 Riikka E. Mäkitie
Riikka E. Mäkitie Alice Costantini
Alice Costantini Anders Kämpe
Anders Kämpe Jessica J. Alm
Jessica J. Alm Outi Mäkitie
Outi Mäkitie- 1Folkhälsan Institute of Genetics and University of Helsinki, Helsinki, Finland
- 2Research Program for Clinical and Molecular Metabolism, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 3Department of Molecular Medicine and Surgery and Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden
- 4Children's Hospital, Pediatric Research Center, University of Helsinki and HUS Helsinki University Hospital, Helsinki, Finland
- 5Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden
Osteoporosis, characterized by deteriorated bone microarchitecture and low bone mineral density, is a chronic skeletal disease with high worldwide prevalence. Osteoporosis related to aging is the most common form and causes significant morbidity and mortality. Rare, monogenic forms of osteoporosis have their onset usually in childhood or young adulthood and have specific phenotypic features and clinical course depending on the underlying cause. The most common form is osteogenesis imperfecta linked to mutations in COL1A1 and COL1A2, the two genes encoding type I collagen. However, in the past years, remarkable advancements in bone research have expanded our understanding of the intricacies behind bone metabolism and identified novel molecular mechanisms contributing to skeletal health and disease. Especially high-throughput sequencing techniques have made family-based studies an efficient way to identify single genes causative of rare monogenic forms of osteoporosis and these have yielded several novel genes that encode proteins partaking in type I collagen modification or regulating bone cell function directly. New forms of monogenic osteoporosis, such as autosomal dominant osteoporosis caused by WNT1 mutations or X-linked osteoporosis due to PLS3 mutations, have revealed previously unidentified bone-regulating proteins and clarified specific roles of bone cells, expanded our understanding of possible inheritance mechanisms and paces of disease progression, and highlighted the potential of monogenic bone diseases to extend beyond the skeletal tissue. The novel gene discoveries have introduced new challenges to the classification and diagnosis of monogenic osteoporosis, but also provided promising new molecular targets for development of pharmacotherapies. In this article we give an overview of the recent discoveries in the area of monogenic forms of osteoporosis, describing the key cellular mechanisms leading to skeletal fragility, the major recent research findings and the essential challenges and avenues in future diagnostics and treatments.
Introduction
Bone Health
Bone is a rigid connective tissue composed mainly of organic components (90% type I collagen, the rest other non-collagenous structural proteins and cells) and inorganic minerals (mostly calcium hydroxyapatite). These combined give bones their sturdiness to withstand an individual's weight and the elasticity to enable movement and resist fractures. Bone comprises dense and compact cortical bone and cancellous, loosely-webbed trabecular bone, and serves as a reservoir for minerals, growth factors, cytokines, and fat. Bone also functions as an endocrine organ by secreting several systemic hormonal factors (1).
Bone is all but a quiescent tissue—it undergoes active renewing and remodeling throughout life. By coupled, successive processes of bone resorption and bone formation, together called “bone turnover,” old or damaged bone is eroded and replaced by new bone to maintain healthy and strong bone tissue. Throughout childhood and adolescent growth, the period of bone mass accrual, bone turnover is formation-favoring, until the highest amount of bone mass, termed “peak bone mass,” is attained by young adulthood. Thereafter, bone mass remains fairly constant until bone resorption begins to dominate by the age of menopause and bone mass slowly declines.
Factors that impede skeletal growth in childhood or accelerate bone loss later in adulthood, such as long-term or chronic illnesses, glucocorticoid treatment and other medications, hypogonadism and menopause, other endocrine disorders and cancers, impose a great risk for low bone mass and osteoporosis (1–3). In childhood, especially glucocorticoids play a major role in secondary osteoporosis. Studies on patients receiving systemic steroids for acute lymphoblastic leukemia (4), juvenile idiopathic arthritis (5, 6), Duchenne muscular dystrophy (7) or asthma (8) all indicate increased peripheral and vertebral fracture rates.
Osteoporosis
Osteoporosis is a chronic skeletal disease with high prevalence and mortality worldwide. It is characterized by low bone mass and bone mineral density (BMD), and by destructed bone microarchitecture that often results from imbalanced bone formation and resorption or from abnormal matrix. Impaired bone quality leads to compromised bone strength and high propensity to low-energy fractures in long bones and vertebrae (9). Osteoporosis, with frequent fractures, pain and physical limitations, causes significant human suffering and burdens the health care system (9). BMD is considered to define osteoporosis and risk of fractures. It is assessed using dual-energy X-ray absorptiometry (DXA), where reduction of more than 2.5 standard deviations from the normal mean for young adults (T-score) is diagnostic of osteoporosis. Of note, osteopenia (T-score 1.0 to −2.5) together with a high probability of fractures, or a fragility fracture without another metabolic bone disease and independent of BMD are also clinically indicative of osteoporosis. Pediatric osteoporosis requires more than mere DXA-determined low BMD, as variation in growth and pubertal maturation make interpretation of BMD values challenging. Therefore, age-, gender-, and body size–adjusted DXA measurements (Z-scores) must be considered together with fracture history. A pathologic fracture history entails (i) ≥2 clinically significant long bone fractures by age 10 years, (ii) ≥3 clinically significant long bone fractures by 19 years, or (iii) one or more vertebral compression fractures in the absence of high-energy trauma, meaning a ≥20% loss in vertebral anterior, middle or posterior height. However, a vertebral compression fracture alone suffices for the diagnosis of pediatric osteoporosis even in the presence of normal BMD (2, 9–12).
Genetics in Bone Health
Genetics in Bone Health
Genetics play a substantial role in determining an individual's skeletal strength, bone microarchitectural properties and risk of osteoporosis. BMD is known to be a highly heritable trait and twin studies have shown genetic factors to determine up to 80% of its variance (13, 14). Genetic factors influence bone health in a polygenic manner and multiple gene variants, or single nucleotide polymorphisms (SNPs), in several different genes each contribute to the overall risk for compromised bone health. Recent research, especially large-scale genome-wide association studies in large cohorts, has elucidated the complexity of genetic networks that are important for bone metabolism but also evidenced limitations in our current knowledge. On the other hand, significant scientific advances have been made by studying rare monogenic forms of osteoporosis in which one mutation in a single gene with a major role in bone metabolism dominates and is alone sufficient to cause osteoporosis. Technical advancements in research methods, especially high through-put sequencing techniques, have made family-based studies an efficient way to identify new genes relevant to osteoporosis. Such studies have enabled recognition of novel molecular mechanisms and given leeway to understanding the intricacies behind bone metabolism (13, 14). In this article we only briefly summarize GWAS methodology and recent advancements while the main focus is on discoveries made from family-based research on patients and families with monogenic forms of osteoporosis.
Genome Wide Studies to Identify Contributing Genetic Factors
Genome wide association studies (GWASs) have proven successful and robust in deciphering the genetic mechanisms underlying complex diseases, including osteoporosis (14, 15). As mentioned, single nucleotide variants (SNVs) in several different genomic sites all contribute to bone quality and strength and risk of osteoporosis but are often very common in the general population and, by themselves, have only a minor effect (16). The current GWAS catalog, released in September 2018, comprises 55 separate studies focusing on bone properties, fractures or osteoporosis. Together they report 425 different lead SNVs, in 118 different genomic regions, that associate with some aspect of bone on a genome-wide significant level (Figure 1). From these, altogether 144 different genes are reported to be directly linked to, or plausible candidate effectors, for the identified signals. Of note, this catalog is not entirely up to date due to the extensive curation required before publication in the GWAS catalog. Recently, Kemp et al. undertook a colossal genome-wide search for genetic factors correlating with BMD, estimated from quantitative ultrasound of the heel (eBMD) (17). The GWAS is the largest to date, encompassing a total of 142,487 individuals from the UK Biobank. The authors were able to identify 203 loci, of which 153 were novel, to be associated with eBMD. These together explained about one third of the total variance in eBMD (17). Although highly successful, none of the previous GWA-studies with DXA-derived BMD have been as successful as the study by Kemp et al.
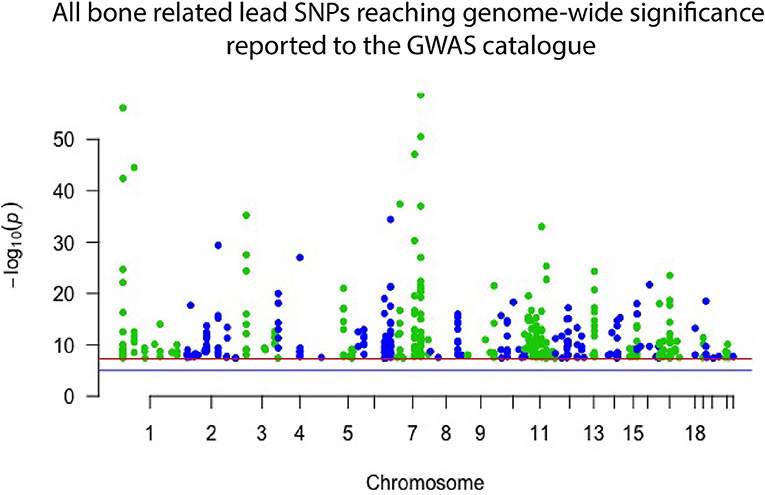
Figure 1. Manhattan plot displaying all lead SNPs independently associated with bone-related traits reported to the GWAS catalog as of September 2018. The associated SNPs highlight genomic regions important to bone. However, they each have only a minor effect on an individual's skeletal qualities and risk of osteoporosis and hence have limited use in clinical practice.
Despite these great advances, thus far, only <10% of the total estimated genetic variance in BMD can be explained by the results of the performed GWA-studies (18–21). Further, GWASs have predominantly been successful in identifying common variants with a small effect size (Figure 1), which, while giving insight into bone biology, have no clear or direct clinical relevance. However, recent GWASs utilizing whole genome sequencing (WGS) data have been able to identify variants with larger effect sizes. In the largest DXA-derived GWAS to date, Zheng et al. showed that rs11692564, a non-coding SNP around 50 kb downstream of EN1, had an estimated effect size of +0.20 SD for lumbar spine BMD (19). Leveraging the Icelandic sequencing initiative with WGS data for >2,000 individuals, rare variants can be imputed and assessed. These data have enabled low frequency variants with large effect sizes for BMD in COL1A2 and LGR4 to be identified (22, 23). Several genomic loci, identified through common genetic variation, have also been linked to genes known to underlie monogenic forms of skeletal pathology. In a large meta-analysis on BMD conducted by Estrada et al. (18), the authors were able to identify 60 genes likely to underlie the association signals. Of these, 13 genes (22%) had been implicated in monogenic skeletal disorders and 27 genes (45%) had a corresponding knockout mouse with a skeletal phenotype (14, 18). This demonstrates that even though the signals picked up by GWASs might indicate a weak effect from the measured variation, it is likely that rare and more damaging genetic variations in the same genomic locus might have a large effect. The genomic areas implicated in these GWASs are therefore likely to be of greater importance than the individual signal divulges (24).
While considering the great success of GWASs, the results need to be interpreted in light of the studied trait. Fracture is the most clinically relevant outcome measured, while BMD represents perhaps the best proxy as it is still considered the main determinant for bone strength, and the main diagnostic measurement for osteoporosis (10, 25). BMD measured by quantitative ultrasound (QUS) of the heel (eBMD) can be used as a cost-effective alternative for BMD and is also independently associated with fractures (ISCD Official positions, 2015). The correlation between BMD and eBMD is, however, not very strong (17, 26). Even the DXA-derived BMD is a blunt measurement for bone health and fracture prediction and needs to be considered with other diagnostic parameters when clinically evaluating a patient's skeletal health (27).
Recent Advances in Genetic Research
As mentioned, several monogenic forms of osteoporosis have been described. Osteogenesis imperfecta (OI) is the best-known form of monogenic osteoporosis and comprises a heterogeneous family of different heritable bone dysplasias with skeletal fragility (28). Parallel to new developments in genetic methodology, new gene discoveries in variable forms of monogenic osteoporosis have been made and, to date, the list of genetic causes of OI and monogenic primary osteoporosis comprises altogether 19 genes (Table 1). The novel genetic findings have considerably enhanced our understanding of the complexities of bone metabolism and uncovered new molecular pathways that regulate bone metabolism and contribute to skeletal pathology. They span beyond the collagen-related pathways to include signaling cascades regulating bone cell function and the extracellular matrix, as described in detail below. The great variability in clinical features and inheritance patterns emphasize the importance of a molecular diagnosis in these patients.
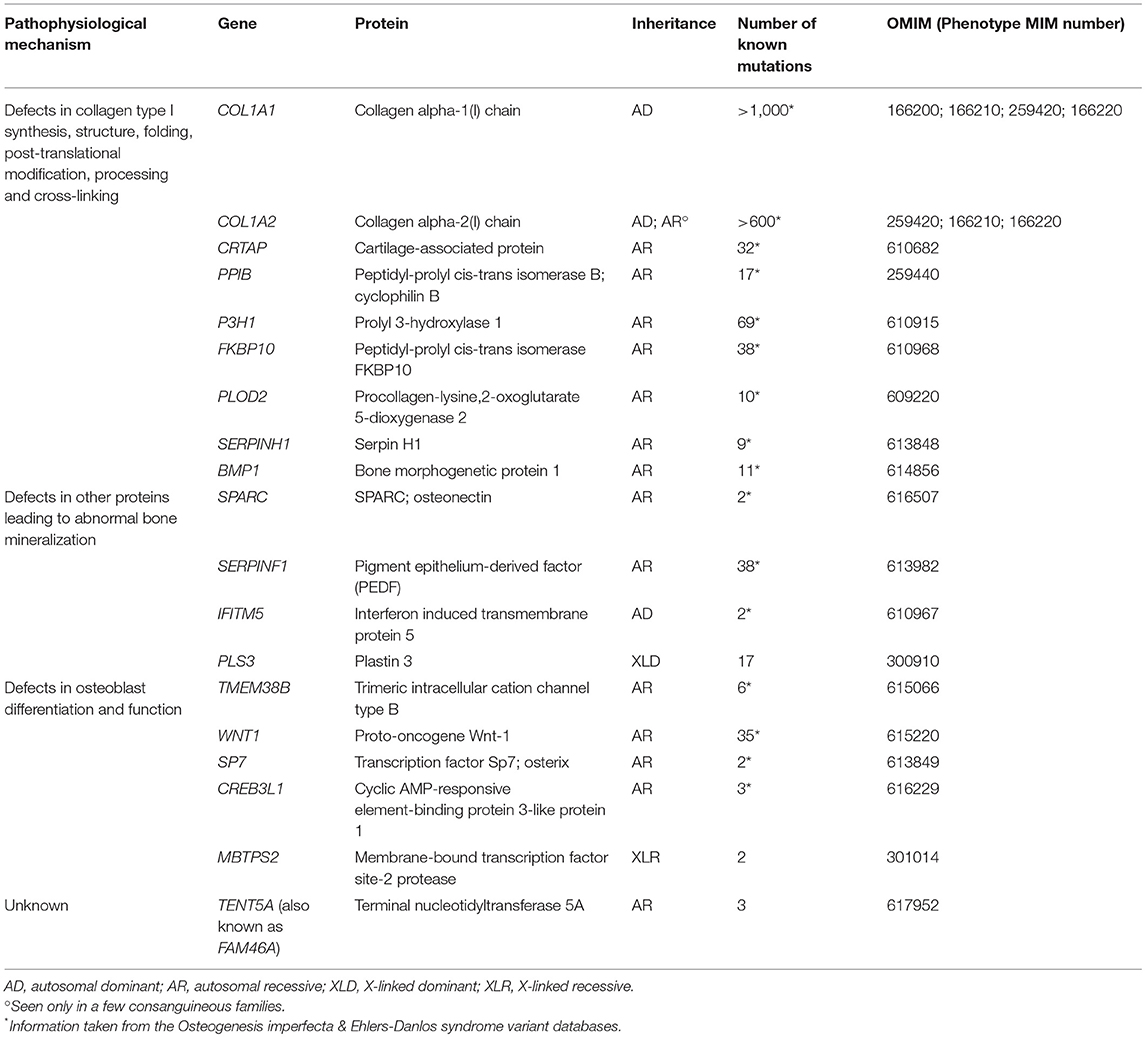
Table 1. Different molecular mechanisms and genes underlying osteogenesis imperfecta.
Paths to Monogenic Osteoporosis
Defects in Bone Cell Function and Bone Remodeling
Normal osteoblast and osteoclast functions are key to sustaining healthy bone tissue. Bone resorption by osteoclasts and formation by osteoblasts are tightly linked in successive repetitive cycles at specific bone sites and the processes are meticulously controlled by several locally produced and circulating systemic factors (29). Communication between the osteoclast and osteoblast is crucial for balanced bone turnover and defects in either cell's function can jeopardize bone health. Osteoblasts express the receptor activator of nuclear factor kappa-B ligand (RANKL), which binds to its conjugate receptor RANK on osteoclast cell surface (Figure 2) (30, 31). This activates osteoclastogenesis and osteoclastic bone resorption. Osteoblasts also secrete osteoprotegerin (OPG) that serves as a decoy receptor for RANKL to inhibit RANKL-RANK–binding, therefore downplaying RANKL's osteoclastogenesis-promoting effect and, as its name implies, protecting bone from over-resorption (Figure 2) (30, 31). Recently, RANK was also noted to relay back by vesicular trafficking from mature osteoclasts to osteoblasts to promote bone formation by reverse signaling (32). The significance of the RANK-RANKL–communication is portrayed in several monogenic conditions with abnormal bone mass resulting from defective RANK-RANKL-OPG–axis: osteoclast-poor osteopetrosis with excessive bone formation due to mutated RANKL, juvenile Paget's disease with osteopenia and progressive skeletal deformity from mutated OPG, and familial expansile osteolysis (FEO) with osteolytic lesions and increased bone remodeling from mutated RANK (33–35).
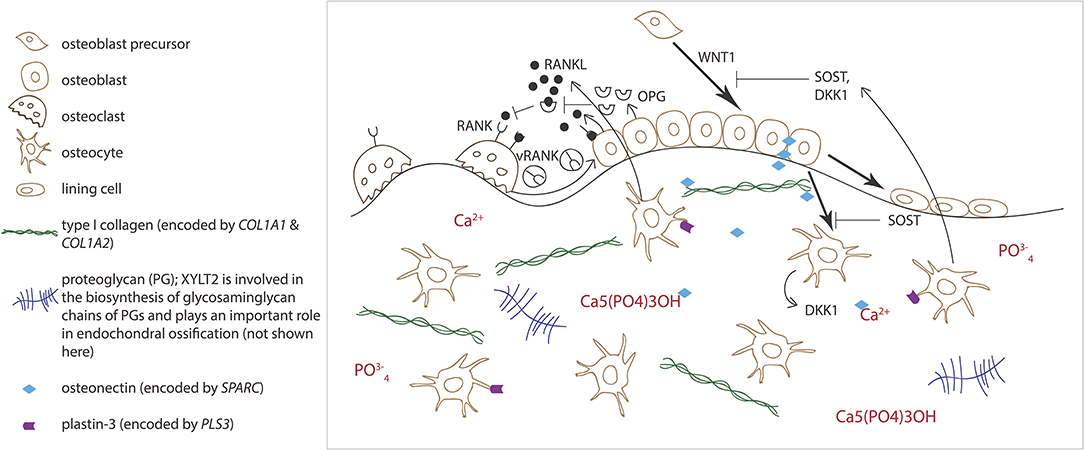
Figure 2. Schematic overview of bone cells and extracellular matrix components involved in regulating bone homeostasis. Receptor activator of nuclear factor kappa-B ligand (RANKL) binds to its conjugate receptor RANK on osteoclast cell surface to stimulate osteoclast differentiation and activity. Osteoprotegerin (OPG) inhibits RANK/RANKL-binding to inhibit bone resorption. WNT signaling pathway stimulates osteoblast function and bone formation. Sclerostin (SOST) and dickkopfs (DKK1), produced by the osteocytes, are two WNT antagonists that promote osteoclasts differentiation. Osteonectin, produced by the osteoblasts, binds calcium, hydroxyapatite and collagen type I and thus regulates bone mineralization. Plastin-3 (PLS3), expressed by the osteocytes, may also be involved in the mineralization of the extracellular matrix but its role in osteoprogenitors and other bone cells is yet to be confirmed.
Alongside osteoblasts and osteoclasts, osteocytes have emerged as key regulators of bone turnover, mineral homeostasis and hematopoiesis (36). Osteocytes are terminally differentiated osteoblasts embedded throughout the mineralized matrix. They communicate with each other and other cells through an extensive network of long cytoplasmic dendritic processes and are thought to orchestrate the interplay between osteoblasts and osteoclasts in bone modeling and remodeling by sensing mechanical loading and responding to endocrine factors, and blood calcium and phosphate concentrations (37). Osteocytes express a range of proteins, such as dentin matric protein 1 (DMP1), phosphate-regulating neutral endopeptidase on chromosome X (PHEX), and matrix extracellular phosphoglycoprotein (MEPE), that are crucial for local matrix mineralization (38). Osteocytes are the primary source of sclerostin, RANKL, and fibroblast growth factor 23 (FGF23), through which osteocytes exert their endocrine functions in bone (Figure 2) (36, 38).
The WNT pathway has a key role in all aspects of bone health—from fetal skeletal development to childhood bone mass accrual to adult bone homeostasis and microarchitectural sustenance (39). WNTs act locally by activating adjacent cells' WNT signaling in a paracrine manner: in developmental stages to partake in the cross-talk between osteoblasts and hematopoietic stem cells (HSCs) in bone marrow and promote bone cell development, differentiation and proliferation, and later in mature adult bone, to induce osteoblastic bone formation (39). WNTs can also act by autocrine means by regulating cells of the same osteoblast or osteoclast lineage (40). The activated pathway is anabolic to bone, leading to increased bone formation and decreased bone resorption. Three different WNT pathways are recognized: the canonical pathway (WNT/β-catenin pathway), the non-canonical planar cell polarity pathway, and the non-canonical WNT/Ca2+ pathway. While the latter two, also known as the β-catenin-independent pathways, participate in a range of development process and in bone metabolism, the canonical WNT/β-catenin pathway is considered the predominant pathway maintaining skeletal health (41).
Dysregulated WNT/β-catenin signaling leads to various skeletal disorders of both high and low bone mass. This was first recognized in 2001 when mutations in low-density lipoprotein 5 (LRP5), encoding a coreceptor for WNT ligands, were found to lead to low bone mass in the autosomal recessive osteoporosis pseudoglioma syndrome (OPPG, MIM 259770), characterized by early-onset severe osteoporosis and blindness (42, 43). The LRP5 mutations inhibit normal WNT signaling and lead to reduced osteoblast proliferation and function and subsequently decreased bone formation (43). Since then, many other mutations in LRP5 have been shown to cause OPPG (44). In addition, functionally significant SNPs in LRP5 have been linked to adolescent bone mass accrual and peak bone mass (45, 46), and genome-wide searches have found common LRP5 polymorphisms that contribute to population-based variance in BMD, confirming its significant role in osteoporosis risk also in the general population (14, 18). The molecular mechanisms by which these missense mutations in LRP5 decrease WNT signaling, however, remain largely unknown (46, 47). Conversely, inadequate WNT inhibition from mutations or deletions in the sclerostin-encoding SOST results in high bone mass phenotypes sclerosteosis (MIM 269500) and van Buchem disease (MIM 239100), respectively (48, 49). In the absence of sufficient sclerostin, WNT signaling is unrestrained, leading to continuous bone formation.
All in all, 19 different WNT proteins are known and together they initiate several intracellular signaling cascades to regulate organogenesis, cell fate determination, primary axis formation, and stem cell renewal (39). Several of the WNT proteins are expressed in bone tissue and regulate bone health at various phases during skeletal growth, development, and e.g., osteoporosis pathogenesis (50). For example, WNT16 is considered an important ligand in bone WNT signaling and has been shown to mediate its bone-specific actions via both canonical and non-canonical WNT pathways (51). Although the specifics behind its mechanisms are unclear, GWASs show that polymorphisms of the WNT16 locus associate with cortical bone thickness, BMD, and osteoporotic fracture risk in large observational studies and variations in WNT16 may also impact individual peak bone mass (18, 52, 53). These findings are echoed in in vivo studies as Wnt16 KO mice have reduced cortical thickness and bone strength leading to spontaneous peripheral fractures (54).
In 2013, several groups identified WNT1 as a key ligand to the WNT pathway in bone; heterozygous WNT1 mutations were reported to cause autosomal dominant osteoporosis, and homozygous mutations, a more severe osteogenesis imperfecta (55). Since then, various other mutations have been found worldwide, all reporting skeletal morbidity with frequent and childhood-onset peripheral and vertebral compression fractures and successive changes in spinal stature (55–61). In our comprehensive clinical analyses of a large cohort of 25 WNT1 mutation-positive subjects with the same heterozygous missense mutation p.C218G, the aberrant WNT1 signaling results in a severe skeletal pathology (62). In addition to prevalent fractures, long bone modeling is altered and BMD low in affected children, while vertebral compression fractures are very common later in adulthood and result in severe kyphotic deformity and loss of adult height soon after the age of 50 years (Figure 3). Bone biopsy histomorphometry demonstrated low-turnover osteoporosis with scarce and inactive bone cells and stagnant bone turnover. Noted extra-skeletal traits included changes in spinal cartilaginous structures, namely vertebral endplate deterioration and frequent Schmorl nodes, and increased reticulin and early-phase–shifted granulopoiesis as signs of abnormal bone marrow function (63, 64).
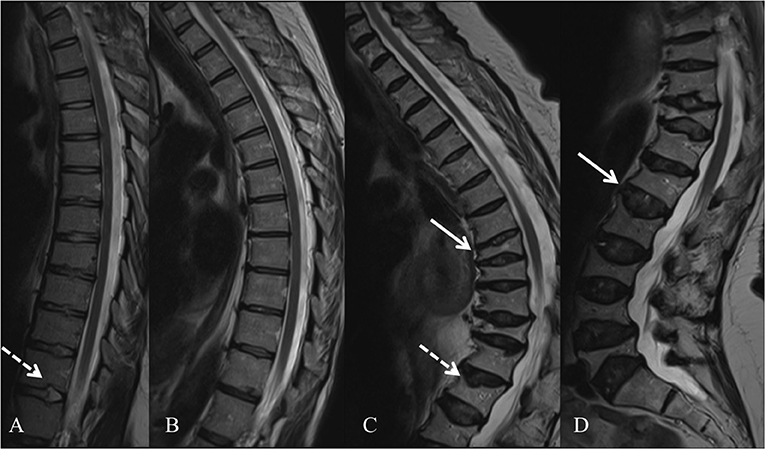
Figure 3. Spinal magnetic resonance images of four WNT1 p.C218G mutation-positive subjects. (A) Thoracic spine of a 17-years-old female showing multiple Schmorl nodes (arrow). (B) Thoracic spine of a 44-years-old female showing exaggerated thoracic kyphosis. (C) Thoracic spine of a 76-years-old male showing several compressed vertebrae, kyphotic stature, and Schmorl hernia (arrow). (D) Lumbar spine of a 74-years-old female showing several compressed vertebrae and enlarged intervertebral discs (arrows). Reprinted from Mäkitie et al. (63) with permission from Elsevier.
The latest finding of dysregulated WNT signaling in monogenic osteoporosis is SFRP4 mutations in Pyle's disease (65). Frizzled-related protein 4 (SFRP4) acts as an WNT inhibitor and biallelic, truncating mutations in its encoding gene SFRP4 result in aberrant regulation of WNT signaling, osteoblasts and osteoclast function and bone remodeling (65). The patients' clinical phenotype is predominated by cortical-bone thinning and fragility and expanded metaphyseal trabecular bone, resulting in limb deformity and high propensity to fracture. Correspondingly, Sfrp4-null mice present with increased trabecular bone, decreased cortical bone and failure in bone modeling (65).
Despite their important functions, known monogenic forms of bone diseases stemming from osteocyte defects are rare and often relate to defective mineral metabolism, especially hypophosphatemia due to disturbed FGF23 regulation. One of the most recently identified monogenic forms of osteoporosis is caused by mutations in the PLS3 gene (66–70), encoding the actin binding, actin bundling protein plastin 3. This X-linked form of primary early-onset osteoporosis is characterized by low BMD, frequent peripheral fractures and vertebral compression fractures, and subsequent severe thoracic kyphosis. Due to its X-chromosomal inheritance, male patients are more severely affected, usually presenting with severe childhood-onset osteoporosis. Clinical manifestations in females with heterozygous PLS3 mutations are variable ranging from subclinical osteopenia to a more severe phenotype resembling that of males' (68). The total number of diagnosed patients is still scarce and hence the comprehension of the clinical and genetic spectrum, the disease progression and appropriate treatment is limited.
While the role of PLS3 in bone fragility is yet unknown, one theory presumes PLS3 to alter osteocyte function through abnormal cytoskeletal microarchitecture. Plastins, in general, are Ca-dependent actin binding and bundling proteins and as such, are involved in cytoskeletal arrangements and partake in regulating cellular morphology, motion, and adherence (71). Despite lack of systematic studies, plastin 3 (also called T-plastin) is supposedly expressed in all solid tissues and through indicated functions in other tissues, such as spinal muscle, inner ear stereocilia, and periodontal ligaments, is suggested to be involved in bone mechano-transduction (72–74). This is supported by the high expression of plastin 3 in chicken osteocyte dendrites, especially during dendrite formation (Figure 2) (75–77). Although this is supported by clinical investigations from biochemical and bone biopsy findings indicating that osteocytes appear affected in PLS3 mutation-positive subjects (78), the observation remains mostly theoretical.
Another suggested role for PLS3 in bone is involvement in mineralization. This is collectively supported by the patients' low BMD and their bone biopsies' histology. We have reported accumulation of non-mineralized osteoid in trabecular bone in patient biopsies (69, 70, 78, 79) and shown that biochemical markers of bone turnover, although not directly echoing the mineralization process, are normal despite altered bone formation (68). The detailed mechanisms of bone tissue mineralization are still debated, but extracellular mineral deposition through budding off of intracellular microvesicles has emerged as one part of the process (80). This process requires dramatic changes in the cell membrane through a complex and well-orchestrated process involving the actin cytoskeleton. Thouverey et al. (81) and Piehl et al. (82) have demonstrated congruently that plastin 3 is involved in the formation of extracellular vesicles. It can thereby be speculated that PLS3 mutations could have deleterious effects on the mineralization process in bone through defective microvesicle formation, although the details behind this too remain undisclosed.
Lastly, a recent experimental animal study presented new findings suggesting involvement of osteoclast malfunction as part of pathophysiology in PLS3 osteoporosis (83). In vivo and in vitro studies using Pls3 knockout and overexpressing mice confirmed the osteoporotic phenotype in the former and thickening cortical bone in the latter. In vitro studies of osteoclasts derived from the animals demonstrated a regulatory role of PLS3 in osteoclastogenesis. Additionally, a dysregulation of osteoclast activity was found in cells from Pls3 knockouts, likely connected to impaired podosome organization due to decreased actin regulation (83). These findings are yet to be confirmed in humans.
Defects in Bone Extracellular Matrix
In addition to bone cells, reduced bone strength and various skeletal disorders can also stem from defects in the extracellular matrix (ECM). The ECM is primarily composed of different collagenous proteins, non-collagenous proteins (in particular glycoproteins and proteoglycans), lipids, minerals and water (84, 85). The most abundant protein is the type I collagen, made of two alpha-1 and one alpha-2 chains intertwined in a triple helical structure. Mutations in the encoding genes, COL1A1 and COL1A2, respectively, lead to qualitative or quantitative defects in the protein and give rise to osteogenesis imperfecta (OI), a skeletal dysplasia characterized by low BMD and enhanced bone fragility, and often extra-skeletal features, such as blue sclerae, dentinogenesis imperfect, and hearing loss (86, 87). Heterozygous glycine substitutions that affect the Gly-Xaa-Yaa pattern in the triple helix are the most common mutations and can cause mild to lethal OI (87). However, multiexonic deletions or deletion of an entire allele have been sporadically found (88–91). Interestingly, mutations that lead to a reduced amount of normal protein give rise to a milder phenotype than missense mutations affecting the primary structure of the triple helix (dominant negative effect) (87). Furthermore, homozygous glycine substitutions in COL1A2 have been identified in a handful of consanguineous families (92–95). Surprisingly, the patients harboring biallelic COL1A2 mutations have a moderate to severe phenotype whereas the mutation carriers are only mildly affected or free from any obvious skeletal impairment. On the other hand, homozygous COL1A1 mutations are likely to be lethal since they have never been reported in humans. Furthermore, some previous reports have indicated that when the COL1A1 or COL1A2 mutation involves the C-propeptide cleavage site, the phenotypic manifestations may include high BMD and mild skeletal fragility (96). A recent study on such cleavage site variants showed that the mutations lead to a distinctive OI phenotype with variable expression, mild to moderate disease severity, moderate fracture rate, high bone mass and increased bone mineral density (97).
Although COL1A1 or COL1A2 mutations are detected in ~85% of OI cases, to date, mutations in altogether 17 other genes are also known to cause OI-like skeletal disorders (Table 1). Some of these genes play a role in the post-translational modification of type I collagen while some are key regulators of osteoblast differentiation and function and/or lead to abnormal bone mineralization (Table 1). One example of severe autosomal recessive OI caused by a mineralization defect is linked to mutations in SPARC (98). The encoded protein Secreted Protein Acidic and Cysteine Rich, better known as osteonectin, is a glycoprotein that is mainly expressed by osteoblasts during bone formation and binds calcium, hydroxyapatite and collagen type I and other proteins in the ECM (Figure 2). Null mutations in SPARC lead to reduced accumulation of type I collagen in the ECM (99). Furthermore, the osteonectin-type I collagen complex is suggested to sequestrate calcium and phosphate in order to initiate bone mineralization (100). An impairment of two other proteins expressed by the osteoblasts, the pigment epithelium-derived factor (encoded by SERPINF1) and the interferon-induced transmembrane protein 5 (encoded by IFITM5), respectively, can also compromise bone mineralization and lead to OI (86, 87, 101, 102). Most recently, mutations in FAM46A, encoding the terminal nucleotidyltransferase 5A, have been detected in four patients with OI. However, the molecular function of this protein and the pathophysiological mechanism by which the mutations lead to OI are not yet known (103).
Besides OI, there are several other skeletal syndromes that feature osteoporosis and are caused by defects in the ECM. For example, mutations in XYLT2 lead to spondyloocular syndrome characterized by childhood-onset osteoporosis, cataract, cardiac defects and hearing impairment (104–106). The mutated protein xylosyltransferase 2 is involved in the biosynthesis of glycosaminoglycan chains and plays an important role in endochondral ossification and chondrocyte differentiation and maturation. Proteoglycans are also important for other tissues and organs, including brain, heart, and retina, which could explain why the clinical manifestations of spondyloocular syndrome are not only restricted to the skeleton (106).
In addition to causing autosomal recessive OI, inadequate folding and post-translational modification of type I collagen can result in another skeletal syndrome characterized by congenital contractures, named Bruck syndrome. Homozygous mutations in FKBP10 and PLOD2 result in Bruck syndrome 1 and 2, respectively (107–110). FKBP10 encodes the immunophilin FKBP65, a molecular chaperon of type I collagen and PLOD2 encodes the procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2, which catalyzes the hydroxylation of lysyl residues in type I collagen. Mutations in both FKBP10 and PLOD2 can also cause autosomal recessive OI (Table 1).
Tools for Diagnosing Monogenic Osteoporosis
Uncovering the Genetics
As discussed, to make the diagnosis of osteoporosis in children two criteria need to be met; (1) low BMD or BMC (Z-score ≤ −2.0 SD) and (2) a clinically significant fracture history. A vertebral fracture indicates severely compromised bone strength and suffices alone for the diagnosis (12). The diagnosis of primary osteoporosis in children can be made when potential causes of secondary osteoporosis, such as other underlying illnesses or medical treatments, have been excluded (2). Most forms of childhood-onset primary osteoporosis are termed osteogenesis imperfecta, although the diagnosis is vague and merely appoints the disease to belong to a heterogeneous group of skeletal disorders with diverse clinical presentation (86, 87). As indicated earlier, the genetic background of OI is heterogeneous and the phenotypic and genetic variability have complicated OI classification. As of yet, there is no consensus indicating which genotype-phenotype combinations should be classified under the umbrella of OI and which should not. The current classification of OI is based on phenotypic features, but the molecular cause is often the key factor determining clinical prognosis, appropriate treatment approach and recurrence risk in the family, and should therefore be emphasized (28). A molecular diagnosis also facilitates the refinement of future treatment and clinical care protocols (87, 111). Although more than 85% of OI cases can still be traced to pathogenic variants in either of the two collagen type I–coding genes COL1A1 or COL1A2 (112, 113), the several other genes identified over the past 12 years in OI or monogenic forms of primary osteoporosis need to be kept in mind (92, 114, 115).
While most clinicians begin by screening COL1A1 and COL1A2 possibly in combination with MLPA, proceeding to a full OI gene panel using massive parallel sequencing is recommended (87). A sequencing-based gene panel will not only capture sequence variants but also possible structural variations including larger deletions and duplications. Although the surge of new genetic findings has facilitated interpretation of sequence variants, deep intronic splice variants or splice variants masked as synonymous variants are still difficult to correctly annotate. Transcriptome analysis using RNA sequencing together with DNA sequencing has proven successful in increasing the diagnostic yield and assessing functional impact of variants that are otherwise hard to interpret (116). This, however, requires that the disease in focus has a readily accessible proxy tissue, where the gene expression reflects the expression in the affected tissue. Unfortunately, tissue accessibility is very difficult in bone diseases and the method cost-restricted in clinical settings.
Regarding structural variants, WGS has provided an advantage in assessing structural variants compared to exome sequencing or other capture-based protocols. However, all short-read sequencing technologies have shortcomings in their ability to detect and identify structural variants, and, as concluded by Telenti et al. (117), after sequencing 10 000 human genomes the interpretation of structural variants on an individual level still remains challenging. Older methods to indirectly detect structural variations, such as array-based comparative genomic hybridization (array-CGH), are still applicable in specific cases and can help clinicians in their search for a molecular diagnosis (91).
Clinical Characterization
Owning to the wide spectrum of genetic causes, the clinical presentation of different OI and primary osteoporosis forms is unsurprisingly miscellaneous (87). The diseases vary in their primary skeletal traits, age-at-onset, natural progression, sensitivity to treatment, and presence and spectrum of extra-skeletal characteristics. Although severely compromised bone strength is usually a unifying finding, the DXA-derived BMD, bone biopsy findings, prevalence and type of fractures, and radiographic findings are inconsistent. The phenotypic severity can vary from mild to severe and disease onset from childhood to early adulthood—at times provoked by pregnancy-related calcium loss. Presentation may vary between patients with different mutations and even between family members with identical mutations (87). Classical OI-related extra-skeletal findings include blue sclerae, increased joint laxity, dentinogenesis imperfecta and impaired hearing (28, 87, 118). Mutations in proteins affecting the collagen-related pathways all seem to exhibit similar traits; only the severity and array of affected skeletal sites vary. Some typical presentations include popcorn epiphyseal plates in CRTAP, calcifications of interosseous membranes and hyperplastic callus formation in IFITM5, and skull ossification defects in SEC24D-related OI (Table 1) (86, 87, 118). The extra-skeletal manifestations of bone cell-related forms are still incompletely defined; with monoallelic WNT1 mutations patients have changes in spinal cartilaginous structures (63) and mild abnormalities in bone marrow hematopoiesis and reticulin formation (119), while in biallelic mutations the phenotype is more severe and OI-like but no bone marrow defects have been reported (55). However, central nervous system manifestations have been reported in some patients with homozygous WNT1 mutations (55, 61). Patients with PLS3 mutations do not exhibit any apparent extra-skeletal traits, though this is still scantily explored.
Novel Biomarkers
In addition to DXA and plain radiography, several factors can be measured from systemic circulation and urine when diagnosing and monitoring patients' disease state, progression and treatment response. The conventional metabolic markers reflect bone turnover and consist of enzymatic and proteinaceous by-products; the most widely used resorption markers include mainly by-products of collagen breakdown, [urinary collagen type 1 cross-linked N-telopeptide (NTX), urinary/serum collagen type 1 cross-linked C-telopeptide (CTX), and collagen fragments from matrix-metalloproteases (ICTP)], and formation markers procollagens from collagen synthesis [serum amino-terminal propeptide (PINP) and carboxyl-terminal propeptide (PICP)] or osteoblast-related proteins (serum osteocalcin (OC) and serum bone isoenzyme of alkaline phosphatase (ALP) (120, 121). While these markers are commonly used and easily analyzed in automated routine laboratories, they do lack specificity and are easily confounded by other patient-related (e.g., body adiposity, inflammation, blood glucose level, time of sampling) and analytical factors. Furthermore, they often respond inadequately to bisphosphonate treatment and correlate poorly with BMD and bone histomorphometric parameters (55, 120–125). None of the monogenic forms of osteoporosis have a specific biomarker profile and these conventional markers are of little value in differentiating between the various genetic forms of osteoporosis.
The limitations of the conventional bone markers have fueled a field-wide search for new potential biomarkers. Zooming into smaller cell-released particles, small microRNAs (miRNAs), as one, have attained much attention and are proposed to hold promise in future diagnostic and treatment in skeletal disorders. These small, non-coding fragments of RNA are highly conserved and comprise, on estimate, 1% of our genome (126, 127). They alter gene expression by RNA silencing and post-transcriptional regulation; each miRNA is predicted to regulate hundreds of different target genes, thus serving important functions in many tissues and biological processes (127, 128). While their exact function in gene regulation is still largely unknown, miRNAs are thought to mediate intercellular communications in various metabolic processes and diseases and a unique imprint of differentially expressed miRNAs is observed in e.g., certain cancers, metabolic diseases and viral infections. In bone, miRNAs contribute to homeostasis and their dysfunctional expression relays to progression of skeletal disorders (129, 130). Their expressions change in result of low BMD, frequent fractures, or menopausal osteoporosis (129, 130).
These findings have encouraged researchers to explore the clinical potential of miRNAs in disease diagnostics and follow-up. Several clinical studies have evaluated miRNA expression in osteoporotic patients and distinguished specific miRNAs correlating with the degree of osteoporosis (131). miR-133a was significantly elevated in postmenopausal Caucasian women with low BMD (132), and miR-194-5p and miR-21-5p negatively correlated with BMD in Chinese osteoporotic women (133, 134). Seeliger et al. (135) also identified miR-21-5p, in addition to four other miRNAs (miR-23a-3p, miR-24-3p, miR-100-5p, and miR-125b-5p) to be differentially expressed in serum and upregulated in bone tissue in patients with osteoporotic fractures. In vitro studies have observed miRNAs that interact with known key regulators of bone metabolism, such as miR-152-3p and miR-335-3p with Dickkopf-1 (136, 137), miR-30e-5p with Lrp6 (138), and the aforementioned miR-133 with Runx2 (139). Furthermore, Anastasilakis et al. (140) reported that serum levels of miRNAs changed in response to anti-osteoporotic treatment. While different studies pinpoint to varying miRNAs depending on cohort size, demographic or other factors, a clear congruency is echoed that a unique miRNA signature is observed in osteoporosis.
We have reported altered miRNA pattern in patients with WNT1 osteoporosis, with two upregulated and six downregulated miRNAs, as compared with age and sex-matched mutation-negative controls from the same family (119). While specific miRNA alterations may be recognized in certain monogenic forms of osteoporosis, the role of miRNAs in complementing or substituting genetic testing remains to be explored in future studies. Further, the utilization of miRNA assessments in clinical practice demands further methodological development but based on present data, they hold great potential for future diagnosis and follow-up, including monogenic forms of osteoporosis.
Options for Treatment
Conventional Osteoporosis Drugs and Implications for Treatment
Conventional osteoporosis drugs, namely bisphosphonates, have been the mainstay of pharmacological treatment in classical, type I collagen-related OI forms. These typically have high bone turnover and thus the osteoclast-targeting and resorption-decreasing bisphosphonates have proven effective in increasing BMD, reducing fractures, and improving VCFs in patients (141–143). Contrary to collagen I-related OI, bisphosphonates have proven insufficient in improving BMD or fracture tendency in several new forms of primary osteoporosis (55, 57, 60). These OI forms often present with low-turnover osteoporosis and hence the benefits of anti-catabolic treatment are not optimal. We have also shown that patients with prior bisphosphonate treatment have abnormal and apoptotic osteocytes, suggesting adverse effects of bisphosphonates in WNT1 osteoporosis (63). However, our longitudinal study on the effects of teriparatide-treatment in WNT1 osteoporosis indicated that exogenous PTH may be efficient in increasing bone formation and BMD during a 24-months-long treatment in adults; however, there may be simultaneous increase in bone marrow adiposity (79). Thus far, the efficacy of anti-sclerostin antibodies have been experimented in mice only; subcutaneous administration of Scl-Ab to the murine model of WNT1 OI Wnt1sw/sw mice significantly improved fracture rate and increased bone mass that seemed to result from increased osteoblast activity (144).
Besides WNT1-related skeletal pathologies, even less is known about the optimal treatments in other new forms of primary osteoporosis and OI, such as PLS3 and XYLT2 (105, 145). Efficacy of bisphosphonates in PLS3 osteoporosis has been evaluated in a handful of cases and indicate positive response (66, 67, 70). Our above-mentioned clinical study on teriparatide also included PLS3 mutation-positive subjects and they showed congruent, although slightly lesser, improvement in bone parameters in 24-months follow-up, as compared with patients with WNT1 osteoporosis (79). Patients with XYLT2 mutations seem to benefit from pamidronate treatment with increase in BMD and improvement in vertebral morphology (104, 105).
Clinical care of OI patients, including both classical and newer forms of OI and monogenic osteoporosis, is often complex and challenging. Means of treatment and pace of clinical follow-up are dependent on the patient's age, clinical manifestations, and degree of impairment, and should be individually tailored and regularly evaluated. Bisphosphonates are still the main treatment option for pediatric patients and are often used to prevent greater decrease in BMD and enable maximum yield in bone mineral throughout childhood and adolescent bone mass accrual. The overall benefits of bisphosphonate treatment in most cases of OI are non-negligible (146). Variable treatment protocols exist. Clinical care and follow-up are advised to be centered in special health care units with abilities to provide multidisciplinary care and expertise.
Novel Target-Drugs
Discoveries through rare, monogenic forms of skeletal disorders have provided new information on the biology of bone health and revealed previously unidentified proteins that take part in key regulatory pathways. Naturally, these proteins also present as appealing target molecules for development of new treatment modalities. In early 2000s, inhibition of RANKL by a monoclonal antibody denosumab brought a novel approach for treatment of osteoporosis (147). The drug has been used to improve skeletal health in some forms of OI. Particularly patients with SERPINF1 mutations show a modest increase in BMD in response to denosumab whereas treatment outcomes with bisphosphonates are poor (148). Due to the coupled nature of osteoblast-osteoclast–activity, blocking osteoclastogenesis through RANKL is also unfavorably accompanied by reduced osteoblast function. The previously mentioned discovery of RANKL reverse signaling could offer a novel solution to avoid this problem (32). Also, inhibition of cathepsin K, an osteoclast-derived lysosomal enzyme, seemed promising due to its coupled bone formation-favoring action, but its development was later discontinued due to increased risk of cardiovascular complications (149). As of recently, the effects of anti-TGF-β antibodies have been studied in Crtap−/− and Col1a1frt/− mice with varying results; while the Crtap−/− showed great improvements in bone mass and biochemical qualities, Col1a1frt/− mice did not show significant changes in bone quality or strength (150).
Along with the discovery of van Buchem disease and sclerosteosis, two human models of sclerostin inhibition, fueled the development of a new anabolic target drug named romosozumab—a monoclonal anti-sclerostin antibody targeting the WNT pathway (151, 152). Its efficacy has been evaluated in several clinical trials with promising results; a placebo-controlled, multicenter, phase II study on 419 postmenopausal women with osteoporosis treated with subcutaneous injections of romosozumab at 3-months intervals showed significant, and superior to those attained by alendronate and teriparatide, increase in areal BMD and a tilt in BTMs reflective of increased bone formation (151), and another phase III study reported a reduction in fracture risk in 7,180 postmenopausal osteoporotic women (153). Anti-DKK1 antibodies act similarly to oppose WNT signaling and are potent as osteoanabolic agents. However, administration of anti-DKK1 is only mildly efficacious as the WNT-neutralizing effect is compensated by upregulation of sclerostin, although the opposite is not seen when given only anti-sclerostin antibodies. Thus, the benefits of anti-DKK1 antibodies manifest only when given in conjunction with anti-sclerostin (154).
Another target of interest for new drug development is Notum. It is a secreted enzyme that inhibits WNTs by removing the palmitoleic acid group that is essential for binding of WNTs to Frizzled receptors, thereby inhibiting WNT signaling. Interestingly, experimental studies in rodents have shown that inhibiting Notum through either knockout, or by oral administration of molecular inhibitors or neutralizing antibodies increase cortical bone formation and strength, but do not affect trabecular bone mass (155, 156).
Possible undesired adverse and extra-skeletal effects of new drugs are inevitable as many of the targeted proteins have tissue-wide expression and key roles in various biological processes. Side effects can be latent and subtle but also challenging and life-threatening. Knowing the WNT pathway's fundamental role in embryonic development, tumorigenesis and pathogenesis of other systemic or chronic diseases, romosozumab has been under careful scrutiny for its clinical safety. In mice receiving different doses, no malignancies were noted over a 98-weeks follow up (157). However, along with the robust and positive skeletal effects, use of romosozumab has been associated with cardiovascular and cerebrovascular events, and the drug is currently under FDA review (Amgen and UCB).
MicroRNAs
Recently, researchers have acknowledged the opportunities in targeting miRNA pathways to develop new therapeutic means and genome editing approaches (128, 158). A few groups have pursued clinical trials to evaluate efficacy of miRNAs in disease target treatment: an on-going clinical trial evaluates the anticancer effect of miRNA lethal-7 in binding to Kirsten rat sarcoma viral oncogene homolog (KRAS) gene in patients suffering from stage III colon cancer, and miR-122 in hepatitis C (159, 160). Bone-specific miRNAs have not been evaluated clinically, but analyses have shown that for example in vitro miR-21 could promote osteogenesis in bone marrow stem cells, and systemic administration of miR-214 induced BMD increase and miR-92a enhance fracture healing in mice (161–163). In fracture healing, also angiogenesis is vital to the repair process and Li et al. (164) were able to demonstrate that implantation of MSCs transfected with an angiogenesis-involved anti-miR-26a showed good bone repair. Further, anti-miR-31-transfected MCSs efficiently repaired bone defects by increasing BMD and new bone volume (165). These findings and the efficacy, safety and possible side effects need to be confirmed and carefully evaluated in clinical settings in vivo.
Conclusions
Recent advances in genetic methodology have resulted in several new discoveries relating to the genetic architecture of bone homeostasis. Not only have the basic clinical and genetic pillars of classical OI been refined, but several new forms of monogenic osteoporosis have also been identified that have pinpointed novel molecular mechanisms contributing to skeletal health and disease. The clinical presentation, inheritance mode, natural course and response to conventional osteoporosis drugs are diverse, often variable and logically dependent on the affected protein. Although uncovering the limitations in our current diagnostic and treatment modalities, they have also provided new signaling pathways that hold promise in new targeted drug development. Future research will hopefully continue expanding the genetics and molecular mechanisms behind bone metabolism and increasing our understanding of the specific skeletal and extra-skeletal characteristics of monogenic osteoporosis, while finding new avenues for improved diagnosis and treatment of patients with severe bone diseases.
Author Contributions
RM and OM initiated the manuscript. RM wrote the first draft. All authors contributed to the writing and approved the final manuscript.
Funding
Our research is supported by research funding from Novo Nordisk Foundation, Academy of Finland, Vetenskapsrådet, Sigrid Jusélius Foundation, Folkhälsan Research Foundation, Foundation for Pediatric Research, Karolinska Institutet's KID funding, Swedish Childhood Cancer Foundation, University of Helsinki and Helsinki University Hospital through the Doctoral Programme in Clinical Research, Finnish Medical Foundation, Biomedicum Helsinki Foundation, Finnish-Norwegian Medical Association, Helsinki Medical Association, Emil Aaltonen Foundation, and Jalmari and Rauha Ahokas Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Florencio-Silva R, Sasso GR, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int. (2015) 2015:421746. doi: 10.1155/2015/421746
2. Mäkitie O. Causes, mechanisms and management of paediatric osteoporosis. Nat Rev Rheumatol. (2013) 9:465–75. doi: 10.1038/nrrheum.2013.45
3. Baxter-Jones AD, Faulkner RA, Forwood MR, Mirwald RL, Bailey DA. Bone mineral accrual from 8 to 30 years of age: an estimation of peak bone mass. J Bone Miner Res. (2011) 26:1729–39. doi: 10.1002/jbmr.412
4. Ward LM, Ma J, Lang B, Ho J, Alos N, Matzinger MA, et al. Steroid-Associated Osteoporosis in the Pediatric Population (STOPP) Consortium. Bone morbidity and recovery in children with acute lymphoblastic leukemia: results of a six-year prospective cohort study. J Bone Miner Res. (2018) 33:1435–43. doi: 10.1002/jbmr.3447
5. Markula-Patjas KP, Valta HL, Kerttula LI, Soini IH, Honkanen VE, Toiviainen-Salo SM, et al. Prevalence of vertebral compression fractures and associated factors in children and adolescents with severe juvenile idiopathic arthritis. J Rheumatol. (2012) 39:365–73. doi: 10.3899/jrheum.110305
6. LeBlanc CM, Ma J, Taljaard M, Roth J, Scuccimarri R, Miettunen P, et al. Incident vertebral fractures and risk factors in the first three years following glucocorticoid initiation among pediatric patients with rheumatic disorders. J Bone Miner Res. (2015) 30:1667–75. doi: 10.1002/jbmr.2511
7. Joseph S, Wang C, Di Marco M, Horrocks I, Abu-Arafeh I, Baxter A, et al. Fractures and bone health monitoring in boys with Duchenne muscular dystrophy managed within the Scottish Muscle Network. Neuromuscul Disord. (2018). doi: 10.1016/j.nmd.2018.09.005. [Epub ahead of print].
8. Gray N, Howard A, Zhu J, Feldman LY, To T. Association between inhaled corticosteroid use and bone fracture in children with asthma. JAMA Pediatr. (2018) 172:57–64. doi: 10.1001/jamapediatrics.2017.3579
9. Camacho PM, Petak SM, Binkley N, Clarke BL, Harris ST, Hurley DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology clinical practice guidelines for the diagnosis and treatment of postmenopausal osteoporosis−2016. Endocr Pract. (2016) 22:1–42. doi: 10.4158/EP161435.GL
10. NIH. (2001). NIH consensus development panel on osteoporosis prevention, diagnosis, and therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 285, 785–95.
11. Mäkitie O, Doria AS, Henriques F, Cole WG, Compeyrot S, Silverman E, et al. Radiographic vertebral morphology: a diagnostic tool in pediatric osteoporosis. J Pediatr. (2005) 146:395–401. doi: 10.1016/j.jpeds.2004.10.052
12. Bishop N, Arundel P, Clark E, Dimitri P, Farr J, Jones G, et al., International Society of Clinical Densitometry. Fracture prediction and the definition of osteoporosis in children and adolescents: the ISCD 2013 Pediatric Official Positions. J Clin Densitom. (2014) 17:275–80. doi: 10.1016/j.jocd.2014.01.004
13. Ralston SH, Uitterlinden AG. Genetics of osteoporosis. Endocr Rev. (2010) 31:629–62. doi: 10.1210/er.2009-0044
14. Rivadeneira F, Mäkitie O. Osteoporosis and bone mass disorders: from gene pathways to treatments. Trends Endocrinol Metab. (2016) 27:262–81. doi: 10.1016/j.tem.2016.03.006
15. Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. (2009) 360:1759–68. doi: 10.1056/NEJMra0808700
16. Clark GR, Duncan EL. The genetics of osteoporosis. Br Med Bull. (2015) 113:73–81. doi: 10.1093/bmb/ldu042
17. Kemp JP, Morris JA, Medina-Gomez C, Forgetta V, Warrington NM, Youlten SE, et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat Genet. (2017) 49:1468–75. doi: 10.1038/ng.3949
18. Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. (2012) 44:491–501. doi: 10.1038/ng.2249
19. Zheng HF, Forgetta V, Hsu YH, Estrada K, Rosello-Diez A, Leo PJ, et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature (2015) 526:112–7. doi: 10.1038/nature14878
20. Peacock M, Turner CH, Econs MJ, Foroud T. Genetics of osteoporosis. Endocr Rev. (2002) 23:303–26. doi: 10.1210/edrv.23.3.0464
21. Hernandez-de Sosa N, Athanasiadis G, Malouf J, Laiz A, Marin A, Herrera S, et al. Heritability of bone mineral density in a multivariate family-based study. Calcif Tissue Int. (2014) 94:590–6. doi: 10.1007/s00223-014-9852-9
22. Styrkarsdottir U, Thorleifsson G, Eiriksdottir B, Gudjonsson SA, Ingvarsson T, Center JR, et al. Two rare mutations in the COL1A2 gene associate with low bone mineral density and fractures in Iceland. J Bone Miner Res. (2016) 31:173–9. doi: 10.1002/jbmr.2604
23. Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature (2013) 497:517–20. doi: 10.1038/nature12124
24. Panagiotou OA, Evangelou E, Ioannidis JP. Genome-wide significant associations for variants with minor allele frequency of 5% or less–an overview: a HuGE review. Am J Epidemiol. (2010) 172:869–89. doi: 10.1093/aje/kwq234
25. Kanis JA, Harvey NC, Johansson H, Oden A, McCloskey EV, Leslie WD. Overview of fracture prediction tools. J Clin Densitom. (2017) 20:444–50. doi: 10.1016/j.jocd.2017.06.013
26. Gonnelli S, Cepollaro C, Gennari L, Montagnani A, Caffarelli C, Merlotti D, et al. Quantitative ultrasound and dual-energy X-ray absorptiometry in the prediction of fragility fracture in men. Osteoporos Int. (2005) 16:963–8. doi: 10.1007/s00198-004-1771-6
27. Marques A, Lucas R, Simoes E, Verstappen SMM, Jacobs JWG, da Silva JAP. Do we need bone mineral density to estimate osteoporotic fracture risk? A 10-year prospective multicentre validation study. RMD Open (2017) 3:e000509. doi: 10.1136/rmdopen-2017-000509
28. Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A (2015) 167A:2869–92. doi: 10.1002/ajmg.a.37365
30. Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology (2001) 142:5050–5. doi: 10.1210/endo.142.12.8536
31. Zuo C, Huang Y, Bajis R, Shih M, Li YP, Dai K, et al. Osteoblastogenesis regulation signals in bone remodeling. Osteoporos Int. (2012) 23:1653–63. doi: 10.1007/s00198-012-1909-x
32. Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature (2018) 561:195–200. doi: 10.1038/s41586-018-0482-7
33. Wright HL, McCarthy HS, Middleton J, Marshall MJ. RANK, RANKL and osteoprotegerin in bone biology and disease. Curr Rev Musculoskelet Med. (2009) 2:56–64. doi: 10.1007/s12178-009-9046-7
34. Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. (2013) 9:522–36. doi: 10.1038/nrendo.2013.137
35. Polyzos SA, Cundy T, Mantzoros CS. Juvenile Paget disease. Metabolism (2018) 80:15–26. doi: 10.1016/j.metabol.2017.10.007
36. Divieti Pajevic P. Recent progress in osteocyte research. Endocrinol Metab. (2013) 28:255–61. doi: 10.3803/EnM.2013.28.4.255
37. Uda Y, Azab E, Sun N, Shi C, Pajevic PD. Osteocyte mechanobiology. Curr Osteoporos Rep. (2017) 15:318–25. doi: 10.1007/s11914-017-0373-0
38. Plotkin LI, Bellido T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat Rev Endocrinol. (2016) 12:593–605. doi: 10.1038/nrendo.2016.71
39. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. (2013) 19:179–92. doi: 10.1038/nm.3074
40. Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. (2012) 18:405–12. doi: 10.1038/nm.2653
41. Sugimura R, Li L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Res C Embryo Today (2010) 90:243–56. doi: 10.1002/bdrc.20195
42. Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R, et al. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12-13. Am J Hum Genet. (1996) 59:146–51.
43. Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al., Osteoporosis-Pseudoglioma Syndrome Collaborative Group. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell (2001) 107:513–23. doi: 10.1016/S0092-8674(01)00571-2
44. Levasseur R, Lacombe D, de Vernejoul MC. LRP5 mutations in osteoporosis-pseudoglioma syndrome and high-bone-mass disorders. Joint Bone Spine (2005) 72:207–14. doi: 10.1016/j.jbspin.2004.10.008
45. Saarinen A, Välimäki VV, Välimäki MJ, Löyttyniemi E, Auro K, Uusen P, et al. The A1330V polymorphism of the low-density lipoprotein receptor-related protein 5 gene (LRP5) associates with low peak bone mass in young healthy men. Bone (2007) 40:1006–12. doi: 10.1016/j.bone.2006.11.010
46. Hartikka H, Mäkitie O, Männikkö M, Doria AS, Daneman A, Cole WG, et al. Heterozygous mutations in the LDL receptor-related protein 5 (LRP5) gene are associated with primary osteoporosis in children. J Bone Miner Res. (2005) 20:783–9. doi: 10.1359/JBMR.050101
47. Saarinen A, Saukkonen T, Kivelä T, Lahtinen U, Laine C, Somer M, et al. Low density lipoprotein receptor-related protein 5 (LRP5) mutations and osteoporosis, impaired glucose metabolism and hypercholesterolaemia. Clin Endocrinol Oxf. (2010) 72:481–8. doi: 10.1111/j.1365-2265.2009.03680.x
48. Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. (2005) 15:928–35. doi: 10.1101/gr.3437105
49. Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. (2001) 10:537–43. doi: 10.1093/hmg/10.5.537
50. Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S. Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns (2009) 9:215–23. doi: 10.1016/j.gep.2008.12.009
51. Movérare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Börjesson AE, et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med. (2014) 20:1279–88. doi: 10.1038/nm.3654
52. Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. (2012) 8:e1002745. doi: 10.1371/journal.pgen.1002745
53. Gori F, Lerner U, Ohlsson C, Baron R. A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. Bonekey Rep. (2015) 4:669. doi: 10.1038/bonekey.2015.36
54. Ohlsson C, Henning P, Nilsson KH, Wu J, Gustafsson KL, Sjögren K, et al. Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice. J Endocrinol. (2018) 237:113–22. doi: 10.1530/JOE-18-0020
55. Laine CM, Joen KS, Laine CM, Joeng KS, Campeau PM, Kiviranta R, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. (2013) 368:1809–16. doi: 10.1056/NEJMoa1215458
56. Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. (2013) 92:590–7. doi: 10.1016/j.ajhg.2013.02.009
57. Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. (2013) 92:565–74. doi: 10.1016/j.ajhg.2013.02.010
58. Faqeih E, Shaheen R, Alkuraya FS. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. J Med Genet. (2013) 50:491–2. doi: 10.1136/jmedgenet-2013-101750
59. Stephen J, Girisha KM, Dalal A, Shukla A, Shah H, Srivastava P, et al. Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. Eur J Med Genet. (2015) 58:21–7. doi: 10.1016/j.ejmg.2014.10.001
60. Liu Y, Song L, Ma D, Lv F, Xu X, Wang J, et al. Genotype-phenotype analysis of a rare type of osteogenesis imperfecta in four Chinese families with WNT1 mutations. Clin Chim Acta (2016) 461:172–80. doi: 10.1016/j.cca.2016.07.012
61. Aldinger KA, Mendelsohn NJ, Chung BH, Zhang W, Cohn DH, Fernandez B, et al. Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations. J Med Genet. (2016) 53:427–30. doi: 10.1136/jmedgenet-2015-103476
62. Mäkitie RE, Haanpää M, Valta H, Pekkinen M, Laine CM, Lehesjoki AE, et al. Skeletal characteristics of WNT1 osteoporosis in children and young adults. J Bone Miner Res. (2016) 31:1734–42. doi: 10.1002/jbmr.2841
63. Mäkitie RE, Niinimäki T, Nieminen MT, Schalin-Jäntti C, Niinimäki J, Mäkitie O. Impaired WNT signaling and the spine–Heterozygous WNT1 mutation causes severe age-related spinal pathology. Bone (2017) 101:3–9. doi: 10.1016/j.bone.2017.04.001
64. Mäkitie RE, Niinimäki R, Kakko S, Honkanen T, Kovanen PE, Mäkitie O. Defective WNT signaling and bone marrow fibrosis–a cross-sectional study on 10 adults with WNT1 osteoporosis. Osteoporosis Int. (2018) 29:479–87. doi: 10.1007/s00198-017-4309-4
65. Kiper POS, Saito H, Gori F, Unger S, Hesse E, Yamana K, et al. Cortical-bone fragility–insights from sFRP4 deficiency in Pyle's disease. N Engl J Med. (2016) 374:2553–62. doi: 10.1056/NEJMoa1509342
66. van Dijk FS, Zillikens MC, Micha D, Riessland M, Marcelis CL, de Die-Smulders CE, et al. PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med. (2013) 369:1529–36. doi: 10.1056/NEJMoa1308223
67. Fahiminiya S, Majewski J, Al-Jallad H, Moffatt P, Mort J, Glorieux FH, et al. Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J Bone Mineral Res. (2014) 29:1805–14. doi: 10.1002/jbmr.2208
68. Laine CM, Wessman M, Toiviainen-Salo S, Kaunisto MA, Mäyränpää MK, Laine T, et al. A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J Bone Miner Res. (2015) 30:510–8. doi: 10.1002/jbmr.2355
69. Kämpe AJ, Costantini A, Levy-Shraga Y, Zeitlin L, Roschger P, Taylan F, et al. PLS3 deletions lead to severe spinal osteoporosis and disturbed bone matrix mineralization. J Bone Miner Res. (2017) 32:2394–404. doi: 10.1002/jbmr.3233
70. Kämpe AJ, Costantini A, Mäkitie RE, Jäntti N, Valta H, Mäyränpää M, et al. PLS3 sequencing in childhood-onset primary osteoporosis identifies two novel disease-causing variants. Osteoporos Int. (2017) 28:3023–32. doi: 10.1007/s00198-017-4150-9
71. Schwebach CL, Agrawal R, Lindert S, Kudryashova E, Kudryashov DS. The roles of actin-binding domains 1 and 2 in the calcium-dependent regulation of actin filament bundling by human plastins. J Mol Biol. (2017) 429:2490–508. doi: 10.1016/j.jmb.2017.06.021
72. Oprea GE, Kroeber S, McWhorter ML, Rossoll W, Mueller S, Krawczak M, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science (2008) 320:524–7. doi: 10.1126/science.1155085
73. Höfer D, Drenckhahn D. Cytoskeletal differences between stereocilia of the human sperm passageway and microvilli/stereocilia in other locations. Anat Rec. (1996) 245:57–64. doi: 10.1002/(SICI)1097-0185(199605)245:1<57::AID-AR10>3.0.CO;2-8
74. van der Pauw MT, Klein-Nulend J, van den Bos T, Burger EH, Everts V, Beertsen W. Response of periodontal ligament fibroblasts and gingival fibroblasts to pulsating fluid flow: nitric oxide and prostaglandin E2 release and expression of tissue non-specific alkaline phosphatase activity. J Periodontal Res. (2000) 35:335–43. doi: 10.1034/j.1600-0765.2000.035006335.x
75. Tanaka-Kamioka K, Kamioka H, Ris H, Lim SS. Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J Bone Miner Res. (1998) 13:1555–68. doi: 10.1359/jbmr.1998.13.10.1555
76. Kamioka H, Sugawara Y, Honjo T, Yamashiro T, Takano-Yamamoto T. Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. J Bone Miner Res. (2004) 19:471–8. doi: 10.1359/JBMR.040128
77. Bacabac RG, Smit TH, Van Loon JJ, Doulabi BZ, Helder M, Klein-Nulend J. Bone cell responses to high-frequency vibration stress: does the nucleus oscillate within the cytoplasm? FASEB J. (2006) 20:858–64. doi: 10.1096/fj.05-4966.com
78. Wesseling-Perry K, Mäkitie RE, Välimäki VV, Laine T, Laine CM, Välimäki MJ, et al. Osteocyte protein expression is altered in low-turnover osteoporosis caused by mutations in WNT1 and PLS3. J Clin Endocrinol Metab. (2017) 102:2340–8. doi: 10.1210/jc.2017-00099
79. Välimäki VV, Mäkitie O, Pereira R, Laine C, Wesseling-Perry K, Määttä J, et al. Teriparatide treatment in patients with WNT1 or PLS3 mutation-related early-onset osteoporosis–a pilot study. J Clin Endocrinol Metab. (2017) 102:535–44. doi: 10.1210/jc.2016-2423
80. Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE, et al. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc Natl Acad Sci USA. (2012) 109:14170–5. doi: 10.1073/pnas.1208916109
81. Thouverey C, Malinowska A, Balcerzak M, Strzelecka-Kiliszek A, Buchet R, Dadlez M, et al. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J Proteomics. (2011) 74:1123–34. doi: 10.1016/j.jprot.2011.04.005
82. Piehl LL, Fischman ML, Hellman U, Cisale H, Miranda PV. Boar seminal plasma exosomes: effect on sperm function and protein identification by sequencing. Theriogenology (2013) 79:1071–82. doi: 10.1016/j.theriogenology.2013.01.028
83. Neugebauer J, Heilig J, Hosseinibarkooie S, Ross BC, Mendoza-Ferreira N, Nolte F, et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum Mol Genet. (2018) 27:4249–62. doi: 10.1093/hmg/ddy318
84. McKee MD, Cole WG. Bone matrix and mineralization. In: Glorieux F, Pettifor JM, Juppner H, editors. Pediatric Bone, Academic Press (2012). p. 9–37.
85. Young MF. Bone matrix proteins: their function, regulation, and relationship to osteoporosis. Osteoporos Int. (2003) 14:35–42. doi: 10.1007/s00198-002-1342-7
86. Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, Paepe A, et al. Osteogenesis imperfecta. Dis Primers (2017) 3:17052. doi: 10.1038/nrdp.2017.52
87. Forlino A, Marini JC. Osteogenesis imperfecta. Lancet (2016) 387:1657–71. doi: 10.1016/S0140-6736(15)00728-X
88. Chessler S, Byers P. Defective folding and stable association with protein disulfide isomerase/prolyl hydroxylase of type I procollagen with a deletion in the ProaZ(1) chain that preserves theG ly-X-Y repeat Pattern (1992) 267:7751–7.
89. Mundlos S, Chan D, Weng Y, Sillence D, Cole W, Bateman J. Multiexon deletions in the type I collagen COL1A2 gene in osteogenesis imperfecta type IB. J Biol Chem. (1996) 271:21068–74.
90. Bardai G, Lemyre E, Moffatt P, Palomo T, Glorieux FH, Tung J, et al. Osteogenesis imperfecta type I caused by COL1A1 deletions. Calcif Tissue Int. (2016) 98:76–84. doi: 10.1007/s00223-015-0066-6
91. Costantini A, Skarp S, Kämpe A, Mäkitie RE, Pettersson M, Männikkö M, et al. Rare copy number variants in array-based comparative genomic hybridization in early-onset skeletal fragility. Front Endocrinol (Lausanne) (2018) 9:380. doi: 10.3389/fendo.2018.00380
92. Costantini A, Tournis S, Kampe A, Ul Ain N, Taylan F, Doulgeraki A, et al. Autosomal recessive osteogenesis imperfecta caused by a novel homozygous COL1A2 mutation. Calcif Tissue Int. (2018) 103:353. doi: 10.1007/s00223-018-0414-4
93. Nicholls AC, Osse G, Schloon HG, Lenard HG, Deak S, Myers JC, et al. The clinical features of homozygous à2(I) collagen deficient osteogenesis imperfecta. J Med Genet. (1984) 21:257–62. doi: 10.1136/jmg.21.4.257
94. De Paepe A, Nuytinck L, Raes M, Fryns JP. Homozygosity by descent for a COL1A2 mutation in two sibs with severe osteogenesis imperfecta and mild clinical expression in the heterozygotes. Hum Genet. (1997) 99:478–83. doi: 10.1007/s004390050392
95. Pihlajaniemi T, Dickson LA, Popell FM, Korhonen VR, Nichollsll A, Prockop DJ, et al. Osteogenesis imperfecta: cloning of a Pro-a2(I) collagen gene with a frameshift mutation. J Biol Chem. (1984) 259:12941–4.
96. Lindahl K, Barnes AM, Fratzl-Zelman N, Whyte MP, Hefferan TE, Makareeva E, et al. COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum Mutat. (2011) 32:598–609. doi: 10.1002/humu.21475
97. Cundy T, Dray M, Delahunt J, Hald JD, Langdahl B, Li C, et al. Mutations that alter the carboxy-terminal-propeptide cleavage site of the chains of type I procollagen are associated with a unique osteogenesis imperfecta phenotype. J Bone Miner Res. 33:1260–71. doi: 10.1002/jbmr.3424
98. Mendoza-Londono R, Fahiminiya S, Majewski J, Care4Rare Canada C, Tetreault M, Nadaf J, et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet. (2015) 96:979–85. doi: 10.1016/j.ajhg.2015.04.021
99. Rosset EM, Bradshaw AD. SPARC/osteonectin in mineralized tissue. Matrix Biol. (2016) 52–54:78–87. doi: 10.1016/j.matbio.2016.02.001
100. Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR. Osteonectin, a bone-specific protein linking mineral to collagen. Cell (1981) 26:99–105. doi: 10.1016/0092-8674(81)90037-4
101. Becker J, Semler O, Gilissen C, Li Y, Bolz Hanno J, Giunta C, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. (2011) 88:362–71. doi: 10.1016/j.ajhg.2011.01.015
102. Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. (2012) 9:343–8. doi: 10.1016/j.ajhg.2012.06.005
103. Doyard M, Bacrot S, Huber C, Di Rocco M, Goldenberg A, Aglan MS, et al. FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta. J Med Genet. (2018) 55:278–84. doi: 10.1136/jmedgenet-2017-104999
104. Taylan F, Costantini A, Coles N, Pekkinen M, Heon E, Siklar Z, et al. Spondyloocular syndrome: novel mutations in XYLT2 gene and expansion of the phenotypic spectrum. J Bone Miner Res. (2016) 31:1577–85. doi: 10.1002/jbmr.2834
105. Munns CF, Fahiminiya S, Poudel N, Munteanu MC, Majewski J, Sillence DO, et al. Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am J Hum Genet. (2015) 96:971–8. doi: 10.1016/j.ajhg.2015.04.017
106. Taylan F, Makitie O. Abnormal proteoglycan synthesis due to gene defects causes skeletal diseases with overlapping phenotypes. Horm Metab Res. (2016) 48:745–54. doi: 10.1055/s-0042-118706
107. Shaheen R, Al-Owain M, Sakati N, Alzayed ZS, Alkuraya FS. FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification? Am J Hum Genet. (2010) 87:306–7. doi: 10.1016/j.ajhg.2010.05.020
108. Schwarze U, Cundy T, Pyott SM, Christiansen HE, Hegde MR, Bank RA, et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet. (2013) 22:1–17. doi: 10.1093/hmg/dds371
109. van der Slot AJ, Zuurmond AM, Bardoel AF, Wijmenga C, Pruijs HE, Sillence DO, et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. (2003) 278:40967–72. doi: 10.1074/jbc.M307380200
110. Ha-Vinh R, Alanay Y, Bank RA, Campos-Xavier AB, Zankl A, Superti-Furga A, et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet. (2004) 131:115–20. doi: 10.1002/ajmg.a.30231
111. van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A (2014) 164A:1470–81. doi: 10.1002/ajmg.a.36545
112. Krassas GE. Idiopathic juvenile osteoporosis. Ann N Y Acad Sci. (2000) 900:409–12. doi: 10.1111/j.1749-6632.2000.tb06253.x
113. Kämpe AJ, Makitie RE, Makitie O. New genetic forms of childhood-onset primary osteoporosis. Horm Res Paediatr. (2015) 84:361–9. doi: 10.1159/000439566
114. Dalgleish R. The human type I collagen mutation database. Nucleic Acids Res. (1997) 25:181–7. doi: 10.1093/nar/25.1.181
115. Dalgleish R. The human collagen mutation database 1998. Nucleic Acids Res. (1998) 26:253–5. doi: 10.1093/nar/26.1.253
116. Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. (2017) 9:eaal5209. doi: 10.1126/scitranslmed.aal5209
117. Telenti A, Pierce LC, Biggs WH, di Iulio J, Wong EH, Fabani MM, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci USA. (2016) 113:11901–6. doi: 10.1073/pnas.1613365113
118. Marini JC, Blissett AR. New genes in bone development: what's new in osteogenesis imperfecta. J Clin Endocrinol Metab. (2013) 98:3095–103. doi: 10.1210/jc.2013-1505
119. Mäkitie RE, Hackl M, Niinimäki R, Kakko S, Grillari J, Mäkitie O. Altered MicroRNA profile in osteoporosis caused by impaired WNT signaling. J Clin Endocrinol Metab. (2018) 103:1985–96. doi: 10.1210/jc.2017-02585
120. Naylor K, Eastell R. Bone turnover markers: use in osteoporosis. Nat Rev Rheumatol. (2012) 8:379–89. doi: 10.1038/nrrheum.2012.86
121. Eastell R, Pigott T, Gossiel F, Naylor KE, Walsh JS, Peel NFA. Diagnosis of endocrine disease: bone turnover markers: are they clinically useful? Eur J Endocrinol. (2018) 178:R19–31. doi: 10.1530/EJE-17-0585
122. Markula-Patjas KP, Ivaska KK, Pekkinen M, Andersson S, Moilanen E, Viljakainen HT, et al. High adiposity and serum leptin accompanied by altered bone turnover markers in severe juvenile idiopathic arthritis. J Rheumatol. (2014) 41:2474–81. doi: 10.3899/jrheum.131107
123. Viljakainen H, Ivaska KK, Paldánius P, Lipsanen-Nyman M, Saukkonen T, Pietiläinen KH, et al. Suppressed bone turnover in obesity: a link to energy metabolism? A case-control study. J Clin Endocrinol Metab. (2014) 99:2155–63. doi: 10.1210/jc.2013-3097
124. Paldanius PM, Ivaska KK, Hovi P, Andersson S, Väänänen HK, Kajantie E, et al. The effect of oral glucose tolerance test on serum osteocalcin and bone turnover markers in young adults. Calcif Tissue Int. (2012) 90:90–5. doi: 10.1007/s00223-011-9551-8
125. Vuorimies I, Toiviainen-Salo S, Hero M, Mäkitie O. Zoledronic acid treatment in children with osteogenesis imperfecta. Horm Res Paediatr. (2011) 75:346–53. doi: 10.1159/000323368
126. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
127. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
128. Kocijan R, Muschitz C, Geiger E, Skalicky S, Baierl A, Dormann R, et al. Circulating microRNA signatures in patients with idiopathic and postmenopausal osteoporosis and fragility fractures. J Clin Endocrinol Metab. (2016) 101:4125–34. doi: 10.1210/jc.2016-2365
129. Hackl M, Heilmeier U, Weilner S, Grillari J. Circulating microRNAs as novel biomarkers for bone diseases–complex signatures for multifactorial diseases? Mol Cell Endocrinol. (2016) 432:83–95. doi: 10.1016/j.mce.2015.10.015
130. Taipaleenmäki H. Regulation of bone metabolism by microRNAs. Curr Osteoporos Rep. (2018) 16, 1–12. doi: 10.1007/s11914-018-0417-0
131. Mandourah AY, Ranganath L, Barraclough R, Vinjamuri S, Hof RV, Hamill S, et al. Circulating microRNAs as potential diagnostic biomarkers for osteoporosis. Sci Rep. (2018) 8:8421. doi: 10.1038/s41598-018-26525-y
132. Wang Y, Li L, Moore BT, Peng XH, Fang X, Lappe JM, et al. MiR-133a in human circulating monocytes: a potential biomarker associated with postmenopausal osteoporosis. PLoS ONE (2012) 7:e34641. doi: 10.1371/journal.pone.0034641
133. Meng J, Zhang D, Pan N, Sun N, Wang Q, Fan J, et al. Identification of miR-194-5p as a potential biomarker for postmenopausal osteoporosis. PeerJ (2015) 3:e971. doi: 10.7717/peerj.971
134. Li H, Wang Z, Fu Q, Zhang J. Plasma miRNA levels correlate with sensitivity to bone mineral density in postmenopausal osteoporosis patients. Biomarkers (2014) 19:553–6. doi: 10.3109/1354750X.2014.935957
135. Seeliger C, Karpinski K, Haug AT, Vester H, Schmitt A, Bauer JS, et al. Five freely circulating miRNAs and bone tissue miRNAs are associated with osteoporotic fractures. J Bone Miner Res. (2014) 29:1718–28. doi: 10.1002/jbmr.2175
136. Xu Y, Chen B, George SK, Liu B. Downregulation of MicroRNA-152 contributes to high expression of DKK1 in multiple myeloma. RNA Biol. (2015) 12:1314–22. doi: 10.1080/15476286.2015.1094600
137. Zhang J, Tu Q, Bonewald LF, He X, Stein G, Lian J, et al. Effects of miR-335-5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J Bone Miner Res. (2011) 26:1953–63. doi: 10.1002/jbmr.377
138. Wang J, Guan X, Guo F, Zhou J, Chang A, Sun B, et al. miR-30e reciprocally regulates the differentiation of adipocytes and osteoblasts by directly targeting low-density lipoprotein receptor-related protein 6. Cell Death Dis. (2013) 4:e845. doi: 10.1038/cddis.2013.356
139. Li Z, Hassan MQ, Volinia S, van Wijnen AJ, Stein JL, Croce CM, et al. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc Natl Acad Sci USA. (2008) 105:13906–11. doi: 10.1073/pnas.0804438105
140. Anastasilakis AD, Makras P, Pikilidou M, Tournis S, Makris K, Bisbinas I, et al. Changes of circulating MicroRNAs in response to treatment with teriparatide or denosumab in postmenopausal osteoporosis. J Clin Endocrinol Metab. (2018) 103:1206–13. doi: 10.1210/jc.2017-02406
141. Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. (2016) 10:CD005088. doi: 10.1002/14651858.CD005088.pub4
142. Biggin A, Munns CF. Long-term bisphosphonate therapy in osteogenesis imperfecta. Curr Osteoporos Rep. (2017) 15:412–8. doi: 10.1007/s11914-017-0401-0
143. Shi CG, Zhang Y, Yuan W. Efficacy of bisphosphonates on bone mineral density and fracture rate in patients with osteogenesis imperfecta: a systematic review and meta-analysis. Am J Ther. (2016) 23:e894–904. doi: 10.1097/MJT.0000000000000236
144. Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, Munivez E, et al. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. (2017) 127:2678–88. doi: 10.1172/JCI92617
145. Kannu P, Mahjoub A, Babul-Hirji R, Carter MT, Harrington J. PLS3 mutations in X-linked osteoporosis: clinical and bone characteristics of two novel mutations. Horm Res Paediatr. (2017) 88:298–304. doi: 10.1159/000477242
146. Trejo P, Rauch F. Osteogenesis imperfecta in children and adolescents-new developments in diagnosis and treatment. Osteoporos Int. (2016) 27:3427–37. doi: 10.1007/s00198-016-3723-3
147. Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, et al., FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. (2009) 361:756–65. doi: 10.1056/NEJMoa0809493
148. Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. (2012) 12:183–8.
149. Mullard A. Merck &Co. drops osteoporosis drug odanacatib. Nat Rev Drug Discov. (2016) 15:669. doi: 10.1038/nrd.2016.207
150. Tauer JT, Abdullah S, Rauch F. Effect of anti-TGF-β treatment in a mouse model of severe osteogenesis imperfecta. J Bone Miner Res. (2018). doi: 10.1002/jbmr.3617. [Epub ahead of print].
151. McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. (2014) 370:412–20. doi: 10.1056/NEJMoa1305224
152. Faienza MF, Chiarito M, D'amato G, Colaianni G, Colucci S, Grano M, et al. Monoclonal antibodies for treating osteoporosis. Expert Opin Biol Ther. (2018) 18:149–57. doi: 10.1080/14712598.2018.1401607
153. Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. (2016) 375:1532–43. doi: 10.1056/NEJMoa1607948
154. Witcher PC, Miner SE, Horan DJ, Bullock WA, Lim KE, Kang KS, et al. Sclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition. JCI Insight. (2018) 3:98673. doi: 10.1172/jci.insight.98673
155. Kakugawa S, Langton PF, Zebisch M, Howell SA, Chang TH, Liu Y, et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature (2015) 519:187–92. doi: 10.1038/nature14259
156. Zhang X, Cheong SM, Amado NG, Reis AH, MacDonald BT, Zebisch M, et al. Notum is required for neural and head induction via Wnt deacylation, oxidation and inactivation. Dev Cell (2015) 32:719–30. doi: 10.1016/j.devcel.2015.02.014
157. Chouinard L, Felx M, Mellal N, Varela A, Mann P, Jolette J, et al. Carcinogenicity risk assessment of romosozumab: a review of scientific weight-of-evidence and findings in a rat lifetime pharmacology study. Regul Toxicol Pharmacol. (2016) 81:212–22. doi: 10.1016/j.yrtph.2016.08.010
158. Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. (2015) 87:3–14. doi: 10.1016/j.addr.2015.05.001
159. Sha D, Lee AM, Shi Q, Alberts SR, Sargent DJ, Sinicrope FA, et al. Association study of the let-7 miRNA-complementary site variant in the 3′ untranslated region of the KRAS gene in stage III colon cancer (NCCTG N0147 clinical trial). Clin Cancer Res. (2014) 20:3319–27. doi: 10.1158/1078-0432.CCR-14-0069
160. van der Ree MH, van der Meer AJ, de Bruijne J, Maan R, van Vliet A, Welzel TM, et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res. (2014) 111:53–9. doi: 10.1016/j.antiviral.2014.08.015
161. Wang Z, Wu G, Feng Z, Bai S, Dong Y, Wu G, et al. Microarc-oxidized titanium surfaces functionalized with microRNA-21-loaded chitosan/hyaluronic acid nanoparticles promote the osteogenic differentiation of human bone marrow mesenchymal stem cells. Int J Nanomed. (2015) 10:6675–87. doi: 10.2147/IJN.S94689
162. Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A, et al. miR-214 targets ATF4 to inhibit bone formation. Nat Med. (2013) 19:93–100. doi: 10.1038/nm.3026
163. Murata K, Ito H, Yoshitomi H, Yamamoto K, Fukuda A, Yoshikawa J, et al. Inhibition of miR-92a enhances fracture healing via promoting angiogenesis in a model of stabilized fracture in young mice. J Bone Miner Res. (2014) 29:316–26. doi: 10.1002/jbmr.2040
164. Li Y, Fan L, Liu S, Liu W, Zhang H, Zhou T, et al. The promotion of bone regeneration through positive regulation of angiogenic-osteogenic coupling using microRNA-26a. Biomaterials (2013) 34:5048–58. doi: 10.1016/j.biomaterials.2013.03.052
Keywords: early-onset osteoporosis, Wnt signaling, osteogenesis imperfecta, PLS3, bone metabolism
Citation: Mäkitie RE, Costantini A, Kämpe A, Alm JJ and Mäkitie O (2019) New Insights Into Monogenic Causes of Osteoporosis. Front. Endocrinol. 10:70. doi: 10.3389/fendo.2019.00070
Received: 26 October 2018; Accepted: 24 January 2019;
Published: 25 February 2019.
Edited by:
David Karasik, Bar-Ilan University, IsraelReviewed by:
Graziana Colaianni, Università degli Studi di Bari, ItalyMaria Felicia Faienza, Università degli Studi di Bari, Italy
Francesca Gori, Harvard School of Dental Medicine, United States
Copyright © 2019 Mäkitie, Costantini, Kämpe, Alm and Mäkitie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Outi Mäkitie, b3V0aS5tYWtpdGllQGhlbHNpbmtpLmZp