 Shokoufeh Mahmoodzadeh1,2*
Shokoufeh Mahmoodzadeh1,2* Elke Dworatzek1,2,3
Elke Dworatzek1,2,3- 1Department of Molecular Muscle Physiology, Max-Delbrueck-Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
- 2DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Berlin, Germany
- 3Institute of Gender in Medicine, Charité Universitaetsmedizin, Berlin, Germany
Numerous epidemiological, clinical, and animal studies showed that cardiac function and manifestation of cardiovascular diseases (CVDs) are different between males and females. The underlying reasons for these sex differences are definitely multifactorial, but major evidence points to a causal role of the sex steroid hormone 17β-estradiol (E2) and its receptors (ER) in the physiology and pathophysiology of the heart. Interestingly, it has been shown that cardiac calcium (Ca2+) ion channels and mitochondrial function are regulated in a sex-specific manner. Accurate mitochondrial function and Ca2+ signaling are of utmost importance for adequate heart function and crucial to maintaining the cardiovascular health. Due to the highly sensitive nature of these processes in the heart, this review article highlights the current knowledge regarding sex dimorphisms in the heart implicating the importance of E2 and ERs in the regulation of cardiac mitochondrial function and Ca2+ ion channels, thus the contractility. In particular, we provide an overview of in-vitro and in-vivo studies using either E2 deficiency; ER deficiency or selective ER activation, which suggest that E2 and ERs are strongly involved in these processes. In this context, this review also discusses the divergent E2-responses resulting from the activation of different ER subtypes in these processes. Detailed understanding of the E2 and ER-mediated molecular and cellular mechanisms in the heart under physiological and pathological conditions may help to design more specifically targeted drugs for the management of CVDs in men and women.
Introduction
Cardiovascular Diseases (CVDs) are one of the top age-associated chronic diseases with growing importance due to the dramatic increase in life expectancy (1) and are the leading cause of mortality in men and women worldwide (2). In the vast majority of CVDs, there are well described sex differences in the incidence, pathophysiology, and outcomes of diseases (3). As result of these observations, research over the last few decades has focused on the contribution of sex steroid hormones, specifically 17β-estradiol (E2), on the cardiovascular system and mechanistic pathways in the diseased heart.
Calcium (Ca2+) is a key player in the regulation of myocardial contraction and the deregulation of Ca2+ signaling due to the alteration of Ca2+ ion channels function in cardiomyocytes is highly associated with the development of cardiac diseases, such as heart failure (4). Just like Ca2+, mitochondria play an essential role in the regulation of energy metabolism of the heart, and defects of mitochondrial function also lead to the development and progression of cardiovascular diseases (5, 6). This review article provides an overview of the current knowledge regarding the sex differences in cardiac health and disease with the focus on the sexually dimorphic effects of E2 and estrogen receptors (ERs) in the regulation of cardiomyocyte's Ca2+ ion channels and mitochondrial function.
The Role of 17β-Estradiol in the Heart
Epidemiological data suggest that premenopausal women are protected from the incidence of CVDs as well as from resulting morbidity and mortality compared with age-matched men, but that this protection is lost after menopause (7–9). This led to the generally accepted conclusion that the sex hormone E2 protects against CVDs in women (10). However, recent large-scale clinical trials revealed conflicting data about the effect of E2 on CVDs, which is still a matter of intense debate. For example, several observational studies such as the Nurse's Health Study showed that postmenopausal women with hormone replacement therapy (HRT) have a lower rate of CVDs and cardiac death, compared to women without HRT (11–14). In contrast, the Women Health Initiative (WHI) and the Heart and Estrogen/Progestin Replacement Study (HERS I and II) showed that HRT has no obvious beneficial effect on CVDs, and may actually increase the risk and events of CVDs in postmenopausal women (15–19). The reasons for this paradox remain unclear and many potential factors, such as the study design and subject characteristics, the form of applied E2 (which type of E2, combination of E2 with progestin), the dosage and pharmacokinetics of the HRT used, and the statistical power to address cardiac risk factors may contribute to the discrepant results and to the adverse outcome of HRT (20–22). In addition, another reason for the contradictory data could be the timing of HRT initiation. Recent studies such as the Kronos Early Estrogen Prevention Study (KEEPS) and the Early vs. Late Intervention Trial with Estradiol (ELITE) addressed the question of the so-called “timing hypothesis.” They showed significant beneficial cardiovascular effects in women who initiated HRT in the early postmenopause vs. late menopause period (19, 23, 24), indicating the importance of the time point of HRT-application.
Modulatory effects of E2 on CVDs in men have also been reported (25, 26). In men with E2 deficiency due to a mutation in the cytochrome P450 aromatase gene (Cyp19a1), which catalyzes the aromatization of androgens to E2, or E2 resistance, caused by a point mutation in the ERα gene (ESR1), the following have been reported: increased total cholesterol level, the development of insulin resistance, impaired glucose tolerance, type 2 diabetes mellitus, and impaired vasodilatation (27–31). These data suggest that the physiological concentrations of E2 might reduce the risk of CVDs in men. Indeed, men with abnormally low (≤13 pg/mL) and abnormally high (≥37 pg/mL) E2-levels have been found to show the highest death rates from congestive heart failure (32). By contrast, individuals with levels of E2 in the range of 22–30 pg/mL had the least number of deaths over a 3-year period. However, the precise role of E2 in men in CVDs remains questionable (33).
Actions of 17β-Estradiol and Estrogen Receptors
E2 belongs together with Estrone (E1) and Estriol (E3) to the group of sex steroids called Estrogens. Thereby, E2 is the predominant and most biologically active form (34). Estrogens have traditionally been associated with the female reproductive development and function, but it is now well-established that they also regulate male reproductive organs and play a physiological role in multiple organs in both sexes (26). In healthy premenopausal women, ovaries are the primary site of E2 production, and in men, E2 is produced in small amounts by the testes. E2 is also synthesized in a number of extragonadal tissues, through the conversion of testosterone by cytochrome p450 aromatase in both sexes, including bone, breast, adipose tissue, and the brain (35). There is increasing evidence that the aromatase is also expressed in the heart tissue and that E2 can also be produced locally in cardiac cells (36–39), suggesting that local cardiac E2 synthesis by aromatase plays a role in the E2-mediated effects on CVDs.
The physiological effects of E2 are predominantly mediated via estrogen receptor alpha (ERα) and beta (ERβ), which are members of the nuclear receptor superfamily (Figure 1) (40). Both receptors carry similar structural domains, however, they differ in their DNA- and ligand-binding regions, which are of crucial importance for their diverse transcriptional actions (41). E2-activated ERs can act as ligand-induced transcription factors inducing changes in transcription of E2 target genes, a process referred to as genomic actions. Here the binding of E2 to the ERs results in homo- or heterodimerization of ER and their translocation into the nucleus of cells. The E2/ER complex either binds to estrogen response elements (ERE) within the promoter of target genes or regulates gene transcription by interacting with other transcription factors, e.g., AP-1 and Sp1 (Figure 1I) (34, 42–44). Additionally, E2-bound ERs can also activate multiple signal transduction pathways, e.g., mitogen-activated protein kinases ERK1/2 and -p38 (ERK1/2-MAPK, p38-MAPK) as well as phosphoinositide 3-kinase-serin/threonine-specific kinase B (PI3K/AKT), which in turn phosphorylate ERs (45–47) or other promoter bound transcription factors that are involved in the regulation of E2-target gene expression (Figure 1II) (48–51). Moreover, through non-genomic actions, E2 rapidly mediates its effects by activation of ERs located in or adjacent to the plasma membrane, which in turn can activate different signal transduction cascades, such as PI3K/AKT and MAPK, leading for example to cytosolic eNOS activation (Figure 1III) (52, 53).
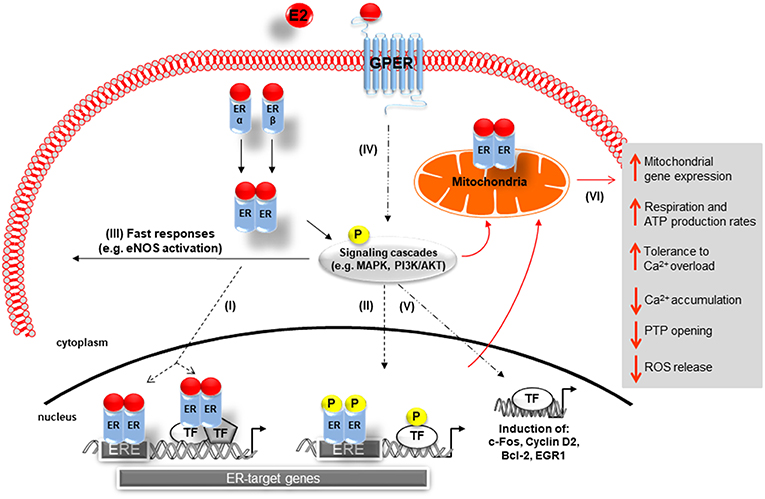
Figure 1. Schematic representation of 17β-Estradiol induced estrogen receptor-alpha, -beta, and G-protein-coupled estrogen receptor signaling. Genomic pathway: (I) The E2/ER complex can bind to estrogen response elements (ERE) within the promoter of target genes or regulates gene transcription by interacting with other transcription factors (TF), e.g., AP-1 and Sp1. (II) In addition, E2/ER activate signaling transduction pathways, leading to phosphorylation of ER or other bound transcription factors modulating gene expression. In the non-genomic action: (III) E2-activated ER lead to rapid tissue responses via phosphorylation of cytosolic signaling cascades. (IV) GPER predominantly mediates rapid, non-genomic E2 signaling by the involvement of several kinases, ion channels, and second messengers. (V) GPER is also involved in gene expression regulation. (VI) E2 initiated cellular and mitochondrial ER/GPER genomic and non-genomic actions modulate mitochondrial respiration, ATP production, and ROS formation (indicated by red arrows). E2, 17β-estradiol; ER, estrogen receptor alpha and beta ERE, estrogen response element; TF, transcription factor; P, phosphorylation; GPER, G-protein-coupled estrogen receptor; Ca2+, calcium, PTP, permeability transition pore; MAPK, mitogen-activated protein kinases; PI3K/AKT, phosphoinositide 3-kinase-serin/threonine-specific kinase B; eNOS: endothelial nitric oxide synthase.
Estrogen Receptors in the Heart
Both ERs are localized in different cardiac cells such as cardiomyocytes, endothelial cells, smooth muscle cells, and cardiac fibroblasts in human hearts from both sexes (54, 55). Studies in rodents also showed that both ER are expressed in whole heart tissue from males and females (36, 39, 56–58). Recent observations from Pugach et al. showed that only ERα, but not ERβ, is expressed in left ventricular heart tissue from mice and isolated rat cardiomyocytes (59). However, there are several other studies that not only showed the expression of both ERs in cardiomyocytes of rodents but also their functional activity on genomic and non-genomic levels (36, 60–67).
Recent reports showed that E2 can signal through a third protein, the G-protein-coupled estrogen receptor (GPER), formerly known as GPR30, a membrane receptor with seven transmembrane spanning domains (68, 69). GPER is strongly expressed in both male and female human and rat cardiac tissue (70–73). Specifically, GPER is present in smooth muscle cells (74, 75), endothelial cells (76), cardiac fibroblasts (77), and cardiomyocytes (70). GPER has been implicated predominantly in the rapid, non-genomic E2 signaling by the involvement of several kinases, ion channels and second messengers in a wide variety of cell types (Figure 1IV) (69, 78–80). However, effects on gene expression, i.e., induction of c-fos, cyclin D2, Egr-1, and Bcl-2 expression, have also been described (81–85).
Association of Genetic Alterations and Polymorphisms of the Estrogen Receptor Genes and Cardiovascular Disease
Studies showed that mutations in the genes coding for ERα and ERβ are associated with differences in heart morphology, such as increased left ventricular mass and wall thickness (86, 87). Furthermore, single nucleotide polymorphisms (SNPs) in both ERα and ERβ have been shown to be associated with the susceptibility for CVDs. Most of the studies analyzing ERα focused on two SNPs: c.454-397T>C (rs2234693) and c.454-351A>G (rs9340799) located in the first intron of the ERα gene and 46 bp apart from each other (88). In fact, the ERα variant rs2234693 was linked to coronary heart disease among Finnish men (89), whereas a study of a Dutch cohort showed that ERα variants, rs2234693, and rs9340799, were associated with increased risk of myocardial infarction (MI) and ischemic heart disease (IHD) only in postmenopausal women, but not in men (90). In contrast, in a prospective study in men and women from the population based offspring cohort of the Framingham Heart Study showed that individuals of both sexes carrying the rs2234693 genotype have substantial increase in risk of MI (91). The authors confirmed their findings in men in a latter study, including 7,000 men in five cohorts from four countries (92). In contrast, other studies found no association between these two SNPs or their haplotypes and MI or risk of CVD in either women or men (88, 93–95). Additionally, the absence of ERα in human vascular smooth muscle cells in premenopausal women (96) or the reduced ERα expression, due to methylation of the receptor with increasing age, is associated with the development of atherosclerosis in the cardiovascular system (97).
For ERβ, the SNP variant rs1271572 was associated with increased risk of MI in Spanish men (98), while Rexrode et al. identified this ERβ variant to be associated with increased risk of MI in women only (99). Additionally, this study showed the linkage of another ERβ variant, the rs1256049, with reduced risk of CVDs or MI in women (99).
The reasons for the inconsistency in data regarding the SNPs within the genes of ERα and ERβ could be due to the limited power within the studies, differences in methodology and study population (93). Despite the inconsistent findings, together these studies provide support for a relationship between ERα and ERβ polymorphisms and the risk of CVDs in men and women. The underlying mechanisms responsible for the phenotype associated with these genetic variants are not yet known. It is recognized that ER-SNPs can cause changes in E2-mediated downstream gene expression and signaling, which can alter the effects of E2 on the heart (100) and may be one possible explanation for the observed effects on the cardiovascular system. In contrast to ERα and ERβ, there are no studies so far regarding the association of polymorphisms within the GPER gene and cardiac risk in humans.
The Role of Estrogen Receptors in Animal Models for Human Cardiovascular Diseases
The physiology of E2-actions through its multiple receptors is diverse and highly complex. The detailed understanding of their effects and underlying molecular mechanisms are essential for future therapeutic applications in humans. In order to clarify remaining questions regarding the functions of each individual receptor within the heart, different mouse models with a deficiency or overexpression of ERα, ERβ, and GPER have been generated (101, 102).
ERα
At the basal level, male and female whole body ERα-deficient (ERKO)-mice are obese and insulin resistant (103). They also exhibit altered cardiac substrate preference with a reduction in glucose uptake indicating that ERα is required to maintain glucose utilization in the mouse heart (104). However, ERKO-mice do not show any cardiac dysfunction under physiological conditions. Following cardiac injuries, such as ischemic-reperfusion (I/R) injury or induced chronic MI, male and female ERKO-mice show increased cardiomyocyte cell death, mitochondrial damage, marked coronary edema, decreased coronary flow rate, and poorer functional recovery of contractility (+dP/dt) and compliance (-dP/dt) in comparison to wild type (WT)-mice (105, 106). These data suggest a cardiac protective role of ERα in both sexes after I/R or MI. In contrast, following pressure overload induced myocardial hypertrophy by transverse aortic constriction, female ERKO-mice developed myocardial hypertrophy to an identical degree as that seen in WT females, indicating that ERα is not essential for the attenuation of pressure overload induced hypertrophy observed in females (107, 108).
Analysis of mice hearts carrying a cardiomyocyte-specific deletion of ERα (cs-ERKO) revealed variations in the expression of genes involved in metabolism, cell growth and differentiation, muscle architecture, and relaxation compared to WT-mice (109). Furthermore, under basal conditions hearts from male and female cs-ERKO-mice showed reduction of left ventricular mass accompanied by decreased left ventricle (LV) diameter compared with WT-mice. These data are in line with published findings in mice with cardiomyocyte specific ERα-overexpression (csERα-OE), showing that constitutive ERα-overexpression in cardiomyocytes resulted in higher left ventricular mass and increased ventricular volumes. In addition, greater cardiomyocyte length, augmented expression of hypertrophy-associated genes such as nppa and nppb, but no fibrosis development was observed (65). In agreement with these data, findings from ovariectomized (OVX) mice also emphasize an E2-dependent role of ERα on regulation of cardiomyocyte size and cardiac growth in healthy mice (110). Overall, these findings indicate that ERα restricted to the cardiomyocytes is associated with the growth in cardiac mass in both sexes.
Interestingly, the use of csERα-OE mice demonstrated that ERα provides cardioprotection in female mice by enhancing neovascularization and impairment of cardiac remodeling in response to cardiac ischemic injury (65). All together, these findings indicate that in the female sex, ERα in cardiomyocytes may have a therapeutic potential in the treatment of ischemic heart disease, leading to more efficient cardiac repair after cardiac injury.
ERβ
In contrast to ERKO-mice, male and female ERβ-deficient (BERKO)-mice show a mild metabolic phenotype characterized by increased cortical bone formation and loss of trabecular bone (111). In addition, ERβ deficiency protects against diet-induced insulin resistance and glucose intolerance (112). However, with increasing age, BERKO-mice show cardiac hypertrophy, hypertension, and pathology in other cell types as they age (113–115). Additionally, BERKO-mice develop severe cardiomyopathy with a disarray of cardiomyocytes, a disruption of intercalated discs, an increase in number and size of gap junctions, and alteration in nuclear structure (114).
Several studies in BERKO-mice demonstrate the relevant role of ERβ in male and female mice after cardiac injury. The lack of ERβ significantly decreased post-ischemic cardiac recovery and therefore myocardial function in female, but not male, mice (116). In OVX mice subjected to MI, E2-treatment did not reduce infarct size in female BERKO-mice, as observed in ERKO- and WT-mice (117). In line with these data, Pelzer et al. reported that OVX BERKO-mice subjected to chronic MI showed increased mortality rates and aggravated signs of heart failure (118). These observations support the protective role of ERβ in response to I/R or MI in females. Following transverse aortic constriction, increase in left ventricular mass was not attenuated by E2-supplementation in OVX BERKO- as observed in WT- and ERKO-mice (108). Indeed, it has been shown that female BERKO-mice responded to transverse aortic constriction, as well as in the deoxycorticosterone acetate-salt mouse model, with a significantly higher increase in myocardial hypertrophy, marked increase in left ventricular diameters, increased cardiomyocyte size and apoptosis compared with female WT-mice (107, 119, 120). Fliegner et al. showed in male mice lacking ERβ significantly higher cardiomyocyte hypertrophy, increased myocyte apoptosis and faster progression toward heart failure (120). Thus, under pressure overload the loss of ERβ is detrimental for both males and females.
In a mouse model with a cardiomyocyte specific ERβ-overexpression (csERβ-OE), under basal conditions there were no observed differences in heart weight, morphology, and function in males and females (66). Interestingly, the overexpressed ERβ was located within the cytoplasm and nuclei of cardiomyocytes (66), while in csERα-OE mice the ERα protein was mainly located within the nuclei of cardiomyocytes (65). In response to MI, csERβ-OE exhibited improved survival in female and male mice compared to the WT counterparts (66). This was due to attenuated increase in heart weight and LV dilatation as well as improved systolic and diastolic function. In addition, both male and female csERβ-OE mice had a lower reduction of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) expression, suggesting less reduction in diastolic Ca2+-reuptake into the sarcoplasmic reticulum post-MI. Most of these functional parameters were improved in both sexes by csERβ-OE; however, the effect on LV volumes and ejection fraction was more pronounced in males than females. This was possibly due to reduced cardiac remodeling with lower cardiac fibrosis and lower expression of fibrosis markers (collagen I and III, periostin and miR-21), which was observed particularly in male csERβ-OE hearts after MI.
GPER
There are several studies stating the phenotype of mice lacking GPER (101). The studies of GPER-KO-mice over the last decade revealed that GPER deficient mice show under basal conditions multiple physiological alterations, including obesity (75), insulin resistance, glucose intolerance, and increase in blood pressure (121). Interestingly, it has been reported that male, but not female, GPER-KO-mice show impaired cardiac function with enlarged LV and decreased +dP/dt and –dP/dt (122) or decreased ejection fraction and fractional shortening with increasing age (123). Under cardiac stress, one study reported in a mouse model of I/R that male WT-, ERKO-, and BERKO-mice respond to E2-treatment with an improved recovery and reduced infract size. However, the application of E2 to male GPER-KO-mice did not lead to observed cardioprotection after I/R (80).
A recent study in mice with a cardiomyocyte-specific GPER-KO (csGPER-KO) revealed under basal conditions adverse alterations in cardiac structure and impaired systolic and diastolic function in both sexes, in comparison to WT-mice, with more profound increases in LV dimensions, and wall-thinning among male KO-mice (124). Using DNA microarray analysis, the authors found differential expression profiles of genes affecting multiple transcriptional networks with marked differences in respect to sex and cardiomyocyte-specific GPER deletion. In detail, mitochondrial genes were enriched in cardiomyocytes from female GPER-KO- compared to female WT-mice, but not in male. In contrast, inflammatory response genes were enriched in GPER-KO- vs. WT-cardiomyocytes from male but not female mice (124, 125).
Although studies with transgenic ER mice failed to provide a clear consensus regarding the physiological and pathological roles of ERs, they suggest that each of the ER subtypes play a protective role in the heart.
The Role of 17β-Estradiol and Estrogen Receptors in Regulation of Ca2+ Channels and Contractility in Cardiomyocytes
Ca2+ is a critical regulator of myocardial function. Ca2+ regulates contraction, and deregulation of Ca2+ signaling has been associated with cardiac dysfunction and pathology such as arrhythmias and heart failure (4). In cardiomyocytes, Ca2+ levels are tightly regulated via the excitation-contraction (EC) coupling pathway (Figure 2). During action potential, in response to depolarization, Ca2+ crosses the sarcolemma and T-tubular membrane through the voltage gated L-type Ca2+ channels. This Ca2+ influx triggers the release of a larger quantity of Ca2+, called Ca2+ sparks, from the sarcoplasmic reticulum (SR), through the opening of SR Ca2+ release channels, known as ryanodine receptors (RyRs, particularly RyR2). This process is termed Ca2+-induced Ca2+ release. The combination of Ca2+ influx via the L-type Ca2+ channels and Ca2+ release from SR leads to the formation of cytosolic Ca2+ transients. The binding of cytosolic Ca2+ to the myofilaments then initiates cardiomyocyte contraction. Subsequent relaxation occurs by removal of Ca2+ from the cytosol mainly via the following mechanisms: (I) The SERCA2a re-uptakes the cytosolic Ca2+ back into the SR; the activity of this channel being modulated by its endogenous inhibitor phospholamban (PLN); (II) The Na+/Ca2+ exchanger (NCX) extrudes the Ca2+ out of the cells; (III) The mitochondrial Ca2+ uniporter transports Ca2+ into the mitochondria (4, 126).
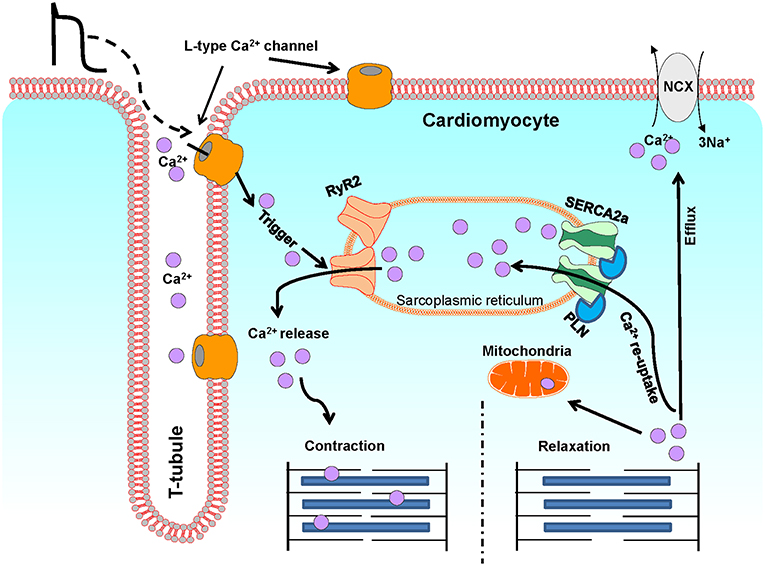
Figure 2. A schematic illustration of Ca2+ fluxes during excitation-contraction in ventricular cardiomyocytes. This diagram depicts the most representative protein complexes and intercellular organelles involved in the cardiac excitation-contraction coupling. Ca2+, calcium; SR, sarcoplasmic reticulum; M, Mitochondria; LTCC, L-type Ca2+ channel; RyR2, Ryanodine receptor 2; SERCA2a, Sarcoplasmic reticulum Ca2+ATPase 2a; PLN, Phospholamban; NCX, Na+/Ca2+ exchanger.
Numerous studies have documented sex differences in cardiac EC coupling (127–129). For example, at rest, women have longer QT intervals and higher left ventricular ejection fraction than men (130–132). Other studies showed that ventricular myocytes in the female human failing heart have significantly greater contractility and enhanced L-type Ca2+ current (ICa, L) compared to male patients (133–135). Studies in animal models also provide convincing evidence of sex differences in contractile function as observed in humans. It has been demonstrated that isolated cardiomyocytes from male rodents exhibit higher contraction than those from females (128, 136, 137). Furthermore, male rat cardiac myocyte and papillary muscle develop higher contractile force as well as significantly greater Ca2+ transient amplitude than females (138–142). In studies, using paced cardiomyocytes at the rates of 0.5–1.0 Hz, cardiac relaxation rate was slower in cardiomyocytes from female rats compared to aged matched males (139, 143).
The expression and function of cardiac L-type Ca2+ channels, which have a direct impact on the functional changes in EC coupling pathway in the heart, also show significant sexual dimorphisms. In adult cardiomyocytes, the Cavα1C or Cav1.2 (cardiac voltage-gated L-type Ca2+ channel) is the most abundant cardiac L-type Ca2+ channel which triggering cardiac contraction by regulation of ICa, L in cardiomyocytes (144–146). Therefore, it represents an important cellular site from which sex-based differences in myocardial intracellular Ca2+ handling and contractility may arise (138). Studies comparing the cardiac L-type Ca2+ Channel expression and ICa, L that have included both female and male animals, are still limited and the existing data are controversial. It has been demonstrated that the levels of L-type Ca2+ channel expression increase or do not change at all in the ventricle of female rats and rabbits in comparison to males (147–149). Similarly, comparative studies using isolated cardiomyocytes from female and male rats, mice, guinea pigs, and dogs showed that compared to males, the ICa, L density is either higher (147, 150–152) or lower in cells from females (153) or that there are no sex differences in ICa, L density at all (137, 140, 141, 154, 155). Even with these discrepancies in the data, which might be due to variations in the experimental protocols, species, and used strains, sex differences in the regulation and expression of L-type Ca2+ channels are apparent, although the underlying signaling mechanisms implicated in these sex differences are poorly understood.
In recent years, several studies provided evidence that the distal part of the C-terminus of the α1C subunit (α1C-dCT) of Cav1.2 channel is proteolytically cleaved and shuttles between the plasma membrane and the nucleus of cardiomyocytes. It serves at the plasma membrane as an auto-inhibitor of Cav1.2 channel activity (156–159), and acts as transcription factor in the nucleus, regulating the expression of different genes, including Cav1.2 gene (CACNA1C) itself (160–163). Schroder et al. have provided evidence that the nuclear import of α1C-dCT in cardiomyocytes depresses Cav1.2 transcription, while nuclear export of α1C-dCT increases Cav1.2 channel activity consistent with a reduction of subsequent increase of Cav1.2 gene transcription rates (161). In a recent study, we observed a remarkable sex-disparity in nuclear shuttling of α1C-dCT in mouse cardiomyocytes (164). Here, the nuclear shuttling was significantly higher in isolated female cardiomyocytes compared to males. Furthermore, we found a significant decrease in nuclear shuttling of α1C-dCT in both female and male cardiomyocytes upon serum withdrawal. However, subsequent E2-treatment normalized the intracellular distribution of α1C-dCT only in male cardiomyocytes. This effect of E2 was reversed by the ER-antagonist ICI 182,780, indicating the involvement of ER in this signaling pathway. These findings provide a possible explanation for the cellular mechanisms responsible for sex differences in the regulation of L-type Ca2+ channel in the heart, revealing the role of E2/ER in this process.
In addition to the L-type calcium channel, sexual dimorphisms in the expression, and activity of other cardiac calcium channels have also been reported. For example, several studies found that the expression and/or current of NCX (INCX) are significantly higher in cardiomyocytes from female humans, rats, and rabbits compared to their male counterparts (135, 147–149, 165). Interestingly, Chen et al. showed that E2 administration increased NCX and INCX in female but not in male cardiomyocytes. These E2 effects appear to be mediated by a genomic mechanism involving the binding of E2 to its receptors, since these E2 effects were blunted by an ER antagonist (ICI 182,780) (165).
On the other hand, several studies have reported contradictory results on sex differences in the regulation of RyR2 expression and activity in the heart. It has been shown that the expression of RyR2 is higher in female rat cardiomyocytes compared to males (148, 149, 166), or that the expression does not differ in male and female rat and mice cardiomyocytes (155, 167). Bell et al. showed, however, that the regulation of RyR2 activity is different in male and female rat cardiomyocytes, with CaMKII (Ca2+/calmodulin-dependent protein kinase II)-mediated phosphorylation of RyR2 being lower in female cardiomyocytes than in male cardiomyocytes (167). This could be a possible explanation for the observed decrease in the gain of EC coupling (measured as SR Ca2+ release/Ca2+ current) in female rat and mice cardiomyocytes, which results from decreased size and duration of Ca2+ sparks by RyR2 (140, 155).
Collectively these findings suggest that the observed sex differences reflect, at least partly, the effects of E2 on myocardial Ca2+ handling, thus on contractility.
In this regard, studies with OVX rodents corroborate the effects of E2 on myocardial Ca2+ handling and contractility. Numerous studies with whole hearts or isolated cardiomyocytes from OVX mice, rats, rabbits, and pigs revealed that the E2 deficiency caused detrimental effects on both Ca2+ regulation and contractility of cardiomyocytes, such as enhanced Ca2+ transients, increased Ca2+ spark amplitudes, decreased myofilament Ca2+ sensitivity, and elevated contractions, in comparison to sham-operated controls (168–179). Remarkably, substitution of E2 effectively prevented the observed adverse effects (168, 169, 172, 174–179) and it could be shown that this is directly mediated via the ER by using the ER-antagonist ICI 182,780 (169).
In this context, several studies suggested that observed E2 effects are mediated by its receptors. Indeed, hearts of male ERKO-mice exhibit increased cardiac L-type Ca2+ channel expression and ICa, L (180), as well as significantly higher Ca2+ accumulation compared to control hearts during I/R (106). In line with these data, a recent study demonstrated that both E2 pre-treatment and/or ERα activation of Tet-on/ERα H9c2 cardiomyoblast cells inhibited isoproterenol-induced cytosolic Ca2+ accumulation in these cells, and this protective effect of the E2/ERα was reversed by treatment with a specific inhibitor of ERα (181). These data indicate that E2/ERα signaling pathway is involved in the regulation of Ca2+ balance in cardiomyocytes, thereby preventing the harmful effects of Ca2+ overload in the pathophysiology of the heart. By contrast, another study using ERKO- and BERKO-mice could not show that the inhibition of ICa, L and decrease in contraction depend on ERα or ERβ action (182). Moreover, it has been shown that in global GPER-KO mice, both left-ventricular contractility, and relaxation capacity were impaired only in males (122).
Furthermore, other studies have confirmed that the specific activation of different ER-isoforms affects cardiac contractility. Pelzer et al. showed that activation of ERα with the subtype-selective ERα agonist 16α-LE2 augments myocardial contractility to a measurable extent in OVX spontaneously hypertensive rats (183). Kulpa et al. showed that activation of ERα using the ERα agonist PPT (4,4',4”-(4-Propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol) depressed actomyosin MgATPase activity and decreased myofilament Ca2+ sensitivity (184). Other studies have demonstrated the respective roles of ERβ and GPER activation in the regulation of SR Ca2+ handling proteins, such as SERCA2a and PLN, leading to improved contractility at the whole heart and single myocyte (66, 185).
These findings reveal that a solid understanding the roles of the various estrogen receptors in the regulation of cardiac contractility are needed in order to be able to find appropriate pharmacological agents that specifically target the receptors of interest.
The Role of 17β-Estradiol and Estrogen Receptors in Cardiac Mitochondrial Function
Mitochondria are the main source of ATP and Reactive Oxygen Species (ROS) in the heart (186). It is considered that mitochondria play an essential role not only in regulation of cardiac contractility by providing ATP and by participating in Ca2+ homeostasis, but also they regulate cell death or apoptosis by ROS formation. Therefore, defects in mitochondrial structure and function are highly associated with CVDs (5, 186). E2 plays an important role in the supporting mitochondrial respiration, ATP production, and reducing ROS formation (Figure 1VI).
Sex differences in mitochondrial structure and function have been described. There is plenty of evidence that mitochondrial morphology and function differ between females and males in several organs and cell types. In the healthy mice hearts, although the female and male hearts displayed similar mitochondrial numbers, the proportion of large mitochondria (≥1 μm2) was significantly higher in female mice compared to males (56). Skeletal muscles from female rats show higher mitochondrial DNA and protein contents, as well as higher capacity of oxidative phosphorylation (OXPHOS) compared to male rats (187). Further, mitochondria in brain and liver from female mice exhibit higher antioxidant gene expression and lower oxidative damage under stress than in male animals (188). Additionally, several studies reported that the rate of ROS production is less in mitochondria from skeletal and cardiac muscle in female compared with aged matched male rats, particularly under stress conditions (187, 189, 190). Moreover, female rat hearts show altered posttranslational modification of several mitochondrial proteins under I/R in comparison to male hearts, including aldehyde dehydrogenase-2 (ALDH2) (189), a protein that has been reported to be involved in cardioprotective processes (191). Whole genome expression profiling performed in hearts of old (78-week) male and female Fischer 344 rats showed that a majority of genes involved in oxidative phosphorylation had higher expression in females compared to male rats (192). These studies suggest that E2 plays a role in the regulation of mitochondrial function, which is supported by evidence from several studies in OVX animals.
In particular, a high throughput quantitative proteomic approach with isolated mitochondria from left ventricles of OVX rat relative to ovary-intact hearts revealed that about 50% of the identified proteins altered in OVX rat cardiac mitochondria are involved in mitochondrial ATP production (193). Indeed, the observed reduction of protein subunits of the electron transport chain complex I (NADH dehydrogenase), II (succinate dehydrogenase), III (cytochrome bc1 complex), IV (cytochrome c oxidase), and V (F0F1 ATP-synthase) in E2-deficint hearts was associated with reduced ATP production that may contribute to increased I/R injury and disease risk with E2 deficiency in aged female rats. Interestingly, in a mouse model of a human hypertrophic cardiomyopathy (cTnT-Q92), E2-supplementation of OVX mice significantly elevated myocardial ATP levels and mitochondrial respiratory function compared to untreated OVX mice, thereby improving diastolic heart function (194). In another model of cardiomyopathy, hearts from OVX rats showed higher Ca2+ accumulation in their mitochondria, lower mitochondrial respiratory function, severely structurally damaged mitochondria, and increased myocardial cell death after I/R injury in comparison to intact animals (195). Again, in this study, E2-treatment of the hearts from OVX animals attenuated cardiac damage by I/R, and thereby maintained the LV function. Furthermore, mitochondria from hearts of OVX rats showed higher expression of apoptotic markers compared to mitochondria of intact animals (196). However, chronic E2-treatment of these animals significantly attenuated mitochondria-dependent apoptotic pathways. These data directly show that alterations in mitochondrial function are a highly selective myocardial response to E2 deficiency, and that E2-mediated cardioprotection at the level of the mitochondria leads to improved cardiac function.
Indeed, several studies demonstrated that E2 through its ERs affects the cardiac mitochondria directly via regulation of mitochondrial gene/protein expression. It has been shown that ERα and ERβ are localized in the mitochondria of cardiac cells (62, 197–199). The presence of ERs in the mitochondria of cardiac cells suggests that they mediate the observed protective effects of E2, at least partly, by regulating mitochondrial structure and function in the heart. In line with the role of ERα and ERβ as transcription factors, distinct evidence supports the notion that mitochondrial DNA (mtDNA) could be one of the major targets for E2 acting via ER in cardiac cells. This is supported, for example, (1) by the presence of putative ERE on the mtDNA (200–202), (2) the E2-induced up-regulation of several mitochondrial-encoded genes, such as COXI and COXII (cytochrome c oxidase subunits I and II) (203, 204), and (3) the E2-induced expression of several nuclear-encoded mitochondrial genes, such as NRF-1 (nuclear respiratory factor 1), NRF-2 (nuclear respiratory factor 2), TFAM (mitochondrial transcription factor), PGC-1α (peroxisome proliferator-activated receptor gamma co-activator-1 alpha), and MEF2a (Myocyte enhancer factor 2A) (56, 202, 205, 206), whose proteins translocate into the mitochondria and thereby influence mitochondrial function. Additionally, it could be shown that in rat myocardium after severe hemorrhage the E2-induced increased expression of these genes was associated with an increase in COX IV (cytochrome c oxidase subunit IV), mtDNA-encoded COX I (cytochrome c oxidase subunit I), ATP synthase β-subunit, and mitochondrial ATP (207, 208). All these effects were abolished with the ER antagonist ICI 182,780, indicating an ER-specific effect.
The role of E2 and ER in the regulation of mitochondrial structure and function is established from studies with ER deficient mouse models. Microarray analysis using ERKO- and BERKO-mice showed that E2/ERβ pathways mediate down-regulation of mRNAs for nuclear-encoded subunits in each of the major complexes of the electron transport chain, whereas ERα is essential for most of the E2-mediated increase in gene expression including electron transport chain proteins and proteins involved in the anti-oxidative stress response (209). In a mouse model of exercise-induced physiological myocardial hypotrophy, we demonstrated that only female WT-mice showed an increase in the expression of key regulators of mitochondrial function e.g., NRF-1,−2, Mef2a, Atp5k (subunit E of mitochondrial F1F0-ATP synthase), and electron transport chain proteins (complexes I, III, and V) after running. Interestingly, ERβ deletion abolished the observed effects (56). Additionally, our study also showed that the activated ERβ significantly increased the expression of MEF2A, NRF-1, and−2 genes in a cardiomyocyte cell line (AC16 cells) (56). In line with these data, the expression of NRF-1 is diminished in BERKO hearts (209). On the other hand, Zhai et al. demonstrated that ERKO-mice hearts showed marked mitochondrial damages (fragmented and swollen mitochondria) and severe impairment of mitochondrial respiratory function compared to control hearts after I/R (106). To our knowledge a direct localization of GPER within the mitochondria has not been documented so far. However, analysis of DNA microarray data followed by Gene Set Enrichment Analysis (GSEA) from female and male cardiomyocytes of WT- and csGPER-KO-mice revealed that mitochondrial genes are enriched only in csGPER-KO females (124, 125), which provided direct evidence that the cardioprotective effects of GPER under physiological and pathological conditions in the female csGPER-KO-mice may be related to enhancements in mitochondrial function.
Several studies demonstrated that E2 also indirectly affects the cardiac mitochondria via regulation of ROS production. Elevated Ca2+ uptake by mitochondria results in the opening of the mitochondrial permeability transition pore (mPTP) and enhanced release of cytochrome c accompanied by dramatic increase in ROS formation, which leads to cell death via the induction of apoptosis pathways (210, 211). It has been shown that in comparison to male, mitochondria from female rat hearts accumulate Ca2+ more slowly (212), which might represent a mechanism that may underlie, at least partly, sex-related differences accounting for females to suffer less injury with I/R. Indeed, several studies demonstrated that E2 administration can acutely attenuate the Ca2+ accumulation in mitochondria, inhibit Ca2+-induced opening of mPTP in isolated heart mitochondria, prevent Ca2+-induced release of cytochrome c from mitochondria, and inhibit ischemia-induced apoptosis in perfused heart (213–215). Interestingly, Feng et al. demonstrated that post-ischemic E2 administration to both male and OVX-female rats preserved mitochondrial structural integrity, which was associated with an increased tolerance to Ca2+ overload or augmented mitochondrial Ca2+ retention capacity (216) which reflects an inhibition of the mPTP opening in both male and OVX-female animals.
Here again, using ER deficient mice could be shown that these E2 effects are mediated by ERs. Male ERKO hearts subjected to I/R showed an accumulated Ca2+ deposition in their mitochondria which led to severe mitochondrial damage (fragmented and swollen mitochondria) in cardiomyocytes, and consequently to the depletion of ATP production (106). Using ERKO-, BERKO-, and ERα and ERβ double knockout (DERKO)-mice, Luo et al. found that both ER subtypes are necessary for E2-mediated cardioprotection during I/R in female hearts. Thereby, E2 and ER upregulate mitochondrial p38β-MAPK activity, with subsequent phosphorylation of the MnSOD (manganese superoxide dismutase), leading to enhanced SOD activity, thereby minimizing mitochondrial-derived ROS production and reduction of myocardial infarct size post I/R (217). By contrast, a systematic analysis of WT-, ERKO-, BERKO-, and GPER-KO-mice subjected to I/R showed that only GPER expression is essential for the acute action of E2 in cardioprotection against I/R injury in male mouse via a cascade involving PKC translocation, ERK1/2/GSK-3β (Glykogensynthase-Kinase 3β)- phosphorylation leading to the inhibition of the mPTP opening, resulting in reduction of harmful mitochondrial ROS generation (80). However, a pre-administration with G15, a specific GPER antagonist, reversed this estrogenic effect. This data indicate that GPER activation mediates E2-induced increase in mitochondrial Ca2+ retention capacity, and the GPER-mediated cardioprotective effect of post-ischaemic E2 is related to a decrease in mPTP sensitivity to Ca2+ overload, a process which is mediated via activation of the MEK/ERK/GSK-3β axis.
These data suggest that depending on the time period of E2-treatment, sex, and species different ERs can be activated by E2, which mediate the mitochondrial-dependent cardioprotective effect of E2 against I/R injury.
Conclusion
In the past, most clinical and animal studies did not include both sexes or differentiate between sexes in the data analysis. This might be the possible reason that our understanding of the molecular and cell-based mechanisms underlying sex-based differences in cardiovascular system are still incomplete so far. A more thorough understanding of underlying sex-dimorphic mechanisms in cardiac health and disease is required to effectively treat patients with CVDs. The presented data in this review support the concept that sex specific regulation of cardiac Ca2+ ion channels and mitochondrial function by E2 and ERs could be, at least partly, responsible for differences in cardiovascular disease incidence and outcomes. However, further attempts toward a more detailed understanding of E2 and ERs roles in the heart are needed to develop new drugs that target the beneficial effects on CVD in both sexes.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
SM was funded by German Research Foundation and Friede Springer Heart Foundation. ED was funded by German Center for Cardiovascular Research (DZHK), German Federal Ministry of Education and Research (BMBF), and German Research Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Dr. Ingo Morano and Dr. Valeria Raparelli for valuable discussions and the critical revision of the manuscript.
References
2. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the american heart association. Circulation. (2019) 139:e56–66. doi: 10.1161/CIR.0000000000000659
3. Regitz-Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. (2017) 97:1–37. doi: 10.1152/physrev.00021.2015
4. Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology. (2006) 21:380–7. doi: 10.1152/physiol.00019.2006
5. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. (2018) 50:121–7. doi: 10.1080/07853890.2017.1417631
6. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15:457–70. doi: 10.1038/s41569-018-0044-6
7. Ren J, Kelley RO. Cardiac health in women with metabolic syndrome: clinical aspects and pathophysiology. Obesity. (2009) 17:1114–23. doi: 10.1038/oby.2009.8
8. Hayward CS, Kelly RP, Collins P. The roles of gender, the menopause and hormone replacement on cardiovascular function. Cardiovasc Res. (2000) 46:28–49. doi: 10.1016/S0008-6363(00)00005-5
9. Yang XP, Reckelhoff JF. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr Opin Nephrol Hypertens. (2011) 20:133–8. doi: 10.1097/MNH.0b013e3283431921
10. Mosca L, Benjamin EJ, Berra K, Bezanson JL, Dolor RJ, Lloyd-Jones DM, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women−2011 update: a guideline from the American Heart Association. J Am Coll Cardiol. (2011) 57:1404–23. doi: 10.1161/CIR.0b013e31820faaf8
11. Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Internal Med. (2000) 133:933–41. doi: 10.7326/0003-4819-133-12-200012190-00008
12. Grodstein F, Stampfer MJ, Colditz GA, Willett WC, Manson JE, Joffe M, et al. Postmenopausal hormone therapy and mortality. N Engl J Med. (1997) 336:1769–75. doi: 10.1056/NEJM199706193362501
13. Schierbeck LL, Rejnmark L, Tofteng CL, Stilgren L, Eiken P, Mosekilde L, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ. (2012) 345:e6409. doi: 10.1136/bmj.e6409
14. Salpeter SR, Walsh JM, Greyber E, Ormiston TM, Salpeter EE. Mortality associated with hormone replacement therapy in younger and older women: a meta-analysis. J Gen Internal Med. (2004) 19:791–804. doi: 10.1111/j.1525-1497.2004.30281.x
15. Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. (2003) 349:523–34. doi: 10.1056/NEJMoa030808
16. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA. (2002) 288:321–33.
17. Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: heart and estrogen/progestin replacement study follow-up (HERS II). JAMA. (2002) 288:49–57. doi: 10.1001/jama.288.1.49
18. Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. (2004) 291:1701–12. doi: 10.1001/jama.291.14.1701
19. Harman SM. Estrogen replacement in menopausal women: recent and current prospective studies, the WHI and the KEEPS. Gender Med. (2006) 3:254–69. doi: 10.1016/S1550-8579(06)80214-7
20. Rosano GM, Vitale C, Fini M. Cardiovascular aspects of menopausal hormone replacement therapy. Climacteric. (2009) 12 (Suppl. 1):41–6. doi: 10.1080/13697130903012306
21. Schnatz PF. Hormonal therapy: does it increase or decrease cardiovascular risk? Obstetrical Gynecol Survey. (2006) 61:673–81. doi: 10.1097/01.ogx.0000238674.98471.bb
22. Haines CJ, Farrell E. Menopause management: a cardiovascular risk-based approach. Climacteric. (2010) 13:328–39. doi: 10.3109/13697130903450154
23. Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, et al. Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med. (2016) 374:1221–31. doi: 10.1056/NEJMoa1505241
24. Hodis HN, Mack WJ, Shoupe D, Azen SP, Stanczyk FZ, Hwang-Levine J, et al. Methods and baseline cardiovascular data from the early versus late intervention trial with estradiol testing the menopausal hormone timing hypothesis. Menopause. (2015) 22:391–401. doi: 10.1097/GME.0000000000000343
25. Sudhir K, Komesaroff PA. Clinical review 110: cardiovascular actions of estrogens in men. J Clin Endocrinol Metab. (1999) 84:3411–5. doi: 10.1210/jc.84.10.3411
26. Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in male physiology. Physiol Rev. (2017) 97:995–1043. doi: 10.1152/physrev.00018.2016
27. Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, et al. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. (1997) 337:91–5. doi: 10.1056/NEJM199707103370204
28. Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. (1994) 331:1056–61. doi: 10.1056/NEJM199410203311604
29. Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. (1995) 80:3689–98. doi: 10.1210/jc.80.12.3689
30. Vikan T, Schirmer H, Njolstad I, Svartberg J. Low testosterone and sex hormone-binding globulin levels and high estradiol levels are independent predictors of type 2 diabetes in men. Eur J Endocrinol. (2010) 162:747–54. doi: 10.1530/EJE-09-0943
31. Sudhir K, Chou TM, Messina LM, Hutchison SJ, Korach KS, Chatterjee K, et al. Endothelial dysfunction in a man with disruptive mutation in oestrogen-receptor gene. Lancet. (1997) 349:1146–7. doi: 10.1016/S0140-6736(05)63022-X
32. Jankowska EA, Rozentryt P, Ponikowska B, Hartmann O, Kustrzycka-Kratochwil D, Reczuch K, et al. Circulating estradiol and mortality in men with systolic chronic heart failure. JAMA. (2009) 301:1892–901. doi: 10.1001/jama.2009.639
33. Vandenplas G, De Bacquer D, Calders P, Fiers T, Kaufman JM, Ouwens DM, et al. Endogenous oestradiol and cardiovascular disease in healthy men: a systematic review and meta-analysis of prospective studies. Heart. (2012) 98:1478–82. doi: 10.1136/heartjnl-2011-301587
34. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev. (2001) 81:1535–65. doi: 10.1152/physrev.2001.81.4.1535
35. Morselli E, Santos RS, Criollo A, Nelson MD, Palmer BF, Clegg DJ. The effects of oestrogens and their receptors on cardiometabolic health. Nat Rev Endocrinol. (2017) 13:352–64. doi: 10.1038/nrendo.2017.12
36. Grohe C, Kahlert S, Lobbert K, Stimpel M, Karas RH, Vetter H, et al. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. (1997) 416:107–12. doi: 10.1016/S0014-5793(97)01179-4
37. Bell JR, Mellor KM, Wollermann AC, Ip WT, Reichelt ME, Meachem SJ, et al. Aromatase deficiency confers paradoxical postischemic cardioprotection. Endocrinology. (2011) 152:4937–47. doi: 10.1210/en.2011-1212
38. Jazbutyte V, Stumpner J, Redel A, Lorenzen JM, Roewer N, Thum T, et al. Aromatase inhibition attenuates desflurane-induced preconditioning against acute myocardial infarction in male mouse heart in vivo. PLoS ONE. (2012) 7:e42032. doi: 10.1371/journal.pone.0042032
39. Iorga A, Li J, Sharma S, Umar S, Bopassa JC, Nadadur RD, et al. Rescue of pressure overload-induced heart failure by estrogen therapy. J Am Heart Assoc. (2016) 5:e002482. doi: 10.1161/JAHA.115.002482
40. Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res. (2016) 118:994–1007. doi: 10.1161/CIRCRESAHA.115.305376
41. Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. (1996) 392:49–53. doi: 10.1016/0014-5793(96)00782-X
42. Mahmoodzadeh S, Pham TH, Kuehne A, Fielitz B, Dworatzek E, Kararigas G, et al. 17beta-Estradiol-induced interaction of ERalpha with NPPA regulates gene expression in cardiomyocytes. Cardiovasc Res. (2012) 96:411–21. doi: 10.1093/cvr/cvs281
43. Duft K, Schanz M, Pham H, Abdelwahab A, Schriever C, Kararigas G, et al. 17beta-Estradiol-induced interaction of estrogen receptor alpha and human atrial essential myosin light chain modulates cardiac contractile function. Basic Res Cardiol. (2017) 112:1. doi: 10.1007/s00395-016-0590-1
44. Dworatzek E, Mahmoodzadeh S, Schriever C, Kusumoto K, Kramer L, Santos G, et al. Sex-specific regulation of collagen I and III expression by 17beta-estradiol in cardiac fibroblasts: role of estrogen receptors. Cardiovasc Res. (2019) 115:315–27. doi: 10.1093/cvr/cvy185
45. Lannigan DA. Estrogen receptor phosphorylation. Steroids. (2003) 68:1–9. doi: 10.1016/S0039-128X(02)00110-1
46. Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. (2008) 22:2116–27. doi: 10.1210/me.2008-0059
47. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. (1995) 270:1491–4. doi: 10.1126/science.270.5241.1491
48. Mahmoodzadeh S, Dworatzek E, Fritschka S, Pham TH, Regitz-Zagrosek V. 17beta-estradiol inhibits matrix metalloproteinase-2 transcription via MAP kinase in fibroblasts. Cardiovasc Res. (2010) 85:719–28. doi: 10.1093/cvr/cvp350
49. Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, et al. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. (2003) 111:1651–64. doi: 10.1172/JCI200317261
50. Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, et al. Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol. (2002) 16:116–27. doi: 10.1210/mend.16.1.0748
51. de Jager T, Pelzer T, Muller-Botz S, Imam A, Muck J, Neyses L. Mechanisms of estrogen receptor action in the myocardium. Rapid gene activation via the ERK1/2 pathway and serum response elements. J Biol Chem. (2001) 276:27873–80. doi: 10.1074/jbc.M010984200
52. Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. (2001) 276:36869–72. doi: 10.1074/jbc.R100029200
53. Simoncini T, Mannella P, Genazzani AR. Rapid estrogen actions in the cardiovascular system. Ann N Y Acad Sci. (2006) 1089:424–30. doi: 10.1196/annals.1386.001
54. Nordmeyer J, Eder S, Mahmoodzadeh S, Martus P, Fielitz J, Bass J, et al. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation. (2004) 110:3270–5. doi: 10.1161/01.CIR.0000147610.41984.E8
55. Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, et al. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. (2006) 20:926–34. doi: 10.1096/fj.05-5148com
56. Dworatzek E, Mahmoodzadeh S, Schubert C, Westphal C, Leber J, Kusch A, et al. Sex differences in exercise-induced physiological myocardial hypertrophy are modulated by oestrogen receptor beta. Cardiovasc Res. (2014) 102:418–28. doi: 10.1093/cvr/cvu065
57. Lizotte E, Grandy SA, Tremblay A, Allen BG, Fiset C. Expression, distribution and regulation of sex steroid hormone receptors in mouse heart. Cell Physiol Biochem. (2009) 23:75–86. doi: 10.1159/000204096
58. Irsik DL, Carmines PK, Lane PH. Classical estrogen receptors and ERalpha splice variants in the mouse. PLoS ONE. (2013) 8:e70926. doi: 10.1371/journal.pone.0070926
59. Pugach EK, Blenck CL, Dragavon JM, Langer SJ, Leinwand LA. Estrogen receptor profiling and activity in cardiac myocytes. Mol Cell Endocrinol. (2016) 431:62–70. doi: 10.1016/j.mce.2016.05.004
60. Lipovka Y, Chen H, Vagner J, Price TJ, Tsao TS, Konhilas JP. Oestrogen receptors interact with the alpha-catalytic subunit of AMP-activated protein kinase. Biosci Rep. (2015) 35:e00264. doi: 10.1042/BSR20150074
61. Huang PC, Kuo WW, Shen CY, Chen YF, Lin YM, Ho TJ, et al. Anthocyanin attenuates doxorubicin-induced cardiomyotoxicity via estrogen receptor-alpha/beta and stabilizes HSF1 to inhibit the IGF-IIR apoptotic pathway. Int J Mol Sci. (2016) 17:1588. doi: 10.3390/ijms17091588
62. Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM Jr, Valencia T, et al. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci USA. (2004) 101:4130–5. doi: 10.1073/pnas.0306948101
63. Grohe C, Kahlert S, Lobbert K, Vetter H. Expression of oestrogen receptor alpha and beta in rat heart: role of local oestrogen synthesis. J Endocrinol. (1998) 156:R1–7. doi: 10.1677/joe.0.156r001
64. Ropero AB, Eghbali M, Minosyan TY, Tang G, Toro L, Stefani E. Heart estrogen receptor alpha: distinct membrane and nuclear distribution patterns and regulation by estrogen. J Mol Cell Cardiol. (2006) 41:496–510. doi: 10.1016/j.yjmcc.2006.05.022
65. Mahmoodzadeh S, Leber J, Zhang X, Jaisser F, Messaoudi S, Morano I, et al. Cardiomyocyte-specific estrogen receptor alpha increases angiogenesis, lymphangiogenesis and reduces fibrosis in the female mouse heart post-myocardial infarction. J Cell Sci Ther. (2014) 5:153. doi: 10.4172/2157-7013.1000153
66. Schuster I, Mahmoodzadeh S, Dworatzek E, Jaisser F, Messaoudi S, Morano I, et al. Cardiomyocyte-specific overexpression of oestrogen receptor beta improves survival and cardiac function after myocardial infarction in female and male mice. Clin Sci. (2016) 130:365–76. doi: 10.1042/CS20150609
67. Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER. Estrogen receptor-beta prevents cardiac fibrosis. Mol Endocrinol. (2010) 24:2152–65. doi: 10.1210/me.2010-0154
68. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
69. Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol. (2014) 389:71–83. doi: 10.1016/j.mce.2014.02.002
70. Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. (2009) 297:H1806–13. doi: 10.1152/ajpheart.00283.2009
71. Filice E, Recchia AG, Pellegrino D, Angelone T, Maggiolini M, Cerra MC. A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J Physiol Pharmacol. (2009) 60:3–10.
72. Patel VH, Chen J, Ramanjaneya M, Karteris E, Zachariades E, Thomas P, et al. G-protein coupled estrogen receptor 1 expression in rat and human heart: protective role during ischaemic stress. Int J Mol Med. (2010) 26:193–9. doi: 10.3892/ijmm_00000452
73. Hutson DD, Gurrala R, Ogola BO, Zimmerman MA, Mostany R, Satou R, et al. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol Sex Differ. (2019) 10:4. doi: 10.1186/s13293-019-0219-9
74. Yu X, Ma H, Barman SA, Liu AT, Sellers M, Stallone JN, et al. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am J Physiol Endocrinol Metab. (2011) 301:E882–8. doi: 10.1152/ajpendo.00037.2011
75. Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. (2009) 104:288–91. doi: 10.1161/CIRCRESAHA.108.190892
76. Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. (2009) 150:3753–8. doi: 10.1210/en.2008-1664
77. Wang H, Zhao Z, Lin M, Groban L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol Cell Biochem. (2015) 405:135–48. doi: 10.1007/s11010-015-2405-3
78. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
79. Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
80. Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3beta pathway after ischemia/reperfusion. PLoS ONE. (2015) 10:e0135988. doi: 10.1371/journal.pone.0135988
81. Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol. (2009) 308:32–8. doi: 10.1016/j.mce.2009.03.026
82. Vivacqua A, Romeo E, De Marco P, De Francesco EM, Abonante S, Maggiolini M. GPER mediates the Egr-1 expression induced by 17beta-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Res Treat. (2012) 133:1025–35. doi: 10.1007/s10549-011-1901-8
83. Kanda N, Watanabe S. 17Beta-estradiol enhances the production of nerve growth factor in THP-1-derived macrophages or peripheral blood monocyte-derived macrophages. J Invest Dermatol. (2003) 121:771–80. doi: 10.1046/j.1523-1747.2003.12487.x
84. Kanda N, Watanabe S. 17beta-estradiol inhibits oxidative stress-induced apoptosis in keratinocytes by promoting Bcl-2 expression. J Invest Dermatol. (2003) 121:1500–9. doi: 10.1111/j.1523-1747.2003.12617.x
85. Kanda N, Watanabe S. 17beta-estradiol stimulates the growth of human keratinocytes by inducing cyclin D2 expression. J Invest Dermatol. (2004) 123:319–28. doi: 10.1111/j.0022-202X.2004.12645.x
86. Leibowitz D, Dresner-Pollak R, Dvir S, Rokach A, Reznik L, Pollak A. Association of an estrogen receptor-alpha gene polymorphism with left ventricular mass. Blood Press. (2006) 15:45–50. doi: 10.1080/08037050500539569
87. Peter I, Shearman AM, Vasan RS, Zucker DR, Schmid CH, Demissie S, et al. Association of estrogen receptor beta gene polymorphisms with left ventricular mass and wall thickness in women. Am J Hypertens. (2005) 18:1388–95. doi: 10.1016/j.amjhyper.2005.05.023
88. Lawlor DA, Timpson N, Ebrahim S, Day IN, Smith GD. The association of oestrogen receptor alpha-haplotypes with cardiovascular risk factors in the British Women's Heart and Health Study. Eur Heart J. (2006) 27:1597–604. doi: 10.1093/eurheartj/ehi833
89. Kunnas T, Silander K, Karvanen J, Valkeapaa M, Salomaa V, Nikkari S. ESR1 genetic variants, haplotypes and the risk of coronary heart disease and ischemic stroke in the Finnish population: a prospective follow-up study. Atherosclerosis. (2010) 211:200–2. doi: 10.1016/j.atherosclerosis.2010.01.026
90. Schuit SC, Oei HH, Witteman JC, Geurts van Kessel CH, van Meurs JB, Nijhuis RL, et al. Estrogen receptor alpha gene polymorphisms and risk of myocardial infarction. JAMA. (2004) 291:2969–77. doi: 10.1001/jama.291.24.2969
91. Shearman AM, Cupples LA, Demissie S, Peter I, Schmid CH, Karas RH, et al. Association between estrogen receptor alpha gene variation and cardiovascular disease. JAMA. (2003) 290:2263–70. doi: 10.1001/jama.290.17.2263
92. Shearman AM, Cooper JA, Kotwinski PJ, Miller GJ, Humphries SE, Ardlie KG, et al. Estrogen receptor alpha gene variation is associated with risk of myocardial infarction in more than seven thousand men from five cohorts. Circ Res. (2006) 98:590–2. doi: 10.1161/01.RES.0000210578.62102.a6
93. Koch W, Hoppmann P, Pfeufer A, Mueller JC, Schomig A, Kastrati A. No replication of association between estrogen receptor alpha gene polymorphisms and susceptibility to myocardial infarction in a large sample of patients of European descent. Circulation. (2005) 112:2138–42. doi: 10.1161/CIRCULATIONAHA.105.545913
94. Kjaergaard AD, Ellervik C, Tybjaerg-Hansen A, Axelsson CK, Gronholdt ML, Grande P, et al. Estrogen receptor alpha polymorphism and risk of cardiovascular disease, cancer, and hip fracture: cross-sectional, cohort, and case-control studies and a meta-analysis. Circulation. (2007) 115:861–71. doi: 10.1161/CIRCULATIONAHA.106.615567
95. Lucas G, Lluis-Ganella C, Subirana I, Senti M, Willenborg C, Musameh MD, et al. Post-genomic update on a classical candidate gene for coronary artery disease: ESR1. Circ Cardiovasc Genet. (2011) 4:647–54. doi: 10.1161/CIRCGENETICS.111.960583
96. Losordo DW, Kearney M, Kim EA, Jekanowski J, Isner JM. Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation. (1994) 89:1501–10. doi: 10.1161/01.CIR.89.4.1501
97. Post WS, Goldschmidt-Clermont PJ, Wilhide CC, Heldman AW, Sussman MS, Ouyang P, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. (1999) 43:985–91. doi: 10.1016/S0008-6363(99)00153-4
98. Domingues-Montanari S, Subirana I, Tomas M, Marrugat J, Senti M. Association between ESR2 genetic variants and risk of myocardial infarction. Clin Chem. (2008) 54:1183–9. doi: 10.1373/clinchem.2007.102400
99. Rexrode KM, Ridker PM, Hegener HH, Buring JE, Manson JE, Zee RY. Polymorphisms and haplotypes of the estrogen receptor-beta gene (ESR2) and cardiovascular disease in men and women. Clin Chem. (2007) 53:1749–56. doi: 10.1373/clinchem.2007.091454
100. Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res. (2010) 16:2702–8. doi: 10.1158/1078-0432.CCR-09-1753
101. Prossnitz ER, Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol. (2015) 153:114–26. doi: 10.1016/j.jsbmb.2015.06.014
102. Dworatzek E, Mahmoodzadeh S. Targeted basic research to highlight the role of estrogen and estrogen receptors in the cardiovascular system. Pharmacol Res. (2017) 119:27–35. doi: 10.1016/j.phrs.2017.01.019
103. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA. (2000) 97:12729–34. doi: 10.1073/pnas.97.23.12729
104. Arias-Loza PA, Kreissl MC, Kneitz S, Kaiser FR, Israel I, Hu K, et al. The estrogen receptor-alpha is required and sufficient to maintain physiological glucose uptake in the mouse heart. Hypertension. (2012) 60:1070–7. doi: 10.1161/HYPERTENSIONAHA.111.190389
105. Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. (2006) 290:H2204–9. doi: 10.1152/ajpheart.01219.2005
106. Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR. Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol. (2000) 278:H1640–7. doi: 10.1152/ajpheart.2000.278.5.H1640
107. Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, et al. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. (2005) 288:H469–76. doi: 10.1152/ajpheart.00723.2004
108. Babiker FA, Lips D, Meyer R, Delvaux E, Zandberg P, Janssen B, et al. Estrogen receptor {beta} protects the murine heart against left ventricular hypertrophy. Arterioscler Thromb Vasc Biol.(2006) 26:1524–30. doi: 10.1161/01.ATV.0000223344.11128.23
109. Devanathan S, Whitehead T, Schweitzer GG, Fettig N, Kovacs A, Korach KS, et al. An animal model with a cardiomyocyte-specific deletion of estrogen receptor alpha: functional, metabolic, and differential network analysis. PLoS ONE. (2014) 9:e101900. doi: 10.1371/journal.pone.0101900
110. Kararigas G, Nguyen BT, Jarry H. Estrogen modulates cardiac growth through an estrogen receptor alpha-dependent mechanism in healthy ovariectomized mice. Mol Cell Endocrinol. (2014) 382:909–14. doi: 10.1016/j.mce.2013.11.011
111. Ohlsson C, Hellberg N, Parini P, Vidal O, Bohlooly YM, Rudling M, et al. Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem Biophys Res Commun. (2000) 278:640–5. doi: 10.1006/bbrc.2000.3827
112. Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, et al. Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet. (2008) 4:e1000108. doi: 10.1371/journal.pgen.1000108
113. Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA. (1998) 95:15677–82. doi: 10.1073/pnas.95.26.15677
114. Forster C, Kietz S, Hultenby K, Warner M, Gustafsson JA. Characterization of the ERbeta-/-mouse heart. Proc Natl Acad Sci USA. (2004) 101:14234–9. doi: 10.1073/pnas.0405571101
115. Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. (2002) 295:505–8. doi: 10.1126/science.1065250
116. Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, et al. Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R972–8. doi: 10.1152/ajpregu.00045.2009
117. Babiker FA, Lips DJ, Delvaux E, Zandberg P, Janssen BJ, Prinzen F, et al. Oestrogen modulates cardiac ischaemic remodelling through oestrogen receptor-specific mechanisms. Acta Physiol. (2007) 189:23–31. doi: 10.1111/j.1748-1716.2006.01633.x
118. Pelzer T, Loza PA, Hu K, Bayer B, Dienesch C, Calvillo L, et al. Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout mice after myocardial infarction. Circulation. (2005) 111:1492–8. doi: 10.1161/01.CIR.0000159262.18512.46
119. Gurgen D, Hegner B, Kusch A, Catar R, Chaykovska L, Hoff U, et al. Estrogen receptor-beta signals left ventricular hypertrophy sex differences in normotensive deoxycorticosterone acetate-salt mice. Hypertension. (2011) 57:648–54. doi: 10.1161/HYPERTENSIONAHA.110.166157
120. Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. (2010) 298:R1597–606. doi: 10.1152/ajpregu.00825.2009
121. Martensson UE, Salehi SA, Windahl S, Gomez MF, Sward K, Daszkiewicz-Nilsson J, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. (2009) 150:687–98. doi: 10.1210/en.2008-0623
122. Delbeck M, Golz S, Vonk R, Janssen W, Hucho T, Isensee J, et al. Impaired left-ventricular cardiac function in male GPR30-deficient mice. Mol Med Rep. (2011) 4:37–40. doi: 10.3892/mmr.2010.402
123. Meoli L, Isensee J, Zazzu V, Nabzdyk CS, Soewarto D, Witt H, et al. Sex- and age-dependent effects of Gpr30 genetic deletion on the metabolic and cardiovascular profiles of diet-induced obese mice. Gene. (2014) 540:210–6. doi: 10.1016/j.gene.2014.02.036
124. Wang H, Sun X, Chou J, Lin M, Ferrario CM, Zapata-Sudo G, et al. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: a sex-specific gene profiling analysis. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1870–82. doi: 10.1016/j.bbadis.2016.10.003
125. Wang H, Sun X, Chou J, Lin M, Ferrario CM, Zapata-Sudo G, et al. Inflammatory and mitochondrial gene expression data in GPER-deficient cardiomyocytes from male and female mice. Data Brief. (2017) 10:465–73. doi: 10.1016/j.dib.2016.11.057
126. Bers DM. Cardiac excitation-contraction coupling. Nature. (2002) 415:198–205. doi: 10.1038/415198a
127. Marsh JD. Turning cardiac excitation into cell contraction: the importance of sex differences. Am J Physiol Heart Circ Physiol. (2010) 299:H16–7. doi: 10.1152/ajpheart.00444.2010
128. Parks RJ, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling. Pflugers Arch. (2013) 465:747–63. doi: 10.1007/s00424-013-1233-0
129. Feridooni HA, Dibb KM, Howlett SE. How cardiomyocyte excitation, calcium release and contraction become altered with age. J Mol Cell Cardiol. (2015) 83:62–72. doi: 10.1016/j.yjmcc.2014.12.004
130. Buonanno C, Arbustini E, Rossi L, Dander B, Vassanelli C, Paris B, et al. Left ventricular function in men and women. Another difference between sexes. Eur Heart J. (1982) 3:525–8. doi: 10.1093/oxfordjournals.eurheartj.a061347
131. Yarnoz MJ, Curtis AB. More reasons why men and women are not the same (gender differences in electrophysiology and arrhythmias). Am J Cardiol. (2008) 101:1291–6. doi: 10.1016/j.amjcard.2007.12.027
132. Wong ND, Gardin JM, Kurosaki T, Anton-Culver H, Sidney S, Roseman J, et al. Echocardiographic left ventricular systolic function and volumes in young adults: distribution and factors influencing variability. Am Heart J. (1995) 129:571–7. doi: 10.1016/0002-8703(95)90287-2
133. Merz CN, Moriel M, Rozanski A, Klein J, Berman DS. Gender-related differences in exercise ventricular function among healthy subjects and patients. Am Heart J. (1996) 131:704–9. doi: 10.1016/S0002-8703(96)90274-4
134. Verkerk AO, Wilders R, Veldkamp MW, de Geringel W, Kirkels JH, Tan HL. Gender disparities in cardiac cellular electrophysiology and arrhythmia susceptibility in human failing ventricular myocytes. Int Heart J. (2005) 46:1105–18. doi: 10.1536/ihj.46.1105
135. Papp R, Bett GCL, Lis A, Rasmusson RL, Baczko I, Varro A, et al. Genomic upregulation of cardiac Cav1.2alpha and NCX1 by estrogen in women. Biol Sex Differ. (2017) 8:26. doi: 10.1186/s13293-017-0148-4
136. Grandy SA, Howlett SE. Cardiac excitation-contraction coupling is altered in myocytes from aged male mice but not in cells from aged female mice. Am J Physiol Heart Circ Physiol. (2006) 291:H2362–70. doi: 10.1152/ajpheart.00070.2006
137. Howlett SE. Age-associated changes in excitation-contraction coupling are more prominent in ventricular myocytes from male rats than in myocytes from female rats. Am J Physiol Heart Circ Physiol. (2010) 298:H659–70. doi: 10.1152/ajpheart.00214.2009
138. Curl CL, Delbridge LM, Wendt IR. Sex differences in cardiac muscle responsiveness to Ca2+ and L-type Ca2+ channel modulation. Eur J Pharmacol. (2008) 586:288–92. doi: 10.1016/j.ejphar.2008.02.053
139. Curl CL, Wendt IR, Kotsanas G. Effects of gender on intracellular. Pflugers Arch. (2001) 441:709–16. doi: 10.1007/s004240000473
140. Farrell SR, Ross JL, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. (2010) 299:H36–45. doi: 10.1152/ajpheart.00299.2010
141. Leblanc N, Chartier D, Gosselin H, Rouleau JL. Age and gender differences in excitation-contraction coupling of the rat ventricle. J Physiol. (1998) 511 (Pt 2):533–48. doi: 10.1111/j.1469-7793.1998.533bh.x
142. Wasserstrom JA, Kapur S, Jones S, Faruque T, Sharma R, Kelly JE, et al. Characteristics of intracellular Ca2+ cycling in intact rat heart: a comparison of sex differences. Am J Physiol Heart Circ Physiol. (2008) 295:H1895–904. doi: 10.1152/ajpheart.00469.2008
143. Schwertz DW, Beck JM, Kowalski JM, Ross JD. Sex differences in the response of rat heart ventricle to calcium. Biol Res Nurs. (2004) 5:286–98. doi: 10.1177/1099800403262615
144. Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. (2005) 115:3306–17. doi: 10.1172/JCI27167
145. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. (2008) 70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455
146. Benitah JP, Alvarez JL, Gomez AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. (2010) 48:26–36. doi: 10.1016/j.yjmcc.2009.07.026
147. Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G. Sex, age, and regional differences in L-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long QT type 2. Circ Res. (2008) 102:e86–100. doi: 10.1161/CIRCRESAHA.108.173740
148. Tappia PS, Dent MR, Aroutiounova N, Babick AP, Weiler H. Gender differences in the modulation of cardiac gene expression by dietary conjugated linoleic acid isomers. Can J Physiol Pharmacol. (2007) 85:465–75. doi: 10.1139/Y06-104
149. Chu SH, Sutherland K, Beck J, Kowalski J, Goldspink P, Schwertz D. Sex differences in expression of calcium-handling proteins and beta-adrenergic receptors in rat heart ventricle. Life Sci. (2005) 76:2735–49. doi: 10.1016/j.lfs.2004.12.013
150. Xiao L, Zhang L, Han W, Wang Z, Nattel S. Sex-based transmural differences in cardiac repolarization and ionic-current properties in canine left ventricles. Am J Physiol Heart Circ Physiol. (2006) 291:H570–80. doi: 10.1152/ajpheart.01288.2005
151. Mason SA, MacLeod KT. Cardiac action potential duration and calcium regulation in males and females. Biochem Biophys Res Commun. (2009) 388:565–70. doi: 10.1016/j.bbrc.2009.08.050
152. Vizgirda VM, Wahler GM, Sondgeroth KL, Ziolo MT, Schwertz DW. Mechanisms of sex differences in rat cardiac myocyte response to beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. (2002) 282:H256–63. doi: 10.1152/ajpheart.2002.282.1.H256
153. James AF, Arberry LA, Hancox JC. Gender-related differences in ventricular myocyte repolarization in the guinea pig. Basic Res Cardiol. (2004) 99:183–92. doi: 10.1007/s00395-003-0451-6
154. Grandy SA, Denovan-Wright EM, Ferrier GR, Howlett SE. Overexpression of human beta2-adrenergic receptors increases gain of excitation-contraction coupling in mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. (2004) 287:H1029–38. doi: 10.1152/ajpheart.00814.2003
155. Parks RJ, Ray G, Bienvenu LA, Rose RA, Howlett SE. Sex differences in SR Ca(2+) release in murine ventricular myocytes are regulated by the cAMP/PKA pathway. J Mol Cell Cardiol. (2014) 75:162–73. doi: 10.1016/j.yjmcc.2014.07.006
156. Crump SM, Andres DA, Sievert G, Satin J. The cardiac L-type calcium channel distal carboxy terminus autoinhibition is regulated by calcium. Am J Physiol Heart Circ Physiol. (2013) 304:H455–64. doi: 10.1152/ajpheart.00396.2012
157. Fu Y, Westenbroek RE, Yu FH, Clark JP 3rd, Marshall MR, Scheuer T, et al. Deletion of the distal C terminus of CaV1.2 channels leads to loss of beta-adrenergic regulation and heart failure in vivo. J Biol Chem. (2011) 286:12617–26. doi: 10.1074/jbc.M110.175307
158. Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, et al. C-terminal fragments of the alpha 1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated alpha 1C subunits. J Biol Chem. (2001) 276:21089–97. doi: 10.1074/jbc.M008000200
159. Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, Ma H, et al. Proteolytic processing of the C terminus of the alpha(1C) subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J Biol Chem. (2000) 275:8556–63. doi: 10.1074/jbc.275.12.8556
160. Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. (2006) 127:591–606. doi: 10.1016/j.cell.2006.10.017
161. Schroder E, Byse M, Satin J. L-type calcium channel C terminus autoregulates transcription. Circ Res. (2009) 104:1373–81. doi: 10.1161/CIRCRESAHA.108.191387
162. Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. (2006) 576 (Pt 1):87–102. doi: 10.1113/jphysiol.2006.111799
163. Bannister JP, Leo MD, Narayanan D, Jangsangthong W, Nair A, Evanson KW, et al. The voltage-dependent L-type Ca2+ (CaV1.2) channel C-terminus fragment is a bi-modal vasodilator. J Physiol. (2013) 591:2987–98. doi: 10.1113/jphysiol.2013.251926
164. Mahmoodzadeh S, Haase H, Sporbert A, Rharass T, Panakova D, Morano I. Nuclear translocation of the cardiac L-type calcium channel C-terminus is regulated by sex and 17beta-estradiol. J Mol Cell Cardiol. (2016) 97:226–34. doi: 10.1016/j.yjmcc.2016.06.004
165. Chen G, Yang X, Alber S, Shusterman V, Salama G. Regional genomic regulation of cardiac sodium-calcium exchanger by oestrogen. J Physiol. (2011) 589 (Pt 5):1061–80. doi: 10.1113/jphysiol.2010.203398
166. Yaras N, Tuncay E, Purali N, Sahinoglu B, Vassort G, Turan B. Sex-related effects on diabetes-induced alterations in calcium release in the rat heart. Am J Physiol Heart Circ Physiol. (2007) 293:H3584–92. doi: 10.1152/ajpheart.00619.2007
167. Bell JR, Raaijmakers AJ, Curl CL, Reichelt ME, Harding TW, Bei A, et al. Cardiac CaMKIIdelta splice variants exhibit target signaling specificity and confer sex-selective arrhythmogenic actions in the ischemic-reperfused heart. Int J Cardiol. (2015) 181:288–96. doi: 10.1016/j.ijcard.2014.11.159
168. Patterson E, Ma L, Szabo B, Robinson CP, Thadani U. Ovariectomy and estrogen-induced alterations in myocardial contractility in female rabbits: role of the L-type calcium channel. J Pharmacol Exp Ther. (1998) 284:586–91.
169. Ren J, Hintz KK, Roughead ZK, Duan J, Colligan PB, Ren BH, et al. Impact of estrogen replacement on ventricular myocyte contractile function and protein kinase B/Akt activation. Am J Physiol Heart Circ Physiol. (2003) 284:H1800–7. doi: 10.1152/ajpheart.00866.2002
170. Fares E, Parks RJ, Macdonald JK, Egar JM, Howlett SE. Ovariectomy enhances SR Ca(2)(+) release and increases Ca(2)(+) spark amplitudes in isolated ventricular myocytes. J Mol Cell Cardiol. (2012) 52:32–42. doi: 10.1016/j.yjmcc.2011.09.002
171. Fares E, Pyle WG, Ray G, Rose RA, Denovan-Wright EM, Chen RP, et al. The impact of ovariectomy on calcium homeostasis and myofilament calcium sensitivity in the aging mouse heart. PLoS ONE. (2013) 8:e74719. doi: 10.1371/journal.pone.0074719
172. Turdi S, Huff AF, Pang J, He EY, Chen X, Wang S, et al. 17-beta estradiol attenuates ovariectomy-induced changes in cardiomyocyte contractile function via activation of AMP-activated protein kinase. Toxicol Lett. (2015) 232:253–62. doi: 10.1016/j.toxlet.2014.11.012
173. Parks RJ, Bogachev O, Mackasey M, Ray G, Rose RA, Howlett SE. The impact of ovariectomy on cardiac excitation-contraction coupling is mediated through cAMP/PKA-dependent mechanisms. J Mol Cell Cardiol. (2017) 111:51–60. doi: 10.1016/j.yjmcc.2017.07.118
174. Yang HY, Firth JM, Francis AJ, Alvarez-Laviada A, MacLeod KT. Effect of ovariectomy on intracellular Ca(2+) regulation in guinea pig cardiomyocytes. Am J Physiol Heart Circ Physiol. (2017) 313:H1031–43. doi: 10.1152/ajpheart.00249.2017
175. Curl CL, Wendt IR, Canny BJ, Kotsanas G. Effects of ovariectomy and 17 beta-oestradiol replacement on [Ca2+]i in female rat cardiac myocytes. Clin Exp Pharmacol Physiol. (2003) 30:489–94. doi: 10.1046/j.1440-1681.2003.03864.x
176. Kravtsov GM, Kam KW, Liu J, Wu S, Wong TM. Altered Ca(2+) handling by ryanodine receptor and Na(+)-Ca(2+) exchange in the heart from ovariectomized rats: role of protein kinase A. Am J Physiol Cell Physiol. (2007) 292:C1625–35. doi: 10.1152/ajpcell.00368.2006
177. Bupha-Intr T, Wattanapermpool J. Regulatory role of ovarian sex hormones in calcium uptake activity of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. (2006) 291:H1101–8. doi: 10.1152/ajpheart.00660.2005
178. Wattanapermpool J, Riabroy T, Preawnim S. Estrogen supplement prevents the calcium hypersensitivity of cardiac myofilaments in ovariectomized rats. Life Sci. (2000) 66:533–43. doi: 10.1016/S0024-3205(99)00623-2
179. Chu SH, Goldspink P, Kowalski J, Beck J, Schwertz DW. Effect of estrogen on calcium-handling proteins, beta-adrenergic receptors, and function in rat heart. Life Sci. (2006) 79:1257–67. doi: 10.1016/j.lfs.2006.03.037
180. Johnson BD, Zheng W, Korach KS, Scheuer T, Catterall WA, Rubanyi GM. Increased expression of the cardiac L-type calcium channel in estrogen receptor-deficient mice. J Gen Physiol. (1997) 110:135–40. doi: 10.1085/jgp.110.2.135
181. Fang HY, Hung MY, Lin YM, Pandey S, Chang CC, Lin KH, et al. 17beta-Estradiol and/or estrogen receptor alpha signaling blocks protein phosphatase 1 mediated ISO induced cardiac hypertrophy. PLoS ONE. (2018) 13:e0196569. doi: 10.1371/journal.pone.0196569
182. Ullrich ND, Krust A, Collins P, MacLeod KT. Genomic deletion of estrogen receptors ERalpha and ERbeta does not alter estrogen-mediated inhibition of Ca2+ influx and contraction in murine cardiomyocytes. Am J Physiol Heart Circ Physiol. (2008) 294:H2421–7. doi: 10.1152/ajpheart.01225.2007
183. Pelzer T, Jazbutyte V, Hu K, Segerer S, Nahrendorf M, Nordbeck P, et al. The estrogen receptor-alpha agonist 16alpha-LE2 inhibits cardiac hypertrophy and improves hemodynamic function in estrogen-deficient spontaneously hypertensive rats. Cardiovasc Res. (2005) 67:604–12. doi: 10.1016/j.cardiores.2005.04.035
184. Kulpa J, Chinnappareddy N, Pyle WG. Rapid changes in cardiac myofilament function following the acute activation of estrogen receptor-alpha. PLoS ONE. (2012) 7:e41076. doi: 10.1371/journal.pone.0041076
185. Alencar AK, da Silva JS, Lin M, Silva AM, Sun X, Ferrario CM, et al. Effect of age, estrogen status, and late-life GPER activation on cardiac structure and function in the fischer344xbrown norway female rat. J Gerontol A Biol Sci Med Sci. (2017) 72:152–62. doi: 10.1093/gerona/glw045
186. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. (2018) 122:1460–78. doi: 10.1161/CIRCRESAHA.118.310082
187. Colom B, Alcolea MP, Valle A, Oliver J, Roca P, Garcia-Palmer FJ. Skeletal muscle of female rats exhibit higher mitochondrial mass and oxidative-phosphorylative capacities compared to males. Cell Physiol Biochem. (2007) 19:205–12. doi: 10.1159/000099208
188. Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med. (2003) 34:546–52. doi: 10.1016/S0891-5849(02)01356-4
189. Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. (2010) 106:1681–91. doi: 10.1161/CIRCRESAHA.109.213645
190. Kander MC, Cui Y, Liu Z. Gender difference in oxidative stress: a new look at the mechanisms for cardiovascular diseases. J Cell Mol Med. (2017) 21:1024–32. doi: 10.1111/jcmm.13038
191. Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. (2008) 321:1493–5. doi: 10.1126/science.1158554
192. Vijay V, Han T, Moland CL, Kwekel JC, Fuscoe JC, Desai VG. Sexual dimorphism in the expression of mitochondria-related genes in rat heart at different ages. PLoS ONE. (2015) 10:e0117047. doi: 10.1371/journal.pone.0117047
193. Lancaster TS, Jefferson SJ, Hunter JC, Lopez V, Van Eyk JE, Lakatta EG, et al. Quantitative proteomic analysis reveals novel mitochondrial targets of estrogen deficiency in the aged female rat heart. Physiol Genomics. (2012) 44:957–69. doi: 10.1152/physiolgenomics.00184.2011
194. Chen Y, Zhang Z, Hu F, Yang W, Yuan J, Cui J, et al. 17beta-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: a preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J Steroid Biochem Mol Biol. (2015) 147:92–102. doi: 10.1016/j.jsbmb.2014.12.011
195. Zhai P, Eurell TE, Cotthaus R, Jeffery EH, Bahr JM, Gross DR. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am J Physiol Heart Circ Physiol. (2000) 279:H2766–75. doi: 10.1152/ajpheart.2000.279.6.H2766
196. Liou CM, Yang AL, Kuo CH, Tin H, Huang CY, Lee SD. Effects of 17beta-estradiol on cardiac apoptosis in ovariectomized rats. Cell Biochem Funct. (2010) 28:521–8. doi: 10.1002/cbf.1687
197. Pelzer T, Shamim A, Wolfges S, Schumann M, Neyses L. Modulation of cardiac hypertrophy by estrogens. Adv Exp Med Biol. (1997) 432:83–9. doi: 10.1007/978-1-4615-5385-4_9
198. Jazbutyte V, Kehl F, Neyses L, Pelzer T. Estrogen receptor alpha interacts with 17beta-hydroxysteroid dehydrogenase type 10 in mitochondria. Biochem Biophys Res Commun. (2009) 384:450–4. doi: 10.1016/j.bbrc.2009.04.139
199. Chen BC, Weng YJ, Shibu MA, Han CK, Chen YS, Shen CY, et al. Estrogen and/or estrogen receptor alpha inhibits BNIP3-induced apoptosis and autophagy in H9c2 cardiomyoblast cells. Int J Mol Sci. (2018) 19:E1298. doi: 10.3390/ijms19051298
200. Demonacos CV, Karayanni N, Hatzoglou E, Tsiriyiotis C, Spandidos DA, Sekeris CE. Mitochondrial genes as sites of primary action of steroid hormones. Steroids. (1996) 61:226–32. doi: 10.1016/0039-128X(96)00019-0
201. Sekeris CE. The mitochondrial genome: a possible primary site of action of steroid hormones. In Vivo. (1990) 4:317–20.
202. Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol. (2008) 22:609–22. doi: 10.1210/me.2007-0029
203. Bettini E, Maggi A. Estrogen induction of cytochrome c oxidase subunit III in rat hippocampus. J Neurochem. (1992) 58:1923–9. doi: 10.1111/j.1471-4159.1992.tb10070.x
204. Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. (2004) 93:358–73. doi: 10.1002/jcb.20178
205. Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, et al. Sex-specific pathways in early cardiac response to pressure overload in mice. J Mol Med. (2008) 86:1013–24. doi: 10.1007/s00109-008-0385-4
206. Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. (2005) 68:959–65. doi: 10.1124/mol.105.014662
207. Hsieh YC, Choudhry MA, Yu HP, Shimizu T, Yang S, Suzuki T, et al. Inhibition of cardiac PGC-1alpha expression abolishes ERbeta agonist-mediated cardioprotection following trauma-hemorrhage. Faseb J. (2006) 20:1109–17. doi: 10.1096/fj.05-5549com
208. Hsieh YC, Yang S, Choudhry MA, Yu HP, Rue LW 3rd, Bland KI, et al. PGC-1 upregulation via estrogen receptors: a common mechanism of salutary effects of estrogen and flutamide on heart function after trauma-hemorrhage. Am J Physiol Heart Circ Physiol. (2005) 289:H2665–72. doi: 10.1152/ajpheart.00682.2005
209. O'Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, et al. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol. (2007) 21:1281–96. doi: 10.1210/me.2006-0497
210. Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2010) 298:H16–23. doi: 10.1152/ajpheart.00588.2009
211. Kurian GA, Rajagopal R, Vedantham S, Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid Med Cell Longev. (2016) 2016:1656450. doi: 10.1155/2016/1656450
212. Arieli Y, Gursahani H, Eaton MM, Hernandez LA, Schaefer S. Gender modulation of Ca(2+) uptake in cardiac mitochondria. J Mol Cell Cardiol. (2004) 37:507–13. doi: 10.1016/j.yjmcc.2004.04.023
213. Morkuniene R, Arandarcikaite O, Borutaite V. Estradiol prevents release of cytochrome c from mitochondria and inhibits ischemia-induced apoptosis in perfused heart. Exp Gerontol. (2006) 41:704–8. doi: 10.1016/j.exger.2006.02.010
214. Morkuniene R, Arandarcikaite O, Ivanoviene L, Borutaite V. Estradiol-induced protection against ischemia-induced heart mitochondrial damage and caspase activation is mediated by protein kinase G. Biochim Biophys Acta. (2010) 1797:1012–7. doi: 10.1016/j.bbabio.2010.03.027
215. Morkuniene R, Jekabsone A, Borutaite V. Estrogens prevent calcium-induced release of cytochrome c from heart mitochondria. FEBS Lett. (2002) 521:53–6. doi: 10.1016/S0014-5793(02)02820-X
216. Feng Y, Madungwe NB, da Cruz Junho CV, Bopassa JC. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol. (2017) 174:4329–44. doi: 10.1111/bph.14033
Keywords: estrogen, estrogen receptor, G-protein-coupled estrogen receptor, cardiomyocytes, sex difference, cardiac mitochondrial function, cardiac Ca2+ ion channel
Citation: Mahmoodzadeh S and Dworatzek E (2019) The Role of 17β-Estradiol and Estrogen Receptors in Regulation of Ca2+ Channels and Mitochondrial Function in Cardiomyocytes. Front. Endocrinol. 10:310. doi: 10.3389/fendo.2019.00310
Received: 14 March 2019; Accepted: 30 April 2019;
Published: 15 May 2019.
Edited by:
Dawn A. Lowe, University of Minnesota Twin Cities, United StatesCopyright © 2019 Mahmoodzadeh and Dworatzek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shokoufeh Mahmoodzadeh, c2hva291ZmVoLm1haG1vb2R6YWRlaEBtZGMtYmVybGluLmRl