 Dongdong Wang1
Dongdong Wang1 Xiaoli Wang
Xiaoli Wang- 1Obstetrics and Gynecology Department of Shengjing Hospital, China Medical University, Shenyang, China
- 2Department of Endocrinology and Metabolism, Institute of Endocrinology, Liaoning Provincial Key Laboratory of Endocrine Diseases, The First Affiliated Hospital of China Medical University, Shenyang, China
Osteogenesis imperfecta (OI) is a hereditary connective tissue disorder, characterized by reduced bone content, fractures and skeletal malformation due to abnormal synthesis or dysfunction of type I collagen protein. Pituitary stalk interruption syndrome (PSIS) is usually associated with environmental and hereditary factors. Here, we report a rare case of OI and PSIS co-occurrence. A 19-year-old male patient sought treatment for growth delay and absent secondary sexual characteristics. Hormone measurements indicated the presence of hypopituitarism (secondary hypothyroidism, growth hormone deficiency, ACTH-cortisol hormone deficiency, hypogonadotropic hypogonadism). Pituitary magnetic resonance imaging indicated reduced morphology of the anterior lobe, absence of the pituitary stalk, and ectopic displacement of the posterior lobe to the infundibulum, supporting a diagnosis of PSIS. In addition, the patient, his monozygotic twin brother (no evidence of PSIS), and their mother all presented blue sclera and susceptibility to bone fractures before adulthood. Next-generation sequencing demonstrated that the family had compound heterozygous mutations in COL1A1 and COL1A2, with no known mutations related to PSIS, pituitary hormone deficiency (PHD), or holoprosencephaly (HPE). The mother experienced breech and natural delivery of the patient and his brother, respectively. Thus, we deduced that the patient's PSIS might have resulted from breech delivery. Although we cannot exclude the possibility that the proband might have an undetected genetic abnormality causing PSIS or increasing his susceptibility to damage to the hypothalamic-pituitary region due to the limitation of exome sequencing, this rare case suggests that breech delivery in the newborn with OI might be related to PSIS.
Background
Osteogenesis imperfecta (OI) is a group of inherited connective tissue disorders. It presents as reduced bone content and increased skeletal fragility, thereby increasing the likelihood of bone fractures (1). The etiology of most patients with autosomal hereditary OI (types I-IV) involves mutations in COL1A1 (MIM 120150) and COL1A2 (MIM 120160). These two genes encode the α1 and α2 chains of type I collagen, respectively. Together, two α1 chains and one α2 chain form the triple-helix structure of type I collagen. Heterozygous null mutations in COL1A1 lead to haploinsufficiency and inadequate proα1 (I) production, thereby leading to type I OI. Type I OI has mild clinical symptoms and presents with an increased likelihood of bone fractures in childhood with or without blue sclera; in adulthood, the likelihood of bone fractures gradually decreases (1). Missense mutations in COL1A1 or COL1A2 usually lead to changes in collagen structure, thereby leading to clinical symptoms of moderate to severe type II-IV OI. Because new pathogenic genes that participate in the post-translational modification of type I collagen are continually being discovered, the number of OI types are also increasing continuously. Thus, studies have recommended reclassifying OI into 5 groups according to their pathogenic mechanism. OI caused by mutations in the COL1A1 or COL1A2 genes are classified under group A, namely, the group with defects in collagen synthesis, structure, or processing (1). Although many cases about OI are available in the literature, co-occurrence of OI with pituitary stalk interruption syndrome (PSIS) has never been reported.
Pituitary stalk interruption syndrome refers to a congenital abnormality of the anatomical structure of the pituitary. It has a typical presentation on magnetic resonance imaging, including a reduced or absent anterior pituitary, interruption, or loss of the pituitary stalk, and displacement of the posterior pituitary (2). Further, it leads to isolated or combined pituitary hormone deficiencies (3). Currently, its pathologic mechanism is still unknown. Pituitary stalk interruption syndrome is believed to be associated with hereditary and environmental factors, particularly perinatal events (3).
Here we report a family with OI that was caused by combined heterozygous mutations in both COL1A1 and COL1A2 genes. The proband showed co-occurrence of PSIS, whereas his older twin did not, suggesting that the proband either has a non-identified mutation or an epigenetic change related to environmental factors such as a perinatal event.
Case Presentation
A 19-year-old male patient who presented with absent secondary sexual characteristics and delayed growth was admitted to the First Affiliated Hospital of China Medical University. The abnormalities in the patient were first noticed by his mother when she compared the patient with his monozygotic twin brother. During puberty, the gap in height between the patient and his twin brother was as much as 20 cm. The twins were born after a 37-week uneventful pregnancy. The mother experienced natural and breech delivery during her labor of the twin brother and the patient, respectively. Past medical history included one fracture on his right ulna when he was 2 years old. The patient's father was healthy. Both the patient's mother and his twin brother had blue sclera, and they both suffered from bone fractures multiple times before adulthood. The patient's parents stood at 172 cm (father) and 160 cm (mother). His twin brother was 170 cm tall.
Physical Exam
The patient showed the following features: height, 158 cm; weight, 53 kg; BMI, 20.70 kg/m2; arm span, 153 cm; upper body height, 67 cm; lower body height, 91 cm; triangular face; light blue sclera (Figure 1C); lack of facial hair; lack of Adam's apple; lack of underarm hair; lack of thyroid swelling; carinatum; enlarged breasts; palpable breast nodules; mild tenderness; lack of galactorrhea; lack of pubic hair; a stretched penis length of 4 cm. Both testes were palpable within the scrotum bilaterally.
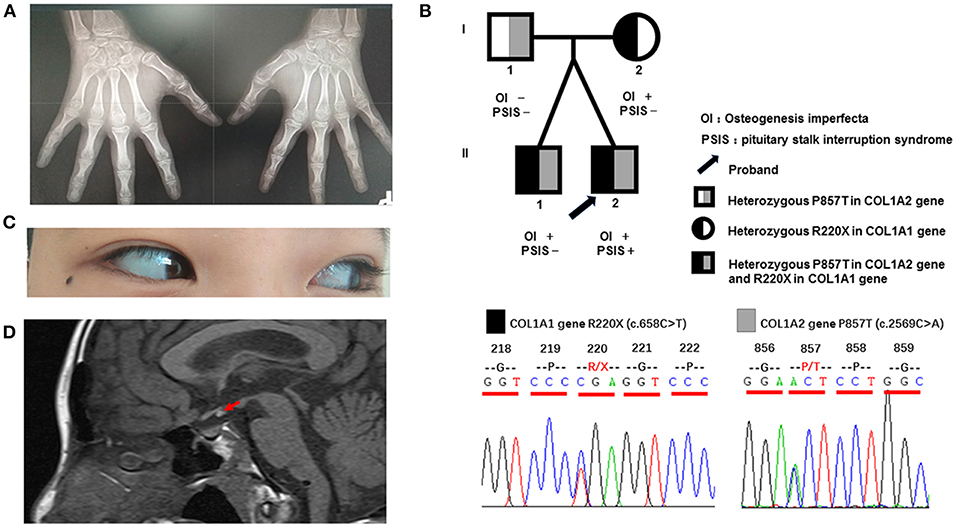
Figure 1. Clinical characteristics and gene sequencing results of the patient and his family members. (A) Digital radiography of the hands. The phalanges and humeral metaphysis are fused, indicating an age of 14–15 years. (B) Pedigree of the patient and gene sequencing results. The father is a heterozygous carrier of the P857T mutation in the COL1A2 gene. The mother is a heterozygous carrier of the R220X mutation of the COL1A1 gene. The twin brothers are heterozygous carriers of the above two mutations. (C) Light blue sclera of the patient. (D) Magnetic resonance imaging of the pituitary of the patient, indicating reduced morphology of the anterior lobe, absence of the pituitary stalk, and displacement of the neurohypophysis to the infundibulum (red arrow).
Work-Up
Testicular ultrasound showed that the right testis had a size of approximately 1.64 × 0.73 × 1.16 cm (0.73 ml), and the left testis had a size of ~1.48 × 0.65 × 1.01 cm (0.51 ml). His sense of smell was also tested formally (T & T olfactometer test), and no olfactory loss or hyposmia was found. Bone mineral density analysis showed decreased bone density in the lumbar vertebrae (Z = −3.1). Digital radiography of both hands indicated his skeletal age to be 14 years (Figure 1A). Magnetic resonance imaging of the pituitary gland showed reduced morphology of the anterior pituitary, absence of a pituitary stalk and ectopic displacement of the posterior lobe to the infundibulum (Figure 1D). These findings together suggested a diagnosis of PSIS. Ophthalmologic and audiologic examinations did not reveal any abnormalities.
Laboratory examinations demonstrated pituitary hypothyroidism, pituitary-adrenal axis dysfunction, hypogonadotropic hypogonadism, slightly increased prolactin level, and presence of insulin resistance with normal blood glucose level (Table 1). The basal GH level of the proband was < 0.5 μg/l and basal IGF-I level was < 25 ng/ml, suggesting a possibility of growth hormone deficiency, but we didn't perform stimulation tests because the patient and his mother refused to take the tests as well as be treated with growth hormone for economic reasons.
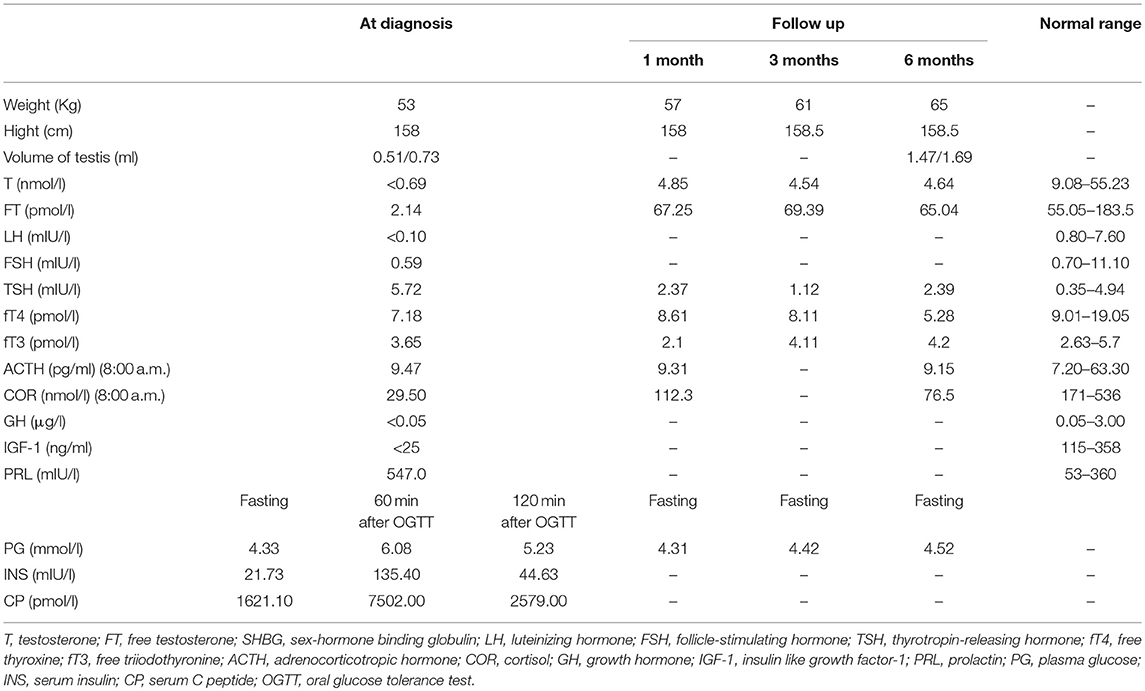
Table 1. Clinical and laboratory data of the patient at diagnosis and follow up.
Genetic Testing
Blood samples were collected from the patient and his family members (father, mother, and twin brother). Genomic DNA was extracted using a blood extraction kit (Tian Jing Biochemical Technology Beijing, Ltd.). Roche Nimblegen SeqCap EZ Choice XL Library was used for exon trapping (including 4,132 genes). Illumina nextseq500 was used for high-throughput sequencing. Burrows-Wheeler Aligner software (BWA, 0.7.12-r1039) was applied to align the sequencing data to the human genome. ANNOVAR was used to annotate the genetic variants detected (based on the recent version of the dbSNP database, Clinvar, ExAC, and 1000 Genomes Project). Emphasis was laid on the analysis of known genes involved in OI, PSIS, PHD, and HPE according to pathology (listed separately in Table 2). The results revealed that the identical twins harbor heterozygous mutations in COL1A1 (c.658C>T, p.Arg220Ter) and COL1A2 (c.2569C>A, p.Pro857Thr). The mother harbors the same COL1A1 mutation (p.Arg220Ter) and the father harbors the COL1A2 (p.Pro857Thr) mutation (Figure 1B). No known pathogenic variants related to PSIS, PHD, or HPE were found in this family. No convincing discordant single nucleotide or indel variants were found in coding or regulatory regions in this phenotypically PSIS-discordant twin.
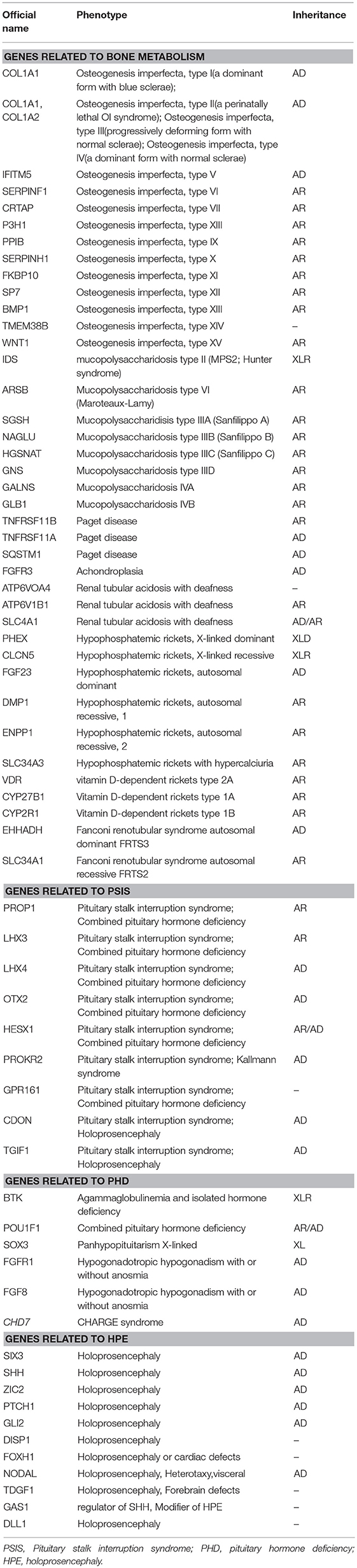
Table 2. List of genes related to bone metabolism, PSIS, PHD, and HPE.
Therapeutic Intervention and Follow-Up
For hypogonadism, 2000 U human chorionic gonadotropin (hCG) was given intramuscularly three times per week, and testosterone and free testosterone levels were re-examined periodically at outpatient visits. The underarm hair and pubic hair of the patient showed significant growth. The testicular volume of the patient increased to 1.47 ml (left) and 1.69 ml (right), and his testosterone levels increased as well but still not reach normal range (Table 1). Human menopausal gonadotropin (hMG) was added to the patient's medications (75 U along with hCG). For secondary hypothyroidism, levothyroxine administration was started at 25 μg once daily, thyroid function was monitored, and the dose was increased to 75 μg/day during follow-up. For secondary adrenal insufficiency, oral hydrocortisone was prescribed initially at 20 mg in the morning (8:00 a. m.) and 10 mg in the afternoon (3:00 p.m.), and the hydrocortisone dose was decreased to 5 mg/day during follow-up because the body weight of the patient increased quickly and he showed no manifestation of adrenal insufficiency. A dose of 15 mg hydrocortisone was recommended for the patient according to the guideline after carefully discussion with the patient and his family (4). For OI, bisphosphonates were recommended.
Discussion
This paper reports a rare case of OI combined with PSIS; this patient was the index case in one OI family. In this family, the patient, his mother and twin brother presented blue sclera and bone fractures, whereas the father seemed clinically normal. Next-generation sequencing revealed that the twins harbor heterozygous mutations in both COL1A1 and COL1A2. Currently, there are no reports on OI cases with such compound heterozygous mutations involving two genes. The COL1A1 mutation (p.Arg220Ter) (rs72667036) was inherited from their mother. It causes termination of the α1 chain, thus resulting in a reduced amount of total type I collagen, which has been reported in many papers to result in type 1 OI (5–9). The COL1A2 mutation (p.Pro857Thr) (rs150179964) was inherited from their father. It has an MAF < 0.01 in the Ensembl database, a SIFT score of 0.02 (deleterious), and a PolyPhen score of 0.036 (benign). A previous paper reported a Vietnamese type IV OI patient carrying p.Pro857Thr associated with a different mutation (p.Gly1012Ser) in COL1A2 (10). The COL1A2 mutation (p.Pro857Thr) was not located at the G (glycine) site of the G-X-X motif, which plays an important role in maintaining the normal protein structure of COL1A1 and COL1A2. Together with a lack of clinical presentation of OI in the patient's father, these results suggest that the COL1A2 mutation (p.Pro857Thr) might have minor effects on the protein structure and function.
Osteogenesis imperfecta is a heterogeneous group of inherited connective tissue disorders that cause bone fragility and deformity (1). About 90% of OI cases are caused by structural or quantitative mutations in the collagen genes COL1A1 and COL1A2 (11, 12). In addition, alterations in more than 13 genes that are linked to collagen processing (BMP1), collagen modification (CRTAP, LEPRE1/P3H1, PPIB, TMEM38B), collagen folding and cross-linking (SERPINH1, FKBP10, PLOD2), bone mineralization (IFITM5, SERPINF1), and osteoblast development with collagen insufficiency (SP7, WNT1, CREB3L1) have been reported to account for OI. These genetic alterations provide further explanations for the pathogenesis, classification, and therapeutic targets of OI (1). Osteogenesis imperfecta is a major cause of skeletal deformities and may lead to secondary damages, such as hearing loss (13), dental abnormalities (14), neurological features including macrocephaly, hydrocephalus, syringomyelia, and basilar invagination (15), and short stature with a normal growth axis (16). Patients with OI may present multiple skeletal deformities, such as flat midface and triangular facies, scoliosis or kyphosis, and chest wall deformities including pectus excavatum, carinatum, and barrel chest (11).
Interestingly, in the present family, the twins shared the same genetic alterations that accounted for their OI, whereas the index case presented evident delay in growth and development. Obviously, OI was not the reason for these differences between the twins, which were attributed to PSIS. Pituitary stalk interruption syndrome is believed to be associated with genetic and environmental factors, especially perinatal events (3). More than 40 candidate genes for PSIS, PHD, and HPE have been reported (17–20), but only 5% of the PSIS patients were identified to harbor these defects (20, 21). Holoprosencephaly is characterized by severely disturbed midline brain development, and genetic studies have shown an association between PSIS and genes involved in HPE, suggesting that PSIS might be a mild form of HPE (20). Recent studies have suggested a polygenic etiology for isolated PSIS (19, 22, 23). Although there might be additional genetic and epigenetic alterations not yet recognized by the current sequencing technologies, it seems that environmental factors play more important roles in the pathogenesis of PSIS. As shown in previous studies, individuals who had a history of perinatal events, such as breech delivery, were more likely to develop PSIS (24, 25). The frequency of breech delivery among PSIS patients is much higher than that in the general population (26). The mechanisms behind the higher risk of PSIS caused by perinatal events remain unclear. Pituitary stalk interruption syndrome is reported to be associated with direct injury to the pituitary stalk, or transient anoxia of hypothalamus (3) during difficult labors. In our case of co-occurrence of OI and PSIS, next-generation sequencing did not show any abnormalities in genes related to PSIS, PHD, or HPE. Considering that the patient was born after a difficult breech delivery, and that his normally delivered twin brother did not present PSIS, we deduced that the PSIS might be a consequence of breech delivery. Studies assessing at-birth health outcomes of neonates with OI reported a high breech delivery rate (24–37%), suggesting that newborns with OI may benefit from additional clinical supervision (27, 28). However, we still cannot exclude the possibility that the proband may have an undetected genetic abnormality causing PSIS or increasing his susceptibility to damage to the hypothalamic-pituitary region, owing to the limitations of exome sequencing.
To the best of our knowledge, the available case reports on PSIS have never mentioned its co-occurrence with OI. The present report reminds us to further investigate the influence of OI in female patients on vaginal deliveries, perinatal events, and the incidence of PSIS among their offspring. If female OI patients unfortunately experience perinatal events, their offspring should be monitored for early identification of any pituitary dysfunction, which may lead to development and growth delay.
Ethics Statement
The hospital ethics committee of China Medical University approved the study, and the patient and his family members provided written informed consent for publication of their clinical details and clinical images.
Author Contributions
DW and XW: study design; MZ: data collection; DW and MZ: manuscript drafting; HG and XW: data interpreting and revision of the manuscript; DW, MZ, HG, and XW: approval of final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our patient and his family members who agreed to participate in this study.
References
1. Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. (2016) 387:1657–71. doi: 10.1016/S0140-6736(15)00728-X
2. Voutetakis A, Sertedaki A, Dacou-Voutetakis C. Pituitary stalk interruption syndrome: cause, clinical manifestations, diagnosis, and management. Curr Opin Pediatr. (2016) 28:545–50. doi: 10.1097/MOP.0000000000000378
3. Bar C, Zadro C, Diene G, Oliver I, Pienkowski C, Jouret B, et al. Pituitary Stalk interruption syndrome from infancy to adulthood: clinical, hormonal, and radiological assessment according to the initial presentation. PLoS ONE. (2015) 10:e0142354. doi: 10.1371/journal.pone.0142354
4. Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, et al. Hormonal replacement in hypopituitarism in adults: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2016) 101:3888–921. doi: 10.1210/jc.2016-2118
5. Korkko J, Ala-Kokko L, De Paepe A, Nuytinck L, Earley J, Prockop DJ. Analysis of the COL1A1 and COL1A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1A1 mutations in 15 patients with osteogenesis imperfecta type I: identification of common sequences of null-allele mutations. Am J Hum Genet. (1998) 62:98–110. doi: 10.1086/301689
6. Roschger P, Fratzl-Zelman N, Misof BM, Glorieux FH, Klaushofer K, Rauch F. Evidence that abnormal high bone mineralization in growing children with osteogenesis imperfecta is not associated with specific collagen mutations. Calcif Tissue Int. (2008) 82:263–70. doi: 10.1007/s00223-008-9113-x
7. Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ, Yu JB, et al. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J Bone Miner Metab. (2012) 30:69–77. doi: 10.1007/s00774-011-0284-6
8. Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, et al. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet. (2015) 23:1042–50. doi: 10.1038/ejhg.2015.81
9. Lin HY, Chuang CK, Su YN, Chen MR, Chiu HC, Niu DM, et al. Genotype and phenotype analysis of Taiwanese patients with osteogenesis imperfecta. Orphanet J Rare Dis. (2015) 10:152. doi: 10.1186/s13023-015-0370-2
10. Ho Duy B, Zhytnik L, Maasalu K, Kandla I, Prans E, Reimann E, et al. Mutation analysis of the COL1A1 and COL1A2 genes in Vietnamese patients with osteogenesis imperfecta. Hum Genomics. (2016) 10:27. doi: 10.1186/s40246-016-0083-1
11. Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. (2011) 7:540–57. doi: 10.1038/nrendo.2011.81
12. Marini JC, Blissett AR. New genes in bone development: what's new in osteogenesis imperfecta. J Clin Endocrinol Metab. (2013) 98:3095–103. doi: 10.1210/jc.2013-1505
13. Pedersen U. Hearing loss in patients with osteogenesis imperfecta. A clinical and audiological study of 201 patients. Scand Audiol. (1984)13:67–74. doi: 10.3109/01050398409043042
14. Levin LS, Salinas CF, Jorgenson RJ. Classification of osteogenesis imperfecta by dental characteristics. Lancet. (1978) 1:332–3. doi: 10.1016/S0140-6736(78)90108-3
15. Charnas LR, Marini JC. Communicating hydrocephalus, basilar invagination, and other neurologic features in osteogenesis imperfecta. Neurology. (1993) 43:2603–8. doi: 10.1212/WNL.43.12.2603
16. Marini JC, Bordenick S, Heavner G, Rose S, Chrousos GP. Evaluation of growth hormone axis and responsiveness to growth stimulation of short children with osteogenesis imperfecta. Am J Med Genet. (1993) 45:261–4. doi: 10.1002/ajmg.1320450223
17. Wang CZ, Guo LL, Han BY, Su X, Guo QH, Mu YM. Pituitary stalk interruption syndrome: from clinical findings to pathogenesis. J Neuroendocrinol. (2017) 29. doi: 10.1111/jne.12451
18. Bashamboo A, Bignon-Topalovic J, Rouba H, McElreavey K, Brauner R. A nonsense mutation in the hedgehog receptor CDON associated with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. (2016) 101:12–5. doi: 10.1210/jc.2015-2995
19. Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, et al. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocrine Rev. (2016) 37:636–75. doi: 10.1210/er.2016-1101
20. Tatsi C, Sertedaki A, Voutetakis A, Valavani E, Magiakou MA, Kanaka-Gantenbein C, et al. Pituitary stalk interruption syndrome and isolated pituitary hypoplasia may be caused by mutations in holoprosencephaly-related genes. J Clin Endocrinol Metab. (2013) 98:E779–84. doi: 10.1210/jc.2012-3982
21. Karaca E, Buyukkaya R, Pehlivan D, Charng WL, Yaykasli KO, Bayram Y, et al. Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. (2015) 100:E140–7. doi: 10.1210/jc.2014-1984
22. Zwaveling-Soonawala N, Alders M, Jongejan A, Kovacic L, Duijkers FA, Maas SM, et al. Clues for polygenic inheritance of pituitary stalk interruption syndrome from exome sequencing in 20 patients. J Clin Endocrinol Metab. (2018) 103:415–28. doi: 10.1210/jc.2017-01660
23. Guo QH, Wang CZ, Wu ZQ, Qin Y, Han BY, Wang AP, et al. Multi-genic pattern found in rare type of hypopituitarism: a whole-exome sequencing study of Han Chinese with pituitary stalk interruption syndrome. J Cell Mol Med. (2017) 21:3626–32. doi: 10.1111/jcmm.13272
24. Guo Q, Yang Y, Mu Y, Lu J, Pan C, Dou J, et al. Pituitary stalk interruption syndrome in Chinese people: clinical characteristic analysis of 55 cases. PLoS ONE. (2013) 8:e53579. doi: 10.1371/journal.pone.0053579
25. Maghnie M, Larizza D, Triulzi F, Sampaolo P, Scotti G, Severi F. Hypopituitarism and stalk agenesis: a congenital syndrome worsened by breech delivery? Horm Res. (1991) 35:104–8. doi: 10.1159/000181883
26. Cruikshank DP. Breech presentation. Clin Obstet Gynecol. (1986) 29:255–63. doi: 10.1097/00003081-198606000-00008
27. Yimgang DP, Brizola E, Shapiro JR. Health outcomes of neonates with osteogenesis imperfecta: a cross-sectional study. J Matern Fetal Neonatal Med. (2016) 29:3889–93. doi: 10.3109/14767058.2016.1151870
Keywords: pituitary stalk interruption syndrome, osteogenesis imperfecta, case report, next generation sequencing, skeletal malformation
Citation: Wang D, Zhang M, Guan H and Wang X (2019) Osteogenesis Imperfecta Due to Combined Heterozygous Mutations in Both COL1A1 and COL1A2, Coexisting With Pituitary Stalk Interruption Syndrome. Front. Endocrinol. 10:193. doi: 10.3389/fendo.2019.00193
Received: 01 August 2018; Accepted: 07 March 2019;
Published: 28 March 2019.
Edited by:
Ursula B. Kaiser, Harvard Medical School, United StatesReviewed by:
Maria Mercedes Pineyro, Universidad de la República, UruguayAna Paula Abreu, Harvard Medical School, United States
Copyright © 2019 Wang, Zhang, Guan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Wang, d2xpdHRsZXBlYXJAMTYzLmNvbQ==
Haixia Guan, aHhndWFuQHZpcC4xMjYuY29t