 Christopher M. Carmean
Christopher M. Carmean Susumu Seino
Susumu Seino- 1Division of Molecular and Metabolic Medicine, Department of Physiology and Cell Biology, Kobe University Graduate School of Medicine, Kobe, Japan
- 2Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, University of Illinois at Chicago, Chicago, IL, United States
Type 2 diabetes mellitus (T2DM) is a serious global health problem, currently affecting an estimated 451 million people worldwide. T2DM is characterized by hyperglycemia and low insulin relative to the metabolic demand. The precise contributing factors for a given individual vary, but generally include a combination of insulin resistance and insufficient insulin secretion. Ultimately, the progression to diabetes occurs only after β-cells fail to meet the needs of the individual. The stresses placed upon β-cells in this context manifest as increased oxidative damage, local inflammation, and ER stress, often inciting a destructive spiral of β-cell death, increased metabolic stress due to further insufficiency, and additional β-cell death. Several pathways controlling insulin resistance and β-cell adaptation/survival are affected by a class of exogenous bioactive compounds deemed endocrine disrupting chemicals (EDCs). Epidemiological studies have shown that, in several regions throughout the world, exposure to the EDC inorganic arsenic (iAs) correlates significantly with T2DM. It has been proposed that a lifetime of exposure to iAs may exacerbate problems with both insulin sensitivity as well as β-cell function/survival, promoting the development of T2DM. This review focuses on the mechanisms of iAs action as they relate to known adaptive and maladaptive pathways in pancreatic β-cells.
Introduction
An estimated 451 million people worldwide have type 2 diabetes (T2DM), with as many as 693 million expected to be affected by the disease in 2045 (1). T2DM is characterized by insufficient insulin production relative to metabolic demand resulting in poor glycemic control. In normal glucose homeostasis, a postprandial increase in circulating glucose concentration initiates a spike in insulin secretion from pancreatic β-cells (2, 3). This circulating insulin then binds to its cognate receptor on muscle, liver, and adipose tissues (among others), inducing glucose uptake to lower the concentration of glucose in the bloodstream. In many cases of T2DM, muscle and liver cells (the major sites of glucose disposal in the body) become insulin resistant, which induces β-cells to compensate by secreting more insulin. As insulin resistance becomes more severe, greater stresses are placed on the β-cells to increase their insulin output. Years of this chronic stress on β-cells eventually causes β-cell dysfunction and/or death. With fewer functional β-cells secreting insulin in the context of severe insulin resistance, an inability to properly maintain glucose homeostasis ensues, manifesting as T2DM (4).
Though there are many factors that contribute to the progression of diabetes, it is important to recognize that ultimately a failure of β-cells to secrete sufficient amounts of insulin is what results in hyperglycemia and the diagnosis of T2DM (5). While tremendous stress may be placed on β-cells from a metabolic standpoint, they also face other insults from environmental factors such as endocrine-disrupting chemicals (EDCs). EDCs may work alone or synergistically to derange the normal compensatory mechanisms enabling β-cells to promote glucose tolerance in insulin-resistant individuals (6). In this sense, EDCs may act as drivers of diabetes risk, becoming more deleterious as they compound with lifestyle factors and genetic susceptibilities. It is therefore especially important to consider that EDCs may impair the functionality of β-cells or increase their susceptibility to the chronic metabolic stressors associated with insulin resistance. The purpose of this review will be to explore the β-cell-specific effects of one such EDC, arsenic, as it poses a substantial and ongoing threat to public health.
Arsenic is widely recognized as a carcinogen and oxidizing agent that damages neuronal, hepatic, cardiovascular, integumentary, and renal organ systems (7). Its health effects vary depending on valence, mixture with other toxins, dosage, route of exposure, and length of exposure. Capable of causing death within 24 h, the acute lethal dose in humans is estimated to be 0.6 mg/kg/day (8). Common routes of exposure include the skin, lungs, and digestive tract. Though inhalation of iAs is a concern during hazardous occupational work and traditional coal-based food preservation practices (9), the greatest number of individuals are at risk of exposure to unsafe levels of iAs from contaminated groundwater (10–12). The most prevalent species of arsenic found naturally in drinking water are inorganic arsenic (iAs) in its trivalent (AsIII) or pentavalent (AsV) forms (13). Organic arsenicals can be found in the food chain as arsenobetaine, arsenocholine, and arsenolipids, and are generally considered relatively non-toxic (14).
Inorganic arsenic is estimated to naturally contaminate the shallow groundwater underneath 140 million people worldwide (12). Among these people at risk, the number actually exposed to iAs is believed to be lower, as not all contaminated groundwater sources are utilized and excellent remediation methods are available in developed countries (12). Despite this fact, the problem of chronic iAs exposure through shallow groundwater consumption persists on an immense scale. As of 2007, an estimated 20 million people in Bangladesh alone were still served by wells naturally contaminated with iAs at a concentration more than 5x higher than the WHO safe limit of 10 μg/L (15). In these areas, where exposure has been pervasive in communities since the digging of shallow groundwater wells in the mid-1900s, it has been estimated that ~21% of all-cause mortality is attributable to iAs exposure (16). The lasting effects of this contamination may represent one of the greatest failures of public health management in recent history.
One observation from this unintended mass-poisoning and other cases of natural exposure is a potential relationship between iAs exposure and diabetes (11). In addition to its role in increasing the risk of several cancers, peripheral neuropathy, and keratinosis, iAs exposure correlates with glucose intolerance or diabetes prevalence in areas with relatively high exposure levels (17–20). The epidemiological analysis regarding the relationship between iAs exposure and diabetes have been reviewed in detail elsewhere (11). For the purposes of this review, an examination of the specific effects of iAs on β-cells will be undertaken, including the evidence from pancreatic endpoints in animal models and mechanistic insights gained from ex vivo and/or interventional studies.
Acute vs. Chronic iAs Exposure
The effects of iAs can be conceptualized on a sliding scale of dosage and time. A single large dose of iAs can cause nausea, vomiting, abdominal pain, diarrhea, and even death (8, 21). Chronic ingestion of lower doses of iAs can occur without any immediate sensory feedback, and yet the ensuing damage over several years may span most organ systems, increasing an individual's risk of cancer, peripheral neuropathy, cardiovascular disease, diabetes, and a multitude of skin problems (7). In trying to better understand the relationship between iAs exposure and diabetes, the effects of chronic, sub-toxic exposure are of the greatest concern and should be delineated from acute toxic effects.
The National Toxicology Program Workshop Review's assessment in 2012 suggested that cell-culture studies utilizing iAs concentrations ≥ 1 mM can be considered acute stress-response studies rather than functional studies of β-cells' role in iAs-associated diabetes, even in cases where physiologically-relevant model systems or physiological endpoints were utilized (11). Given that the highest circulating plasma concentration of iAs ever recorded in a human population drinking iAs-contaminated water was 0.6 μM, and clear evidence of reduced cell growth or survival ex vivo has been reported for iAs exposures ≥5 μM (22, 23), 1 mM appears to be a generous and appropriate cutoff (24, 25).
Although studies have repeatedly shown that exposure to ≤1 μM iAs significantly decreases glucose-induced insulin secretion in clonal β-cells, it should be noted that the use of somewhat higher concentrations for short periods has spurned fruitful follow-up at lower, more physiological concentrations of iAs. For instance, Pi et al reported that 5 μM iAs significantly induced antioxidant gene expression (26), eventually leading to deeper investigation of the role of nuclear factor (erythroid-derived)-like 2 (Nrf2), a major antioxidant-regulating transcription factor, in the adaptive response to iAs. This launched a series of investigations that expanded our understanding of iAs's mechanism(s) of action (27, 28). Given the fine line between the concentration of iAs capable of inhibiting insulin secretion and cytotoxicity or the induction of apoptosis, this course of studies might be taken as an excellent example of how to successfully exploit shorter time courses and higher dosages to generate novel, environmentally-relevant findings.
Pancreatic iAs Accumulation, Speciation, Disposal
IAs undergoes several stages of metabolism once ingested (29). IAs enters cells via ion transporters, including aquaporin proteins 3, 7, and 9 and glucose transporter 1 (30–33). Although a reduction of these transporters protects cell lines against iAs toxicity (34), efficient cellular import of arsenicals is critical for normal in vivo iAs detoxification and urinary excretion (35). A single sub-lethal bolus of iAs (1 mg/kg) administered intraperitoneally to rats, mice, hamsters, and guinea pigs, can be cleared from the bloodstream in ~24 h (36). Once inside the cell, iAs may be conjugated to glutathione or methylated multiple times (37, 38). Once modified, methylated or glutathione-conjugated arsenicals are exported from the cell by ABC transporters, including multidrug resistance-associated proteins 1a, 1b, and 2 (39, 40), enabling more efficient renal excretion and minimizing internal exposure. Arsenical efflux activity partially determines an organism's sensitivity to iAs, as the activities and expression levels of these ABC transporters are critical for adaptation to iAs (31).
IAs is taken up by pancreatic tissue and β-cells dose-dependently (41). Of the in vivo rodent studies utilizing chronic iAs exposure considered here, several studies specifically measured iAs accumulation in the pancreas (Table 1). In all these cases except one, iAs accumulated significantly in the pancreas (41, 43, 44, 46, 57–60, 64, 74). In the one exception, with low levels of iAs used in a cocktail of other dilute toxins, no appreciable pancreatic iAs accumulation was observed (77). The concentration of iAs used in this study by Radike et al was lower than the World Health Organization's safe limit of 10 μg/L.
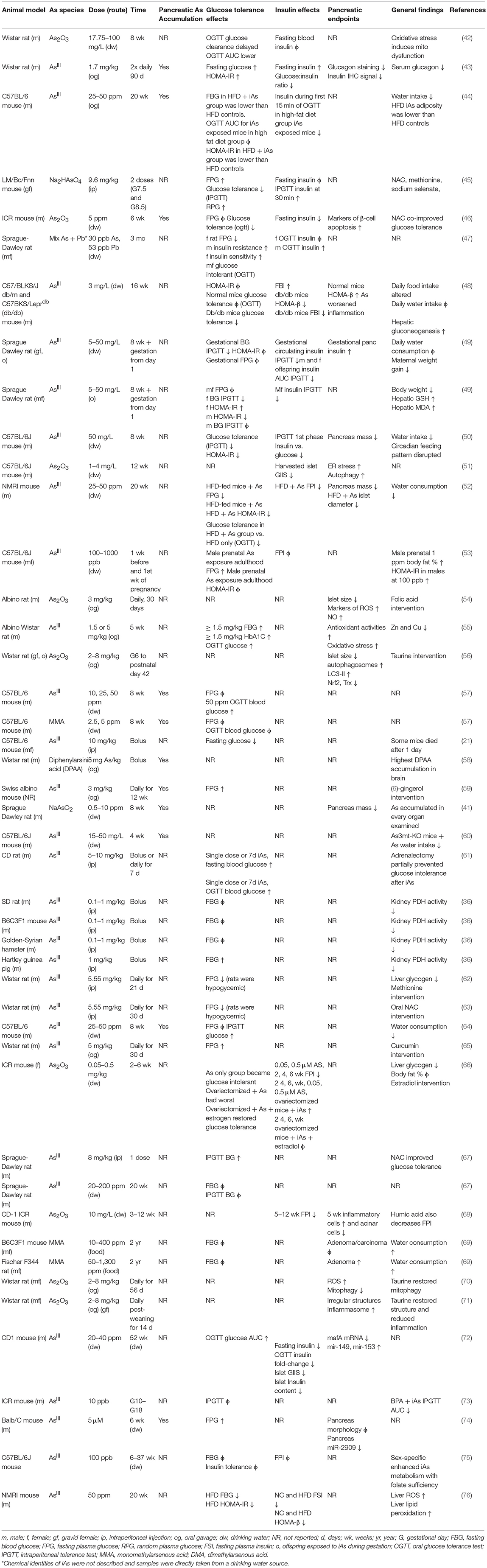
Table 1. Rodent models of iAs exposure with pancreatic endpoints.
Chronic administration of iAs in drinking water results in iAs accumulation as both iAs and methylated arsenicals in the pancreas, with the majority stored as monomethyl arsenous acid (MMA) or dimethylarsenous acid (DMA) (43, 44, 57, 60, 64). Isolated islet and β-cell cell line studies have demonstrated the ability of β-cells to methylate iAs intracellularly (78, 79). The physiological effects of these methylated arsenicals appear to be different from those of iAs. MMA can inhibit mitochondrial function and decrease glucose-induced insulin secretion, even at 5-fold lower concentrations than other arsenicals (80). Since MMA is necessarily created prior to repeated bouts of methylation resulting in dimethylation and trimethylation, it is noteworthy that this intermediate may be more toxic than its precursor or end-products.
The relationship between exposure and tissue-level accumulation as well as the propensity to induce DNA damage vary across organ systems. One study measured iAs accumulation and cytosine methylation in several tissues following 24 weeks of iAs administration (41). Relative to the lung, kidney, heart, and spleen, the pancreas accumulated less iAs as a function of exposure level and displayed a resistance to the iAs-induced 5-hydroxymethylation events observed in these other tissues. Despite this apparent resistance, the pancreas itself was smaller after adjusting for body weight in iAs-exposed mice, a phenomenon also observed by other groups (50, 52), raising the possibility that the organ may possess a unique resistance to iAs accumulation, but also a unique susceptibility to the effects of iAs exposure.
Model System Evidence for Pancreatic β-Cell Involvement
The data are mixed in animal models of iAs-induced metabolic dysfunction regarding the relative contributions of insulin-secretory vs. insulin-sensitivity factors in the development of glucose intolerance (Table 1). Impairments in pancreatic (41, 50–52, 54–56, 68, 69) and hepatic (48, 55, 59, 61–63, 66) function have been implicated. Insulin sensitivity, however, has been reported to increase (44), decrease (43, 47, 49, 53), or remain unchanged (44, 47, 50) in sex-specific or diet-specific manners, making the integration of this particular endpoint across studies more difficult. It is worth noting, however, that where insulin sensitivity was reported to increase, this was on a background of already-impaired diet-induced glucose intolerance in which Paul et al also reported both lower circulating insulin, lower adiposity, and lower HOMA-IR in the high-fat diet, iAs-treated group vs. high-fat diet controls. In this study as well as others, a reduction in circulating insulin was reported either following fasting or during a glucose tolerance test (44, 46, 50, 57–60, 64, 74, 77–80). In consideration of the studies examined here, it appears that a primary defect in β-cell function precedes the development of glucose intolerance, which may or may not include a component of insulin resistance. IAs may be protective against insulin resistance in diet-induced obesity while simultaneously impairing pancreatic β-cells, a model that deviates from the canonical type I, type II, or gestational forms of diabetes (81). Replication will be critical for reconciling the differences between insulin sensitivity outcomes in these recent animal models of iAs exposure.
In β-cell lines there is a consistently observed reduction in GIIS associated with chronic, sub-toxic iAs exposure (Table 2), although there is some disagreement in the literature about whether basal insulin secretion is altered by iAs exposure (23, 28, 42, 82, 83). Some of these differences may be related to the model systems employed. It is notable that, in cell line studies, the dosages and times used to study the effects of iAs exposure have varied dramatically. Timeframes utilized for studying the effects of GIIS have ranged from 1 to 144 h, with higher concentrations evaluated on shorter time courses, such as 5 mM iAs exposure for 60 min (22) or over 100 μM for 90 min (86, 87), and lower doses for longer time courses, such as 50 nM for 96 h (28). The lowest concentration thus far reported to significantly affect GIIS in cell culture studies is 0.1 μM (23).
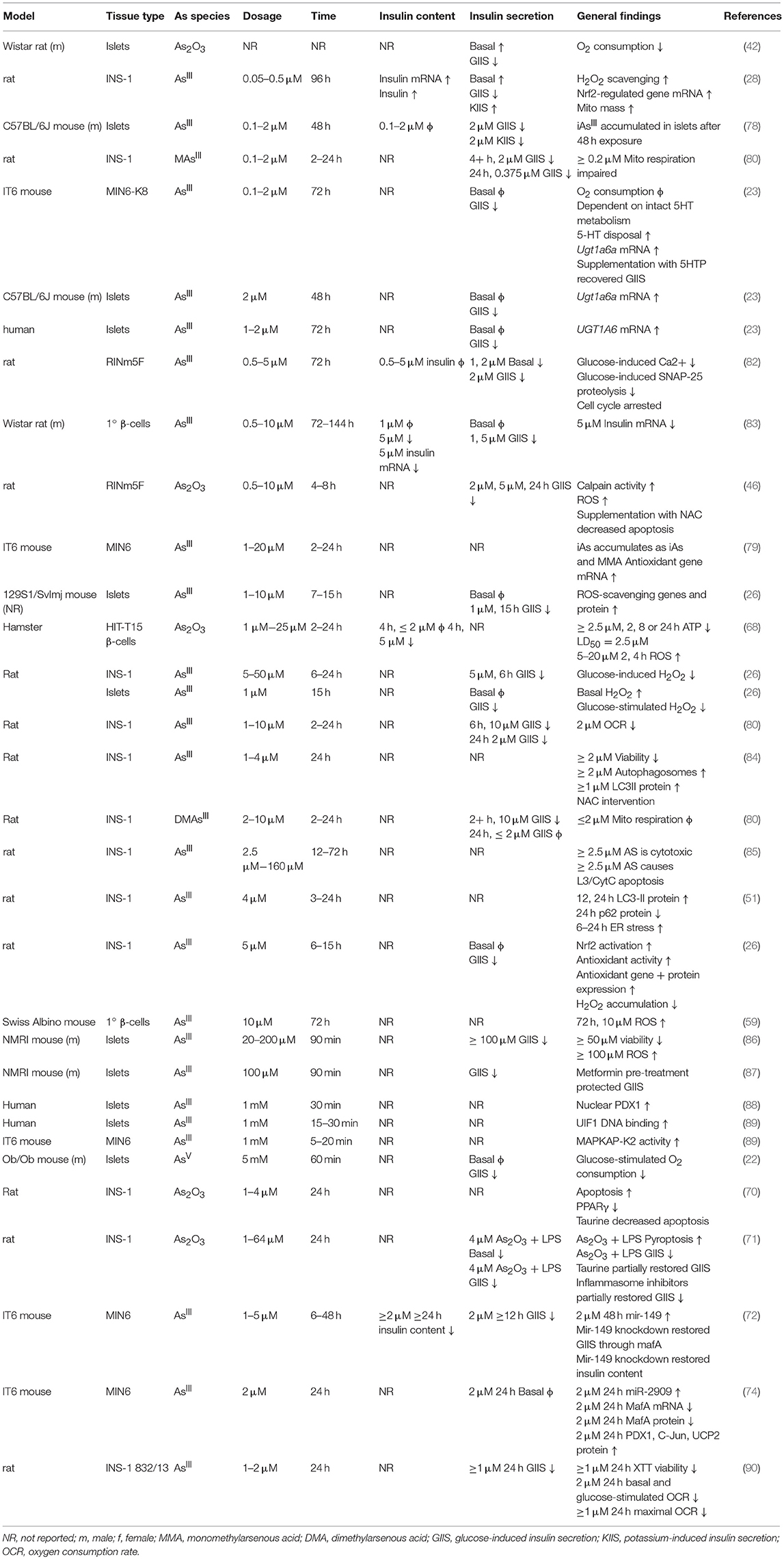
Table 2. Ex vivo models of iAs exposure.
Significant effects of iAs on insulin gene expression and transcription factor activities have also been reported. A decrease of MafA transcriptional activity regulated by miR-149 may contribute to the iAs-induced impairment of β-cell function (72). Such decreases in MafA, Pdx1, or Nkx6.1 are generally considered indications of β-cell failure or de-differentiation (91). Other model systems of iAs exposure, in which gene expression levels of these transcription factors were measured, did not report such a de-differentiation phenotype (23). One study even observed an increase in nuclear PDX1 following exposure to iAs, suggestive of increased insulin gene expression (88). DNA binding of the β-cell specific transcription factor UIF1, which promotes insulin gene expression, has also been observed to increase in response to iAs, suggesting a mechanism by which iAs may affect insulin content (89). Additional replication may therefore be warranted to identify which features of transcription factor activities are robust and translatable to human exposure.
Inflammation and Reactive-Oxygen Species (ROS)
ROS accumulation is a hallmark of iAs toxicity. There is strong evidence from both in vivo and in vitro studies suggesting that iAs damages pancreatic tissue, observed as elevated pro-inflammatory genes (48), pancreatic nitric oxide (54, 55) glutathione levels (43, 55), endoplasmic reticulum stress (51), and autophagy (51, 56). More severe phenotypes have been observed with increased apoptosis (46), decreased islet size (54), accumulation of pro-inflammatory cells (68), and detection of pancreatic adenomas (69). The generalized cellular responses to iAs-induced ROS have been reviewed in detail elsewhere (92). Markers of ROS have been observed at the lowest concentrations that also affect GIIS (28). Interestingly, arsenic trioxide (As2O3) appears to increase ROS production, apoptosis, and TUNEL staining while decreasing PPARγ in the INS-1 cell line. Restoration of normal ROS production by taurine administration or rescue of PPARγ expression ameliorates the apoptotic and DNA-damaging effects of As2O3 exposure (70). This is in line with similar observations for liver cells lines by the same group (93).
The ROS produced as a result of iAs exposure induce a compensatory increase in gene expression levels for genes regulated by antioxidant response elements (26–28, 94). These genes, which include catalase, superoxide dismutase 1, and superoxide dismutase 2, are critical for reducing otherwise toxic accumulation of ROS, and are positively regulated at the level of transcription by Nrf2 (94, 95). In β-cells specifically, induction of the Nrf2-mediated antioxidant-response program has been shown to protect against iAs-induced toxicity (94). Deletion of the major transcription factor regulating this pathway, Nrf2, in β-cells has been shown to enhance susceptibility to iAs toxicity (79). In the context of β-cell function, this antioxidant activity may actually suppress the normal physiological changes in ROS that β-cells depend on to induce insulin secretion in response to a rise in extracellular glucose (26). In this way, a tradeoff may occur in which β-cells' survival improves by adaptive upregulation of antioxidant activity, while at the same time glucose-induce insulin secretion is suppressed by the same mechanism (96). That several interventional studies focused on suppressing the antioxidant response successfully ameliorated some of the effects of iAs supports the hypothesis that ROS may be one of the salient, translatable, and addressable features of low-dose, chronic iAs exposure (Table 3) (45, 46, 53, 59, 63, 65, 67, 71, 75, 84, 97).
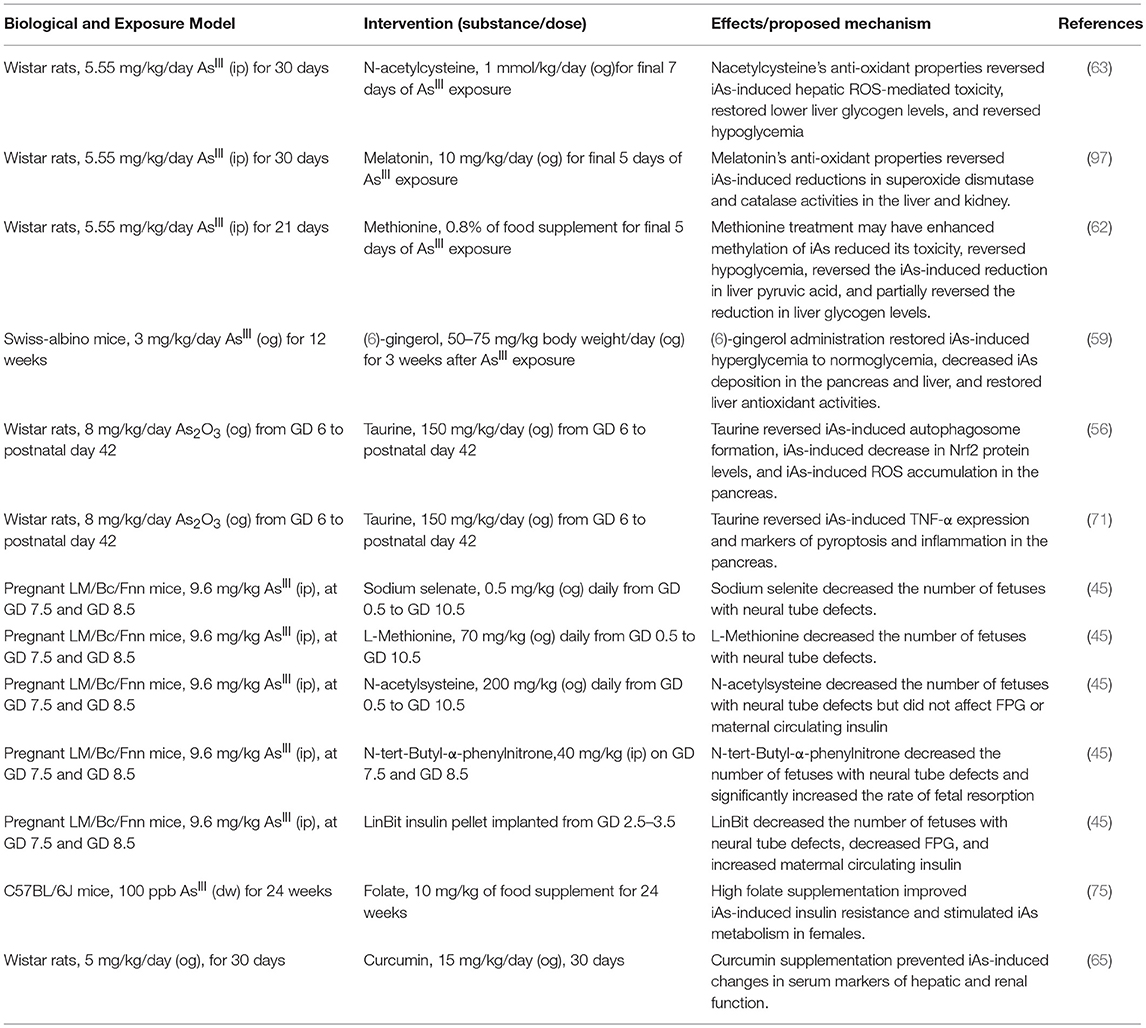
Table 3. In vivo interventional studies.
Although oxidative damage is a common observation following iAs exposure in many different tissues, the implications for ROS accumulation in β-cells may be unique. For instance, a recent meta-analysis of iAs-exposure studies in mice and rats indicated that iAs tends to decrease expression levels of key antioxidant genes (92). These include superoxide dismutase, catalase, and glutathione-peroxidase, among others. This is inverted compared to the response to iAs in β-cells, which manifests as increased expression of antioxidant genes, presumably to limit changes to the cellular redox state (27). This may be because glucose-induced insulin secretion in β-cells is partly mediated by relatively small changes in redox status (26). These cells are so sensitive to ROS that incubation with just 1 μM H2O2 affects basal insulin secretion (26), and just 100 μM H2O2 significantly decreases viability (98). In stark contrast, 250 μM H2O2 is used as a moderate positive control to quantify accumulation of ROS in liver cell lines (99). Thus, this unique sensitivity highlights the need to study the effects of iAs on β-cells directly, and not to rely too heavily on studies in other tissues.
Cytotoxicity, Autophagy, and the Cell Cycle
Indications of cytotoxicity, disrupted autophagy, or apoptosis have been observed in β-cell lines using concentrations of iAs as low as 1 μM, although the minimum threshold for toxic effects vary with cell line and duration of exposure (46, 70, 84). Reduced viability as measured by reducing potential has been observed in the MIN6 cell line following 24 h exposure to ≥1 μM arsenite (although reducing potential may also be affected by changes to cellular energetics independent of toxicity), with a 50% reduction in viability at approximately 5 μM and activation of the antioxidant-response gene expression program (79). The INS-1 line exposed to iAs for just 24 h showed significantly decreased proliferation at ≥2.5 μM, with decreased mitochondrial membrane potential and increased cytoplasmic cytochrome c, indicative of autophagy (85). This cell line at 1–2 μM iAs exposure also exhibits reduced oxygen consumption capacity and viability (90). Pan et al estimated the IC50 for INS-1 cells to be about 30 μM (85). By comparison, isolated islets exposed to 20–50 μM iAs for 24 h exhibited >50% islet destruction, suggesting that islets, INS-1 cells, and MIN6 cells may be similarly sensitive to the cytotoxic effects of iAs (100).
There is substantial evidence that chronic in vivo iAs exposure disrupts autophagy in other tissues. In one such study, 20 weeks of exposure to 50 ppm iAs in drinking water during high-fat diet administration significantly induced hepatic expression of 17 out of 21 autophagy-related genes examined, with the remaining 4 genes trending toward an increase. This was also accompanied by a significant increase in hepatic lipid peroxidation and ROS accumulation (76). In β-cell lines as well, investigators have observed iAs-induced changes in autophagy (51, 56, 84), often noting enhanced levels of the autophagy marker LC3-II.
There is some debate about whether autophagy induced by iAs in β-cells is mediated by ROS. Some investigators have found that iAs induces autophagy in an ROS-dependent fashion (84). Other groups using non-β-cell lines have shown that autophagy can be activated in the absence of excessive ROS generation, and have therefore concluded that iAs-induced autophagy is ROS-independent (99). That autophagy induction occurs at comparable doses of iAs in other tissues without significant ROS accumulation suggests that perhaps β-cells, while susceptible to ROS, may also be affected by parallel, ROS-independent iAs-induced autophagy. This may be considered an unresolved topic in the field and additional mechanistic investigations at environmentally-relevant concentrations of iAs are warranted.
Serotonin Metabolism
In mouse β-cells and islets, and to a lesser extent in human islets, serotonin regulates glucose-induced insulin secretion and proliferation (101, 102). Several parameters determine the concentration and effects of serotonin in β-cells, including serotonin production, serotonin disposal, and the specific distribution of serotonin receptors (103, 104). IAs exposure was recently observed to enhance serotonin disposal in the MIN6-K8 line by upregulation of the serotonin disposal gene Ugt1a6a. The upregulation phenomenon in response to iAs exposure was replicated in mouse islets, and the same pattern was observed for the human homolog of this gene, UGT1A6, in human islets upon chronic exposure to iAs (23). It is not clear what pathways are responsible for induction of Ugt1a6a, however Ugt1a6a expression is known in other tissues to be regulated by Nrf2 and the aryl hydrocarbon receptor. As Ugt1a6a was previously unappreciated as a regulator of β-cell function, this study highlights how EDCs such as iAs can be utilized to identify novel regulators of glucose-induced insulin secretion. Further study is warranted to evaluate the translatability of this work to animal models and cases of human exposure.
Considerations and Future Directions
Importantly, these studies intersect two major public health crises: chronic arsenic exposure and diabetes. In the past 20 years, substantial supporting evidence for the involvement of disrupted autophagy and oxidative damage as the major mediators of iAs-induced pancreatic β-cell dysfunction, manifesting as altered cell survival and impaired insulin secretion, has been reported both in vivo and in vitro (Figure 1). In more recent years, the concentrations used to study the phenomenon have decreased dramatically, and the lengths of exposure time have increased. These are positive trends for the field and are largely possible as a result of more sensitive analytical methods that are now more widespread. The use of these techniques has enabled the relatively recent discovery that MMA and DMA have different activities in β-cells and should be further explored. Now that human pancreatic islets are more widely available for research purposes throughout the world, replication of animal model findings in human islets is a more practical and reasonable option.
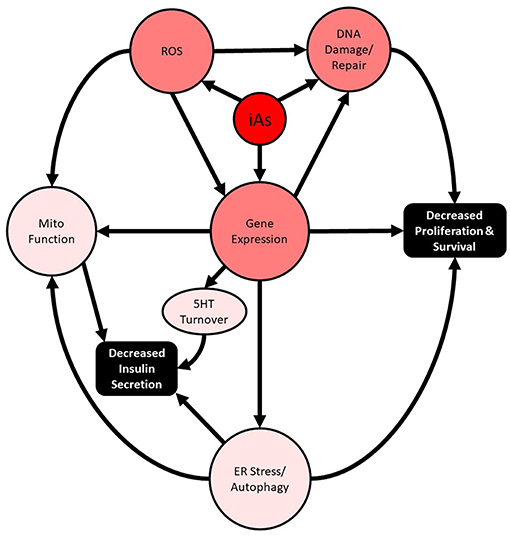
Figure 1. Summary of routes to iAs-induced toxicity and insulin-secretory effects.
With the largest population-scale exposures of arsenic ongoing in developing or impoverished nations, it is less likely that synthetic therapeutic interventions targeting β-cells (without broader applicability to arsenic-independent β-cell function) may find traction at the levels of commercial development and clinical use. Appropriately, interventional studies aimed at improving β-cell resistance to arsenic exposure appear designed in consideration of this fact, mostly utilizing relatively inexpensive nutritional supplementation that may also have more systemic benefits to arsenic-exposed individuals (Table 3). Thus, the model system research presented here provide evidence that optimal nutrition rich in natural antioxidants may improve β-cells' resistance to chronic arsenic exposure in vivo.
The problem of arsenic exposure through contaminated drinking water is ultimately addressable at the level of public policy. Though diabetes itself may feel less immediate than acutely-life-threatening afflictions associated with arsenic exposure (such as cancer, cardiovascular disease, and nephropathy), highlighting the diabetes link provides yet another mechanism by which the scientific community can provide lawmakers and policy officials with justification to prioritize access to clean and safe water. That population-level arsenic exposure has been a known problem for more than 30 years in Bangladesh alone, with other pockets of exposure around the globe, reveals a global failure of institutions to address the needs of the impoverished and exposed. As water treatment technology and infrastructure are developed to address the pressing dangers of arsenic exposure, they will undoubtedly reduce economic and inertial barriers to the further improvement of water quality. Future studies and research communications might emphasize the preventability of chronic arsenic exposure as a point of discussion in the hopes that the issue can be continually brought to the forefront of public concern.
Author Contributions
CC and SS conceived, authored, revised, and approved the manuscript and take responsibility for its publication.
Funding
CC received financial support from the Japan Society for the Promotion of Science Standard Term Postdoctoral Fellowship for Overseas Researchers. SS received financial support from the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (S) 24229007 and MSD K.K. Japan.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Robert M. Sargis and Daniel Ruiz for their productive feedback and insights.
References
1. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
2. Seino S, Shibasaki T, Minami K. Pancreatic beta-cell signaling: toward better understanding of diabetes and its treatment. Proc Jpn Acad Ser B Phys Biol Sci. (2010) 86:563–77. doi: 10.2183/pjab.86.563
3. Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. (2009) 52:739–51. doi: 10.1007/s00125-009-1314-y
4. Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, et al. β-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. (2014) 37:1751–8. doi: 10.2337/dc14-0396
5. Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest. (2011) 121:2118–25. doi: 10.1172/JCI45680
6. Bonini MG, Sargis RM. Environmental toxicant exposures and type 2 diabetes mellitus: two interrelated public health problems on the rise. Curr Opin Toxicol. (2018) 7:52–9. doi: 10.1016/j.cotox.2017.09.003
7. Singh AP, Goel RK, Kaur T. Mechanisms pertaining to arsenic toxicity. Toxicol Int. (2011) 18:87–93. doi: 10.4103/0971-6580.84258
8. Ratnaike RN. Acute and chronic arsenic toxicity. Postgrad Med J. (2003) 79:391–6. doi: 10.1136/pmj.79.933.391
9. Finkelman RB, Belkin HE, Zheng B. Health impacts of domestic coal use in China. Proc Natl Acad Sci USA. (1999) 96:3427–31. doi: 10.1073/pnas.96.7.3427
10. Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, et al. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect. (2013) 121:295–302. doi: 10.1289/ehp.1205875
11. Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, et al. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect. (2012) 120:1658–70. doi: 10.1289/ehp.1104579
12. Ravenscroft P, Brammer H, Richards K. Arsenic Pollution: A Global Synthesis. Wiley-Blackwell (2009).
13. Frumkin H. Toxicological Profile for Arsenic. Agency for Toxic Substances and Disease Registry, Agency for Toxic Substances and Disease Registry (2007).
14. Navas-Acien A, Francesconi KA, Silbergeld EK, Guallar E. Seafood intake and urine concentrations of total arsenic, dimethylarsinate and arsenobetaine in the US population. Environ Res. (2011) 111:110–8. doi: 10.1016/j.envres.2010.10.009
15. Johnston RB, Sarker MH. Arsenic mitigation in Bangladesh: national screening data and case studies in three upazilas. J Environ Sci Health A Tox Hazard Subst Environ Eng. (2007) 42:1889–96. doi: 10.1080/10934520701567155
16. Karagas MR. Arsenic-related mortality in Bangladesh. Lancet. (2010) 376:213–4. doi: 10.1016/S0140-6736(10)61002-1
17. Tseng CH, Tai TY, Chong CK, Tseng CP, Lai MS, Lin BJ, et al. Long-term arsenic exposure and incidence of non-insulin-dependent diabetes mellitus: a cohort study in arseniasis-hyperendemic villages in Taiwan. Environ Health Perspect. (2000) 108:847–51. doi: 10.1289/ehp.00108847
18. Lai M-S, Hsueh Y-M, Chen C-J, Shyu M-P, Chen S-Y, Kuo T-L, et al. Ingested inorganic arsenic and prevalence of diabetes mellitus. Am J Epidemiol. (1994) 139:484–92. doi: 10.1093/oxfordjournals.aje.a117031
19. Pan WC, Seow WJ, Kile ML, Hoffman EB, Quamruzzaman Q, Rahman M, et al. Association of low to moderate levels of arsenic exposure with risk of type 2 diabetes in Bangladesh. Am J Epidemiol. (2013) 178:1563–70. doi: 10.1093/aje/kwt195
20. Wang SL, Chiou JM, Chen CJ, Tseng CH, Chou WL, Wang CC, et al. Prevalence of non-insulin-dependent diabetes mellitus and related vascular diseases in southwestern arseniasis-endemic and nonendemic areas in Taiwan. Environ Health Perspect. (2003) 111:155–9. doi: 10.1289/ehp.5457
21. Boquist L, Boquist S, Ericsson I. Structural beta-cell changes and transient hyperglycemia in mice treated with compounds inducing inhibited citric acid cycle enzyme activity. Diabetes. (1988) 37:89–98. doi: 10.2337/diabetes.37.1.89
22. Ortsäter H, Liss P, Akerman KE, Bergsten P. Contribution of glycolytic and mitochondrial pathways in glucose-induced changes in islet respiration and insulin secretion. Pflugers Arch. (2002) 444:506–12. doi: 10.1007/s00424-002-0842-9
23. Carmean CM, Yokoi N, Takahashi H, Oduori OS, Kang C, Kanagawa A, et al. Arsenic modifies serotonin metabolism through glucuronidation in pancreatic β-cells. Am J Physiol Endocrinol Metab. (2019) 316:E464–74. doi: 10.1152/ajpendo.00302.2018
24. C.-Tseng H, The potential biological mechanisms of arsenic-induced diabetes mellitus. Toxicol Appl Pharmacol. (2004) 197:67–83. doi: 10.1016/j.taap.2004.02.009
25. Wu MM, Chiou HY, Ho IC, Chen CJ, Lee TC. Gene expression of inflammatory molecules in circulating lymphocytes from arsenic-exposed human subjects. Environ Health Perspect. (2003) 111:1429–38. doi: 10.1289/ehp.6396
26. Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. (2007) 56:1783–91. doi: 10.2337/db06-1601
27. Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, et al. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. (2010) 244:77–83. doi: 10.1016/j.taap.2009.05.025
28. Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, Collins S, et al. Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect. (2010) 118:864–70. doi: 10.1289/ehp.0901608
29. Thomas DJ. Molecular processes in cellular arsenic metabolism. Toxicol Appl Pharmacol. (2007) 222:365–73. doi: 10.1016/j.taap.2007.02.007
30. Liu Z, Shen J, Carbrey JM, Mukhopadhyay R, Agre P, Rosen BP. Arsenite transport by mammalian aquaglyceroporins AQP7 and AQP9. Proc Natl Acad Sci USA. (2002) 99:6053–8. doi: 10.1073/pnas.092131899
31. Lee TC, Ho IC, Lu WJ, Huang JD. Enhanced expression of multidrug resistance-associated protein 2 and reduced expression of aquaglyceroporin 3 in an arsenic-resistant human cell line. J Biol Chem. (2006) 281:18401–7. doi: 10.1074/jbc.M601266200
32. Liu Z, Sanchez MA, Jiang X, Boles E, Landfear SM, Rosen BP. Mammalian glucose permease GLUT1 facilitates transport of arsenic trioxide and methylarsonous acid. Biochem Biophys Res Commun. (2006) 351:424–30. doi: 10.1016/j.bbrc.2006.10.054
33. Shinkai Y, Sumi D, Toyama T, Kaji T, Kumagai Y. Role of aquaporin 9 in cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. Toxicol Appl Pharmacol. (2009) 237:232–6. doi: 10.1016/j.taap.2009.03.014
34. Leung J, Pang A, Yuen WH, Kwong YL, Tse EW. Relationship of expression of aquaglyceroporin 9 with arsenic uptake and sensitivity in leukemia cells. Blood. (2007) 109:740–6. doi: 10.1182/blood-2006-04-019588
35. Carbrey JM, Song L, Zhou Y, Yoshinaga M, Rojek A, Wang Y, et al. Reduced arsenic clearance and increased toxicity in aquaglyceroporin-9-null mice. Proc Natl Acad Sci USA. (2009) 106:15956–60. doi: 10.1073/pnas.0908108106
36. Mitchell RD, Ayala-Fierro F, Carter DE. Systemic indicators of inorganic arsenic toxicity in four animal species. J Toxicol Environ Health A. (2000) 59:119–34. doi: 10.1080/009841000157014
37. Drobna Z, Styblo M, Thomas DJ. An overview of arsenic metabolism and toxicity. Curr Protoc Toxicol. (2009) 42:4.31.1–6. doi: 10.1002/0471140856.tx0431s42
38. Watanabe T, Hirano S. Metabolism of arsenic and its toxicological relevance. Arch Toxicol. (2013) 87:969–79. doi: 10.1007/s00204-012-0904-5
39. Kala SV, Neely MW, Kala G, Prater CI, Atwood DW, Rice JS, et al. The MRP2/cMOAT transporter and arsenic-glutathione complex formation are required for biliary excretion of arsenic. J Biol Chem. (2000) 275:33404–8. doi: 10.1074/jbc.M007030200
40. Kala SV, Kala G, Prater CI, Sartorelli AC, Lieberman MW. Formation and urinary excretion of arsenic triglutathione and methylarsenic diglutathione. Chem Res Toxicol. (2004) 17:243–9. doi: 10.1021/tx0342060
41. Zhang J, Mu X, Xu W, Martin FL, Alamdar A, Liu L, et al. Exposure to arsenic via drinking water induces 5-hydroxymethylcytosine alteration in rat. Sci Total Environ. (2014) 497–8:618–25. doi: 10.1016/j.scitotenv.2014.08.009
42. Cobo JM, Castiñeira M. Oxidative stress, mitochondrial respiration, and glycemic control: clues from chronic supplementation with Cr3+ or As3+ to male Wistar rats. Nutrition. (1997) 13:965–70. doi: 10.1016/S0899-9007(97)00338-9
43. Izquierdo-Vega JA, Soto CA, Sanchez-Peña LC, De Vizcaya-Ruiz A, Del Razo LM. Diabetogenic effects and pancreatic oxidative damage in rats subchronically exposed to arsenite. Toxicol Lett. (2006) 160:135–42. doi: 10.1016/j.toxlet.2005.06.018
44. Paul DS, Walton FS, Saunders RJ, Stýblo M. Characterization of the impaired glucose homeostasis produced in C57BL/6 mice by chronic exposure to arsenic and high-fat diet. Environ Health Perspect. (2011) 119:1104–9. doi: 10.1289/ehp.1003324
45. Hill DS, Wlodarczyk BJ, Mitchell LE, Finnell RH. Arsenate-induced maternal glucose intolerance and neural tube defects in a mouse model. Toxicol Appl Pharmacol. (2009) 239:29–36. doi: 10.1016/j.taap.2009.05.009
46. Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, et al. Arsenic induces pancreatic β-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol Lett. (2011) 201:15–26. doi: 10.1016/j.toxlet.2010.11.019
47. Palacios J, Roman D, Cifuentes F. Exposure to low level of arsenic and lead in drinking water from Antofagasta city induces gender differences in glucose homeostasis in rats. Biol Trace Elem Res. (2012) 148:224–31. doi: 10.1007/s12011-012-9355-3
48. Liu S, Guo X, Wu B, Yu H, Zhang X, Li M. Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci Rep. (2014) 4:6894. doi: 10.1038/srep06894
49. Bonaventura MM, Bourguignon NS, Bizzozzero M, Rodriguez D, Ventura C, Cocca C, et al. Arsenite in drinking water produces glucose intolerance in pregnant rats and their female offspring. Food Chem Toxicol. (2017) 100:207–16. doi: 10.1016/j.fct.2016.12.025
50. Kirkley AG, Carmean CM, Ruiz D, Ye H, Regnier SM, Poudel A, et al. Arsenic exposure induces glucose intolerance and alters global energy metabolism. Am J Physiol Regul Integr Comp Physiol. (2017). doi: 10.1152/ajpregu.00522.2016
51. Wu W, Yao X, Jiang L, Zhang Q, Bai J, Qiu T, et al. Pancreatic islet-autonomous effect of arsenic on insulin secretion through endoplasmic reticulum stress-autophagy pathway. Food Chem Toxicol. (2018) 111:19–26. doi: 10.1016/j.fct.2017.10.043
52. Ahangarpour A, Alboghobeish S, Rezaei M, Khodayar MJ, Oroojan AA, Zainvand M. Evaluation of diabetogenic mechanism of high fat diet in combination with arsenic exposure in male mice. Iran J Pharm Res. (2018) 17:164–83.
53. Huang MC, Douillet C, Dover EN, Stýblo M. Prenatal arsenic exposure and dietary folate and methylcobalamin supplementation alter the metabolic phenotype of C57BL/6J mice in a sex-specific manner. Arch Toxicol. (2018) 92:1925–37. doi: 10.1007/s00204-018-2206-z
54. Mukherjee S, Das D, Mukherjee M, Das AS, Mitra C. Synergistic effect of folic acid and vitamin B12 in ameliorating arsenic-induced oxidative damage in pancreatic tissue of rat. J Nutr Biochem. (2006) 17:319–27. doi: 10.1016/j.jnutbio.2005.08.003
55. Patel HV, Kalia K. Role of hepatic and pancreatic oxidative stress in arsenic induced diabetic condition in Wistar rats. J Environ Biol. (2013) 34:231–6.
56. Bai J, Yao X, Jiang L, Qiu T, Liu S, Qi B, et al. Taurine protects against As2O3-induced autophagy in pancreas of rat offsprings through Nrf2/Trx pathway. Biochimie. (2016) 123:1–6. doi: 10.1016/j.biochi.2016.01.002
57. Paul DS, Devesa V, Hernandez-Zavala A, Adair BM, Walton FS, Drobnâ Z, et al. Environmental arsenic as a disruptor of insulin signaling. Met Ions Biol Med. (2008) 10:1–7.
58. Naranmandura H, Suzuki N, Takano J, McKnight-Whitford T, Ogra Y, Suzuki KT, et al. Systemic distribution and speciation of diphenylarsinic acid fed to rats. Toxicol Appl Pharmacol. (2009) 237:214–20. doi: 10.1016/j.taap.2009.03.023
59. Chakraborty D, Mukherjee A, Sikdar S, Paul A, Ghosh S, Khuda-Bukhsh AR. [6]-Gingerol isolated from ginger attenuates sodium arsenite induced oxidative stress and plays a corrective role in improving insulin signaling in mice. Toxicol Lett. (2012) 210:34–43. doi: 10.1016/j.toxlet.2012.01.002
60. Currier JM, Douillet C, Drobná Z, Stýblo M. Oxidation state specific analysis of arsenic species in tissues of wild-type and arsenic (+3 oxidation state) methyltransferase-knockout mice. J Environ Sci. (2016) 49:104–12. doi: 10.1016/j.jes.2016.06.018
61. Ghafghazi T, Ridlington JW, Fowler BA. The effects of acute and subacute sodium arsenite administration on carbohydrate metabolism. Toxicol Appl Pharmacol. (1980) 55:126–30. doi: 10.1016/0041-008X(80)90228-8
62. Pal S, Chatterjee AK. Protective effect of methionine supplementation on arsenic-induced alteration of glucose homeostasis. Food Chem Toxicol. (2004) 42:737–42. doi: 10.1016/j.fct.2003.12.009
63. Pal S, Chatterjee AK. Protective effect of N-acetylcysteine against arsenic-induced depletion in vivo of carbohydrate. Drug Chem Toxicol. (2004) 27:179–89. doi: 10.1081/DCT-120037501
64. Paul DS, Hernández-Zavala A, Walton FS, Adair BM, Dedina J, Matousek T, et al. Examination of the effects of arsenic on glucose homeostasis in cell culture and animal studies: development of a mouse model for arsenic-induced diabetes. Toxicol Appl Pharmacol. (2007) 222:305–14. doi: 10.1016/j.taap.2007.01.010
65. Yousef MI, El-Demerdash FM, Radwan FM. Sodium arsenite induced biochemical perturbations in rats: ameliorating effect of curcumin. Food Chem Toxicol. (2008) 46:3506–11. doi: 10.1016/j.fct.2008.08.031
66. Huang CF, Yang CY, Chan DC, Wang CC, Huang KH, Wu CC, et al. Arsenic exposure and glucose intolerance/insulin resistance in estrogen-deficient female mice. Environ Health Perspect. (2015) 123:1138–44. doi: 10.1289/ehp.1408663
67. Rezaei M, Khodayar MJ, Seydi E, Soheila A, Parsi IK. Acute, but not chronic, exposure to arsenic provokes glucose intolerance in rats: possible roles for oxidative stress and the adrenergic pathway. Can J Diabetes. (2017) 41:273–80. doi: 10.1016/j.jcjd.2016.10.008
68. Yen CC, Lu FJ, Huang CF, Chen WK, Liu SH, Lin-Shiau SY. The diabetogenic effects of the combination of humic acid and arsenic: in vitro and in vivo studies. Toxicol Lett. (2007) 172:91–105. doi: 10.1016/j.toxlet.2007.05.008
69. Arnold LL, Eldan M, van Gemert M, Capen CC, Cohen SM. Chronic studies evaluating the carcinogenicity of monomethylarsonic acid in rats and mice. Toxicology. (2003) 190:197–219. doi: 10.1016/S0300-483X(03)00165-3
70. Zhang Q, Bai J, Yao X, Jiang L, Wu W, Yang L, et al. Taurine rescues the arsenic-induced injury in the pancreas of rat offsprings and in the INS-1 cells. Biomed Pharmacother. (2019) 109:815–22. doi: 10.1016/j.biopha.2018.10.134
71. Pei P, Yao X, Jiang L, Qiu T, Wang N, Yang L, et al. Inorganic arsenic induces pyroptosis and pancreatic β cells dysfunction through stimulating the IRE1α/TNF-α pathway and protective effect of taurine. Food Chem Toxicol. (2019) 125:392–402. doi: 10.1016/j.fct.2019.01.015
72. Sun Q, Yang Q, Xu H, Xue J, Chen C, Yang X, et al. miR-149 negative regulation of mafA is involved in the arsenite-induced dysfunction of insulin synthesis and secretion in pancreatic beta cells. Toxicol Sci. (2019) 167:116–25. doi: 10.1093/toxsci/kfy150
73. Wang D, Zhu W, Yan S, Meng Z, Yan J, Teng M, et al. Impaired lipid and glucose homeostasis in male mice offspring after combined exposure to low-dose bisphenol A and arsenic during the second half of gestation. Chemosphere. (2018) 210:998–1005. doi: 10.1016/j.chemosphere.2018.07.094
74. Ramdas M, Sharma S, Kaul D, Bhatia A. Possible role of miR-2909 RNomics in arsenic mediated pancreatic β-cell dysfunction. J Trace Elem Med Biol. (2018) 50:263–7. doi: 10.1016/j.jtemb.2018.07.006
75. Huang MC, Douillet C, Dover EN, Zhang C, Beck R, Tejan-Sie A, et al. Metabolic phenotype of wild-type and As3mt-knockout C57BL/6J mice exposed to inorganic arsenic: the role of dietary fat and folate intake. Environ Health Perspect. (2018) 126:127003. doi: 10.1289/EHP3951
76. Zeinvand-Lorestani M, Kalantari H, Khodayar MJ, Teimoori A, Saki N, Ahangarpour A, et al. Autophagy upregulation as a possible mechanism of arsenic induced diabetes. Sci Rep. (2018) 8:11960. doi: 10.1038/s41598-018-30439-0
77. Radike M, Warshawsky D, Caruso J, Goth-Goldstein R, Reilman R, Collins T, et al. Distribution and accumulation of a mixture of arsenic, cadmium, chromium, nickel, and vanadium in mouse small intestine, kidneys, pancreas, and femur following oral administration in water or feed. J Toxicol Environ Health A. (2002) 65:2029–52. doi: 10.1080/00984100290071324
78. Douillet C, Currier J, Saunders J, Bodnar WM, Matoušek T, Stýblo M. Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol Appl Pharmacol. (2013) 267:11–5. doi: 10.1016/j.taap.2012.12.007
79. Cui Q, Fu J, Hu Y, Li Y, Yang B, Li L, et al. Deficiency of long isoforms of Nfe2l1 sensitizes MIN6 pancreatic β cells to arsenite-induced cytotoxicity. Toxicol Appl Pharmacol. (2017) 329:67–74. doi: 10.1016/j.taap.2017.05.013
80. Dover EN, Beck R, Huang MC, Douillet C, Wang Z, Klett EL, et al. Arsenite and methylarsonite inhibit mitochondrial metabolism and glucose-stimulated insulin secretion in INS-1 832/13 β cells. Arch Toxicol. (2018) 92:693–704. doi: 10.1007/s00204-017-2074-y
81. DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. (2015) 1:15019. doi: 10.1038/nrdp.2015.19
82. Díaz-Villaseñor A, Burns AL, Salazar AM, Sordo M, Hiriart M, Cebrián ME, et al. Arsenite reduces insulin secretion in rat pancreatic beta-cells by decreasing the calcium-dependent calpain-10 proteolysis of SNAP-25. Toxicol Appl Pharmacol. (2008) 231:291–9. doi: 10.1016/j.taap.2008.05.018
83. Díaz-Villaseñor A, Sánchez-Soto MC, Cebrián ME, Ostrosky-Wegman P, Hiriart M. Sodium arsenite impairs insulin secretion and transcription in pancreatic beta-cells. Toxicol Appl Pharmacol. (2006) 214:30–4. doi: 10.1016/j.taap.2005.11.015
84. Zhu XX, Yao XF, Jiang LP, Geng CY, Zhong LF, Yang G, et al. Sodium arsenite induces ROS-dependent autophagic cell death in pancreatic β-cells. Food Chem Toxicol. (2014) 70:144–50. doi: 10.1016/j.fct.2014.05.006
85. Pan X, Jiang L, Zhong L, Geng C, Jia L, Liu S, et al. Arsenic induces apoptosis by the lysosomal-mitochondrial pathway in INS-1 cells. Environ Toxicol. (2016) 31:133–41. doi: 10.1002/tox.22027
86. Ahangarpour A, Oroojan AA, Rezae M, Khodayar MJ, Alboghobeish S, Zeinvand M. Effects of butyric acid and arsenic on isolated pancreatic islets and liver mitochondria of male mouse. Gastroenterol Hepatol Bed Bench. (2017) 10:44–53.
87. Ahangarpour A, Zeidooni L, Rezaei M, Alboghobeish S, Samimi A, Oroojan AA. Protective effect of metformin on toxicity of butyric acid and arsenic in isolated liver mitochondria and langerhans islets in male mice: an in vitro study. Iran J Basic Med Sci. (2017) 20:1297–305. doi: 10.22038/IJBMS.2017.9567
88. Macfarlane WM, McKinnon CM, Felton-Edkins ZA, Cragg H, James RF, Docherty K. Glucose stimulates translocation of the homeodomain transcription factor PDX1 from the cytoplasm to the nucleus in pancreatic beta-cells. J Biol Chem. (1999) 274:1011–6. doi: 10.1074/jbc.274.2.1011
89. Macfarlane WM, Smith SB, James RF, Clifton AD, Doza YN, Cohen P, et al. The p38/reactivating kinase mitogen-activated protein kinase cascade mediates the activation of the transcription factor insulin upstream factor 1 and insulin gene transcription by high glucose in pancreatic beta-cells. J Biol Chem. (1997) 272:20936–44. doi: 10.1074/jbc.272.33.20936
90. Dover EN, Patel NY, Stýblo M. Impact of in vitro heavy metal exposure on pancreatic β-cell function. Toxicol Lett. (2018) 299:137–44. doi: 10.1016/j.toxlet.2018.09.015
91. Shirakawa J, Terauchi Y. Selective and sequential loss of transcriptional factors: a hallmark of β-cell failure in type 2 diabetes? J Diabetes Investig. (2014) 5:359–61. doi: 10.1111/jdi.12212
92. Xu M, Rui D, Yan Y, Xu S, Niu Q, Feng G, et al. Oxidative damage induced by arsenic in mice or rats: a systematic review and meta-analysis. Biol Trace Elem Res. (2017) 176:154–75. doi: 10.1007/s12011-016-0810-4
93. Bai J, Yao X, Jiang L, Zhang Q, Guan H, Liu S, et al. Taurine protects against As2O3-induced autophagy in livers of rat offsprings through PPARγ pathway. Sci Rep. (2016) 6:27733. doi: 10.1038/srep27733
94. Fu J, Zheng H, Wang H, Yang B, Zhao R, Lu C, et al. Protective role of nuclear factor E2-related factor 2 against acute oxidative stress-induced pancreatic β -cell damage. Oxid Med Cell Longev. (2015) 2015:639191. doi: 10.1155/2015/639191
95. Wang H, Zhu J, Li L, Li Y, Lv H, Xu Y, et al. Effects of Nrf2 deficiency on arsenic metabolism in mice. Toxicol Appl Pharmacol. (2017) 337:111–9. doi: 10.1016/j.taap.2017.11.001
96. Fu J, Hou Y, Xue P, Wang H, Xu Y, Qu W, et al. Nrf2 in type 2 diabetes and diabetic complications: Yin and Yang. Curr Opin Toxicol. (2016) 1:9–19. doi: 10.1016/j.cotox.2016.08.001
97. Pal S, Chatterjee AK. Possible beneficial effects of melatonin supplementation on arsenic-induced oxidative stress in Wistar rats. Drug Chem Toxicol. (2006) 29:423–33. doi: 10.1080/01480540600837993
98. Meares GP, Fontanilla D, Broniowska KA, Andreone T, Lancaster JR, Corbett JA. Differential responses of pancreatic β-cells to ROS and RNS. Am J Physiol Endocrinol Metab. (2013) 304:E614–22. doi: 10.1152/ajpendo.00424.2012
99. Dodson M, de la Vega MR, Harder B, Castro-Portuguez R, Rodrigues SD, Wong PK, et al. Low-level arsenic causes proteotoxic stress and not oxidative stress. Toxicol Appl Pharmacol. (2018) 341:106–13. doi: 10.1016/j.taap.2018.01.014
100. Yang B, Fu J, Zheng H, Xue P, Yarborough K, Woods CG, et al. Deficiency in the nuclear factor E2-related factor 2 renders pancreatic β-cells vulnerable to arsenic-induced cell damage. Toxicol Appl Pharmacol. (2012) 264:315–23. doi: 10.1016/j.taap.2012.09.012
101. Ohta Y, Kosaka Y, Kishimoto N, Wang J, Smith SB, Honig G, et al. Convergence of the insulin and serotonin programs in the pancreatic β-cell. Diabetes. (2011) 60:3208–16. doi: 10.2337/db10-1192
102. Ohara-Imaizumi M, Kim H, Yoshida M, Fujiwara T, Aoyagi K, Toyofuku Y, et al. Serotonin regulates glucose-stimulated insulin secretion from pancreatic β cells during pregnancy. Proc Natl Acad Sci USA. (2013) 110:19420–5. doi: 10.1073/pnas.1310953110
103. Almaça J, Molina J, Menegaz D, Pronin AN, Tamayo A, Slepak V, et al. Human beta cells produce and release serotonin to inhibit glucagon secretion from alpha cells. Cell Rep. (2016) 17:3281–91. doi: 10.1016/j.celrep.2016.11.072
Keywords: arsenic, diabetes, pancreas, β-cells, reactive oxygen species, glucose tolerance, endocrine disrupters, insulin secretion
Citation: Carmean CM and Seino S (2019) Braving the Element: Pancreatic β-Cell Dysfunction and Adaptation in Response to Arsenic Exposure. Front. Endocrinol. 10:344. doi: 10.3389/fendo.2019.00344
Received: 03 October 2018; Accepted: 13 May 2019;
Published: 14 June 2019.
Edited by:
Yann Gibert, University of Mississippi Medical Center, United StatesReviewed by:
Alessandro Cavarape, University of Udine, ItalyDaniel Hesselson, Garvan Institute of Medical Research, Australia
Copyright © 2019 Carmean and Seino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. Carmean, Y2hyaXN0b3BoZXIuY2FybWVhbkBnbWFpbC5jb20=; Susumu Seino, c2Vpbm9AbWVkLmtvYmUtdS5hYy5qcA==