 Hussein Majdoub1Serge Amselem2Marie Legendre2Shoshana Rath3Dani Bercovich4
Hussein Majdoub1Serge Amselem2Marie Legendre2Shoshana Rath3Dani Bercovich4 Yardena Tenenbaum-Rakover3,5*
Yardena Tenenbaum-Rakover3,5*- 1Pediatric Endocrine Clinic, Clalit Health Services, Northern region, Haifa, Israel
- 2Sorbonne Université, Inserm U933 and Assistance Publique de Hopitaux de Paris, Hôpital Trousseau, Paris, France
- 3Ha'Emek Medical Center, Pediatric Endocrine Institute, Afula, Israel
- 4Tel Hai College and GGA - Galilee Genetic Analysis Lab, Katzrin, Israel
- 5Rappaport Faculty of Medicine, Technion, Haifa, Israel
Background: POU1F1 is an essential transcription factor for the differentiation, proliferation and survival of somatotrophs, lactotrophs, and thyrotrophs. Mutations in the POU1F1 gene are characterized by growth hormone (GH), thyrotropin, and prolactin deficiencies, commonly presenting with growth retardation and central hypothyroidism. Since the first report in 1992, more than 25 mutations have been identified in POU1F1.
Case Description: We describe a 17-year-old male who presented to our Pediatric Endocrinology clinic with extreme short stature (height 81.7 cm, −9.3 SD), cognitive impairment, deaf-mutism, and neurological disabilities. L-thyroxine supplemental therapy, which had been initiated at the age of 6 months but ceased due to non-compliance, was reintroduced at presentation. GH therapy was initiated at 19 years of age, resulting in 42 cm linear growth, to a final height of 124 cm. Sequencing of POU1F1 revealed a previously described homozygous insertion mutation—c.580_581insT, p (Thr194Ilefs*7)—in exon 4 causing a frameshift that introduces a stop codon 7 amino acids downstream, leading to a severely truncated protein lacking the homeodomain.
Conclusion: This case report sheds light on the natural history of untreated patients with POU1F1 mutations and raises awareness for early diagnosis and adequate treatment of central congenital hypothyroidism and GH deficiency.
Introduction
Combined pituitary hormone deficiency (CPHD) is a rare heterogeneous condition in which there are at least two anterior pituitary hormone deficiencies. Anterior pituitary-derived hormones require multiple transcription factors for pituitary morphogenesis, differentiation and hormones synthesis. Mutations in the genes PROP1, POU1F1 (PIT1), LHX3, GLI2, HESX1, LHX3, LHX4, OTX2, SOX2, ROBO1, and SOX3 (1–5), which encode transcription factors essential for pituitary development, have been described in human CPHD with an incidence of between 1:4,000 and 1:8,000.
POU class 1 homeobox 1 (POU1F1), also known as pituitary transcription factor 1 (PIT-1), was the first pituitary-specific transcription factor to be identified in humans and mice (6, 7). It is expressed in the anterior pituitary and is essential for differentiation, proliferation and survival of somatotrophic, lactotrophic, and thyrotrophic cells.
The human POUIFI gene (OMIM 601538) is located on chromosome 3p11, consists of 6 exons and encodes a 317-amino acid protein (NM_001122757) that has three functional domains: a transactivation domain, a POU-specific (POU-S) domain and a POU-homeo (POU-H) domain. The N-terminal part of POU1F1 is involved in the transcriptional activation of several pituitary-expressed genes (7). The first mutations of POU1F1 in humans were described in 1992 (8–10), involving growth hormone (GH), prolactin (PRL), and thyrotropin (TSH) deficiencies. Since those first reports, more than 25 POU1F1 mutations characterized as having clinical significance and presenting with CPHD have been described (https://www.ncbi.nlm.nih.gov/clinvar?term=173110[MIM]); these mutations are responsible for dominant or recessive CPHD, with 3 small insertion mutations among them (11–32) The clinical characteristics of patients with POU1F1 mutations include severe growth retardation in infancy due to GH deficiency (GHD) with distinctive facial features characterized by prominent forehead, marked mid-facial hypoplasia, and depressed nasal bridge. In addition, central hypothyroidism of variable severity has been reported, and brain magnetic resonance imaging (MRI) demonstrates either normal or hypoplastic anterior pituitary with unaffected posterior pituitary.
Here we describe a 17-year-old male who presented with extreme short stature, mental retardation, deaf-mutism, neurological impairment, and dysmorphic features, in whom CPHD (GH, TSH, and PRL deficiencies) caused by a previously described homozygous frameshift mutation of the POU1F1 gene (30) was identified. Marked clinical improvement occurred with GH and levothyroxine (LT4) therapy. This description extends the recognized phenotype of POU1F1 mutations, shedding light on the severity of presentation in the untreated state.
Case Presentation
The proband, a male born to consanguineous parents (first cousins) of Arab descent, was born after pregnancy with polyhydramnios and delivered at term, weighing 2,900 g and his length was 45 cm. Both parents were healthy; paternal height was 170 cm and maternal height 160 cm. There was one healthy brother of normal stature. There was no family history of short stature, hypothyroidism, deafness, or neurological impairment. In the first week of life, the proband had recurrent vomiting and prolonged neonatal jaundice with indirect hyperbilirubinemia of 15 mg/dL that resolved spontaneously. At age 6 months, severe failure to thrive and dehydration were diagnosed, resulting in hospital admission. Delayed psychomotor development, growth retardation, and dysmorphic facial features were noted. On physical examination, he had enlarged fontanelle, severe hypotonia, umbilical hernia, and dry skin consistent clinically with hypothyroidism. Laboratory evaluation revealed low TSH (0.08 mIU/L; normal range 0.27–4.2) and free thyroxine (FT4) (5.4 pmol/L; normal range 12–22), indicating central hypothyroidism. Further investigation demonstrated mild bilateral frontal and interhemispheric cortical atrophy (on brain computed tomography (CT) scan and sensorineural (SN) hearing loss (Brainstem Auditory Evoked Response test). Comprehensive metabolic investigations were normal, as were echocardiography and ophthalmic examination. The karyotype was 46, XY, and no chromosomal abnormalities were identified. LT4 treatment was initiated at a dose of 30 μg daily, but was discontinued by the parents after 2 months. At the age of 4 years, he was diagnosed with severe mental retardation and was fully disabled. At 5 years of age, the proband presented to a different Endocrine clinic for investigation of severe growth retardation. At that time, TSH was 0.01 mU/L, FT4 1.34 pmol/L and FT3 < 0.44 nmol/L (normal range 1.3–3.1). Thyrotropin-releasing hormone (TRH) test confirmed the diagnosis of central hypothyroidism (Table 1) and LT4 therapy was recommended. In addition, insulin growth factor-1 (IGF-1) and insulin growth factor binding protein-3 (IGFBP-3) levels were undetectable with no GH response to arginine stimulation test (Table 1). Unfortunately, although the family was informed on several occasions as to the importance of supplemental LT4 therapy and the need for GH treatment, compliance remained poor and the family failed to attend follow-up appointments so GH therapy was not initiated. The patient was seen for the first time in our Pediatric Endocrine clinic when he presented at age 17.5 years for evaluation of extreme short stature. At that time, his appearance resembled that of a 2-year-old child. His height was 81.7 cm (−9.3 SD) and weight 11.5 kg (−9.3 SD). He had dysmorphic facial features with a deep nasal bridge, a prominent forehead, micrognathia, flat nose, big, prominent auricles, multiple preauricular skin tags, and widely spaced teeth (Figures 1A,B). He had acromicria and a prominent abdomen without organomegaly. His pubertal stage was Tanner 1 with testicular volume of 2 mL bilaterally. Severe neurodevelopmental delay was noted, with general hypotonia; he was unable to stand unsupported. He was deaf and mute, communicating with sign language at a basic level. Investigations revealed a bone age of 8.5 years (at chronological age 17.5 years), undetectable TSH, FT4 3.8 pmol/L, PRL < 8 mIU/L (normal range 45–375), IGF-1 < 3.25 ng/mL, and IGFBP-3 < 500 ng/mL. Cortisol and adrenocorticotropic hormone (ACTH) levels were within normal limits and gonadotropin levels were pre-pubertal (Table 1). As previous investigations were unavailable at that time, the diagnoses of central hypothyroidism and GHD were reconfirmed by stimulation tests. Brain MRI demonstrated a hypoplastic anterior pituitary, a normally located posterior pituitary bright spot, white matter demyelination and cervicomedullary kinking. Biochemistry, celiac screen, and complete blood count were within normal limits. LT4 therapy was recommenced with dose adjusted to maintain FT4 within a normal range. GH therapy was initiated with dose adjusted to weight (0.03 mg/kg per day) and growth velocity. Gonadotropin-releasing hormone (GnRH) analog treatment (3.75 mg every 28 days) was initiated at the onset of puberty (Tanner P2, G2, age 21 years) to facilitate maximal linear growth. Calcium supplement and vitamin D3 were added due to low serum levels of vitamin D. In addition, physiotherapy and speech therapy were implemented. At the age of 24 years, growth velocity decreased to <2 cm per year and therefore GH and GnRH analog therapy were terminated. At 28 years of age, height had increased by 42.3 cm (+3 SD) to a final adult height of 124 cm (−6.2 SD; Figure 2), and weight was 28 kg (−7.5 SD). Pubertal stage Tanner P5 was attained with testicular volume of 15 mL bilaterally. Considerable improvement in tone and psychomotor function was observed consequent to implementation of appropriate therapy; he learned to stand independently, walk several steps unsupported, and communicate vocally with family and medical staff. Overall, he had a happier demeanor, but unfortunately remained non-verbal.
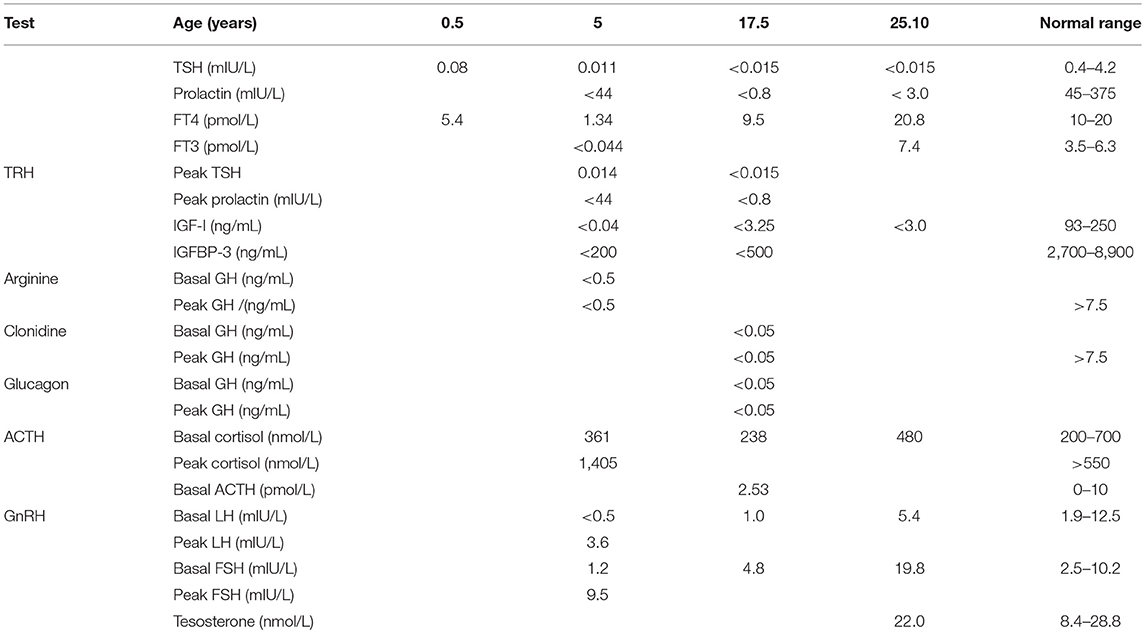
Table 1. Summary of hormonal results.
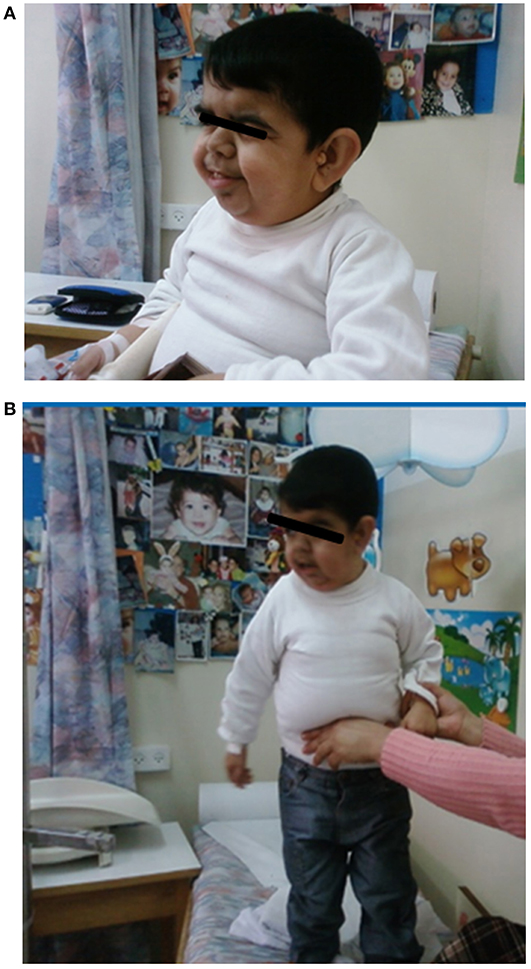
Figure 1. The patient at age 17 years. He had a deep nasal bridge, prominent forehead, micrognathia, flat nose, prominent and large auricles, multiple pre-auricular skin tags, and widely spaced teeth (With permission from the family).
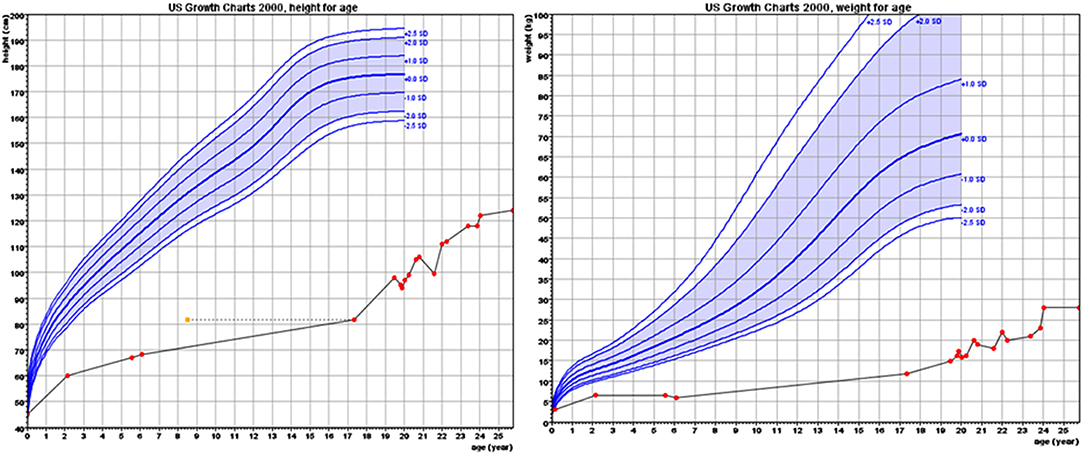
Figure 2. Growth charts of height and weight.
A blood sample was collected for genetic analysis after obtaining parental informed consent with approval of the Institutional Review Board of Ha'Emek Medical Center. Additional, informed consent was obtained from the parents for publication of this case report and any potentially identification images/information. The exons and flanking intronic boundaries of the POU1F1 gene were amplified and sequenced directly using primer sets that are available upon request. A homozygous frameshift mutation, c.580_581insT p.(Thr194Ilefs*7), formerly c.502insT (p.Thr168IlefsX7) as previously described by our group (30), was identified. This insertion results in a frameshift that introduces an early termination codon, leading to a severely truncated protein lacking the entire homeodomain. Both parents were heterozygous for the identified mutation. There is no known familial connection to the patient in whom this mutation was previously described.
Discussion
We report a 17-year-old male with untreated CPHD (GH, TSH, and PRL deficiencies), who presented with extreme short stature, deaf-mutism, and neurological disabilities (both cognitive and physical) with a homozygous POU1F1 mutation c.580_581insT p.(Thr194llefs*7). CPHD is classically divided into two subgroups according to the transcription factor gene types affected: early expressed pituitary transcription factor genes, defined as syndromic CPHD, and late expressed pituitary developmental genes, defined as non-syndromic CPHD. Whereas, mutations in early developmental genes are commonly involved in organogenesis and therefore present clinically with brain, ophthalmic, and/or skeletal anomalies, mutations in late developmental genes generally present with isolated pituitary hormone deficiencies in the absence of other anomalies (3). POU1F1 mutations belong to the non-syndromic CPHD subgroup. Patients with POU1F1 gene mutations are generally characterized by GH, PRL, and TSH deficiencies, without extrapituitary abnormalities (1–5). Variability exists in time of onset and severity of hormonal deficiencies; whilst GHD commonly presents in the neonatal period or early childhood, hypothyroidism may not occur at all or may present at any time from the neonatal period to the second decade of life. POU1F1 mutations may in fact present with isolated GHD (28).
Neonatal thyroid screening in Israel aims to identify primary congenital hypothyroidism (CH), such that only abnormally elevated levels of TSH are reported. Central hypothyroidism (in which TSH is low) is thus not reported on neonatal screening, potentially resulting in delayed diagnosis. Furthermore, TSH is also frequently relied upon for screening when hypothyroidism is suspected later in life (with FT4 measured only if it is elevated), such that a diagnosis of central hypothyroidism may again be missed. In this case, diagnosis was delayed until 6 months of age with limited subsequent treatment compliance. LT4 therapy was ceased within 2 months of the initial diagnosis, briefly reinitiated at 5 years of age, but ceased again shortly thereafter until presentation to our clinic at age 17.5 years.
Descriptions of patients with CH before the widespread implementation of neonatal screening in the 1980s, as well as reports of children with endemic cretinism due to iodine deficiency, describe clinical phenotypes with severe cognitive and neurological impairment including spasticity, particularly in the lower extremities, shuffling gait, discoordination, jerky movements, tremor, hypotonia, and extrapyramidal disorders (33). In addition, deaf-mutism, hearing loss, dysarthria, and extreme short stature are reported (33, 34). It is therefore likely that the cognitive impairment, hypotonia, neurological deficits, and deaf-mutism seen in this case are consequences of delayed diagnosis and poor compliance with LT4 treatment. Mental retardation, microcephaly, sensorineural deafness, and severe prenatal and postnatal growth failure were described in patients with IGF-1 mutation (35) but not in patients with IGF-1 deficiency secondary to GH deficiency despite the occurrence of recurrent hypoglycemic events in infancy. This may indicate that the severe mental retardation in our case is attributable primarily to prolonged untreated hypothyroidism rather than unrecognized hypoglycemic events in infancy. Early diagnosis and initiation of LT4 therapy as well as GH treatment might have prevented neurodevelopmental deterioration and improved final height. No previous descriptions of POU1F1 mutations describe the untreated natural history of this condition. This case therefore extends the phenotypic spectrum associated with POU1F1 mutations, highlighting the importance of appropriate treatment and follow-up.
Although GH therapy resulted in a 42 cm (+3 SD) height increment, final height was only 124 cm (−6.2 SD). In our previously reported patient with the same POU1F1 mutation, GH therapy was initiated in the first year of life but final height was only 154 cm (−3.15 SD) (30). Whilst in the latter case, there may also have been difficulties with treatment compliance, severe adult short stature has been reported in other subjects with POU1F1 mutations [131 cm (24), 119 cm (21), and 116 cm (27)].
Sequencing of the candidate gene, POU1F1, revealed a homozygous mutation c.580_581 insT p.(Thr194llefs*7) and heterozygous parents. This mutation abolishes the transactivation properties of POU1F1 on three target promoters, resulting in severe TSH, PRL and GH deficiencies (30) which may be additional cause for the extreme short final height in the proband.
Conclusion
This case of a 17-year-old male with a frameshift mutation in the POU1F1 gene illustrates the consequences of inadequate CPHD treatment. The remarkable presentation of this patient, who looked 2 years old at age 17.5 years, and suffered from deafness and extensive neurological impairment, extends the recognized phenotype of POU1F1 mutations and emphasizes the great importance of early and adequate thyroid and GH supplemental therapy.
Data Availability
No datasets were generated or analyzed for this study.
Ethics Statement
This study was approved by the Institutional Review Board of Ha'Emek Medical Center. The patient is described anonymously and gave written informed consent with the publication.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Camille Vainstein for professional language editing and the patient and his family for participating in the study.
References
1. Amselem S. Current approaches for deciphering the molecular basis of combined anterior pituitary hormone deficiency in humans. Mol Cell Endocrinol. (2002) 197:47–56. doi: 10.1016/S0303-7207(02)00278-2
2. Castinetti F, Reynaud R, Saveanu A, Jullien N, Quentien MH, Rochette C, et al. An update in the genetic aetiologies of combined pituitary hormone deficiency. Eur J Endocrinol. (2016) 174:R239–47. doi: 10.1530/EJE-15-1095
3. Mehta A, Dattani MT. Developmental disorders of the hypothalamus and pituitary gland associated with congenital hypopituitarism. Best Pract Res Clin Endocrinol Metabol. (2008) 22:191–206. doi: 10.1016/j.beem.2007.07.007
4. Pfäffle R, Klammt J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metabol. (2011) 25:43–60. doi: 10.1016/j.beem.2010.10.014
5. Romero CJ, Nesi-França S, Radovick S. The molecular basis of hypopituitarism. Trends Endocrinol Metabol. (2009) 20:506–16. doi: 10.1016/j.tem.2009.06.005
6. Andersen B, Pearse RV, Jenne K, Sornson M, Lin SC, Bartke A, et al. The ames dwarf gene is required for Pit-1 gene activation. Dev Biol. (1995) 172:495–503. doi: 10.1006/dbio.1995.8040
7. Jacobson EM, Li P, Leon-del-Rio A, Rosenfeld MG, Aggarwal AK. Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev. (1997) 11:198–212. doi: 10.1101/gad.11.2.198
8. Pfäffle RW, DiMattia GE, Parks JS, Brown MR, Wit JM, Jansen M, et al. Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia. Science. (1992) 257:1118–21.
9. Radovick S, Nations M, Du Y, Berg LA, Weintraub BD, Wondisford FE. A mutation in the POU-homeodomain of Pit-1 responsible for combined pituitary hormone deficiency. Science. (1992) 257:1115–8.
10. Tatsumi K, Miyai K, Notomi T, Kaibe K, Amino N, Mizuno Y, et al. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet. (1992) 1:56–8. doi: 10.1038/ng0492-56
11. Birla S, Khadgawat R, Jyotsna VP, Jain V, Garg MK, Bhalla AS, et al. Identification of novel PROP1 and POU1F1 mutations in patients with combined pituitary hormone deficiency. Hormone Metabol Res. (2016) 48:822–7. doi: 10.1055/s-0042-117112
12. Carlomagno Y, Salerno M, Vivenza D, Capalbo D, Godi M, Mellone S, et al. A novel recessive splicing mutation in the POU1F1 gene causing combined pituitary hormone deficiency. J Endocrinol Invest. (2009) 32:653–8. doi: 10.1007/BF03345736
13. Cohen LE, Zanger K, Brue T, Wondisford FE, Radovick S. Defective retinoic acid regulation of the Pit-1 gene enhancer: a novel mechanism of combined pituitary hormone deficiency. Mol Endocrinol. (1999) 13:476–84. doi: 10.1210/mend.13.3.0251
14. Fofanova OV, Takamura N, Kinoshita E, Yoshimoto M, Tsuji Y, Peterkova VA, et al. Rarity of PIT1 involvement in children from Russia with combined pituitary hormone deficiency. Am J Med Genet. (1998) 77:360–5. doi: 10.1002/(SICI)1096-8628(19980605)77:5<360::AID-AJMG4>3.0.CO;2-R
15. Gat-Yablonski G, Lazar L, Pertzelan A, Phillip M. A novel mutation in PIT-1: phenotypic variability in familial combined pituitary hormone deficiencies. J Pediatr Endocrinol Metabol. (2002) 15:325–30. doi: 10.1515/JPEM.2002.15.3.325
16. Hashimoto Y, Cisternino M, Cohen LE. A novel nonsense mutation in the Pit-1 gene: evidence for a gene dosage effect. J Clin Endocrinol Metabol. (2003) 88:1241–7. doi: 10.1210/jc.2002-021510
17. Hendriks-Stegeman BI, Augustijn KD, Bakker B, Holthuizen P, van der Vliet PC, Jansen M. Combined pituitary hormone deficiency caused by compound heterozygosity for two novel mutations in the POU domain of the PIT1/POU1F1 gene. J Clin Endocrinol Metabol. (2001) 86:1545–50. doi: 10.1210/jcem.86.4.7371
18. Holl RW, Pfäffle R, Kim C, Sorgo W, Teller WM, Heimann G. Combined pituitary deficiencies of growth hormone, thyroid stimulating hormone and prolactin due to Pit-1 gene mutation: a case-report. Eur J Pediatr. (1997) 156:835–7. doi: 10.1007/s004310050723
19. Irie Y, Tatsumi K, Ogawa M, Kamijo T, Preeyasombat C, Suprasongsin C, et al. A novel E250X mutation of the PIT1 gene in a patient with combined pituitary hormone deficiency. Endocr J. (1995) 42:351–4. doi: 10.1507/endocrj.42.351
20. Malvagia S, Poggi GM, Pasquini E, Donati MA, Pela I, Morrone A, et al. The de novo Q167K mutation in the POU1F1 gene leads to combined pituitary hormone deficiency in an italian patient. Pediatr Res. (2003) 54:635–40. doi: 10.1203/01.PDR.0000084113.41375.1E
21. Rodrigues Martineli AM, Braga M, De Lacerda L, Raskin S, Graf H. Description of a brazilian patient bearing the R271W Pit-1 gene mutation. Thyroid. (1998) 8:299–304. doi: 10.1089/thy.1998.8.299
22. McLennan K, Jeske Y, Cotterill A, Cowley D, Penfold J, Jones T, et al. Combined pituitary hormone deficiency in australian children: clinical and genetic correlates. Clin Endocrinol. (2003) 58:785–94. doi: 10.1046/j.1365-2265.2003.01781.x
23. Ohta K, Nobukuni Y, Mitsubuchi H, Fujimoto S, Matsuo N, Inagaki H, et al. Mutations in the PIT-1 gene in children with combined pituitary hormone deficiency. Biochem Biophys Res Commun. (1992) 189:851–5. doi: 10.1016/0006-291X(92)92281-2
24. Pellegrini-Bouiller I, Bélicar P, Barlier A, Gunz G, Charvet JP, Jaquet P, et al. A new mutation of the gene encoding the transcription factor Pit-1 is responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metabol. (1996) 81:2790–6. doi: 10.1210/jc.81.8.2790
25. Pernasetti F, Milner RD, al Ashwal AA, de Zegher F, Chavez VM, Muller M, et al. Pro239Ser: a novel recessive mutation of the Pit-1 gene in seven middle eastern children with growth hormone, prolactin, and thyrotropin deficiency. J Clin Endocrinol Metabol. (1998) 83:2079–83. doi: 10.1210/jcem.83.6.4901
26. Salemi S, Besson A, Eblé A, Gallati S, Pfäffle RW, Mullis PE. New N-terminal located mutation (Q4ter) within the POU1F1-gene (PIT-1) causes recessive combined pituitary hormone deficiency and variable phenotype. Growth Hormone IGF Res. (2003) 13:264–8. doi: 10.1016/S1096-6374(03)00015-7
27. Snabboon T, Plengpanich W, Buranasupkajorn P, Khwanjaipanich R, Vasinanukorn P, Suwanwalaikorn S, et al. A novel germline mutation, IVS4+1G>A, of the POU1F1 gene underlying combined pituitary hormone deficiency. Hormone Res Paediatr. (2008) 69:60–4. doi: 10.1159/000111797
28. Sobrier ML, Tsai YC, Pérez C, Leheup B, Bouceba T, Duquesnoy P, et al. Functional characterization of a human POU1F1 mutation associated with isolated growth hormone deficiency: a novel etiology for IGHD. Hum Mol Genet. (2016) 25:472–83. doi: 10.1093/hmg/ddv486
29. Takagi M, Kamasaki H, Yagi H, Fukuzawa R, Narumi S, Hasegawa T. A novel heterozygous intronic mutation in POU1F1 is associated with combined pituitary hormone deficiency. Endocr J. (2017) 64:229–34. doi: 10.1507/endocrj.EJ16-0361
30. Tenenbaum-Rakover Y, Sobrier ML, Amselem S. A novel POU1F1 mutation (p.Thr168IlefsX7) associated with an early and severe form of combined pituitary hormone deficiency: functional analysis and follow-up from infancy to adulthood. Clin Endocrinol. (2011) 75:214–9. doi: 10.1111/j.1365-2265.2011.04028.x
31. Turton JP, Reynaud R, Mehta A, Torpiano J, Saveanu A, Woods KS, et al. Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency. The J Clin Endocrinol Metabol. (2005) 90:4762–70. doi: 10.1210/jc.2005-0570
32. Ward L, Chavez M, Huot C, Lecocq P, Collu R, Décarie JC, et al. Severe congenital hypopituitarism with low prolactin levels and age-dependent anterior pituitary hypoplasia: a clue to a PIT-1 mutation. J Pediatr. (1998) 132:1036–8. doi: 10.1016/S0022-3476(98)70405-6
33. Hulse A. Congenital hypothyroidism and neurological development. J Child Psychol Psychiatry. (1983) 24:629–35. doi: 10.1111/j.1469-7610.1983.tb00139.x
34. DeLong GR, Stanbury JB, Fierro-Benitez R. Neurological signs in congenital iodine-deficiency disorder (Endemic Cretinism). Dev Med Child Neurol. (2008) 27:317–24. doi: 10.1111/j.1469-8749.1985.tb04542.x
Keywords: POU1F1 gene, GH deficiency, GHD, central hypothyroidism, congenital hypothyroidism, CH
Citation: Majdoub H, Amselem S, Legendre M, Rath S, Bercovich D and Tenenbaum-Rakover Y (2019) Extreme Short Stature and Severe Neurological Impairment in a 17-Year-Old Male With Untreated Combined Pituitary Hormone Deficiency Due to POU1F1 Mutation. Front. Endocrinol. 10:381. doi: 10.3389/fendo.2019.00381
Received: 28 March 2019; Accepted: 28 May 2019;
Published: 27 June 2019.
Edited by:
Monica Livia Gheorghiu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Semra Çetinkaya, Dr Sami Ulus Child Health and Diseases Training and Research Hospital, TurkeyAlberto M. Pereira, Leiden University Medical Center, Netherlands
Copyright © 2019 Majdoub, Amselem, Legendre, Rath, Bercovich and Tenenbaum-Rakover. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yardena Tenenbaum-Rakover, cmFrb3Zlcl95QGNsYWxpdC5vcmcuaWw=