 Urša Kovač
Urša Kovač Cene Skubic
Cene Skubic Laura Bohinc
Laura Bohinc Damjana Rozman
Damjana Rozman Tadeja Režen
Tadeja Režen- Centre for Functional Genomics and Bio-Chips, Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
This review focuses on the role of oxidized sterols in three major gastrointestinal cancers (hepatocellular carcinoma, pancreatic, and colon cancer) and how the circadian clock affects the carcinogenesis by regulating the lipid metabolism and beyond. While each field of research (cancer, oxysterols, and circadian clock) is well-studied within their specialty, little is known about the intertwining mechanisms and how these influence the disease etiology in each cancer type. Oxysterols are involved in pathology of these cancers, but final conclusions about their protective or damaging effects are elusive, since the effect depends on the type of oxysterol, concentration, and the cell type. Oxysterol concentrations, the expression of key regulators liver X receptors (LXR), farnesoid X receptor (FXR), and oxysterol-binding proteins (OSBP) family are modulated in tumors and plasma of cancer patients, exposing these proteins and selected oxysterols as new potential biomarkers and drug targets. Evidence about how cholesterol/oxysterol pathways are intertwined with circadian clock is building. Identified key contact points are different forms of retinoic acid receptor related orphan receptors (ROR) and LXRs. RORs and LXRs are both regulated by sterols/oxysterols and the circadian clock and in return also regulate the same pathways, representing a complex interplay between sterol metabolism and the clock. With this in mind, in addition to classical therapies to modulate cholesterol in gastrointestinal cancers, such as the statin therapy, the time is ripe also for therapies where time and duration of the drug application is taken as an important factor for successful therapies. The final goal is the personalized approach with chronotherapy for disease management and treatment in order to increase the positive drug effects.
Introduction
Cholesterol and Oxysterols
Cholesterol is an essential molecule that participates in many cellular processes. It enables proper functioning of cellular membrane, is a precursor for synthesis of steroid hormones, oxysterols and bile acid, and functions as a signaling molecule regulating cell cycle, modifying proteins, and affecting its own synthesis (1–3). Most of the cholesterol in cells resides in the cellular membranes, where it plays a crucial role in stabilization of membranes, affects its fluidity and has an important role in lipid rafts (3). Oxysterols also have multiple functions, such as affecting membrane fluidity, regulating SREBP (sterol regulatory element binding transcription protein) signaling pathway through regulation of INSIGs (insulin induced genes) and by this sterol synthesis, interacting with NPC1 (NPC intracellular cholesterol transporter 1) and OSBP/OSBPL [oxysterol-binding proteins (like)] and, most importantly, are ligands and activators of several nuclear receptors, such as RORs (NR1F1-3, retinoic acid receptor related orphan receptors A, B, C), FXR (NR1H4, farnesoid X receptor alpha), PXR (NR1I2, pregnane X receptor), ESR1/2 (NR3A1/2, estrogen receptor alpha/beta), and LXR (NR1H3, liver X receptor alpha) (4, 5). The term oxysterol usually means oxidized sterols that are produced from cholesterol enzymatically or by auto oxidation (Figure 1). However, also other sterols, including intermediates of cholesterol synthesis can be oxidized at least enzymatically by cytochrome P450 (CYP) enzymes (6). Auto oxidation of cholesterol usually happens in the presence of reactive oxygen species and oxidation occurs on the B ring of sterol nucleus, mainly at positions C7 or C6. With this process 7α/β-hydroxycholesterol, 7-ketocholesterol and 6-hydroxycholesterol are formed (7, 8). Enzymatic synthesis includes the side chain oxidation by CYP or non-CYP. For example, 24- hydroxycholesterol is synthesized by CYP46A1, 25-hydroxycholesterol by 25-hydroxylase (non-heme iron-containing protein) and 27-hydroxycholesterol by CYP27A1 (7, 9). Other oxysterols are formed by oxidation of the sterol nucleus, like 4β-hydroxycholesterol synthesized by CYP3A4 or 7α-hydroxycholesterol by CYP7A1 (10). The concentration of oxysterols in normal healthy tissue and blood is 104- to 106-fold lower compared to cholesterol (11). In addition to endogenous synthesis, oxysterols can also derive from the diet. The cholesterol-rich food contains 10 to 100 μM concentration of oxysterols. Most common are 7α- and 7β-hydroxycholesterol, 7-ketocholesterol, cholestane-3ß,5α,6ß-triol, 5α,6α-epoxycholesterol, and 5β,6β-epoxycholesterol (12). When oxysterols were fed to humans, they were found in chylomicrons and lipoproteins (13).
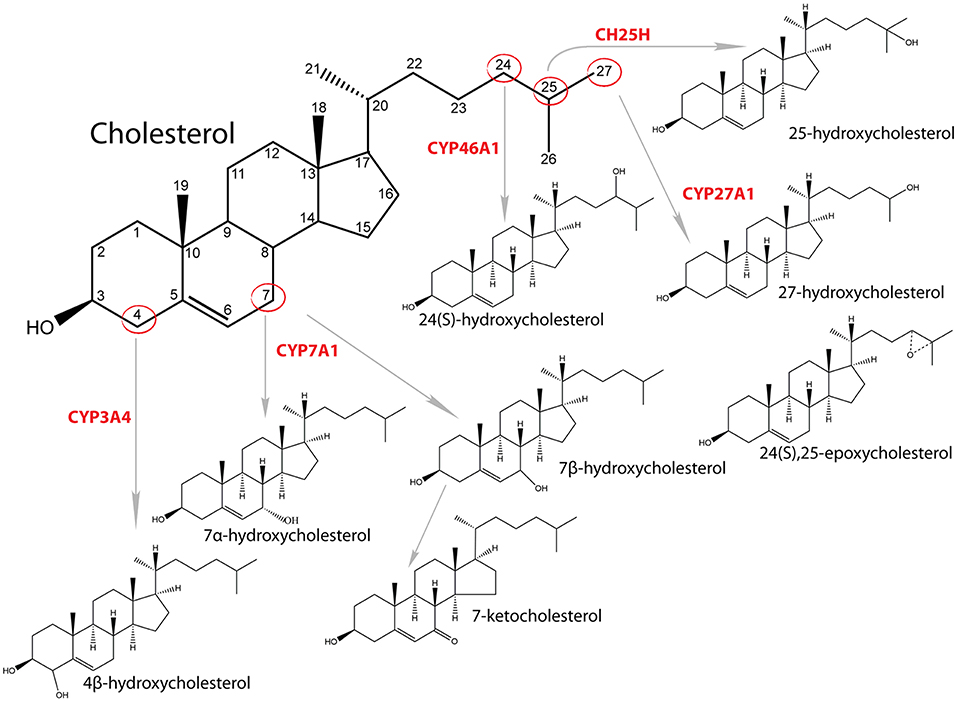
Figure 1. Synthesis of oxysterols and catalyzing enzymes.
SREBPs are transcription factors that activate transcription of genes necessary for cholesterol synthesis and uptake. Mammalian cells express three SREBP isoforms, SREBP-1a, SREBP-1c, and SREBP-2, which are responsible for expression of different lipid associated genes. SREBPs are located in endoplasmic reticulum (ER) together with SCAP (SREBP cleavage-activating protein) the escort protein, and INSIGs, the inhibitors of translocation. When cholesterol level declines, SREBPs are translocated to Golgi apparatus together with SCAP where they are proteolytic cleaved. A smaller SREBP is translocated to nucleus where it induces transcription of target genes (14). The major regulator of cholesterol homeostasis is SREBP-2 (15) as shown by the early mouse knockout experiments as well as in follow-up studies (16, 17). When SCAP is bound to INSIG, the vesicular transport of SREBP from ER is disabled. The cholesterol and oxysterols level regulate the INSIG inhibition of SCAP/SREBP transport (18, 19). Interestingly, cholesterol and oxysterols both induce the SCAP-INSIG interaction but by different mechanisms. Cholesterol acts by binding to SCAP and causing its conformational change that promotes binding to INSIG. Oxysterols, on the other hand, act directly on INSIG. For INSIG-2 it was shown that hydroxyl group on the side chain of sterols (22, 24, 25, or 27 position) is necessary for successful binding. A study confirmed these results, when 27-hydroxycholesterol levels were upregulated by CYP27A1 overexpression in mice or when primary hepatocytes were treated with 27-hydroxycholesterol, Insig-2 expression was induced and SREBP-1 translocation was prevented (20). In this way, oxysterols regulate cholesterol synthesis and uptake through regulation of SREBP signaling pathway (18).
All these pathways are regulated by cholesterol and cholesterol synthesis intermediates but also by oxysterols themselves. Oxysterol synthesis itself is regulated in a similar manner as cholesterol and bile acid synthesis since many enzymes, transporters, and transcription factors are common. The excess of oxysterols is toxic for cells; therefore, oxysterols are transported to the liver, where they are metabolized to bile acid products and excreted from the body (21). FXR is a nuclear receptor of bile acids and is also the major regulator of bile acid synthesis. Hence, FXR regulates directly or indirectly expression and activity of oxysterol producing enzymes (CYP7A1, CYP27A1, CYP3A) (22). Also 22(R)-hydroxycholesterol has been shown to regulate expression of ABCB11 (ATP binding cassette subfamily B member 11, alternatively also BSEP- bile salt export protein) through FXR in hepatocytes (23). Since bile acids can act through different signaling pathways not connected to oxysterols, we will focus only on FXR direct involvement in selected cancers.
Circadian Rhythm
Circadian clock as the inner rhythm in mammals is recognized as a cell-autonomous and self-sustaining mechanism, which controls almost every aspect of our life. The period of these rhythms is ~24 h long, thus circadian (from latin circa—approximately; diem—day) (24). The circadian rhythmicity is known to be a crucial endogenous process of organisms, described in almost every live species from cyanobacteria to human, and is capable to adapt to the environmental rhythm of the day. Circadian homeostasis in mammals is maintained by the central clock located in the suprachiasmatic nucleus (SCN) of the hypothalamus which orchestrates numerous clocks in peripheral tissues. The peripheral clocks have been observed in cells and tissues all over the mammalian body. Light, however, is not the only signal for entrainment of internal clocks. Systemic cues including hormones, body temperature, feeding/fasting cycles also influence the circadian rhythm in tissues throughout the body (25). The molecular basis of clock is constituted of periodical expression of clock genes driven by the autoregulatory transcription-translation feedback loops involving cis-regulatory elements such as E-boxes, D-boxes, and ROREs (ROR-elements) (26). The positive transcriptional loop is formed by transcriptional activators ARNTL/BMAL1 (Aryl Hydrocarbon Receptor Nuclear Translocator Like) and CLOCK (Clock Circadian Regulator, NPAS2 in neuronal tissue, Neuronal PAS Domain Protein). The CLOCK:BMAL1 heterodimer binds to conserved E-box sequences in target gene promotors of PER1,2,3 (Period Circadian Regulator 1, 2, 3), CRY1,2 (Cryptochrome 1, 2), and DEC1,2 (Deleted In Esophageal Cancer 1, 2) genes contributing to the activation of their expression (27). PER and CRY proteins dimerize in the cytoplasm and after translocation to the nucleus inhibit further CLOCK:BMAL1 transcription, forming the negative feedback loop. Also DEC1 and DEC2 can interact with the component of the SCN core clock genes. DEC1 and DEC2 proteins form dimers and translocate to the nucleus where they inhibit the activity of CLOCK:BMAL1 heterodimer (28). The degradation of PER, CRY and DEC proteins is essential for the restart of a new cycle of the transcription with ~24 h periodicity. An additional regulatory loop of BMAL1 and CLOCK dimer activity is through interaction with RORs and REVERBA (Nuclear Receptor Subfamily 1 Group D Member 1, NR1D1). RORs and REVERBA compete for ROR response element in the promotor region of BMAL1, where REVERBA acts as inhibitor and RORs as activators of BMAL1 transcription. In turn, CLOCK:BMAL1 heterodimers activate REVERBA transcription. Besides E-boxes and ROREs, D-boxes play an important role as well. Transcription regulation through D-boxes goes via different transcription factors, such as DBP (albumin gene D-site binding protein) expressed in the SCN with a clear rhythm in the light-dark or constant dark conditions. The role of TIMELESS (timeless circadian regulator gene), which is mammalian ortholog of Drosophila TIM, remains ambiguous (29). In Drosphila TIM protein plays an important role and entrains the internal clock through light. After heterodimerization with PER, translocation to the nucleus occurs and transcription of core clock cycle genes is inhibited.
Regulation of circadian rhythm, however does not stop with transcription-translation regulatory loops. Post-translational changes such as phosphorylation, ubiquitination and acetylation as well as epigenetic changes have been proven to play an important role in regulation of circadian rhythms (30–32). PER1-3 proteins are subjects to temporal changes in phosphorylation by CKIε (casein kinase Iε) and CKIδ, reaching a peak right before its destruction. Their reversible phosphorylation is a dynamic process with kinases and phosphatases as counterparts, their net effect leading to altered PER2 protein stability and cellular location, ultimately affecting the length of circadian period (33). Additionally, CLOCK has been found to have HAT (histone acetyltransferase) activity (34). While HAT activity results in transcription chromatin states, its counterpart, histone deacetylase, would condense chromatin and silence gene expression. Indeed, SIRT1 (sirtuin 1), a NAD+ (nicotinamide adenine dinucleotide) dependent protein deacetylase, is a representative of HDAC (histone deacetylase) family, whose deacetylation is under circadian control. In the liver, SIRT1 deacetylates BMAL1 and PER2 as well as histone H3 in promotors of clock controlled genes (31, 35). With its reaction depending on NAD+, SIRT1 scopes far beyond its circadian regulation. Primarily known physiological effects of SIRT1 deacetylation were regulation of metabolism and response to oxidative stress. It therefore represents a functional link between cellular metabolic activity and stress response, cell proliferation and genome stability, standing at crossroads of the processes we are tackling (31, 36–38).
Cholesterol and Oxysterols Around the Clock
Disorders of lipid metabolism are responsible for many adverse pathologies, including tumor development. It is already well-established that circadian clock controls every aspect of our life, also the lipid metabolism (Figure 2). The core circadian machinery is placed in the hypothalamus and orchestrates other peripheral clocks in tissues like liver, colon, and pancreas. Peripheral clocks can be synchronized trough different channels. They can be directly synchronized by SCN (neural and hormones signals), trough food entrainment or body temperature (39). Lipid metabolism focusing on cholesterol metabolism is under a daytime specific regulation. Recently it was shown that adropin, identified as a protein that has a role in maintaining the energy homeostasis, could represent a link between circadian clock and cholesterol metabolism. Expression of adropin is diminished by cholesterol and 7-oxygenated sterols that can modulate the RORA/C signaling. The RORA/C and REVERB receptors might thus link adropin and its synthesis to circadian rhythm of lipid metabolism which is a novel and important connection (40).
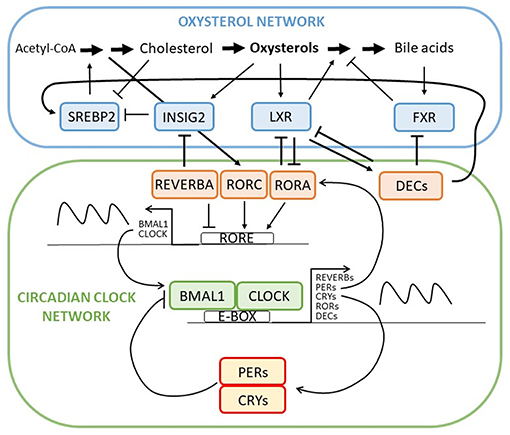
Figure 2. Intertwined oxysterol and circadian clock networks with multiple negative feedback loops and several contact points between oxysterol network and circadian clock regulation through RORA-LXR, REVERBA-INSIG, and DEC-FXR/LXR/SREBP interactions. Through contact points circadian network regulates circadian expression of oxysterol network genes and metabolites and vice versa.
Liver X receptors whose natural ligands are oxysterols have been found to regulate Dec1, a transcription factor involved in hepatic clock system and metabolism (41). In vivo observation showed reciprocal suppression between RORA and LXR (42). Research on Reverbα mutant mice showed a circadian controlled expression of Cyp7a1. Furthermore, Martelot et al. also demonstrated that REVERBA cooperates in modulation of cholesterol and bile acid synthesis through control of SREBP. Thus, REVERBA control the rhythmic abundance of Insig2 and is further responsible for diurnal translocation of SREBP to the nucleus. The circadian activation of SREBP drives the circadian transcription of cholesterol biosynthesis genes and hence the oxysterols, which activate LXR. The cyclically activated LXR is responsible for cyclic expression of CYP7A1 (43). Both, Cyp7a1 and Cyp27a1 showed circadian expression where sex differences in circadian variation were observed, indicting the importance of sex when planning the therapy (44). In the Clock mutant mice on a diet containing cholesterol and cholic acid showed reduced and disrupted Cyp7a1 expression and high liver cholesterol accumulation (45). Timed high fat diet in mice, on the other hand, resulted in changed Cyp7a1 expression via PPARα (peroxisome proliferator activated receptor alpha) as well as increased hepatic cholesterol (46). Cyp7a1 anomalies were found in Reverbα deficient mice as well as Per1−/− and Per2−/− mice, where bile acid homeostasis was disrupted (47, 48). Other CYP enzymes involved in cholesterol biosynthesis have been reported in association with circadian clock metabolism. CYP3A has a circadian pattern, but researched did not find it to be important for drug therapy due to small changes observed (49). mRNA levels and metabolic activity of CYP3A4 in serum-shocked HepG2 cells showed a 24 h rhythmicity. DBP was responsible for CYP3A4 activation (50). CYP8B1, an important player in bile acid and liver metabolism, is also under the RORA regulation resulting in its diurnal rhythm and fasting induction (51). Furthermore, DEC2 was shown as an important regulator that convey the circadian signal to the liver that has role in CYP7A, CYP8B, and CYP51A1 expression (52).
Hepatocellular Carcinoma (HCC)
Hepatocellular carcinoma (HCC) is one of the most common causes of death worldwide and represents the most common primary malignancy in the liver. It is commonly associated with chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, aflatoxins and alcoholic cirrhosis (53). It is a global health problem and over 80% of HCC cases are seen in developing countries with the highest incidence in areas endemic for HCV and HBV (54). Most patients with HCC have liver cirrhosis but there is also a number of cases where patients with non-alcoholic fatty liver disease (NAFLD) and without cirrhosis develop HCC, demonstrating the role of NAFLD in its pathogenesis (55, 56). The incidence is increasing rapidly and is higher in men compared to women (57). Other types of liver cancers like intrahepatic cholangiocarcinoma, hepatoblastoma, and angiosarcoma are not so common in comparison to HCC. The molecular pathogenesis of HCC is extremely complex where circadian clock and lipid metabolism play an important role (58). Genetic and epigenetic alterations have been shown to also drive hepatocarcinogenesis. Knowing the crucial signaling pathways in HCC will enable us to provide better therapies to treat this cancer (59). The overview of factors associated with HCC are presented in Table 1.
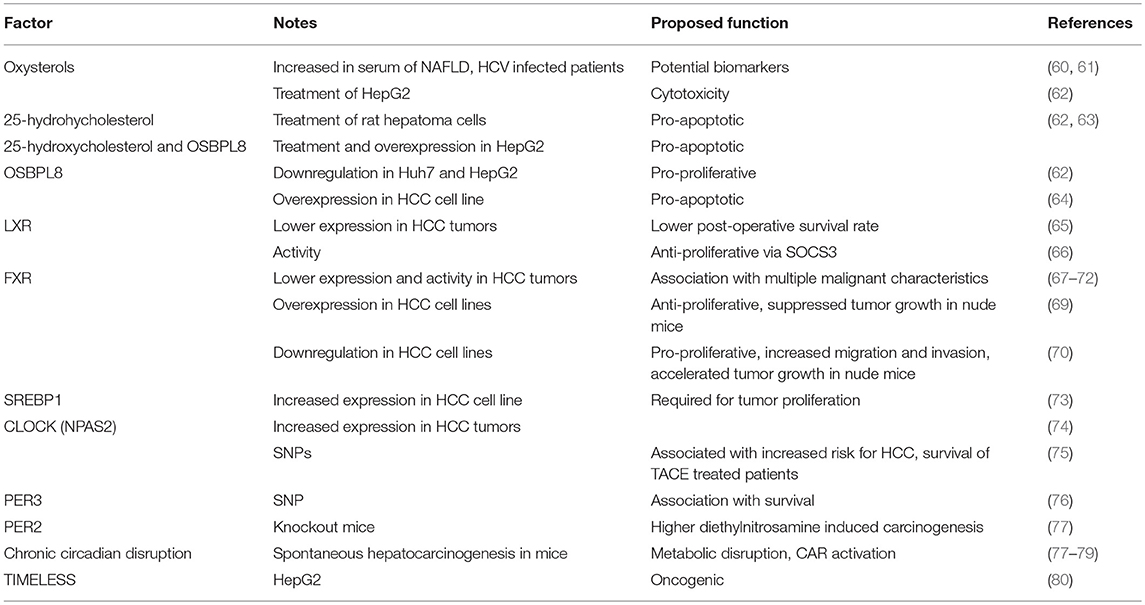
Table 1. Oxysterol-circadian factors influencing HCC development.
In patients infected with HCV, the cholesterol transport in the liver is modified and serum concentration of cholesterol was lower compared to healthy individuals. Interestingly, the serum levels of oxysterols 4β-hydroxycholesterol, 25-hydroxycholesterol, and 7α-hydroxycholesterol were elevated despite lower cholesterol concentration. When patients with HCV were treated with anti-viral therapy the oxysterols where reduced back to normal values. Further analyses indicated that these oxysterols were probably not elevated because of the activity of liver enzymes CYP3A4 and CYP7A1. CYP7A1 expression was unchanged in patients with HCV (61) and CYP3A4 activity was even downregulated (81). Direct effect of higher oxysterol concentrations on the development of HCC remains unknown, but oxysterols may be potential biomarkers and even potential novel targets for inhibiting disease progression (61). One of the major primary risk factors for HCC is NAFLD. With increasing control over hepatitis infections, it is predicted that NAFLD will soon become the major risk factor for HCC development. NAFLD is a complex multi systemic disease, caused by many factors such as genetics, dietary, environmental, and others (82, 83). In NAFLD patients, serum cholesterol is elevated and consequently some oxysterols (4β-, 25-, and 27-hydroxycholesterol) are also elevated. These oxysterols can affect the resorption of cholesterol in some tissues, through LXR activation, which regulates ATP-binding cassette transporters. In this way oxysterols affect the cholesterol concentration in NAFLD, which can evolve in HCC (60). In rat model with induced hepatoma tumor in the liver, the measurements showed elevated levels of following oxysterols: 24S-, 25- and 27-hydroxycholesterol, and 24S,25-epoxycholesterol, which are all known to bind to LXR. In these tumors, the ABCA1 (ATP binding cassette subfamily A member 1) and ABCG1 (ATP binding cassette subfamily G member 1) cholesterol transporters were elevated on mRNA and protein levels, most likely through LXR activation. There is some data indicating that 25-hydroxycholesterol has a pro-apoptotic function in rat hepatoma cells and is able to induce sub-Gl apoptosis (63). Authors suggested that local addition of 25-hydroxycholesterol in combination with other drugs could be potentially used in treatment of hepatoma patients. HCC cells have changed metabolism to sustain the fast proliferation. Because of this, many cellular functions are altered, including cholesterol metabolism and transport. It was shown that oxysterols are cytotoxic, can induce cell death in HepG2 cells, and can suppress the growth of HCC cells by inhibiting ACAT2 (Acyl-coenzyme A:cholesterol acyltransferase) enzyme, which results in intracellular unesterified oxysterol accumulation (62). When OSBPL8 (also known as ORP8) is downregulated, cell proliferation is promoted. When ORP8 was overexpressed in HepG2 cells and 25-hydroxycholesterol was added, the ER stress and apoptosis were promoted (62). ORP8 overexpression was already enough to trigger apoptosis in primary HCC cells isolated from human liver and cell lines (64). Lower LXR expression was associated with lower post-operative survival rate (65). LXR regulated HCC response to TGFβ1 (transforming growth factor beta 1), which acts as cytostatic and pro-apoptotic (84). LXR suppressed HCC proliferation through activation of SOCS3 (suppressor of cytokine signaling 3) (66).
FXR's role in HCC development was indicated when whole body Fxr knockout mice developed spontaneous HCC by the age of 14 months (85, 86). These mice had abnormal level of bile acids in the serum and administration of cholestyramine decreased HCC incidence (85). Many studies confirmed a significant decrease in FXR expression and activity in human HCC tumors and this was associated with multiple malignant characteristics (67–72). However, intestine-specific Fxr reactivation in Fxr knockout mice restored bile acid enterohepatic circulation, limiting hepatic inflammation and proliferation while preventing spontaneous hepatocarcinogenesis (87). Also only 20 and 5% incidence of hepatic tumors was observed in hepatocyte specific and enterocyte specific Fxr knockout mice, respectively. No serum or hepatic increase in bile acid level was observed in either of cell specific knockout models and no change in expression of cell-cycle regulators (88). These results lead to a hypothesis that high bile acid levels and FXR deregulation are needed for HCC development. There is, however, a mounting data pointing to a direct involvement of FXR in tumor suppression.
Overexpression of FXR decreased proliferation of HCC cell line and suppressed tumor growth in nude mice (69). Inhibition of FXR expression resulted in enhanced cell proliferation, migration and invasion in HCC cell lines and accelerated tumor growth in nude mice. FXR was shown to directly interact with β-catenin and repress its transcriptional activity (70). FXR also directly upregulates NDRG2 (N-myc downregulated gene family member 2) (72). NDRG2 is a known tumor suppressor, whose expression is reduced in HCC samples and correlates with aggressive tumor characteristics (72, 89). FXR also directly inhibited gankyrin expression via C/EBPβ-HDAC1 (CCAAT enhancer binding protein beta/ histone deacetylase 1) complex (90). Gankyrin is a known oncogene and is increased in HCC (91). In this study, they also showed that long-lived Little mice, which have high FXR liver expression, do not develop liver cancer after diethylnitrosoamine injection in comparison to wild type mice (90). FXR upregulated mir-122 which suppressed proliferation of HCC cells and the growth of HCC xenografts in vivo (71). In HCC human tumors miR-122 was significantly downregulated and this correlated with FXR expression (71). FXR and its agonist waltonitone repressed HCC cell proliferation by activating mir-22 repression of CCNA2 (Cyclin A2) (92). In HCC tumors FXR and miR-22 were downregulated while CCNA2 expression was opposite (92, 93). FXR also repressed inflammation via SOCS3 and via inhibiting NFκB (nuclear factor Kappa B) signaling (94, 95). FXR is a direct regulator of SOCS3, which enabled FXR-mediated cell growth repression, and their expression is correlated in HCC human samples (96). In summary, a direct role of FXR in protection against hepatocarcinogenesis could also be via defense against bile acid-induced injury, prevention of liver injury and apoptosis, prevention of ROS generation, promoting liver repair and generation after injury, suppressing cancer cell proliferation, etc. (97). There are some potential explanations available for the mechanisms behind FXR downregulation in HCC. In HCC cell lines miR-421 downregulated FXR and by this promoted cell proliferation, migration and invasion (98). Increased expression of miR-421 and its correlation with patient's survival was confirmed in HCC patients (99). Another factor decreasing the expression and activity of FXR is inflammation and inflammatory cytokines were shown to inhibit HNF1A (HNF1 homeobox A) binding to FXR promotor (68). SIRT1 overexpression leads to deacetylation of FXR in the mouse liver, and inverse expression of SIRT1 and FXR was confirmed in human HCC samples (100). In spite of many tumor suppressor actions discovered, there is also some caution needed. FXR also upregulates FGF19 (fibroblast growth factor 19) which is anti-cholestatic and anti-fibrotic factor in the liver and was proposed for the treatment of NASH. However, FGF19, but not its rodent counterpart FGF15, is potentially pro-tumorigenic as shown by ectopic expression in mice (101). FGF19 expression is upregulated in HCC samples and correlates with poor prognosis (102). This is an important factor to consider when translating results from studies in rodent models to humans. However, new engineered FGF19 variants have been developed excluding pro-tumorigenic activity and are currently in evaluation for the treatment of NASH (103).
More and more results show the importance of SREBPs, especially SREBP-1, as a link between oncogenic signaling and tumor metabolism. It was shown that cancerous cells have higher de novo lipogenesis, which is required for rapid tumor proliferation. In the cancer cells, SREBP-1 is highly activated and it was shown, that pharmacological targeting of SREBP-1 greatly inhibits tumor growth. This rises hope for SREBP-1 to be potential anti-cancerous target (73). Higher expression of SREBP-1 was shown in different cancer cell lines (104) and higher expression of SREBP-1 is associated with many metabolic diseases; including NAFLD and NASH. One of the factors activating SREBP-1 in HCC is hepatoma-derived growth factor (HDGF). Co-expression of HDGF and SREBP-1 is an indicator of poor HCC prognosis. HDGR knockdown or its mutation in HepG2 cells resulted in decreased expression of SREBP-1 targeted genes (105). A study on a mouse model of HCC showed that lipid biosynthesis (fatty acids and cholesterol) is crucial for HCC development and that targeting SREBP pathway can be used as an anti-tumor strategy (106). Study on hepatic stellate cells, which have an important role in liver fibrosis, showed that in these cell line INSIG-1 and INSIG-2 expression is downregulated. By downregulation of INSIGs, SREPB signaling is promoted, which is the characteristic of HCC and crucial for cancer proliferation (107). Bioinformatics analysis showed that two miRNA (has-miR-221 and has-miR-29c) are potential HCC diagnostic markers and that they have one common target, which is INSIG1 (108). All these data show the importance of SREBP signaling and its potential as drug target.
Data on human and mice reported a connection between circadian rhythms and liver disease where alterations in rhythmicity had a profound effect on metabolic pathways resulting also in adverse outcomes, like HCC. NPAS2 or CLOCK was shown to have a critical role in HCC and expression of the protein was significantly upregulated in HCC patients (74). In other study, single nucleotide polymorphisms (SNPs) in NPAS2 were associated with the increased risk for HCC or with overall survival in HCC patients treated with transcatheter arterial chemoembolization (TACE) (75). The rs2640908 polymorphism within PER3 was also associated with overall survival in HCC patients treated with TACE (76). SNPs from circadian negative feedback loop genes were further studied and were suggested to have an independent role as prognostic biomarkers for prediction of HCC clinical outcome (109). Study on mice showed that circadian disruption due to Per2 mutation or jet lag has a profound role in liver carcinogenesis induced by diethhylnitrosamine (77). Other research addressed the importance of the circadian clock metabolism in hepatocarcinogenesis where the changes in expression of several core clock genes were downregulated (BMAL1, CLOCK, PER1, PER2, CRY1, CRY2), while upregulation of clock related genes (NR1D1 and DBP) was observed (110). Circadian metabolism and carcinogenesis are highly interconnected and the connection is complicated. Kettner et al. demonstrated that chronic circadian disruption induces NAFLD and spontaneous hepatocarcinogenesis, promotes genome-wide deregulation and metabolic disruption, and activates CAR (NR1I3, constitutive androstane receptor) which drives NAFLD to NASH, fibrosis and HCC progression (78). Chronic circadian disruption in mice had a profound effect on metabolism, what was also observed in humans and circadian disruption with or without steroid receptor Coactivator-2 (SRC-2) ablation showed association with human HCC gene signature (79). The TIMELESS protein has oncogenic function in human HCC as well (80). Studies on HepG2 cell line demonstrated the importance of RORA in different steps in liver carcinogenesis (111).
Pancreatic Cancer (PC)
Pancreatic cancer is one of the most malignant cancers, estimated to cause 432 242 deaths in 2018 and ranking seventh among estimated cancer deaths in the world (112). Estimated 5-year survival rate is extremely low and only 4% of patients will live 5 years after diagnosis. An important risk factor is smoking, since around 20% of tumors are attributable to smoking. Other risk factors include family history of chronic pancreatitis or pancreatic cancer, male sex, advancing age, diabetes mellitus, obesity and occupational exposure (113, 114). Novel epidemiological studies propose that dietary factors also impact pancreatic cancer risk. In a large population saturated fatty acid intake was linked to higher pancreatic cancer risk, while omega-3 fatty acids, increased vitamin C and vitamin E intake lowered cancer risk (115). Pancreatic cancer patients have unspecific signs, making the diagnosis difficult, often discovering already advanced stages of the disease. The overview of factors associated with PC are presented in Table 2.
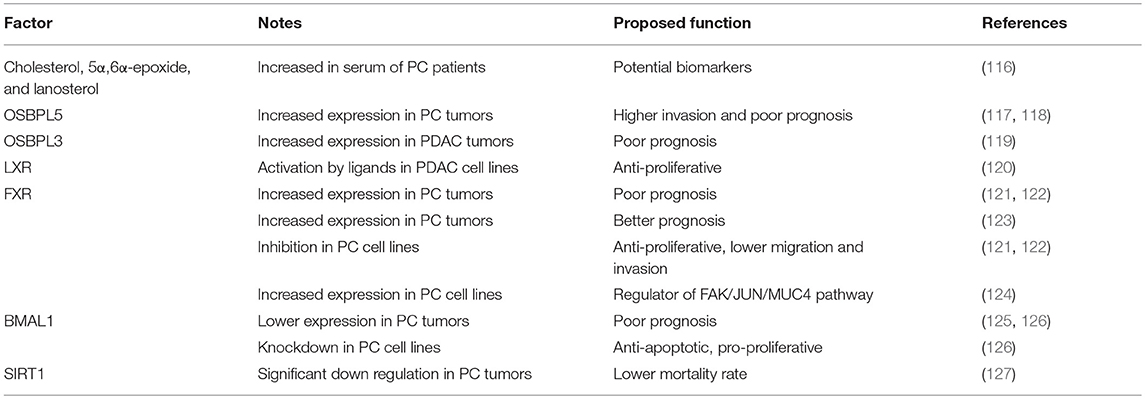
Table 2. Oxysterol-circadian factors influencing PC development.
Very little is known about the oxysterol role in the development of pancreatic cancer. Serum cholesterol, 5α,6α-epoxide, and lanosterol were identified as highly discriminating between pancreatic cancer patients and healthy subjects (116). Two meta-analyses showed that dietary cholesterol may be associated with pancreatic cancer in worldwide populations (128, 129). OSBPL have been shown to be involved in pathology of PC. Increased expression of OSBPL5 was associated with higher invasion and poor prognosis of pancreatic cancer patients (117, 118). In pancreatic ductal adenocarcinoma (PDAC) patients, OSBPL3 was found as differentially expressed in four studies and was correlated with poor prognosis of these patients (119). The role of nuclear receptors also remains unknown. Activation of LXR by synthetic ligands had anti-proliferative effects on PDAC cells (120). Very few studies measured the expression FXR and its role in pancreatic cancer. Moreover, there are conflicting results available. The increase in FXR mRNA and protein expression was confirmed in pancreatic tumors in comparison to adjacent tissue, and high FXR expression was correlated with the poor prognosis. FXR inhibition reduced cell proliferation, migration and invasion in pancreatic cancer cell lines (121, 122). It was proposed that FXR induced phosphorylation of SP1 (Sp1 transcription factor) and by this promoted cancer progression (122). While another study found correlation between high FXR expression with less aggressive phenotype, smaller tumors, absence of metastases and better prognosis (123). The difference in FXR expression and correlation between studies can simply be attributed to a low sample number, different groups of patients, different length of time in survival analyses, correlation with other factors etc. However, bile acids were increased in plasma and pancreatic juice in PC patients, and the mouse model confirmed the correlation between the increase in serum bile acids level with the severity of the disease (124). FXR was significantly increased in pancreatic cancer cell lines in comparison to normal pancreatic cells and FXR was found as the regulator of FAK (PTK2, protein tyrosine kinase 2)/c-Jun (JUN, Jun proto-oncogene, AP-1 transcription factor subunit) / MUC4 (mucin 4) signaling pathway (124). Similar to HCC, SREBP1 pathway and de novo lipogenesis is upregulated in PC. Study on 60 PC patients showed high correlation between elevated SREBP1 expression and poor disease prognosis. Inhibition of SREBP1 in a mouse model lead to decreased in vivo weight of tumor, indication the importance of SREBP1 upregulation for cancer cell growth (130). Study on PC cell model showed that inhibitors of SREBP1 decrease PC cell viability and proliferation. This indicate that targeting SREBP1 pathway is potential target for PC disease management and should be further explored (131). One of the potential molecules for targeting SREBP1 is resveratrol. Recent study on mouse model of PC and human cell lines treated with gemcitabine (chemotherapy medication) showed that resveratrol suppressed stemness induced by gemcitabine. Resveratrol should be considered as an additive for chemotherapeutic drugs (132).
Circadian activity in pancreas is also controlled by SCN (133, 134). The clock genes expression (BMAL1, PER1, PER2, PER3, CRY2, TIMELESS, and CK1ε) is altered in PDAC which is the most common type of pancreatic malignant tumors (135, 136). In humans, lower BMAL1 expression was associated with poorer survival rate, and correlated with higher tumor stage, poorer histological differentiation, and increased vascular invasion in PDAC (125, 126). Since knockdown of Bmal1 resulted in an anti-apoptotic and pro-proliferative profile, while its overexpression had the opposite effect, Bmal1 can act as an anti-oncogene by binding directly to p53 promoter region (126). Since circadian rhythms and metabolic pathways go both ways, and pancreatic cancer risk is increased in people with metabolic syndrome (137), the role of metabolism and its disruption plays an important role in pancreatic cancerogenesis. A recent study evaluating dietary influence on cancer risk in women confirmed the role of obesity (138). Bmal1 and its downstream regulation seems to play a crucial role in pancreatic regulation of metabolic processes, as Bmal1 knock out in mouse β-cells led to glucose intolerance and development of diabetes (139). Since glucose metabolism is also under the circadian control, it is not surprising that alterations in clock genes (Clock, Bmal1) lead to impaired glucose tolerance (140), with a profound role in pathogenesis of pancreatic cancer (141). Deregulation of SIRT1, which regulates the central and peripheral clock, has an important role in PDAC as well (127).
Colorectal Cancer (CRC)
Colorectal cancer is the third most commonly diagnosed cancer, the second most common cause of oncological death in the world estimated in 2018 (112) and as such represents a major public health issue worldwide. About 6% of CRCs are predetermined by a defined hereditary syndrome, while around 25% of CRCs are familial, the latter being tightly intertwined with many of the modifiable risk factors as well as genetic factors. Non-modifiable risk factors for CRCs include increasing age, male gender, African American race and residence in high-income countries (142), while modifiable factors include obesity, moderate to heavy alcohol consumption, smoking, high consumption of red or processed meat and a diet with low fiber, fruit, and vegetables intake (112, 142). Also, genetics plays an important role in its pathogenesis (142). In CRC one of the most commonly dysregulated pathway is the Wnt/β-catenin signaling, leading to β-catenin accumulation, ultimately resulting in activation of several gene targets, including oncogenes that contribute to the development of cancerous phenotype (143). Its component APC (APC regulator of WNT signaling pathway) mutations are present in majority of sporadic CRCs (144). Since onset and progression of CRC are tightly intertwined with its mutational pathways, detailed knowledge of their regulation is crucial for obtaining new markers in order to better understand risk factors as well as treatment and prognosis. The overview of factors associated with CRC are presented in Table 3.
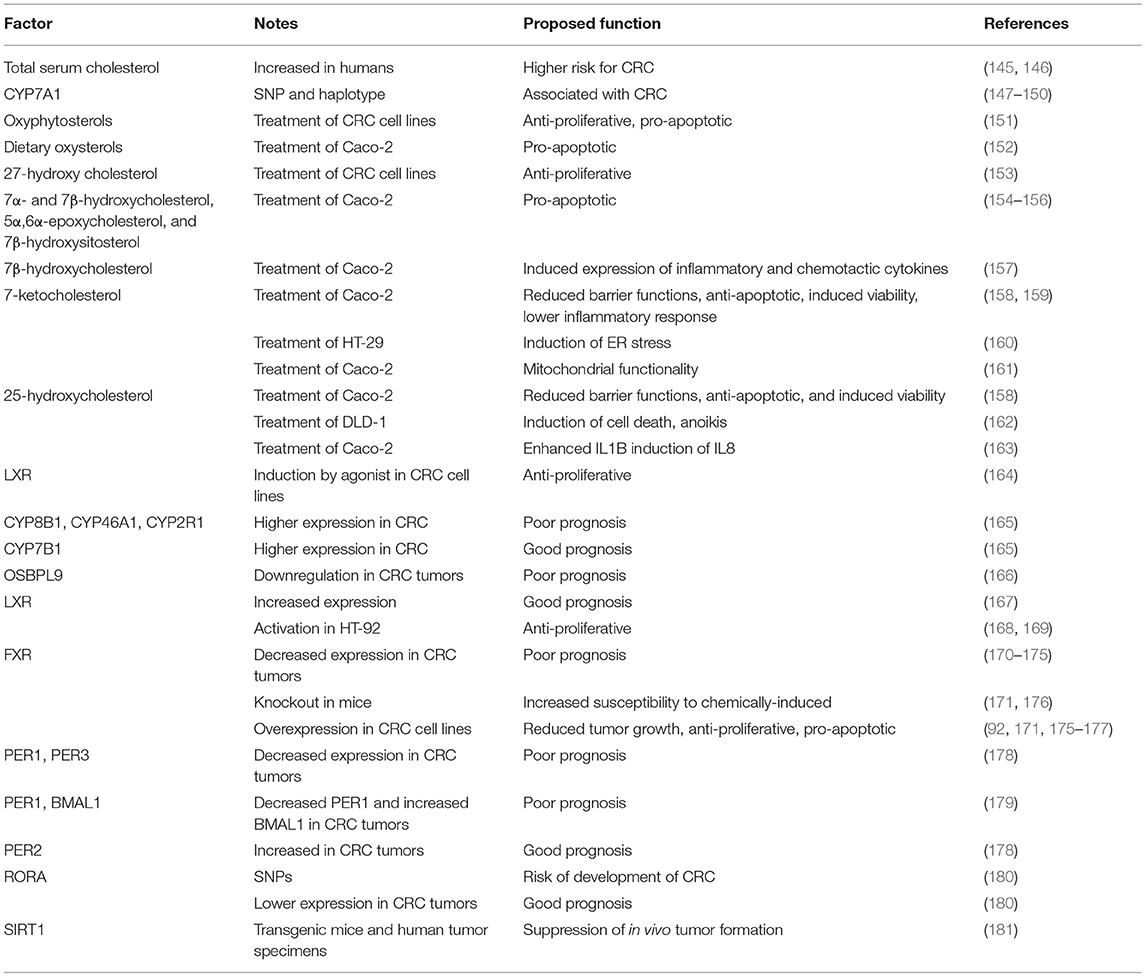
Table 3. Oxysterol-circadian factors influencing CRC development.
High total cholesterol level is associated with a higher risk for colon cancer in men (145). Another study taking into an account also genetic factors again confirmed a link between hypercholesterolemia and colorectal cancer risk (146). A CYP7A1 haplotype and SNPs were significantly associated with colorectal cancer and it was proposed to be the effect of fecal bile acids (147–150). Whole grain food significantly reduces colon cancer risk and phytochemicals that are involved in this protection are especially interesting (182).
Studies in colon cancer cell lines indicate that different oxysterols are cytotoxic and induce apoptosis. A study of wheat bran showed that it contains several different oxyphytosterols with anti-proliferative effect on colon cancer cell lines, some could also induce apoptosis (151). Dietary-representative oxysterol mixture induced apoptosis in differentiated colonic CaCo-2 cells (152). A study in major cell line models for CRC, Caco2 and SW620 cells, showed that 27-hydroxycholesterol decreased proliferation of these cells (153). This effect was not due to cellular cytotoxicity or induction of apoptosis and it was independent of nuclear receptors LXR and ESRs. 7α- and 7β-hydroxycholesterol, 5α,6α-epoxycholesterol and 7β-hydroxysitosterol were all able to induce apoptosis in human colon cancer cell line Caco-2 (154, 155). 7β- hydroxycholesterol was cytotoxic to colon cancer cell lines in concentrations from 3 to 10 μM (156). 7-ketocholesterol and 25-hydroxycholesterol reduced barrier functions, apoptosis and induced viability of Caco-2 cells (158). 7-ketocholesterol besides decreasing epithelial barrier also induced inappropriate development of inflammatory response to food (159). 7-ketocholesterol induced ER stress in HT-29 colon cancer cell line (160) and affected mitochondrial functionality in Caco-2 cells, while co-treatment with 7-ketostigmasterol reduced the toxic effect (161). Only 25-hydroxycholesterol, but not the 22-(R)-hydroxycholesterol and other oxysterols (7 beta-hydroxycholesterol and 5-cholesten-3beta-ol-7-one) induced anoikis, a type of programmed cell death, in DLD-1 cells (162). Some of these effects could be through activation of LXRs, since LXR induction by GW3965 had anti-proliferative effects on colon cancer cells (164).
On the other hand, oxysterols also affect expression of inflammatory molecules in colon cancer cells. Inflammatory bowel disease is an important risk factor for development of CRC and chronic inflammation and oxidative stress are part of pathogenesis (183). 7β-hydroxycholesterol induced expression of key inflammatory and chemotactic cytokines in CaCo-2 cell line (157). 25-hydroxycholesterol pre-treatment enhanced IL1B (interleukin 1 beta) induced IL8 (interleukin 8) production in Caco-2 cells (184). Representative mixture of oxysterols increased oxidative stress in differentiated Caco-2 cells and was followed by the production of cytokines IL6 (interleukin 6) and IL8 (163). In CRC patients, serum level of IL8 was increasing with the progression of cancer (185). Expression of enzymes and oxysterol receptors is modulated in CRC. Human tissue microarray analyses revealed significantly higher protein expression of CYP2R1, CYP7B1, CYP8B1, CYP46A1, CYP51A1, while CYP27A1 and CYP39A1 had no significant change, in primary colorectal tumor compared to normal colonic mucosa (165). In this study, they also showed a significant association between patient prognosis and survival with CYP expression. For example, higher expression of CYP8B1, CYP2R1, CYP27A1, and CYP46A1 in tumor was associated with poor prognosis, while high CYP7B1 expression correlated with good prognosis. OSBPL1A short transcript variant was down-regulated in colon cancer tumors, but the long variant was unchanged in comparison to normal samples (186). OSBPL9 was included in a gene expression signature as a predictor of survival in colon cancer and was downregulated in poor prognosis patients (166).
LXR expression was proposed to be a prognostic indicator for CRC and its expression was associated with favorable clinical outcome. Positive LXR expression was associated with better survival rate and there was a significant negative association between LXR expression and vascular invasion, but no association was found between LXR expression and the patient's age, sex, tumor size, grade or TNM (tumor/node/metastasis) stage (167). A genome wide analyses revealed regulatory programs of LXR activation which lead to inhibition of HT29, a colorectal cancer cell line, proliferation (168). LXR was shown to directly bind to β-catenin and suppress its activity and cellular proliferation (169). A recent review has already summarized FXR and bile acids role in the development of colon cancer (187). Several studies showed that FXR expression is reduced in intestinal tumors in humans and that it is inversely correlated with the degree of malignancy and clinical outcome (170–175). The FXR knockout mice have increased susceptibility to chemically-induced colorectal carcinogenesis, while FXR overexpression in gut cells reduced tumor development and growth (171, 176). The activation of FXR was shown to suppress proliferation and induce apoptosis in colon cancer cell lines (171, 177). The observed downregulation of FXR expression is due to increased methylation of FXR promotor by the loss of APC function, which was confirmed in CRC cell lines, animal models and colonic tumors from patients (174, 188, 189). However, FXR is also downregulated by intestinal inflammation, western diet, and microRNA (187, 190). The current hypothesis is that the decreased FXR regulation in combination with Western diet and hence higher levels of secondary bile acids results in pro-tumorigenic colon environment leading to the development of colon cancer. Moreover, recent studies showed that FXR exhibits anti-cancerogenesis effects beyond regulation of bile acid level, by also affecting other cellular signaling pathways in colon cells. A study in mouse and organoid models, showed that FXR regulated proliferation of intestinal cancer stem cells (191). FXR repressed proliferation of colon cells by inhibiting the MMP7 (matrix metallopeptidase 7), a known intestinal tumor promotor expression; by activating mir-22 repression of CCNA2 (Cyclin A2); and by activating the EGFR/SRC (epidermal growth factor receptor/SRC proto-oncogene) pathway in colon cells (92, 175, 177). Upregulation of SREBP1 pathway is also present in CRC (192). Inhibition of SREBP1 in CRC shows promising result for cancer management. It was shown that ordonin (diterpenoid isolated from Rabdosia rubescens) reduced expression of SREBP1 and induce apoptosis in CRC cells cultures (193).
Epidemiological studies have shed light on connection between circadian disruption and elevated risk of cancer, including colorectal cancer (128, 194, 195). Meta-analysis showed increased incidence of colorectal cancer in people with long-term exposure to night light (shift work). The proposed mechanism for circadian disruption on oncogenesis is melatonin suppression and loss of its protective effect against cancer via apoptosis, anti-angiogenesis, anti-oxidation and regulation of the immune system. One of the proposed additional factors is low level of 25-hydroxyvitamin D due to lower sun exposure (128). In mouse models, physical destruction of the SCN as well as functional disturbance of circadian rhythms (chronic jet lag) resulted in accelerated tumor growth in transplantable tumor models (Glasgow osteosarcoma and Pancreatic adenocarcinoma), indicating a role of circadian system in controlling malignant growth (196). In human CRC, there are multiple studies reporting abnormal expression of circadian genes including altered expressions of CLOCK, BMAL1, PER1, PER2, PER3, and CK1ε (179, 197–200). Whether the disruption originates in core clock genes and drives tumorous transformation or measured disruption is a consequence of cancer, remains unclear. However, studies suggest that clock genes have an important role in tumor suppression (201). Although clinical correlations between specific mechanisms of clock gene disruption and colorectal cancer phenotype and prognosis have not been conclusive, some pathways show typical clinical and pathological features. Decreased expression of PER1 and PER3 in tumor tissue as such indicate poorer survival rate (178), decreased PER1 and high BMAL1 expression correlate with poorer outcome and liver metastasis (179), and high PER2 expression correlate with significantly better disease outcome (178). In recent systematic evaluation of genetic variants in the circadian pathways connected with CRC, examining 119 SNPs in RORA was proposed as potentially important marker for CRC risk and prognosis. While people carrying SNP in RORA were much more inclined to developing CRC, lower RORA correlated with better differentiated tumors and better disease outcome (180). On the other hand, changes in cellular metabolism may cause circadian disruption, further influencing colorectal cancer phenotype (202). It has been shown that SIRT1 suppresses colorectal tumor formation in vivo by β-catenin deacetylation (181). A recent study showed that both metabolic and circadian dysregulation progressed during cancer progression. Their findings suggested that clock-related glycolysis genes alterations might add to a clock-driven rewiring of metabolism, connected to cancer progression and altering response to cancer therapy (203).
New Therapeutic Strategies
Targeting Oxysterol-Cholesterol Network
Statins are widely used lipid-lowering drugs which inhibit HMGCR, a rate limiting enzyme from cholesterol synthesis. Statins have been proposed in several studies as potential drugs used for reducing the risk of development and mortality in gastrointestinal cancers. Statin use was associated with a decreased risk of mortality in several cancers also colorectal cancer (204). However, another study showed no association with colorectal cancer incidence in United States cohort (205). Longer statin use was connected to a reduction in all-cause mortality in patients after colorectal diagnosis (206). However, overall conclusion of meta-analyses of 42 studies was that statin use was associated with a modest reduction in risk of CRC (207). This association was confirmed for lipophilic statins but not for hydrophilic statins. In addition, long-term statin use (>5 years) did not significantly affect the CRC risk.
HCC occurs mainly in cirrhotic liver and statins may affect the risk of HCC by their anti-fibrotic effect. Three meta-analyses confirmed that statin use is associated with lower risk of HCC in different populations (58, 208, 209). Fluvastatin was pointed out in one of these studies. A registry-based study also confirmed an association between statin use and the risk of HCC and additionally showed a dose response relationship (210). Stain use also significantly decreased the risk of HCC in patients with HBV (211, 212). Studies suggest that statins lower the risk of cancer in general in chronic hepatitis patients (213). Overview of studies on statin effect on mean survival rate revealed a link to extended survival, but the length of survival was variable among studies (214). Perioperative statins have been associated with improved recurrence-free survival in HCC patients (215, 216). Meta-analysis confirmed an association between statin use and decreased risk in mortality in pancreatic patients, but other studies found no association (217, 218). A review of studies revealed that observational studies found the association but randomized controlled trials did not. Statins have anti-neoplastic properties through anti-proliferative, pro-apoptotic, anti-angiogenic and immunomodulatory effects and can affect multiple signaling pathways in cells (219). Most important is the inhibition of HMGCR and by this synthesis of mevalonic acid. HMGCR genetic variant significantly modified the protective association between statins and CRC risk (220). Mevalonate pathway is upregulated in several cancers, also pancreatic and hepatic, and is responsible for activation of small G proteins (221). All these data make statins compelling therapy for decreasing the risk of cancer and lowering mortality of patients, but due to conflicting results, it is difficult to form final recommendations. An important issue in prescribing statins are the side effects of statins such as hepatotoxicity and drug-induced myopathy.
Since SREBPs target genes are upregulated in many cancers, SREBPs could have therapeutic potential (106). Several studies showed a successful targeting of SREBP pathway and suppression of lipid metabolism with substances like TAK1, Emodin and using different miRNA (222–225). Since oxysterols bind to INSIG and affect SREBP signaling pathway, oxysterols also have a potential to be used in cancer proliferation inhibition, but more studies on oxysterol-INSIG interaction are needed.
Oxysterols activate several nuclear receptors most importantly LXR, which is hypothesized to exert their anti-cancerogenic effects. Targeting LXR for prevention and therapy of cancers is already evaluated in clinical studies. LXR is an interesting target, because it is activated by phytosterols, which can reduce the incidence of colon cancer (226). LXR is connected to TGFB1 actions and was also proposed as a potential target for treatment of HCC (84). Bergapten, a LXR agonist, was already evaluated for HCC treatment (227). LXR agonists were proposed also for treatment of CRC since LXR activation reduced intestinal tumor formation in a mouse (APCmin/+) CRC model and also blocked proliferation of human colorectal cells (228). LXR is a promising target, but majority of evidence was gathered through studying the effects of LXR activation in cell lines (4).
FXR is also a promising target for cancer treatment. Activation of FXR would prevent toxic bile acids build up, but also repress other tumorigenic proteins. In preclinical and clinical trials FXR agonists show a potential for treatment of different liver disease among them also HCC (175, 229, 230). In the mouse (APCmin/+) CRC model on high fat diet, treatment with FexD, a deuterated analog of fexaramine with the gut-restricted activity, resulted in slower tumor progression, improved bile acid homeostasis and improved survival (191). However, FXR's role in pancreatic cancer is conflicting and the fact that in humans it activates a potentially pro-tumorigenic FGF19 emphasizes the needed for more studies that will confirm the positive role of FXR agonists.
Chronotherapy as a New Therapeutic Strategy
Cancer represents the second leading cause of dead worldwide (231). The available treatment is not always the most efficient thus new therapeutic strategies are needed to be developed. In addition to standard therapy (chemotherapy, radiation, and surgery), additional factors need to be taken into consideration, such as lifestyle and biology, when providing integrative treatment for different cancer types (232). Since the circadian clock metabolism has a profound role in pathogenesis of cancers, chronotherapy might be a better therapeutic strategy. We need to adjust the pattern of drug delivery to improve the treatment efficiency, by reducing the drug at the time point where tissue is most susceptible to toxicity, and increasing the dose at times with most susceptibility to the positive drug effect (233).
Therapeutic strategy for HCC is well-established. Since the incidence is rising drastically, early diagnosis and definitive treatment is currently the only way to increase the survival rate of HCC patients. Great research breakthroughs in chronobiology led to the development of this field. Recently it was demonstrated that isoform of the HNF4A (nuclear factor 4 alpha) plays a crucial role in HCC progression. Forced expression of BMAL1 in HCC that is positive for HNF4A stops the growth of tumors in vivo (234). Furthermore, SULT1A1 (sulfotransferase 1a1) that has a circadian pattern at the mRNA and protein level and is responsible for detoxifications of various drugs in the liver is regulated trough BMAL1. Knockdown of Bmal1 resulted in changed rhythmicity in Hepa-1c1c7 cells (235). The role of IFNA (interferon alpha) was assessed in circadian manner in HepG2 cells as well. It was proposed that IFNA could have pharmacological role, since its continuous administration resulted in significantly decreased levels of CLOCK and BMAL1 protein (236).
Pancreatic cancer is one of the most aggressive tumors, responding poorly to therapy; therefore, new therapeutic strategies are in high demand. Circadian gene Per2 overexpression increases the sensitivity to cisplatin, possibly by inducing a reduction in of the BCL (B-cell lymphoma) proteins (237). In search of cancer cell growth inhibitors downstream circadian controlled pathways, ligands of PPARG (peroxisome proliferator activated receptor gamma) showed to be reducing tumor aggressiveness and enhancing cytotoxic action of anti-cancer agents (238, 239). Another mechanism showing promise in pancreatic cancer treatment might be TIMELESS, involved in DNA damage response, whose expression was found to be altered in pancreatic cancer (135).
Chronotherapy has also proven to be effective in CRC. In metastatic CRC patients chemotherapy induced circadian disruption correlated with poorer disease outcome, suggesting its prevention could improve treatment results (240). Furthermore, a meta-analysis of five randomized controlled trials showed a significant improvement in overall survival in metastatic CRC patients treated with chronobiologically timed chemotherapy compared to conventional chemotherapy (241). Chronomodulated hepatic arterial infusion also showed promise as a possible drug administration strategy in heavily pretreated patients with CRC liver metastases (242). Recent study examining the role of PER3 in CRC found that its overexpression enhanced fluorouracil sensitivity in CRC cells, proposing it as a potential target in CRC treatment (243). Moreover, another study showed sex-specific expression and sex-specific prognostic value of clock and clock-controlled genes, shedding light on colorectal cancer and patient characteristics that have to be taken into consideration in order to provide optimal treatment (244).
Circadian-Oxysterol Network in Cancerogenesis
Members of the oxysterol and circadian clock networks were exposed as the new promising prognostic biomarkers, genetic risk factors and potential therapeutics in gastrointestinal cancers. The roles of oxysterols in various cancers have been reviewed previously and mechanisms by which oxysterol can affect cancer pathogenesis and disease development were pointed out (8). Oxysterols activate different signaling pathways in cells which can either promote or inhibit cancer development. For example, 22(R)-, 24-, 25-, and 27-hydroxycholesterol are in vivo ligands of LXR and act as tumor suppressor in selected cancers. Increased LXR activity was shown to be beneficial in all three presented cancers exposing LXR as a promising drug target in oncology. However, one oxysterol can have a different role depending on the tissue. Current hypothesis is that the dual role of oxysterols is due to activation of different signaling pathways resulting in tumor suppressor LXR-dependent or oncogenic LXR-independent actions (245). Oxysterols also interact with OSBP/L proteins and these have been connected to many human diseases, such as dyslipidemia and cancers (246). Studies indicate that OSBPL family members could also have a dual role. In HCC and CRC their expression is downregulated and this is correlated with a poor prognosis. While in PC their expression is increased and correlates with a poor prognosis. The opposite roles could be explained by the fact that members of OSBPL family have tissue specific expression, which is disturbed in tumors, and also have different roles in cell physiology. Enzymes involved in oxysterol synthesis are also potential new prognostic biomarkers and drug targets in gastrointestinal cancers. The expression and the SNPs in cytochromes P450 enzymes are associated with the risk for development and prognosis of selected cancers. The level of serum cholesterol itself is a prognostic biomarker associated with a higher risk for development of numerous cancers. The association is consolidated by the fact that statins are emerging novel therapies in gastrointestinal cancers. Cholesterol is the key player not only due to being the oxysterol progenitor molecule but also one of the key cellular ingredients needed in growing cancerous cells. The oxysterol metabolites, the bile acids, and their receptor FXR are emerging new biomarkers and therapeutic targets in gastrointestinal cancers. The role of FXR in these cancers is two faced. FXR downregulation is observed with progression of cancerogenesis and FXR role in suppression of proliferation, migration and invasion in colon cells and hepatocytes was confirmed. However, FXR role in pancreatic cancer seems to be the opposite. These data indicate that FXR activity is essential in tissues with high FXR expression such as liver and colon, while FXR activity in tissues where FXR and bile acids are not common is potentially tumorigenic.
Disruptions of circadian rhythm are clearly tumorigenic in gastrointestinal cancers as indicated by rodent models. The circadian molecular clock genes are also new emerging prognostic biomarkers and therapeutic targets. Several genes involved in regulation of cellular circadian expression have direct oncogenic or tumor suppressor roles. Not only their expression correlates with prognosis in patients, knockouts in rodent models lead to spontaneous cancerogenesis and SNPs are associated with the risk of cancer development and prognosis. They are interesting therapeutic targets, moreover, the time of the therapy must also be considered and chronotherapy has already been shown to be effective in gastrointestinal cancers.
Summary
Summary of reviewed data revealed that circadian regulatory network, cytochromes P450, nuclear receptors, cholesterol biosynthesis and oxysterols have overlapping roles in gastrointestinal cancers. Several factors from sterol homeostasis and circadian rhythm have been identified as potential novel prognostic biomarkers, genetic risk factors and drug targets. However, there are still several issues that remain open. The role of oxysterols in carcinogenesis is far from being conclusive. The cells are dealing with a variety of oxysterol molecules that promote their actions through modulation of transcription factors, such as LXR, RORs and others. Depending on the type of cancer, an oxysterol modulated pathway can have beneficial or damaging effect on carcinogenesis. This is one of the obstacles in developing new therapeutic strategies, besides statins, where it is clear that a more personalized approach is essential to increase the positive drug effects. The link between cholesterol, oxysterol synthesis and circadian rhythm was until recently mostly unidirectional: the clock controlled the expression of lipogenic genes. It now becomes evident that both pathways are interconnected by energy metabolism where RORA/C and LXR are at the crossroad. The ROR receptors are circadian regulators, being co-responsible for the rhythmic expression of output metabolic genes. However, their own transcriptional activity depends on sterols and oxysterols whose endogenous synthesis is regulated by the clock. This is again a new field of translational research termed chronotherapy which was proven to be successful particularly in treatments of colorectal cancer.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by Slovenian Research Agency program grant P1-0390 and research project J1-9176. UK was supported by young researcher scholarship. LB is a masters' student of medicine at the Faculty of Medicine University of Ljubljana.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ačimovič J, Rozman D. Steroidal triterpenes of cholesterol synthesis. Molecules. (2013) 18:4002–17. doi: 10.3390/molecules18044002
2. Singh P, Saxena R, Srinivas G, Pande G, Chattopadhyay A. Cholesterol biosynthesis and homeostasis in regulation of the cell cycle. PloS ONE. (2013) 8:e58833. doi: 10.1371/journal.pone.0058833
3. Simons K, Ikonen E. How cells handle cholesterol. Science. (2000) 290:1721–6. doi: 10.1126/science.290.5497.1721
4. De Boussac H, Alioui A, Viennois E, Dufour J, Trousson A, Vega A, et al. Oxysterol receptors and their therapeutic applications in cancer conditions. Exp Opin Ther Targets. (2013) 17:1029–38. doi: 10.1517/14728222.2013.820708
5. Björkhem I, Diczfalusy U. Oxysterols: friends, foes, or just fellow passengers? Arterioscler Thromb Vasc Biol. (2002) 22:734–42. doi: 10.1161/01.ATV.0000013312.32196.49
6. Ačimovič J, Goyal S, Košir R, Goličnik M, Perše M, Belič A, et al. Cytochrome P450 metabolism of the post-lanosterol intermediates explains enigmas of cholesterol synthesis. Sci Rep. (2016) 6:28462. doi: 10.1038/srep28462
7. Brown AJ, Jessup W. Oxysterols: sources, cellular storage and metabolism, and new insights into their roles in cholesterol homeostasis. Mol Aspects Med. (2009) 30:111–22. doi: 10.1016/j.mam.2009.02.005
8. Kloudova A, Peter Guengerich F, Soucek P. The role of oxysterols in human cancer. Trends Endocrinol Metab. (2017) 28:485–96. doi: 10.1016/j.tem.2017.03.002
9. Russell DW. Oxysterol biosynthetic enzymes. Biochim Biophys Acta. (2000) 1529:126–35. doi: 10.1016/S1388-1981(00)00142-6
10. Gill S, Chow R, Brown AJ. Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res. (2008) 47:391–404. doi: 10.1016/j.plipres.2008.04.002
11. DeBose-Boyd RA. Feedback regulation of cholesterol synthesis. Cell Res. (2008) 18:609–21. doi: 10.1038/cr.2008.61
12. Kanner J. Dietary advanced lipid oxidation endproducts are risk factors to human health. Mol Nutr Food Res. (2007) 51:1094–101. doi: 10.1002/mnfr.200600303
13. Staprans I, Pan X-M, Rapp JH, Feingold KR. Oxidized cholesterol in the diet is a source of oxidized lipoproteins in human serum. J Lipid Res. (2003) 44:705–15. doi: 10.1194/jlr.M200266-JLR200
14. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. (2006) 124:35–46. doi: 10.1016/j.cell.2005.12.022
15. Shimano H. Sterol Regulatory Element-Binding Proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res. (2001) 40:439–52. doi: 10.1016/S0163-7827(01)00010-8
16. Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS, et al. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Investig. (1997) 100:2115–24. doi: 10.1172/JCI119746
17. Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. (2002) 277:9520–8. doi: 10.1074/jbc.M111421200
18. Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of srebps from endoplasmic reticulum to golgi : oxysterols block transport by binding to insights. PNAS. (2007) 104:6511–18. doi: 10.1073/pnas.0700899104
19. Dong XY, Tang SQ, Chen JD. Dual functions of insig proteins in cholesterol homeostasis. Lipids Health Dis. (2012) 11:173. doi: 10.1186/1476-511X-11-173
20. Li D, Long W, Huang R, Chen Y, Xia M. 27-Hydroxycholesterol inhibits sterol regulatory element-binding protein 1 activation and hepatic lipid accumulation in mice. Obesity. (2018) 26:713–22. doi: 10.1002/oby.22130
21. Lu M, Hu XH, Li Q, Xiong Y, Hu GJ, Xu JJ, et al. A specific cholesterol metabolic pathway is established in a subset of hccs for tumor growth. J Mol Cell Biol. (2013) 5:404–15. doi: 10.1093/jmcb/mjt039
22. Rezen T, Rozman D, Pascussi JM, Monostory K. Interplay between cholesterol and drug metabolism. Biochim Biophys Acta. (2011) 1814:146–60. doi: 10.1016/j.bbapap.2010.05.014
23. Deng R, Yang D, Yang J, Yan B. Oxysterol 22 (R) -hydroxycholesterol induces the expression of the bile salt export pump through nuclear receptor farsenoid X receptor but not liver X receptor. J Pharmacol Exp Ther. (2006) 317:317–25. doi: 10.1124/jpet.105.097758
24. Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. (2002) 418:935–41. doi: 10.1038/nature00965
25. Zmrzljak UP, Rozman D. Circadian regulation of the hepatic endobiotic and xenobitoic detoxification pathways: the time matters. Chem Res Toxicol. (2012) 25:811–24. doi: 10.1021/tx200538r
26. Korencic A, Bordyugov G, Kosir R, Rozman D, Golicnik M, Herzel H. The interplay of cis-regulatory elements rules circadian rhythms in mouse liver. PloS ONE. (2012) 7:e46835. doi: 10.1371/journal.pone.0046835
27. Dumbell R, Matveeva O, Oster H. Circadian clocks, stress, and immunity. Front Endocrinol. (2016) 7:37. doi: 10.3389/fendo.2016.00037
28. Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. (2002) 419:841–4. doi: 10.1038/nature01123
29. Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Ann Rev Genomics Hum Genet. (2004) 5:407–41. doi: 10.1146/annurev.genom.5.061903.175925
30. Ripperger JA, Merrow M. Perfect timing: epigenetic regulation of the circadian clock. FEBS Lett. (2011) 585:1406–11. doi: 10.1016/j.febslet.2011.04.047
31. Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, et al. SIRT1 Regulates circadian clock gene expression through PER2 deacetylation. Cell. (2008) 134:317–28. doi: 10.1016/j.cell.2008.06.050
32. Hirano A, Fu YH, Ptáek LJ. The intricate dance of post-translational modifications in the rhythm of life. Nat Struct Mol Biol. (2016) 23:1053–60. doi: 10.1038/nsmb.3326
33. Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, et al. Control of mammalian circadian rhythm by CKI -regulated proteasome-mediated PER2 degradation. Mol Cell Biol. (2005) 25:2795–807. doi: 10.1128/MCB.25.7.2795-2807.2005
34. Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. (2006) 125:497–508. doi: 10.1016/j.cell.2006.03.033
35. Bellet MM, Masri S, Astarita G, Sassone-Corsi P, Della Fazia MA, Servillo G. Histone deacetylase SIRT1 controls proliferation, circadian rhythm, and lipid metabolism during liver regeneration in mice. J Biol Chem. (2016) 291:23318–29. doi: 10.1074/jbc.M116.737114
36. Grimaldi B, Nakahata Y, Kaluzova M, Masubuchi S, Sassone-Corsi P. Chromatin remodeling, metabolism and circadian clocks: the interplay of CLOCK and SIRT1. Int J Biochem Cell Biol. (2009) 41:81–6. doi: 10.1016/j.biocel.2008.08.035
37. Utani K, Fu H, Jang SM, Marks AB, Smith OK, Zhang Y, et al. Phosphorylated SIRT1 associates with replication origins to prevent excess replication initiation and preserve genomic stability. Nucleic Acids Res. (2017) 45:7807–24. doi: 10.1093/nar/gkx468
38. Guarente L. Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol. (2011) 76:81–90. doi: 10.1101/sqb.2011.76.010629
39. Gnocchi D, Pedrelli M, Hurt-Camejo E, Parini P, Gnocchi D, Pedrelli M, et al. Lipids around the Clock: focus on circadian rhythms and lipid metabolism. Biology. (2015) 4:104–32. doi: 10.3390/biology4010104
40. Ghoshal S, Stevens JR, Billon C, Girardet C, Sitaula S, Leon AS, et al. Adropin: an endocrine link between the biological clock and cholesterol homeostasis. Mol Metab. (2018) 8:51–64. doi: 10.1016/j.molmet.2017.12.002
41. Cho Y, Noshiro M, Choi M, Morita K, Kawamoto T, Fujimoto K, et al. The basic helix-loop-helix proteins Differentiated Embryo Chondrocyte (DEC) 1 and DEC2 function as corepressors of retinoid X receptors. Mol Pharmacol. (2009) 76:1360–9. doi: 10.1124/mol.109.057000
42. Wada T, Kang HS, Angers M, Gong H, Bhatia S, Khadem S, et al. Identification of oxysterol 7 -hydroxylase (Cyp7b1) as a novel retinoid-Related Orphan Receptor (ROR) (NR1F1) target gene and a functional cross-talk between ROR and liver X receptor (NR1H3). Mol Pharmacol. (2007) 73:891–9. doi: 10.1124/mol.107.040741
43. Martelot GL, Claudel T, Gatfield D, Schaad O, Kornmann B, Sasso GL, et al. REV-ERBα participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. (2009) 7:1–12. doi: 10.1371/journal.pbio.1000181
44. Lu YF, Jin T, Xu Y, Zhang D, Wu Q, Zhang Y-K, et al. Sex differences in the circadian variation of cytochrome P450 genes and corresponding nuclear receptors in mouse liver. Chronobiol Int. (2013) 30:1135–43. doi: 10.3109/07420528.2013.805762
45. Kudo T, Kawashima M, Tamagawa T, Shibata S. Clock mutation facilitates accumulation of cholesterol in the liver of mice fed a cholesterol and/or cholic acid diet. Am J Physiol Endocrinol Metab. (2008) 294:E120–30. doi: 10.1152/ajpendo.00061.2007
46. Wang X, Xue J, Yang J, Xie M. Timed high-fat diet in the evening affects the hepatic circadian clock and PPARα-mediated lipogenic gene expressions in mice. Genes Nutr. (2013) 8:457–63. doi: 10.1007/s12263-013-0333-y
47. Duez HJ, van der Veen N, Duhem C, Pourcet B, Touvier T, Fontaine C, et al. Regulation of bile acid synthesis by the nuclear receptor rev-Erbα. Gastroenterology. (2008) 135:689–98. doi: 10.1053/j.gastro.2008.05.035
48. Ma K, Xiao R, Tseng HT, Shan L, Fu L, Moore DD. Circadian dysregulation disrupts bile acid homeostasis. PLoS ONE. (2009) 4:e6843. doi: 10.1371/journal.pone.0006843
49. Tomalik-Scharte D, Suleiman AA, Frechen S, Kraus D, Kerkweg U, Rokitta D, et al. Population pharmacokinetic analysis of circadian rhythms in hepatic CYP3A activity using midazolam. J Clin Pharmacol. (2014) 54:1162–9. doi: 10.1002/jcph.318
50. Takako T, Tomita M, Matsunaga N, Nakagawa H, Koyanagi S, Ohdo S. Molecular basis for rhythmic expression of Cyp3a4 in serum-shocked Hepg2 cells. Pharmacogenet Genomics. (2007) 17:1047–56. doi: 10.1097/FPC.0b013e3282f12a61
51. Pathak P, Li T, Chiang JYL. Retinoic acid-related orphan receptor α regulates diurnal rhythm and fasting induction of sterol 12α-hydroxylase in bile acid synthesis. J Biol Chem. (2013) 288:37154–65. doi: 10.1074/jbc.M113.485987
52. Noshiro M, Kawamoto T, Furukawa M, Fujimoto K, Yoshida Y, Sasabe E, et al. Rhythmic expression of DEC1 and DEC2 in peripheral tissues: DEC2 is a potent suppressor for hepatic cytochrome P450s opposing DBP. Genes Cells. (2004) 9:317–29. doi: 10.1111/j.1356-9597.2004.00722.x
53. Bartosch B. Hepatitis B and C viruses and hepatocellular carcinoma. Viruses. (2010) 2:1504–9. doi: 10.3390/v2081504
54. El-Serag HB. Hepatocellular carcinoma. N Eng J Med. (2011) 365:1118–27. doi: 10.1056/NEJMra1001683
55. Kovac U, Rozman D. Genetics of non-alcoholic fatty liver disease. In: eLS. American Cancer Society (2015). p. 1–8. doi: 10.1002/9780470015902.a0025335
56. Naik A, Košir R, Rozman D. Genomic aspects of NAFLD pathogenesis. Genomics. (2013) 102:84–95. doi: 10.1016/j.ygeno.2013.03.007
57. Petrick JL, Kelly SP, Altekruse SF, McGlynn KA, Rosenberg PS. Future of hepatocellular carcinoma incidence in the united states forecast through 2030. J Clin Oncol. (2016) 34:1787–94. doi: 10.1200/JCO.2015.64.7412
58. Zhou YY, Zhu GQ, Wang Y, Zheng JN, Ruan LY, Cheng Z, et al. Systematic review with network meta-analysis: statins and risk of hepatocellular carcinoma. Oncotarget. (2016) 7:21753–62. doi: 10.18632/oncotarget.7832
59. Blagotinšek K, Rozman D. Targeting signalling pathways in hepatocellular carcinoma. Curr Pharmaceut Design. (2016) 22:1–1. doi: 10.2174/1381612822666161006160005
60. Ikegami T, Hyogo H, Honda A, Miyazaki T, Tokushige K, Hashimoto E, et al. Increased serum liver X receptor ligand oxysterols in patients with non-alcoholic fatty liver disease. J Gastroenterol. (2012) 47:1257–66. doi: 10.1007/s00535-012-0585-0
61. Ikegami T, Honda A, Miyazaki T, Kohjima M, Nakamuta M, Matsuzaki Y. Increased serum oxysterol concentrations in patients with chronic hepatitis C virus infection. Biochem Biophys Res Commun. (2014) 446:736–40. doi: 10.1016/j.bbrc.2014.01.176
62. Li J, Zheng X, Lou N, Zhong W, Yan D. Oxysterol binding protein-related protein 8 mediates the cytotoxicity of 25-hydroxycholesterol. J Lipid Res. (2016) 57:1845–53. doi: 10.1194/jlr.M069906
63. Yokoyama S, Nozawa F, Tanaka S, Muneyuki S, Ogawa M. Anti-tumor effects of 25-hydroxycholesterol and low-dose recombinant tumor necrosis factor-alpha on rat liver tumorigenesis: modulated differentiation therapy for hepatocellular carcinoma. Int J Oncol. (2000) 16:1029–33. doi: 10.3892/ijo.16.5.1029
64. Zhong W, Qin S, Zhu B, Pu M, Liu F, Wang L, et al. Oxysterol-binding Protein-Related Protein 8 (ORP8) increases sensitivity of hepatocellular carcinoma cells to fas-mediated apoptosis. J Biol Chem. (2015) 290:8876–87. doi: 10.1074/jbc.M114.610188
65. Long H, Guo X, Qiao S, Huang Q. Tumor LXR expression is a prognostic marker for patients with hepatocellular carcinoma. Pathol Oncol Res. (2018) 24:339–44. doi: 10.1007/s12253-017-0249-8
66. Xiong H, Zhang Y, Chen S, Ni Z, He J, Li X, et al. Induction of SOCS3 by liver X receptor suppresses the proliferation of hepatocellular carcinoma cells. Oncotarget. (2017) 8:64083–94. doi: 10.18632/oncotarget.19321
67. Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL. Increased activation of the wnt-/- NLcatenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. (2011) 2011:jpet.111.179390. doi: 10.1124/jpet.111.179390
68. Liu N, Meng Z, Lou G, Zhou W, Wang X, Zhang Y, et al. Hepatocarcinogenesis in FXR-/- mice mimics human hcc progression that operates through hnf1α regulation of fxr expression. Mol Endocrinol. (2012) 26:775–85. doi: 10.1210/me.2011-1383
69. Su H, Ma C, Liu J, Li N, Gao M, Huang A, et al. Downregulation of nuclear receptor FXR Is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G1245–53. doi: 10.1152/ajpgi.00439.2011
70. Liu X, Zhang X, Ji L, Gu J, Zhou M, Chen S. Farnesoid X receptor associates with β-catenin and inhibits its activity in hepatocellular carcinoma. Oncotarget. (2015) 6:4226–38. doi: 10.18632/oncotarget.2899
71. He J, Zhao K, Zheng L, Xu Z, Gong W, Chen S, et al. Upregulation of microRNA-122 by farnesoid X receptor suppresses the growth of hepatocellular carcinoma cells. Mol Cancer. (2015) 4:163. doi: 10.1186/s12943-015-0427-9
72. Schulz A, Schlu T, Kinzel O, Abel U, Deuschle U, Schu J, et al. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS ONE. (2012) 7:e43044. doi: 10.1371/journal.pone.0043044
73. Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharma Design. (2014) 20:2619–26. doi: 10.2174/13816128113199990486
74. Yuan P, Li J, Zhou F, Huang Q, Zhang J, Guo X, et al. NPAS2 promotes cell survival of hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis. (2017) 8:e2704. doi: 10.1038/cddis.2017.131
75. Yuan P, Wang S, Zhou F, Wan S, Yang Y, Huang X, et al. Functional polymorphisms in the NPAS2Gene are associated with overall survival in transcatheter arterial chemoembolization-treated hepatocellular carcinoma patients. Cancer Sci. (2014) 105:825–32. doi: 10.1111/cas.12428
76. Zhao B, Lu J, Yin J, Liu H, Guo X, Yang Y, et al. a functional polymorphism in per3 gene is associated with prognosis in hepatocellular carcinoma. Liver Int. (2012) 32:1451–9. doi: 10.1111/j.1478-3231.2012.02849.x
77. Filipski E, Subramanian P, Carrière J, Guettier C, Barbason H, Lévi F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat Res Genet Toxicol Environ Mutagenesis. (2009) 680:95–105. doi: 10.1016/j.mrgentox.2009.10.002
78. Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, et al. Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell. (2016) 30:909–24. doi: 10.1016/j.ccell.2016.10.007
79. Fleet T, Stashi E, Zhu B, Rajapakshe K, Marcelo KL, Kettner NM, et al. Genetic and Environmental models of circadian disruption link src-2 function to hepatic pathology. Journal of Biol Rhythms. (2016) 31:443–60. doi: 10.1177/0748730416657921
80. Elgohary N, Pellegrino R, Neumann O, Elzawahry HM, Saber MM, Zeeneldin AA, et al. Protumorigenic role of timeless in hepatocellular carcinoma. Int J Oncol. (2015) 46:597–606. doi: 10.3892/ijo.2014.2751
81. Morcos PN, Moreira SA, Brennan BJ, Blotner S, Shulman NS, Smith PF. Influence of chronic hepatitis C infection on cytochrome P450 3A4 activity using midazolam as an in vivo probe substrate. Eur J Clin Pharmacol. (2013) 69:1777–84. doi: 10.1007/s00228-013-1525-5
82. Jepsen P, Turati F, La Vecchia C. NAFLD and cancer: more cause for concern? J Hepatol. (2018) 68:10–2. doi: 10.1016/j.jhep.2017.10.008
83. Skubic C, Drakulić Ž, Rozman D. Pharmacogenetic considerations for personalized therapy when tackling nonalcoholic fatty liver disease: the perspective of genes, drugs, and the sex. Expert Opin Drug Metab Toxicol. (2018) 14:831–41. doi: 10.1080/17425255.2018.1492552
84. Bellomo C, Caja L, Fabregat I, Mikulits W, Kardassis D, Heldin CH, et al. Snail mediates crosstalk between TGFβ and LXRα in hepatocellular carcinoma. Cell Death Differ. (2018) 25:885–903. doi: 10.1038/s41418-017-0021-3
85. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. (2007) 67:863–7. doi: 10.1158/0008-5472.CAN-06-1078
86. Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. (2007) 28:940–6. doi: 10.1093/carcin/bgl249
87. Degirolamo C, Modica S, Vacca M, Di Tullio G, Morgano A, Orazio AD, et al. Prevention of spontaneous hepatocarcinogenesis n farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology. (2014) 61:161–70. doi: 10.1002/hep.27274
88. Takahashi S, Tanaka N, Fukami T, Xie C, Yagai T, Kim D, et al. Role of farnesoid X receptor and bile acids in hepatic tumor development. Hepatol Commun. (2018) 2:1567–82. doi: 10.1002/hep4.1263
89. Lee DC, Kang YK, Kim WH, Jang YJ, Kim DJ, Park IY, et al. Functional and Clinical evidence for NDRG2 as a candidate suppressor of liver cancer metastasis. Cancer Res. (2008) 11:4210–21. doi: 10.1158/0008-5472.CAN-07-5040
90. Jiang Y, Iakova P, Jin J, Sullivan E, Sharin VI, Hong H, et al. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology. (2013) 57:1098–106. doi: 10.1002/hep.26146
91. Dawson SP. Hepatocellular carcinoma and the ubiquitin - proteasome system. Biochim Biophys Acta. (2008) 1782:775–84. doi: 10.1016/j.bbadis.2008.08.003
92. Yang F, Hu Y, Liu HX, Wan YJY. MiR-22-silenced cyclin a expression in colon and liver cancer cells is regulated by bile acid receptor. J Biol Chem. (2015) 290:6507–15. doi: 10.1074/jbc.M114.620369
93. Yang F, Gong J, Wang G, Chen P, Yang L. Waltonitone inhibits proliferation of hepatoma cells and tumorigenesis via FXR-MiR-22-CCNA2 signaling pathway. Oncotarget. (2016) 7:75165–75. doi: 10.18632/oncotarget.12614
94. Xu Z, Huang G, Gong W, Zhou P, Zhao Y, Zhang Y, et al. FXR ligands protect against hepatocellular in Fl ammation via SOCS3 induction. Cell Signal. (2012) 24:1658–64. doi: 10.1016/j.cellsig.2012.04.015
95. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology. (2008) 48:1632–43. doi: 10.1002/hep.22519
96. Guo F, Xu Z, Zhang Y, Jiang P, Huang G, Chen S. FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget. (2015) 6:34606–16. doi: 10.18632/oncotarget.5314
97. Wang X, Fu X, Van Ness C, Meng Z, Ma X, Huang W. Bile acid receptors and liver cancer. Curr Pathobiol Rep. (2013) 1:29–35. doi: 10.1007/s40139-012-0003-6
98. Zhang Y, Gong W, Dai S, Huang G, Shen X, Gao M. Downregulation of human farnesoid X receptor by MiR-421 promotes proliferation and migration of hepatocellular carcinoma cells. Mol Cancer Res. (2012) 10:516–23. doi: 10.1158/1541-7786.MCR-11-0473
99. Lu M, Kong X, Wang H, Huang G, Ye C, He Z. A novel MicroRNAs expression signature for hepatocellular carcinoma diagnosis and prognosis. Oncotarget. (2017) 8:8775–84. doi: 10.18632/oncotarget.14452
100. Garcia-Rodriguez JL, Barbier-Torres L, Fernandez-Alvarez S, Gutierrez-de Juan V, Monte MJ, Halilbasic E, et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology. (2014) 59:1972–83. doi: 10.1002/hep.26971
101. Zhou M, Luo J, Chen M, Yang H, Learned RM, Depaoli AM, et al. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J Hepatol. (2017) 66:1182–92. doi: 10.1016/j.jhep.2017.01.027
102. Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. (2012) 12:56. doi: 10.1186/1471-2407-12-56
103. Piglionica M, Cariello M, Moschetta A. The gut-liver axis in hepatocarcinoma: a focus on the nuclear receptor fxr and the enterokine fgf19. Curr Opin Pharmacol. (2018) 43:93–8. doi: 10.1016/j.coph.2018.08.005
104. Li C, Yang W, Zhang J, Zheng X, Yao Y, Tu K, et al. SREBP-1 has a prognostic role and contributes to invasion and metastasis in human hepatocellular carcinoma. Int J Mol Sci. (2014) 15:7124–38. doi: 10.3390/ijms15057124
105. Min X, Wen J, Zhao L, Wang K, Li Q, Huang G, et al. Role of hepatoma-derived growth factor in promoting de novo lipogenesis and tumorigenesis in hepatocellular carcinoma. Mol Oncol. (2018) 12:1480–97. doi: 10.1002/1878-0261.12357
106. Li NZ, Zhou S, Shen Y, Xu J, Miao HH, Xiong Y, et al. Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology. (2017) 65:1936–47. doi: 10.1002/hep.29018
107. Tomita K, Toshiaki T, Suzuki T, Shimizu M, Sato H, Narimatsu K, et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. (2014) 59:154–69. doi: 10.1002/hep.26604
108. Ji J, Chen H, Liu XP, Wang YH, Luo CL, Zhang W, Xie W, et al. A MiRNA combination as promising biomarker for hepatocellular carcinoma diagnosis: a study based on bioinformatics analysis. J Cancer. (2018) 9:3435–46. doi: 10.7150/jca.26101
109. Zhang Y, Qiu Z, Wei L, Tang R, Lian B, Zhao Y, et al. Integrated analysis of mutation data from various sources identifies key genes and signaling pathways in hepatocellular carcinoma. PloS ONE. (2014) 9:e100854. doi: 10.1371/journal.pone.0100854
110. Li H, Lu YF, Chen H, Liu J. Dysregulation of metallothionein and circadian genes in human hepatocellular carcinoma. Chronobiol Int. (2017) 34:192–202. doi: 10.1080/07420528.2016.1256300
111. Chauvet C, Vanhoutteghem A, Duhem C, Saint-Auret G, Bois-Joyeux B, Djian P, et al. Control of gene expression by the retinoic acid-related orphan receptor alpha in HepG2 Human hepatoma cells. PloS ONE. (2011) 6:e22545. doi: 10.1371/journal.pone.0022545
112. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
113. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, et al. Recent progress in pancreatic cancer. Cancer J Clin. (2013) 63:318–48. doi: 10.3322/caac.21190
114. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. (2011) 378:607–20. doi: 10.1016/S0140-6736(10)62307-0
115. Gong Z, Holly EA, Wang F, Chan JM, Bracci PM. Intake of fatty acids and antioxidants and pancreatic cancer in a large population-based case-control study in the San Francisco Bay Area. Int J Cancer. (2010) 127:1893–904. doi: 10.1002/ijc.25208
116. Di Gangi IM, Mazza T, Fontana A, Copetti M, Fusilli C, Ippolito A, et al. Metabolomic Profile in pancreatic cancer patients: a consensus-based approach to identify highly discriminating metabolites. Oncotarget. (2016) 7:5815–29. doi: 10.18632/oncotarget.6808
117. Ishikawa S, Nagai Y, Masuda T, Koga Y, Nakamura T, Imamura Y, et al. The role of oxysterol binding protein-related protein 5 in pancreatic cancer. Cancer Sci. (2010) 101:898–905. doi: 10.1111/j.1349-7006.2009.01475.x
118. Koga Y, Ishikawa S, Nakamura T, Masuda T, Nagai Y, Takamori H, et al. Oxysterol Binding protein-related protein-5 is related to invasion and poor prognosis in pancreatic cancer. Cancer Sci. (2008) 99:2387–94. doi: 10.1111/j.1349-7006.2008.00987.x
119. Li H, Wang X, Fang Y, Huo Z, Lu X, Zhan X, et al. Integrated expression profiles analysis reveals novel predictive biomarker in pancreatic ductal adenocarcinoma. Oncotarget. (2017) 8:52571–83. doi: 10.18632/oncotarget.16732
120. Candelaria NR, Addanki S, Zheng J, Nguyen-Vu T, Karaboga H, Dey P, et al. Antiproliferative effects and mechanisms of liver X receptor ligands in pancreatic ductal adenocarcinoma cells. PLoS ONE. (2014) 9:e106289. doi: 10.1371/journal.pone.0106289
121. Lee JY, Lee KT, Lee JK, Lee KH, Jang KT, Heo JS, et al. Farnesoid X receptor, overexpressed in pancreatic cancer with lymph node metastasis promotes cell migration and invasion. Br J Cancer. (2011) 104:1027–37. doi: 10.1038/bjc.2011.37
122. Hu H, Wu L, Han T, Zhuo M, Lei W, Cui J. Correlated high expression of FXR and Sp1 in cancer cells confers a poor prognosis for pancreatic cancer : a study based on TCGA and tissue microarray. Oncotarget. (2017) 8:33265–75. doi: 10.18632/oncotarget.16633
123. Giaginis C, Koutsounas I, Alexandrou P, Patsouris E, Kouraklis G, Theocharis S. Elevated Farnesoid X Receptor (FXR) and Retinoid X Receptors (RXRs) expression is associated with less tumor aggressiveness and favourable prognosis in patients with pancreatic adenocarcinoma. Neoplasma. (2015) 62:332–41. doi: 10.4149/neo_2015_040
124. Joshi S, Cruz E, Rachagani S, Guha S, Brand RE, Ponnusamy MP, et al. Bile acids-mediated overexpression of MUC4 via FAK-dependent c-Jun activation in pancreatic cancer. Mol Oncol. (2016) 10:1063–77. doi: 10.1016/j.molonc.2016.04.007
125. Li W, Liu L, Liu D, Jin S, Yang Y, Tang W, et al. Decreased circadian component Bmal1 predicts tumor progression and poor prognosis in human pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun. (2016) 472:156–62. doi: 10.1016/j.bbrc.2016.02.087
126. Jiang W, Zhao S, Jiang X, Zhang E, Hu G, Hu B, et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the P53 tumor suppressor pathway. Cancer Lett. (2016) 371:314–25. doi: 10.1016/j.canlet.2015.12.002
127. Tavano F, Pazienza V, Fontana A, Burbaci FP, Panebianco C, Saracino C, et al. SIRT1 and circadian gene expression in pancreatic ductal adenocarcinoma: effect of starvation. Chronobiol Int. (2015) 32:497–512. doi: 10.3109/07420528.2014.1003351
128. Wang X, Ji A, Zhu Y, Liang Z, Wu J, Li S, et al. A meta-analysis including dose-response relationship between night shift work and the risk of colorectal cancer. Oncotarget. (2015) 6:25046–60. doi: 10.18632/oncotarget.4502
129. Chen H, Qin S, Wang M, Zhang T, Zhang S. Association between cholesterol intake and pancreatic cancer risk: evidence from a meta-analysis. Sci Rep. (2015) 5:8243. doi: 10.1038/srep08243
130. Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol. (2015) 36:4133–41. doi: 10.1007/s13277-015-3047-5
131. Siqingaowa SS, Gopalakrishnan V, Taghibiglou C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem Biophys Res Commun. (2017) 488:136–40. doi: 10.1016/j.bbrc.2017.05.023
132. Zhou C, Qian W, Ma J, Cheng L, Jiang Z, Yan B, et al. Resveratrol enhances the chemotherapeutic response and reverses the stemness induced by gemcitabine in pancreatic cancer cells via targeting SREBP1. Cell Proliferat. (2019) 52:e12514. doi: 10.1111/cpr.12514
133. Rong W, Jiang W, Wan R. It is about time: circadian clock in the pancreas. Pancreatic Disord Ther. (2015) 5:2–5. doi: 10.4172/2165-7092.1000155
134. Mühlbauer E, Wolgast S, Finckh U, Peschke D, Peschke E. Indication of circadian oscillations in the rat pancreas. FEBS Lett. (2004) 564:91–6. doi: 10.1016/S0014-5793(04)00322-9
135. Relles D, Sendecki J, Chipitsyna G, Hyslop T, Yeo CJ, Arafat HA. Circadian gene expression and clinicopathologic correlates in pancreatic cancer. J Gastrointest Surg. (2013) 17:443–50. doi: 10.1007/s11605-012-2112-2
136. Luchini C, Capelli P, Scarpa A. Pancreatic ductal adenocarcinoma and its variants. Surg Pathol Clin. (2016) 9:547–60. doi: 10.1016/j.path.2016.05.003
137. Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. (2012) 35:2402–11. doi: 10.2337/dc12-0336
138. Jiao L, Chen L, White DL, Tinker L, Chlebowski RT, Van Horn LV, et al. Low-fat dietary pattern and pancreatic cancer risk in the women's health initiative dietary modification randomized controlled trial. J Natl Cancer Inst. (2018) 110:49–56. doi: 10.1093/jnci/djx117
139. Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, et al. Bmal1 and -cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced -cell failure in mice. Mol Cell Biol. (2013) 33:2327–38. doi: 10.1128/MCB.01421-12
140. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. (2010) 466:627–31. doi: 10.1038/nature09253
141. Rosato V, Tavani A, Bosetti C, Pelucchi C, Talamini R, Polesel J, et al. Metabolic syndrome and pancreatic cancer risk: a case-control study in italy and meta-analysis. Metabolism. (2011) 60:1372–8. doi: 10.1016/j.metabol.2011.03.005
142. Obuch JC, Ahnen DJ. Colorectal cancer: genetics is changing everything. Gastroenterol Clin N Am. (2016) 45:459–76. doi: 10.1016/j.gtc.2016.04.005
143. Karantanos T, Theodoropoulos G, Pektasides D, Gazouli M. Clock genes: their role in colorectal cancer. World J Gastroenterol. (2014) 20:1986–92. doi: 10.3748/wjg.v20.i8.1986
144. Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. (2007) 56:417–25. doi: 10.1136/gut.2006.093310
145. Kitahara CM, Berrington de González A, Freedman ND, Huxley R, Mok Y, Jee SH, et al. Total cholesterol and cancer risk in a large prospective study in korea. J Clin Oncol. (2011) 29:1592–8. doi: 10.1200/JCO.2010.31.5200
146. Rodriguez-Broadbent H, Law PJ, Sud A, Palin K, Tuupanen S, Gylfe A, et al. Mendelian randomisation implicates hyperlipidaemia as a risk factor for colorectal cancer. Int J Cancer. (2017) 140:2701–8. doi: 10.1002/ijc.30709
147. Ito H, Matsuo K, Hosono S, Watanabe M, Kawase T, Suzuki T, et al. Association between CYP7A1 and the risk of proximal colon cancer in japanese. Int J Mol Epidemiol Genet. (2010) 1:35–46. doi: 10.1158/1940-6207.PREV-09-B116
148. Wertheim BC, Smith JW, Fang C, Alberts DS, Lance P, Thompson PA. Risk modification of colorectal adenoma by CYP7A1 polymorphisms and the role of bile acid metabolism in carcinogenesis. Cancer Prev Res. (2012) 5:197–204. doi: 10.1158/1940-6207.CAPR-11-0320
149. Tabata S, Yin G, Ogawa S, Yamaguchi K, Mineshita M, Kono S. Genetic polymorphism of cholesterol 7α-hydroxylase (CYP7A1) and colorectal adenomas: self defense forces health study. Cancer Sci. (2006) 97:406–10. doi: 10.1111/j.1349-7006.2006.00182.x
150. Hagiwara T, Kono S, Yin G, Toyomura K, Nagano J, Mizoue T, et al. Genetic polymorphism in cytochrome P450 7A1 and risk of colorectal cancer: the fukuoka colorectal cancer study. Cancer Res. (2005) 65:2979–82. doi: 10.1158/0008-5472.CAN-04-3872
151. Zhu Y, Soroka D, Sang S. Oxyphytosterols as active ingredients in wheat bran suppress human colon cancer cell growth: identification, chemical synthesis, and biological evaluation. J Agric Food Chem. (2015) 63:2267–76. doi: 10.1021/jf506361r
152. Biasi F, Mascia C, Astegiano M, Chiarpotto E, Nano M, Vizio B, et al. Pro-oxidant and proapoptotic effects of cholesterol oxidation products on human colonic epithelial cells: a potential mechanism of inflammatory bowel disease progression. Free Radic Biol Med. (2009) 47:1731–41. doi: 10.1016/j.freeradbiomed.2009.09.020
153. Warns J, Marwarha G, Freking N, Ghribi O. 27-Hydroxycholesterol decreases cell proliferation in colon cancer cell lines. Biochimie. (2018) 153:171–80. doi: 10.1016/j.biochi.2018.07.006
154. Biasi F, Chiarpotto E, Sottero B, Maina M, Mascia C, Guina T, et al. Evidence of cell damage induced by major components of a diet-compatible mixture of oxysterols in human colon cancer CaCo-2 cell line. Biochimie. (2013) 95:632–40. doi: 10.1016/j.biochi.2012.10.011
155. Roussi S, Winter A, Gosse F, Werner D, Zhang X, Marchioni E, et al. Different apoptotic mechanisms are involved in the antiproliferative effects of 7β-hydroxysitosterol and 7β-hydroxycholesterol in human colon cancer cells. Cell Death Differ. (2005) 12:128–35. doi: 10.1038/sj.cdd.4401530
156. Maier G, Bing G, Wagner E, Unger C. Antitumor activity and induction of apoptosis by water-soluble derivatives of 7β-hydroxycholesterol in human colon carcinoma cell lines. Anticancer Res. (1999) 19:4251–56.
157. Mascia C, Maina M, Chiarpotto E, Leonarduzzi G, Poli G, Biasi F. Proinflammatory effect of cholesterol and its oxidation products on CaCo-2 human enterocyte-like cells: effective protection by epigallocatechin-3-gallate. Free Radic Biol Med. (2010) 49:2049–57. doi: 10.1016/j.freeradbiomed.2010.09.033
158. Chalubinski M, Zemanek K, Skowron W, Wojdan K, Gorzelak P, Broncel M. The effect of 7-ketocholesterol and 25-hydroxycholesterol on the integrity of the human aortic endothelial and intestinal epithelial barriers. Inflamm Res. (2013) 62:1015–23. doi: 10.1007/s00011-013-0660-x
159. Chalubinski M, Wojdan K, Gorzelak P, Borowiec M, Broncel M. The effect of oxidized cholesterol on barrier functions and IL-10 MRNA expression in human intestinal epithelium co-cultured with dendritic cells in the transwell system. Food Chem Toxicol. (2014) 69:289–93. doi: 10.1016/j.fct.2014.04.005
160. Lee WH, Lee CS, Kwon K, Kwon YS, Kim SW, Goo TW, et al. 7-Ketocholesterol induces endoplasmic reticulum stress in HT-29 cells. Z Naturforsch C. (2009) 64:307–10. doi: 10.1515/znc-2009-3-425
161. Alemany L, Laparra JM, Barberá R, Alegría A. Evaluation of the cytotoxic effect of 7keto-stigmasterol and 7keto-cholesterol in human intestinal (Caco-2) cells. Food Chem Toxicol. (2012) 50:3106–13. doi: 10.1016/j.fct.2012.06.036
162. Tanaka AR, Noguchi K, Fukazawa H, Igarashi Y, Arai H, Uehara Y. P38MAPK and Rho-dependent kinase are involved in anoikis induced by anicequol or 25-hydroxycholesterol in DLD-1 colon cancer cells. Biochem Biophys Res Commun. (2013) 430:1240–5. doi: 10.1016/j.bbrc.2012.12.067
163. Serra G, Incani A, Serreli G, Porru L, Melis MP, Tuberoso CIG, et al. Olive oil polyphenols reduce oxysterols -induced redox imbalance and pro-inflammatory response in intestinal cells. Redox Biol. (2018) 17:348–54. doi: 10.1016/j.redox.2018.05.006
164. Vedin LL, Gustafsson JA, Steffensen KR. The oxysterol receptors LXRα and LXRβ suppress proliferation in the colon. Mol Carcinogenesis. (2013) 52:835–44. doi: 10.1002/mc.21924
165. Swan R, Alnabulsi A, Cash B, Alnabulsi A, Murray GI. Characterisation of the oxysterol metabolising enzyme pathway in mismatch repair proficient and deficient colorectal cancer. Oncotarget. (2016) 7:46509–27. doi: 10.18632/oncotarget.10224
166. Abdul Aziz NA, Mokhtar NM, Harun R, Mollah MMH, Mohamed Rose I, Sagap I, et al. A 19-gene expression signature as a predictor of survival in colorectal cancer. BMC Med Genomics. (2016) 9:58. doi: 10.1186/s12920-016-0218-1
167. Yun SH, Park MG, Kim YM, Roh MS, Park JI. Expression of chicken ovalbumin upstream promoter-transcription factor II and liver X receptor as prognostic indicators for human colorectal cancer. Oncol Lett. (2017) 14:4011–20. doi: 10.3892/ol.2017.6659
168. Savic D, Ramaker RC, Roberts SB, Dean EC, Burwell TC, Meadows SK, et al. Distinct gene regulatory programs define the inhibitory effects of liver X receptors and PPARG on cancer cell proliferation. Genome Med. (2016) 8:74. doi: 10.1186/s13073-016-0328-6
169. Uno S, Endo K, Jeong Y, Kawana K, Miyachi H, Hashimoto Y, et al. Suppression of β-catenin signaling by liver X receptor ligands. Biochem Pharmacol. (2009) 77:186–95. doi: 10.1016/j.bcp.2008.10.007
170. De Gottardi ADE, Touri F, Maurer CA, Perez A, Maurhofer O, Ventre G, et al. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig Dis Sci. (2004) 49:982–9. doi: 10.1023/B:DDAS.0000034558.78747.98
171. Modica S, Murzilli S, Salvatore L, Schmidt DR, Moschetta A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res. (2008) 23:9589–95. doi: 10.1158/0008-5472.CAN-08-1791
172. Modica S, Gofflot F, Murzilli S, Orazio AD, Salvatore L, Pellegrini F, et al. The intestinal nuclear receptor signature with epithelial localization patterns and expression modulation in tumors. YGAST. (2010) 138:636–48.e12. doi: 10.1053/j.gastro.2009.09.060
173. Lax S, Schauer G, Prein K, Kapitan M, Silbert D, Berghold A, et al. Expression of the nuclear bile acid receptor / farnesoid x receptor is reduced in human colon carcinoma compared to nonneoplastic mucosa independent from site and may be associated with adverse prognosis. Int J Cancer. (2012) 2239:2232–9. doi: 10.1002/ijc.26293
174. Bailey AM, Zhan L, Maru D, Shureiqi I, Pickering CR, Kiriakova G, et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am J Physiol Gastrointest Liver Physiol. (2014) 306:G48–58. doi: 10.1152/ajpgi.00234.2013
175. Peng Z, Chen J, Drachenberg CB, Raufman JP, Xie G. Farnesoid X receptor represses matrix metalloproteinase 7 expression, revealing this regulatory axis as a promising therapeutic target in colon cancer. J Biol Chem. (2019) 294:8529–42. doi: 10.1074/jbc.RA118.004361
176. Maran RRM, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther. (2009) 328:469–77. doi: 10.1124/jpet.108.145409
177. Peng Z, Raufman JP, Xie G. Src-mediated cross-talk between farnesoid X and epidermal growth factor receptors inhibits human intestinal cell proliferation and tumorigenesis. PLoS ONE. (2012) 7:e48461. doi: 10.1371/journal.pone.0048461
178. Mazzoccoli G, Panza A, Valvano MR, Palumbo O, Carella M, Pazienza V, et al. Clock gene expression levels and relationship with clinical and pathological features in colorectal cancer patients. Chronobiol Int. (2011) 28:841–51. doi: 10.3109/07420528.2011.615182
179. Oshima T, Takenoshita S, Akaike M, Kunisaki C, Fujii S. Expression of circadian genes correlates with liver metastasis and outcomes in colorectal cancer. Oncol Rep. (2011) 25:1439–46. doi: 10.3892/or.2011.1207
180. Gu D, Li S, Ben S, Du M, Chu H, Zhang Z, et al. Circadian clock pathway genes associated with colorectal cancer risk and prognosis. Arch Toxicol. (2018) 92:2681–9. doi: 10.1007/s00204-018-2251-7
181. Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. (2008) 3:e2020. doi: 10.1371/journal.pone.0002020
182. Aune D, Chan DSM, Lau R, Vieira R, Greenwood DC, Kampman E, et al. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ. (2011) 343:d6617. doi: 10.1136/bmj.d6617
183. Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J Gastroenterol. (2014) 20:9872. doi: 10.3748/wjg.v20.i29.9872
184. Bai B, Yamamoto K, Sato H, Sugiura H, Tanaka T. Combined effect of 25-hydroxycholesterol and IL-1β on IL-8 production in human colon carcinoma cell line (Caco-2). Inflammation. (2005) 29:141–6. doi: 10.1007/s10753-006-9009-8
185. Biasi F, Guina T, Maina M, Nano M, Falcone A, Aroasio E, et al. Progressive increase of matrix metalloprotease-9 and interleukin-8 serum levels during carcinogenic process in human colorectal tract. PLoS ONE. (2012) 7:e41839. doi: 10.1371/journal.pone.0041839
186. Thorsen K, Schepeler T, Øster B, Rasmussen MH, Vang S, Wang K, et al. Tumor-specific usage of alternative transcription start sites in colorectal cancer identified by genome-wide exon array analysis. BMC Genomics. (2011) 12:505. doi: 10.1186/1471-2164-12-505
187. Gadaleta RM, Oldenburg B, Willemsen ECL, Spit M, Murzilli S, Salvatore L, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating nf- κ b signaling in the intestine. BBA Mol Basis Dis. (2011) 1812:851–8. doi: 10.1016/j.bbadis.2011.04.005
188. Selmin OI, Fang C, Lyon AM, Doetschman TC, Thompson PA, Martinez JD, et al. Inactivation of adenomatous polyposis coli reduces bile acid / farnesoid X receptor expression through fxr gene cpg methylation in mouse colon tumors and human colon cancer. J Nutr. (2016) 146:236–42. doi: 10.3945/jn.115.216580
189. Romagnolo DF, Donovan MG, Doetschman TC, Selmin OI. n-6 linoleic acid induces epigenetics alterations associated with colonic inflammation and cancer. Nutrients. (2019) 11:171. doi: 10.3390/nu11010171
190. Krattinger R, Boström A, Schiöth HB, Thasler WE, Mwinyi J, Kullak-ublick GA. MicroRNA-192 suppresses the expression of the farnesoid X receptor. Am J Physiol Gastrointest Liver Physiol. (2019) 310:1044–51. doi: 10.1152/ajpgi.00297.2015
191. Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F, et al. FXR regulates intestinal cancer stem cell proliferation. Cell. (2019) 176:1098–112.e18. doi: 10.1016/j.cell.2019.01.036
192. Li JN, Mahmoud MA, Han WF, Ripple M, Pizer ES. Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia. Exp Cell Res. (2000) 261:159–65. doi: 10.1006/excr.2000.5054
193. Kwan HY, Yang Z, Fong WF, Hu YM, Yu ZL, Hsiao WL. The anticancer effect of oridonin is mediated by fatty acid synthase suppression in human colorectal cancer cells. J Gastroenterol. (2013) 48:182–92. doi: 10.1007/s00535-012-0612-1
194. Erren TC, Morfeld P, Foster RG, Reiter RJ, Groß JV, Westermann IK. Sleep and cancer: synthesis of experimental data and meta-analyses of cancer incidence among some 1,500,000 study individuals in 13 countries. Chronobiol Int. (2016) 33:325–50. doi: 10.3109/07420528.2016.1149486
195. Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, et al. Night-shift work and risk of colorectal cancer in the nurses' health study. J Natl Cancer Inst. (2003) 95:825–8. doi: 10.1093/jnci/95.11.825
196. Filipski E, Li XM, Lévi F. Disruption of circadian coordination and malignant growth. Cancer Causes Control. (2006) 17:509–14. doi: 10.1007/s10552-005-9007-4
197. Mazzoccoli G, Colangelo T, Panza A, Rubino R, De Cata A, Tiberio C, et al. Deregulated expression of cryptochrome genes in human colorectal cancer. Mol Cancer. (2016) 15:1–20. doi: 10.1186/s12943-016-0492-8
198. Karantanos T, Theodoropoulos G, Gazouli M, Vaiopoulou A, Karantanou C, Lymberi M, et al. Expression of clock genes in patients with colorectal cancer. Int J Biol Mark. (2013) 28:280–5. doi: 10.5301/JBM.5000033
199. Yang X, Wood PA, Ansell CM, Ohmori M, Hrushesky WJM. B -catenin induces b -trcp-mediated per2 degradation altering circadian clock gene expression in intestinal mucosa of apc min / + mice. J Biol Chem. (2009) 145:289–97. doi: 10.1093/jb/mvn167
200. Momma T, Okayama H, Saitou M, Sugeno H, Yoshimoto N, Takebayashi Y, et al. Expression of circadian clock genes in human colorectal adenoma and carcinoma. Oncol Lett. (2017) 14:5319–25. doi: 10.3892/ol.2017.6876
201. Chen-Goodspeed M, Lee CC. Tumor suppression and circadian function. J Biol Rhythms. (2007) 22:291–8. doi: 10.1177/0748730407303387
202. Ishida N. Circadian clock, cancer and lipid metabolism. Neurosci Res. (2007) 57:483–90. doi: 10.1016/j.neures.2006.12.012
203. Fuhr L, El-Athman R, Scrima R, Cela O, Carbone A, Knoop H, et al. The circadian clock regulates metabolic phenotype rewiring via HKDC1 and modulates tumor progression and drug response in colorectal cancer. EBiomedicine. (2018) 33:105–21. doi: 10.1016/j.ebiom.2018.07.002
204. Zhong S, Zhang X, Chen L, Ma T, Tang J, Zhao J. Statin use and mortality in cancer patients: systematic review and meta-analysis of observational studies. Cancer Treat Rev. (2015) 41:554–67. doi: 10.1016/j.ctrv.2015.04.005
205. Jacobs EJ, Newton CC, Thun MJ, Gapstur SM. Long-term use of cholesterol-lowering drugs and cancer incidence in a large united states cohort. Cancer Res. (2011) 71:1763–71. doi: 10.1158/0008-5472.CAN-10-2953
206. Cardwell CR, Hicks BM, Hughes C, Murray LJ. Statin use after colorectal cancer diagnosis and survival: a population-based cohort study. J Clin Oncol. (2014) 32:3177–83. doi: 10.1200/JCO.2013.54.4569
207. Liu Y, Tang W, Wang J, Xie L, Li T, He Y, et al. Association between statin use and colorectal cancer risk: a meta-analysis of 42 studies. Cancer Causes Control. (2014) 25:237–49. doi: 10.1007/s10552-013-0326-6
208. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. (2013) 144:323–32. doi: 10.1053/j.gastro.2012.10.005
209. Shi M, Zheng H, Nie B, Gong W, Cui X. Statin use and risk of liver cancer: an update meta-analysis. BMJ Open. (2014) 4:e005399. doi: 10.1136/bmjopen-2014-005399
210. Tsan YT, Lee CH, Ho WC, Lin MH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J Clin Oncol. (2013) 31:1514–21. doi: 10.1200/JCO.2012.44.6831
211. Tsan YT, Lee CH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis b virus infection. J Clin Oncol. (2012) 30:623–30. doi: 10.1200/JCO.2011.36.0917
212. Hsiang JC, Wong GL, Tse YK, Wong VW, Yip TC, Chan HL. Statin and the risk of hepatocellular carcinoma and death in a hospital-based hepatitis B-infected population: a propensity score landmark analysis. J Hepatol. (2015) 63:1190–7. doi: 10.1016/j.jhep.2015.07.009
213. Chen CI, Kuan CF, Fang YA, Liu SH, Liu JC, Wu LL, et al. Cancer risk in HBV patients with statin and metformin use: a population-based cohort study. Medicine. (2015) 94:e462. doi: 10.1097/MD.0000000000000462
214. Janicko M, Drazilova S, Pella D, Fedacko J, Jarcuska P. Pleiotropic effects of statins in the diseases of the liver. World J Gastroenterol. (2016) 22:6201–13. doi: 10.3748/wjg.v22.i27.6201
215. Nishio T, Taura K, Nakamura N, Seo S, Yasuchika K, Kaido T, et al. Impact of statin use on the prognosis of patients with hepatocellular carcinoma undergoing liver resection: a subgroup analysis of patients without chronic hepatitis viral infection. Surgery. (2018) 163:264–9. doi: 10.1016/j.surg.2017.09.026
216. Kawaguchi Y, Sakamoto Y, Ito D, Ito K, Arita J, Akamatsu N, et al. Statin use is associated with a reduced risk of hepatocellular carcinoma recurrence after initial liver resection. BioSci Trends. (2017) 11:574–80. doi: 10.5582/bst.2017.01191
217. Archibugi L, Arcidiacono PG, Capurso G. Statin use is associated to a reduced risk of pancreatic cancer: a meta-analysis. Digest Liver Dis. (2018) 51:28–37. doi: 10.1016/j.dld.2018.09.007
218. Hamada T, Khalaf N, Yuan C, Babic A, Morales-Oyarvide V, Qian ZR, et al. Statin use and pancreatic cancer risk in two prospective cohort studies. J Gastroenterol. (2018) 53:959–66. doi: 10.1007/s00535-018-1430-x
219. Singh S, Singh PP, Roberts LR, Sanchez W. chemopreventive strategies in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2014) 11:45–54. doi: 10.1038/nrgastro.2013.143
220. Lipkin SM, Chao EC, Moreno V, Rozek LS, Rennert H, Pinchev M, et al. Genetic variation in 3-hydroxy-3-methylglutaryl coa reductase modifies the chemopreventive activity of statins for colorectal cancer. Cancer Prev Res. (2010) 3:597–603. doi: 10.1158/1940-6207.CAPR-10-0007
221. Amin S, Boffetta P, Lucas AL. The role of common pharmaceutical agents on the prevention and treatment of pancreatic cancer. Gut Liver. (2016) 10:665–71. doi: 10.5009/gnl15451
222. Kim YS, Lee YM, Oh TI, Shin DHG, Kim HS, Kan Y, et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism. Int J Mol Sci. (2018) 19:E3127. doi: 10.3390/ijms19103127
223. Morioka S, Sai K, Omori E, Ikeda Y, Matsumoto K, Ninomiya-Tsuji J. TAK1 Regulates hepatic lipid homeostasis through SREBP. Oncogene. (2016) 35:3829–38. doi: 10.1038/onc.2015.453
224. Zhang H, Feng Z, Huang R, Xia Z, Xiang G, Zhang J. MicroRNA-449 suppresses proliferation of hepatoma cell lines through blockade lipid metabolic pathway related to SIRT1. Int J Oncol. (2014) 45:2143–52. doi: 10.3892/ijo.2014.2596
225. Gabitova L, Gorin A, Astsaturov I. Molecular pathways: sterols and receptor signaling in cancer. Clin Cancer Res. (2014) 20:28–34. doi: 10.1158/1078-0432.CCR-13-0122
226. Chuu C-P. Modulation of liver X receptor signaling as a prevention and therapy for colon cancer. MedHypotheses. (2011) 76:697–9. doi: 10.1016/j.mehy.2011.01.037
227. Pattanayak SP, Bose P, Sunita P, Siddique MUM, Lapenna A. Bergapten inhibits liver carcinogenesis by modulating LXR/PI3K/Akt and IDOL/LDLR pathways. Biomed Pharmacother. (2018) 108:297–308. doi: 10.1016/j.biopha.2018.08.145
228. Sasso GLO, Bovenga F, Murzilli S, Salvatore L, Tullio GDI, Martelli N, et al. Liver X receptors inhibit proliferation of human colorectal cancer cells and growth of intestinal tumors in mice. YGAST. (2013) 144:1497–1507.e13. doi: 10.1053/j.gastro.2013.02.005
229. Alawad AS, Levy C. FXR agonists: from bench to bedside, a guide for clinicians. Digest Dis Sci. (2016) 61:3395–404. doi: 10.1007/s10620-016-4334-8
230. Kowdley KV, Luketic V, Chapman R, Hirschfield GM, Poupon R, Schramm C, et al. A randomized trial of obeticholic acid biliary cholangitis. Hepatology. (2018) 67:1890–902. doi: 10.1002/hep.29569
231. Ortiz R, Melguizo C, Prados J, Alvarez PJ, Caba O, Rodriguez-Serrano F, et al. New gene therapy strategies for cancer treatment: a review of recent patents. Recent Patents Anti Cancer Drug Discov. (2012) 7:297–312. doi: 10.2174/157489212801820093
232. Block KI, Block PB, Gyllenhaal C. Integrative treatment for colorectal cancer: a comprehensive approach. J Altern Complement Med. (2018) 24:890–901. doi: 10.1089/acm.2018.0125
233. Davidson AJ, Straume M, Block GD, Menaker M. Daily timed meals dissociate circadian rhythms in hepatoma and healthy host liver. Int J Cancer. (2006) 118:1623–7. doi: 10.1002/ijc.21591
234. Fekry B, Ribas-Latre A, Baumgartner C, Deans JR, Kwok C, Patel P, et al. Incompatibility of the circadian protein BMAL1 and HNF4α in hepatocellular carcinoma. Nat Commun. (2018) 9:4349. doi: 10.1038/s41467-018-06648-6
235. Guo L, Yu F, Zhang T, Wu B. The clock protein BMAL1 regulates circadian expression and activity of sulfotransferase 1A1 in mice. Drug Metab Disposit. (2018) 46:1403–10. doi: 10.1124/dmd.118.082503
236. Koyanagi S, Ohdo S. Alteration of intrinsic biological rhythms during interferon treatment and its possible mechanism. Mol Pharmacol. (2002) 62:1393–9. doi: 10.1124/mol.62.6.1393
237. Akira ODA, Katayose YU, Yabuuchi S, Yamamoto K, Mizuma M, Shirasou S, et al. Clock gene mouse period2 overexpression inhibits growth of human pancreatic cancer cells and has synergistic effect with cisplatin. Anticancer Res. (2009) 29:1201–10.
238. Pazienza V, Tavano F, Francavilla M, Fontana A, Pellegrini F, Benegiamo G, et al. Time-qualified patterns of variation of ppar γ, dnmt1, and dnmt3b expression in pancreatic cancer cell lines. PPAR Res. (2012) 2012:1–8. doi: 10.1155/2012/890875
239. Hashimoto K, Ethridge RT, Evers BM. Peroxisome proliferator-activated receptor γ ligand inhibits cell growth and invasion of human pancreatic cancer cells. Int J Gastrointest Cancer. (2002) 32:7–22. doi: 10.1385/IJGC:32:1:7
240. Innominato PF, Giacchetti S, Moreau T, Bjarnason GA, Smaaland R, Focan C, et al. Fatigue and weight loss predict survival on circadian chemotherapy for metastatic colorectal cancer. Cancer. (2013) 119:2564–73. doi: 10.1002/cncr.28072
241. Liao C, Li J, Bin Q, Cao Y, Gao F. Chronomodulated chemotherapy versus conventional chemotherapy for advanced colorectal cancer: a meta-analysis of five randomized controlled trials. Int J Colorectal Dis. (2010) 25:343–50. doi: 10.1007/s00384-009-0838-4
242. Bouchahda M, Adam R, Giacchetti S, Castaing D, Brezault-Bonnet C, Hauteville D, et al. Rescue chemotherapy using multidrug chronomodulated hepatic arterial infusion for patients with heavily pretreated metastatic colorectal cancer. Cancer. (2009) 115:4990–9. doi: 10.1002/cncr.24549
243. Zhang F, Sun H, Zhang S, Yang X, Zhang G, Su T. Overexpression of PER3 inhibits self-renewal capability and chemoresistance of colorectal cancer stem-like cells via inhibition of notch and β-catenin signaling. Oncol Res Featuring Preclin Clin Cancer Ther. (2017) 25:709–19. doi: 10.3727/096504016X14772331883976
244. Hasakova K, Vician M, Reis R, Zeman M, Herichova I. The expression of clock genes cry1 and cry2 in human colorectal cancer and tumor adjacent tissues correlates differently dependent on tumor location. Neoplasma. (2018) 65:986–92. doi: 10.4149/neo_2018_180122N47
245. Rossin D, Calfapietra S, Sottero B, Poli G, Biasi F. HNE and cholesterol oxidation products in colorectal inflammation and carcinogenesis. Free Radic Biol Med. (2017) 111:186–95. doi: 10.1016/j.freeradbiomed.2017.01.017
Keywords: oxysterols, circadian rhythm, hepatocellular carcinoma, pancreatic cancer, colorectal cancer, ROR, LXR, FXR
Citation: Kovač U, Skubic C, Bohinc L, Rozman D and Režen T (2019) Oxysterols and Gastrointestinal Cancers Around the Clock. Front. Endocrinol. 10:483. doi: 10.3389/fendo.2019.00483
Received: 08 January 2019; Accepted: 03 July 2019;
Published: 17 July 2019.
Edited by:
Vincenzo Pezzi, University of Calabria, ItalyReviewed by:
Carsten Carlberg, University of Eastern Finland, FinlandThomas Burris, Washington University in St. Louis, United States
Jean-Marc A. Lobaccaro, Université Clermont Auvergne, France
Copyright © 2019 Kovač, Skubic, Bohinc, Rozman and Režen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tadeja Režen, dGFkZWphLnJlemVuQG1mLnVuaS1sai5zaQ==
†These authors have contributed equally to this work