerc. (1993) 25:1126–34. doi: 10.1249/00005768-199310000-00008
94. Bruunsgaard H, Hartkopp A, Mohr T, Konradsen H, Heron I, Mordhorst CH, et al. In vivo cell-mediated immunity and vaccination response following prolonged, intense exercise. Med Sci Sports Exerc. (1997) 29:1176–81. doi: 10.1097/00005768-199709000-00009
95. Baj Z, Kantorski J, Majewska E, Zeman K, Pokoca L, Fornalczyk E, et al. Immunological status of competitive cyclists before and after the training season. Int J Sports Med. (1994) 15:319–24. doi: 10.1055/s-2007-1021067
96. Gleeson M, McDonald WA, Pyne DB, Clancy RL, Cripps AW, Francis JL, et al. Immune status and respiratory illness for elite swimmers during a 12-week training cycle. Int J Sports Med. (2000) 21:302–7. doi: 10.1055/s-2000-313
97. Horn PL, Pyne DB, Hopkins WG, Barnes CJ. Lower white blood cell counts in elite athletes training for highly aerobic sports. Eur J Appl Physiol. (2010) 110:925–32. doi: 10.1007/s00421-010-1573-9
98. Hough J, Corney R, Kouris A, Gleeson M. Salivary cortisol and testosterone responses to high-intensity cycling before and after an 11-day intensified training period. J Sports Sci. (2013) 31:1614–23. doi: 10.1080/02640414.2013.792952
99. Simpson RJ, Lowder TW, Spielmann G, Bigley AB, LaVoy EC, Kunz H. Exercise and the aging immune system. Ageing Res Rev. (2012) 11:404–20. doi: 10.1016/j.arr.2012.03.003
100. Simpson RJ, Bosch JA. Special issue on exercise immunology: current perspectives on aging, health and extreme performance. Brain Behav Immun. (2014) 39:1–7. doi: 10.1016/j.bbi.2014.03.006
101. Kohut ML, Arntson BA, Lee W, Rozeboom K, Yoon KJ, Cunnick JE, et al. Moderate exercise improves antibody response to influenza immunization in older adults. Vaccine (2004) 22:2298–306. doi: 10.1016/j.vaccine.2003.11.023
102. Woods JA, Keylock KT, Lowder T, Vieira VJ, Zelkovich W, Dumich S, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc. (2009) 57:2183–91. doi: 10.1111/j.1532-5415.2009.02563.x
103. Perna FM, LaPerriere A, Klimas N, Ironson G, Perry A, Pavone J, et al. Cardiopulmonary and CD4 cell changes in response to exercise training in early symptomatic HIV infection. Med Sci Sports Exerc. (1999) 31:973–9. doi: 10.1097/00005768-199907000-00009
104. Lowder T, Padgett DA, Woods JA. Moderate exercise protects mice from death due to influenza virus. Brain Behav Immun. (2005) 19:377–80. doi: 10.1016/j.bbi.2005.04.002
105. Fairey AS, Courneya KS, Field CJ, Bell GJ, Jones LW, Mackey JR. Randomized controlled trial of exercise and blood immune function in postmenopausal breast cancer survivors. J Appl Physiol. (2005) 98:1534–40. doi: 10.1152/japplphysiol.00566.2004
106. Hojman P, Dethlefsen C, Brandt C, Hansen J, Pedersen L, Pedersen BK. Exercise-induced muscle-derived cytokines inhibit mammary cancer cell growth. Am J Physiol Endocrinol Metab. (2011) 301:E504–10. doi: 10.1152/ajpendo.00520.2010
107. Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer (2011) 11:886–95. doi: 10.1038/nrc3174
108. Yan H, Kuroiwa A, Tanaka H, Shindo M, Kiyonaga A, Nagayama A. Effect of moderate exercise on immune senescence in men. Eur J Appl Physiol. (2001) 86:105–11. doi: 10.1007/s004210100521
109. Shinkai S, Kohno H, Kimura K, Komura T, Asai H, Inai R, et al. Physical activity and immune senescence in men. Med Sci Sports Exerc. (1995) 27:1516–26. doi: 10.1249/00005768-199511000-00008
110. Woods JA, Ceddia MA, Wolters BW, Evans JK, Lu Q, McAuley E. Effects of 6 months of moderate aerobic exercise training on immune function in the elderly. Mech Ageing Dev. (1999) 109:1–19. doi: 10.1016/S0047-6374(99)00014-7
111. Pedersen BK, Bruunsgaard H. Possible beneficial role of exercise in modulating low-grade inflammation in the elderly. Scand J Med Sci Sports (2003) 13:56–62. doi: 10.1034/j.1600-0838.2003.20218.x
112. Drela N, Kozdron E, Szczypiorski P. Moderate exercise may attenuate some aspects of immunosenescence. BMC Geriatr. (2004) 4:8. doi: 10.1186/1471-2318-4-8
113. Spielmann G, McFarlin BK, O'Connor DP, Smith PJ, Pircher H, Simpson RJ. Aerobic fitness is associated with lower proportions of senescent blood T-cells in man. Brain Behav Immun. (2011) 25:1521–9. doi: 10.1016/j.bbi.2011.07.226
114. Phillips MD, Flynn MG, McFarlin BK, Stewart LK, Timmerman KL. Resistance training at eight-repetition maximum reduces the inflammatory milieu in elderly women. Med Sci Sports Exerc. (2010) 42:314–25. doi: 10.1249/MSS.0b013e3181b11ab7
115. McFarlin BK, Flynn MG, Phillips MD, Stewart LK, Timmerman KL. Chronic resistance exercise training improves natural killer cell activity in older women. J Gerontol A Biol Sci Med Sci. (2005) 60:1315–8. doi: 10.1093/gerona/60.10.1315
116. Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev. (2010) 16:105–18.
117. Mastorakos G, Pavlatou M, Diamanti-Kandarakis E, Chrousos GP. Exercise and the stress system. Hormones (Athens) (2005) 4:73–89.
118. Shaikh SR, Edidin M. Polyunsaturated fatty acids, membrane organization, T cells, and antigen presentation. Am J Clin Nutr. (2006) 84:1277–89. doi: 10.1093/ajcn/84.6.1277
119. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
120. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
121. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
122. Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G, Weidner J, et al. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1 beta precursor at two distinct sites and does not cleave 31-kDa IL-1 alpha. J Immunol. (1991) 147:2964–9.
123. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386:619–23. doi: 10.1038/386619a0
124. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science (1997) 275:206–9. doi: 10.1126/science.275.5297.206
125. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
126. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481:278–86. doi: 10.1038/nature10759
127. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
128. Ringseis R, Eder K, Mooren FC, Kruger K. Metabolic signals and innate immune activation in obesity and exercise. Exerc Immunol Rev. (2015) 21:58–68.
129. Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, et al. Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci USA. (1997) 94:9360–5. doi: 10.1073/pnas.94.17.9360
130. Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Foger-Samwald U, Ellinger I. Immunology of osteoporosis: a mini-review. Gerontology (2016) 62:128–37. doi: 10.1159/000431091
131. Fougere B, Boulanger E, Nourhashemi F, Guyonnet S, Cesari M. Chronic inflammation: accelerator of biological aging. J Gerontol A Biol Sci Med Sci. (2017) 72:1218–25. doi: 10.1093/gerona/glw240
132. Mansoori MN, Shukla P, Kakaji M, Tyagi AM, Srivastava K, Shukla M, et al. IL-18BP is decreased in osteoporotic women: prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci Rep. (2016) 6:33680. doi: 10.1038/srep33680
133. Snouwaert JN, Nguyen M, Repenning PW, Dye R, Livingston EW, Kovarova M, et al. An NLRP3 mutation causes arthropathy and osteoporosis in humanized mice. Cell Rep. (2016) 17:3077–88. doi: 10.1016/j.celrep.2016.11.052
134. Wang L, Chen K, Wan X, Wang F, Guo Z, Mo Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem Biophys Res Commun. (2017) 484:871–7. doi: 10.1016/j.bbrc.2017.02.007
135. Alippe Y, Wang C, Ricci B, Xiao J, Qu C, Zou W, et al. Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci Rep. (2017) 7:6630. doi: 10.1038/s41598-017-07014-0
136. Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. (2012) 8:457–65. doi: 10.1038/nrendo.2012.49
137. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. (2001) 280:E745–51. doi: 10.1152/ajpendo.2001.280.5.E745
138. Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes (2003) 52:1799–805. doi: 10.2337/diabetes.52.7.1799
139. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. (2004) 25:4–7. doi: 10.1016/j.it.2003.10.013
140. Eckardt K, Gorgens SW, Raschke S, Eckel J. Myokines in insulin resistance and type 2 diabetes. Diabetologia (2014) 57:1087–99. doi: 10.1007/s00125-014-3224-x
141. Hojbjerre L, Rosenzweig M, Dela F, Bruun JM, Stallknecht B. Acute exercise increases adipose tissue interstitial adiponectin concentration in healthy overweight and lean subjects. Eur J Endocrinol. (2007) 157:613–23. doi: 10.1530/EJE-07-0213
142. Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science (1992) 257:88–91. doi: 10.1126/science.1621100
143. Le Goff B, Blanchard F, Berthelot JM, Heymann D, Maugars Y. Role for interleukin-6 in structural joint damage and systemic bone loss in rheumatoid arthritis. Joint Bone Spine (2010) 77:201–5. doi: 10.1016/j.jbspin.2010.03.002
144. Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. (2002) 169:3353–62. doi: 10.4049/jimmunol.169.6.3353
145. Saidenberg-Kermanac'h N, Cohen-Solal M, Bessis N, De Vernejoul MC, Boissier MC. Role for osteoprotegerin in rheumatoid inflammation. Joint Bone Spine (2004) 71:9–13. doi: 10.1016/S1297-319X(03)00131-3
146. Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. (2009) 60:2747–56. doi: 10.1002/art.24781
147. De Benedetti F, Pignatti P, Vivarelli M, Meazza C, Ciliberto G, Savino R, et al. In vivo neutralization of human IL-6 (hIL-6) achieved by immunization of hIL-6-transgenic mice with a hIL-6 receptor antagonist. J Immunol. (2001) 166:4334–40. doi: 10.4049/jimmunol.166.7.4334
148. Wong PK, Quinn JM, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum. (2006) 54:158–68. doi: 10.1002/art.21537
149. Li L, Chen X, Lv S, Dong M, Zhang L, Tu J, et al. Influence of exercise on bone remodeling-related hormones and cytokines in ovariectomized rats: a model of postmenopausal osteoporosis. PLoS ONE (2014) 9:e112845. doi: 10.1371/journal.pone.0112845
150. Park JH, Park KH, Cho S, Choi YS, Seo SK, Lee BS, et al. Concomitant increase in muscle strength and bone mineral density with decreasing IL-6 levels after combination therapy with alendronate and calcitriol in postmenopausal women. Menopause (2013) 20:747–53. doi: 10.1097/GME.0b013e31827cabca
151. Mezil YA, Allison D, Kish K, Ditor D, Ward WE, Tsiani E, et al. Response of bone turnover markers and cytokines to high-intensity low-impact exercise. Med Sci Sports Exerc. (2015) 47:1495–502. doi: 10.1249/MSS.0000000000000555
152. Aguirre L, Napoli N, Waters D, Qualls C, Villareal DT, Armamento-Villareal R. Increasing adiposity is associated with higher adipokine levels and lower bone mineral density in obese older adults. J Clin Endocrinol Metab. (2014) 99:3290–7. doi: 10.1210/jc.2013-3200
153. Marques EA, Mota J, Viana JL, Tuna D, Figueiredo P, Guimaraes JT, et al. Response of bone mineral density, inflammatory cytokines, and biochemical bone markers to a 32-week combined loading exercise programme in older men and women. Arch Gerontol Geriatr. (2013) 57:226–33. doi: 10.1016/j.archger.2013.03.014
154. Broholm C, Laye MJ, Brandt C, Vadalasetty R, Pilegaard H, Pedersen BK, et al. LIF is a contraction-induced myokine stimulating human myocyte proliferation. J Appl Physiol. (2011) 111:251–9. doi: 10.1152/japplphysiol.01399.2010
155. Broholm C, Mortensen OH, Nielsen S, Akerstrom T, Zankari A, Dahl B, et al. Exercise induces expression of leukaemia inhibitory factor in human skeletal muscle. J Physiol. (2008) 586:2195–201. doi: 10.1113/jphysiol.2007.149781
156. Malaval L, Aubin JE. Biphasic effects of leukemia inhibitory factor on osteoblastic differentiation. J Cell Biochem Suppl. (2001) 36:63–70. doi: 10.1002/jcb.1086
157. Sims NA, Johnson RW. Leukemia inhibitory factor: a paracrine mediator of bone metabolism. Growth Factors (2012) 30:76–87. doi: 10.3109/08977194.2012.656760
158. Haugen F, Norheim F, Lian H, Wensaas AJ, Dueland S, Berg O, et al. IL-7 is expressed and secreted by human skeletal muscle cells. Am J Physiol Cell Physiol. (2010) 298:C807–16. doi: 10.1152/ajpcell.00094.2009
159. Zhao R. Immune regulation of osteoclast function in postmenopausal osteoporosis: a critical interdisciplinary perspective. Int J Med Sci. (2012) 9:825–32. doi: 10.7150/ijms.5180
160. Lee SK, Surh CD. Role of interleukin-7 in bone and T-cell homeostasis. Immunol Rev. (2005) 208:169–80. doi: 10.1111/j.0105-2896.2005.00339.x
161. Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R. Interleukin-7 stimulates osteoclast formation by up-regulating the T-cell production of soluble osteoclastogenic cytokines. Blood (2000) 96:1873–8.
162. Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci USA. (2003) 100:125–30. doi: 10.1073/pnas.0136772100
163. Andersson H, Bohn SK, Raastad T, Paulsen G, Blomhoff R, Kadi F. Differences in the inflammatory plasma cytokine response following two elite female soccer games separated by a 72-h recovery. Scand J Med Sci Sports (2010) 20:740–7. doi: 10.1111/j.1600-0838.2009.00989.x
164. Kraemer WJ, Hatfield DL, Comstock BA, Fragala MS, Davitt PM, Cortis C, et al. Influence of HMB supplementation and resistance training on cytokine responses to resistance exercise. J Am Coll Nutr. (2014) 33:247–55. doi: 10.1080/07315724.2014.911669
165. Gorgens SW, Eckardt K, Jensen J, Drevon CA, Eckel J. Exercise and regulation of adipokine and myokine production. Prog Mol Biol Transl Sci. (2015) 135:313–36. doi: 10.1016/bs.pmbts.2015.07.002
166. Nieman DC, Davis JM, Henson DA, Walberg-Rankin J, Shute M, Dumke CL, et al. Carbohydrate ingestion influences skeletal muscle cytokine mRNA and plasma cytokine levels after a 3-h run. J Appl Physiol. (2003) 94:1917–25. doi: 10.1152/japplphysiol.01130.2002
167. Rinnov A, Yfanti C, Nielsen S, Akerstrom TC, Peijs L, Zankari A, et al. Endurance training enhances skeletal muscle interleukin-15 in human male subjects. Endocrine (2014) 45:271–8. doi: 10.1007/s12020-013-9969-z
168. Tamura Y, Watanabe K, Kantani T, Hayashi J, Ishida N, Kaneki M. Upregulation of circulating IL-15 by treadmill running in healthy individuals: is IL-15 an endocrine mediator of the beneficial effects of endurance exercise? Endocr J. (2011) 58:211–5. doi: 10.1507/endocrj.K10E-400
169. Riechman SE, Balasekaran G, Roth SM, Ferrell RE. Association of interleukin-15 protein and interleukin-15 receptor genetic variation with resistance exercise training responses. J Appl Physiol. (2004) 97:2214–9. doi: 10.1152/japplphysiol.00491.2004
170. Nielsen AR, Mounier R, Plomgaard P, Mortensen OH, Penkowa M, Speerschneider T, et al. Expression of interleukin-15 in human skeletal muscle effect of exercise and muscle fibre type composition. J Physiol. (2007) 584(Pt 1):305–12. doi: 10.1113/jphysiol.2007.139618
171. Ogata Y, Kukita A, Kukita T, Komine M, Miyahara A, Miyazaki S, et al. A novel role of IL-15 in the development of osteoclasts: inability to replace its activity with IL-2. J Immunol. (1999) 162:2754–60.
172. Okabe I, Kikuchi T, Mogi M, Takeda H, Aino M, Kamiya Y, et al. IL-15 and RANKL play a synergistically important role in osteoclastogenesis. J Cell Biochem. (2017) 118:739–47. doi: 10.1002/jcb.25726
173. Amarasekara DS, Yun H, Kim S, Lee N, Kim H, Rho J. Regulation of osteoclast differentiation by cytokine networks. Immune Netw. (2018) 18:e8. doi: 10.4110/in.2018.18.e8
174. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature (1997) 387:83–90. doi: 10.1038/387083a0
175. Hamrick MW. A role for myokines in muscle-bone interactions. Exerc Sport Sci Rev. (2011) 39:43–7. doi: 10.1097/JES.0b013e318201f601
176. Nam J, Perera P, Gordon R, Jeong YH, Blazek AD, Kim DG, et al. Follistatin-like 3 is a mediator of exercise-driven bone formation and strengthening. Bone (2015) 78:62–70. doi: 10.1016/j.bone.2015.04.038
177. Harber MP, Crane JD, Dickinson JM, Jemiolo B, Raue U, Trappe TA, et al. Protein synthesis and the expression of growth-related genes are altered by running in human vastus lateralis and soleus muscles. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R708–14. doi: 10.1152/ajpregu.90906.2008
178. Kim JS, Cross JM, Bamman MM. Impact of resistance loading on myostatin expression and cell cycle regulation in young and older men and women. Am J Physiol Endocrinol Metab. (2005) 288:E1110–9. doi: 10.1152/ajpendo.00464.2004
179. Dalbo VJ, Roberts MD, Sunderland KL, Poole CN, Stout JR, Beck TW, et al. Acute loading and aging effects on myostatin pathway biomarkers in human skeletal muscle after three sequential bouts of resistance exercise. J Gerontol A Biol Sci Med Sci. (2011) 66:855–65. doi: 10.1093/gerona/glr091
180. Dieli-Conwright CM, Spektor TM, Rice JC, Sattler FR, Schroeder ET. Hormone therapy and maximal eccentric exercise alters myostatin-related gene expression in postmenopausal women. J Strength Cond Res. (2012) 26:1374–82. doi: 10.1519/JSC.0b013e318251083f
181. Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM, Kraus WE. Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc. (2010) 42:2023–9. doi: 10.1249/MSS.0b013e3181e0b9a8
182. Jaworska J, Micielska K, Kozłowska M, Wnorowski K, Skrobecki J, Radzimin L, et al. A 2-week specific volleyball training supported by the whole body cryostimulation protocol induced an increase of growth factors and counteracted deterioration of physical performance. Front Physiol. (2018) 9:1711. doi: 10.3389/fphys.2018.01711
183. Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R, et al. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol. (2004) 270:19–30. doi: 10.1016/j.ydbio.2004.01.046
184. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell (2003) 113:685–700. doi: 10.1016/S0092-8674(03)00432-X
185. Miura T, Kishioka Y, Wakamatsu J, Hattori A, Hennebry A, Berry CJ, et al. Decorin binds myostatin and modulates its activity to muscle cells. Biochem Biophys Res Commun. (2006) 340:675–80. doi: 10.1016/j.bbrc.2005.12.060
186. Kanzleiter T, Rath M, Gorgens SW, Jensen J, Tangen DS, Kolnes AJ, et al. The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem Biophys Res Commun. (2014) 450:1089–94. doi: 10.1016/j.bbrc.2014.06.123
187. Heinemeier KM, Bjerrum SS, Schjerling P, Kjaer M. Expression of extracellular matrix components and related growth factors in human tendon and muscle after acute exercise. Scand J Med Sci Sports (2013) 23:e150–61. doi: 10.1111/j.1600-0838.2011.01414.x
188. Gorgens SW, Raschke S, Holven KB, Jensen J, Eckardt K, Eckel J. Regulation of follistatin-like protein 1 expression and secretion in primary human skeletal muscle cells. Arch Physiol Biochem. (2013) 119:75–80. doi: 10.3109/13813455.2013.768270
189. Norheim F, Raastad T, Thiede B, Rustan AC, Drevon CA, Haugen F. Proteomic identification of secreted proteins from human skeletal muscle cells and expression in response to strength training. Am J Physiol Endocrinol Metab. (2011) 301:E1013–21. doi: 10.1152/ajpendo.00326.2011
190. Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med. (2015) 21:1085–90. doi: 10.1038/nm.3917
191. Hamrick MW, Shi X, Zhang W, Pennington C, Thakore H, Haque M, et al. Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone (2007) 40:1544–53. doi: 10.1016/j.bone.2007.02.012
192. Bettis T, Kim BJ, Hamrick MW. Impact of muscle atrophy on bone metabolism and bone strength: implications for muscle-bone crosstalk with aging and disuse. Osteoporos Int. (2018) 29:1713–20. doi: 10.1007/s00198-018-4570-1
193. Kerschan-Schindl K, Thalmann MM, Weiss E, Tsironi M, Foger-Samwald U, Meinhart J, et al. Changes in serum levels of myokines and wnt-antagonists after an ultramarathon race. PLoS ONE (2015) 10:e0132478. doi: 10.1371/journal.pone.0132478
194. Kawao N, Morita H, Obata K, Tatsumi K, Kaji H. Role of follistatin in muscle and bone alterations induced by gravity change in mice. J Cell Physiol. (2018) 233:1191–201. doi: 10.1002/jcp.25986
195. Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia (2009) 52:1409–18. doi: 10.1007/s00125-009-1364-1
196. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. (2001) 24:677–736. doi: 10.1146/annurev.neuro.24.1.677
197. Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, et al. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol. (2009) 94:1062–9. doi: 10.1113/expphysiol.2009.048512
198. Gold SM, Schulz KH, Hartmann S, Mladek M, Lang UE, Hellweg R, et al. Basal serum levels and reactivity of nerve growth factor and brain-derived neurotrophic factor to standardized acute exercise in multiple sclerosis and controls. J Neuroimmunol. (2003) 138:99–105. doi: 10.1016/S0165-5728(03)00121-8
199. Ferris LT, Williams JS, Shen CL. The effect of acute exercise on serum brain-derived neurotrophic factor levels and cognitive function. Med Sci Sports Exerc. (2007) 39:728–34. doi: 10.1249/mss.0b013e31802f04c7
200. Schmidt-Kassow M, Schadle S, Otterbein S, Thiel C, Doehring A, Lotsch J, et al. Kinetics of serum brain-derived neurotrophic factor following low-intensity versus high-intensity exercise in men and women. Neuroreport (2012) 23:889–93. doi: 10.1097/WNR.0b013e32835946ca
201. Griffin EW, Mullally S, Foley C, Warmington SA, O'Mara SM, Kelly AM. Aerobic exercise improves hippocampal function and increases BDNF in the serum of young adult males. Physiol Behav. (2011) 104:934–41. doi: 10.1016/j.physbeh.2011.06.005
202. Goekint M, De Pauw K, Roelands B, Njemini R, Bautmans I, Mets T, et al. Strength training does not influence serum brain-derived neurotrophic factor. Eur J Appl Physiol. (2010) 110:285–93. doi: 10.1007/s00421-010-1461-3
203. Gmiat A, Micielska K, Kozlowska M, Flis DJ, Smaruj M, Kujach S, et al. The impact of a single bout of high intensity circuit training on myokines' concentrations and cognitive functions in women of different age. Physiol Behav. (2017) 179:290–7. doi: 10.1016/j.physbeh.2017.07.004
204. Gmiat A, Jaworska J, Micielska K, Kortas J, Prusik K, Prusik K, et al. Improvement of cognitive functions in response to a regular Nordic walking training in elderly women - A change dependent on the training experience. Exp Gerontol. (2018) 104:105–12. doi: 10.1016/j.exger.2018.02.006
205. Yamashiro T, Fukunaga T, Yamashita K, Kobashi N, Takano-Yamamoto T. Gene and protein expression of brain-derived neurotrophic factor and TrkB in bone and cartilage. Bone (2001) 28:404–9. doi: 10.1016/S8756-3282(01)00405-7
206. Kilian O, Hartmann S, Dongowski N, Karnati S, Baumgart-Vogt E, Hartel FV, et al. BDNF and its TrkB receptor in human fracture healing. Ann Anat. (2014) 196:286–95. doi: 10.1016/j.aanat.2014.06.001
207. Camerino C, Zayzafoon M, Rymaszewski M, Heiny J, Rios M, Hauschka PV. Central depletion of brain-derived neurotrophic factor in mice results in high bone mass and metabolic phenotype. Endocrinology (2012) 153:5394–405. doi: 10.1210/en.2012-1378
208. Coniglio SJ. Role of tumor-derived chemokines in osteolytic bone metastasis. Front Endocrinol. (2018) 9:313. doi: 10.3389/fendo.2018.00313
209. Qin S, LaRosa G, Campbell JJ, Smith-Heath H, Kassam N, Shi X, et al. Expression of monocyte chemoattractant protein-1 and interleukin-8 receptors on subsets of T cells: correlation with transendothelial chemotactic potential. Eur J Immunol. (1996) 26:640–7. doi: 10.1002/eji.1830260320
210. Vella L, Caldow MK, Larsen AE, Tassoni D, Della Gatta PA, Gran P, et al. Resistance exercise increases NF-kappaB activity in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. (2012) 302:R667–73. doi: 10.1152/ajpregu.00336.2011
211. Della Gatta PA, Garnham AP, Peake JM, Cameron-Smith D. Effect of exercise training on skeletal muscle cytokine expression in the elderly. Brain Behav Immun. (2014) 39:80–6. doi: 10.1016/j.bbi.2014.01.006
212. Peake JM, Suzuki K, Hordern M, Wilson G, Nosaka K, Coombes JS. Plasma cytokine changes in relation to exercise intensity and muscle damage. Eur J Appl Physiol. (2005) 95:514–21. doi: 10.1007/s00421-005-0035-2
213. Ogawa K, Sanada K, Machida S, Okutsu M, Suzuki K. Resistance exercise training-induced muscle hypertrophy was associated with reduction of inflammatory markers in elderly women. Mediators Inflamm. (2010) 2010:171023. doi: 10.1155/2010/171023
214. Greiwe JS, Cheng B, Rubin DC, Yarasheski KE, Semenkovich CF. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J. (2001) 15:475–82. doi: 10.1096/fj.00-0274com
215. Gielen S, Adams V, Mobius-Winkler S, Linke A, Erbs S, Yu J, et al. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol. (2003) 42:861–8. doi: 10.1016/S0735-1097(03)00848-9
216. Febbraio MA, Steensberg A, Starkie RL, McConell GK, Kingwell BA. Skeletal muscle interleukin-6 and tumor necrosis factor-alpha release in healthy subjects and patients with type 2 diabetes at rest and during exercise. Metabolism (2003) 52:939–44. doi: 10.1016/S0026-0495(03)00105-7
217. Plomgaard P, Nielsen AR, Fischer CP, Mortensen OH, Broholm C, Penkowa M, et al. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia (2007) 50:2562–71. doi: 10.1007/s00125-007-0834-6
218. Golbidi S, Laher I. Exercise induced adipokine changes and the metabolic syndrome. J Diabetes Res. (2014) 2014:726861. doi: 10.1155/2014/726861
219. Paulsen G, Mikkelsen UR, Raastad T, Peake JM. Leucocytes, cytokines and satellite cells: what role do they play in muscle damage and regeneration following eccentric exercise? Exerc Immunol Rev. (2012) 18:42–97.
220. Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol. (2006) 24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646
221. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. (2000) 275:4858–64. doi: 10.1074/jbc.275.7.4858
222. Komine M, Kukita A, Kukita T, Ogata Y, Hotokebuchi T, Kohashi O. Tumor necrosis factor-alpha cooperates with receptor activator of nuclear factor kappaB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone (2001) 28:474–83. doi: 10.1016/S8756-3282(01)00420-3
223. Park ES, Choi S, Shin B, Yu J, Yu J, Hwang JM, et al. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-sphingosine 1-phosphate (S1P) interaction. J Biol Chem. (2015) 290:9660–73. doi: 10.1074/jbc.M114.609685
224. Yao Z, Lei W, Duan R, Li Y, Luo L, Boyce BF. RANKL cytokine enhances TNF-induced osteoclastogenesis independently of TNF receptor associated factor (TRAF) 6 by degrading TRAF3 in osteoclast precursors. J Biol Chem. (2017) 292:10169–79. doi: 10.1074/jbc.M116.771816
225. Kitaura H, Kimura K, Ishida M, Kohara H, Yoshimatsu M, Takano-Yamamoto T. Immunological reaction in TNF-alpha-mediated osteoclast formation and bone resorption in vitro and in vivo. Clin Dev Immunol. (2013) 2013:181849. doi: 10.1155/2013/181849
226. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science (2005) 307:426–30. doi: 10.1126/science.1097243
227. Lombardi G, Banfi G. Effects of sample matrix and storage conditions on full-length visfatin measurement in blood. Clin Chim Acta (2015) 440:140–2. doi: 10.1016/j.cca.2014.11.006
228. Sommer G, Garten A, Petzold S, Beck-Sickinger AG, Bluher M, Stumvoll M, et al. Visfatin/PBEF/Nampt: structure, regulation and potential function of a novel adipokine. Clin Sci. (2008) 115:13–23. doi: 10.1042/CS20070226
229. Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, et al. Skeletal muscle NAMPT is induced by exercise in humans. Am J Physiol Endocrinol Metab. (2010) 298:E117–26. doi: 10.1152/ajpendo.00318.2009
230. Ziemann E, Olek RA, Grzywacz T, Antosiewicz J, Kujach S, Luszczyk M, et al. Whole-body cryostimulation as an effective method of reducing low-grade inflammation in obese men. J Physiol Sci. (2013) 63:333–43. doi: 10.1007/s12576-013-0269-4
231. Brandauer J, Vienberg SG, Andersen MA, Ringholm S, Risis S, Larsen PS, et al. AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J Physiol. (2013) 591:5207–20. doi: 10.1113/jphysiol.2013.259515
232. Frydelund-Larsen L, Akerstrom T, Nielsen S, Keller P, Keller C, Pedersen BK. Visfatin mRNA expression in human subcutaneous adipose tissue is regulated by exercise. Am J Physiol Endocrinol Metab. (2007) 292:E24–31. doi: 10.1152/ajpendo.00113.2006
233. Ghanbari-Niaki A, Saghebjoo M, Soltani R, Kirwan JP. Plasma visfatin is increased after high-intensity exercise. Ann Nutr Metab. (2010) 57:3–8. doi: 10.1159/000313936
234. Thommesen L, Stunes AK, Monjo M, Grosvik K, Tamburstuen MV, Kjobli E, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. (2006) 99:824–34. doi: 10.1002/jcb.20915
235. Xie H, Tang SY, Luo XH, Huang J, Cui RR, Yuan LQ, et al. Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int. (2007) 80:201–10. doi: 10.1007/s00223-006-0155-7
236. Moschen AR, Geiger S, Gerner R, Tilg H. Pre-B cell colony enhancing factor/NAMPT/visfatin and its role in inflammation-related bone disease. Mutat Res. (2010) 690:95–101. doi: 10.1016/j.mrfmmm.2009.06.012
237. Li Y, He X, Li Y, He J, Anderstam B, Andersson G, et al. Nicotinamide phosphoribosyltransferase (Nampt) affects the lineage fate determination of mesenchymal stem cells: a possible cause for reduced osteogenesis and increased adipogenesis in older individuals. J Bone Miner Res. (2011) 26:2656–64. doi: 10.1002/jbmr.480
238. Sliwicka E, Nowak A, Zep W, Leszczynski P, Pilaczynska-Szczesniak L. Bone mass and bone metabolic indices in male master rowers. J Bone Miner Metab. (2015) 33:540–6. doi: 10.1007/s00774-014-0619-1
239. Iacobellis G, Iorio M, Napoli N, Cotesta D, Zinnamosca L, Marinelli C, et al. Relation of adiponectin, visfatin and bone mineral density in patients with metabolic syndrome. J Endocrinol Invest. (2011) 34:e12–5. doi: 10.1007/BF03346703
240. Zhang H, Xie H, Zhao Q, Xie GQ, Wu XP, Liao EY, et al. Relationships between serum adiponectin, apelin, leptin, resistin, visfatin levels and bone mineral density, and bone biochemical markers in post-menopausal Chinese women. J Endocrinol Invest. (2010) 33:707–11. doi: 10.1007/BF03346674
241. Tohidi M, Akbarzadeh S, Larijani B, Kalantarhormozi M, Ostovar A, Assadi M, et al. Omentin-1, visfatin and adiponectin levels in relation to bone mineral density in Iranian postmenopausal women. Bone (2012) 51:876–81. doi: 10.1016/j.bone.2012.08.117
242. Gruodyte R, Jurimae J, Cicchella A, Stefanelli C, Passariello C, Jurimae T. Adipocytokines and bone mineral density in adolescent female athletes. Acta Paediatr. (2010) 99:1879–84. doi: 10.1111/j.1651-2227.2010.01905.x
243. Yamauchi T, Iwabu M, Okada-Iwabu M, Kadowaki T. Adiponectin receptors: a review of their structure, function and how they work. Best Pract Res Clin Endocrinol Metab. (2014) 28:15–23. doi: 10.1016/j.beem.2013.09.003
244. Han SH, Quon MJ, Kim JA, Koh KK. Adiponectin and cardiovascular disease: response to therapeutic interventions. J Am Coll Cardiol. (2007) 49:531–8. doi: 10.1016/j.jacc.2006.08.061
245. Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. (2007) 178:1748–58. doi: 10.4049/jimmunol.178.3.1748
246. Santos MJ, Fonseca JE. Metabolic syndrome, inflammation and atherosclerosis - the role of adipokines in health and in systemic inflammatory rheumatic diseases. Acta Reumatol Port. (2009) 34:590–8.
247. Delaigle AM, Jonas JC, Bauche IB, Cornu O, Brichard SM. Induction of adiponectin in skeletal muscle by inflammatory cytokines: in vivo and in vitro studies. Endocrinology (2004) 145:5589–97. doi: 10.1210/en.2004-0503
248. Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, et al. Functional adiponectin resistance at the level of the skeletal muscle in mild to moderate chronic heart failure. Circ Heart Fail. (2010) 3:185–94. doi: 10.1161/CIRCHEARTFAILURE.109.885525
249. Bluher M, Bullen JW Jr, Lee JH, Kralisch S, Fasshauer M, Kloting N, et al. Circulating adiponectin and expression of adiponectin receptors in human skeletal muscle: associations with metabolic parameters and insulin resistance and regulation by physical training. J Clin Endocrinol Metab. (2006) 91:2310–6. doi: 10.1210/jc.2005-2556
250. Saunders TJ, Palombella A, McGuire KA, Janiszewski PM, Despres JP, Ross R. Acute exercise increases adiponectin levels in abdominally obese men. J Nutr Metab. (2012) 2012:148729. doi: 10.1155/2012/148729
251. Christiansen T, Bruun JM, Paulsen SK, Olholm J, Overgaard K, Pedersen SB, et al. Acute exercise increases circulating inflammatory markers in overweight and obese compared with lean subjects. Eur J Appl Physiol. (2013) 113:1635–42. doi: 10.1007/s00421-013-2592-0
252. Ferguson MA, White LJ, McCoy S, Kim HW, Petty T, Wilsey J. Plasma adiponectin response to acute exercise in healthy subjects. Eur J Appl Physiol. (2004) 91:324–9. doi: 10.1007/s00421-003-0985-1
253. Jamurtas AZ, Theocharis V, Koukoulis G, Stakias N, Fatouros IG, Kouretas D, et al. The effects of acute exercise on serum adiponectin and resistin levels and their relation to insulin sensitivity in overweight males. Eur J Appl Physiol. (2006) 97:122–6. doi: 10.1007/s00421-006-0169-x
254. Bobbert T, Wegewitz U, Brechtel L, Freudenberg M, Mai K, Mohlig M, et al. Adiponectin oligomers in human serum during acute and chronic exercise: relation to lipid metabolism and insulin sensitivity. Int J Sports Med. (2007) 28:1–8. doi: 10.1055/s-2006-924028
255. Bouassida A, Lakhdar N, Benaissa N, Mejri S, Zaouali M, Zbidi A, et al. Adiponectin responses to acute moderate and heavy exercises in overweight middle aged subjects. J Sports Med. Phys Fitness (2010) 50:330–5.
256. Numao S, Katayama Y, Hayashi Y, Matsuo T, Tanaka K. Influence of acute aerobic exercise on adiponectin oligomer concentrations in middle-aged abdominally obese men. Metabolism (2011) 60:186–94. doi: 10.1016/j.metabol.2009.12.011
257. Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab. (2006) 290:E961–7. doi: 10.1152/ajpendo.00506.2005
258. Fatouros IG, Tournis S, Leontsini D, Jamurtas AZ, Sxina M, Thomakos P, et al. Leptin and adiponectin responses in overweight inactive elderly following resistance training and detraining are intensity related. J Clin Endocrinol Metab. (2005) 90:5970–7. doi: 10.1210/jc.2005-0261
259. Fatouros IG, Chatzinikolaou A, Tournis S, Nikolaidis MG, Jamurtas AZ, Douroudos II, et al. Intensity of resistance exercise determines adipokine and resting energy expenditure responses in overweight elderly individuals. Diabetes Care (2009) 32:2161–7. doi: 10.2337/dc08-1994
260. Biver E, Salliot C, Combescure C, Gossec L, Hardouin P, Legroux-Gerot I, et al. Influence of adipokines and ghrelin on bone mineral density and fracture risk: a systematic review and meta-analysis. J Clin Endocrinol Metab. (2011) 96:2703–13. doi: 10.1210/jc.2011-0047
261. Sucunza N, Barahona MJ, Resmini E, Fernandez-Real JM, Ricart W, Farrerons J, et al. A link between bone mineral density and serum adiponectin and visfatin levels in acromegaly. J Clin Endocrinol Metab. (2009) 94:3889–96. doi: 10.1210/jc.2009-0474
262. Ruscica M, Steffani L, Magni P. Adiponectin interactions in bone and cartilage biology and disease. Vitam Horm. (2012) 90:321–39. doi: 10.1016/B978-0-12-398313-8.00012-9
263. Berner HS, Lyngstadaas SP, Spahr A, Monjo M, Thommesen L, Drevon CA, et al. Adiponectin and its receptors are expressed in bone-forming cells. Bone (2004) 35:842–9. doi: 10.1016/j.bone.2004.06.008
264. Shinoda Y, Yamaguchi M, Ogata N, Akune T, Kubota N, Yamauchi T, et al. Regulation of bone formation by adiponectin through autocrine/paracrine and endocrine pathways. J Cell Biochem. (2006) 99:196–208. doi: 10.1002/jcb.20890
265. Zhang Y, Zhou P, Kimondo JW. Adiponectin and osteocalcin: relation to insulin sensitivity. Biochem Cell Biol. (2012) 90:613–20. doi: 10.1139/o2012-022
266. Sadie-Van Gijsen H, Crowther NJ, Hough FS, Ferris WF. The interrelationship between bone and fat: from cellular see-saw to endocrine reciprocity. Cell Mol Life Sci. (2013) 70:2331–49. doi: 10.1007/s00018-012-1211-2
267. Munzberg H, Morrison CD. Structure, production and signaling of leptin. Metabolism (2015) 64:13–23. doi: 10.1016/j.metabol.2014.09.010
268. Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature (1998) 393:684–8. doi: 10.1038/31474
269. Upadhyay J, Farr OM, Mantzoros CS. The role of leptin in regulating bone metabolism. Metabolism (2015) 64:105–13. doi: 10.1016/j.metabol.2014.10.021
270. Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell (2002) 111:305–17. doi: 10.1016/S0092-8674(02)01049-8
271. Zaidi M, Buettner C, Sun L, Iqbal J. Minireview: the link between fat and bone: does mass beget mass? Endocrinology (2012) 153:2070–5. doi: 10.1210/en.2012-1022
272. Sienkiewicz E, Magkos F, Aronis KN, Brinkoetter M, Chamberland JP, Chou S, et al. Long-term metreleptin treatment increases bone mineral density and content at the lumbar spine of lean hypoleptinemic women. Metabolism (2011) 60:1211–21. doi: 10.1016/j.metabol.2011.05.016
273. Kim YS, Nam JS, Yeo DW, Kim KR, Suh SH, Ahn CW. The effects of aerobic exercise training on serum osteocalcin, adipocytokines and insulin resistance on obese young males. Clin Endocrinol. (2015) 82:686–94. doi: 10.1111/cen.12601
274. Yang C, Chen J, Wu F, Li J, Liang P, Zhang H, et al. Effects of 60-day head-down bed rest on osteocalcin, glycolipid metabolism and their association with or without resistance training. Clin Endocrinol. (2014) 81:671–8. doi: 10.1111/cen.12535
275. Keller P, Keller C, Steensberg A, Robinson LE, Pedersen BK. Leptin gene expression and systemic levels in healthy men: effect of exercise, carbohydrate, interleukin-6, and epinephrine. J Appl Physiol. (2005) 98:1805–12. doi: 10.1152/japplphysiol.00592.2004
276. Essig DA, Alderson NL, Ferguson MA, Bartoli WP, Durstine JL. Delayed effects of exercise on the plasma leptin concentration. Metabolism (2000) 49:395–9. doi: 10.1016/S0026-0495(00)90396-2
277. Olive JL, Miller GD. Differential effects of maximal- and moderate-intensity runs on plasma leptin in healthy trained subjects. Nutrition (2001) 17:365–9. doi: 10.1016/S0899-9007(01)00522-6
278. Phillips MD, Patrizi RM, Cheek DJ, Wooten JS, Barbee JJ, Mitchell JB. Resistance training reduces subclinical inflammation in obese, postmenopausal women. Med Sci Sports Exerc. (2012) 44:2099–110. doi: 10.1249/MSS.0b013e3182644984
279. Yang CB, Chuang CC, Kuo CS, Hsu CH, Tsao TH. Effects of an acute bout of exercise on serum soluble leptin receptor (sOB-R) levels. J Sports Sci. (2014) 32:446–51. doi: 10.1080/02640414.2013.828848
280. Shah K, Armamento-Villareal R, Parimi N, Chode S, Sinacore DR, Hilton TN, et al. Exercise training in obese older adults prevents increase in bone turnover and attenuates decrease in hip bone mineral density induced by weight loss despite decline in bone-active hormones. J Bone Miner Res. (2011) 26:2851–9. doi: 10.1002/jbmr.475
281. Varady KA, Bhutani S, Church EC, Phillips SA. Adipokine responses to acute resistance exercise in trained and untrained men. Med Sci Sports Exerc. (2010) 42:456–62. doi: 10.1249/MSS.0b013e3181ba6dd3
282. Codoner-Franch P, Alonso-Iglesias E. Resistin: insulin resistance to malignancy. Clin Chim Acta (2015) 438:46–54. doi: 10.1016/j.cca.2014.07.043
283. Oh KW, Lee WY, Rhee EJ, Baek KH, Yoon KH, Kang MI, et al. The relationship between serum resistin, leptin, adiponectin, ghrelin levels and bone mineral density in middle-aged men. Clin Endocrinol. (2005) 63:131–8. doi: 10.1111/j.1365-2265.2005.02312.x
284. Peng XD, Xie H, Zhao Q, Wu XP, Sun ZQ, Liao EY. Relationships between serum adiponectin, leptin, resistin, visfatin levels and bone mineral density, and bone biochemical markers in Chinese men. Clin Chim Acta (2008) 387:31–5. doi: 10.1016/j.cca.2007.08.012
285. Pluskiewicz W, Adamczyk P, Marek B, Czekajlo A, Drozdzowska B, Kajdaniuk D, et al. Adiponectin and resistin in relationship with skeletal status in women from the RAC-OST-POL study. Endokrynol Pol. (2012) 63:427–31.
286. Mohiti-Ardekani J, Soleymani-Salehabadi H, Owlia MB, Mohiti A. Relationships between serum adipocyte hormones (adiponectin, leptin, resistin), bone mineral density and bone metabolic markers in osteoporosis patients. J Bone Miner Metab. (2014) 32:400–4. doi: 10.1007/s00774-013-0511-4
287. Sowers MR, Wildman RP, Mancuso P, Eyvazzadeh AD, Karvonen-Gutierrez CA, Rillamas-Sun E, et al. Change in adipocytokines and ghrelin with menopause. Maturitas (2008) 59:149–57. doi: 10.1016/j.maturitas.2007.12.006
288. Fisher A, Southcott E, Li R, Srikusalanukul W, Davis M, Smith P. Serum resistin in older patients with hip fracture: relationship with comorbidity and biochemical determinants of bone metabolism. Cytokine (2011) 56:157–66. doi: 10.1016/j.cyto.2011.06.023
289. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature (2012) 481:463–8. doi: 10.1038/nature10777
290. Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, et al. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Metab. (2015) 22:734–40. doi: 10.1016/j.cmet.2015.08.001
291. Mahgoub MO, D'Souza C, Al Darmaki R, Baniyas M, Adeghate E. An update on the role of irisin in the regulation of endocrine and metabolic functions. Peptides (2018) 104:15–23. doi: 10.1016/j.peptides.2018.03.018
292. Tsuchiya Y, Ando D, Goto K, Kiuchi M, Yamakita M, Koyama K. High-intensity exercise causes greater irisin response compared with low-intensity exercise under similar energy consumption. Tohoku J Exp Med. (2014) 233:135–40. doi: 10.1620/tjem.233.135
293. Miyamoto-Mikami E, Sato K, Kurihara T, Hasegawa N, Fujie S, Fujita S, et al. Endurance training-induced increase in circulating irisin levels is associated with reduction of abdominal visceral fat in middle-aged and older adults. PLoS ONE (2015) 10:e0120354. doi: 10.1371/journal.pone.0120354
294. Gmiat A, Mieszkowski J, Prusik K, Prusik K, Kortas J, Kochanowicz A, et al. Changes in pro-inflammatory markers and leucine concentrations in response to Nordic Walking training combined with vitamin D supplementation in elderly women. Biogerontology (2017) 18:535–48. doi: 10.1007/s10522-017-9694-8
295. Dulian K, Laskowski R, Grzywacz T, Kujach S, Flis DJ, Smaruj M, et al. The whole body cryostimulation modifies irisin concentration and reduces inflammation in middle aged, obese men. Cryobiology (2015) 71:398–404. doi: 10.1016/j.cryobiol.2015.10.143
296. Bluher S, Panagiotou G, Petroff D, Markert J, Wagner A, Klemm T, et al. Effects of a 1-year exercise and lifestyle intervention on irisin, adipokines, and inflammatory markers in obese children. Obesity (2014) 22:1701–8. doi: 10.1002/oby.20739
297. Szumilewicz A, Worska A, Piernicka M, Kuchta A, Kortas J, Jastrzebski Z, et al. The exercise-induced irisin is associated with improved levels of glucose homeostasis markers in pregnant women participating in 8-week prenatal group fitness program: a pilot study. Biomed Res Int. (2017) 2017:9414525. doi: 10.1155/2017/9414525
298. Sanchis-Gomar F, Alis R, Lippi G. Circulating irisin detection: does it really work? Trends Endocrinol Metab. (2015) 26:335–6. doi: 10.1016/j.tem.2015.05.004
299. Moreno-Navarrete JM, Ortega F, Serrano M, Guerra E, Pardo G, Tinahones F, et al. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J Clin Endocrinol Metab. (2013) 98:E769–78. doi: 10.1210/jc.2012-2749
300. Roca-Rivada A, Castelao C, Senin LL, Landrove MO, Baltar J, Belen Crujeiras A, et al. FNDC5/irisin is not only a myokine but also an adipokine. PLoS ONE (2013) 8:e60563. doi: 10.1371/journal.pone.0060563
301. Wrann CD, White JP, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, et al. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. (2013) 18:649–59. doi: 10.1016/j.cmet.2013.09.008
302. Tabak O, Simsek G, Erdenen F, Sozer V, Hasoglu T, Gelisgen R, et al. The relationship between circulating irisin, retinol binding protein-4, adiponectin and inflammatory mediators in patients with metabolic syndrome. Arch Endocrinol Metab. (2017) 61:515–23. doi: 10.1590/2359-3997000000289
303. Colaianni G, Cuscito C, Mongelli T, Oranger A, Mori G, Brunetti G, et al. Irisin enhances osteoblast differentiation in vitro. Int J Endocrinol. (2014) 2014:902186. doi: 10.1155/2014/902186
304. Anastasilakis AD, Polyzos SA, Makras P, Gkiomisi A, Bisbinas I, Katsarou A, et al. Circulating irisin is associated with osteoporotic fractures in postmenopausal women with low bone mass but is not affected by either teriparatide or denosumab treatment for 3 months. Osteoporos Int. (2014) 25:1633–42. doi: 10.1007/s00198-014-2673-x
305. Palermo A, Strollo R, Maddaloni E, Tuccinardi D, D'Onofrio L, Briganti SI, et al. Irisin is associated with osteoporotic fractures independently of bone mineral density, body composition or daily physical activity. Clin Endocrinol. (2015) 82:615–9. doi: 10.1111/cen.12672
306. Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, Liu P, et al. The myokine irisin increases cortical bone mass. Proc Natl Acad Sci USA. (2015) 112:12157–62. doi: 10.1073/pnas.1516622112
307. Colaianni G, Mongelli T, Cuscito C, Pignataro P, Lippo L, Spiro G, et al. Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Sci Rep. (2017) 7:2811. doi: 10.1038/s41598-017-02557-8
308. Takayanagi H. New immune connections in osteoclast formation. Ann NY Acad Sci. (2010) 1192:117–23. doi: 10.1111/j.1749-6632.2009.05303.x
309. Alnaeeli M, Penninger JM, Teng YT. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J Immunol. (2006) 177:3314–26. doi: 10.4049/jimmunol.177.5.3314
310. Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. (2008) 29:403–40. doi: 10.1210/er.2007-0038
311. Lee SH, Kim TS, Choi Y, Lorenzo J. Osteoimmunology: cytokines and the skeletal system. BMB Rep. (2008) 41:495–510. doi: 10.5483/BMBRep.2008.41.7.495
312. Garrett RW, Emerson SG. Bone and blood vessels: the hard and the soft of hematopoietic stem cell niches. Cell Stem Cell (2009) 4:503–6. doi: 10.1016/j.stem.2009.05.011
313. Weitzmann MN, Ofotokun I. Physiological and pathophysiological bone turnover - role of the immune system. Nat Rev Endocrinol. (2016) 12:518–32. doi: 10.1038/nrendo.2016.91
314. Yun TJ, Chaudhary PM, Shu GL, Frazer JK, Ewings MK, Schwartz SM, et al. OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J Immunol. (1998) 161:6113–21.
315. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood (2007) 109:3839–48. doi: 10.1182/blood-2006-07-037994
316. Weitzmann MN. Bone and the immune system. Toxicol Pathol. (2017) 45:911–24. doi: 10.1177/0192623317735316
317. Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, et al. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci USA. (2003) 100:10405–10. doi: 10.1073/pnas.1533207100
318. Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest. (2000) 106:1229–37. doi: 10.1172/JCI11066
319. Pacifici R, Brown C, Puscheck E, Friedrich E, Slatopolsky E, Maggio D, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci USA. (1991) 88:5134–8. doi: 10.1073/pnas.88.12.5134
320. Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. (2006) 116:1186–94. doi: 10.1172/JCI28550
321. Ryan MR, Shepherd R, Leavey JK, Gao Y, Grassi F, Schnell FJ, et al. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc Natl Acad Sci USA. (2005) 102:16735–40. doi: 10.1073/pnas.0505168102
322. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS ONE (2012) 7:e44552. doi: 10.1371/journal.pone.0044552
323. Tyagi AM, Mansoori MN, Srivastava K, Khan MP, Kureel J, Dixit M, et al. Enhanced immunoprotective effects by anti-IL-17 antibody translates to improved skeletal parameters under estrogen deficiency compared with anti-RANKL and anti-TNF-alpha antibodies. J Bone Miner Res. (2014) 29:1981–92. doi: 10.1002/jbmr.2228
324. DeSelm CJ, Takahata Y, Warren J, Chappel JC, Khan T, Li X, et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem. (2012) 113:2895–902. doi: 10.1002/jcb.24165
325. Sjogren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, et al. The gut microbiota regulates bone mass in mice. J Bone Miner Res. (2012) 27:1357–67. doi: 10.1002/jbmr.1588
326. Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J Clin Invest. (2016) 126:2049–63. doi: 10.1172/JCI86062
327. Pacifici R. T cells: critical bone regulators in health and disease. Bone (2010) 47:461–71. doi: 10.1016/j.bone.2010.04.611
328. Verdelli C, Corbetta S. MECHANISMS IN ENDOCRINOLOGY: kidney involvement in patients with primary hyperparathyroidism: an update on clinical and molecular aspects. Eur J Endocrinol. (2017) 176:R39–52. doi: 10.1530/EJE-16-0430
329. Riggs BL, Melton LJ III. Involutional osteoporosis. N Engl J Med. (1986) 314:1676–86. doi: 10.1056/NEJM198606263142605
330. Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. (2007) 357:905–16. doi: 10.1056/NEJMra067395
331. Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, et al. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab. (2008) 8:132–45. doi: 10.1016/j.cmet.2008.07.001
Keywords: exercise, training, biomechanical loading, inflammatory mediators, inflammasomes
Citation: Lombardi G, Ziemann E and Banfi G (2019) Physical Activity and Bone Health: What Is the Role of Immune System? A Narrative Review of the Third Way. Front. Endocrinol. 10:60. doi: 10.3389/fendo.2019.00060
Received: 17 December 2018; Accepted: 22 January 2019;
Published: 07 February 2019.
Edited by:
Patrizia D'Amelio, University of Turin, ItalyReviewed by:
Graziana Colaianni, Università degli Studi di Bari, ItalyJan Josef Stepan, Charles University, Czechia
Copyright © 2019 Lombardi, Ziemann and Banfi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Lombardi, Z2lvdmFubmkubG9tYmFyZGlAZ3J1cHBvc2FuZG9uYXRvLml0