 Sara Penna
Sara Penna Valentina Capo
Valentina Capo Eleonora Palagano
Eleonora Palagano Cristina Sobacchi
Cristina Sobacchi Anna Villa
Anna Villa- 1San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), San Raffaele Hospital, Milan, Italy
- 2Translational and Molecular Medicine (DIMET), University of Milano-Bicocca, Monza, Italy
- 3The National Research Council (CNR) Institute for Genetic and Biomedical Research (IRGB)- CNR-IRGB, Milan Unit, Milan, Italy
- 4Humanitas Research Hospital, Rozzano, Italy
Osteopetrosis is a condition characterized by increased bone mass due to defects in osteoclast function or formation. In the last decades, the molecular dissection of osteopetrosis has unveiled a plethora of molecular players responsible for different forms of the disease, some of which present also primary neurodegeneration that severely limits the therapy. Hematopoietic stem cell transplantation can cure the majority of them when performed in the first months of life, highlighting the relevance of an early molecular diagnosis. However, clinical management of these patients is constrained by the severity of the disease and lack of a bone marrow niche that may delay immune reconstitution. Based on osteopetrosis genetic heterogeneity and disease severity, personalized therapies are required for patients that are not candidate to bone marrow transplantation. This review briefly describes the genetics of osteopetrosis, its clinical heterogeneity, current therapy and innovative approaches undergoing preclinical evaluation.
Introduction
The term osteopetrosis derives from the Greek “osteo,” bone, and “petros,” stone, to define a genetically heterogenous group of diseases affecting the skeletal tissue, ranging in severity from benign to fatal in early childhood (1). Osteopetrosis is characterized by increased bone mass due to defective resorption activity or differentiation of osteoclasts (2), causing a disequilibrium of bone turnover, deformities, dental abnormalities and impaired mineral homeostasis, and giving rise to structural fragility that causes frequent fractures. Moreover, osteopetrotic patients are characterized by reduction of marrow cavity, affecting hematologic function; related phenotypes are severe anemia, pancytopenia, frequent infections and hepatosplenomegaly (1, 2) and increased frequency of circulating CD34+ cells in the peripheral blood (3). The overly dense cranial nerve foramina lead to impairment of neurologic functions with progressive deafness, blindness and nerve palsies (1, 2). Three different forms of osteopetrosis have been described, based on the pattern of inheritance: autosomal recessive osteopetrosis (ARO), autosomal dominant osteopetrosis (ADO) and X-linked osteopetrosis (2, 4). The only cure for osteopetrosis is allogeneic hematopoietic stem cell transplantation (HSCT), that has greatly improved its outcome overtime (5–7). In this review, we describe the different forms of the disease and therapeutic options, highlighting advances in the setting of safer conditioning regimens and alternative therapies to overcome the limited donor availability.
Autosomal Recessive Osteopetrosis (ARO)
The autosomal recessive form of osteopetrosis (ARO), also known as infantile malignant osteopetrosis (IMO), has an incidence of 1:250000 live births, with higher rates in specific geographic areas because of geographic isolation, high frequency of parental consanguinity or the presence of a founder effect (8). Unless treated with HSCT, ARO is usually fatal within the first 10 years of life (8). Children present with failure to thrive, skull abnormalities (macrocephaly, frontal bossing, choanal stenosis), hydrocephalus, hypocalcemia due to defective calcium mobilization activity of osteoclasts (1) and abnormal tooth eruption with frequent development of dental caries (9). ARO is caused by mutations in different genes that are implicated in osteoclast function (osteoclast-rich osteopetrosis) or differentiation (osteoclast-poor osteopetrosis) (Figure 1).
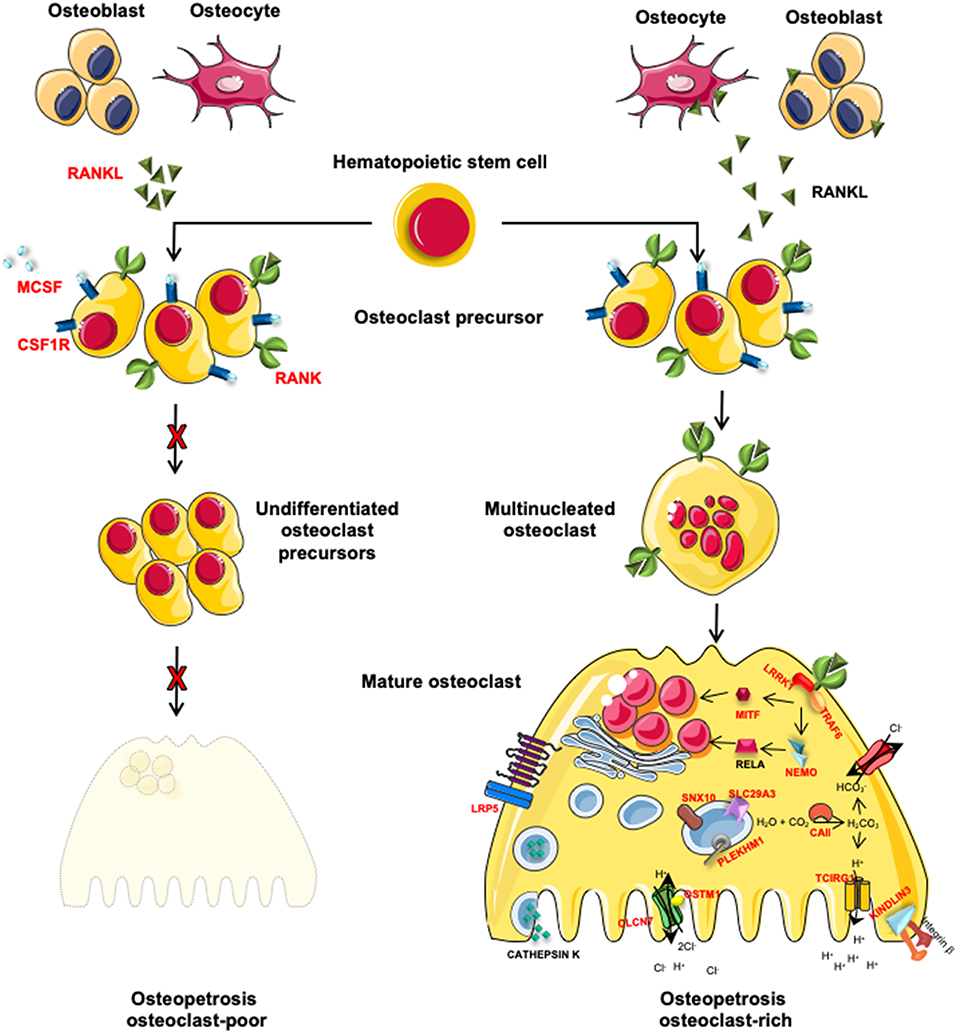
Figure 1. Schematic representation of genes involved in osteoclast-poor and osteoclast-rich osteopetrosis. In red are indicated genes involved in the pathogenesis of ARO. MCSF and RANKL, cytokines secreted by osteoblasts and osteocytes, are necessary for the differentiation of osteoclast precursors into mature and resorbing osteoclasts. When these signals are absent (TNFSF11 gene mutations) or the pathway is interrupted by the lack of cytokine receptors (TNFRSF11A and CSF1R gene mutations), osteoclast precursors are not able to differentiate into mature osteoclast causing osteoclast-poor forms of osteopetrosis. Alternatively, if osteopetrosis is caused by mutations in genes encoding for protein necessary for bone resorption, the disease is defined as osteoclast-rich osteopetrosis. On the right of the figure, are indicated genes involved in bone resorption activity with different roles: i.e., acidification of resorption lacunae and pH regulation (TCIRG1, CLCN7, OSTM1, and CAII), vesicular trafficking and sorting of protein complex to the membrane (SNX10 and PLEKHM1), cytoskeletal rearrangement for ruffle border formation (FERMT3 and LRRK1). Other molecules involved in different signal transductions, essential for osteoclast functions (MITF, LRP5, and IKBKG) are reported.
Osteoclast-Rich Osteopetrosis
The most frequent form is caused by mutations in the TCIRG1 (T cell immune regulator 1) gene, accounting for more than 50% of ARO cases. TCIRG1 encodes for the a3 subunit of V0 complex of the V-ATPase proton pump, mainly expressed by osteoclasts and gastric parietal cells on apical membrane. The V-ATPase pump acidifies the resorption lacuna in the bone for the dissolution of the hydroxyapatite crystals, that form the bone mineral fraction, and the degradation of the matrix (10). The a3 subunit has also been implicated in the interaction between actin cytoskeleton and microtubules, fundamental for the osteoclast ruffled border formation (8, 11). Accordingly, TCIRG1-mutated osteoclasts show defective ruffled border and markedly reduced resorptive activity (11, 12). Moreover, the V-ATPase maintains the low pH in the stomach for the dietary Ca2+ absorption (13), and, since gastric acidification is also relevant for calcium uptake, this form of osteopetrosis is characterized by rickets or osteomalacia. The second most frequent form of ARO (17% of the cases) is caused by loss of function mutations in the CLCN7 (chloride voltage-gated channel 7) gene (2, 14). This gene codes a 2Cl−/H+ antiporter regulated by voltage-gating mechanism, expressed on the osteoclast ruffled border and on the membrane of late endosomes and lysosomes (15). This channel cooperates with the V-ATPase in the acid pH maintenance of the resorption lacuna. CLCN7 is involved in vesicle trafficking in early and recycling endosomes by regulating the luminal Cl− concentration (16). Mutations in the CLCN7 gene are responsible for a wide spectrum of clinical manifestations. Biallelic mutations cause a very severe form in which bone defects and hematological failure are associated in some patients with primary neurodegeneration, resembling lysosomal storage disease, cerebral atrophy, spasticity, axial hypotonia and peripheral hypertonia (8, 14, 17). Carrier individuals do not show any overt bone phenotype. CLCN7-deficient osteoclasts have been reported to display impaired endolysosomal trafficking (8). In rare intermediate forms of TCIRG1- and CLCN7-deficient ARO, milder presentation or later onset and slower progression have been recently reported (18–21).
OSTM1 (osteopetrosis-associated transmembrane protein 1) mutations are reported in 5% of ARO cases (4, 22, 23) and invariably cause osteopetrosis and severe primary neurodegeneration, with a life expectancy lower than 2 years (22, 24–26). OSTM1 has a highly glycosylated N-terminus that has been reported to stabilize CLCN7 protein and to be required, together with its transmembrane region, for CLCN7 Cl−/H+ transport activity (15). OSTM1 acts also as an E3 ubiquitin ligase for the heterotrimeric G-protein Gαi3 and potentiates WNT canonical signaling by modulating β-catenin/Lef1 interaction (27, 28).
Less than 5% of ARO cases are caused by mutations in the SNX10 gene, encoding for the sortin nexin 10 protein, one of the major interactors of the V-ATPase. It is involved in the vesicular sorting of the V-ATPase complex from the Golgi network and in its targeting to the ruffled border (8, 29). In the original work, SNX10-dependent osteopetrosis was reported to show few and small osteoclasts (30), while in a more recent paper SNX10-deficient osteoclasts were larger and pale at tartrate-resistant acid phosphatase (TRAP) staining (31). Overall, the severity of clinical manifestations is variable (29, 31, 32).
Rare cases of osteoclast-rich osteopetrosis caused by mutations in other genes have also been reported. For example, osteopetrosis caused by carbonic anhydrase II (CA-II) deficiency appears in less than one in a million live births and is associated with cerebral calcification and renal tubular acidosis (2, 33). Carbonic anhydrase II enzyme provides protons to the vacuolar proton pump. Since renal defects are more severe than bone abnormalities, CA-II deficiency generally is not considered a classic form of ARO (34).
Loss-of-function mutations in the PLEKHM1 (pleckstrin homology domain–containing family M member 1) gene cause mild osteopetrosis in the ia (incisors absent) rat, as well as an intermediate form of human osteopetrosis (35). PLEKHM1 is a cytosolic protein involved in lysosomal trafficking likely acting as an effector of Rab7 (36, 37). Patient-derived PLEKHM1-deficient osteoclasts displayed altered morphology and abnormal podosome distribution (35).
Mutations in FERMT3 (fermitin family member 3) gene have been reported to cause osteopetrosis in association with leukocyte adhesion deficiency type III (LAD III). FERMT3 gene is expressed in hematopoietic cells and encodes kindlin-3 protein, necessary for integrin signaling and platelet aggregation (38). Patients affected with FERMT3-deficiency are characterized by frequent bleeding and recurrent infections (39, 40).
LRRK1 (leucine-rich repeat kinase 1) gene mutation was found in a single patient affected by osteosclerotic metaphyseal dysplasia, that specifically compromises the methaphyses of long bones, vertebral endplates, costal ends and margin of flat bones (41).
Another mutated gene associated with osteopetrosis is MITF (microphtalmia-associated growth factor) that encodes for a transcription factor acting downstream RANK/RANKL pathway (42). MITF deficiency is responsible for COMMAD (coloboma, osteopetrosis, microphthalmia, macrocephaly, albinism, and deafness) syndrome in two unrelated patients, suggesting a role for MITF in regulating various processes beside bone development and homeostasis (43).
Finally, a homozygous mutation in C16orf57 has been described in poikiloderma and neutropenia associated with osteopetrosis (44). This gene encodes for a phosphodiesterase responsible for modification and stabilization of the U6 small nuclear RNA, fundamental element of the spliceosome machinery (45), however, its pathophysiologic function in osteoclast still has to be elucidated.
Osteoclast-Poor Osteopetrosis
The complete absence of osteoclasts is the key feature of the osteoclast-poor form of osteopetrosis (46). Patients are characterized by absence of TRAP-positive osteoclasts in bone biopsies. The defective osteoclastogenesis is caused by either the lack of RANKL (receptor activator of nuclear kappa B ligand) cytokine (2% of all ARO cases) or of its receptor RANK (4.5% of ARO forms) (47–50). RANKL is encoded by the TNFSF11 gene and the binding to its receptor RANK, encoded by the TNFRSF11A gene, determines the activation of the downstream pathway that drives osteoclast differentiation and activation (51). In bone, RANKL is produced mainly by the stromal compartment in physiological condition, while other cell sources are more important in pathological context (52). Recent evidence suggests that RANKL has also an osteogenic role through an autocrine loop in mesenchymal stem cells (53) and through reverse signaling from the osteoclasts to the osteoblasts (54). In addition, in patients RANKL absence leads to a partial defect in T cell proliferation and cytokine production (50), while RANK-deficiency perturbs B cell memory subset and immunoglobulin production (48, 49).
A rare osteoclast-poor form of osteopetrosis, called dysosteosclerosis (DOS), accompanied by red violet macular atrophy, platyspondyly and metaphyseal osteosclerosis, is caused by mutations of the SLC29A3 (solute carrier family 29 member 3) gene encoding for a lysosomal nucleoside transporter highly expressed in myeloid cells (21, 55). More recently a novel splice-site mutation in the intron 6 of TNFRSF11A has been described in one patient indicating TNFRSF11A as additional gene responsible for DOS (56).
A recent report described two affected siblings presenting osteopetrosis associated with severe combined immunodeficiency (SCID) caused by a large deletion on chromosome 11 encompassing RAG1 and RAG2 genes and the 5′ region of TRAF6 (TNF receptor-associated factor 6 gene), the most important adaptor for the RANK/RANKL signaling pathway (57).
Lastly, a heterozygous truncating mutation in the CSF1R gene, which encodes for MCSF (macrophage colony-stimulating factor) receptor, was reported in the consanguineous parents of two deceased siblings, showing osteopetrosis and brain malformations (58). This mutation could not be assessed in the probands, however based on this report, this genetic variant could be responsible for the disease in this family (59).
Autosomal Dominant Osteopetrosis
Autosomal Dominant Osteopetrosis (ADO) has an incidence of 1:20,000 live births with clinical onset typically in adolescence or adulthood (4) and cases diagnosed in pediatric age are reported too (18, 60). It is characterized by diffuse osteosclerosis, primarily involving the axial skeleton, and symmetrical defects of the long bones, with no or little modeling defects. ADO form, also known as benign form, is caused by heterozygous missense mutations of the CLCN7 gene with dominant negative characteristic, in which the mutant subunit is able to dimerize, functionally impairing the protein (12, 17). Patients affected with ADO present a wide range of symptoms: radiographic alterations, frequent atraumatic fractures, osteonecrosis or osteomyelitis, vision and hearing impairment due to cranial nerve compression and occasional bone marrow failure (4, 8, 61). Although CLCN7 is widely expressed in the body and the biallelic loss of function causes neurodegeneration in some CLCN7-deficient ARO patients, only sporadic cases of cognitive failure have been reported in ADO patients (4, 12).
X-linked Osteopetrosis
Osteopetrosis caused by mutations of the IKBKG (inhibitor of nuclear factor kappa B kinase subunit gamma) gene, located on the X chromosome, occurs as a moderate complication of the OL-EDA-ID syndrome, lymphedema, anhidrotic ectodermal dysplasia and immunodeficiency (hence, the acronym) (62–65). The IKBKG gene encodes NEMO, the regulatory subunit of IKK complex, fundamental for the activation of NF-kB transcription factor to induce osteoclastogenesis (62). Consistently, inhibition of NF-kappaB signaling in mouse models of inflammation showed amelioration of osteolysis and inflammation (66). Bone biopsy evaluation in a patient revealed that osteoclasts were present in normal numbers and showed no morphological abnormalities (63). Thus, OL-EDA-ID is classified as an osteoclast-rich osteopetrosis (67).
Current Therapies and Management of Osteopetrosis
The majority of osteopetrotic forms are caused by osteoclast dysfunction, while a lower proportion of cases are caused by impaired osteoclastogenesis (8). Table 1 summarizes the main clinical features in various forms of osteopetrosis. Since osteoclasts derive from the myeloid lineage, HSCT is the recommended treatment. A successful HSCT allows the engraftment of donor-derived osteoclast precursors, which further differentiate and give rise to functional mature osteoclasts, resulting in bone remodeling and haematopoiesis (9). However, HSCT is contraindicated in patients with primary neurodegenerative disease (Table 1).
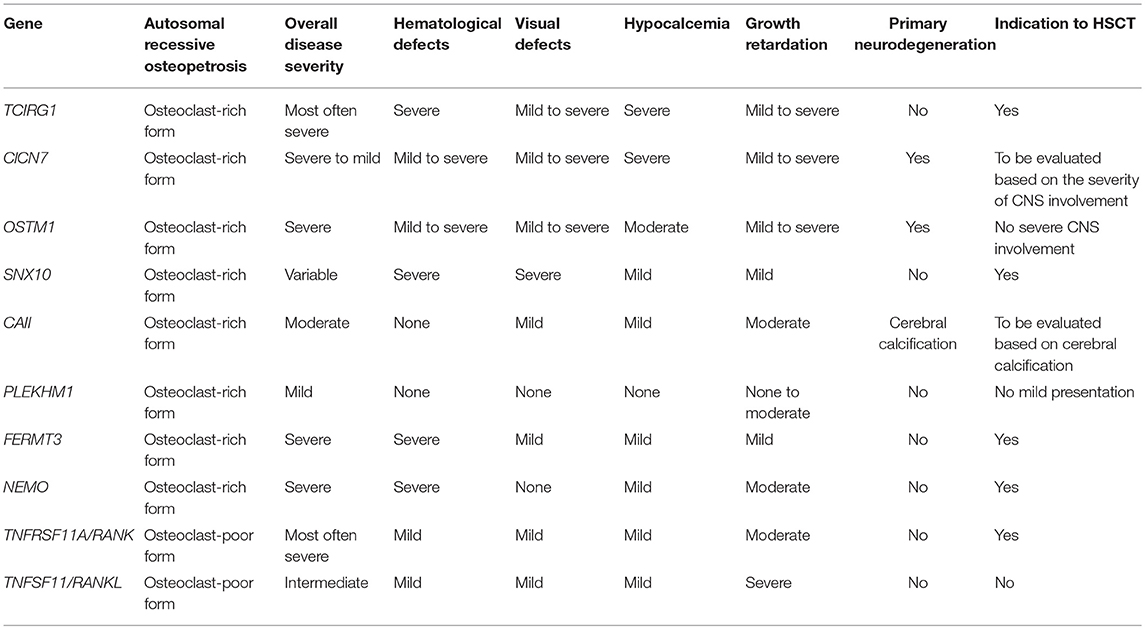
Table 1. Main clinical features and indications for treatment in osteopetrosis.
Since secondary neurological defects are not rescued by transplant, genetic diagnosis and HSCT need to be performed as soon as possible (7, 68, 69). To this end, in utero HSC transplantation might represent in the future a therapeutic option as demonstrated by successful preclinical studies performed in the oc/oc mouse model (70, 71). Multicentre studies reported that patients undergoing HLA-haploidentical HSCT before the age of 10 months, survived with a full donor engraftment. On the contrary, almost all patients receiving HSCT after the age of 10 months underwent graft rejection or autologous reconstitution, even when an haploidentical donor source was used (7). Taken together, these evidences suggest that the fast diagnosis and timing of treatment, play a fundamental role in the long-term efficacy of HSCT (8). The degree of donor compatibility is another key point to obtain a high rate of 5-years disease-free survival (DFS) after allogenic transplant. Data collected during the years on transplant outcomes, proved that the early diagnosis, the constant monitoring and prompt intervention for the associate comorbidities, the optimization of the donor source in term of HLA-matching and the choice of reduced intensity conditioning regimens allowed higher efficacy and safety of HSCT (9, 69, 72). The most recent report of transplants performed by Ulm and Paris Transplant Units highlighted the improved outcomes of HSCT with 93% of survival using T cell replete matched donor and 80% of survival using T cell depleted haploidentical donor (7). Unrelated cord blood is not recommended because its use is associated to high risk of primary engraftment failure (73). Fludarabine-based conditioning, performed better than the conventional cyclophosphamide-based one, in terms of higher engraftment and reduced toxicity with a higher 5-years DFS. In a selected cohort of 31 patients transplanted from related or unrelated fully matched donors, reduced intensity conditioning (RIC), based on fludarabine, treosulfan and thiotepa with proximal serotherapy dosing using anti-thymocyte globulin, allowed 100% overall survival (69).
The most frequent post-transplant complication is the engraftment failure caused by a delayed hematological reconstitution, due to limited or nearly absent bone marrow space (7) and graft vs. host disease (GvHD) (69). T-cell replete haploidentical graft with the administration of cyclophosphamide after HSCT has been proposed in patients older than 10 months (74). Frequently, transplanted ARO patients showed liver and pulmonary venous-occlusive disease (VOD) (75). Respiratory problems, such as choanal stenosis with upper airway obstructions, capillary leak syndrome, primary pulmonary infections and primary pulmonary hypertension were also frequent. When feasible, the use of a RIC regimen may reduces significantly the incidence of pulmonary hypertension (9, 69).
In addition, central nervous system complications may occur in terms of hydrocephalus, hypocalcaemic convulsions or deterioration of preexisting symptoms. Lastly a recurrent post-transplant risk was the onset of hypercalcemia, that can be treated by the use of Denosumab (76).
Alternative Treatments and Future Therapies
Despite recent improvement in the HSCT outcome, the availability of HLA-matched donors remains an open issue. For individuals lacking compatible donor, a strategy based on gene therapy (GT) has been proposed. The protocol would exploit the use of genetically modified CD34+ cells, isolated from peripheral blood without the need of pharmacological HSC mobilization (3). The efficacy and the feasibility of GT have been studied in the oc/oc murine model, to evaluate neonatal transplantation of genetically corrected HSC in the context of TCIRG1-dependent osteopetrosis. Retroviral vectors were able to improve bone resorption and survival of oc/oc mice (77). Unfortunately, clinical trials in which immunodeficient patients were treated with this type of vector showed the risk of leukemia (78). In recent years, lentiviral vector GT has proven to provide clinical benefit in patients affected by a number of diseases, avoiding the leukemic side effects (79, 80). Moreover, transduction of CD34+ cells from the blood of TCIRG1-deficient patients with a lentiviral vector achieved the correction of the osteoclast dysfunction in vitro (81).
ARO caused by osteoclast extrinsic deficiency, such as TNFSF11 mutations, requires a different approach. In particular, a replacement therapy has been evaluated at the preclinical level: the product of the TNFSF11 gene, RANKL cytokine, has been administered pharmacologically to Tnfsf11 knockout mice, rescuing bone defects and hematopoietic organ architecture (82). Additional strategies could be considered, for example, mesenchymal stem cell (MSC) transplantation to replace the osteoblast precursor population (83); however clinical application still raises doubts and challenges, thus this is far from a mature therapeutic option. The second method exploited the use of biotechnological devices, implanted subcutaneously, to release soluble RANKL and allowing osteoclastogenesis in Tnfsf11 knockout mice (84). More recently, a promising biomimetic scaffold, seeded with Tnfsf11 knockout MSC, overexpressing human soluble RANKL after transduction with lentiviral vector has been developed. When implanted subcutaneously, the 3D system was well tolerated and was able to drive the differentiation of TRAP positive cells (85).
Regarding new approaches for the treatment of ADO2, small interfering RNA has been demonstrated to silence specifically the mutated CLCN7 allele, and to be effective and safe in vitro on human cells and in vivo, in an ADO2 mouse model (86). Therefore, efforts have been undertaken to move into the clinic (87). Alternatively, the administration of different doses of IFN-γ partially reduced whole-body bone mineral density of ADO2 mice, although further studies for clinical applications are needed (88).
Conclusions
Genetic dissection of osteopetrosis has unveiled the complex scenario of molecules involved in the pathogenesis of this disease. Early genetic diagnosis is important to establish treatment and thus prevent worsening of the clinical signs. However, despite new molecular techniques have defined ARO molecular complexity, there is the need to further understand their clinical heterogeneity and design novel and suitable cure to these patients. To this end, significant progress has been made in the treatment of ARO thanks to the improvement of novel conditioning regimens and source of donor HSPC, however additional work remains to be done to overcome the limited availability of donors or lack of a therapy for patients carrying RANKL defects or presenting with neurodegenerative osteopetrosis. On this basis, efforts are currently ongoing to further extend the number of molecular players causative of the disease in parallel with the design of novel clinical strategies to be offered as curative treatment for different forms of osteopetrosis.
Author Contributions
SP, VC, and AV wrote the manuscript. CS and EP critically revised the manuscript and contributed to design the figure.
Funding
The original work was supported by the Italian Telethon Grant C5, the European Community's Seventh Framework Program (FP7/2007-2013, SYBIL Project), PRIN Project (2015F3JHMB_004) and by Programma Nazionale per la Ricerca-Consiglio Nazionale delle Ricerche Aging Project to AV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADO, autosomal dominant osteopetrosis; ARO, autosomal recessive osteopetrosis; DOS, dysosteosclerosis; DSF, disease-free survival; GT, gene therapy; HLA, human leukocyte antigen; HSCT, hematopoietic stem cell transplantation; HSPC, hematopoietic stem progenitor cells; MSC, mesenchymal stem cell; RIC, reduced intensity conditioning; TRAP, tartrate-resistant acid phosphatase.
References
1. Wu CC, Econs MJ, DiMeglio LA, Insogna KL, Levine MA, Orchard PJ , et al. Diagnosis and management of osteopetrosis: consensus guidelines from the osteopetrosis working group. J Clin Endocrinol Metab. (2017) 102:3111–23. doi: 10.1210/jc.2017-01127
2. Palagano E, Menale C, Sobacchi C, Villa A. Genetics of osteopetrosis. Curr Osteoporos Rep. (2018) 16:13–25. doi: 10.1007/s11914-018-0415-2
3. Steward CG, Blair A, Moppett J, Clarke E, Virgo P, Lankester A , et al. High peripheral blood progenitor cell counts enable autologous backup before stem cell transplantation for malignant infantile osteopetrosis. Biol Blood Marrow Transplant. (2005) 11:115–21. doi: 10.1016/j.bbmt.2004.11.001
4. Teti A, Econs MJ. Osteopetroses, emphasizing potential approaches to treatment. Bone (2017) 102:50–9. doi: 10.1016/j.bone.2017.02.002
5. Ballet JJ, Griscelli C, Coutris C, Milhaud G, Maroteaux P. Bone marrow transplantation in osteopetrosis. Lancet (1977) 310:1137. doi: 10.1016/S0140-6736(77)90592-X
6. Coccia PF, Krivit W, Cervenka J, Clawson C, Kersey JH, Kim TH , et al. Successful bone-marrow transplantation for infantile malignant osteopetrosis. N Engl J Med. (1980) 302:701–8. doi: 10.1056/NEJM198003273021301
7. Schulz AS, Moshous D, Steward CG, Villa A, Sobacchi C. Osteopetrosis. Consensus Guidelines for Diagnosis, Therapy and Follow-Up (2015). Available online at: https://Esid.Org/.2015.654
8. Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. (2013) 9:522–36. doi: 10.1038/nrendo.2013.137
9. Natsheh J, Drozdinsky G, Simanovsky N, Lamdan R, Erlich O, Gorelik N , et al. Improved outcomes of hematopoietic stem cell transplantation in patients with infantile malignant osteopetrosis using fludarabine-based conditioning. Pediatric Blood Cancer (2015) 63:535–40. doi: 10.1002/pbc.25801
10. Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP , et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. (2000) 25:343–6. doi: 10.1038/77131
11. Nakamura I, Takahashi N, Udagawa N, Moriyama Y, Kurokawa T, Jimi E , et al. Lack of vacuolar proton ATPase association with the cytoskeleton in osteoclasts of osteosclerotic (Oc/Oc) mice. FEBS Lett. (1997) 401:207–12. doi: 10.1016/S0014-5793(96)01454-8
12. Del Fattore A, Peruzzi B, Rucci N, Recchia I, Cappariello A, Longo M , et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implications for diagnosis and treatment. J Med Genet. (2006) 43:315–25. doi: 10.1136/jmg.2005.036673
13. Schinke T, Schilling AF, Baranowsky A, Seitz S, Marshall RP, Linn T , et al. Impaired gastric acidification negatively affects calcium homeostasis and bone mass. Nat Med. (2009) 15:674–81. doi: 10.1038/nm.1963
14. Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A , et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell (2001) 104:205–15. doi: 10.1016/S0092-8674(01)00206-9
15. Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T. ClC-7 is a slowly voltage-gated 2Cl-/1H+-exchanger and requires Ostm1 for transport activity. EMBO J. (2011) 30:2140–52. doi: 10.1038/emboj.2011.137
16. Novarino G, Weinert S, Rickheit G, Jentsch TJ. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science (2010) 328:1398–402. doi: 10.1126/science.1188070
17. Pang Q, Chi Y, Zhao Z, Xing X, Li M, Wang O , et al. Novel mutations of CLCN7 cause autosomal dominant osteopetrosis Type II (ADO-II) and intermediate autosomal recessive osteopetrosis (IARO) in Chinese patients. Osteoporos Int. (2016) 27:1047–55. doi: 10.1007/s00198-015-3320-x
18. Pangrazio A, Pusch M, Caldana E, Frattini A, Lanino E, Tamhankar PM , et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations. Hum Mutat. (2010) 31:1071–80. doi: 10.1002/humu.21167
19. Palagano E, Blair HC, Pangrazio A, Tourkova I, Strina D, Angius A , et al. Buried in the middle but guilty: intronic mutations in the TCIRG1 gene cause human autosomal recessive osteopetrosis. J Bone Miner Res. (2015) 30:1814–21. doi: 10.1002/jbmr.2517
20. Sobacchi C, Pangrazio A, Lopez AG, Gomez DP, Caldana ME, Susani L , et al. As little as needed: the extraordinary case of a mild recessive osteopetrosis owing to a novel splicing hypomorphic mutation in the TCIRG1 gene. J Bone Miner Res. (2014) 29:1646–50. doi: 10.1002/jbmr.2203
21. Howaldt A, Nampoothiri S, Quell LM, Ozden A, Fischer-Zirnsak B, Collet C , et al. Sclerosing bone dysplasias with hallmarks of dysosteosclerosis in four patients carrying mutations in SLC29A3 and TCIRG1. Bone (2018) 120:495–503. doi: 10.1016/j.bone.2018.12.002
22. Chalhoub N, Benachenhou N, Rajapurohitam V, Pata M, Ferron M, Frattini A , et al. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med. (2003) 9:399–406. doi: 10.1038/nm842
23. Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC. ClC-7 requires Ostm1 as a β-subunit to support bone resorption and lysosomal function. Nature (2006) 440:220–3. doi: 10.1038/nature04535
24. Pangrazio A, Poliani PL, Megarbane A, Lefranc G, Lanino E, Di Rocco M , et al. Mutations in OSTM1 (Grey Lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement. J Bone Miner Res. (2006) 21:1098–105. doi: 10.1359/jbmr.060403
25. Ott CE, Fischer B, Schröter P, Richter R, Gupta N, Verma N , et al. Severe neuronopathic autosomal recessive osteopetrosis due to homozygous deletions affecting OSTM1. Bone (2013) 55:292–7. doi: 10.1016/j.bone.2013.04.007
26. Overholt KM, Rose MJ, Joshi S, Herman GE, Bajwa R, Abu-Arja R , et al. Hematopoietic cell transplantation for a child with OSTM1 osteopetrosis. Blood Adv. (2017) 1:279–81. doi: 10.1182/bloodadvances.2016002345
27. Fischer T, De Vries L, Meerloo T, Farquhar MG. Promotion of G Alpha I3 subunit down-regulation by GIPN, a putative E3 ubiquitin ligase that interacts with RGS-GAIP. Proc Natl Acad Sci USA. (2003) 100:8270–5. doi: 10.1073/pnas.1432965100
28. Feigin ME, Malbon CC. OSTM1 regulates β-Catenin/Lef1 interaction and is required for Wnt/β-catenin signaling. Cell Signal. (2008) 20:949–57. doi: 10.1016/j.cellsig.2008.01.009
29. Pangrazio A, Fasth A, Sbardellati A, Orchard PJ, Kasow KA, Raza J , et al. SNX10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity. J Bone Miner Res. (2013) 28:1041–9. doi: 10.1002/jbmr.1849
30. Aker M, Rouvinski A, Hashavia S, Ta-Shma A, Shaag A, Zenvirt S , et al. An SNX10 mutation causes malignant osteopetrosis of infancy. J Med Genet. (2012) 49:221–6. doi: 10.1136/jmedgenet-2011-100520
31. Stattin EL, Henning P, Klar J, McDermott E, Stecksen-Blicks C, Sandström PE , et al. SNX10 gene mutation leading to osteopetrosis with dysfunctional osteoclasts. Sci Rep. (2017) 7:1–16. doi: 10.1038/s41598-017-02533-2
32. Mégarbané A, Pangrazio A, Villa A, Chouery E, Maarawi J, Sabbagh S , et al. Homozygous stop mutation in the SNX10 gene in a consanguineous Iraqi boy with osteopetrosis and corpus callosum hypoplasia. Eur J Med Genet. (2013) 56:32–5. doi: 10.1016/j.ejmg.2012.10.010
33. Alsharidi A, Al-Hamed M, Alsuwaida A. Carbonic anhydrase II deficiency: report of a novel mutation. CEN Case Reports (2016) 5:108–12. doi: 10.1007/s13730-015-0205-y
34. Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. (2009) 4:5. doi: 10.1186/1750-1172-4-5
35. Van Wesenbeeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B , et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Cell Invest. (2007) 117:919–30. doi: 10.1172/JCI30328
36. Fujiwara T, Ye S, Castro-Gomes T, Winchell CG, Andrews NW, Voth DE , et al. plekhm1/def8/rab7 complex regulates lysosome positioning and bone homeostasis. JCI Insight (2016) 1:e86330. doi: 10.1172/jci.insight.86330
37. Marwaha R, Arya SB, Jagga D, Kaur H, Tuli A, Sharma M. The Rab7 effector PLE KHM1 binds Arl8b to promote cargo traffic to lysosomes. J Cell Biol. (2017) 216:1051–70. doi: 10.1083/jcb.201607085
38. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. (2008) 14:325–30. doi: 10.1038/nm1722.
39. McDowall A, Svensson L, Stanley P, Patzak I, Chakravarty P, Howarth K , et al. Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood (2010) 115:4834–42. doi: 10.1182/blood-2009-08-238709
40. Palagano E, Slatter MA, Uva P, Menale C, Villa A, Abinun M , et al. Hematopoietic stem cell transplantation corrects osteopetrosis in a child carrying a novel homozygous mutation in the FERMT3 gene. Bone (2017) 97:126–9. doi: 10.1016/j.bone.2017.01.012
41. Iida A, Xing W, Docx MK, Nakashima T, Wang Z, Kimizuka M , et al. Identi Fi cation of Biallelic LRRK1 mutations in osteosclerotic metaphyseal dysplasia and evidence for locus heterogeneity. J Med Genet. (2016) 53:568–74. doi: 10.1136/jmedgenet-2016-103756
42. Lu SY, Li M, Lin YL. Mitf regulates osteoclastogenesis by modulating NFATc1 activity. Exp Cell Res. (2014) 328:32–43. doi: 10.1016/j.yexcr.2014.08.018
43. George A, Zand DJ, Hufnagel RB, Sharma R, Sergeev YV, Legare JM , et al. Biallelic mutations in MITF cause coloboma, osteopetrosis, microphthalmia, macrocephaly, albinism, and deafness. Am J Human Genet. (2016) 99:1388–94. doi: 10.1016/j.ajhg.2016.11.004
44. Colombo EA, Bazan JF, Negri G, Gervasini C, Elcioglu NH, Yucelten D , et al. Novel C16orf57 mutations in patients with poikiloderma with neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis. (2012) 7:7. doi: 10.1186/1750-1172-7-7
45. Mroczek S, Krwawicz J, Kutner J, Lazniewski M, Kucinski I, Ginalski K , et al. C16orf57, a gene mutated in poikiloderma with neutropenia, encodes a putative phosphodiesterase responsible for the U6 SnRNA 3′ end modification. Genes Dev. (2012) 26:1911–25. doi: 10.1101/gad.193169.112
46. Villa A, Guerrini MM, Cassani B, Pangrazio A, Sobacchi C. Infantile malignant, autosomal recessive osteopetrosis: the rich and the poor. Calcif Tissue Int. (2009) 84:1–12. doi: 10.1007/s00223-008-9196-4
47. Lo Iacono N, Pangrazio A, Abinun M, Bredius R, Zecca M, Blair HC , et al. RANKL cytokine: from pioneer of the osteoimmunology era to cure for a rare disease. Clin Dev Immunol. (2013) 2013:412768. doi: 10.1155/2013/412768
48. Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A , et al. Human Osteoclast-Poor Osteopetrosis with Hypogammaglobulinemia Due to TNFRSF11A (RANK) Mutations. Am J Hum Genet. (2008) 83:64–76. doi: 10.1016/j.ajhg.2008.06.015
49. Pangrazio A, Cassani B, Guerrini MM, Crockett JC, Marrella V, Zammataro L , et al. RANK-dependent autosomal recessive osteopetrosis: characterization of five new cases with novel mutations. J Bone Miner Res. (2012) 27:342–51. doi: 10.1002/jbmr.559
50. Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L , et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. (2007) 39:960–2. doi: 10.1038/ng2076
51. Leibbrandt A, Penninger JM. RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci. (2008) 1143:123–50. doi: 10.1196/annals.1443.016
52. Liu W, Zhang X. Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (Review). Mol Med Rep. (2015) 11:3212–8. doi: 10.2119/molmed.2015.00022
53. Schena F, Menale C, Caci E, Diomede L, Palagano E, Recordati C , et al. Murine Rankl–/– mesenchymal stromal cells display an osteogenic differentiation defect improved by a RANKL-expressing lentiviral vector. Stem Cells (2017) 35:1365–77. doi: 10.1002/stem.2574
54. Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M , et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature (2018) 561:195–200. doi: 10.1038/s41586-018-0482-7
55. Campeau PM, Lu JT, Sule G, Jiang MM, Bae Y, Madan S , et al. Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Human Mol Genet. (2012) 21:4904–9. doi: 10.1093/hmg/dds326
56. Guo L, Elcioglu NH, Karalar OK, Topkar MO, Wang Z, Sakamoto Y , et al. Dysosteosclerosis is also caused by TNFRSF11A mutation. J Hum Genet. (2018) 769–74. doi: 10.1038/s10038-018-0447-6
57. Weisz Hubshman M, Basel-Vanagaite L, Krauss A, Konen O, Levy Y, Garty BZ , et al. Homozygous deletion of RAG1, RAG2 and 5′ region TRAF6 causes severe immune suppression and atypical osteopetrosis. Clin Genet. (2017) 91:902–7. doi: 10.1111/cge.12916
58. Monies D, Maddirevula S, Kurdi W, Alanazy MH, Alkhalidi H, Al-Owain M , et al. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet Med. (2017) 19:1144–50. doi: 10.1038/gim.2017.22
59. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S , et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99:111–20. doi: 10.1182/blood.V99.1.111
60. Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M , et al. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res. (2009) 18:1740–7. doi: 10.1359/jbmr.2003.18.10.1740
61. Waguespack SG, Hui SL, Dimeglio LA, Econs MJ. Autosomal dominant osteopetrosis : clinical severity and natural history of 94 subjects with a chloride channel 7 gene mutation. J Clin Endocrinol Metab. (2007) 92:771–8. doi: 10.1210/jc.2006-1986
62. Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A , et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat. Genet. (2001) 27:277–85. doi: 10.1038/85837
63. Dupuis-Girod S, Corradini N, Hadj-Rabia S, Fournet JC, Faivre L, Le Deist F , et al. Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics (2002) 109:e97. doi: 10.1542/peds.109.6.e97
64. Roberts CM, Angus JE, Leach IH, McDermott EM, Walker DA, Ravenscroft JC. A novel NEMO gene mutation causing osteopetrosis, lymphoedema, hypohidrotic ectodermal dysplasia and immunodeficiency (OL-HED-ID). Eur J Pediatr. (2010) 169:1403–7. doi: 10.1007/s00431-010-1206-7
65. Carlberg VM, Lofgren SM, Mann JA, Austin JP, Nolt D, Shereck EB , et al. Hypohidrotic ectodermal dysplasia, osteopetrosis, lymphedema, and immunodeficiency in an infant with multiple opportunistic infections. Pediatr Dermatol. (2013) 31:716–21. doi: 10.1111/pde.12103
66. Abu-Amer Y, Darwech I, Otero J. Role of the NF-κB axis in immune modulation of osteoclasts and bone loss. Autoimmunity (2008) 41:204–11. doi: 10.1080/08916930701694543
67. Steward CG. Hematopoietic stem cell transplantation for osteopetrosis. Pediatr Clin North Am. (2010) 57:171–80. doi: 10.1016/j.pcl.2009.11.006
68. Behfar M, Dehghani SS, Hosseini AS, Jalali A, Hamidieh AA, Ghavamzadeh A. Non-total body irradiation myeloablative conditioning with intravenous busulfan and cyclophosphamide in hematopoietic stem cell transplantation for malignant infantile osteopetrosis. Pediatr Transplant. (2015) 19:422–7. doi: 10.1111/petr.12476
69. Shadur B, Zaidman I, NaserEddin A, Lokshin E, Hussein F, Oron HC , et al. Successful hematopoietic stem cell transplantation for osteopetrosis using reduced intensity conditioning. Pediatric Blood Cancer (2018) 2017:e27010. doi: 10.1002/pbc.27010
70. Frattini A, Blair HC, Sacco MG, Cerisoli F, Faggioli F, Catò EM , et al. Rescue of ATPa3-deficient murine malignant osteopetrosis by hematopoietic stem cell transplantation in utero. Proc Natl Acad Sci USA. (2005) 102:14629–34. doi: 10.1073/pnas.0507637102
71. Tondelli B, Blair HC, Guerrini M, Patrene KD, Cassani B, Vezzoni P , et al. Fetal liver cells transplanted in utero rescue the osteopetrotic phenotype in the Oc/Oc mouse. Am J Pathol. (2009) 174:727–35. doi: 10.2353/ajpath.2009.080688
72. Orchard PJ, Fasth AL, Le Rademacher J, He W, Boelens JJ, Horwitz EM , et al. Hematopoietic stem cell transplantation for infantile osteopetrosis. Blood (2015) 126:270–6. doi: 10.1182/blood-2015-01-625541
73. Chiesa R, Ruggeri A, Paviglianiti A, Zecca M, Gónzalez-Vicent M, Bordon V , et al. Outcomes after unrelated umbilical cord blood transplantation for children with osteopetrosis. Biol Blood Marrow Transplant. (2016) 22:1997–2002. doi: 10.1016/j.bbmt.2016.07.015
74. Fuchs EJ. Human leukocyte antigen-haploidentical stem cell transplantation using T-cell-replete bone marrow grafts. Curr Opin Hematol. (2012) 19:440–7. doi: 10.1097/MOH.0b013e32835822dc
75. Corbacioglu S, Kernan N, Lehmann L, Brochstein J, Revta C, Grupp S , et al. Defibrotide for the treatment of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Expert Rev Hematol. (2012) 5:291–302. doi: 10.1586/ehm.12.18
76. Shroff R, Beringer O, Rao K, Hofbauer LC, Schulz A. Denosumab for post-transplantation hypercalcemia in osteopetrosis. New Engl J Med. (2012) 367:1766–7. doi: 10.1056/NEJMc1206193
77. Johansson MK, de Vries TJ, Schoenmaker T, Ehinger M, Brun AC, Fasth A , et al. Hematopoietic stem cell – targeted neonatal gene therapy reverses lethally progressive osteopetrosis in Oc / Oc mice. Blood (2007) 109:5178–85. doi: 10.1182/blood-2006-12-061382
78. Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. (2011) 12:301–15. doi: 10.1038/nrg2985
79. Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C , et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet (2016) 388:476–87. doi: 10.1016/S0140-6736(16)30374-9
80. Thrasher AJ, Williams DA. Evolving gene therapy in primary immunodeficiency. Mol Ther. (2017) 25:1132–41. doi: 10.1016/j.ymthe.2017.03.018
81. Moscatelli I, Löfvall H, Schneider Thudium C, Rothe M, Montano C, Kertész Z , et al. Targeting NSG mice engrafting cells with a clinically applicable lentiviral vector corrects osteoclasts in infantile malignant osteopetrosis. Hum. Gene Ther. (2017) 29:938–49. doi: 10.1089/hum.2017.053.
82. Lo Iacono N, Blair HC, Poliani PL, Marrella V, Ficara F, Cassani B , et al. Osteopetrosis rescue upon RANKL administration to Rankl-/-Mice: a new therapy for human RANKL-dependent ARO. J Bone Miner Res. (2012) 27:2501–10. doi: 10.1002/jbmr.1712
83. Bianco P, Robey PG. (2004). Skeletal stem cells. Handjournal Stem Cells. 2:415–24. doi: 10.1016/B978-012436643-5/50129-2
84. Cappariello A, Paone R, Maurizi A, Capulli M, Rucci N, Muraca M , et al. Biotechnological approach for systemic delivery of membrane receptor activator of NF-κB ligand (RANKL) active domain into the circulation. Biomaterials (2015) 46:58–69. doi: 10.1016/j.biomaterials.2014.12.033
85. Menale C, Campodoni E, Palagano E, Mantero S, Erreni M, Inforzato A , et al. MSC-seeded biomimetic scaffolds as a factory of soluble RANKL in rankl-deficient osteopetrosis. Stem Cells Transl Med. (2018) 8:22–34. doi: 10.1002/sctm.18-0085
86. Capulli M, Maurizi A, Ventura L, Rucci N, Teti A. Effective small interfering RNA therapy to treat CLCN7-dependent autosomal dominant osteopetrosis Type 2. Mol Ther. (2015) 4:e248. doi: 10.1038/mtna.2015.21
87. Maurizi A, Capulli M, Patel R, Curle A, Rucci N, Teti A. RNA interference therapy for autosomal dominant osteopetrosis type 2. towards the preclinical development. Bone (2018) 110:343–54. doi: 10.1016/j.bone.2018.02.031
Keywords: bone disease, osteopetrosis, osteoclasts, hematopoietic stem cell transplantation, gene therapy
Citation: Penna S, Capo V, Palagano E, Sobacchi C and Villa A (2019) One Disease, Many Genes: Implications for the Treatment of Osteopetroses. Front. Endocrinol. 10:85. doi: 10.3389/fendo.2019.00085
Received: 14 December 2018; Accepted: 31 January 2019;
Published: 19 February 2019.
Edited by:
Giacomina Brunetti, Università degli Studi di Bari, ItalyReviewed by:
Nadia Rucci, University of L'Aquila, ItalyEmanuela Galliera, University of Milan, Italy
Copyright © 2019 Penna, Capo, Palagano, Sobacchi and Villa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Villa, dmlsbGEuYW5uYUBoc3IuaXQ=
†Co-first authorship