 Miaolong Lu
Miaolong Lu Ya Wang
Ya Wang Xianquan Zhan
Xianquan Zhan- 1Key Laboratory of Cancer Proteomics of Chinese Ministry of Health, Xiangya Hospital, Central South University, Changsha, China
- 2Hunan Engineering Laboratory for Structural Biology and Drug Design, Xiangya Hospital, Central South University, Changsha, China
- 3State Local Joint Engineering Laboratory for Anticancer Drugs, Xiangya Hospital, Central South University, Changsha, China
- 4National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
Mitogen-activated protein kinases (MAPKs) include ERK, p38, and JNK MAPK subfamilies, which are crucial regulators of cellular physiology, cell pathology, and many diseases including cancers. For the MAPK signaling system in pituitary adenomas (PAs), the activation of ERK signaling is generally thought to promote cell proliferation and growth; whereas the activations of p38 and JNK signaling are generally thought to promote cell apoptosis. The role of MAPK in treatment of PAs is demonstrated through the effects of currently used medications such as somatostatin analogs such as SOM230 and OCT, dopamine agonists such as cabergoline and bromocriptine, and retinoic acid which inhibit the MAPK pathway. Further, there are potential novel therapies based on putative molecular targets of the MAPK pathway, including 18beta-glycyrrhetinic acid (GA), dopamine-somatostatin chimeric compound (BIM-23A760), ursolic acid (UA), fulvestrant, Raf kinase inhibitory protein (RKIP), epidermal growth factor pathway substrate number 8 (Eps8), transmembrane protein with EGF-like and two follistatin-like domains (TMEFF2), cold inducible RNA-binding protein (CIRP), miR-16, and mammaliansterile-20-like kinase (MST4). The combined use of ERK inhibitor (e.g., SOM230, OCT, or dopamine) plus p38 activator (e.g., cabergoline, bromocriptine, and fulvestrant) and/or JNK activator (e.g., UA), or the development of single drug (e.g., BIM-23A760) to target both ERK and p38 or JNK pathways, might produce better anti-tumor effects on PAs. This article reviews the advances in understanding the role of MAPK signaling in pituitary tumorigenesis, and the MAPK pathway-based potential therapeutic drugs for PAs.
Introduction
Pituitary adenomas (PAs) are commonly benign tumors, accounting for about ten percent of intracranial tumors (1, 2), and are clinically divided into functioning PAs (FPAs) and non-functioning PAs (NFPAs) (3, 4). It can cause significant morbidity and mortality (5). The molecular mechanisms in tumorigenesis and functional regulation of PAs have been extensively studied. This review article focuses on the roles of mitogen-activated protein kinase (MAPK) in PA tumorigenesis and the MAPK pathway-based potential therapeutic targets for PAs. MAPKs mainly include three subfamilies based on the conserved Thr-Xaa-Tyr motif signature: ERK1/2, p38, and JNK (Jun N-terminal kinase) (6), which are activated by multiple factors such as growth factors and stress. The activation of ERK promotes cell proliferation; whereas, the activations of p38 and JNK promote cell apoptosis. Studies demonstrate that MAPKs are involved in multiple cellular processes, such as cell differentiation, proliferation, apoptosis, inflammation, stress responses, and immune defense (7–9).
The MAPK signaling pathways play important roles in cell dissemination, survival, and drug resistance of human cancers including PAs (2, 10–12). With the in-depth studies of the MAPK signaling pathway network, MAPK pathways-based target-specific drugs have been developed, and some drugs has been used for clinical trials; and the relevance of MAPK in response and resistance to antitumor drugs has also been recognized (Figure 1 and Table 1). Because of the important roles of MAPK signaling pathways in tumorigenesis, the use of the MAPK signaling pathways as therapeutic targets has continuously been considered as a promising strategy for cancer therapy. This review highlights new advances in the role of MAPK signaling in pituitary tumorigenesis and development, the key molecules in this pathway network, and anti-pituitary tumor drugs targeting MAPK signaling pathway.
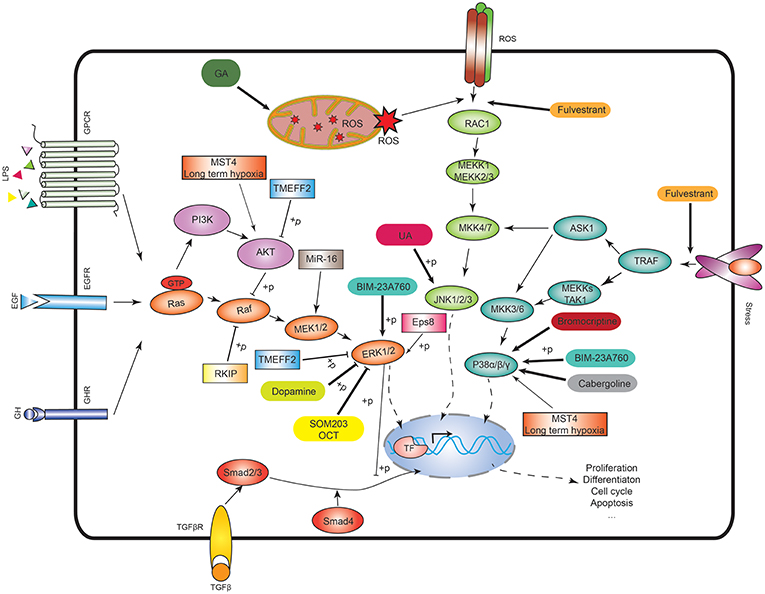
Figure 1. MAPK signaling pathways and the potential therapeutic targets. In the ERK signaling, Ras activates the serine/threonine protein kinase Raf to activate MEK1/2, then MEK1/2 phosphorylates the ERK1/2. In p38 signaling, TNF receptor-associated factor (TRAF) activates ASK1, TAK1, or MEKK1, which activates MKK3/6, and then MKK3/6 phosphorylates p38 isoforms. In JNK signaling, RAC1 activates MEKK1 or MEKK2/3 to activate MKK4/7, and then MKK4/7 phosphorylates JNK1/2/3. The ASK1 in the p38 signaling also activates MKK4/7 to crosstalk with JNK signaling. ROS means reactive oxygen species. GA means 18beta-glycyrrhetinic acid. BIM-23A760 is a dopamine–somatostatin chimeric compound. OCT means octreotide. SOM230 and OCT are somatostatin analogs. Rectangle means the potential drug targets.
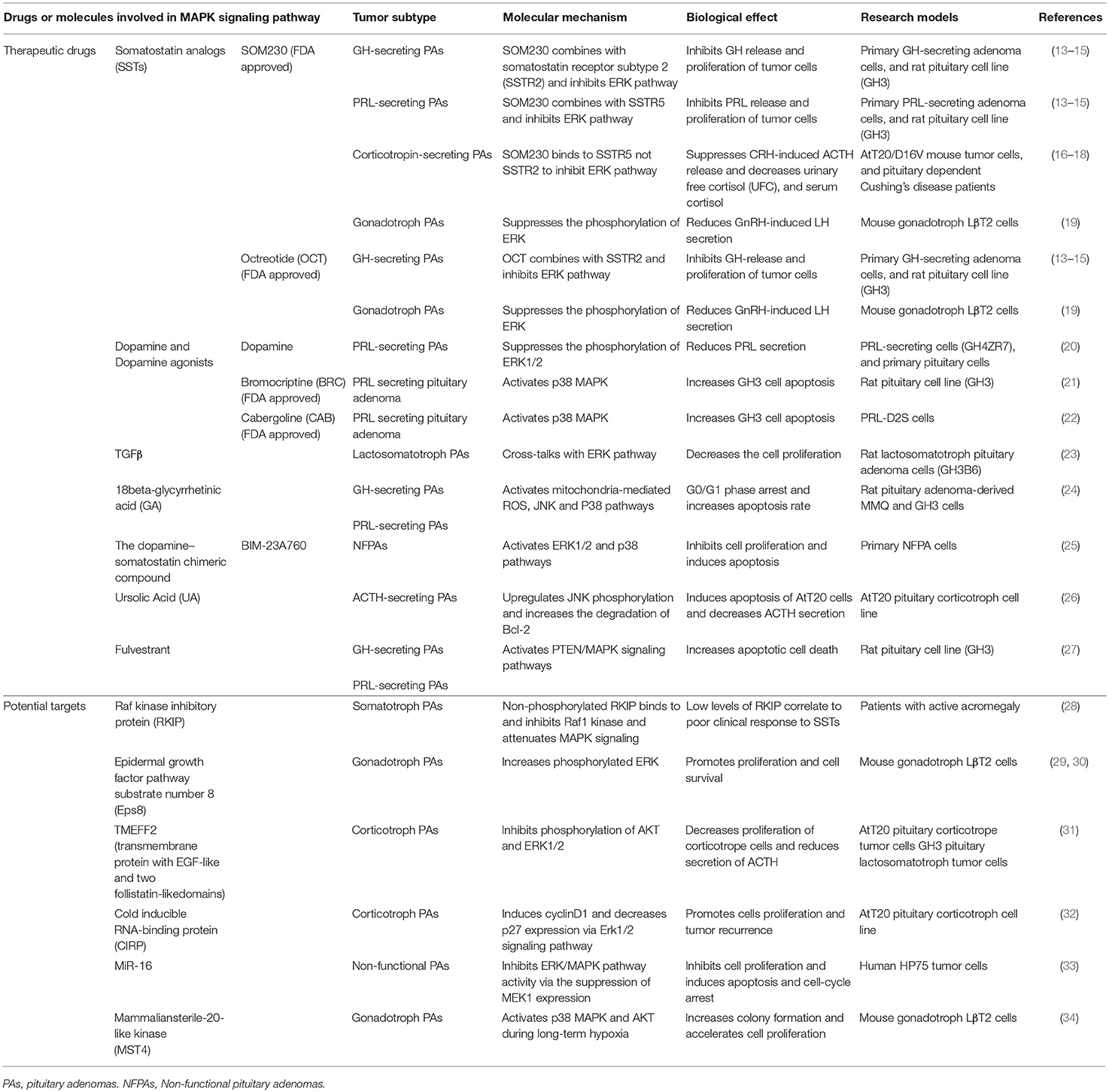
Table 1. Drugs or molecules involved in MAPK signaling pathway in PAs.
The ERK Pathway in PAs
The ERK/MAPK pathway delivers signals from cellular surface receptors via ERK pathway. Briefly, different cellular surface receptors such as EGFR, GPCR, and RKT are activated by the corresponding extracellular factors (e.g., growth factors, hormones, and stresses) to activate Ras and small GTPase. The activated Ras-small GTPase complex recruits Raf kinase to the cell membrane and activates it. Then, Raf activates MEK (MAPK and ERK kinase) through phosphorylation. The phosphorylated MEK subsequently activates ERK through phosphorylation (35) (Figure 1). Rafs include Raf-1, A-Raf, and B-Raf. Raf-1 can bind to the pro-apoptotic kinases, such as mammalian sterile-twenty-like-2 (MST2) and apoptosis signal-regulating kinase (ASK1), to involve in cell apoptosis (36). Raf-1 also exerts scaffolding function in regulation of the Rho pathway (37). Typically, cytokines and growth factors binding to TKR activate ERK1/2, which transduces the signals into its upstream Ras/Raf/MEK pathway. In PAs, H-Ras mutations have been identified in two cases of prolactinomas, which indicates that Ras/ERK takes part in regulation of PAs (38, 39). Overexpression of B-Raf is pre-dominantly observed in NFPAs (40). The downstream kinases of B-Raf in ERK MAPK pathway are also over-activated in NFPAs, growth hormone (GH)-secreting PAs, ACTH-secreting PAs, and prolactinomas. The phosphorylation levels at pSer217/221 of MEK 1/2 and pThr183 of ERK1/2 are significantly increased in these PAs compared to controls (41), which indicates that Raf/MEK/ERK pathway acts as a pro-proliferative role in PAs.
The Effects of ERK MAPK on Different-Origin PAs
The effect of ERK signaling on PAs depends on the PA subtypes. (i) In lactotroph cells, the ERK signaling exerts different effect on cell proliferation based on the exposure time. Short-time activation of the ERK (24–96 h) leads to increased proliferation in rat pituitary lactotroph or somatolactotroph cell lines in vitro (42, 43). However, long-time activation of ERK (over 6 days) promotes somatolactotroph cell differentiation into a lactotroph cell phenotype, and then decreases proliferation and tumorigenicity with time (44). Thus, persistent activation of ERK signaling produces anti-proliferative and anti-tumorigenic effects in somatolactotroph cells. (ii) In somatotroph cells, ERK signaling produces pro-proliferative effects. Protein kinase A (PKA) and C (PKC) pathways regulate ERK signaling. PKA pathway activates ERK signaling, and leads to improved proliferation in GH-secreting cells. PKC stimulates ERK signaling and increases cell proliferation through regulating GH-releasing hormone (GHRH) (45). ERK pathway is necessary for somatotrophs to produce GH. In somatotroph PAs, GH-releasing hormone (GHRH) can promote cell proliferation through activating ERK signaling (46). In addition to regulation of cell proliferation, ERK signaling also contributes to GH secretion by somatotrophs (47). Somatostatin (SST) analogs are used in clinical treatment of GH-secreting PAs due to its anti-proliferative effect on somatotroph cells. SST treatment results in a reduction of pERK1/2 expression and a significant increase in p27 protein expression. In addition, cell proliferation is driven by cell cycle which is regulated by a series of cyclins and cyclin dependent kinases (CDKs). A cyclin-dependent kinase (CDK) inhibitor has a negative effect on cell-cycle progression which has a synergistic effect with SST analogs (13). (iii) In gonadotroph cells, gonadotropin-releasing hormone (GnRH) can activate ERK, p38, and JNK signaling in the LβT2 gonadotroph cell lines to contribute to production of luteinizing hormone (LH), and GnRH phosphorylated ERK via PKC-dependent pathways (48). Most of NFPAs originate from gonadotroph cells where B-Raf is upregulated and ERK is over-activated relative to control pituitary tissues (40, 49). (iv) In thyrotroph cells, ERK cascade has anti-proliferative effects. The ERK pathway is activated to cause growth arrest after thyrotroph adenomas are treated with thyroid hormone (50). And (v) In corticotroph cells, ERK signaling is activated to produce pro-proliferative effects (51).
ERK MAPK Pathway-Targeted Pharmacological Treatments of PAs
Somatostatin (SST) Analogs Treatment
SST inhibits cell growth, through G protein-coupled receptors to inhibit the release of growth factors and angiogenesis, and increases apoptosis. The majority of NFPAs express SST receptors on cell membranes. An appropriate concentration of SST analogs (octreotide or SOM230) can inhibit the release of GH, prolactin (PRL), and their α-subunit in GH-secreting PAs, PRL-secreting PAs, ACTH-secreting PAs, and NFPAs, respectively (14–18). Their anti-tumor effects are in that SST analogs can inactivate ERK signaling pathways; for example, octreotide acts on both ERK and PI3K/Akt signaling pathways, and SOM230 acts on ERK signaling pathway (13, 52). Octreotide can bind to and activate SST receptor subtype-2 (SSTR2) and SSTR5, while pasireotide (SOM230) can activate SSTR1, 2, 3, and 5 (53, 54). A study shows that octreotide or SOM230 reduces cell proliferation and pERK1/2 expression in rat somatotroph cell line GH3 (13). Octreotide also blocks the transient G0/G1 cell cycle to produce a cytostatic effect on GH3 cell proliferation (55). SST analogs (octreotide and pasireotide) also decrease secretion of LH induced by GnRH in LβT2 cells (19), and inhibit NFPA cell viability in vitro (56–59). The SST analogs are the primary medical therapy to treat acromegaly for maintenance of GH homeostasis and shrinkage of tumor size (60–62). Moreover, octreotide and lanreotide bind with high affinity to SSTR2, and with low affinity to SSTR3 and SSTR5. The decreased expression of SSTR2 in tumor is associated with lack of response to SST analogs (63, 64). Some studies demonstrate that SST analogs exert their anti-proliferative effects on somatotroph cells through inhibition of ERK signaling (13).
Dopamine and Dopamine Agonists Treatment
Hypothalamic dopamine suppresses the production of pituitary PRL (65). Dopamine acts via the D2 receptor to inhibit cAMP/PKA and MAPK signaling pathways to control PRL-secretion and lactotroph proliferation (20). Dopamine agonists such as bromocriptine (BRC) and cabergoline (CAB) are primary medical therapy drugs for prolactinomas and idiopathic hyperprolactinemia and prolactinomas (66). Dopamine agonists target the dopamine D2 receptor (D2R) subtype to exert its anti-tumor effects. D2R-activated ERK signaling cascades inhibit the synthesis and release of PRL in the pituitary. D2R includes D2L and D2S isoforms. Overexpression of D2L elevates PRL, and overexpression of D2S reduces PRL. The ratio of D2L to D2S affects PRL-secretion of lactotroph cells (67). A study shows that when pituitary tumor cells are treated with dopamine, D2S activation stimulates ERK signaling to inhibit lactotroph cell proliferation (68).
TGFβ Treatment
TGFβ is widely considered to be a tumor suppressor (69). However, TGFβ1 produces weak growth inhibitory effect on pituitary tumor cells. TGFβ mainly uses Smad signaling pathway to convey signals from cytosol to nucleus to regulate expression of genes that control cell cycle progression (70). In addition, TGFβ can also use non-Smad signaling pathway to convey signals, such as MAPK pathway (71). TGFβ1 treatment decreases pituitary tumor cell proliferation, and this inhibitory effect is amplified by MEK inhibitors, because TGFβ1/Smad pathway cross-talks with MEK/ERK1/2 pathway (23). It clearly demonstrates that inhibition of MEK/ERK1/2 pathway synergizes with TGFβ1 to inhibit pituitary tumor cell proliferation.
Potential Targets Related to ERK MAPK Signaling Pathway for PA Treatment
Components and regulators of the ERK MAPK pathway are all potential targets for treating pituitary tumors. (i) Raf kinase inhibitory protein (RKIP). RKIP is a modulator of MAPK signaling, which inhibits Raf-1 phosphorylation to inhibit Ras/Raf-1/MEK/ERK signaling pathway (72–74). RKIP interferes with Raf-1 in several mechanisms. One is that Raf-1 binding to RKIP causes the conformational change of Raf-1 (75). Another is that RKIP inhibits phosphorylation of MAPK-MEK-1 to interfere with the interaction between two kinases (76). Moreover, protein kinase C (PKC) can negatively regulate the roles of RKIP because PKC phosphorylates RKIP to cause the separation of RKIP from Raf-1 (73). Studies demonstrate that the low expression level of RKIP in GH-secreting adenomas is correlated with less GH and IGF-1 reduction with SST analog therapy because RKIP can inhibit the phosphorylation of Raf1 kinase to attenuate the activity of MAPK signaling pathway (28).
(ii) Epidermal growth factor pathway substrate number 8 (Eps8). Over-expression of Eps8 and over-activation of Raf, MEK, and ERK in the ERK signaling pathway promote cell proliferation and survival in PAs (41). Also, Eps8 is a substrate of receptor tyrosine kinases (RTKs) in the ERK signaling pathway (Figure 1), which can enhance EGF-dependent mitogenic signaling (29). Eps8 expression is significantly higher in human gonadotroph adenomas relative to controls. Upregulation of EGFR protein and phosphorylation of ERK are demonstrated in Eps8-overexpressing LβT2 cells. EGF ligand stimulation leads to increased proliferation in Eps8-overexpressing LβT2 cells. MAPK kinase inhibitor (PD98059) can abrogate the proliferative effects. Silence of Eps8 also inhibits cell proliferation, which suggests that Eps8 promotes pituitary tumor cell proliferation through enhancing the Raf/MEK/ERK signaling (30). Therefore, Eps8 is a potential drug target for PA treatment.
(iii) Retinoic acid (RA). RA has antiproliferative effect in corticotroph cell, and long-term treatment with RA has some clinical efficacy in patients with Cushing's disease (77). The mechanism of RA anti-tumor effects is shown with the expression of TMEFF2 (transmembrane protein with EGF-like and two follistatin-likedomains) that inhibits phosphorylation of AKT and ERK1/2 (31). TMEFF2 is significantly downregulated in corticotropinomas relative to control tissues, which suggests that TMEFF2 might be a tumor suppressor. Silence of TMEFF2 in pituitary corticotroph cell line AtT20 promotes cell proliferation, while over-expression of TMEFF2 inhibits cell proliferation (31). Thus, TMEFF2 is a potential therapeutic target for ACTH-secreting adenomas.
(iv) Cold inducible RNA binding protein (CIRP). The underlying mechanism of cold-shock protein (CIRP) in its role in tumorigenesis is through its induction of cyclinD1 which decreases p27 expression via ERK1/2 signaling (32, 78). CIRP is significantly upregulated at the mRNA and protein levels in multiple cancers (79, 80), including human corticotroph adenomas relative to normal pituitary tissues. CIRP over-expression is associated with recurrence of corticotroph adenomas in murine models (32). CIRP-overexpressing AtT20 cells have increased cell proliferative abilities. Tumor xenografts generated by CIRP-overexpressed AtT20 cells are significantly larger than AtT20 cells with normal level of CIRP expression.
The p38 Pathway in PAs
The p38 MAPK includes isoforms p38α, p38β, p38γ, and p38δ, with ~60% of sequence similarity among four isoforms (81). Of these, p38α (MAPK14) and p38β (MAPK11) are highly expressed in various tissues, p38γ (MAPK12/ERK6) in muscle, and p38δ (MAPK13/SAPK4) in lung and kidney (34, 82). The p38 MAPK plays vital roles in cell responses to stimulators, including proinflammatory cytokines and environmental stresses such as ultraviolet irradiation and heat shock; and is involved in cellular differentiation, cell migration, and inflammation. Activation of p38 kinases are due to phosphorylations at Thr180 and Tyr182 within Thr-Gly-Try motif (83). This canonical phosphorylation is regulated by MKK3 and MKK6, which are highly selective for p38 MAPKs (84–86) (Figure 1). There exist a large body of substrates of p38 MAPKs both in cytoplasm and nucleus, such as transcription factors (p53, MEF2, CHOP, and ATF2), and other protein kinases (MNK1/MNK2, and MSK1/MSK2) which in turn phosphorylate other important proteins (Hsp27, and eIF-4E) (81). In PAs, p38 MAPK plays an important role in immune escape. Tumor immune escape means that tumor cells escape from the body's immune system recognition and attacking to survive and proliferate in the body. When tumor cells appear in the healthy body, the body's immune surveillance system can recognize and specifically remove these “non-self” tumor cells through natural and acquired immunity to prevent the development of tumors (87). However, in some cases, malignant cells can escape the immune surveillance of the body through various mechanisms to rapidly proliferate and form tumors. Studies demonstrate that phosphorylated p38 stimulates the expression of matrix metalloproteinase 9 (MMP9), which is involved in accelerating the process of tumor immune escape (88). In addition, studies based on murine gonadotroph cells LβT2 reveal that mammalian sterile-20-like kinase (MST4) is upregulated in the levels of mRNA and protein to promote cell proliferation by activating p38 MAPK and AKT during long-term hypoxia (89).
PA Treatment Related to p38 MAPK Signaling
Previous studies demonstrate that p38 MAPK is associated with apoptosis, and drugs that activate this pathway can thereby induce apoptosis in pituitary tumor cells. A study found that dopamine agonists such as BRC and CAB activate p38 pathway to induce cell apoptosis; for example, when BRC is used to treat rat lactosomatotroph GH3 cells, BRC activates p38 MAPK and promotes cell apoptosis. Moreover, p38 MAPK inhibitors (SB202190, SB203580) completely inhibit BRC-induced p38 MAPK activation and cell apoptosis (21). Similarly, CAB also activates p38 MAPK and induces apoptosis in PRL-D2S cells (22). In addition to dopamine agonists, the natural compound 18beta-glycyrrhetinic acid (GA) extracted from liquorice can induce several types of tumor cell apoptosis (24, 90). In rat PA cells MMQ and GH3, GA can induce cellular damage, decrease cell viability, and cause G0/G1 phase arrest to contribute to cell apoptosis (91). GA can enhance the phosphorylation of JNK and p38, and these effects are abrogated through pretreatment with JNK inhibitor (SP60125) or p38 inhibitor (SB203580). Furthermore, the fact that ROS inhibitor (NAC) abolished the activation of JNK and P38 suggests that GA exerts the anti-PA effects by activating ROS/MAPKs (JNK and P38)-dependent pathway (91). BIM-23A760, a dopamine-somatostatin chimeric compound, by activating p38 and ERK1/2, inhibits cell proliferation and demonstrates cytotoxic effects in primary culture of NFPAs (25).
Potential Targets Related to p38 MAPK Signaling Pathway for PA Treatment
In recent years, with the development of next-generation sequencing (NGS), a large number of non-coding RNAs have been identified (92, 93). Non-coding RNAs not only deepen our understanding of tumorigenesis and development, but also provide new directions for the diagnosis and treatment of tumors. Studies demonstrate that microRNA-16 (miR-16) is significantly downregulated in PAs compared to the healthy controls, and overexpression of miR-16 reduces the protein expressions of phosphorylated p38, VEGFR2, MMP-9, and NF-kB in HP75 cells, which suggests that miR-16 is involved in PA cell proliferation and angiogenesis via VEGFR2/p38/NF-κB pathway (94). Studies show that miR-16 suppresses MEK1 expressions thereby inhibiting ERK/MAPK pathway activity, leading to inhibition of cell proliferation, cell-cycle arrest, and apoptosis in PAs (33). Therefore, miR-16, as a regulator of p38 MAPK, might be a diagnostic biomarker and a target of PAs. In addition, based on miR-6-related studies, miR-6-protein and/or miR-6-lncRNA interactions are also worth further exploring for discovery of potential therapeutic targets. Further, MST4 promotes cell proliferation by activating p38 MAPK under long-term hypoxia. Therefore, MST4 might be a target for PA treatment (34).
The JNK MAPK Pathway in PAs
The JNK MAPK pathway is mainly activated by various stress stimuli, including oxidative stress, UV irradiation, osmotic shock, heat shock, and proinflammatory cytokines (95), and plays vital roles in controlling proliferation, cell growth, apoptosis, inflammatory, and immune responses (96–98). JNK includes three isoforms: JNK1 and JNK2 are extensively distributed in different tissues, and JNK3 is mainly expressed in testis, heart, and brain (99, 100). JNK is activated by a cascade reaction: stress signals are delivered by small GTPases (Rac, Rho, and cdc42) to a series of kinase cascades, and eventually MKK4/7 activates JNKs (101). In addition, MKK4/7 can also be activated by a member of the germinal center kinase (GCK) family to activate JNKs (95). Moreover, MKK4 might also activate p38 MAPKs (p38α and p38δ), which lets JNK pathway cross-talk with p38 MAPK pathway (34). The activated JNKs are translocated from cytoplasm to nucleus where it can regulate the activity of multiple transcription factors (ATF-2, Elk-1, Smad4, p53, NFAT4, and Stat3) (102).
JNK pathway has been reported to be involved in many kinds of cancers, including retinoblastoma, melanoma, colorectal cancer, breast cancer, and ovarian cancer; and these cancers exhibit the elevated JNK activities (103–107). While there are limited studies on the role of the JNK pathway in the initiation and progression of pituitary tumors, several studies have demonstrated that alteration of JNK in the pituitary gland could be associated with pituitary tumoregensis. For instance, mice with a conditionally inactivated JNK1 in nestin-expressed cells (JNK1ΔNES) are used to study the effects of JNK1 signaling on glucose metabolism. Unexpectedly, the decreased somatic growth and increased thyroid axis activities are observed in JNK1ΔNES mice with decreased levels of circulating GH and IGF1 (108). Another study shows that ablation of Jnk genes in anterior pituitary gland of mice leads to increased energy expenditure, and decreased obesity compared to control mice; and pituitary thyroid-stimulating hormone (TSH) and blood thyroid hormone (T4) are increased (109). Thus, JNK signaling might be involved in pituitary tumorigenesis (110).
JNK MAPK Pathway-Targeted Pharmacological Treatments of PAs
The first one is ursolic acid (UA), a triterpenoid compound found in food, medical herbs, and other plants (111), which has antitumor effects in a number of tumors such as hepatocellular carcinoma, melanoma, breast cancer, colorectal cancer, bladder cancer, and prostate cancer (112–117). In the treatment of PAs, UA decreases cell viability and induces apoptosis in AtT20 cells by upregulating JNK phosphorylation. JNK signaling can also cross-talk with UA-induced mitochondrial apoptotic signaling transduction through phosphorylation and degradation of Bcl-2 (26). Moreover, GA, as described above, can induce cellular cytotoxicity and apoptosis by enhancing the phosphorylation of JNK. With further research progress, it is strongly believed that more drugs will be developed to target JNK MAPK pathway.
The MAPK Pathway Network in PAs
ERK, p38, and JNK signaling pathways both independently and concordantly contribute to pituitary tumorigenesis. Thus, some chemotherapeutic drugs for PAs may target several subfamilies of the MAPK signaling pathway at the same time. For example, fulvestrant is an estrogen receptor antagonist without agonist effects (118), which is approved in the EU and USA to treat post-menopausal women who have hormone-sensitive advanced breast cancer, after prior antiestrogen therapy (119). In the treatment of PAs, recent studies reveal that fulvestrant significantly suppresses the cell viability and invasion of rat PA GH3 cells by simultaneous regulation of ERK1/2, JNK1/2, and p38 signaling pathways (27). GA exerts anti-tumor effects against PAs through enhancing the activations of JNK and p38 MAPK signaling pathways (91). BIM-23A76 that is a dopamine-somatostatin chimeric compound demonstrates the function of inhibiting cell proliferative and cytotoxic effects by activating p38 and ERK1/2 (25).
The comprehensive pathway-network analysis of multiple sets of proteomic data in PAs (120–124) reveals that MAPK signaling abnormalities, including ERK-MAPK signaling pathway, are significantly associated with PAs (125), and that some important molecules such as ERK, p38, JNK, Ras, Akt, NF-kB, TNF, and TGFb1 in MAPK signaling pathway network are identified in human PAs. In addition, the regulatory effect of MAPK cascades on cell differentiation, proliferation, survival and apoptosis interact with other transduction pathways (126). For instance, both PI3K-Akt and Raf/MEK/ERK pathways synergistically promote cell proliferation at the initial stage of PAs (127). Another study demonstrates the critical role of ERK1/2 and cAMP in determination of tumoural phenotype in PAs (128). Thus, a combination of drugs that target pathways which cross-talk with MAPK signaling may produce a more effective treatment for PAs.
In the MAPK network system in PAs, the activation of ERK is generally thought to promote cell proliferation and growth; whereas the activations of p38 and JNK are generally thought to promote cell apoptosis. The MAPK signaling pathway can be targeted by several mechanisms. Two types of combination strategies can be used: (i) A single drug to target ERK pathway and p38 or JNK pathway. For example, BIM-23A76 inhibits cell proliferation through targeting ERK1/2 pathway, and promotes cytotoxic effects through targeting p38 pathway (25). And (ii) multiple drugs to target different ERK and p38 or JNK pathways, such as ERK inhibitor (e.g., SOM230, OCT, or dopamine) plus p38 activator (e.g., cabergoline, bromocriptine, and fulvestrant) and/or JNK activator (e.g., UA). Also, fulvestrant can target both p38 and JNK pathways to promote the cell apoptosis for cell cytotoxic effects. These MAPK pathway-based combination therapies might produce better anti-cancer effects on PAs.
Conclusion
This review summarized the studies of MAPK signaling in pituitary tumorigenesis. We discussed some important molecules involving in MAPK signaling pathway and potential drugs targeting the MAPK signaling (Figure 1 and Table 1). The ERK-MAPK signaling, p38-MAPK signaling, and JNK signaling all play important roles in PAs. Some therapeutic drugs exert anti-tumor effects by targeting one of these pathways or all these three pathways at the same time. MAPK signaling is a very complex network, and always interacts with other pathways such as PI3K and cAMP pathway to affect tumor progression. The latest development of MAPK signaling in PAs and the related anti-tumor drugs targeting MAPK signaling pathways would provide new insights on PA pathogenic mechanisms and pre-clinical data for treatment.
Author Contributions
ML and YW collected and analyzed literature, prepared figures and tables, and wrote and revised manuscript. XZ conceived the concept, designed, coordinated, wrote and critically revised manuscript, and was responsible for its financial supports and the corresponding works. All authors approved the final manuscript.
Funding
This work was supported by the grants from the China 863 plan Project (Grant No. 2014AA020610-1 to XZ), National Natural Science Foundation of China (Grant No. 81572278 and 81272798 to XZ), the Hunan Provincial Hundred Talent Plan (to XZ), the Xiangya Hospital Funds for Talent Introduction (to XZ), and the Hunan Provincial Natural Science Foundation of China (Grant No. 14JJ7008 to XZ).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACTH, Adrenocorticotropin; CIRP, Cold inducible RNA binding protein; CRH, Corticotropin-releasing hormone; CSAIDs, Cytokine-suppressive anti-inflammatory drugs; Eps8, Epidermal growth factor pathway substrate number 8; ERK, Extracellular signal-related kinase; GA, 18beta-glycyrrhetinic acid; GH, Growth hormone; GnRH, gonadotropin-releaseing hormone; GPCR, G protein-coupled receptor; IGF1, Insulin-like growth factor 1; JNK, Jun N-terminal kinase; LPS, Lipopolysaccharide; MAPKs, Mitogen-activated protein kinases; MKK, MAPK kinases; MKKK, MAPK kinase kinases; MKPs, MAPK phosphatases; MST4, Mammalian sterile-20-like kinase; PRL, Prolactin; RA, Retinoic acid; RKIP, Raf kinase inhibitory protein; RTK, Receptor tyrosine kinase; SAPKs, Stress-activated protein kinases; SSTs, Somatostatin analogs; SST, Somatostatin; TSH, Thyroid-stimulating hormone.
References
1. Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. (2002) 2:836–49. doi: 10.1038/nrc926
2. Zhan X, Wang X, Cheng T. Human pituitary adenoma proteomics: new progresses and perspectives. Front Endocrinol. (2016) 7:54. doi: 10.3389/fendo.2016.00054
3. Asa SL, Kovacs K. Clinically non-functioning human pituitary adenomas. Can J Neurol Sci. (1992) 19:228–35.
4. Zhan X, Desiderio DM, Wang X, Zhan X, Guo T, Li M, et al. Identification of the proteomic variations of invasive relative to non-invasive non-functional pituitary adenomas. Electrophoresis. (2014) 35:2184–94. doi: 10.1002/elps.201300590
5. Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S. Clonal origin of pituitary adenomas. J Clin Endocrinol Metab. (1990) 71:1427–33. doi: 10.1210/jcem-71-6-1427
6. Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. (1996) 8:402–11. doi: 10.1016/S0952-7915(96)80131-2
7. Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. (2008) 60:261–310. doi: 10.1124/pr.107.00106
8. Duan W, Wong WS. Targeting mitogen-activated protein kinases for asthma. Curr Drug Targets. (2006) 7:691–8. doi: 10.2174/138945006777435353
9. Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. (2007) 6:391–403. doi: 10.1038/nrd2289
10. De Luca A, Maiello MR, D'Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. (2012) 16(Suppl. 2):S17–27. doi: 10.1517/14728222.2011.639361
11. Zhan X, Wang X, Long Y, Desiderio DM. Heterogeneity analysis of the proteomes in clinically nonfunctional pituitary adenomas. BMC Med Genomics. (2014) 7:69. doi: 10.1186/s12920-014-0069-6
12. Zhan X, Long Y. Exploration of Molecular network variations in different subtypes of human non-functional pituitary adenomas. Front Endocrinol. (2016) 7:13. doi: 10.3389/fendo.2016.00013
13. Hubina E, Nanzer AM, Hanson MR, Ciccarelli E, Losa M, Gaia D, et al. Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur J Endocrinol. (2006) 155:371–9. doi: 10.1530/eje.1.02213
14. Murray RD, Kim K, Ren SG, Lewis I, Weckbecker G, Bruns C, et al. The novel somatostatin ligand (SOM230) regulates human and rat anterior pituitary hormone secretion. J Clin Endocrinol Metab. (2004) 89:3027–32. doi: 10.1210/jc.2003-031319
15. Hofland LJ, van der Hoek J, van Koetsveld PM, de Herder WW, Waaijers M, Sprij-Mooij D, et al. The novel somatostatin analog SOM230 is a potent inhibitor of hormone release by growth hormone- and prolactin-secreting pituitary adenomas in vitro. J Clin Endocrinol Metab. (2004) 89:1577–85. doi: 10.1210/jc.2003-031344
16. van der Hoek J, Waaijers M, van Koetsveld PM, Sprij-Mooij D, Feelders RA, Schmid HA, et al. Distinct functional properties of native somatostatin receptor subtype 5 compared with subtype 2 in the regulation of ACTH release by corticotroph tumor cells. Am J Physiol Endocrinol Metab. (2005) 289:E278–87. doi: 10.1152/ajpendo.00004.2005
17. Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med. (2012) 366:914–24. doi: 10.1056/NEJMoa1105743
18. Boscaro M, Ludlam WH, Atkinson B, Glusman JE, Petersenn S, Reincke M, et al. Treatment of pituitary-dependent Cushing's disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. J Clin Endocrinol Metab. (2009) 94:115–22. doi: 10.1016/S0084-3741(09)79383-7
19. Toma K, Otsuka F, Oguni K, Terasaka T, Komatsubara M, Tsukamoto-Yamauchi N, et al. BMP-6 modulates somatostatin effects on luteinizing hormone production by gonadrotrope cells. Peptides. (2016) 76:96–101. doi: 10.1016/j.peptides.2016.01.011
20. Liu JC, Baker RE, Sun C, Sundmark VC, Elsholtz HP. Activation of Go-coupled dopamine D2 receptors inhibits ERK1/ERK2 in pituitary cells. A key step in the transcriptional suppression of the prolactin gene. J Biol Chem. (2002) 277:35819–25. doi: 10.1074/jbc.M202920200
21. Kanasaki H, Fukunaga K, Takahashi K, Miyazaki K, Miyamoto E. Involvement of p38 mitogen-activated protein kinase activation in bromocriptine-induced apoptosis in rat pituitary GH3 cells. Biol Reprod. (2000) 62:1486–94. doi: 10.1095/biolreprod62.6.1486
22. Radl DB, Ferraris J, Boti V, Seilicovich A, Sarkar DK, Pisera D. Dopamine-induced apoptosis of lactotropes is mediated by the short isoform of D2 receptor. PLoS ONE. (2011) 6:e18097. doi: 10.1371/journal.pone.0018097
23. Petiti JP, Sosa Ldel V, Sabatino ME, Vaca AM, Gutierrez S, De Paul AL, et al. Involvement of MEK/ERK1/2 and PI3K/Akt pathways in the refractory behavior of GH3B6 pituitary tumor cells to the inhibitory effect of TGFbeta1. Endocrinology. (2015) 156:534–47. doi: 10.1210/en.2014-1070
24. Lee CS, Yang JC, Kim YJ, Jang ER, Kim W, Myung SC. 18beta-Glycyrrhetinic acid potentiates apoptotic effect of trichostatin A on human epithelial ovarian carcinoma cell lines. Eur J Pharmacol. (2010) 649:354–61. doi: 10.1016/j.ejphar.2010.09.047
25. Peverelli E, Olgiati L, Locatelli M, Magni P, Fustini MF, Frank G, et al. The dopamine–somatostatin chimeric compound BIM-23A760 exerts antiproliferative and cytotoxic effects in human non-functioning pituitary tumors by activating ERK1/2 and p38 pathways. Cancer Lett. (2010) 288:170–6. doi: 10.1016/j.canlet.2009.06.034
26. Gong YY, Liu YY, Yu S, Zhu XN, Cao XP, Xiao HP. Ursolic acid suppresses growth and adrenocorticotrophic hormone secretion in AtT20 cells as a potential agent targeting adrenocorticotrophic hormone-producing pituitary adenoma. Mol Med Rep. (2014) 9:2533–9. doi: 10.3892/mmr.2014.2078
27. Gao H, Xue Y, Cao L, Liu Q, Liu C, Shan X, et al. ESR1 and its antagonist fulvestrant in pituitary adenomas. Mol Cell Endocrinol. (2017) 443:32–41. doi: 10.1016/j.mce.2016.12.029
28. Fougner SL, Bollerslev J, Latif F, Hald JK, Lund T, Ramm-Pettersen J, et al. Low levels of raf kinase inhibitory protein in growth hormone-secreting pituitary adenomas correlate with poor response to octreotide treatment. J Clin Endocrinol Metab. (2008) 93:1211–6. doi: 10.1210/jc.2007-2272
29. Fazioli F, Minichiello L, Matoska V, Castagnino P, Miki T, Wong WT, et al. Eps8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J. (1993) 12:3799–808. doi: 10.1002/j.1460-2075.1993.tb06058.x
30. Xu M, Shorts-Cary L, Knox AJ, Kleinsmidt-DeMasters B, Lillehei K, Wierman ME. Epidermal growth factor receptor pathway substrate 8 is overexpressed in human pituitary tumors: role in proliferation and survival. Endocrinology. (2009) 150:2064–71. doi: 10.1210/en.2008-1265
31. Labeur M, Wolfel B, Stalla J, Stalla GK. TMEFF2 is an endogenous inhibitor of the CRH signal transduction pathway. J Mol Endocrinol. (2015) 54:51–63. doi: 10.1530/JME-14-0225
32. Jian F, Chen Y, Ning G, Fu W, Tang H, Chen X, et al. Cold inducible RNA binding protein upregulation in pituitary corticotroph adenoma induces corticotroph cell proliferation via Erk signaling pathway. Oncotarget. (2016) 7:9175–87. doi: 10.18632/oncotarget.7037
33. Wang DW, Wang YQ, Shu HS. MiR-16 inhibits pituitary adenoma cell proliferation via the suppression of ERK/MAPK signal pathway. Eur Rev Med Pharmacol Sci. (2018) 22:1241–8.
34. Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, et al. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. (1997) 272:30122–8. doi: 10.1074/jbc.272.48.30122
35. Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. (2005) 6:827–37. doi: 10.1038/nrm1743
36. O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. (2004) 306:2267–70. doi: 10.1126/science.1103233
37. Ehrenreiter K, Piazzolla D, Velamoor V, Sobczak I, Small JV, Takeda J, et al. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. (2005) 168:955–64. doi: 10.1083/jcb.200409162
38. Karga HJ, Alexander JM, Hedley-Whyte ET, Klibanski A, Jameson JL. Ras mutations in human pituitary tumors. J Clin Endocrinol Metab. (1992) 74:914–9. doi: 10.1210/jc.74.4.914
39. Cai WY, Alexander JM, Hedley-Whyte ET, Scheithauer BW, Jameson JL, Zervas NT, et al. ras mutations in human prolactinomas and pituitary carcinomas. J Clin Endocrinol Metab. (1994) 78:89–93. doi: 10.1210/jcem.78.1.8288721
40. Ewing I, Pedder-Smith S, Franchi G, Ruscica M, Emery M, Vax V, et al. A mutation and expression analysis of the oncogene BRAF in pituitary adenomas. Clin Endocrinol. (2007) 66:348–52. doi: 10.1111/j.1365-2265.2006.02735.x
41. Dworakowska D, Wlodek E, Leontiou CA, Igreja S, Cakir M, Teng M, et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr Relat Cancer. (2009) 16:1329–38. doi: 10.1677/ERC-09-0101
42. Chaturvedi K, Sarkar DK. Mediation of basic fibroblast growth factor-induced lactotropic cell proliferation by Src-Ras-mitogen-activated protein kinase p44/42 signaling. Endocrinology. (2005) 146:1948–55. doi: 10.1210/en.2004-1448
43. Oomizu S, Chaturvedi K, Sarkar DK. Folliculostellate cells determine the susceptibility of lactotropes to estradiol's mitogenic action. Endocrinology. (2004) 145:1473–80. doi: 10.1210/en.2003-0965
44. Booth A, Trudeau T, Gomez C, Lucia MS, Gutierrez-Hartmann A. Persistent ERK/MAPK activation promotes lactotrope differentiation and diminishes tumorigenic phenotype. Mol Endocrinol. (2014) 28:1999–2011. doi: 10.1210/me.2014-1168
45. Lania A, Filopanti M, Corbetta S, Losa M, Ballare E, Beck-Peccoz P, et al. Effects of hypothalamic neuropeptides on extracellular signal-regulated kinase (ERK1 and ERK2) cascade in human tumoral pituitary cells. J Clin Endocrinol Metab. (2003) 88:1692–6. doi: 10.1210/jc.2002-021207
46. Pombo CM, Zalvide J, Gaylinn BD, Dieguez C. Growth hormone-releasing hormone stimulates mitogen-activated protein kinase. Endocrinology. (2000) 141:2113–9. doi: 10.1210/endo.141.6.7513
47. Cuny T, Gerard C, Saveanu A, Barlier A, Enjalbert A. Physiopathology of somatolactotroph cells: from transduction mechanisms to cotargeting therapy. Ann N Y Acad Sci. (2011) 1220:60–70. doi: 10.1111/j.1749-6632.2010.05924.x
48. Liu F, Austin DA, Mellon PL, Olefsky JM, Webster NJ. GnRH activates ERK1/2 leading to the induction of c-fos and LHbeta protein expression in LbetaT2 cells. Mol Endocrinol. (2002) 16:419–34. doi: 10.1210/mend.16.3.0791
49. Rubinfeld H, Shimon I. PI3K/Akt/mTOR and Raf/MEK/ERK signaling pathways perturbations in non-functioning pituitary adenomas. Endocrine. (2012) 42:285–91. doi: 10.1007/s12020-012-9682-3
50. Woodmansee WW, Kerr JM, Tucker EA, Mitchell JR, Haakinson DJ, Gordon DF, et al. The proliferative status of thyrotropes is dependent on modulation of specific cell cycle regulators by thyroid hormone. Endocrinology. (2006) 147:272–82. doi: 10.1210/en.2005-1013
51. Zhang D, Bergsneider M, Wang MB, Heaney AP. Targeting the ERK pathway for the treatment of Cushing's disease. Oncotarget. (2016) 7:69149–58. doi: 10.18632/oncotarget.12381
52. Theodoropoulou M, Zhang J, Laupheimer S, Paez-Pereda M, Erneux C, Florio T, et al. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res. (2006) 66:1576–82. doi: 10.1158/0008-5472.CAN-05-1189
53. Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. (2002) 146:707–16. doi: 10.1530/eje.0.1460707
54. Weckbecker G, Briner U, Lewis I, Bruns C. SOM230: a new somatostatin peptidomimetic with potent inhibitory effects on the growth hormone/insulin-like growth factor-I axis in rats, primates, and dogs. Endocrinology. (2002) 143:4123–30. doi: 10.1210/en.2002-220219
55. Cheung NW, Boyages SC. Somatostatin-14 and its analog octreotide exert a cytostatic effect on GH3 rat pituitary tumor cell proliferation via a transient G0/G1 cell cycle block. Endocrinology. (1995) 136:4174–81. doi: 10.1210/en.136.10.4174
56. Florio T, Thellung S, Arena S, Corsaro A, Spaziante R, Gussoni G, et al. Somatostatin and its analog lanreotide inhibit the proliferation of dispersed human non-functioning pituitary adenoma cells in vitro. Eur J Endocrinol. (1999) 141:396–408. doi: 10.1530/eje.0.1410396
57. Padova H, Rubinfeld H, Hadani M, Cohen ZR, Nass D, Taylor JE, et al. Effects of selective somatostatin analogs and cortistatin on cell viability in cultured human non-functioning pituitary adenomas. Mol Cell Endocrinol. (2008) 286:214–8. doi: 10.1016/j.mce.2007.12.011
58. Zatelli MC, Piccin D, Vignali C, Tagliati F, Ambrosio MR, Bondanelli M, et al. Pasireotide, a multiple somatostatin receptor subtypes ligand, reduces cell viability in non-functioning pituitary adenomas by inhibiting vascular endothelial growth factor secretion. Endocr Relat Cancer. (2007) 14:91–102. doi: 10.1677/ERC-06-0026
59. Colao A, Di Somma C, Pivonello R, Faggiano A, Lombardi G, Savastano S. Medical therapy for clinically non-functioning pituitary adenomas. Endocr Relat Cancer. (2008) 15:905–15. doi: 10.1677/ERC-08-0181
60. Kauppinen-Makelin R, Sane T, Reunanen A, Valimaki MJ, Niskanen L, Markkanen H, et al. A nationwide survey of mortality in acromegaly. J Clin Endocrinol Metab. (2005) 90:4081–6. doi: 10.1210/jc.2004-1381
61. Bates PR, Carson MN, Trainer PJ, Wass JA, Group UKNARS. Wide variation in surgical outcomes for acromegaly in the UK. Clin Endocrinol. (2008) 68:136–42. doi: 10.1111/j.1365-2265.2007.03012.x
62. Melmed S, Sternberg R, Cook D, Klibanski A, Chanson P, Bonert V, et al. A critical analysis of pituitary tumor shrinkage during primary medical therapy in acromegaly. J Clin Endocrinol Metab. (2005) 90:4405–10. doi: 10.1210/jc.2004-2466
63. Fougner SL, Borota OC, Berg JP, Hald JK, Ramm-Pettersen J, Bollerslev J. The clinical response to somatostatin analogues in acromegaly correlates to the somatostatin receptor subtype 2a protein expression of the adenoma. Clin Endocrinol. (2008) 68:458–65. doi: 10.1111/j.1365-2265.2007.03065.x
64. Hubina E, Ruscica M, Nanzer AM, Czirjak S, Goth MI, Grossman AB, et al. Novel molecular aspects of pituitary adenomas. J Endocrinol Invest. (2005) 28(11 Suppl. International):87–92.
65. Stack J, Surprenant A. Dopamine actions on calcium currents, potassium currents and hormone release in rat melanotrophs. J Physiol. (1991) 439:37–58. doi: 10.1113/jphysiol.1991.sp018655
66. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. (2011) 96:273–88. doi: 10.1210/jc.2010-1692
67. Iaccarino C, Samad TA, Mathis C, Kercret H, Picetti R, Borrelli E. Control of lactotrop proliferation by dopamine: essential role of signaling through D2 receptors and ERKs. Proc Natl Acad Sci USA. (2002) 99:14530–5. doi: 10.1073/pnas.222319599
68. Radl D, De Mei C, Chen E, Lee H, Borrelli E. Each individual isoform of the dopamine D2 receptor protects from lactotroph hyperplasia. Mol Endocrinol. (2013) 27:953–65. doi: 10.1210/me.2013-1008
69. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. (2001) 29:117–29. doi: 10.1038/ng1001-117
70. Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. (2009) 19:385–94. doi: 10.1016/j.tcb.2009.05.008
71. Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res. (2012) 347:11–20. doi: 10.1007/s00441-011-1201-y
72. Trakul N, Rosner MR. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res. (2005) 15:19–23. doi: 10.1038/sj.cr.7290258
73. Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. (2005) 280:24931–40. doi: 10.1074/jbc.M413929200
74. Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. (1999) 401:173–7. doi: 10.1038/43686
75. Eves EM, Shapiro P, Naik K, Klein UR, Trakul N, Rosner MR. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell. (2006) 23:561–74. doi: 10.1016/j.molcel.2006.07.015
76. Lee SJ, Lee SH, Yoon MH, Park BJ. A new p53 target gene, RKIP, is essential for DNA damage-induced cellular senescence and suppression of ERK activation. Neoplasia. (2013) 15:727–37. doi: 10.1593/neo.121862
77. Pecori Giraldi F, Ambrogio AG, Andrioli M, Sanguin F, Karamouzis I, Corsello SM, et al. Potential role for retinoic acid in patients with Cushing's disease. J Clin Endocrinol Metab. (2012) 97:3577–83. doi: 10.1210/jc.2012-2328
78. Nishiyama H, Itoh K, Kaneko Y, Kishishita M, Yoshida O, Fujita J. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol. (1997) 137:899–908. doi: 10.1083/jcb.137.4.899
79. Artero-Castro A, Callejas FB, Castellvi J, Kondoh H, Carnero A, Fernandez-Marcos PJ, et al. Cold-inducible RNA-binding protein bypasses replicative senescence in primary cells through extracellular signal-regulated kinase 1 and 2 activation. Mol Cell Biol. (2009) 29:1855–68. doi: 10.1128/MCB.01386-08
80. Guo X, Wu Y, Hartley RS. Cold-inducible RNA-binding protein contributes to human antigen R and cyclin E1 deregulation in breast cancer. Mol Carcinog. (2010) 49:130–40. doi: 10.1002/mc.20582
81. Zarubin T, Jiahuai H. Activation and signaling of the p38 MAP kinase pathway. Cell Res. (2005) 15:11–8. doi: 10.1038/sj.cr.7290257
82. Wang XS, Diener K, Manthey CL, Wang S, Rosenzweig B, Bray J, et al. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J Biol Chem. (1997) 272:23668–74. doi: 10.1074/jbc.272.38.23668
83. Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. (1995) 270:7420–6. doi: 10.1074/jbc.270.13.7420
84. Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. (1995) 267:682–5. doi: 10.1126/science.7839144
85. Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, et al. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. (1995) 268:286–90. doi: 10.1126/science.7716521
86. Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. (1996) 16:1247–55. doi: 10.1128/MCB.16.3.1247
87. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med. (2016) 94:509–22. doi: 10.1007/s00109-015-1376-x
88. Han X, Geng X, Li Z, Chen Z, Liu Y, Liu P, et al. The relationship between phospho-p38, MMP-9, and MICA expression in pituitary adenomas demonstrates a new mechanism of pituitary adenoma immune escape. World Neurosurg. (2018) 123:e116–24. doi: 10.1016/j.wneu.2018.11.077
89. Xiong W, Knox AJ, Xu M, Kiseljak-Vassiliades K, Colgan SP, Brodsky KS, et al. Mammalian Ste20-like kinase 4 promotes pituitary cell proliferation and survival under hypoxia. Mol Endocrinol. (2015) 29:460–72. doi: 10.1210/me.2014-1332
90. Sharma G, Kar S, Palit S, Das PK. 18beta-glycyrrhetinic acid induces apoptosis through modulation of Akt/FOXO3a/Bim pathway in human breast cancer MCF-7 cells. J Cell Physiol. (2012) 227:1923–31. doi: 10.1002/jcp.22920
91. Wang D, Wong HK, Feng YB, Zhang ZJ. 18beta-glycyrrhetinic acid induces apoptosis in pituitary adenoma cells via ROS/MAPKs-mediated pathway. J Neuro Oncol. (2014) 116:221–30. doi: 10.1007/s11060-013-1292-2
92. Lu M, Zhan X. The crucial role of multiomic approach in cancer research and clinically relevant outcomes. EPMA J. (2018) 9:77–102. doi: 10.1007/s13167-018-0128-8
93. Cheng T, Zhan X. Pattern recognition for predictive, preventive, and personalized medicine in cancer. EPMA J. (2017) 8:51–60. doi: 10.1007/s13167-017-0083-9
94. Lu B, Liu GL, Yu F, Li WJ, Xiang XX, Xiao HZ. MicroRNA16/VEGFR2/p38/NFkappaB signaling pathway regulates cell growth of human pituitary neoplasms. Oncol Rep. (2018) 39:1235–44. doi: 10.3892/or.2018.6227
95. Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. (2010) 1804:463–75. doi: 10.1016/j.bbapap.2009.11.002
96. Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann N Y Acad Sci. (1998) 851:139–46. doi: 10.1111/j.1749-6632.1998.tb08987.x
97. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. (2007) 19:142–9. doi: 10.1016/j.ceb.2007.02.001
98. Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int J Biochem Cell Biol. (2001) 33:1047–63. doi: 10.1016/S1357-2725(01)00093-0
99. Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. (1996) 15:2760–70. doi: 10.1002/j.1460-2075.1996.tb00636.x
100. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. (2000) 103:239–52. doi: 10.1016/S0092-8674(00)00116-1
101. Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol. (2011) 90:536–44. doi: 10.1016/j.ejcb.2010.11.008
102. Mizukami Y, Yoshioka K, Morimoto S, Yoshida K. A novel mechanism of JNK1 activation. Nuclear translocation and activation of JNK1 during ischemia and reperfusion. J Biol Chem. (1997) 272:16657–62. doi: 10.1074/jbc.272.26.16657
103. Gulmann C, Sheehan KM, Conroy RM, Wulfkuhle JD, Espina V, Mullarkey MJ, et al. Quantitative cell signalling analysis reveals down-regulation of MAPK pathway activation in colorectal cancer. J Pathol. (2009) 218:514–9. doi: 10.1002/path.2561
104. Chen Z, Yang A, Xu C, Xing Y, Gong W, Li J. c-Jun N-terminal kinase is involved in the regulation of proliferation and apoptosis by integrin-linked kinase in human retinoblastoma cells. Graefes Arch Clin Exp Ophthalmol. (2011) 249:1399–407. doi: 10.1007/s00417-010-1607-3
105. Jorgensen K, Davidson B, Florenes VA. Activation of c-jun N-terminal kinase is associated with cell proliferation and shorter relapse-free period in superficial spreading malignant melanoma. Mod Pathol. (2006) 19:1446–55. doi: 10.1038/modpathol.3800662
106. Wang X, Chao L, Li X, Ma G, Chen L, Zang Y, et al. Elevated expression of phosphorylated c-Jun NH2-terminal kinase in basal-like and “triple-negative” breast cancers. Hum Pathol. (2010) 41:401–6. doi: 10.1016/j.humpath.2009.08.018
107. Odegaard E, Staff AC, Abeler VM, Kopolovic J, Onsrud M, Lazarovici P, et al. The activated nerve growth factor receptor p-TrkA is selectively expressed in advanced-stage ovarian carcinoma. Hum Pathol. (2007) 38:140–6. doi: 10.1016/j.humpath.2006.06.027
108. Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci USA. (2010) 107:6028–33. doi: 10.1073/pnas.1001796107
109. Vernia S, Cavanagh-Kyros J, Barrett T, Jung DY, Kim JK, Davis RJ. Diet-induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes Dev. (2013) 27:2345–55. doi: 10.1101/gad.223800.113
110. Chen XH, Ling XM, Shi S. microRNA-106a induces the proliferation and apoptosis of glioma cells through regulating JNK/MAPK pathway. Eur Rev Med Pharmacol Sci. (2015) 19:3412–7.
111. Liu J. Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol. (1995) 49:57–68. doi: 10.1016/0378-8741(95)90032-2
112. Shyu MH, Kao TC, Yen GC. Oleanolic acid and ursolic acid induce apoptosis in HuH7 human hepatocellular carcinoma cells through a mitochondrial-dependent pathway and downregulation of XIAP. J Agric Food Chem. (2010) 58:6110–8. doi: 10.1021/jf100574j
113. Harmand PO, Duval R, Delage C, Simon A. Ursolic acid induces apoptosis through mitochondrial intrinsic pathway and caspase-3 activation in M4Beu melanoma cells. Int J Cancer. (2005) 114:1–11. doi: 10.1002/ijc.20588
114. De Angel RE, Smith SM, Glickman RD, Perkins SN, Hursting SD. Antitumor effects of ursolic acid in a mouse model of postmenopausal breast cancer. Nutr Cancer. (2010) 62:1074–86. doi: 10.1080/01635581.2010.492092
115. Prasad S, Yadav VR, Sung B, Reuter S, Kannappan R, Deorukhkar A, et al. Ursolic acid inhibits growth and metastasis of human colorectal cancer in an orthotopic nude mouse model by targeting multiple cell signaling pathways: chemosensitization with capecitabine. Clin Cancer Res. (2012) 18:4942–53. doi: 10.1158/1078-0432.CCR-11-2805
116. Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH, Ai X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Biophys Res Commun. (2012) 419:741–7. doi: 10.1016/j.bbrc.2012.02.093
117. Kassi E, Papoutsi Z, Pratsinis H, Aligiannis N, Manoussakis M, Moutsatsou P. Ursolic acid, a naturally occurring triterpenoid, demonstrates anticancer activity on human prostate cancer cells. J Cancer Res Clin Oncol. (2007) 133:493–500. doi: 10.1007/s00432-007-0193-1
118. Vergote I, Abram P. Fulvestrant, a new treatment option for advanced breast cancer: tolerability versus existing agents. Ann Oncol. (2006) 17:200–4. doi: 10.1093/annonc/mdj047
119. McCormack P, Sapunar F. Pharmacokinetic profile of the fulvestrant loading dose regimen in postmenopausal women withhormone receptor-positive advanced breast cancer. Clin Breast Cancer. (2008) 8:347–51. doi: 10.3816/CBC.2008.n.040
120. Zhan X, Desiderio DM. A reference map of a human pituitary adenoma proteome. Proteomics. (2003) 3:699–713. doi: 10.1002/pmic.200300408
121. Moreno CS, Evans CO, Zhan X, Okor M, Desiderio DM, Oyesiku NM. Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res. (2005) 65:10214–22. doi: 10.1158/0008-5472.CAN-05-0884
122. Evans CO, Moreno CS, Zhan X, McCabe MT, Vertino PM, Desiderio DM, et al. Molecular pathogenesis of human prolactinomas identified by gene expression profiling, RT-qPCR, and proteomic analyses. Pituitary. (2008) 11:231–45. doi: 10.1007/s11102-007-0082-2
123. Zhan X, Desiderio DM. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. (2006) 354:279–89. doi: 10.1016/j.ab.2006.05.024
124. Zhan X, Desiderio DM. The human pituitary nitroproteome: detection of nitrotyrosyl-proteins with two-dimensional Western blotting, and amino acid sequence determination with mass spectrometry. Biochem Biophys Res Commun. (2004) 325:1180–6. doi: 10.1016/j.bbrc.2004.10.169
125. Zhan X, Desiderio DM. Signaling pathway networks mined from human pituitary adenoma proteomics data. BMC Med Genomics. (2010) 3:13. doi: 10.1186/1755-8794-3-13
126. Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. (2007) 26:3122–42. doi: 10.1038/sj.onc.1210407
127. Cakir M, Grossman AB. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin Ther Targets. (2009) 13:1121–34. doi: 10.1517/14728220903170675
128. Fernández M, Sanchez-Franco F, Palacios N, Sanchez I, Cacicedo L. IGF-I and vasoactive intestinal peptide (VIP) regulate cAMP-response element-binding protein (CREB)-dependent transcription via the mitogen-activated protein kinase (MAPK) pathway in pituitary cells: requirement of Rap1. J Mol Endocrinol. (2005) 34:699–712. doi: 10.1677/jme.1.01703
Keywords: MAPK, ERK, p38, JNK, signaling pathway, pituitary tumor, biomarker, therapeutic drug
Citation: Lu M, Wang Y and Zhan X (2019) The MAPK Pathway-Based Drug Therapeutic Targets in Pituitary Adenomas. Front. Endocrinol. 10:330. doi: 10.3389/fendo.2019.00330
Received: 08 January 2019; Accepted: 07 May 2019;
Published: 22 May 2019.
Edited by:
Hidenori Fukuoka, Kobe University, JapanReviewed by:
Odelia Cooper, Cedars-Sinai Medical Center, United StatesSergei I. Bannykh, Cedars-Sinai Medical Center, United States
Zhousheng Xiao, The University of Tennessee Health Science Center, United States
Copyright © 2019 Lu, Wang and Zhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianquan Zhan, eWp6aGFuMjAxMUBnbWFpbC5jb20u