 Ferenc A. Antoni
Ferenc A. Antoni- Centre for Discovery Brain Sciences, Deanery of Biomedical Sciences, University of Edinburgh, Edinburgh, United Kingdom
It is now widely accepted that magnocellular vasopressinergic neurons in the supraoptic and paraventricular nuclei participate in the control of adrenocorticotropin secretion by the anterior pituitary gland. However, it remains to be explored in further detail, when and how these multifunctional neurons are involved in the control of anterior pituitary function. This paper highlights the role of magnocellular vasopressin in the hypothalamic pituitary adrenocortical axis with special reference to escape from glucocorticoid feedback inhibition. The signaling mechanisms underlying glucocorticoid escape by pituitary corticotrope cells, as well as the wider physiologic and pathologic contexts in which escape is known to occur—namely strenuous exercise, and autoimmune inflammation will be considered. It is proposed that by inducing escape from glucocorticoid feedback inhibition at the pituitary level, magnocellular vasopressin is critically important for the anti-inflammatory, and immunosuppressant actions of endogenous corticosteroids.
Introduction
This paper is the consequence of a symposium held in the honor of the late Dr. Kevin J. Catt (1932–2017) at Semmelweis University in Budapest. As a first year student at this institution, which has a tradition of teaching medicine for 250 years, I was handed the course textbook, “Functional Anatomy” by János Szentágothai (Schimmer) (1). First published as a single author book in 1971, this tome managed a readable, if in places decidedly subjective synthesis of classic gross anatomy and histology with modern studies of ultrastructure, physiology, and biochemistry. The didactic approach was unashamedly teleologic in emphasizing the unity of form and function. However, it went beyond Aristotelian metaphysics. It was an almost poetic celebration of the harmony of the human body, of how, from single cells to complex organs, structure reflected purpose, and function.
The scientific oeuvre of Kevin Catt heralds the beginning of a new era where the structures and regulatory properties of cells are pared back to their ultimate molecular constituents. Now in full-swing, this era of discovery is best symbolized by the complex analysis of individual cells, involving the combination of multi-parameter imaging with transcriptomics, and proteomics (2). We are able to map how the seemingly stochastic expression of genes underpins the functional plasticity of cell populations to support macroscopic physiologic reflexes and control mechanisms. This review aims to follow the path of Szentágothai in the quest for achieving the synthesis of structure and function, also incorporating the molecular insights of cellular signaling pathways pioneered by Kevin Catt.
An Outline of the Hypothalamic-Pituitary-Adrenocortical Axis
The hypothalamic-pituitary-adrenocortical system comprises a classic neuroendocrine regulatory feedback circuit. Neuroendocrine neurons in the hypothalamus drive the secretion of adrenocorticotropin (ACTH) by releasing neuropeptides, namely 41-residue corticotropin releasing-factor (CRF) and vasopressin (VP), into hypophysial portal blood-vessels that irrigate the anterior pituitary gland (3). These neurons receive a large number of diverse neural inputs from various parts of the brain that contribute to the hormonal response to stress (4–8). The production and secretion of steroid hormones—glucocorticoids, as well as mineralocorticoids—by the adrenal cortex is stimulated by ACTH secreted by the pituitary gland. An important effect of the steroids is to provide inhibitory feedback at the level of hypothalamic neroendocrine neurons, adenohypophysial corticotropes, as well as the higher brain centers involved in the control of neuroendocrine neurons (9, 10). The relative importance of the site(s) of feedback inhibition that govern the size and duration of the HPA response to stress may be dependent on the biological paradigm analyzed. In clinical practice, HPA responsiveness is usually assessed by some form of the dexamethasone suppression-test (11), utilizing a synthetic steroid largely selective for Type II glucocorticoid receptors (10). The main site of feedback tested by this paradigm appears to be the anterior pituitary corticotrope cell (12, 13).
Vasopressin
Vasopressin (VP) is a small, COOH-terminally amidated peptide chiefly synthesized by nerve cells, hence designated as a neuropeptide. Arginine-vasopressin is the most common form in mammals, but in some species Arg is replaced by Lys see review by Bichet (14). Vasopressin is elaborated upon the processing of a larger protein precursor—preprovasopressin (15), which is packaged and processed in dense-core secretory granules that are transported to sites of release along the axon as well as the dendrites (15, 16). Note that neurophysin II and copeptin, the COOH-terminal segment of the precursor, are also co-secreted with VP. The biological role of copeptin is unknown. However, its stability in human plasma and immunogenicity has been exploited to use copeptin as surrogate for the activity of VP-secreting neurons with intriguing results (17, 18). The cellular actions of VP are mediated by G protein-coupled 7-transmembrane domain receptors. Of these, the V1b isoform appears most relevant for the stress response (19, 20).
Functional Neuroanatomy of Vasopressinergic Neurons of the Hypothalamus
The Magnocellular System
The magnocellular neurosecretory system of the hypothalamus was discovered a relatively long time ago, on the basis of the distinct histochemical reaction produced by the high amounts of neurohormone elaborated by these neurons reviewed by Scharrer (21). The cell bodies are found in the supraoptic and paraventricular hypothalamic nuclei and send their axons to terminate in the neurohemal interface zone located at the bottom of the hypothalamus. The neurohemal interface is comprised of the median eminence and the posterior lobe of the pituitary gland also referred to as the neurohypophysis. Magnocellular neurons constitute the classic antidiuretic system that is activated by hypernatremia or hypovolemia and releases VP into the systemic circulation (14). Subsequent work has shown that the axons of magnocellular neurons also release VP en-passant in the internal zone of the median eminence (22–24). The VP released by this process appears in pituitary portal blood that irrigates the anterior pituitary gland [(25), also see Box 1]. It is of note, that magnocellular VP neurons also release vasopressin from their dendrites (16). Whether or not dendritic release contributes to the control of the pituitary secretion of ACTH remains to be clarified. However, it is well-established that magnocellular neurons make various forms of contact with CRF producing cells in the medial parvicellular paraventricular nucleus of the hypothalamus (26, 27). Moreover, Plotsky and co-workers found that in rats VP given into the 3rd cerebral ventricle attenuated the release of CRF into hypophysial portal blood and, conversely, inhibition of AVP action increased the levels of CRF (28). These results indicate that there may be a functional link between the magnocellular VP neurons and parvicellular neurons producing CRF.
Box 1. Six decades of dogma-busting: The magnocellular vasopressinergic neuron as a modulator of anterior pituitary ACTH secretion.
1. Neurosecretion into the bloodstream-non-conforming to neuron doctrine.
2. Negates single hypothalamic releasing hormone for each pituitary hormone hypothesis.
3. VP is not just the antidiuretic hormone (ADH).
4. Secretory path from internal zone of the median eminence into pituitary portal blood-“en passant” neuropeptide release.
5. Negates the hyophyseotrophic area = the mediobasal hypothalamus concept.
6. VP alone has no ACTH releasing effect at physiologic concentrations i.e., it is a new type of neuroendocrine modulator, a major action of which is to induce glucocorticoid resistance at the level of the corticotrope cell.
Parvicellular Neurons Producing CRF and VP
A second population of VP-ergic neurons projecting to the median eminence is located in the medial parvicellular part of the hypothalamic paraventricular nucleus. These are small neurons that also co-produce and co-package CRF with VP (29, 30) and are the key neuroendocrine neurons that regulate the pituitary secretion of ACTH (31, 32). With respect to the stress response two functional differences between the magnocellular and parvicellular neurons are of note. First, under normal conditions, magnocellular neurons produce up to 10-fold more VP than parvicellular neurons (30, 33). Second, the production and release of VP by parvicellular neurons is under prominent inhibitory control by adrenal corticosteroids (34), while that in magnocellular neurons is not (35).This is despite the fact that magnocellular VP neurons express Type II glucocorticoid receptors (36). In the absence of adrenal corticosteroids, VP release from the parvicellular neurons is massively increased and becomes comparable to that from magnocellular neurons (33, 37).
The Relevance of Magno- and Parvi-Cellular VP-Ergic Neurons to the Stress Response
The role of VP and its pituitary V1b receptor in the stress response has been analyzed in considerable detail in various models, including genetically vasopressin-deficient Brattleboro rats (38) and V1b receptor gene-deleted mice (20). These studies, and many others see (39) for review, support an important role for VP in the stimulatory control of stress-induced ACTH secretion. However, very few studies have attempted to distinguish between the effects of parvicellular and magnocellular VP. Plotsky and Thivikraman first produced evidence showing that glucocorticoid negative feedback appeared to be absent in conscious rats subjected to repeated moderate hemorrhage (40). Subsequently, these workers showed that HPA activation by air-puff startle, a psychological stressor, was sensitive to inhibition by corticosterone whereas that caused by small or moderate hemorrhage was not (41). Moreover, they found early immediate gene activation in magnocellular neurons of the supraoptic and paraventricular hypothalamic nuclei during hemorrhage and activation in the dorsomedial part of the hypothalamus by air-puff startle. Thus, this work differentiated feedback prone and feedback resistant mild stressors and observed signs of activation in magnocellular hypothalamic neurons in the case of the latter.
Signal Integration by Corticotrope Cells of the Anterior Pituitary Gland and the VP-Induced Escape From Inhibition by Glucocorticoids
The stimulatory input to corticotrope cells is comprised of CRF and VP. A hypothalamic ACTH-inhibiting factor, atrial natriuretic peptide (ANP) has also been identified (39, 42). Finally, corticotrope cells are a well-established site of glucocorticoid feedback-inhibition (9, 10).
CRF
The widely accepted mechanism of action in the case of CRF is activation of CRF1 receptors and conservative coupling to the stimulatory G protein Gs. The consequent activation of transmembrane adenylyl cyclase(s) leads to increase of intracellular cAMP levels (43). The scenario of intracellular signaling downstream of cAMP is complex, as reviewed in detail elsewhere (44, 45). However, it is clear that intracellular Ca2+ signals are generated by CRF, and that the electrical activity of the cells ultimately determines the secretion of ACTH.
VP
In the case of VP, the consensus is the activation of V1b receptors coupled to Gq and consequent activation of PLC, with ensuing increases of IP3 levels and PKC activity. Exactly which isoform(s) of PKC is (are) involved, is not entirely clear, but as phorbol esters can mimic most of the known actions of VP including the potentiation of the cAMP signal and the stimulation of ACTH secretion, the alpha and beta PKC-isoforms are the likely candidates. Once more, events after IP3 and PKC activation are too complex to be considered here (44, 46).
ANP
There is a signaling pathway to inhibit stimulated ACTH secretion through 3′5′guanosine monophosphate (cGMP), that is not shared with glucocorticoids (47). This pathway can be activated by ANP (48, 49), and may become functionally more prominent under conditions where the pituitary corticotropes are in “glucocorticoid escape” mode.
Glucocorticoids
Feedback inhibition in the HPA axis occurs in three time-domains; rapid <15 min, early delayed—within 20 min and up to 2 h, and delayed, beyond 2 h (9, 10, 50). With respect to the time-domains of feedback manifested in corticotropes: in contrast to a recent study (51), we have failed to observe a rapid inhibitory effect of glucocorticoids in dispersed rat anterior pituitary (52) or AtT-20D16:16 mouse corticotrope tumor cells (53). Thus, the focus of the studies outlined here was early-delayed feedback, which is the main inhibitory effect in the first 2 h after exposure to glucocorticoids. The properties of this inhibition have been reviewed extensively (9, 54, 55). In brief, it is mediated by Type II glucocorticoid receptors and is sensitive to inhibitors of transcription and translation, indicating a requirement of mRNA synthesis/processing and protein synthesis (52). What is the molecular and cellular basis of early- delayed inhibition? In AtT-20 cells, the balance of cAMP-dependent phosphorylation on the stress-axis-regulated-insert (STREX) type α-subunit of the large conductance Ca2+-dependent K+-channel (BK-channel) is shifted toward dephosphorylation. As a result, the channel is not inhibited by cAMP-dependent phosphorylation induced by CRF (56). Hence, the enhancement of intracellular free Ca2+ transients and the consequent stimulation of ACTH secretion by CRF are suppressed, reviewed in refs (54, 57). Subsequent work has implicated a protein phosphatase 2A-like enzyme as the mediator of glucocorticoid action (58), but definitive evidence is still outstanding. In contrast to AtT20 cells, blockade of BK-channels had no appreciable effect on the efficacy of corticosterone to inhibit CRF-induced ACTH secretion in cultured rat anterior pituitary cells (59). This is despite the fact, that BK channels play a prominent role in governing the firing pattern and the intracellular Ca2+ transients of anterior pituitary cells in culture (57) and that corticosterone has an inhibitory effect on the bursting activity of mouse corticotropes cells (44). Moreover, inhibition of agonist-induced ACTH release by ANP/cGMP, could be reversed by blockers of BK as well as other K+ channels (60). In contrast, studies with a variety of K+ inhibitors failed to identify conclusively the K+ channel key for early delayed inhibition by corticosteroids (61). Others, using a similar panel of K+ channel blockers, reported a role for ether-á-go-go-related gene K+-channels as the mediators of glucocorticoid inhibition (62). However, these findings have not been confirmed by other, more direct methods, such as patch-clamp electrophysiology or gene-knock down. In summary, current evidence indicates that early-delayed corticosteroid feedback inhibition at the pituitary level is by glucocorticoid-induced protein(s) that facilitate the activation of K+-channels. A similar conclusion was reached in a study of hippocampal pyramidal neurons (63). Significantly, this scenario implies that glucocorticoid inhibition is sensitive to depolarization of the membrane potential (59, 64).
One of the complicating factors in understanding signaling processes in corticotropes is the variety of the in vitro models used. It is clear that cultured cells, acutely dispersed cells, and pituitary slices may have different properties. Moreover, the temporal dynamics in perifused tissue are different to that seen in static systems. Finally, the concentrations of the effectors employed tend to vary from physiologic (CRF up to 0.3 nM, VP up to 3 nM) to the grossly aphysiologic.
With these considerations in mind, a detailed analysis of CRF-induced cAMP levels was carried out in the context of the inhibition of cAMP synthesis by intracellular free Ca2+, and the efficacy of corticosteroid feedback inhibition of stimulated ACTH release. It transpired that CRF-induced cAMP synthesis is under feedback inhibition by Ca2+ derived from a ryanodine/caffeine sensitive intracellular pool (65). However, above 1 nM CRF, the effect of Ca2+ was no longer apparent. A similar “escape” of CRF-induced cAMP synthesis from Ca2+ inhibition could be induced by physiologic concentrations of VP, an effect also mimicked by phorbol-ester activation of protein kinase C (65). Analysis of cAMP hydrolysis indicated that while PDE4 isoforms were active when low concentrations of CRF were used, inhibitors of PDE1 were required for inhibiting cAMP breakdown when CRF was applied in combination with VP. When interpreted in the context of the diversity of cAMP signaling proteins (66, 67), the results indicate that physiologic concentrations of CRF activate a Ca2+ inhibited adenylyl cyclase such as AC5, 6, or 9, while in the presence of VP an adenylyl cyclase activated by protein kinase C such as AC2 or AC7 enters the fray (Figure 1). This latter cyclase is responsible for the markedly increased cAMP levels that require high-capacity, low-affinity phosphodiesterases to control cAMP signaling. By the combination of immunocytochemistry, Western blotting and RT-PCR, we concluded that AC7, PDE1a, and PDE1b were the best candidates underlying the switching of corticotropes to high cAMP levels in the context of stimulation by CRF and VP. Importantly, VP was present at concentrations that are only produced by magnocellular neurons under physiologic conditions.
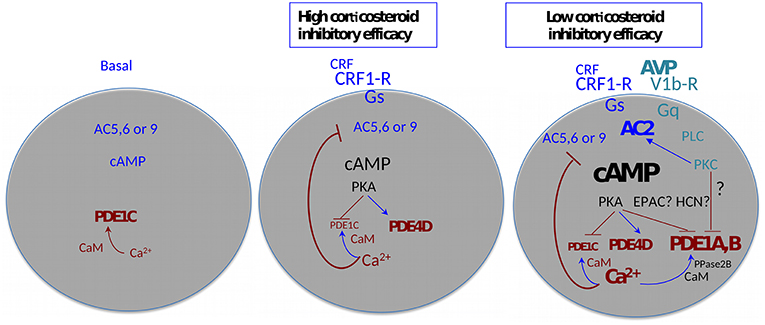
Figure 1. Agonist-induced plasticity of cAMP signaling—multiple functional states of the adenohypophysial corticotroph cell. AC, adenylyl cyclase; CaM, calmodulin; EPAC, exchange factors directly activated by cAMP; HCN, Hyperpolarization activated cyclic nucleotide gated channels; PDE- cyclic nucleotide phosphodiesterase; PKA, cAMP dependent protein kinase; PKC, protein kinase C; PLC, phospholipase C; PPase2B, protein phosphatase 2B (alias calcineurin). Arrows indicate facilitation, T-bars indicate inhibition. The size and thickness of the lettering is proportional to the amplitude/activity of the denoted entity.
It is pertinent to highlight here that the “amplitude modulation” of the cAMP signal outlined above has potentially important consequences: whilst PKA is maximally active at 1 μM cAMP, other effectors such as EPACS, hyperpolarization-activated cyclic nucleotide–gated (HCN) channels, and cyclic nucleotide gated channels all require >10 μM cAMP for activity. Thus, a whole plethora of cAMP signaling systems is potentially mobilized in corticotrope cells in the presence of high (magnocellular) concentrations of VP that are not active at levels of stimulation that can be achieved via parvicellular CRF/VP. It is also important to reiterate here that the effects of VP on cAMP synthesis are conditional to the presence of CRF.
Several of the inferences in the aforementioned studies were made on the basis of the application of pharmacologic agents or the use of antibodies. It later transpired, that the antibodies used to localize AC2 in the anterior pituitary gland were unreliable, similarly, an antibody against AC5/6 gave also inconsistent results (68). More recently, mRNA-seq analyses of single anterior pituitary cells have been published (69). The data show selective enrichment of AC2 in corticotrope cells, in contrast, the levels of AC7 mRNA in anterior pituitary cells are very low. All three Ca2+-inhibited ACs, AC5, 6, and 9 appear to be expressed in corticotropes. Moreover, the highest relative levels of PDE1a and PDE1b appear to be associated with cells identified as corticotropes.
Taken together, the molecular machinery of Ca2+ feedback on cAMP biosynthesis and for achieving the VP-mediated boost of cellular cAMP to levels that address EPACS, HCNs and CNGs is present in corticotropes. Finally, is attractive to speculate here, that high VP overrides early feedback through depolarization of the membrane potential by activation of an HCN channel (70, 71).
The analysis of the efficacy of corticosterone to inhibit CRF-induced ACTH release showed interesting parallels with the characteristics of the inhibition of cAMP synthesis by Ca2+(61). In the physiologic range for CRF, corticosterone inhibition was highly efficient, while at supraphysiological CRF the inhibitory efficacy was markedly reduced. Application of the PDE4 blocker rolipram or high, physiologic levels of VP with physiologic concentrations of CRF also markedly reduced the inhibitory efficacy of corticosterone. In essence, irrespective of how intracellular cAMP levels were elevated above 20 μM, glucocorticoid efficacy was progressively reduced. Thus, if the agonists by-pass the Ca2+ inhibitory feedback on adenylyl cyclase, the efficacy of glucocorticoid inhibition is dramatically reduced. These observations link the VP mediated switch of low cAMP signaling induced by CRF to high cAMP, with the escape from glucocorticoid feedback at the pituitary level. A summary cartoon of the corticosteroid sensitive and insensitive states of anterior pituitary corticotrope cells is shown in Figure 1.
When Is Glucocorticoid Escape of Biological Importance?
The pronounced deleterious effects of long-term corticosteroid excess on body homeostasis indicate that glucocorticoid escape is encountered in life-threatening situations and is likely to be of relatively short duration. The classic clinical examples of persistent glucocorticoid non-suppression are Cushing's disease (72) and melancholic depression (12).
Autoimmune Inflammation
In preclinical models of autoimmune inflammation, such as experimental autoimmune encephalomyelitis and adjuvant-induced arthritis, a sustained rise of plasma corticosteroids lasting several days is crucial for survival (73). Indeed, it is widely recognized that adrenal corticosteroids are important facilitators of endogenous immunosuppression (74). The only way this can occur is by escape from glucocorticoid feedback inhibition. Several lines of evidence indicate, that the sustained and vital increase of corticosteroids in preclinical autoimmune models is mediated by VP (75, 76). Moreover, the data of Suzuki etal. (76), indicated at the level of VP expression rather than mRNA, that magnocellular vasopressinergic neurons become activated during autoimmune inflammation. It is relevant to recall here that VP synthesis is not closely controlled by adrenal corticosteroids in magnocellular neurons (35) and that the effects of low levels of CRF on corticotropes are amplified dramatically by VP (65, 77). Thus, activation of magnocellular VP neurons can bypass glucocorticoid feedback in the CNS and induce escape from glucocorticoid feedback at the pituitary level to support the sustained secretion of corticosteroids. The trigger for the activation of hypothalamic neurons during autoimmune inflammation is unknown. It is plausibly a cytokine, above all, interleukin-6 (IL-6) (78) known to activate the HPA axis in humans as well as rodents (79). However, the exact mechanism by which this occurs remains to be clarified (80). Intriguingly, magnocellular VP neurons also produce IL-6 (81). Moreover, increases of IL-6 levels in magnocellular neurons were observed after various types of stress paradigms (82). Whether or not this IL-6 is involved in the regulation VP release e.g., as a paracrine mediator, is unknown. A summary diagram of the by-pass of glucocorticoid feedback in the HPA axis through the activation of magnocellular VP neurons by immune mediators is shown in Figure 2.
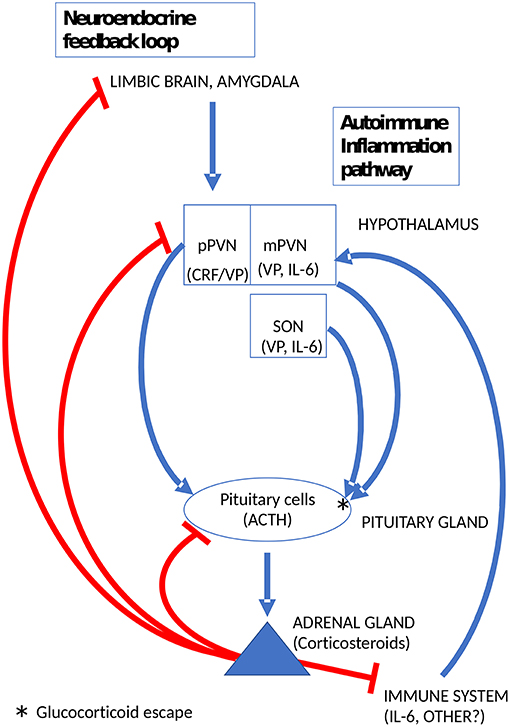
Figure 2. Glucocorticoid escape at the pituitary gland induced by magnocellular VP to mobilize the endogenous immunosuppressant activity of adrenal corticosteroids. pPVN, parvicellular medial dorsal part of the hypothalamic paraventricular nucleus; mPVN lateral magnocellular part of the PVN; SON, supraoptic hypothalamic nucleus.
High Intensity Exercise and Hyponatremia
Work by Deuster et al. (83) reported that high-intensity exercise results in dexamethasone non-suppression in men as well as women. One of the threats of high-intensity exercise is hyponatremia (84), which may be lethal due to the development of hypotonic encephalopathy (85). Increases of plasma VP are prominent under these conditions and it has been argued that exercise-associated hypernatremia is a case of inappropriate VP secretion (SIADH). It is likely, that IL-6 released from muscle cells during exercise is a trigger for VP secretion. Additionally, hyperhydration by the subjects during exercise may contribute to hyponatremia. In the pituitary portal system, where VP levels are considerably higher than in the periphery, the levels of vasopressin are sufficient to induce glucocorticoid escape at the pituitary level. The ensuing higher levels of ACTH would enhance aldosterone secretion by zona glomerulosa cells and improve sodium retention. In addition, VP has a direct action to stimulate aldosterone secretion in rat (86) as well as human zona glomerulosa cells (87). Thus, while exercise may symptomatically mimic SIADH in that plasma levels of VP increase despite hypoosmolarity, it may be a fluid-volume preservation reflex that simultaneously promotes sodium retention by bringing about glucocorticoid non-suppression at the pituitary level.
Summary
In a day-to-day setting, the activity of the HPA axis is under powerful feedback inhibitory control. The situation during certain types of stress may be different. In a seminal review, Munck et al. (88) proposed that the physiologic rationale of stress-induced glucocorticoid production was to dampen and reset the production of powerful immune mediators induced by the stress response. Three decades, two dozen cytokines and a few microbiomes later, this concept is stronger than ever. In particular, the emergence of IL-6 as a pleiotropic stress-responsive protein and its activation of the hypothalamic CRF and VP systems is of particular note. If corticosteroids are controlled by strict neuroendocrine feedback tailored for stress-free and/or casual mild stress conditions, they could not be deployed to combat stress with a substantial inflammatory component. Increasingly, it appears that most types of stress also mobilize inflammatory mediators (89, 90). Similarly, given the threat of hyponatremia in severe hypovolemia, overriding inhibitory feedback to allow sodium retention through aldosterone may be key to survival. Therefore, feedback inhibition has to be plastic. Indeed, the functional neuroanatomy, as well as the molecular and cellular elements underpinning the plasticity of corticosteroid feedback inhibition are clearly apparent in the hypothalamo-hypophysial system. Where sustained secretion of adrenal corticosteroids is vital for survival, the evidence points to a critical role of magnocellular VP to induce glucocorticoid escape at the pituitary level. Whether or not this insight can be exploited therapeutically remains to be explored.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Dr. Paul LeTissier and Dr. Nicola Romano for helpful discussions regarding pituitary gene-expression.
References
1. Réthelyi M, Szentágothai. Functional Anatomy: Anatomy, Histology and Embryology for Medical and Dental Students. Budapest: Medicina (2018).
2. Budnik B, Levy E, Harmange G, Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. (2018) 19:161. doi: 10.1186/s13059-018-1547-5
3. Fink G. 60 years of neuroendocrinology: memoir: harris' neuroendocrine revolution: of portal vessels and self-priming. J Endocrinol. (2015) 226:T13–24. doi: 10.1530/JOE-15-0130
4. Kiss JZ, Palkovits M, Záborszky L, Tribollet E, Szabó D, Makara GB. Quantitative histological studies on the hypothalamic paraventricular nucleus in rats. II. number of local and certain afferent nerve terminals. Brain Res. (1983) 265:11–20. doi: 10.1016/0006-8993(83)91328-8
5. Sawchenko PE, Swanson LW. The organization of forebrain afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol. (1983) 218:121–44. doi: 10.1002/cne.902180202
6. Ziegler DR, Herman JP. Neurocircuitry of stress integration: anatomical pathways regulating the hypothalamo-pituitary-adrenocortical axis of the rat. Integr Comp Biol. (2002) 42:541–51. doi: 10.1093/icb/42.3.541
7. Mcklveen JM, Myers B, Herman JP. The medial prefrontal cortex: coordinator of autonomic, neuroendocrine and behavioural responses to stress. J Neuroendocrinol. (2015) 27:446–56. doi: 10.1111/jne.12272
8. Myers B, Scheimann JR, Franco-Villanueva A, Herman JP. Ascending mechanisms of stress integration: Implications for brainstem regulation of neuroendocrine and behavioral stress responses. Neurosci Biobehav Rev. (2017) 74:366–75. doi: 10.1016/j.neubiorev.2016.05.011
9. Keller-Wood MB, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. (1984) 5:1–23. doi: 10.1210/edrv-5-1-1
10. De Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. (2005) 6:463–75. doi: 10.1038/nrn1683
11. Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression–a potential biomarker? Biol Psychiatry. (2007) 62:47–54. doi: 10.1016/j.biopsych.2006.07.039
12. Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res. (1994) 28:341–56. doi: 10.1016/0022-3956(94)90017-5
13. Cole MA, Kim PJ, Kalman BA, Spencer RL. Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology. (2000) 25:151–67. doi: 10.1016/S0306-4530(99)00045-1
14. Bichet DG. The posterior pituitary. In: Melmed S, editor. The Pituitary. San Diego, CA: Academic Press (2017). p. 251–88.
15. Brownstein MJ, Russell JT, Gainer H. Synthesis, transport, and release of posterior pituitary hormones. Science. (1980) 207:373–8. doi: 10.1126/science.6153132
16. Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci. (2006) 7:126–36. doi: 10.1038/nrn1845
17. Morgenthaler NG, Struck J, Jochberger S, Dunser MW. Copeptin: clinical use of a new biomarker. Trends Endocrinol Metab. (2008) 19:43–9. doi: 10.1016/j.tem.2007.11.001
18. Christ-Crain M, Fenske W. Copeptin in the diagnosis of vasopressin-dependent disorders of fluid homeostasis. Nat Rev Endocrinol. (2016) 12:168–76. doi: 10.1038/nrendo.2015.224
19. Antoni FA. Novel ligand specificity of pituitary vasopressin receptors in the rat. Neuroendocrinology. (1984) 39:186–8. doi: 10.1159/000123976
20. Roper JA, O'carroll AM, Young WS III, Lolait SJ. The vasopressin AVP1b receptor: Molecular and pharmacological studies. Stress. (2011) 14:98–115. doi: 10.3109/10253890.2010.512376
21. Scharrer B. Neurosecretion: beginnings and new directions in neuropeptide research. Annu Rev Neurosci. (1987) 10:1–17. doi: 10.1146/annurev.neuro.10.1.1
22. Holmes MC, Antoni FA, Aguilera G, Catt KJ. Magnocellular axons in passage through the median eminence release vasopressin. Nature. (1986) 319:326–9. doi: 10.1038/319326a0
23. Morris JF, Pow DV. Capturing and quantifying the exocytotic event. J Exp Biol. (1988) 139:81–103.
24. Engelmann M, Landgraf R, Wotjak CT. The hypothalamic-neurohypophysial system regulates the hypothalamic-pituitary-adrenal axis under stress: an old concept revisited. Front Neuroendocrinol. (2004) 25:132–49. doi: 10.1016/j.yfrne.2004.09.001
25. Antoni FA, Fink G, Sheward WJ. Corticotrophin-releasing peptides in rat hypophysial portal blood after paraventricular lesions: a marked reduction in the concentration of 41-residue corticotrophin releasing factor, but no change in vasopressin. J Endocrinol. (1990) 125:175–83. doi: 10.1677/joe.0.1250175
26. Léránth C, Antoni FA, Palkovits M. Ultrastructural demonstration of ovine CRF-like immunoreactivity (oCRF-LI) in the rat hypothalamus: processes of magnocellular neurons establish membrane specializations with parvocellular neurons containing oCRF-LI. Regulatory Peptides. (1983) 6:179–88. doi: 10.1016/0167-0115(83)90011-3
27. Liposits Z, Paull WK, Sétáló G, Vigh S. Evidence for local corticotropin releasing factor (CRF)-immunoreactive neuronal circuits in the paraventricular nucleus of the rat hypothalamus. Histochemistry. (1985) 83:5–16. doi: 10.1007/BF00495294
28. Plotsky PM, Bruhn TO, Vale W. Central modulation of immunoreactive corticotropin-releasing factor secretion by arginine vasopressin. Endocrinology. (1985) 115:1639–41. doi: 10.1210/endo-115-4-1639
29. Whitnall MH, Mezey E, Gainer H. Co-localization of corticotropin-releasing factor and vasopressin in median eminence neurosecretory vesicles. Nature. (1985) 317:248–50. doi: 10.1038/317248a0
30. Kiss JZ, Bertini LT. The CRF neurosecretory vesicle: vasopressin-dependent changes in vesicle size after adrenalectomy. Brain Res. (1992) 597:353–7. doi: 10.1016/0006-8993(92)91495-Z
31. Makara GB, Antoni FA, Stark E, Kárteszi M. Hypothalamic organization of CRF containing structures. Neuroendocrine Perspect. (1984) 3:71–110. doi: 10.1016/B978-0-444-90377-8.50006-5
32. Antoni FA. Hypothalamic regulation of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin-releasing factor. Endocr Rev. (1986) 7:351–78. doi: 10.1210/edrv-7-4-351
33. Knepel W, Przewlocki R, Nutto D, Herz A. Foot shock stress-induced release of vasopressin in adenohypophysectomized and hypophysectomized rats. Endocrinology. (1985) 117:292–9. doi: 10.1210/endo-117-1-292
34. Kovács KJ, Földes A, Sawchenko PE. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J Neurosci. (2000) 20:3843–52. doi: 10.1523/JNEUROSCI.20-10-03843.2000
35. Raff H. Glucocorticoid inhibition of neurohypophysial vasopressin secretion. Am J Physiol. (1987) 252:R635–44. doi: 10.1152/ajpregu.1987.252.4.R635
36. Kiss JZ, Van Eekelen JA, Reul JM, Westphal HM, De Kloet ER. Glucocorticoid receptor in magnocellular neurosecretory cells. Endocrinology. (1988) 122:444–9. doi: 10.1210/endo-122-2-444
37. Holmes MC, Antoni FA, Catt KJ, Aguilera G. Predominant release of vasopressin vs. corticotropin releasing factor from the isolated median eminence after adrenalectomy. Neuroendocrinology. (1986) 43:245–51. doi: 10.1159/000124533
38. Makara GB, Mergl Z, Zelena D. The role of vasopressin in hypothalamo-pituitary-adrenal axis activation during stress: an assessment of the evidence. Ann N Y Acad Sci. (2004) 1018:151–61. doi: 10.1196/annals.1296.018
39. Antoni FA. Vasopressinergic control of anterior pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol. (1993) 14:76–122. doi: 10.1006/frne.1993.1004
40. Thrivikraman KV, Plotsky PM. Absence of glucocorticoid negative feedback to moderate hemorrhage in conscious rats. Am J Physiol. (1993) 264:E497–503. doi: 10.1152/ajpendo.1993.264.4.E497
41. Thrivikraman KV, Nemeroff CB, Plotsky PM. Sensitivity to glucocorticoid-mediated fast-feedback regulation of the hypothalamic-pituitary-adrenal axis is dependent upon stressor specific neurocircuitry. Brain Res. (2000) 870:87–101. doi: 10.1016/S0006-8993(00)02405-7
42. Engler D, Redei E, Kola I. The corticotropin-release inhibitory factor hypothesis: a review of the evidence for the existence of inhibitory as well as stimulatory hypophysiotropic regulation of adrenocorticotropin secretion and biosynthesis. Endocr Rev. (1999) 20:460–500. doi: 10.1210/edrv.20.4.0376
43. Dautzenberg FM, Grigoriadis DE, Hauger RL, Steckler T, Vale WW. Corticotropin-releasing factor receptors: CRF1 receptor. IUPHAR/BPS Guide to Pharmacology. Available online at: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=212andfamilyId=219andfamilyType=GPCR~(2019).
44. Duncan PJ, Tabak J, Ruth P, Bertram R, Shipston MJ. Glucocorticoids inhibit CRH/AVP-evoked bursting activity of male murine anterior pituitary corticotrophs. Endocrinology. (2016) 157:3108–21. doi: 10.1210/en.2016-1115
45. Fletcher PA, Zemkova H, Stojilkovic SS, Sherman A. Modeling the diversity of spontaneous and agonist-induced electrical activity in anterior pituitary corticotrophs. J Neurophysiol. (2017) 117:2298–311. doi: 10.1152/jn.00948.2016
46. Lee AK, Tse FW, Tse A. Arginine vasopressin potentiates the stimulatory action of CRH on pituitary corticotropes via a protein kinase C-dependent reduction of the background TREK-1 current. Endocrinology. (2015) 156:3661–72. doi: 10.1210/en.2015-1293
47. Antoni FA, Dayanithi G. Evidence for distinct glucocorticoid and guanine 3′,5′ monophosphate effected inhibition of secretagogue stimulated ACTH release in vitro. Endocrinology. (1990) 126:1355–60. doi: 10.1210/endo-126-3-1355
48. Dayanithi G, Antoni FA. Atriopeptins are potent inhibitors of ACTH secretion by rat anterior pituitary cells in vitro: involvement of the atriopeptin receptor domain of membrane bound guanylyl cyclase. J Endocrinol. (1990) 125:39–44. doi: 10.1677/joe.0.1250039
49. Antoni FA, Hunter EFM, Lowry PJ, Noble JM, Seckl JR. Atriopeptin: an endogenous corticotropin-release inhibiting hormone. Endocrinology. (1992) 130:1753–5. doi: 10.1210/endo.130.3.1311248
50. Osterlund CD, Rodriguez-Santiago M, Woodruff ER, Newsom RJ, Chadayammuri AP, Spencer RL. Glucocorticoid fast feedback inhibition of stress-induced ACTH secretion in the male rat: rate independence and stress-state resistance. Endocrinology. (2016) 157:2785–98. doi: 10.1210/en.2016-1123
51. Deng Q, Riquelme D, Trinh L, Low MJ, Tomic M, Stojilkovic S, et al. Rapid glucocorticoid feedback inhibition of acth secretion involves ligand-dependent membrane association of glucocorticoid receptors. Endocrinology. (2015) 156:3215–27. doi: 10.1210/EN.2015-1265
52. Dayanithi G, Antoni FA. Rapid as well as delayed inhibitory effects of glucocorticoid hormones on pituitary adrenocorticotropic hormone release are mediated by type II glucocorticoid receptors and require newly synthesized messenger ribonucleic acid as well as protein. Endocrinology. (1989) 125:308–13. doi: 10.1210/endo-125-1-308
53. Woods MD, Shipston MJ, Mullens EL, Antoni FA. Pituitary corticotrope tumor (AtT20) cells as a model system for the study of early inhibition by glucocorticoids. Endocrinology. (1992) 131:2873–80. doi: 10.1210/endo.131.6.1332850
54. Antoni FA. Calcium checks cyclic AMP — mechanism of corticosteroid feedback in adenohypophysial corticotrophs. J Neuroendocrinol. (1996) 8:659–72. doi: 10.1046/j.1365-2826.1996.05152.x
55. Osterlund C, Spencer RL. Corticosterone pretreatment suppresses stress-induced hypothalamic-pituitary-adrenal axis activity via multiple actions that vary with time, site of action, and de novo protein synthesis. J Endocrinol. (2011) 208:311–22. doi: 10.1530/JOE-10-0413
56. Shipston MJ, Kelly JS, Antoni FA. Glucocorticoids block protein kinase A inhibition of calcium-activated potassium channels. J Biol Chem. (1996) 271:9197–200. doi: 10.1074/jbc.271.16.9197
57. Shipston MJ. Control of anterior pituitary cell excitability by calcium-activated potassium channels. Mol Cell Endocrinol. (2018) 463:37–48. doi: 10.1016/j.mce.2017.06.003
58. Tian L, Knaus HG, Shipston MJ. Glucocorticoid regulation of calcium-activated potassium channels mediated by serine/threonine protein phosphatase. J Biol Chem. (1998) 273:13531–6. doi: 10.1074/jbc.273.22.13531
59. Lim MC, Shipston MJ, Antoni FA. Depolarisation counteracts glucocorticoid inhibition of adenohypophysial corticotroph cells. Br J Pharmacol. (1998) 124:1735–43. doi: 10.1038/sj.bjp.0702024
60. Antoni FA, Dayanithi G. Blockage of K+ channels reverses the inhibition of secretagogue stimulated ACTH release by atriopeptin. J Endocrinol. (1990) 126:183–91. doi: 10.1677/joe.0.1260183
61. Lim MC, Shipston MJ, Antoni FA. Posttranslation al modulation of glucocorticoid inhibition at the pituitary level. Endocrinology. (2002) 143:3796–801. doi: 10.1210/en.2002-220489
62. Yamashita M, Oki Y, Iino K, Hayashi C, Matsushita F, Faje A, et al. The role of ether-à-go-go-related gene K(+) channels in glucocorticoid inhibition of adrenocorticotropin release by rat pituitary cells. Regul Pept. (2009) 152:73–8. doi: 10.1016/j.regpep.2008.09.002
63. Joëls M, De Kloet ER. Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science. (1989) 245:1502–5. doi: 10.1126/science.2781292
64. Woods MD, Shipston MJ, Mcferran B, Guild SB, Antoni FA. Early glucocorticoid inhibition of hormone release in pituitary corticotrope cells is voltage dependent. Ann N Y Acad Sci. (1994) 746:456–9. doi: 10.1111/j.1749-6632.1994.tb39284.x
65. Antoni FA, Sosunov AA, Haunso A, Paterson JM, Simpson J. Short-term plasticity of cyclic adenosine 3′,5′-monophosphate signaling in anterior pituitary corticotrope cells: the role of adenylyl cyclase isotypes. Mol Endocrinol. (2003) 17:692–703. doi: 10.1210/me.2002-0369
66. Antoni FA. Molecular diversity of cyclic AMP signaling. Front Neuroendocrinol. (2000) 21:103–32. doi: 10.1006/frne.1999.0193
67. Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International union of basic and clinical pharmacology. ci structures and small molecule modulators of mammalian adenylyl cyclases Pharmacol Rev. (2017) 69:93–139. doi: 10.1124/pr.116.013078
68. Antoni FA, Wiegand UK, Black J, Simpson J. Cellular localisation of adenylyl cyclase: a post-genome perspective. Neurochem Res. (2006) 32:287–95. doi: 10.1007/s11064-005-9019-1
69. Cheung LYM, George AS, McGee SR, Daly AZ, Brinkmeier ML, Ellsworth BS, et al. Single-cell RNA sequencing reveals novel markers of male pituitary stem cells and hormone-producing cell types. Endocrinology. (2018) 159:3910–24. doi: 10.1210/en.2018-00750
70. Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a 'depolarization reserve' and is not required for heart rate acceleration in mice. EMBO J. (2007) 26:4423–32. doi: 10.1038/sj.emboj.7601868
71. Raggenbass M. Overview of cellular electrophysiological actions of vasopressin. Eur J Pharmacol. (2008) 583:243–54. doi: 10.1016/j.ejphar.2007.11.074
72. Lonser RR, Nieman L, Oldfield EH. Cushing's disease: pathobiology, diagnosis, and management. J Neurosurg. (2017) 126:404–17. doi: 10.3171/2016.1.JNS152119
73. Mason D. Genetic variation in the stress response susceptibility to experimental allergic encephalomyelitis and implications for human inflammatory disease. Immunol Today. (1991) 12:57–60. doi: 10.1016/0167-5699(91)90158-P
74. Morand EF, Leech M. Hypothalamic-pituitary-adrenal axis regulation of inflammation in rheumatoid arthritis. Immunol Cell Biol. (2001) 79:395–9. doi: 10.1046/j.1440-1711.2001.01028.x
75. Chowdrey HS, Larsen PJ, Harbuz MS, Jessop DS, Aguilera G, Eckland DJ, et al. Evidence for arginine vasopressin as the primary activator of the HPA axis during adjuvant-induced arthritis. Br J Pharmacol. (1995) 116:2417–24. doi: 10.1111/j.1476-5381.1995.tb15089.x
76. Suzuki H, Onaka T, Kasai M, Kawasaki M, Ohnishi H, Otsubo H, et al. Response of arginine vasopressin-enhanced green fluorescent protein fusion gene in the hypothalamus of adjuvant-induced arthritic rats. J Neuroendocrinol. (2009) 21:183–90. doi: 10.1111/j.1365-2826.2009.01841.x
77. Gillies GE, Linton EA, Lowry PJ. Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature. (1982) 299:355–7. doi: 10.1038/299355a0
78. Kishimoto T, Kang S, Tanaka T. IL-6: A New Era for the Treatment of Autoimmune Inflammatory Diseases. Tokyo: Springer (2015). doi: 10.1007/978-4-431-55651-0_11
79. Mastorakos G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. (1993) 77:1690–4. doi: 10.1210/jc.77.6.1690
80. Swart RM, Hoorn EJ, Betjes MG, Zietse R. Hyponatremia and inflammation: the emerging role of interleukin-6 in osmoregulation. Nephron Physiol. (2011) 118:45–51. doi: 10.1159/000322238
81. Ghorbel MT, Sharman G, Leroux M, Barrett T, Donovan DM, Becker KG, et al. Microarray Analysis Reveals Interleukin-6 as a novel secretory product of the hypothalamo-neurohypophyseal system. J Biol Chem. (2003) 278:19280–5. doi: 10.1074/jbc.M209902200
82. Jankord R, Zhang R, Flak JN, Solomon MB, Albertz J, Herman JP. Stress activation of IL-6 neurons in the hypothalamus. Am J Physiol Regul Integr Comp Physiol. (2010) 299:R343–351. doi: 10.1152/ajpregu.00131.2010
83. Deuster PA, Petrides JS, Singh A, Lucci EB, Chrousos GP, Gold PW. High intensity exercise promotes escape of adrenocorticotropin and cortisol from suppression by dexamethasone: sexually dimorphic responses. J Clin Endocrinol Metab. (1998) 83:3332–8. doi: 10.1210/jc.83.9.3332
84. Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol. (2007) 2:151–61. doi: 10.2215/CJN.02730806
85. Siegel AJ, Verbalis JG, Clement S, Mendelson JH, Mello NK, Adner M, et al. Hyponatremia in marathon runners due to inappropriate arginine vasopressin secretion. Am J Med. (2007) 120: e411–67. doi: 10.1016/j.amjmed.2006.10.027
86. Balla T, Enyedi P, Spät A, Antoni FA. Pressor-type vasopressin receptors in the adrenal cortex: properties of binding, effects on phosphoinositide metabolism and aldosterone secretion. Endocrinology. (1985) 117:421–3. doi: 10.1210/endo-117-1-421
87. Guillon G, Trueba M, Joubert D, Grazzini E, Chouinard L, Cote M, et al. Vasopressin stimulates steroid secretion in human adrenal glands: comparison with angiotensin-II effect. Endocrinology. (1995) 136:1285–95. doi: 10.1210/endo.136.3.7867583
88. Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. (1984) 5:25–44. doi: 10.1210/edrv-5-1-25
89. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. (2000) 21:55–89. doi: 10.1210/er.21.1.55
Keywords: vasopressin, adrenal corticosteroids, ACTH, dexamethasone non-suppression, interleukin-6, agonist-induced plasticity
Citation: Antoni FA (2019) Magnocellular Vasopressin and the Mechanism of “Glucocorticoid Escape”. Front. Endocrinol. 10:422. doi: 10.3389/fendo.2019.00422
Received: 15 March 2019; Accepted: 12 June 2019;
Published: 26 June 2019.
Edited by:
László Hunyady, Semmelweis University, HungaryReviewed by:
Hana Zemkova, Institute of Physiology (ASCR), CzechiaMaristela Oliveira Poletini, Federal University of Minas Gerais, Brazil
Copyright © 2019 Antoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ferenc A. Antoni, ZmVyZW5jLmFudG9uaUBlZC5hYy51aw==