 Meital Charni-Natan
Meital Charni-Natan Ronit Aloni-Grinstein
Ronit Aloni-Grinstein Etty Osher3
Etty Osher3- 1Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel
- 2Department of Biochemistry and Molecular Genetics, Israel Institute for Biological Research, Ness-Ziona, Israel
- 3Sackler Faculty of Medicine, Tel Aviv-Sourasky Medical Center, Institute of Endocrinology Metabolism and Hypertension, Tel Aviv University, Tel Aviv, Israel
The liver is the main metabolic organ in the body, serving as a significant hormonal secretory gland and functioning to maintain hormone balance and homeostasis. Steroid hormones regulate various biological pathways, mainly in the reproductive system and in many metabolic processes. The liver, as well as steroid hormones, contribute significantly, through functional intertwine, to homeostasis maintenance, and proper responses during stress. Malfunction of either has a significant impact on the other and may lead to severe liver diseases as well as to several endocrine syndromes. Thus, the regulation on liver functions as on steroid hormones levels and activities is well-controlled. p53, the well-known tumor suppressor gene, was recently found to regulate metabolism and general homeostasis processes, particularly within the liver. Moreover, p53 was shown to be involved in steroid hormones regulation. In this review, we discuss the bi-directional regulation of the liver and the steroid hormones pointing to p53 as a novel regulator in this axis. A comprehensive understanding of the molecular mechanisms of this axis may help to prevent and treat related disease, especially with the increasing exposure of the population to environmental steroid hormones and steroid hormone-based medication.
Introduction
Steroid hormones are involved in regulation of different biological pathways mainly in the reproductive system and in metabolic homeostasis maintenance. Steroid hormones are derived from cholesterol and are synthesized primarily by endocrine glands such as the adrenal cortex, gonads (testes and ovaries), and placenta. They are classified to five main groups, based on their corresponding nuclear receptor: mineralocorticoids, glucocorticoids, androgens, estrogens, and progestogens (1–5). The nuclear steroid receptors act as ligand-activated transcriptional activators, which may regulate different genes expression (6). Steroid hormones affect the entire body homeostasis. Thus, it is not surprising that their regulation is complex and based on several parameters including production and degradation, activation vs. inactivation and the proportion between free and bound circulating steroid compounds (7, 8).
Furthermore, this multi-level regulation takes place not only in steroidogenic tissues but also in different peripheral organs, mainly in the liver. The liver, as a central metabolic organ, plays a crucial role in steroid hormones homeostasis (9, 10) and in the elimination of toxic metabolites, which may be destructive to the tissue and in the end to the whole body, leading to ongoing stress and liver diseases. The fact that there is a tight relationship between liver pathologies and steroids hormones (Table 1), together with the enormous potential held in the use of hormone therapy for different diseases, raises the necessity of a better understanding the mechanisms underlying the liver regulation of steroid hormone balance and function. Interestingly the transcription factor p53, the well-known guardian of the genome, which reacts upon stress signals to maintain genome fidelity, was also suggested to control the entire organism homeostasis (11, 12), and to regulate many processes in the liver including steroid hormone regulation (13, 14).
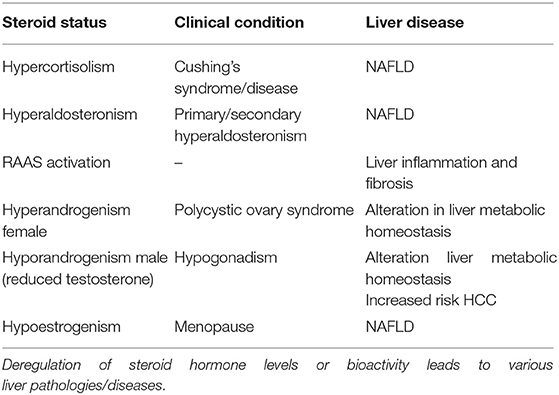
Table 1. Steroid hormone deregulation and liver pathologies.
In this review, we discuss the interesting bidirectional relationship between the different steroid hormones and the liver, independently and dependently on p53, in the context of health and liver diseases.
Steroid Hormones Homeostasis Regulation by the Liver
The liver participates in most steps of steroid hormone regulation starting from biosynthesis of cholesterol, which is the main source for steroids biosynthesis. Cholesterol is obtained either by de-novo production, hydrolysis of stored cholesterol, interiorization of plasma membrane cholesterol, or from LDL and HDL, which are secreted from the liver to the plasma (15). Subsequently, the cholesterol is processed to steroids by several enzymatic steps, which occur mainly in steroidogenic organs. Further, metabolism of these steroids also occurs in the liver. For example, the liver enzyme 5α-reductase 1 (5αR1) regulates metabolic processes of both androgens and glucocorticoids (GC) (16, 17). Studies have shown that 5αR1-KO mice exhibited augmented mRNA levels of various hepatic metabolic regulators genes (e.g., Acc1, Agpat2, Cpt2, and Dgat2) in comparison to WT mice, suggesting a crucial role of the liver enzyme, 5αR1, in the regulation of GC and androgens accumulation and effects (18).
Another example is the cytochromes P450 enzymes (CYP), which are responsible for the metabolism of many drugs and lipophilic compounds (19). CYP3A4, CYP19, CYP2C2B1, and CYP2C11 are the liver CYPs that take part in steroid hormones hydroxylation and processing. CYP3A4 hydroxylases several steroids such as cortisol, androstenedione, testosterone, and progesterone (20, 21), CYP19 (Aromatase) transforms androgens to estrogens by the removal of C19 carbon and the aromatization of the steroid A ring, while CYP2C11 and CYP2B1 regulate hydroxylation of testosterone (10, 22). Accumulating data suggest that different mechanisms underlay liver regulation on CYP expression levels. Several nuclear receptors complexes such as PXR, VDR, RXR were also found to bind CYP3A4 chromatin and affect its expression (23, 24).
Additional processes such as steroids conjugation, are exerted by specific enzymes such as sulfotransferases and the uridine diphosphate-glucuronosyltransferases (UGT) that transfer the steroid hormones into higher polarity metabolites that are better suited to be excreted from the body (10). The sulfotransferases that are expressed by hepatic cells and are related to steroids conjugations are HSST, EST, SULT 2A1, and SULT 1E1 (25). Studies have shown that sulfotransferase inhibition, as well as EST KO, led to the acceleration of free steroids and thus to sexual abnormalities (10, 26). Interestingly, the sulfotransferase expression was found to be regulated by androgens, GCs, and nuclear receptors such as PXR (27), suggesting a possible regulatory feedback between the hepatic enzymes that are part of the steroid hormones processing and the steroid hormones themselves.
UGT enzymes consist of two subfamilies; the UGT2B subfamily is mainly expressed in the liver and is related to the processing of steroid hormones (28). UGT enzymes induce glucuronidation of steroids, a process that interrupts steroids activity, and enables their elimination. UGTs enzymes are regulated by several xenobiotics compounds (e.g., PCN, PB), which were reported to increase their mRNA expression levels in rats' livers (29).
As steroids, these hormones are lipophilic thus, when secreted into the blood stream they need to be bound to carrier proteins such as Sex hormone binding globulin (SHBG), Corticosteroid binding globulin (CBG) and to lesser extent albumin, which facilitate their transportation to their target organs. These carriers are glycoproteins which are secreted mainly by the liver and bind with high affinity cortisol, testosterone, and estradiol (30, 31). In addition to their role as carrier proteins, they serve as a buffer for steroid hormones homeostasis and balance. The steroid hormones binding proteins have a principal role in hormone regulation. On the one hand, based on the “free hormone hypothesis,” the carrier proteins bind steroids and turn them to be biologically inactive (32). On the other hand, it was shown that both SHBG and CBG could bind to their target cells receptors and effect different molecular pathways and signaling such as apoptosis (33, 34). Several factors regulate SHBG expression in the liver including metabolic and hormonal factors such as glucose (35), thyroid hormone (36), and other factors such as the hepatocyte nuclear factor 4α (HNF-4α) (37), PPAR-γ (38), and p53 (14). Together, these findings point out that the liver, as the primary organ that produces and secrets these carrier proteins to the plasma (39), is the central regulator of steroid hormones bioavailability.
Glucocorticoids
Glucocorticoids are vital endocrine regulators of homeostasis and adaptation to environmental variations. GC is widely used clinically as a potent anti-inflammatory and immunosuppressive agents (40). They act via genomic (transcriptional) and non-genomic mechanisms. Upon binding to their cognate intracellular receptor (GR), the complex is translocated into the nucleus, and regulates various genes transcription (40, 41). Cortisol, the main glucocorticoid secreted by the adrenal cortex, is essential for various cellular functions including the immune system, vascular tone maintenance, and more (42). Cortisol is a major player during stress and severe illness mainly by increasing cardiac output and vascular tonus and decreasing pro-inflammatory cytokines release (43, 44). Cortisol levels are under the control of the hypothalamus-pituitary-adrenal (HPA) axis through the adrenocorticotropic hormone (ACTH) and the corticotropin-releasing hormone (CRH) (42). Eighty percent of the circulating cortisol is synthesized mainly from the liver- secreted high-density lipoproteins (HDL) cholesterol. Thus, in severe low-density lipoproteins (LDL) cholesterol insufficiency, due to enzymatic metabolic errors or acquired cases, cortisol production can be impaired (45).
Glucocorticoids and Liver Diseases
Non-alcoholic fatty liver disease (NAFLD) is a metabolic disorder characterized by hepatic steatosis, namely, the presence of free fatty acids or triglycerides in the liver (46). Studies in animal models have shown that rodents on a high fat diet and chronically elevated GC had a dramatic exacerbation in the development of NAFLD (47). Similarly, patients with elevated GC levels, as in Cushing's syndrome, are prone to develop features of NAFLD (48). NAFLD, as well as other forms of liver diseases, can develop into cirrhosis, which is a late stage of scarring (fibrosis) of the liver. Cirrhosis characterized by low arterial pressure, circulatory failure, vasodilation, and increased production of cytokines (45). Cirrhotic patients were reported to suffer from adrenal insufficiency (AI), a condition where the adrenal glands cannot synthesize the adequate amount of cortisol (49). This is mainly due to their low cholesterol levels and to increased cytokines production such as TNF α, IL-6, IL-1, and endotoxin like lipopolysaccharide, that over stimulate and exhaust the HPA axis (50–53). The accumulating observations of the high prevalence (40–60%) of AI (54) in various stages of cirrhosis suggests that this disease may have a predisposition to AI, which may be a feature of liver disease per se (55–58). Indeed, using “conservative” diagnostic criteria, Marik et al. (59) reported a surprisingly high incidence of AI in a large cohort of critically ill patients with liver disease and coined the term “hepatoadrenal syndrome” to describe the association between adrenal dysfunction and liver disease. Rauschecker et al. demonstrated that AI is common, in patient with cirrhosis. This was done by using serum free cortisol for diagnosis, as protein abnormalities might affect interpretation of total serum cortisol levels (60).
It seems that end stage of liver disease may coexist with AI and on the contrary, excess glucocorticoids have been implicated in the pathogenesis of liver disease. Patients with NAFLD seem to have a subtle chronic activation of the hypothalamic pituitary adrenal axis leading to a state of subclinical hypercortisolism. Suggesting a bidirectional relationship between liver and the adrenal (61).
Mineralocorticoids
Mineralocorticoids are hormones synthesized by the adrenal cortex that influence salt and water balance. Among the mineralocorticoids, the renin-angiotensin-aldosterone system (RAAS) is the principal hormonal system responsible for regulating cardiovascular, renal, and adrenal function and maintaining blood pressure and electrolyte balance (62).
Hepatocytes synthesis angiotensinogen (AGT) which is released into the bloodstream and transformed into angiotensin I and then to angiotensin II (ANG II), the main effector of this system. The significant biological actions of ANG II are mediated by binding two types of G protein coupled receptors, the angiotensin type 1 (AT1), and AT2 receptors. They can induce vasoconstriction and the release of aldosterone, which causes sodium and water reabsorption (63, 64). The AT1 and AT2 receptors are abundant in different tissues. AT1 receptors are expressed in hepatocytes, bile duct cells, hepatic stellate cells (HSC), myofibroblasts, and vascular endothelial cells (65). ANG II activates the AT1 receptors and induces HSC contraction and proliferation, oxidative stress, and inflammation responses, and endothelial cells dysfunction and growth (66). Moreover, in fibroblasts it upregulates TGF-β and collagen 1 gene expression (67–69). AT2 receptors were also found to be expressed in the liver (70) with possible anti-fibrogenic effects (71).
Mineralocorticoids and Liver Diseases
Renin-angiotensin-aldosterone system has an important role not only in the vascular system but also in different organs such as the liver. RAAS is implicated in various liver pathologies. Results of a prospective study suggest that patients with Hyperaldosteronism, a condition in which there is excessive secretion of aldosterone, frequently exhibit NAFLD (72). Indeed, ANG II was shown to be involved in cirrhosis-related portal hypertension (73, 74) and in the pathogenesis of insulin resistance and NAFLD, through its role in liver inflammation and fibrosis development (75, 76). Infusion of ANG II into mice was shown to upregulate TGF-β in normal rat livers (77), suggesting that upregulation of RAAS is sufficient to cause damage even in the absence of an underlying liver disease process (78). Thus, it is not surprising that due to the involvement of the RAAS system in liver disease, there is a growing interest in using RAAS inhibitors to treat NAFLD. Indeed, blocking RAAS by angiotensin-converting enzyme inhibitors and angiotensin receptor blockers reduced fibrosis in an experimental model of hepatic fibrosis (79).
Transcriptional Regulation of RAAS in Liver Diseases
Angiotensinogen has been suggested to be a key genetic determinant of RAAS. In experimental animal models a change, as little as 20%, in AGT expression levels are reflected by high blood pressure phenotype. In vivo studies have revealed that direct-repeat motifs (AGGTCA) within the human AGT gene promoter are functionally required for its expression in the liver (80). Examination of the human AGT gene-regulatory sequence revealed a single nucleotide polymorphism, resulting in two haplotypes. Haplotype I is associated with increased blood pressure, whereas, haplotype II is related to normal blood pressure. Interestingly, physiological changes, such as high fat diet, might change the transcriptional environment, and cause a modulation in AGT gene expression in a polymorphism dependent manner. Moreover, following high fat diet, increased levels of the transcription factors GR, HNF-1, STAT-3, and C/EBPβ were noticed in the liver and adipose tissues, leading to an increased expression of the AGT gene in liver tissue of haplotype I compared with haplotype II. These findings may have a significant clinical impact allowing the early identification and treatment of patients with the unfavorable haplotype (haplotype I) (81).
Androgens
Androgens are the principal male sex hormones that regulate masculinizing effects and male sexual behavior. While the major circulating androgens are dehydroepiandrosterone (DHT), androstenedione, testosterone, and dihydrotestosterone, only testosterone and DHT can bind to the androgen receptor (AR) (82). Testosterone is considered to be the most significant androgen in humans (82), playing a significant role in controlling metabolism processes of carbohydrate, fat, glycogen, lipids, and cholesterol (83). Thus, it is not surprising that testosterone deficiency usually is characterized with liver diseases (83).
Androgens and Liver Diseases
Studies in men have shown an association between hepatic steatosis and low levels of serum testosterone (84). Moreover, mice with non-functional 5αR1, the enzyme that converts testosterone to DHT, exhibit hepatic steatosis (85, 86). Interestingly, ARs in male liver tissue have a critical role in maintaining lipid metabolism compared to female (83, 87). However, testosterone can also participate in the hepatic lipid deposition, independently of the classic AR, operating by regulating several critical lipogenic enzymes activity (88).
Androgens play a role in glucose and cholesterol homeostasis of the liver. Androgens regulate liver glucose homeostasis with gender differences; while in males high testosterone favors hepatic glucose metabolism in females it impairs (89). Several longitudinal studies have shown that low levels of testosterone independently predict the later development of type 2 diabetes or metabolic syndrome (89). Moreover, prostate cancer patients that are undergoing androgen deprivation therapy are under increased risk to develop diabetes (90, 91). Testosterone treatment in men with hypogonadism is associated with a significant reduction in fasting plasma glucose, HbA1c, fat mass, and triglycerides (92).
Low levels of testosterone have been associated with increased levels of LDL cholesterol, triglycerides, and with decreased HDL levels (83). Androgen replacement treatments resulted in decrease in serum LDL levels by enhancing liver cholesterol uptake, suppressing cholesterol removal, and promoting cholesterol storage (93).
Liver malfunction, manifested by the alteration in its metabolic homeostasis regulation, can pave the way to liver tumor development (94). Since hepatocellular carcinoma (HCC) is more prevalent in men rather than women, sex hormones might be involved in this malignant process. Indeed, higher levels of androgen signaling, reflected by higher testosterone levels, were found to increase the risk for HBV-related HCC in men (95). Moreover, long-term use of oral contraceptives and anabolic androgenic steroids can induce both benign and malignant hepatocellular tumors (96). Additional evidence comes from the fact that individuals with HCC express augmented levels of ARs in their tumor tissue and in the surrounding liver (97). These data support the notion that AR could affect HCC progression and that a combination of sorafenib, (kinase inhibitor drug approved for cancer treatment), together with AR inhibitors, might be a potentially improve treatment for patients with advanced HCC (98).
Estrogens
Estrogens are the female principal sex hormones that regulate female reproductive, physiology, and sexual behavior. In humans, the most essential biologically relevant form of estrogens is the 17β estradiol (E2). While in premenopausal women E2 is mainly produced from cholesterol, in postmenopausal women it is primarily converted from testosterone by aromatase (99). E2 exerts its functions by binding to both the nuclear (ER) (ER-α and ER-β) and the membrane estrogen receptors (100). In addition to their expression in reproductive organs, ERs are also expressed to a lower extent in the liver (101–103).
Estrogens and Liver Diseases
Several studies have demonstrated that mimicking of menopause and reduction in estrogen signaling lead to insulin resistance, fatty liver, and dyslipidemia, as determined by decreased HDL and increased LDL and triglycerides (104–106). Indeed, while treatment with a specific ER-α agonist decreases fat mass and triglycerides, ER-α KO mice accumulate liver triglycerides and diglycerides (107–110). Furthermore, while estrogen deficiency causes steatosis, estrogen replacement decreases steatosis (111–113). Thus, it seems that estrogen together with ER-α plays a role in preventing liver malfunctioning. These data are in line with the observation that women in menopause are in a higher risk to develop NAFLD (114). Moreover, hormone treatment can reduce liver damage, as manifest by reduced plasma levels of liver enzymes (115).
Transcriptional Regulation Mediated by ER
Upon binding of E2 to ERs, they translocate into the nucleus where the receptors bind to specific genomic sequences. Interestingly, estrogen responsive elements (REs) are found in promoters and enhancers of liver genes. Genetic analysis revealed that more than 1,000 human liver genes have an expression based on sex bias (116). More than 40 lipids-related genes transcriptionally regulated by ER-α (117). Fascinatingly, this regulation is in tight coordination with the reproductive needs (118).
p53 As a Major Factor in Hepatic Responses Regulation
p53 is a well-known tumor suppressor which plays a central role in cell fate determination through cell-cycle arrest, senescence, differentiation, and apoptosis, in response to various stress signals (119–121). Novel data suggest that p53 governs additional biological pathways besides its traditional role as a tumor suppressor. This includes the regulation of metabolism, as well as pathways that affect the cell microenvironment and regulation of general homeostasis (122). Therefore, it is not surprising that the liver, an organ that regulates many metabolic processes, and coordinates homeostasis, serves as a unique platform for p53 to perform its classical and non-classical activities, both in health and in disease (94). Indeed, changes in the ratio of the hepatic enzymes ALP, ALT, and AST which serves as indicators of liver failure (123), were observed in p53-KO mice. These observations suggest that physiological p53 is a main regulator of liver homeostasis (124). Upon stress, hepatic p53 is activated and acts as a double-edged sword. As a tumor suppressor, p53 functions locally by preventing the development of HCC (94), and distally by inducing apoptosis of breast cancer cells by the secretion of SHBG (14). In addition, activated p53 can induce the secretion of senescence-associated secretory phenotype (SASP) in hepatic cells. This induction leads to a reduction in the accumulation of fibrotic tissue and to the stimulation of immune surveillance, which maintains tissue homeostasis and suppresses cancer development (125, 126).
On the other hand, the p53 induced apoptosis in the liver may cause infiltration of inflammatory cells and may lead, in the long run, to steatohepatitis, cirrhosis, and even to HCC (94). Moreover, the hepatic p53 acts in non-cell autonomous fashion by affecting and altering the liver secretome in response to different signals. Thus, presenting a novel function for p53 in homeostatic regulation of metabolic processes within the liver (124). Some of these secreted factors are mainly related to cell migration, implying a cross-talk between a distal tumor and the liver (124). Furthermore, p53 was also found to be an important regulator of lipid homeostasis (127). Hepatic p53 was shown to bind directly to p53 REs within the chromatin and to induce the transcription of mainly three genes that represent the different aspects of lipid metabolism (Pltp, Abca12, and Cel) (12). Interestingly, it was suggested that p53 also binds p53 REs in the promoter of cytochrome P450 enzymes and regulates their expression. These include CYP3A43, CYP3A5, CYP3A7, CYP4F2, CYP4F3, CYP4F11, CYP4F12, CYP19A1, CYP21A2, CYP24A1, and CYP3A4 (13). The latter is known to be the main enzyme which participates in the biosynthesis of steroid hormones (20, 21). In a proceeding study, it was found that CYP21A2 that catalyzes the conversion of 17 alphahydroxyprogesterone to 11-deoxycortisol, the immediate precursor of cortisol (128), is also transcriptionally regulated by p53 (14).
Moreover, p53 was shown to induce the expression and secretion of both CBG and SHBG by directly binding to p53 REs in their promoter (14). These data are in line with the observation that p53-WT mice exhibit higher CBG expression compared with their p53-KO counterparts. It should be noted that p53-KO female mice are known to have difficulties in reproduction (129). Hence, it is intriguing to speculate whether the lower levels of CBG in p53-KO mice may influence the level of active or free androgens and lead to fertility difficulties.
p53 was shown to transcriptionally regulate aromatase, a key enzyme that converts androgens to estrogen (see also section Estrogen). A putative p53-binding element was found in intron 1 of the aromatase gene. Under high fat diet conditions, p53 KO male mice produced dramatically more testosterone than WT p53 mice, while the serum testosterone levels were not significantly different. The level of E2 was low in both groups of mice resulting in higher testosterone/E2 level in p53 KO male mice (130). Others have shown data suggesting that p53 is globally required for GC receptor nuclear export (131). As GCs exert their function in the nucleus this may place p53 as an important regulator of GCs function.
In sum, these data suggest that p53 is associated with the regulation of the endocrine system at large, and particularly within the liver, with high impact on steroid hormones.
Concluding Remarks
Nowadays, population exposure to environmental steroid hormones, endocrine disruptor chemicals, and the increasing repertoire of medicines used by the public has an increasing impact on steroid hormones levels and activities as well as liver functions, leading to various liver diseases and endocrine syndromes. A comprehensive understanding of the molecular mechanisms underlying these diseases may help to prevent and treat them. An apparent bidirectional link exists between steroid hormones over secretion or insufficiency and liver metabolic or functional disease. Moreover, the liver and p53 are involved in steroidogenesis and steroid hormones homeostasis and balance. This notion may place p53 as a potential regulator in the liver/steroid hormone axis, maintaining homeostasis and preventing diseases (Figure 1).
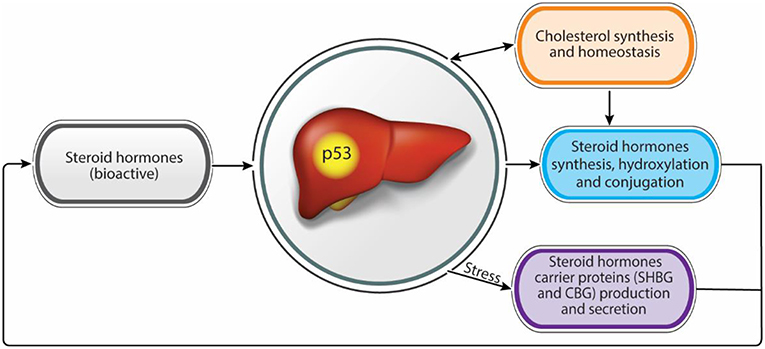
Figure 1. Steroid hormones homeostasis regulation by the liver. Functional intertwine exists between the liver and the steroid hormones. While the liver contributes to adequate levels of bioactive steroid hormones, through the modulation of synthesis and bioactivity, steroid hormones contribute to proper liver functions. p53 has a dual effect in regulating both steroid hormone levels and bioactivity as well as in maintaining liver hemostasis suggesting a role as a potential regulator in this axis.
Author Contributions
MC-N, RA-G, and EO wrote the manuscript. VR edited and reviewed the manuscript.
Funding
This publication was supported by the Center of Excellence from Flight Attendant Medical Research Institute (FAMRI), Israel Cancer Research Fund (ICRF), and from the Israel Science Foundation (ISF). This publication reflects the authors' views and not necessarily those of the European Community. VR is the incumbent of the Norman and Helen Asher Professorial Chair Cancer Research at the Weizmann institute.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones–a focus on rapid, non-genomic effects. Pharmacol Rev. (2000) 52:513–56.
2. Becker KL. Principles and Practice of Endocrinology and Metabolism. Lippincott Williams & Wilkins (2001).
3. Holst JP, Soldin OP, Guo T, Soldin SJ. Steroid hormones: relevance and measurement in the clinical laboratory. Clin Lab Med. (2004) 24:105–18. doi: 10.1016/j.cll.2004.01.004
4. Frye CA. Steroids, reproductive endocrine function, and affect. A review. Minerva Ginecol. (2009) 61:541–62.
5. Birzniece V. Hepatic actions of androgens in the regulation of metabolism. Curr Opin Endocrinol Diabetes Obes.(2018) 25:201–8. doi: 10.1097/MED.0000000000000405
6. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. (1995) 83:835–9. doi: 10.1016/0092-8674(95)90199-X
7. Witt BR, Thorneycroft IH. Reproductive steroid hormones: generation, degradation, reception, and action. Clin Obstet Gynecol. (1990) 33:563–75. doi: 10.1097/00003081-199009000-00023
8. Lee SR, Lee SY, Kim SY, Ryu SY, Park BK, Hong EJ. Hydroxylation and sulfation of sex steroid hormones in inflammatory liver. J Biomed Res. (2017) 31:437–44. doi: 10.1155/2017/3505784
9. Russell DW, Berman DM, Bryant JT, Cala KM, Davis DL, Landrum CP, et al. The molecular genetics of steroid 5α-reductases. Recent Prog Horm Res. (1994) 49:275–84. doi: 10.1016/B978-0-12-571149-4.50018-0
10. You L. Steroid hormone biotransformation and xenobiotic induction of hepatic steroid metabolizing enzymes. Chem Biol Interact. (2004) 147:233–46. doi: 10.1016/j.cbi.2004.01.006
11. Armata HL, Golebiowski D, Jung DY, Ko HJ, Kim JK, Sluss HK. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol Cell Biol. (2010) 30:5787–94. doi: 10.1128/MCB.00347-10
12. Goldstein I, Ezra O, Rivlin N, Molchadsky A, Madar S, Goldfinger N, et al. p53, a novel regulator of lipid metabolism pathways. J Hepatol. (2012) 56:656–62. doi: 10.1016/j.jhep.2011.08.022
13. Goldstein I, Rivlin N, Shoshana OY, Ezra O, Madar S, Goldfinger N, et al. Chemotherapeutic agents induce the expression and activity of their clearing enzyme CYP3A4 by activating p53. Carcinogenesis. (2013) 34:190–8. doi: 10.1093/carcin/bgs318
14. Charni M, Molchadsky A, Goldstein I, Solomon H, Tal P, Goldfinger N, et al. Novel p53 target genes secreted by the liver are involved in non-cell-autonomous regulation. Cell Death Differ. (2016) 23:509–20. doi: 10.1038/cdd.2015.119
15. Hu J, Zhang Z, Shen WJ, Azhar S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr Metab. (2010) 7:47. doi: 10.1186/1743-7075-7-47
16. Thigpen AE, Silver RI, Guileyardo JM, Casey ML, Mcconnell JD, Russell DW. Tissue distribution and ontogeny of steroid 5α-reductase isozyme expression. J Clin Invest. (1993) 92:903–10. doi: 10.1172/JCI116665
17. El-Awady MK, El-Garf W, El-Houssieny L. Steroid 5α reductase mRNA type 1 is differentially regulated by androgens and glucocorticoids in the rat liver. Endocr J. (2004) 51:37–46. doi: 10.1507/endocrj.51.37
18. Livingstone DE, Di Rollo EM, Mak TC, Sooy K, Walker BR, Andrew R. Metabolic dysfunction in female mice with disruption of 5α-reductase 1. J Endocrinol. (2017) 232:29–36. doi: 10.1530/JOE-16-0125
19. Zimniak P, Waxman DJ. Liver cytochrome P450 metabolism of endogenous steroid hormones, bile acids, and fatty acids. In: Schenkman JB, Greim H, editors. Cytochrome P450. Berlin, Heidelberg: Springer Berlin Heidelberg (1993) p. 123–44. doi: 10.1007/978-3-642-77763-9_8
20. Waxman DJ, Attisano C, Guengerich FP, Lapenson DP. Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 β-hydroxylase cytochrome P-450 enzyme. Arch Biochem Biophys. (1988) 263:424–36. doi: 10.1016/0003-9861(88)90655-8
21. Niwa T, Murayama N, Imagawa Y, Yamazaki H. Regioselective hydroxylation of steroid hormones by human cytochromes P450. Drug Metab Rev. (2015) 47:89–110. doi: 10.3109/03602532.2015.1011658
22. Arlotto MP, Trant JM, Estabrook RW. Measurement of steroid hydroxylation reactions by high-performance liquid chromatography as indicator of P450 identity and function. Methods Enzymol. (1991) 206:454–62. doi: 10.1016/0076-6879(91)06114-I
23. Wang K, Chen S, Xie W, Wan YJ. Retinoids induce cytochrome P450 3A4 through RXR/VDR-mediated pathway. Biochem Pharmacol. (2008) 75:2204–13. doi: 10.1016/j.bcp.2008.02.030
24. Istrate MA, Nussler AK, Eichelbaum M, Burk O. Regulation of CYP3A4 by pregnane X receptor: The role of nuclear receptors competing for response element binding. Biochem Biophys Res Commun. (2010) 393:688–93. doi: 10.1016/j.bbrc.2010.02.058
25. Strott CA. Sulfonation and molecular action. Endocr Rev. (2002) 23:703–32. doi: 10.1210/er.2001-0040
26. Qian YM, Sun XJ, Tong MH, Li XP, Richa J, Song WC. Targeted disruption of the mouse estrogen sulfotransferase gene reveals a role of estrogen metabolism in intracrine and paracrine estrogen regulation. Endocrinology. (2001) 142:5342–50. doi: 10.1210/endo.142.12.8540
27. Runge-Morris M, Wu W, Kocarek TA. Regulation of rat hepatic hydroxysteroid sulfotransferase (SULT2-40/41) gene expression by glucocorticoids: evidence for a dual mechanism of transcriptional control. Mol Pharmacol. (1999) 56:1198–206. doi: 10.1124/mol.56.6.1198
28. Girard C, Barbier O, Veilleux G, El-Alfy M, Belanger A. Human uridine diphosphate-glucuronosyltransferase UGT2B7 conjugates mineralocorticoid and glucocorticoid metabolites. Endocrinology. (2003) 144:2659–68. doi: 10.1210/en.2002-0052
29. Vansell NR, Klaassen CD. Increase in rat liver UDP-glucuronosyltransferase mRNA by microsomal enzyme inducers that enhance thyroid hormone glucuronidation. Drug Metab Dispos. (2002) 30:240–6. doi: 10.1124/dmd.30.3.240
30. Avvakumov GV, Cherkasov A, Muller YA, Hammond GL. Structural analyses of sex hormone-binding globulin reveal novel ligands and function. Mol Cell Endocrinol. (2010) 316:13–23. doi: 10.1016/j.mce.2009.09.005
31. Lin HY, Muller YA, Hammond GL. Molecular and structural basis of steroid hormone binding and release from corticosteroid-binding globulin. Mol Cell Endocrinol. (2010) 316:3–12. doi: 10.1016/j.mce.2009.06.015
32. Mendel CM. The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev. (1989) 10:232–74. doi: 10.1210/edrv-10-3-232
33. Fortunati N, Catalano MG, Boccuzzi G, Frairia R. Sex hormone-binding globulin (SHBG), estradiol and breast cancer. Mol Cell Endocrinol. (2010) 316:86–92. doi: 10.1016/j.mce.2009.09.012
34. Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocrinol. (2016) 230:R13–25. doi: 10.1530/JOE-16-0070
35. De Moor P, Joossens JV. An inverse relation between body weight and the activity of the steroid binding -globulin in human plasma. Steroidologia. (1970) 1:129–36.
36. Javitt NB. Hep G2 cells as a resource for metabolic studies: lipoprotein, cholesterol, and bile acids. FASEB J. (1990) 4:161–8. doi: 10.1096/fasebj.4.2.2153592
37. Selva DM, Hogeveen KN, Innis SM, Hammond GL. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. J Clin Invest. (2007) 117:3979–87. doi: 10.1172/JCI32249
38. Selva DM, Hammond GL. Peroxisome-proliferator receptor γ represses hepatic sex hormone-binding globulin expression. Endocrinology. (2009) 150:2183–9. doi: 10.1210/en.2008-1289
39. Rosner W, Hryb DJ, Khan MS, Singer CJ, Nakhla AM. Are corticosteroid-binding globulin and sex hormone-binding globulin hormones? Ann N Y Acad Sci. (1988) 538:137–45. doi: 10.1111/j.1749-6632.1988.tb48859.x
40. Vandewalle J, Luypaert A, De Bosscher K, Libert C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol Metab. (2018) 29:42–54. doi: 10.1016/j.tem.2017.10.010
41. Godowski PJ, Rusconi S, Miesfeld R, Yamamoto KR. Glucocorticoid receptor mutants that are constitutive activators of transcriptional enhancement. Nature. (1987) 325:365–8. doi: 10.1038/325365a0
42. Arlt W, Stewart PM. Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinol Metab Clin North Am. (2005) 34:293–313. doi: 10.1016/j.ecl.2005.01.002
43. Snijdewint FG, Kapsenberg ML, Wauben-Penris PJ, Bos JD. Corticosteroids class-dependently inhibit in vitro Th1- and Th2-type cytokine production. Immunopharmacology. (1995) 29:93–101. doi: 10.1016/0162-3109(94)00048-K
44. Yang S, Zhang L. Glucocorticoids and vascular reactivity. Curr Vasc Pharmacol. (2004) 2:1–12. doi: 10.2174/1570161043476483
45. Cicognani C, Malavolti M, Morselli-Labate AM, Zamboni L, Sama C, Barbara L. Serum lipid and lipoprotein patterns in patients with liver cirrhosis and chronic active hepatitis. Arch Intern Med. (1997) 157:792–6. doi: 10.1001/archinte.1997.00440280120012
46. Tsochatzis EA, Newsome PN. Non-alcoholic fatty liver disease and the interface between primary and secondary care. Lancet Gastroenterol Hepatol. (2018) 3:509–17. doi: 10.1016/S2468-1253(18)30077-3
47. D'souza AM, Beaudry JL, Szigiato AA, Trumble SJ, Snook LA, Bonen A, et al. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G850–63. doi: 10.1152/ajpgi.00378.2011
48. Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, et al. Hepatic steatosis in Cushing's syndrome: a radiological assessment using computed tomography. Eur J Endocrinol. (2003) 149:543–8. doi: 10.1530/eje.0.1490543
49. Marik PE. Adrenal-exhaustion syndrome in patients with liver disease. Intensive Care Med. (2006) 32:275–80. doi: 10.1007/s00134-005-0005-5
50. Deviere J, Content J, Denys C, Vandenbussche P, Schandene L, Wybran J, et al. Excessive in vitro bacterial lipopolysaccharide-induced production of monokines in cirrhosis. Hepatology. (1990) 11:628–34. doi: 10.1002/hep.1840110416
51. Khoruts A, Stahnke L, Mcclain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. (1991) 13:267–76. doi: 10.1002/hep.1840130211
52. Sekiyama KD, Yoshiba M, Thomson AW. Circulating proinflammatory cytokines (IL-1 β, TNF-α, and IL-6) and IL-1 receptor antagonist (IL-1Ra) in fulminant hepatic failure and acute hepatitis. Clin Exp Immunol. (1994) 98:71–7. doi: 10.1111/j.1365-2249.1994.tb06609.x
53. Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. (2010) 4:659–72. doi: 10.1007/s12072-010-9219-x
54. Paz-Delgadillo J, Monreal-Robles R, Villarreal-Perez JZ, Maldonado-Garza HJ, Bosques-Padilla FJ, Lavalle-Gonzalez FJ. Algorithm for screening of adrenal function in stable patients with cirrhosis. Ann Hepatol. (2017) 16:788–96. doi: 10.5604/01.3001.0010.2797
55. Tan T, Chang L, Woodward A, Mcwhinney B, Galligan J, Macdonald GA, et al. Characterising adrenal function using directly measured plasma free cortisol in stable severe liver disease. J Hepatol. (2010) 53:841–8. doi: 10.1016/j.jhep.2010.05.020
56. Fede G, Spadaro L, Tomaselli T, Privitera G, Piro S, Rabuazzo AM, et al. Assessment of adrenocortical reserve in stable patients with cirrhosis. J Hepatol. (2011) 54:243–50. doi: 10.1016/j.jhep.2010.06.034
57. Triantos CK, Marzigie M, Fede G, Michalaki M, Giannakopoulou D, Thomopoulos K, et al. Critical illness-related corticosteroid insufficiency in patients with cirrhosis and variceal bleeding. Clin Gastroenterol Hepatol. (2011) 9:595–601. doi: 10.1016/j.cgh.2011.03.033
58. Singh RR, Walia R, Sachdeva N, Bhalla A, Singh A, Singh V. Relative adrenal insufficiency in cirrhotic patients with ascites (hepatoadrenal syndrome). Dig Liver Dis. (2018). doi: 10.1016/j.dld.2018.05.011
59. Marik PE, Gayowski T, Starzl TE, Hepatic Cortisol R, Adrenal Pathophysiology Study G. The hepatoadrenal syndrome: a common yet unrecognized clinical condition. Crit Care Med. (2005) 33:1254–9. doi: 10.1097/01.CCM.0000164541.12106.57
60. Rauschecker M, Abraham SB, Abel BS, Wesley R, Saverino E, Trivedi A, et al. Cosyntropin-stimulated serum free cortisol in healthy, adrenally insufficient, and mildly cirrhotic populations. J Clin Endocrinol Metab. (2016) 101:1075–81. doi: 10.1210/jc.2015-2285
61. Papanastasiou L, Fountoulakis S, Vatalas IA. Adrenal disorders and non-alcoholic fatty liver disease. Minerva Endocrinol. (2017) 42:151–63. doi: 10.23736/S0391-1977.16.02583-9
62. Hall JE, Guyton AC, Mizelle HL. Role of the renin-angiotensin system in control of sodium excretion and arterial pressure. Acta Physiol Scand Suppl. (1990) 591:48–62.
63. Gunther S, Alexander RW, Atkinson WJ, Gimbrone MA Jr. Functional angiotensin II receptors in cultured vascular smooth muscle cells. J Cell Biol. (1982) 92:289–98. doi: 10.1083/jcb.92.2.289
64. Lumbers ER. Angiotensin and aldosterone. Regul Pept. (1999) 80:91–100. doi: 10.1016/S0167-0115(99)00026-9
65. Warner FJ, Lubel JS, Mccaughan GW, Angus PW. Liver fibrosis: a balance of ACEs? Clin Sci. (2007) 113:109–18. doi: 10.1042/CS20070026
66. Moreira De Macedo S, Guimaraes TA, Feltenberger JD, Sousa Santos SH. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides. (2014) 62:189–96. doi: 10.1016/j.peptides.2014.10.005
67. Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J Clin Invest. (1994) 93:2431–7. doi: 10.1172/JCI117251
68. Marshall RP, Mcanulty RJ, Laurent GJ. Angiotensin II is mitogenic for human lung fibroblasts via activation of the type 1 receptor. Am J Respir Crit Care Med. (2000) 161:1999–2004. doi: 10.1164/ajrccm.161.6.9907004
69. Tharaux PL, Chatziantoniou C, Fakhouri F, Dussaule JC. Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension. (2000) 36:330–6. doi: 10.1161/01.HYP.36.3.330
70. Bataller R, Sancho-Bru P, Gines P, Lora JM, Al-Garawi A, Sole M, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. (2003) 125:117–25. doi: 10.1016/S0016-5085(03)00695-4
71. Harkness RE, Braun V. Colicin M inhibits peptidoglycan biosynthesis by interfering with lipid carrier recycling. J Biol Chem. (1989) 264:6177–82.
72. Fallo F, Dalla Pozza A, Tecchio M, Tona F, Sonino N, Ermani M, et al. Nonalcoholic fatty liver disease in primary aldosteronism: a pilot study. Am J Hypertens. (2010) 23:2–5. doi: 10.1038/ajh.2009.206
73. Rockey DC, Weisiger RA. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: implications for regulation of portal pressure and resistance. Hepatology. (1996) 24:233–40. doi: 10.1002/hep.510240137
74. Schneider AW, Kalk JF, Klein CP. Effect of losartan, an angiotensin II receptor antagonist, on portal pressure in cirrhosis. Hepatology. (1999) 29:334–9. doi: 10.1002/hep.510290203
75. Bataller R, Gines P, Nicolas JM, Gorbig MN, Garcia-Ramallo E, Gasull X, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. (2000) 118:1149–56. doi: 10.1016/S0016-5085(00)70368-4
76. Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. (2001) 34:745–50. doi: 10.1053/jhep.2001.28231
77. Ishizaka N, Saito K, Noiri E, Sata M, Ikeda H, Ohno A, et al. Administration of ANG II induces iron deposition and upregulation of TGF-β1 mRNA in the rat liver. Am J Physiol Regul Integr Comp Physiol. (2005) 288:R1063–70. doi: 10.1152/ajpregu.00281.2004
78. Goto M, Hoxha N, Osman R, Wen J, Wells RG, Dell KM. Renin-angiotensin system activation in congenital hepatic fibrosis in the PCK rat model of autosomal recessive polycystic kidney disease. J Pediatr Gastroenterol Nutr. (2010) 50:639–44. doi: 10.1097/MPG.0b013e3181cc80e4
79. Paschos P, Tziomalos K. Nonalcoholic fatty liver disease and the renin-angiotensin system: Implications for treatment. World J Hepatol. (2012) 4:327–31. doi: 10.4254/wjh.v4.i12.327
80. Shimizu T, Oishi T, Omori A, Sugiura A, Hirota K, Aoyama H, et al. Identification of cis-regulatory sequences in the human angiotensinogen gene by transgene coplacement and site-specific recombination. Mol Cell Biol. (2005) 25:2938–45. doi: 10.1128/MCB.25.8.2938-2945.2005
81. Rana A, Jain S, Puri N, Kaw M, Sirianni N, Eren D, et al. The transcriptional regulation of the human angiotensinogen gene after high-fat diet is haplotype-dependent: novel insights into the gene-regulatory networks and implications for human hypertension. PLoS ONE. (2017) 12:e0176373. doi: 10.1371/journal.pone.0176373
82. Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol. (2015) 2015:294278. doi: 10.1155/2015/294278
83. Kelly DM, Jones TH. Testosterone: a metabolic hormone in health and disease. J Endocrinol. (2013) 217:R25–45. doi: 10.1530/JOE-12-0455
84. Volzke H, Aumann N, Krebs A, Nauck M, Steveling A, Lerch MM, et al. Hepatic steatosis is associated with low serum testosterone and high serum DHEAS levels in men. Int J Androl. (2010) 33:45–53. doi: 10.1111/j.1365-2605.2009.00953.x
85. Dowman JK, Hopkins LJ, Reynolds GM, Armstrong MJ, Nasiri M, Nikolaou N, et al. Loss of 5α-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice. Endocrinology. (2013) 154:4536–47. doi: 10.1210/en.2013-1592
86. Livingstone DE, Barat P, Di Rollo EM, Rees GA, Weldin BA, Rog-Zielinska EA, et al. 5α-Reductase type 1 deficiency or inhibition predisposes to insulin resistance, hepatic steatosis, and liver fibrosis in rodents. Diabetes. (2015) 64:447–58. doi: 10.2337/db14-0249
87. Lin HY, Yu IC, Wang RS, Chen YT, Liu NC, Altuwaijri S, et al. Increased hepatic steatosis and insulin resistance in mice lacking hepatic androgen receptor. Hepatology. (2008) 47:1924–35. doi: 10.1002/hep.22252
88. Kelly DM, Nettleship JE, Akhtar S, Muraleedharan V, Sellers DJ, Brooke JC, et al. Testosterone suppresses the expression of regulatory enzymes of fatty acid synthesis and protects against hepatic steatosis in cholesterol-fed androgen deficient mice. Life Sci. (2014) 109:95–103. doi: 10.1016/j.lfs.2014.06.007
89. Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. (2006) 295:1288–99. doi: 10.1001/jama.295.11.1288
90. Grossmann M, Zajac JD. Androgen deprivation therapy in men with prostate cancer: how should the side effects be monitored and treated? Clin Endocrinol. (2011) 74:289–93. doi: 10.1111/j.1365-2265.2010.03939.x
91. Keating NL, O'malley A, Freedland SJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. J Natl Cancer Inst. (2012) 104:1518–23. doi: 10.1093/jnci/djp404
92. Corona G, Monami M, Rastrelli G, Aversa A, Sforza A, Lenzi A, et al. Type 2 diabetes mellitus and testosterone: a meta-analysis study. Int J Androl. (2011) 34:528–40. doi: 10.1111/j.1365-2605.2010.01117.x
93. Moverare-Skrtic S, Venken K, Andersson N, Lindberg MK, Svensson J, Swanson C, et al. Dihydrotestosterone treatment results in obesity and altered lipid metabolism in orchidectomized mice. Obesity Silver Spring. (2006) 14:662–72. doi: 10.1038/oby.2006.75
94. Charni M, Rivlin N, Molchadsky A, Aloni-Grinstein R, Rotter V. p53 in liver pathologies-taking the good with the bad. J Mol Med. (2014) 92:1229–34. doi: 10.1007/s00109-014-1223-5
95. Yu MW, Cheng SW, Lin MW, Yang SY, Liaw YF, Chang HC, et al. Androgen-receptor gene CAG repeats, plasma testosterone levels, and risk of hepatitis B-related hepatocellular carcinoma. J Natl Cancer Inst. (2000) 92:2023–8. doi: 10.1093/jnci/92.24.2023
96. Giannitrapani L, Soresi M, La Spada E, Cervello M, D'alessandro N, Montalto G. Sex hormones and risk of liver tumor. Ann N Y Acad Sci. (2006) 1089:228–36. doi: 10.1196/annals.1386.044
97. Ohnishi S, Murakami T, Moriyama T, Mitamura K, Imawari M. Androgen and estrogen receptors in hepatocellular carcinoma and in the surrounding noncancerous liver tissue. Hepatology. (1986) 6:440–3. doi: 10.1002/hep.1840060320
98. Kanda T, Takahashi K, Nakamura M, Nakamoto S, Wu S, Haga Y, et al. Androgen receptor could be a potential therapeutic target in patients with advanced hepatocellular carcinoma. Cancers. (2017) 9:1–6. doi: 10.3390/cancers9050043
99. Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. (2003) 86:225–30. doi: 10.1016/S0960-0760(03)00360-1
100. Sharma G, Prossnitz ER. G-protein-coupled estrogen receptor (GPER) and sex-specific metabolic homeostasis. Adv Exp Med Biol. (2017) 1043:427–53. doi: 10.1007/978-3-319-70178-3_20
101. Iavarone M, Lampertico P, Seletti C, Francesca Donato M, Ronchi G, Del Ninno E, et al. The clinical and pathogenetic significance of estrogen receptor-β expression in chronic liver diseases and liver carcinoma. Cancer. (2003) 98:529–34. doi: 10.1002/cncr.11528
102. Miceli V, Cocciadiferro L, Fregapane M, Zarcone M, Montalto G, Polito LM, et al. Expression of wild-type and variant estrogen receptorα in liver carcinogenesis and tumor progression. OMICS. (2011) 15:313–7. doi: 10.1089/omi.2010.0108
103. Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med. (2013) 19:197–209. doi: 10.1016/j.molmed.2012.12.007
104. Romero-Aleshire MJ, Diamond-Stanic MK, Hasty AH, Hoyer PB, Brooks HL. Loss of ovarian function in the VCD mouse-model of menopause leads to insulin resistance and a rapid progression into the metabolic syndrome. Am J Physiol Regul Integr Comp Physiol. (2009) 297:R587–92. doi: 10.1152/ajpregu.90762.2008
105. Barrera J, Chambliss KL, Ahmed M, Tanigaki K, Thompson B, Mcdonald JG, et al. Bazedoxifene and conjugated estrogen prevent diet-induced obesity, hepatic steatosis, and type 2 diabetes in mice without impacting the reproductive tract. Am J Physiol Endocrinol Metab. (2014) 307:E345–54. doi: 10.1152/ajpendo.00653.2013
106. De Oliveira MC, Gilglioni EH, De Boer BA, Runge JH, De Waart DR, Salgueiro CL, et al. Bile acid receptor agonists INT747 and INT777 decrease oestrogen deficiency-related postmenopausal obesity and hepatic steatosis in mice. Biochim Biophys Acta. (2016) 1862:2054–62. doi: 10.1016/j.bbadis.2016.07.012
107. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci USA. (2000) 97:12729–34. doi: 10.1073/pnas.97.23.12729
108. Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, et al. Evidence that oestrogen receptor-α plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. (2006) 49:588–97. doi: 10.1007/s00125-005-0105-3
109. Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, et al. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am J Physiol Endocrinol Metab. (2010) 298:E304–319. doi: 10.1152/ajpendo.00504.2009
110. Pedram A, Razandi M, O'mahony F, Harvey H, Harvey BJ, Levin ER. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci Signal. (2013) 6:ra36. doi: 10.1126/scisignal.2004013
111. Kim JH, Meyers MS, Khuder SS, Abdallah SL, Muturi HT, Russo L, et al. Tissue-selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol Metab. (2014) 3:177–90. doi: 10.1016/j.molmet.2013.12.009
112. Wang X, Lu Y, Wang E, Zhang Z, Xiong X, Zhang H, et al. Hepatic estrogen receptor α improves hepatosteatosis through upregulation of small heterodimer partner. J Hepatol. (2015) 63:183–90. doi: 10.1016/j.jhep.2015.02.029
113. Chambliss KL, Barrera J, Umetani M, Umetani J, Kim SH, Madak-Erdogan Z, et al. Non-nuclear estrogen receptor activation improves hepatic steatosis in female mice. Endocrinology. (2016) 157:3731–41. doi: 10.1210/en.2015-1629
114. Ryu S, Suh BS, Chang Y, Kwon MJ, Yun KE, Jung HS, et al. Menopausal stages and non-alcoholic fatty liver disease in middle-aged women. Eur J Obstet Gynecol Reprod Biol. (2015) 190:65–70. doi: 10.1016/j.ejogrb.2015.04.017
115. Mckenzie J, Fisher BM, Jaap AJ, Stanley A, Paterson K, Sattar N. Effects of HRT on liver enzyme levels in women with type 2 diabetes: a randomized placebo-controlled trial. Clin Endocrinol. (2006) 65:40–4. doi: 10.1111/j.1365-2265.2006.02543.x
116. Zhang Y, Klein K, Sugathan A, Nassery N, Dombkowski A, Zanger UM, et al. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS ONE. (2011) 6:e23506. doi: 10.1371/journal.pone.0023506
117. Gao H, Falt S, Sandelin A, Gustafsson JA, Dahlman-Wright K. Genome-wide identification of estrogen receptor α-binding sites in mouse liver. Mol Endocrinol. (2008) 22:10–22. doi: 10.1210/me.2007-0121
118. Villa A, Della Torre S, Stell A, Cook J, Brown M, Maggi A. Tetradian oscillation of estrogen receptor α is necessary to prevent liver lipid deposition. Proc Natl Acad Sci USA. (2012) 109:11806–11. doi: 10.1073/pnas.1205797109
120. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. (2000) 408:307–10. doi: 10.1038/35042675
121. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. (2009) 9:749–58. doi: 10.1038/nrc2723
122. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. (2014) 14:359–70. doi: 10.1038/nrc3711
123. Sulava E, Bergin S, Long B, Koyfman A. Elevated liver enzymes: emergency department-focused management. J Emerg Med. (2016). doi: 10.1016/j.jemermed.2016.10.016
124. Charni-Natan M, Solomon H, Molchadsky A, Jacob-Berger A, Goldfinger N, Rotter V. Various stress stimuli rewire the profile of liver secretome in a p53-dependent manner. Cell Death Dis. (2018) 9:647. doi: 10.1038/s41419-018-0697-4
125. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
126. Raj N, Attardi LD. Tumor suppression: p53 alters immune surveillance to restrain liver cancer. Curr Biol. (2013) 23:R527–530. doi: 10.1016/j.cub.2013.04.076
127. Goldstein I, Rotter V. Regulation of lipid metabolism by p53–fighting two villains with one sword. Trends Endocrinol Metab. (2012) 23:567–75. doi: 10.1016/j.tem.2012.06.007
128. Wedell A. Molecular genetics of 21-hydroxylase deficiency. Endocr Dev. (2011) 20:80–7. doi: 10.1159/000321223
129. Hu W. The role of p53 gene family in reproduction. Cold Spring Harb Perspect Biol. (2009) 1:a001073. doi: 10.1101/cshperspect.a001073
130. Wang X, Zhao X, Gao X, Mei Y, Wu M. A new role of p53 in regulating lipid metabolism. J Mol Cell Biol. (2013) 5:147–50. doi: 10.1093/jmcb/mjs064
Keywords: p53, steroid hormones, liver, NAFLD, cirrhosis, estrogen, testosterone
Citation: Charni-Natan M, Aloni-Grinstein R, Osher E and Rotter V (2019) Liver and Steroid Hormones—Can a Touch of p53 Make a Difference? Front. Endocrinol. 10:374. doi: 10.3389/fendo.2019.00374
Received: 22 October 2018; Accepted: 28 May 2019;
Published: 12 June 2019.
Edited by:
Lars Grøntved, University of Southern Denmark, DenmarkReviewed by:
Consolato Sergi, University of Alberta Hospital, CanadaOfir Hakim, Bar-Ilan University, Israel
Copyright © 2019 Charni-Natan, Aloni-Grinstein, Osher and Rotter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Varda Rotter, dmFyZGEucm90dGVyQHdlaXptYW5uLmFjLmls
†These authors have contributed equally to this work