 Du Soon Swee1,2*
Du Soon Swee1,2* Richard Quinton1,3
Richard Quinton1,3- 1Department of Endocrinology, Royal Victoria Infirmary, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
- 2Department of Endocrinology, Singapore General Hospital, Singapore, Singapore
- 3Institute of Genetic Medicine, University of Newcastle upon Tyne, Newcastle upon Tyne, United Kingdom
Congenital hypogonadotrophic hypogonadism (CHH) is a rare but important etiology of pubertal failure and infertility, resulting from impaired gonadotrophin-releasing hormone secretion or action. Despite the availability of effective hormonal therapies, the majority of men with CHH experience unsatisfactory outcomes, including chronic psychosocial and reproductive sequelae. Early detection and timely interventions are crucial to address the gaps in medical care and improve the outlook for these patients. In this paper, we review the clinical implications of missing minipuberty in CHH and therapeutic strategies that can modify the course of disease, as well as explore a targeted approach to identifying affected male infants by integrating clinical and biochemical data in the early postnatal months.
Introduction
Congenital hypogonadotrophic hypogonadism (CHH) is a rare genetic condition characterized by reproductive disorder due to deficiency in secretion or action of gonadotrophin-releasing hormone (GnRH). It is traditionally considered to be a male-predominant condition, with a male: female gender ratio 3.6: 1 that remains unexplained (1). The major clinical consequences of CHH are pubertal failure and infertility.
Although generally considered a rare disorder, accurate determination of the prevalence of CHH has not been possible because of scarce literature. Based on a French study of potential military conscripts who attended medical examination (2) and, more recently, a retrospective study of nationwide hospital records in Finland (3), (both of which were methodologically prone to under-ascertainment), male CHH prevalence of 1 in 4,415–15,000 is currently estimated.
The genetic defects underpinning CHH broadly fall into two principle groups, comprising (a) those causing neurodevelopmental defects of GnRH neuron migration frequently associated with non-reproductive defects, particularly anosmia/hyposmia from olfactory axon misrouting (i.e., Kallmann syndrome–KS), and (b) those causing pure neuroendocrine impairment of GnRH secretion or action (normosmic CHH). Belying this apparently simple dichotomy is the huge diversity of genetic mutations, with over 30 gene loci implicated to date, despite nearly half of CHH cases remaining unaccounted for. Moreover, some genes are implicated in both normosmic CHH and KS (4). This complexity is also reflected in the various modes of transmission possible, including oligogenicity as well as all forms of classical Mendelian inheritance (4, 5).
Furthermore, CHH is phenotypically heterogeneous. Besides the variable association with non-reproductive features, such as deafness, synkinesis (mirror movements), renal agenesis, digital and dental anomalies, and clefting, reproductive manifestation range from absent puberty, pubertal arrest, to even spontaneous reversal of hypogonadism in a small minority (5). CHH males frequently present with cryptorchidism and/or micropenis, which are important features of severe fetal-infancy GnRH deficiency (absent minipuberty) (6). However, patient experiences indicate that these early presentations only rarely signpost timely disease recognition and treatment-initiation in later life.
Despite medical advances in genetics, diagnostics and treatment, health outcomes of CHH men remain disappointing, with a significant proportion bearing the long-term consequences of suboptimal care (7). In this chapter, we will review the factors contributing to poor outcomes of men with CHH, and the strategies that can improve the quality of life and fertility potential, with special focus on harnessing the window of minipuberty for early diagnosis, and intervention.
Delayed Diagnosis and Treatment
Delayed puberty is the main mode of presentation in CHH males. Approximately 2/3 of CHH males adolescents do not show any sign of spontaneous puberty at >17 years of age (testis volume <4 mL), with the remainder exhibiting arrested early puberty (8).
Unfortunately, CHH is biochemically indistinguishable from constitutional delay in growth and puberty (CDGP), the latter accounting for up to 65% of delayed puberty in younger teenage boys (9), but obviously declining precipitously with advancing age at presentation. Both conditions are characterized by low sex steroids in association with low or inappropriately normal follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels. In terms of their height development, the baseline height SD scores and growth velocities do not appear to differ significantly between CDGP and CHH adolescents, unlike in functional hypogonadotrophic hypogonadism where there is a tendency for lower height SD scores and reduced growth velocity of <3 cm/year (9, 10). It is however worth noting that progressive reduction in height SDs has been observed in a proportion of CDGP boys during the pre-pubertal years, with final attained height falling short of their genetic potential (11). On the other hand, preservation of height relative to parental height in CHH patients has been suggested (12). Hence, careful interrogation of the growth history can yield useful information.
Although a variety of stimulation tests (such as GnRH stimulation test and hCG test) and, recently, inhibin B (IB) concentrations (marker of Sertoli cell function), have been proposed, there is still lack of consensus on the “gold standard” test to reliably distinguish them (13). Hence, making a diagnosis of CHH remains a challenging task and clinicians frequently default to the classic dogma of expectant management, i.e., allowing adolescents enough time for those with CDGP to undergo spontaneous initiation of puberty, as a means to identify those with CHH (14). It is noteworthy that in a recent large single center retrospective series, the combination of testicular volume and basal IB level–both of which are obtainable with relative ease without the involvement of complicated dynamic tests–demonstrated utility in discriminating between CHH and CDGP, and therefore could have the potential for wider application (10).
However, there seems to be consistent misapplication of “wait & see & reassure” guidance (intended for individuals with undifferentiated pubertal delay) to those with red flag markers of CHH who should, instead, be presumed hypogonadal until proven otherwise and receive sex hormone replacement from mean age of pubertal-onset in that population i.e., 12 years in boys (6, 15), whereas treatment should not be unduly withheld in those without CHH features but have turned 14 years old (16).
As a result, the diagnosis of CHH and initiation of clinically-meaningful treatment to induce puberty is typically unduly delayed until late adolescence or early adulthood. Despite different survey techniques across different European countries, data on the mean ages of diagnosis and initiation of clinically-meaningful1 treatment for CHH men have been remarkably consistent, with Dwyer et al (web-based, pan-European; n = 101) finding these to be 18 ± 6 and 19 ± 5 years, respectively (7); Raivio et al (nationwide Finnish cohort study) finding the median age of starting treatment to be 18.3 years (range, 11–34 years) (17), and Quinton (case notes based survey; n = 200) finding it to be 18.9 ± 9 years (18).
A surprisingly common pitfall is the failure to recognize the significance of cryptorchidism in an adolescent with pubertal delay. In retrospective CHH series, between 30 and 50% of CHH males have a history of cryptorchidism, of whom up to 2/3 have bilateral undescended testes (8, 19–21). By contrast, cryptorchidism is very rare in CDGP. In a large series of boys seen at a specialized center referred from primary care for delayed puberty, only 2% of CDGP boys had a history of cryptorchidism, compared with 36% of those with CHH (10). Hence, a history of cryptorchidism should alert clinicians to presume the diagnosis of CHH until proven otherwise (22). A concomitant family history of cryptorchidism, micropenis, infertility and/or non-reproductive features such as anosmia, renal agenesis and cleft palate/lip could provide valuable clues to an underlying diagnosis of CHH, though it is absent in majority of cases, in part due to variable disease penetrance and phenotypic expression, as well as oligogenic inheritance with unaffected parents (4).
Likewise, the significance of bilateral undescended testes in neonates as a possible indicator of CHH is typically underappreciated. Persistent bilateral cryptorchidism is found in a quarter of CHH infants, as compared to <0.7% among infants in the general population (Table 1) In a large single-center retrospective series, only one-third of CHH males with history of bilateral orchidopexy in childhood were referred to pediatric endocrinology for evaluation, with most cases re-presenting much later in life with absent puberty (19). Current clinical guidelines have largely focused on evaluation for possible congenital adrenal hyperplasia (CAH) and disorder of sex development (DSD), and neglected to provide sufficient guidance on the investigations necessary in cryptorchid boys to rule out CHH (36, 37), resulting in missed opportunity for early recognition.
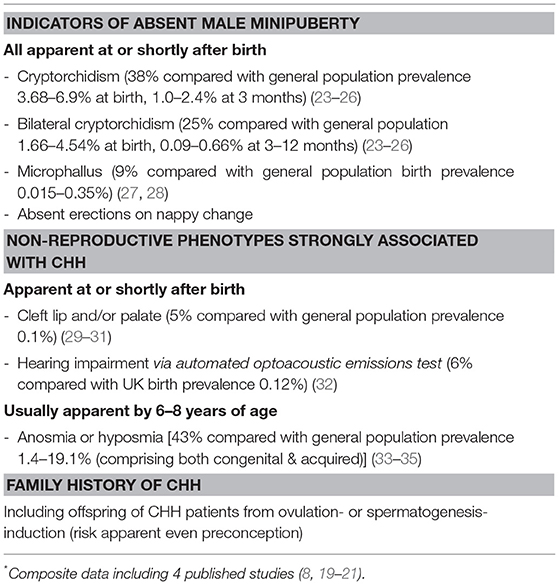
Table 1. “Red Flag” clinical markers for congenital GnRH deficiency*.
As a result of late diagnosis and delayed treatment, the optimal standard of care is not delivered with patients with CHH, leading to significant psychosocial and reproductive sequelae.
Unmet Health Needs
Poor Psychological Well-Being
Psychological morbidities and antidepressant usage are highly prevalent among CHH men (17). In an international study comprising of participants from North and South America, Europe and Australasia, nearly two-thirds suffered from depression, with majority of them exhibiting moderate to severe symptoms (38). An important consequence arising from chronic affective disorders is poor adherence to long-term hormone replacement. It is concerning that more than one-third of the survey respondents reported long gaps (>1 year) in treatment, which could potentially exacerbate affective symptoms in turn as part of a vicious cycle.
The psychosexual impact of the disease is tremendous. A significant number of patients suffer from anxiety and low self-esteem, resulting in inability to form intimate relationships and social isolation. Consequently, in the aforementioned study cohort (38), approximately 50% of the men were not in a stable relationship at the time of survey, and many never had a sexual partner.
On the positive side, the majority of men surveyed had received tertiary level education and were in gainful employment, reflecting reasonable socioeconomic circumstances, although this did not negate the psychological effects of their disorder. However, CHH men with adopted or biological children were less likely to report depressive symptoms, suggesting the positive effect of family companionship, and the vicious circle that socially-withdrawn CHH men may be trapped in.
A major factor that contributes to poor mental health and impaired quality of life is the delay in diagnosis and the failure to administer age-appropriate induction of secondary sexual characteristic in many CHH males. As alluded to earlier, most patients only start to receive meaningful treatment in late adolescence (median age 18–19 years) typically after a prolonged and frustrating diagnostic odyssey (frequently shared by their female counterparts too) (1). Consequently, they are at risk of developing a multitude of psychosocial problems associated with pubertal delay, including low self-esteem, social withdrawal, poor school performance and even higher rates of substance use disorder (16, 39, 40). Furthermore, inadequately treated cryptorchidism and/or micropenis can lead to long-lasting adverse impact on their sexuality (17).
Compromised Fertility Potential
In young adult males with CHH, gonadotrophin treatment achieves a significantly greater positive impact than direct testosterone (T) replacement on health-related quality of life (41). While both treatments are effective in improving physical function and general health, patients receiving gonadotrophins perform better in psychological domains, including emotional and mental health, particularly if sperms are found in ejaculate. This strongly suggests that patients psyches are deeply affected by their perceived chances of achieving paternity.
The infertility in CHH patients is due to spermatogenic failure, which is potentially amenable to GnRH or gonadotrophin treatment. Unfortunately, classic spermatogenic treatment–human chorionic gonadotrophin (hCG) monotherapy or combined gonadotropin treatment (hCG+FSH)–is much less successful in men with severe CHH (testes <4 mL), particularly those with history of bilateral cryptorchidism, than in men with HH of postpubertal-onset, e.g., due to acquired pituitary disease (42). Nonetheless, in centers experienced in the care of CHH patients, up to three-quarters can achieve spermatogenesis during hormonal induction treatment (6, 43), and pregnancy rates can be further enhanced with assisted reproductive techniques (6, 44).
Patients with rare medical disorders, defined by a prevalence of <5 in 10,000 in the population, often face challenges due to the lack of knowledge of their healthcare providers (45, 46). As such, specialized centers with expertise in diagnosis and interdisciplinary treatment of rare diseases are vital in the provision of care to these patients (47). Similarly, patients with CHH should ideally receive tertiary-level care to avoid gaps in treatment, and benefit from the latest technologies and advances in the research field. Early diagnosis would provide the opportunity for patients to receive appropriate and consistent care and support at specialized centers without delay. Treatment can also be tailored according to the needs and goals in different stages of life (6, 48).
But in real-life setting, according to one survey, it appears that only a minority of CHH men seeking fertility are able to achieve desired outcomes on fertility-inducing treatment (7). It is unclear how many of these men were treated at centers with the necessary expertise, but given that only half of the whole study cohort being followed-up at specialized/academic centers, access to such resources is likely to be correspondingly restricted.
Elevated Risk for Low Bone Mineral Density
Chronic sex steroid deficiency is a major risk factor for osteoporosis and fragility fractures that affects both genders. As men with CHH have early-onset of T insufficiency, a delay in and/or lack of adequate androgen replacement would result in poor bone mass accretion and accelerated bone loss (49). Indeed, patients who are initiated on T replacement at older age appear to accrue less bone mineral compared to younger age, further supporting the importance of timely treatment (50). Nonetheless, even for those who are diagnosed and started on treatment only later in life, encouraging improvement in bone mineral density, particularly at trabecular-rich lumber spine, has been observed (51).
Minipuberty–Critical Period of Genitalia Development and the Window to Early Diagnosis
High incidence of cryptorchidism and microphallus is observed in CHH males because of the absence of minipuberty, which is a critical period in the ontogenesis of the male reproductive tract, characterized by activation of the GnRH axis in the initial months postnatal. During this developmental phase, serum T and gonadotropin levels rise rapidly and peak at age 3 months–remarkably approaching adult male levels–before to mid-childhood quiescence by about 6 months of age (52, 53).
This robust hormonal activity is necessary to complete the processes of inguinoscrotal testicular descent and anchoring in the scrotum as well as penile growth, which had begun earlier in utero during the third trimester. Specifically, LH-stimulated secretion of T and INSL3 peptide by Leydig cells are the key factors involved in driving these physical changes (54, 55). There is a concurrent exponential increase in FSH-stimulated IB and anti-Müllerian hormone (AMH) secretion, signifying active proliferation of Sertoli cells. Expansion of Sertoli and germ cells and seminiferous tubule formation are key determinants of future fertility potential and are responsible for 90% of testicular volume (56). Despite the brisk gonadotrophic activity and T secretion during this proliferative phase, germ cell maturation and spermatogenesis do not occur, because androgen receptors are not expressed on Sertoli cells until 5 years of age (57).
Following minipuberty, the hypothalamic-pituitary-testicular (HPT) axis retreats into quiescence for the rest of childhood. Serum T, LH, and FSH levels decline to low levels until reactivation of gonadal axis occurs in early adolescence, heralding the onset of puberty and marked by testicular enlargement (≥4 mL), followed by penile and pubic hair growth. At this stage, Sertoli cell maturation occurs, evidenced by a rise in IB and decline in AMH levels, and spermatogenesis is achieved by the concerted actions of FSH and intra-testicular T (58). Therefore, pulsatile GnRH secretion in the neonatal period appears to be paramount for the normal development of male genitalia, with a far-reaching impact on male reproductive phenotype and fertility potential later in adult life.
Another important clinical implication of minipuberty is that it potentially provides a window-of-opportunity to facilitate detection of children with congenital GnRH deficiency, who would demonstrate abnormally low FSH, LH, and T levels if measured, thereby offering the advantage of a definitive prepubertal diagnosis and sign-posting them to pre-planned pubertal-induction with sex hormones at median age of pubertal-onset, rather than expectative management.
Early Diagnosis for Avoiding Delay in Pubertal Induction
Diagnosis of CHH in early life facilitates the structuring of long-term surveillance and treatment plans, as well as ensuring that counseling and psychological support to patients and families are made available. When patients reach early adolescence, age-appropriate pubertal induction treatment will ensure that secondary sexual characteristics develop in tandem with peers (15), thereby avoiding the delay that has been traditionally experienced by most CHH men. As such, uncertainties are minimized and anxieties can be allayed.
Potential Benefit of Early Diagnosis in Improving Prospect of Fertility
Early identification of boys with CHH could also plausibly provide the opportunity for intervention to optimize fertility potential. Although GnRH or gonadotrophin combination treatment are effective for most CHH men in spermatogenesis-induction, sperm outcomes are generally suboptimal (43, 59, 60). Moreover, nearly one-third with severe CHH remain azoospermic even with prolonged combined gonadotropin therapy, and hCG-monotherapy has proved to be despairingly ineffective.
Clinical features (consistent with long-standing severe GnRH deficiency minipuberty) that are predictive of poor treatment response, include: complete absence of puberty at presentation, cryptorchidism (especially if bilateral), low serum IB concentrations (indicating depleted Sertoli cells) and prepubertal testicular volume (indicating depletion of Sertoli and germ cells and seminiferous tubules) (43, 59). On the other hand, CHH men with partial GnRH deficiency (testicular volume ≥4 mL) respond much better to combined gonadotrophin treatment, with around 80% achieving sperm in the ejaculate (61).
Therefore, in men with complete CHH, it would be theoretically advantageous to first maximize proliferation of Sertoli and germ cell and growth of seminiferous tubules by administering FSH-monotherapy prior to the introduction of hCG, so as to prevent premature maturation of a depleted pool of Sertoli cells under the influence of intra-testicular T.
Pathfinder studies of FSH-monotherapy in children and adolescents with HH of prepubertal-onset demonstrated its efficacy in promoting testicular growth and circulating IB concentrations (62, 63). In particular, there was an encouraging spermatogenesis response in a subgroup of CHH adolescents, in whom FSH-priming before the combination with hCG successfully induced spermatogenesis in 3 out of 4 patients (63).
The potential benefit of unopposed FSH treatment was further studied in a randomized, open-label trial of 13 treatment-naive adult CHH men with prepubertal testes (<4 mL) (64). Seven men were randomized to recombinant FSH-pretreatment for 4 months before embarking on a 24-month GnRH treatment protocol. During the FSH-only phase, testicular volume doubled and IB levels rose to adult levels, and all subjects in this arm subsequently developed sperm in ejaculate on GnRH therapy, compared to 4 of 6 men in the 24-month GnRH-only arm. There were also trends to larger testicular volume, higher maximal sperm counts and shorter time to first appearance of sperm in the ejaculate in the FSH-pretreated group.
Therefore, the findings of the benefits of FSH-priming and the potential deleterious effect of premature hCG therapy would be important to inform clinicians on the choice of treatment in adolescents with CHH, which would be greatly facilitated by timely diagnosis.
Potential of Neonatal Gonadotrophin Treatment to Further Optimize Outcomes
Recognizing the critical role of minipuberty in the development of external genitalia and Sertoli cell proliferation and its subsequent influence on future reproductive function, the feasibility and benefits of recreating the physiological hormonal milieu in male CHH infants has been studied.
In the first published report of a boy with CHH and micropenis who received short-term recombinant human LH and FSH from age 7.9–13.7 months, the penile length successfully increased by 50% and the testicular volume nearly tripled by the end of treatment (65). In another report of 2 male infants, one case each of combined pituitary hormone deficiency (CPHD) and CHH, 6-month gonadotrophin combination therapy via subcutaneous pump infusion initiated at age 8 and 20 weeks, respectively, led to 4-fold increase in both penile length and testicular volume (66). More recently, 3–6 months of continuous subcutaneous infusion of recombinant human gonadotrophins in 5 male infants [4 CHH, 1 CPHD] produced several-fold increase in IB concentrations, testicular volume, and T secretion (67).
Besides these encouraging responses to combined gonadotrophin treatment during infancy, it could also aid in the management of undescended testes. Cryptorchidism is present in approximately one-half of boys with severe CHH (19), and it is an independent predictor of infertility. Delay in orchidopexy has been associated with dramatic decline in germ cells (68), and as such it is generally recommended that surgical treatment take place by about 1 year of age (37, 69). However, small testes render surgical manipulation technically challenging and would result in excess risk of testicular trauma and tissue loss (70). By administering a period of presurgical gonadotrophin therapy, it allows the enlargement of testicular volume to facilitate the procedure.
Indeed, there are emerging evidence that spontaneous testicular descent could be successfully induced by gonadotrophin treatment in infants with central hypogonadism, and thus obviate the need for surgery. In a cohort of eight infants with maldescended testes due to underlying hypogonadotrophic hypogonadism (5 CHH, 3 CPHD), gonadotrophin infusion induced full testicular descent in 6 boys and partial descent in 2 boys, such that only 1 had to undergo orchidopexy nearly a year later because of re-ascent of both testes (71). Another study showed that combined recombinant FSH and LH in the first 6 months of life successfully induced spontaneous testicular descent in 2 of 4 bilateral-cryptorchid CHH/CPHD boys (67).
Therefore, short-term neonatal gonadotrophin treatment in ascertained cases of hypogonadotrophic hypogonadism appears to be effective in replicating the effects of minipuberty, by correcting micropenis, promoting testicular growth due to Sertoli cell expansion, and inducing spontaneous descent of malpositioned testes. Importantly, treatment was well-tolerated in all reported cases. Although definitive evidence is currently lacking, the prospect of early childhood hormonal intervention in CHH boys in augmenting sexual and reproductive function in adult life is worth serious consideration, and hopefully will provide impetus for larger clinical trials.
A Proposed Strategy to Improve Detection of Male Infants With Severe CHH
The phase of male minipuberty provides an extraordinary useful diagnostic window to confirm (or refute) the diagnosis of congenital hypogonadism with a relatively straightforward biochemical hormone profiling without the need for complex dynamic testing. In a cohort of CPHD male infants (predominantly presenting with hypoglycaemia), findings of low circulating FSH, LH, and T concentrations reliably identified concomitant hypogonadotrophic hypogonadism in 14 of 15 infants with genitalia anomalies, whereas all other boys with normal genitalia demonstrated intact pituitary-gonadal axis function (72). This contrasts starkly with the conundrum of differentiating CHH from CDGP in adolescence.
However, unlike CPHD, neonates with CHH without cryptorchidism do not necessarily have clinical manifestations that would trigger referral to pediatric endocrinologists for pituitary hormonal evaluation. In addition, there is a lack of awareness and clinical guidance to consider CHH as a differential diagnosis of male infants with cryptorchidism, particularly if bilateral or associated with micropenis, so that appropriate evaluation can be undertaken (22, 37, 69). Indeed, the presence of “red flag” markers (Table 1) should warrant further investigations to exclude such a possibility.
Another challenge would be the interpretation of less robust serum gonadotrophin and testosterone results in the male infants. Of relevance, normative data of several important reproductive hormone values during minipuberty–including LH, FSH, T (by both radioimmunoassay and tandem mass spectrometry), AMH and IB-have recently been derived from a large cohort of healthy Danish infants, with cut-off values discriminating between sexes determined (53). By extension, values above the cut-off levels–generally lower than adult ranges–could be regarded as intactness of HPT axis function, whereas equivocal biochemical results that lie below the cut-off values may warrant repeat testing. Furthermore, expanding screening panel to include AMH and IB could improve diagnostic confidence. Crucially, this study also demonstrated that the various sex hormones peak just before 3.5 months of age, suggesting that diagnostic performance would be most optimal when evaluation is undertaken around this time.
Bilateral Cryptorchidism as Potential Screening Criterion
While routine screening for minipuberty is impractical, a targeted approach to evaluate male infants with suspicious signs of CHH could be cost-effective. Among the associated clinical features, bilateral cryptorchidism is particularly important because of its high prevalence (13.9–34.5%) among CHH males and the association with severe GnRH deficiency that has both prognostic and therapeutic implications (8, 19, 21).
Although cryptorchidism is a common congenital urogenital abnormality in newborns, most would undergo spontaneous descent in the absence of hypogonadism or other organic disorders. Data from prospective studies shows that the prevalence of bilateral cryptorchidism decreases substantially from 1.66 to 4.54% at time of birth to 0.09–0.66% by 3–12 months of age (23–26). In addition, spontaneous descent is unlikely to occur beyond 3 months postnatal (73), which coincides closely with the expected peak of minipuberty, making it an ideal time point to undertake testing of reproductive hormones in boys with persistent bilateral cryptorchidism. Moreover, although non-CHH cryptorchid boys may also exhibit hormonal abnormalities, they tend to have higher FSH, similar T and slightly lower IB values compared to healthy infants, which are different from the biochemical pattern expected of in CHH male infants (74).
Therefore, testing male infants with bilateral cryptorchidism with or without micropenis at 3 months of age for absent minipuberty could potentially be a feasible approach to facilitate the early diagnosis of CHH. The diagnostic yield of such a selective screening strategy is explored here, using Britain's birth data as an example for mathematical illustration:
• With an average male live birth rate of 390,070 per annum in Britain (75), between 26 and 88 boys born each year could be affected by CHH (based on estimated prevalence of 1 in 4,415–15,000).
• On the other hand, bilateral cryptorchidism could affect between 6,475 and 17,709 (1.66–4.54%) of all boys at birth, regardless of the presence of CHH. Among them, between 4 and 30 infants could have underlying CHH (as bilateral cryptorchidism affects 13.9–34.5% of CHH population), thus representing 0.02–0.46% of all bilateral-cryptorchid boys at birth.
• By 3 months of age, following spontaneous testicular descent expected in majority of non-CHH infants, the total number of infants with persistent bilateral cryptorchidism would be expected to reduce substantially to between 351 and 2,574 (0.09–0.66%).
• As spontaneous testicular descent is not expected in CHH-affected infants, they now account for greater proportion of all persistently bilateral-cryptorchid boys: 0.16–8.55% (in contrast to 0.02–0.46% at birth). Hence, screening at 3 months of age appears to be most cost-effective by avoiding investigations in vast majority of infants without persistent bilateral cryptorchidism.
Based on this hypothesis, possibly up to 1 in 11–12 male infants with persistent bilateral cryptorchidism at 3 months of age could have underlying CHH. Interestingly, this estimate is consistent with the findings from an historical surgical series of patients who had been treated with orchidopexy. In this study, of the 98 patients evaluated for possible underlying endocrine cause of cryptorchidism, 2 were found to have CHH in adult life and both of them had bilateral undescended testes (76). That represents 6.1% (2/33) of all individuals with bilateral cryptorchidism in the series.
However, it is worth reiterating that, at present, there is no literature to suggest that CHH screening in infancy has been systematically studied by any research group, and thus is a working concept that remains in early exploratory phase. Further long-term multicentre research is necessary to validate the utility of any screening strategy that seeks to establish early diagnosis of this rare condition, as well as the effectiveness and safety of hormonal treatment in CHH infants. Protocols should also be developed collaboratively by pediatric and adult endocrinologists to ensure that these children are placed in a structured follow-up and transition programme, thereby ensuring that no one falls through the crack in the health system.
A Short Note on Pre-pubertal Acquired Hypogonadotrophic Hypogonadism
Pubertal failure in male adolescents with known history of acquired hypopituitarism generally avoids the diagnostic conundrum of CHH, hence allowing pubertal-induction therapy to be planned for at around 12 years of age, with gradual increase in testosterone dose before reaching adult replacement dose in about 3 years (15). Importantly, because of preserved minipuberty, normal Sertoli cell proliferation is expected in early childhood, and testicular maldescent would be less likely to occur (63). Therefore, these individuals tend to have a more optimistic fertility outlook, with greater spermatogenic response to gonadotrophin treatment anticipated (77), unless testicular tissue has been compromised e.g., prior gonadotoxic chemotherapy treatment.
Conclusion
Early detection of CHH through detection of absent minipuberty can potentially modify patients' experience by facilitating timely interventions at different stages of life. This is achievable with a systematic approach in place to identify male infants with “red flag” markers of CHH, particularly bilateral cryptorchidism, so that biochemical testing can be undertaken within the narrow diagnostic window.
Neonatal gonadotrophin treatment appears to be beneficial in correcting micropenis and even cryptorchidism. In adolescents, age-appropriate pubertal induction is the main goal, and a brief course of FSH monotherapy should be considered to optimize future fertility potential. Close collaborations between pediatric care providers and adult endocrinologists would ensure that these patients transit smoothly to adulthood, during which the aims of treatment would shift to fertility-induction and long-term androgen replacement. Equally important are the psychological support and genetic counseling that should be provided along the way to empower patients, and families, so that they can cope with their conditions confidently.
Future Directions
Proactive screening of male infants with bilateral cryptorchidism (and/or micropenis) is anticipated to have a direct impact on the quality of care delivered to affected children by establishing early diagnosis. However, this issue has not been addressed by major clinical guidelines developed for the management of infants with abnormal genitalia, thereby leading to missed opportunities for patients and clinicians alike in accessing the most appropriate treatment possible. To overcome this barrier, it is imperative that collaborative research by key stakeholder centers seek to clarify the feasibility of such an approach systematically, as well as expanding the work on neonatal gonadotrophin treatment, so as to advance the agenda for wider adoption among endocrinologists, pediatricians, and pediatric surgeons.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^For instance, 50mg intramuscular testosterone monthly would not remotely constitute appropriate or meaningful replacement therapy for a 20 year-old, 1.7m and 70kg prepubertal male with Kallmann syndrome.
References
1. Dzemaili S, Tiemensma J, Quinton R, Pitteloud N, Morin D, Dwyer AA. Beyond hormone replacement: quality of life in women with congenital hypogonadotropic hypogonadism. Endocr Connect. (2017) 6:404–12. doi: 10.1530/EC-17-0095
2. Fromantin M, Gineste J, Didier A, Rouvier J. [Impuberism and hypogonadism at induction into military service. Statistical study]. Probl Actuels Endocrinol Nutr. (1973) 16:179–99.
3. Laitinen E-M, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, et al. Incidence, phenotypic features and molecular genetics of kallmann syndrome in Finland. Orphanet J Rare Dis. (2011) 6:41. doi: 10.1186/1750-1172-6-41
4. Maione L, Dwyer AA, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, et al. Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. (2018) 178:R55–80. doi: 10.1530/EJE-17-0749
5. Mitchell AL, Dwyer A, Pitteloud N, Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab. (2011) 22:249–58. doi: 10.1016/j.tem.2011.03.002
6. Boehm U, Bouloux PM, Dattani MT, De Roux N, Dodé C, Dunkel L, et al. Expert consensus document: european consensus statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
7. Dwyer AA, Tiemensma J, Quinton R, Pitteloud N, Morin D. Adherence to treatment in men with hypogonadotrophic hypogonadism. Clin Endocrinol. (2017) 86:377–83. doi: 10.1111/cen.13236
8. Bonomi M, Vezzoli V, Krausz C, Guizzardi F, Vezzani S, Simoni M. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. (2018) 178:23–32. doi: 10.1530/EJE-17-0065
9. Sedlmeyer IL, Palmert MR, Hospital CNS. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metab. (2002) 87:1613–20. doi: 10.1210/jcem.87.4.8395
10. Varimo T, Miettinen PJ, Känsäkoski J, Raivio T, Hero M. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center. Hum Reprod. (2017) 32:147–53. doi: 10.1093/humrep/dew294
11. Wehkalampi K, Vangonen K, Laine T, Dunkel L. Progressive reduction of relative height in childhood predicts adult stature below target height in boys with constitutional delay of growth and puberty. Horm Res. (2007) 68:99–104. doi: 10.1159/000101011
12. Young J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2012) 97:707–18. doi: 10.1210/jc.2011-1664
13. Harrington J, Palmert MR. Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: Critical appraisal of available diagnostic tests. J Clin Endocrinol Metab. (2012) 97:3056–67. doi: 10.1210/jc.2012-1598
14. Christie D, Viner R. Adolescent development. BMJ (2005) 330:301–4. doi: 10.1136/bmj.330.7486.301
15. Dunkel L, Quinton R. Transition in endocrinology: induction of puberty. Eur J Endocrinol. (2014) 170:R229–39. doi: 10.1530/EJE-13-0894
16. Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med. (2012) 366:443–53. doi: 10.1056/NEJMcp1109290
17. Varimo T, Hero M, Laitinen EM, Sintonen H, Raivio T. Health-related quality of life in male patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol (Oxf). (2015) 83:141–3. doi: 10.1111/cen.12701
18. Quinton R. Phenotypic Aspect of Kallmann's Syndrome, MD dissertation, University of Cambridge. (2000)
19. Quinton R, Duke VM, Robertson A, Kirk JMW, Matfin G, De Zoysa PA, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf). (2001) 55:163–74. doi: 10.1046/j.1365-2265.2001.01277.x
20. Stamou MI, Varnavas P, Kentrou M, Adamidou F, Voutetakis A, Jing J, et al. Isolated GNRH deficiency: genotypic and phenotypic characteristics of the genetically heterogeneous Greek population. Eur J Endocrinol. (2017) 176:L1–5. doi: 10.1530/EJE-16-0505
21. Pitteloud N, Hayes FJ, Boepple PA, Decruz S, Seminara SB, MacLaughlin DT, et al. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2002) 87:152–60. doi: 10.1210/jcem.87.1.8131
22. Quinton R, Mamoojee Y, Jayasena CN, Young J, Howard S, Dunkel L, et al. Society for Endocrinology UK guidance on the evaluation of suspected disorders of sexual development: emphasizing the opportunity to predict adolescent pubertal failure through a neonatal diagnosis of absent minipuberty. Clin Endocrinol (Oxf). (2017) 86:305–6. doi: 10.1111/cen.13257
23. Acerini CL, Miles HL, Dunger DB, Ong KK, Hughes IA. The descriptive epidemiology of congenital and acquired cryptorchidism in a UK infant cohort. Arch Dis Child. (2009) 94:868–72. doi: 10.1136/adc.2008.150219
24. Berkowitz GS, Lapinski RH, Dolgin SE, Gazella JG, Bodian CA, Holzman IR. Prevalence and natural history of cryptorchidism. Pediatrics (1993) 92:44–9.
25. John Radcliffe Hospital Cryptorchidism Study Group. Cryptorchidism: a prospective study of 7500 consecutive male births, 1984-8. John Radcliffe Hospital Cryptorchidism Study Group. Arch Dis Child. (1992) 67:892–9. doi: 10.1136/adc.67.7.892
26. Ghirri P, Ciulli C, Vuerich M, Cuttano A, Faraoni M, Guerrini L, et al. Incidence at birth and natural history of cryptorchidism: a study of 10,730 consecutive male infants. J Endocrinol Invest. (2002) 25:709–15. doi: 10.1007/BF03345105
27. Nelson CP, Park JM, Wan J, Bloom DA, Dunn RL, Wei JT. The increasing incidence of congenital penile anomalies in the United States. J Urol. (2005) 174:1573–6. doi: 10.1097/01.ju.0000179249.21944.7e
28. Gaspari L, Paris F, Jandel C, Kalfa N, Orsini M, Daurs JP, et al. Prenatal environmental risk factors for genital malformations in a population of 1442 French male newborns: a nested casecontrol study. Hum Reprod. (2011) 26:3155–62. doi: 10.1093/humrep/der283
29. Public Health England. National Congenital Anomaly and Rare Disease Registration Service. Congenital anomaly statistics 2016 PHE publications. (2018) Available online at: https://www.gov.uk/government/publications/ncardrs-congenital-anomaly-annual-data (Accessed November 26, 2018).
30. EUROCAT Association. European Surveillance of Congenital Anomalies. (2018) Available online at: http://www.eurocat-network.eu/ (Accessed November 25, 2018).
31. Centers for Disease Control and Prevention. Updated National Birth Prevalence Estimates for Selected Birth Defects in the United States, 2004-2006. (2018) Available online at: https://www.cdc.gov/ncbddd/birthdefects/features/birthdefects-keyfindings.html
32. Davis A, Wood S, Healy R, Webb H, Rowe S. Risk factors for hearing disorders: epidemiologic evidence of change over time in the UK. J Am Acad Audiol. (1995) 6:365–70.
33. Hoffman HJ, Ishii EK, Macturk RH. Age-related changes in the prevalence of smell/taste problems among the United States adult population. Results of the 1994 disability supplement to the National Health Interview Survey (NHIS). Ann N Y Acad Sci. (1998) 855:716–22. doi: 10.1111/j.1749-6632.1998.tb10650.x
34. Lin SH, Chu ST, Yuan BC, Shu CH. Survey of the frequency of olfactory dysfunction in Taiwan. J Chin Med Assoc. (2009) 72:68–71. doi: 10.1016/S1726-4901(09)70025-5
35. Brämerson A, Johansson L, Ek L, Nordin S, Bende M. Prevalence of Olfactory Dysfunction: the Skövde Population-Based Study. Laryngoscope (2004) 114:733–7. doi: 10.1097/00005537-200404000-00026
36. Ahmed SF, Achermann JC, Arlt W, Balen A, Conway G, Edwards Z, et al. Society for Endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development 84, Clin Endocrinol. (2016) 84:771–88. doi: 10.1111/cen.12857
37. Kolon TF, Herndon CDA, Baker LA, Baskin LS, Baxter CG, Cheng EY, et al. Evaluation and treatment of Cryptorchidism: AUA guideline. J Urol. (2014) 192:337–45. doi: 10.1016/j.juro.2014.05.005
38. Dwyer AA, Quinton R, Morin D, Pitteloud N. Identifying the unmet health needs of patients with congenital hypogonadotropic hypogonadism using a web-based needs assessment: implications for online interventions and peer-to-peer support. Orphanet J Rare Dis. (2014) 9:83. doi: 10.1186/1750-1172-9-83
39. Graber JA, Seeley JR, Brooks-Gunn J, Lewinsohn PM. Is pubertal timing associated with psychopathology in young adulthood? J Am Acad Child Adolesc Psychiatry (2004) 43:718–26. doi: 10.1097/01.chi.0000120022.14101.11
41. Shiraishi K, Oka S, Matsuyama H. Assessment of quality of life during gonadotrophin treatment for male hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). (2014) 81:259–65. doi: 10.1111/cen.12435
42. Kirk JM, Savage MO, Grant DB, Bouloux PM, Besser GM. Gonadal function and response to human chorionic and menopausal gonadotrophin therapy in male patients with idiopathic hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf) (1994) 41:57–63. doi: 10.1111/j.1365-2265.1994.tb03785.x
43. Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2002) 87:4128–36. doi: 10.1210/jc.2002-020518
44. Fahmy I, Kamal A, Shamloul R, Mansour R, Serour G, Aboulghar M. ICSI using testicular sperm in male hypogonadotrophic hypogonadism unresponsive to gonadotrophin therapy. Hum Reprod. (2004) 19:1558–61. doi: 10.1093/humrep/deh243
46. Budych K, Helms TM, Schultz C. How do patients with rare diseases experience the medical encounter? Exploring role behavior and its impact on patient-physician interaction. Health Policy (2012) 105:154–64. doi: 10.1016/j.healthpol.2012.02.018
47. Hannemann-Weber H, Kessel M, Schultz C. Research performance of centers of expertise for rare diseases-The influence of network integration, internal resource access and operational experience. Health Policy (2012) 105:138–45. doi: 10.1016/j.healthpol.2012.02.008
48. Howard SR, Dunkel L. Management of hypogonadism from birth to adolescence. Best Pract Res Clin Endocrinol Metab. (2018) 32:355–72. doi: 10.1016/j.beem.2018.05.011
49. Laitinen EM, Hero M, Vaaralahti K, Tommiska J, Raivio T. Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism. Int J Androl. (2012) 35:534–40. doi: 10.1111/j.1365-2605.2011.01237.x
50. Finkelstein JS, Klibanski A, Neer RM, Doppelt SH, Rosenthal DI, Segre G V, et al. Increases in bone density during treatment of men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (1989) 69:776–83.
51. Pazderska A, Mamoojee Y, Artham S, Miller M, Ball SG, Cheetham T QR. Safety and tolerability of one-year intramuscular testosterone regime to induce puberty in older men with CHH. Endocr Connect. (2018) 7:133–8. doi: 10.1530/EC-17-0241
52. Forest MG, Cathiard AM, Bertrand JA. Evidence of testicular activity in early infancy. J Clin Endocrinol Metab. (1973) 37:148–51. doi: 10.1210/jcem-37-1-148
53. Johannsen TH, Main KM, Ljubicic ML, Jensen TK, Andersen HR, Andersen MS, et al. Sex-differences in reproductive hormones during mini-puberty in infants with normal and disordered sex development. J Clin Endocrinol Metab. (2018) 103:3028–37. doi: 10.1210/jc.2018-00482
54. Bay K, Main KM, Toppari J, SkakkebÆk NE. Testicular descent: INSL3, testosterone, genes and the intrauterine milieu. Nat Rev Urol. (2011) 8:187–96. doi: 10.1038/nrurol.2011.23
55. Koskenniemi JJ, Virtanen HE, Wohlfahrt-Veje C, Löyttyniemi E, Skakkebaek NE, Juul A, et al. Postnatal changes in testicular position are associated with IGF-I and function of sertoli and leydig cells. J Clin Endocrinol Metab. (2018) 103:1429–37. doi: 10.1210/jc.2017-01889
56. Russell LD, Ren HP, Hikim IS, Schulze W, Hikim APS. A comparative study in twelve mammalian species of volume densities, volumes, and numerical densities of selected testis components, emphasizing those related to the sertoli cell. Am J Anat. (1990) 188:21–30. doi: 10.1002/aja.1001880104
57. Rey RA, Musse M, Venara M, Chemes HE. Ontogeny of the androgen receptor expression in the fetal and postnatal testis: its relevance on sertoli cell maturation and the onset of adult spermatogenesis. Microsc Res Tech. (2009) 72:787–95. doi: 10.1002/jemt.20754
58. Andersson AM, Müller J, Skakkebæk NE. Different roles of prepubertal and postpubertal germ cells and sertoli cells in the regulation of serum inhibin B levels. J Clin Endocrinol Metab. (1998) 83:4451–8. doi: 10.1210/jc.83.12.4451
59. Liu PY, Baker HWG, Jayadev V, Zacharin M, Conway AJ, Handelsman DJ. Induction of spermatogenesis and fertility during gonadotropin treatment of Gonadotropin-Deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. (2009) 94:801–8. doi: 10.1210/jc.2008-1648
60. Liu L, Banks SM, Barnes KM, Sherins RJ. Two-year comparison of testicular responses to pulsatile gonadotropin-releasing hormone and exogenous gonadotropins from the inception of therapy in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (1988) 67:1140–5. doi: 10.1210/jcem-67-6-1140
61. Burris AS, Rodbard HW, Winters SJ, Sherins RJ. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. (1988) 66:1144–51. doi: 10.1210/jcem-66-6-1144
62. Raivio T, Toppari J, Perheentupa A, McNeily AS, Dunkel L. Treatment of prepubertal gonadotrophin-deficient boys with recombinant human follicle-stimulating hormone. Lancet (1997) 350:263–4. doi: 10.1016/S0140-6736(05)62227-1
63. Raivio T, Wikström AM, Dunkel L. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol. (2007) 156:105–11. doi: 10.1530/eje.1.02315
64. Dwyer AA, Sykiotis GP, Hayes FJ, Boepple PA, Lee H, Loughlin KR, et al. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2013) 98:E1790-5. doi: 10.1210/jc.2013-2518
65. Main KM, Schmidt IM, Toppari J, Skakkebæk NE. Early postnatal treatment of hypogonadotropic hypogonadism with recombinant human FSH and LH. Eur J Endocrinol. (2002) 146:75–9. doi: 10.1530/eje.0.1460075
66. Bougnères P, François M, Pantalone L, Rodrigue D, Bouvattier C, Demesteere E, et al. Effects of an early postnatal treatment of hypogonadotropic hypogonadism with a continuous subcutaneous infusion of recombinant follicle-stimulating hormone and luteinizing hormone. J Clin Endocrinol Metab. (2008) 93:2202–5. doi: 10.1210/jc.2008-0121
67. Stoupa A, Samara-Boustani D, Flechtner I, Pinto G, Jourdon I, González-Briceño L, et al. Efficacy and safety of continuous subcutaneous infusion of recombinant human gonadotropins for congenital micropenis during early infancy. Horm Res Paediatr. (2017) 87:103–10. doi: 10.1159/000454861
68. Kollin C, Stukenborg JB, Nurmio M, Sundqvist E, Gustafsson T, Söder O, et al. Boys with undescended testes: endocrine, volumetric and morphometric studies on testicular function before and after orchidopexy at nine months or three years of age. J Clin Endocrinol Metab. (2012) 97:4588–95. doi: 10.1210/jc.2012-2325
69. Martin Ritzén E, Bergh A, Bjerknes R, Christiansen P, Cortes D, Haugen SE, et al. Nordic consensus on treatment of undescended testes. Acta Paediatr. (2007) 96:638–43. doi: 10.1111/j.1651-2227.2006.00159.x
70. Bouvattier C, Maione L, Bouligand J, Dodé C, Guiochon-Mantel A, Young J. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2012) 8:172–82. doi: 10.1038/nrendo.2011.164
71. Lambert A-S, Bougneres P. Growth and descent of the testes in infants with hypogonadotropic hypogonadism receiving subcutaneous gonadotropin infusion. Int J Pediatr Endocrinol (2016) 2016:13. doi: 10.1186/s13633-016-0031-9
72. Braslavsky D, Grinspon RP, Ballerini MG, Bedecarras P, Loreti N, Bastida G, et al. Hypogonadotropic hypogonadism in infants with congenital hypopituitarism: a challenge to diagnose at an early stage. Horm Res Paediatr. (2015) 84:289–97. doi: 10.1159/000439051
73. Sijstermans K, Hack WWM, Meijer RW, Voort-Doedens LMV Der. The frequency of undescended testis from birth to adulthood: a review. Int J Androl. (2008) 31:1–11. doi: 10.1111/j.1365-2605.2007.00770.x
74. Suomi AM, Main KM, Kaleva M, Schmidt IM, Chellakooty M, Virtanen HE, et al. Hormonal changes in 3-month-old cryptorchid boys. J Clin Endocrinol Metab. (2006) 91:953–8. doi: 10.1210/jc.2004-2318
75. Birth Ratio Statistics Department of Health UK. Gender ratios at birth in Great Britain, 2011 to 2015 2017. (2018) Available online at: https://www.gov.uk/government/statistics/gender-ratios-at-birth-in-great-britain-2011-to-2015 (Accessed November 25, 2018).
76. Hortling H, Chapelle A, Johansson CJ, Niemi M, Sulamaa M. An endocrinologic follow-up study of operated cases of cryptorchism. J Clin Endocrinol Metab. (1967) 27:120–9. doi: 10.1210/jcem-27-1-120
77. Rohayem J, Hauffa BP, Zacharin M, Kliesch S, Zitzmann M, Wüsthof A, et al. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? -a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence-. Clin Endocrinol (Oxf). (2017) 86:75–87. doi: 10.1111/cen.13164
Keywords: congenital hypogonadotropic hypogonadism, kallmann syndrome, puberty delay, minipuberty of infancy, infertility–male, cryptorchidism, gonadotrophin releasing hormone deficiency
Citation: Swee DS and Quinton R (2019) Congenital Hypogonadotrophic Hypogonadism: Minipuberty and the Case for Neonatal Diagnosis. Front. Endocrinol. 10:97. doi: 10.3389/fendo.2019.00097
Received: 30 November 2018; Accepted: 01 February 2019;
Published: 21 February 2019.
Edited by:
Ranjith Ramasamy, University of Miami, United StatesReviewed by:
Sasha Howard, Queen Mary University of London, United KingdomNannan Thirumavalavan, Baylor College of Medicine, United States
Copyright © 2019 Swee and Quinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Du Soon Swee, c3dlZS5kdS5zb29uQHNpbmdoZWFsdGguY29tLnNn