 Lan Xiao
Lan Xiao Yin Xiao
Yin Xiao- 1Key Laboratory of Oral Medicine, Guangzhou Institute of Oral Disease, Stomatology Hospital of Guangzhou Medical University, Guangzhou, China
- 2Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, QLD, Australia
- 3The Australia-China Centre for Tissue Engineering and Regenerative Medicine (ACCTERM), Queensland University of Technology, Brisbane, QLD, Australia
It has been long realized that the immune and skeletal systems are closely linked. This crosstalk, also known as osteoimmunology, is a primary process required for bone health. For example, the immune system acts as a key regulator in osteoclasts-osteoblasts coupling to maintain the balanced bone remodeling. Osteoimmunology is achieved through many cellular and molecular processes, among which autophagy has recently been found to play an indispensable role. Autophagy is a highly conserved process in eukaryotic cells, by which the cytoplasm components such as dysfunctional organelles are degraded through lysosomes and then returned to the cytosol for reuse. Autophagy is present in all cells at basal levels to maintain homeostasis and to promote cell survival in response to cellular stress conditions such as nutrition deprivation and hypoxia. Autophagy is a required process in immune cell activation/polarization and osteoclast differentiation, which protecting cells from oxidative stress. The essential of autophagy in osteogenesis is its involvement in osteoblast differentiation and mineralization, especially the role of autophagosome in extracellular calcium transportation. The modulatory feature of autophagy in both immune and skeleton systems suggests its crucial roles in osteoimmunology. Furthermore, autophagy also participates in the maintenance of bone marrow hematopoietic stem cell niche. The focus of this review is to highlight the role of autophagy in the immune-skeleton interactions and the effects on bone physiology, as well as the future application in translational research.
Introduction
The skeletal bone is a dynamic tissue with a life-long continuous renovation termed bone remodeling (1). This remodeling consists of bone resorption and formation and plays a fundamental role in the maintenance of bone homeostasis (2). Bone remodeling is kept in balance under physiological conditions, as the amount of bone resorption equals to that of formation (3). This balance is achieved via sophisticated regulations originated from the immune system (4). The link between the immune and skeleton systems has been identified for almost fifty years and termed as “osteoimmunology” (5). Further research into osteoimmunology has recognized the complex mutual regulations between immune cells and bone cells, that at one level, immune response determines the balance of bone remodeling, whereas on another level, bone cells mediate the polarization and function of immune cells (3, 4, 6). This interaction consists of multiple factors such as cytokines, receptors, signaling pathways (4); and it has been recently indicated that autophagy plays elementary roles in both immune (7) and skeletal (8) systems. Autophagy is defined as the delivery of cytoplasmic materials to the lysosome in animal cells or the vacuole in plant and yeast cells (9). Especially in eukaryotic cells, autophagy plays a key role in homeostasis maintenance (10, 11). Moreover, autophagy promotes cell survival in response to stress conditions such as nutrition deprivation and hypoxia (12). Autophagy is required in the differentiation of osteoclast and osteoblast (13, 14); meanwhile, it participates in the immune cell polarization/function and therefore regulates immune response (7, 15), suggesting a complex and rather intriguing role in osteoimmunology. This review highlights the effects of autophagy in the immune-skeleton interactions and proposes the regulation of autophagy for future application in bone regeneration.
Autophagy Mechanism
There are mainly three types of autophagy, known as macroautophagy, microautophagy, and chaperone-mediated autophagy (9). The current review will focus on macroautophagy (hereafter referred to as autophagy), a highly conserved “self-eating” lysosomal degradation pathway in eukaryotic cells to clear intracellular waste (10, 11). Autophagy initiates with the sequestration of cytoplasmic organelles within double-membrane vesicles known as autophagosomes, which then fuse with lysosomes to form autolysosomes to degrade or recycle the autoghagic contents, such as damaged organelles, intracellular pathogens, glycogens, lipids, and nucleotides proteins (9, 16). Concomitantly, the cytosolic form of microtubule-associated protein 1A/1B-light chain 3 (LC3-I) is converted to form LC3-phosphatidylethanolamine conjugate (LC3-II), which is attached to the autophagosome membrane and then degraded (17, 18). This conversion from LC3-I to LC3-II is identified as one of the autophagy hallmarks. On the other hand, microautophagy is defined as the direct engulfment of small cytoplasmic portions by inward deformation of the lysosomal or late endosomal membrane (9, 19). Chaperone-mediated autophagy does not require membrane reorganization: the substrate proteins containing a KFERQ-like pentapeptide are selectively targeted by cytosolic heat shock cognate 70 (Hsc70) protein, then directly translocated into the lysosomal lumen (9, 20).
Autophagy is maintained at basal levels in all cell types, which plays a “quality control” role to maintain cellular homeostasis (11). On the other hand, autophagy is induced in response to stress conditions such as nutrient deprivation, oxidative stress, hypoxia and infection, which is one of the main strategies to promote cell survival (12, 21). Autophagy facilitates the “recycle” of cellular components and therefore provides energy for cells under starvation (22). Another important function of autophagy is to scavenge malfunctioning/damaged proteins and organelles (22). For example, autophagy-mediated clearance of damaged mitochondria, also termed as “mitophagy,” inhibits reactive oxygen species (ROS) accumulation and thereby protecting cell from oxidative stress and apoptosis (15, 23). This has been identified as a required process during bone cell differentiation and immune cell polarization, making a central role in osteoimmunology.
Autophagy in the Skeletal System
General Bone Biology
As the main constituent of the vertebrate skeletal system, bone serves as supporters and protectors of organs in the body (24). Bone consists of cellular components such as osteoclast, osteoblast, and osteocyte, as well as collagen, osteoid and inorganic mineral deposits (25). Osteoclast and osteoblast are the major players in bone remodeling (26). Originated from the hematopoietic stem cells (HSCs), osteoclast is considered as the major cell type responsible for bone resorption (25). The macrophage–monocyte lineage-derived osteoclast precursors fuse with each other to form a giant, multinucleated cell—the osteoclast (27). On the other hand, the mesenchymal stem cells (MSCs)-derived osteoblast is the major bone formation cell (25), which builds the osteoid matrix and eventually differentiate into osteocyte, the most abundant cell type in bone (28). The fact that osteoclastogenesis is dependent on osteoblast-derived receptor activator of nuclear factor factor-kappa B ligand (RANKL) (29, 30), suggests “coupling” between osteoclast and osteoblast, therefore linking osteoclastogenesis to osteogenesis (31). RANKL binds with receptor activator of nuclear factor factor-kappa B (RANK) on osteoclast-precursors, therefore initiating the differentiation of osteoclast. On the other hand, osteoblast produces osteoprotegerin (OPG), a decoy receptor of RANKL, to interrupt osteoclastogenesis (32). Hence, the balance between RANKL and OPG determines the outcome of bone remodeling (33). Furthermore, osteoblast secrets other factors to regulate osteoclastogenesis and osteogenesis in a paracrine or endocrine manner (34). For example, osteoblast is one of the major source of macrophage colony-stimulating factor (M-CSF) —a major factor for osteoclast differentiation—in the bone microenvironment (31, 35–37). Osteoblast-derived semaphorin 3A (Sema3A) and Wnt16 have been found to reduce osteoclastogenesis via interrupting the RANKL-RANK signaling (38, 39), while osteoblast-originated Wnt5 induces osteoclast differentiation by enhancing RANK expression in osteoclast-precursors (40, 41). Osteoblast also produces factors such as vascular endothelial growth factor A (VEGF-A) to induce osteogenesis (34, 42). Beside osteoblast, osteocyte is considered as another critical producer of RANKL, which also produces sclerostin (SOST) to reduce osteogenesis, therefore acting as the orchestrator of bone remodeling (43–46).
Autophagy in the Differentiation/Function of Osteoclast, Osteoblast, and Osteocyte
Recent studies have identified the importance of autophagy in osteoclast differentiation and function. Autophagy activation has been reported during the osteoclastogenic process. During the RANKL-induced osteoclast differentiation, the autophagic protein levels (such as autophagy related (ATG) 5/7/12) and the LC3-II/LC3-I ratio have been reported to increase in accompany with degradation of p62 (also known as SQSTM1/sequestome1) (47). This degradation plays an essential role in the generation of filamentous actin (F-actin) ring, a key feature of osteoclatogenesis (47, 48). Mutant p62 results in abnormal osteoclasts with increased size, number, multinuclearity, and activity (49). The autophagic proteins Atg5/7/4B and LC3 have also been reported to play decisive roles in regulating the osteoclast-ruffled border (RB) generation and the lysosomal secretion (Figure 1), thereby determining osteoclast function in vitro and in vivo (50). Especially, in rheumatoid arthritis (RA) patients, autophagy is found activated by the pro-inflammatory cytokine tumor necrosis factor α (TNF- α) in osteoclasts, which results in induced osteoclastogenesis and bone resorption in vitro and in vivo, suggesting a central role of autophagy in the pathogenesis of inflammatory bone loss (51).
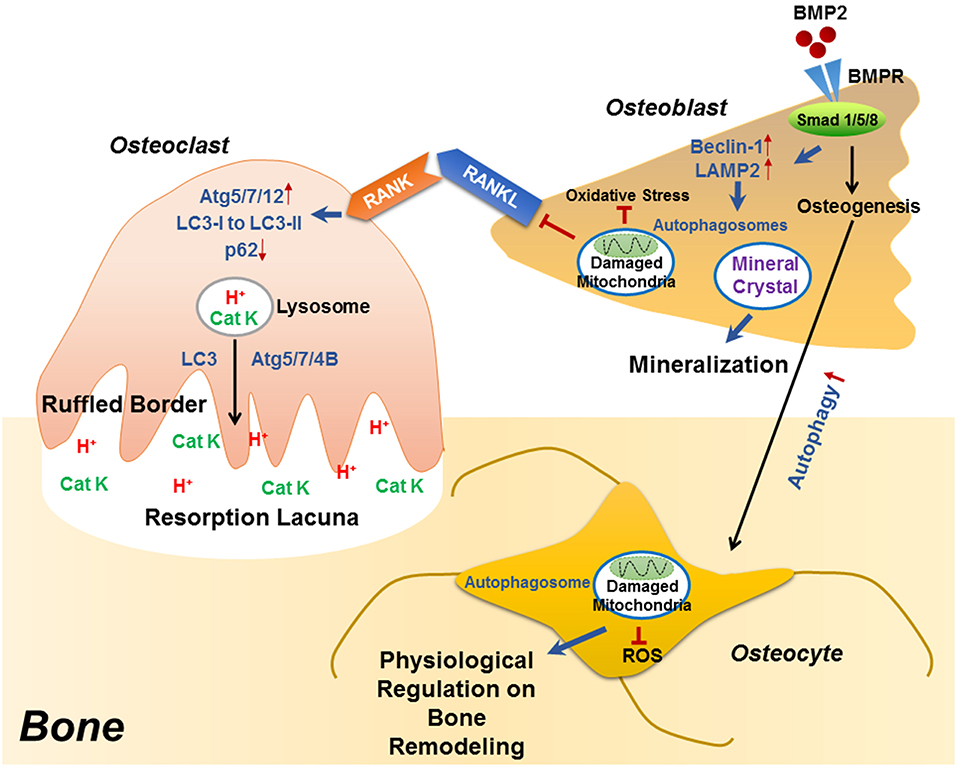
Figure 1. The role of autophagy in the differentiation/function of osteoclast, osteoblast and osteocyte. During RANKL-RANK induced osteoclast differentiation, the protein levels of ATG5/7/12 are increased, accompanied with enhanced conversion from LC3-I to LC3-II and p62 degradation, which plays an essential role in the generation of F-actin ring. Besides differentiation, autophagy also plays decisive roles in osteoclast function, that the physiological levels of Atg5/7/4B are required for lysosomal [containing H+ and cathepsin K (CatK)] trafficking and fusion with the plasma membrane to generate mature ruffled border, as well as to release H+ and cathepsin K to resorb bone. During osteoblast differentiation, the binding of BMP2 to its receptors (BMPR) activates Smad signaling pathway to initiate osteogenesis, which also induced the expression of beclin 1 and LAMP2 (autophagy-related proteins) as well as autophagy pathway. Autophagosomes are utilized for transporting mineral crystals to extracellular matrix and thereby facilitating mineralization. Autophagy reduces oxidative stress during osteoblast differentiation via clearance of damaged mitochondria, which also suppresses RANKL production and hence inhibiting osteoclastogenesis. Compared with osteoblast, the autophagy level is increased in osteocyte, which not only maintains homeostasis of osteocyte, but also guarantees a physiological osteocyte-derived regulation on bone remodeling.
Autophagy has long been considered as a necessary part in cell differentiation (22). In a recent study, induced autophagy has been found during osteoblast differentiation and mineralization in vitro; autophagosomes act as cargos to transport the intracellular mineral crystal-like structures to facilitate the extracellular mineralization (52). Autophagy inhibition can result in impaired mineralization in vitro and decreased bone mass/volume in vivo (Table 1), which is usually followed by induced oxidative stress and RANKL production (52). These results suggest the fundamental roles of autophagy during osteoblast differentiation and mineralization, which serves as mineralization vehicles, protects osteoblast from increased oxidative stress and moreover, reduces osteoblast-derived RANKL production and thereby inhibits osteoclastogenesis during bone formation (52). This is in accordance with the phenomenon that autophagy deficiency/inhibition in osteoblast leads to an osteoporotic-like phenotype with induced osteoclast differentiation (62). Another study found out that bone morphogenetic protein 2 (BMP2), an osteoinductive agent for clinical-use, leaded to increased protein levels of beclin-1 and lysosome-associated membrane protein 2 (LAMP2) (63), both of which are known as autophagy markers (64, 65). On the other hand, autophagy inhibition has been found to affect the differentiation and immunoregulatory capacities of MSCs in ovariectomy-induced osteoporosis model mice (66). In the field of bone tissue-engineering and biomaterials development, the autophagy-inductive bioactive silica nanoparticles have been found to promote osteogenesis (67), suggesting that autophagy could be a potential therapeutic target for bone repair (Figure 1).

Table 1. Effects of Atg gene-knockout on osteoimmunology in vivo.
Autophagy is particularly critical for terminally-differentiated cells such as neurons and osteocytes, which provides these cells with “intracellular refreshment” to enable the cellular homeostasis and function during their long-life periods (13, 22). Osteocyte is terminally-differentiated osteoblast embedded in bone matrix. The long dendritic processes of osteocyte facilitate the communication within osteocytes as well as the connection with bone surface, making osteocyte capable of sensing mechanical or bio-chemical stimulus from the microenvironment (14). Osteocyte in response to the stimulation therefore acts as director in bone remodeling such as producing RANKL and sclerostin (43, 44, 46, 68). Living in a hypoxic and potentially nutrient poor environment, osteocyte has been reported to keep higher levels of autophagy than the bone surface osteoblast in vivo (13, 69). Accordingly, it is found that the terminally-differentiated osteocytes show induced autophagy, as compared with the pre-osteocytes in vitro (69). Genetic autophagy suppression (selective Atg7-deletion) in murine osteocytes results in skeletal changes in young adult mice (Table 1), such as decreased bone mass and volume, reduced osteoclastogenesis and impaired bone formation, as well as induced ROS in the bone marrow; a phenotype similar to the aging bone (70). Another study has also found that autophagy-activation/inhibition is involved in glucocorticoids-related bone loss (71). All these studies suggest that autophagy at a certain level maintains the homeostasis and function osteocyte to facilitate the physiological balance of bone remodeling (Figure 1).
Autophagy in Osteoimmunology
Regulations of the Immune System on Bone Remodeling
The crosstalk between skeletal and immune systems was initially identified by the finding that immune cells-originated interleukin-1 (IL-1) could induce osteoclastogenesis (5). Since then, more evidence has revealed the regulatory role of immune system on bone remodeling (4). T-helper cells, derived from the adaptive immune system, have been found to play a critical role in inflammatory bone loss in arthritis via producing RANKL (72). Besides RANKL, the type 17 helper T (Th17) cell-originated IL-17 has long been identified as a key pre-inflammatory cytokine that promotes osteoclastogenesis (73, 74); while the Th2 cell-originated IL-4 and IL-10 are considered as inhibitors for osteoclasts (75–78). On the other hand, the immune-suppressive regulatory T (Treg) cells (79), inhibit osteoclastogenesis either in a direct cell-to-cell contact-dependent manner (cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) on Treg cells binding with CD80 and CD86 on osteoclast precursors), or via production of IL-4, IL-10, and TGF-β (80, 81). Cells from the innate immune system, such as macrophages, not only serve as osteoclast precursors (82), but also participate in the osteoclastogenesis regulation. Macrophages are a population of cells with three subsets: (1) non-activated M0 macrophage; (2) pro-inflammatory M1 macrophage, which is classically activated by microbe-derived lipopolysaccharide (LPS) or Th1 cells-derived IFNγ; and (3) anti-inflammatory M2 macrophage, which is alternatively activated by Th2 cells-derived IL-4 or IL-13 (83–86). M1 macrophage induces osteoclastogenesis by producing cytokines such as IL-1α/β (87, 88), IL-6 (89–91), TNF-α (92–95); while M2 macrophage reduces osteoclast differentiation via secretion of IL-10 and TGF-β (85, 96, 97).
The regulation of immune system on osteogenesis is not so clear-cut. There are conflicting results regarding the positive/negative effects of inflammatory/anti-inflammatory cytokines on osteoblast differentiation, known as IL-1 (98–101), IL-17 (102–104), TNF-α (100, 101, 105, 106) and IFNγ (107, 108). An interesting finding is that the pro-inflammatory cytokine IL-6 could induce osteogenesis through the oncostatin M (OSM)-STAT3 signaling pathway, suggests the inflammatory response, at a certain level, could initiate osteoblast differentiation (109–115). This is in accordance with the phenomenon that early stage inflammation with macrophage infiltration is regarded as indispensable in bone fracture healing (116). However, this inflammation will be gradually quenched, as the M1 to M2 macrophages conversion happens along with bone repair. This conversion has been found to improve bone formation (116, 117). The M2 macrophage-derived factors, such as BMP2 and TGF-β, are identified to promote osteoblast differentiation and functions, as well as to enhance mineralization (118, 119). These findings suggest that the transformation from the M1 macrophage-mediated inflammatory microenvironment, to the M2 macrophage-mediated regenerative one, should be a required part in bone formation; modulation of M2 polarization should be considered as a potential therapeutic approach for bone regeneration.
The Role of Autophagy-Mediated Immunomodulation in Bone Remodeling
Autophagy is now identified as a multifunctional pathway in immunity such as lymphocyte differentiation (22), pathogen elimination (120), antigen presentation and inflammation regulation (7, 15). T cell-specific ATG genes deletion (such as Atg7 or Atg5) results in decreased T lymphocyte counts, mitochondria accumulation, and induced apoptosis in mature T cells (121, 122). This is due to the critical role of autophagy-mediated mitochondria clearance in the development of thymocytes into circulating mature T cells (123). Beside differentiation, autophagy also provides barriers against invading pathogens, that autophagosome and autophagolysosome are utilized for selective-detection and elimination of intracellular pathogens (120, 124–127). Especially, the immunomodulatory roles of autophagy have been found in both innate and adaptive immune responses, making autophagy a potential key regulator in osteoimmunology.
The importance of autophagy has long been addressed in macrophage polarization and inflammatory response. Although autophagy is induced by toll-like receptor 4 (TLR4) signaling during M1 macrophage polarization (128), further research has suggested that autophagy plays an immunosuppressive role in macrophage inflammatory response (129). Atg5- or Atg16L1-deficiency on macrophage is found to direct M2 macrophage to polarize toward a M1-like phenotype with induced secretion of pro-inflammatory cytokines (55, 58). Mice with Atg5-knockout macrophages showed induced systemic inflammation (56). Primary bone marrow-derived macrophages (BMDMs) obtained from this mice type exhibited abnormal polarization, that the M1 polarization was increased while the M2 polarization was impaired (Table 1), which further indicating that autophagy-deficiency would induce inflammatory response in macrophages (56). In mice with Atg 7 gene deletion in the hematopoietic system (vav-Atg7-/- mice), monocytes failed to differentiated into macrophages under M-CSF stimulation (130). Moreover, macrophages obtained from vav-Atg7–/– mice were found to have a phenotype similar to aged macrophages, which showed reduced abilities of phagocytosis and nitrite burst, while induced inflammatory response (Table 1); suggesting that autophagy maintained at a certain level would correct the abnormities in immune system to prevent aging associated chronical inflammation (57). It has been demonstrated that autophagy inhibition (either by Atg gene deletion or pharmacological intervention) results in induced IL-1β secretion of macrophage, suggesting autophagy limits the inflammatory response of macrophage (15, 131, 132). Further studies have found that inflammatory stimulus causes mitochondrial damage, and then consequently results in induced ROS release/apoptosis in macrophage. ROS interact with NF-κB signaling pathway and then activate the NLRP3 inflammasome to trigger the secretion of IL-1β and IL-18, therefore eventually initiating the inflammatory cascade (132–134). During this process, autophagy scavenges the damaged mitochondria through a collaboration between p62 and LC3, that p62 selectively recognizes damaged mitochondria by its UBA domain, which, collectively, combines with LC3 and ensures the lysosomal degradation of damaged mitochondria, thereby interrupts the inflammatory cascade (134–136). As mentioned before, the macrophage inflammation has been demonstrated to induce osteoclastogenesis and bone loss, while the conversion from pro-inflammatory M1 toward anti-inflammatory M2 phenotypes has been suggested to improve bone repair (116, 117). Therefore, this autophagy-mediated regulation on macrophage response should be considered as beneficial for bone regeneration. The nanomaterials-derived autophagy induction has been found to potentially introduce a polarization toward M2 macrophage and thereby improve osteogenesis (137), which further suggests that autophagy could be a potential immunomodulation target in regenerative medicine, especially for therapies against disorders with inflammatory bone loss, such as arthritis (138), periodontitis (139), periapical lesions (140).
Besides its role in the innate immunity, autophagy also acts as a key regulator in the adaptive immune response, such as T cell activation and polarization. Autophagy promotes major histocompatibility complex (MHC) class II-mediated antigen presentation via inducing the fusion of antigens to LC3 in CD4+ T cells (141), which facilitates the elimination of autoreactive CD4+ T cells (123). On the other hand, although autophagy-dependent antigen presentation is required in in antimicrobial response of dendritic cells (DCs) (141, 142), the autophagy-deficient DCs show hyper-stable interactions with T cells and thereby enhance T cell activation, suggesting the modulatory role of autophagy to prevent excessive T cell response (143). Consistent with this idea, a study has found that graphene quantum dots (GQDs) induce the tolerogenic phenotype of DCs in an autophagy-dependent manner, which show reduced capacity in antigen-presenting and thereby reduce T cell inflammatory response via introducing the polarization of Th1 and Th17 cells toward Th2 and Treg cells, respectively (144). Autophagy also directly inhibits nuclear factor-κB (NF-κB) activation in in antigen-activated T cells and thereby suppresses inflammation (145). On the other hand, autophagy prevents the secretion of macrophage-derived IL-1β (132–134), a cytokine known as promoting Th17 cell response via collaboration with IL-6 and TGF-β (59). In mice with Atg5-deficient myeloid cells, CD4+ Th17 cell response is induced (59), further suggesting the role of autophagy in preventing inflammation (Table 1). Although inflammatory cytokines such as IL-17 has been reported to induce osteogenesis, excessive IL-17 production results in enhanced RANKL secretion and osteoclastogenesis (73, 74) and therefore is still considered as detrimental for bone regeneration. Especially, autophagy-mediated conversion from Th1 to Th2 cells would in turn induce the polarization from M1 to M2 macrophages (146, 147), a central part in bone regeneration (116, 117). Hence, it could be presumed that autophagy-derived immunomodulation on T cells creates a microenvironment favoring bone repair.
The role of autophagy in osteoimmunology has been further demonstrated in the pathogenesis of autoimmune rheumatic diseases, such as rheumatoid arthritis (RA), a disease with abnormities in organs including joints, heart, vascular system, lungs, and skin (148, 149). Chronical inflammation, as well as bone and cartilage destruction are typical syndromes in RA (150), which mainly due to the interactions within local cells, known as immune cells (such as T and B cells, macrophages), synovial fibroblasts, chondrocytes, as well as osteoclasts and osteoblasts. As autophagy plays decisive roles in osteoclastogenesis, the inhibition of autophagy successfully reduces bone destruction and osteoclast formation in experimental arthritis mouse models (151), suggesting drugs with autophagy inhibition could be used to prevent bone loss in RA patients (148). In addition, autophagy protects cells from apoptosis—a crucial mechanism to extinguish excessive inflammation (132–134, 148), therefore playing essential roles in the pathogenesis and progression of RA, via regulating the balance between immune cell survival and death (148). Induced autophagy/reduced apoptosis have been observed in synovial fibroblasts and synovial tissues obtained from RA patients (152–154), while autophagy inhibition has been found to reduce synovial inflammation in a collagen induced arthritis (CIA) rat model (148, 155). Autophagy hyper-activation has been found in CD4+ T cells obtained from CIA mouse, and autophagy is considered to regulate T and B lymphocytes homeostasis to maintain the RA chronical inflammatory response (148, 156). Besides, autophagy dysregulation in endothelial cells is considered as responsible for atherosclerosis in RA (157). Especially, autophagy participates in the pathogenesis of RA via inducing the generation of citrullinated peptides, which consequently interrupting immune tolerance (150). The anti-cyclic citrullinated peptide (anti-CCP), an autoantibody (against citrullinated peptides) produced by immune cells upon activation of self-antigens, is a general marker for clinical RA diagnosis (150, 158, 159). The RA patient-derived anti-CCP antibody (Ab) has been found to induce osteoclastogenesis and bone loss (160). Autophagy is recently considered to participate in both the presentation of citrullinated peptides and the generation of anti-CCP Ab (148). Autophagy is required for the antigen presenting cells (APCs) to perform the presentation of citrullinated proteins (161). Furthermore, autophagy is involved in the citrullination processes of Normal Human Bronchial Epithelial (NHBE) cells and human synovial fibroblasts (150, 162). Increased citrullinated peptide production has been observed following autophagy-induction in human synovial fibroblasts from RA patients, and the autophagy level is significantly associated with that of anti-CCP Ab in early-stage RA patients (150), suggesting the fundamental role of autophagy in RA establishment via inducing the generation of citrullinated peptides (150, 163). All these studies indicate that autophagy-derived modulation on osteoimmunology plays a central part not only in physiological bone homeostasis but also in pathological bone diseases, which needs further study in the future.
Future Remarks & Conclusion
Many questions still remain un-resolved regarding the role of autophagy in osteoimmunology. For example, although autophagy is indispensable in osteoclastogenesis, rapamycin (also named as sirolimus), an autophagy inducer via inhibition of the Ser/Thr protein kinase mTOR (mammalian target of rapamycin) (164, 165), has been found to reduce osteoclastogenesis and bone resorption in a mouse model of arthritis, an effect similar to anti-TNF (by Infliximab) treatment (166). It is also found that rapamycin reduces osteoclastogenesis in young rats (167) and post-transplant bone resorption in renal transplant patients (168). This is quite contrary to the positive effect of autophagy in osteoclast differentiation and function as mentioned before. It is presumed that autophagy plays a more maintenance than regulatory role in the differentiation of osteoclast, which is induced and kept in a certain level in response to energy/metabolism variations or intracellular accumulation of damaged organelles such as mitochondria. Whereas, in immune response, autophagy might act more as a regulator to quench the inflammation fire (7), which in turn reduce bone resorption (Figure 2). The rapamycin-mediated inhibition of bone loss might be achieved via immunomodulation, suggests that autophagy is an attractive target for osteoimmunology regulation to improve bone tissue regeneration (Figure 2). Autophagy might also participate in the mutual regulations between immune-skeletal systems. Previous studies have suggested that the immunosuppressive role of MSCs is achieved through the programmed death 1/ programmed death-ligand 1 (PD-1/PDL1) (169), an autophagy-related signaling pathway (170). Another study has found that the autophagy regulator p62 plays a central role in maintenance of the “macrophage-osteoblast niche,” which is indispensable for the retention of HSCs in bone marrow (171). It is also found that compared with the undifferentiated MSCs, the osteogenically differentiated MSCs not only induce the recruitment of macrophages, but also regulate local macrophage response in a VEGFA–C-X-C motif chemokine 12/C-X-C chemokine receptor type 4 (VEGFA-CXCL12/CXCR4) axis dependent manner (172). As CXCR4 signaling has been reported to regulate autophagy via the cross-talk with mTOR (173–175), hence, autophagy should be involved in MSC-mediated regulation on immune cells. Furthermore, It could be predicted that immune cells, especially macrophage, regulate bone remodeling in an autophagy-dependent manner, as both the M1 and M2 macrophage-derived cytokines have been found to modulate autophagy, such as IL-1 (176), TNF-α (177–179), IL-10 (180–182), TGF-β (183, 184); the fundamental role of M1-M2 conversion in osteogenesis might be partially due to different autophagy levels and metabolism states during the differentiation from osteoblast to osteocyte.
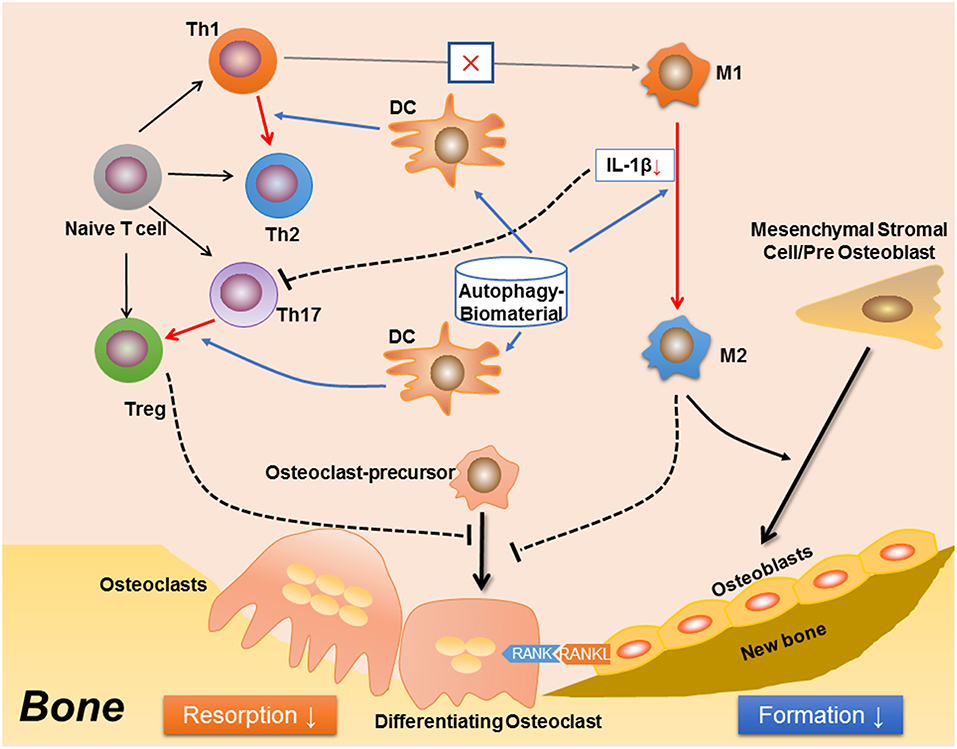
Figure 2. A proposed effect in osteoimmunology regarding biomaterial with the property of inducing autophagy (autophagy-biomaterial). The autophagy-biomaterial should induce bone repair by suppressing the inflammatory response. On one hand, autophagy-induction directly reduces macrophage inflammation and IL-1β secretion. The decreased IL-1β also impedes the polarization and function of Th17 cell (Th17). On the other hand, autophagy-induction in dendritic cell (DC) interrupts its interaction with T cells, thereby inhibiting T cell inflammatory response via introducing the polarization from Th1 toward Th2 cells, as well as that from Th17 to Treg cells. The Th1 to Th2 conversion would in turn reduce M1 polarization while induce M2 polarization. Hence, the autophagy-biomaterial creates an immune microenvironment favoring bone regeneration: the limited inflammatory responses of T cell and M1 macrophage reduce osteoclastogenesis, while the conversion of M1 to M2 macrophages improves osteogenesis.
In summary, autophagy, a conserved “self-eating” pathway present in all mammalian cells, plays a complex role in osteoimmunology, that at one level, autophagy maintains the cellular homeostasis during differentiation of osteoclast and osteoblast, facilitates the survival and function of osteocyte, and more importantly, direct the immune response to avoid the damage of excessive inflammation. Therefore, the definition of autophagy as “self-eating” should be more appropriate as “self-editing” in maintaining bone homeostasis. This regulatory role in osteoimmunology suggests autophagy could be a novel therapeutic target (e.g., autophagy-inductive biomaterial) to improve bone regeneration in the future translational medicine (Figure 2).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. LX: involved in the concept and design of the article and wrote the manuscript. YX: involved in the conception and design of the article and reviewed the manuscript.
Funding
Funding for this study was provided by the National Natural Science Foundation of China (NSFC, Grant No. 31771025).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. (2010) 285:25103–8. doi: 10.1074/jbc.R109.041087
2. Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci. (2006) 1092:385–96. doi: 10.1196/annals.1365.035
3. Arron JR, Choi Y. Osteoimmunology: bone versus immune system. Nature. (2000) 408:535–6. doi: 10.1038/35046196
4. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. (2007) 7:292. doi: 10.1038/nri2062
5. Dewhirst FE, Stashenko PP, Mole JE, Tsurumachi T. Purification and partial sequence of human osteoclast-activating factor: identity with interleukin 1 beta. J Immunol. (1985) 135:2562–8.
6. Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. (2007) 110:3499–06. doi: 10.1182/blood-2007-02-069716
7. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. (2013) 13:722–37. doi: 10.1038/nri3532
8. Srinivas V, Bohensky J, Zahm AM, Shapiro IM. Autophagy in mineralizing tissues microenvironmental perspectives. Cell Cycle. (2009) 8:391–93. doi: 10.4161/cc.8.3.7545
9. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
10. Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. (2010) 2:a006734. doi: 10.1101/cshperspect.a006734
11. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. (2010) 29:969–80. doi: 10.1038/emboj.2009.405
12. Mortimore GE, Pösö AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. (1987) 7:539–68. doi: 10.1146/annurev.nu.07.070187.002543
13. Hocking LJ, Whitehouse C, Helfrich MH. Autophagy: a new player in skeletal maintenance? J Bone Miner Res. (2012) 27:1439–47. doi: 10.1002/jbmr.1668
14. Shapiro IM, Layfield R, Lotz M, Settembre C, Whitehouse C. Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy. (2014) 10:7–19. doi: 10.4161/auto.26679
15. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. (2011) 469:323. doi: 10.1038/nature09782
16. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Cur Opin Cell Biol. (2010) 22:124–31. doi: 10.1016/j.ceb.2009.11.014
17. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. (2000) 19:5720–28. doi: 10.1093/emboj/19.21.5720
18. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. (2004) 117:2805–12. doi: 10.1242/jcs.01131
19. Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. (2011) 7:673–82. doi: 10.4161/auto.7.7.14733
20. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. (2014) 24:92. doi: 10.1038/cr.2013.153
21. Shen, H-M. Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. (2011) 7:457–65. doi: 10.4161/auto.7.5.14226
22. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. (2010) 12:823. doi: 10.1038/ncb0910-823
23. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. (2015) 524:309. doi: 10.1038/nature14893
24. Steele DG, Bramblett CA. The Anatomy and Biology of the Human Skeleton. College Station: A&M University Press (1988).
25. Kini U, Nandeesh B. Physiology of bone formation, remodeling, and metabolism. In: Radionuclide and Hybrid Bone Imaging. Berlin; Heidelberg: Springer (2012). p. 29–57. doi: 10.1007/978-3-642-02400-9_2
26. Hernández-Gil I, Gracia MAA, Pingarrón M, Jerez L. Physiological bases of bone regeneration II. The remodeling process. Med Oral Patol Oral Cir Bucal. (2006) 11:E151–215.
27. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. (2003) 423:337–42. doi: 10.1038/nature01658
28. Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn. (2006) 235:176–90. doi: 10.1002/dvdy.20603
29. Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Ann Rev Immunol. (2002) 20:795–23. doi: 10.1146/annurev.immunol.20.100301.064753
30. Kong Y-Y, Yoshida H, Sarosi I, Tan H-L, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. doi: 10.1038/16852
31. Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci USA. (1990) 87:7260–4. doi: 10.1073/pnas.87.18.7260
32. Simonet W, Lacey D, Dunstan C, Kelley M, Chang M-S, Lüthy R, et al, Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. (1997) 89:309–19. doi: 10.1016/S0092-8674(00)80209-3
33. Fonseca J, Cortez-Dias N, Francisco A, Sobral M, Canhao H, Resende C, et al. Inflammatory cell infiltrate and RANKL/OPG expression in rheumatoid synovium: comparison with other inflammatory arthropathies and correlation with outcome. Clin Exp Rheumatol. (2004) 23:185–92. Available online at: https://www.clinexprheumatol.org/article.asp?a=2565
34. Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and endocrine actions of bone—the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. (2018) 6:16. doi: 10.1038/s41413-018-0019-6
35. Teitelbaum SL. Bone resorption by osteoclasts. Science. (2000) 289:1504–08. doi: 10.1126/science.289.5484.1504
36. Glantschnig H, Fisher J, Wesolowski G, Rodan G, Reszka A. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. (2003) 10:1165. doi: 10.1038/sj.cdd.4401285
37. Felix R, Halasy-Nagy J, Wetterwald A, Cecchini MG, Fleisch H, Hofstetter W. Synthesis of membrane-and matrix-bound colony-stimulating factor-1 by cultured osteoblasts. J Cell Physiol. (1996) 166:311–22. doi: 10.1002/(SICI)1097-4652(199602)166:2<311::AID-JCP9>3.0.CO;2-S
38. Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature. (2012) 485:69. doi: 10.1038/nature11000
39. Movérare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Börjesson AE, et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med. (2014) 20:1279. doi: 10.1038/nm.3654
40. Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. (2012) 18:405. doi: 10.1038/nm.2653
41. Takada I, Mihara M, Suzawa M, Ohtake F, Kobayashi S, Igarashi M, et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-γ transactivation. Nat Cell Biol. (2007) 9:1273. doi: 10.1038/ncb1647
42. Hu K, Olsen BR. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J Clin Invest. (2016) 126:509–26. doi: 10.1172/JCI82585
43. O'Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. (2013) 54:258–63. doi: 10.1016/j.bone.2012.08.121
44. Xiong J, Piemontese M, Thostenson JD, Weinstein RS, Manolagas SC, O'Brien CA. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone. (2014) 66:146–54. doi: 10.1016/j.bone.2014.06.006
45. van Bezooijen RL, ten Dijke P, Papapoulos SE, Löwik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. (2005) 16:319–27. doi: 10.1016/j.cytogfr.2005.02.005
46. Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. (2003) 22:6267–76. doi: 10.1093/emboj/cdg599
47. Li R-F, Chen G, Ren J-G, Zhang W, Wu Z-X, Liu B, et al. The adaptor protein p62 is involved in RANKL-induced autophagy and osteoclastogenesis. J Histochem Cytochem. (2014) 62:879–88. doi: 10.1369/0022155414551367
48. Akisaka T, Yoshida H, Inoue S, Shimizu K, Organization of cytoskeletal F-actin, G-actin, and gelsolin in the adhesion structures in cultured osteoclast. J Bone Miner Res. (2001) 16:1248–55. doi: 10.1359/jbmr.2001.16.7.1248
49. Kurihara N, Hiruma Y, Zhou H, Subler MA, Dempster DW, Singer FR, et al. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest. (2007) 117:133–42. doi: 10.1172/JCI28267
50. DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. (2011) 21:966–74. doi: 10.1016/j.devcel.2011.08.016
51. Lin N-Y, Stefanica A, Distler JH. Autophagy: a key pathway of TNF-induced inflammatory bone loss. Autophagy. (2013) 9:1253–55. doi: 10.4161/auto.25467
52. Nollet M, Santucci-Darmanin S, Breuil V, Al-Sahlanee R, Cros C, Topi M, et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy. (2014) 10:1965–77. doi: 10.4161/auto.36182
53. Chung Y-H, Choi B, Song D-H, Song Y, Kang S-W, Yoon S-Y, et al. Interleukin-1β promotes the LC3-mediated secretory function of osteoclast precursors by stimulating the Ca2+-dependent activation of ERK. Int J Biochem Cell Biol. (2014) 54:198–207. doi: 10.1016/j.biocel.2014.07.018
54. Piemontese M, Onal M, Xiong J, Han L, Thostenson JD, Almeida M, et al. Low bone mass and changes in the osteocyte network in mice lacking autophagy in the osteoblast lineage. Sci Rep. (2016) 6:24262. doi: 10.1038/srep24262
55. Chang C, Su Y, Hu C, Lei H. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. (2013) 20:515. doi: 10.1038/cdd.2012.146
56. Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. (2015) 11:271–84. doi: 10.1080/15548627.2015.1009787
57. Stranks AJ, Hansen AL, Panse I, Mortensen M, Ferguson DJ, Puleston DJ, et al. Autophagy controls acquisition of aging features in macrophages. J Innate Immun. (2015) 7:375–91. doi: 10.1159/000370112
58. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B.-G., Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. (2008) 456:264. doi: 10.1038/nature07383
59. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA. (2012) 109:E3168–76. doi: 10.1073/pnas.1210500109
60. Amersfoort J, Douna H, Schaftenaar FH, Foks AC, Kröner MJ, van Santbrink PJ, et al. Defective autophagy in T cells impairs the development of diet-induced hepatic steatosis and atherosclerosis. Front Immunol. (2018) 9:2937. doi: 10.3389/fimmu.2018.02937
61. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. (2016) 17:277. doi: 10.1038/ni.3365
62. Li H, Li D, Ma Z, Qian Z, Kang X, Jin X, et al. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy. (2018) 14:1726–41. doi: 10.1080/15548627.2018.1483807
63. Cao Y, Yang W, Tyler MA, Gao X, Duan C, Kim SO, et al. Noggin attenuates cerulein-induced acute pancreatitis and the impaired autophagy. Pancreas. (2013) 42:301. doi: 10.1097/MPA.0b013e31825b9f2c
64. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. (1999) 402:672. doi: 10.1038/45257
65. Saftig P, Beertsen W, Eskelinen E-L. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. (2008) 4:510–2. doi: 10.4161/auto.5724
66. Qi M, Zhang L, Ma Y, Shuai Y, Li L, Luo K, et al. Autophagy maintains the function of bone marrow mesenchymal stem cells to prevent estrogen deficiency-induced osteoporosis. Theranostics. (2017) 7:4498. doi: 10.7150/thno.17949
67. Ha S-W, Weitzmann MN, Beck GR Jr. Bioactive silica nanoparticles promote osteoblast differentiation through stimulation of autophagy and direct association with LC3 and p62. ACS Nano. (2014) 8:5898–10. doi: 10.1021/nn5009879
68. Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Löwik CW, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. (2005) 19:1842–4. doi: 10.1096/fj.05-4221fje
69. Zahm AM, Bohensky J, Adams CS, Shapiro IM, Srinivas V. Bone cell autophagy is regulated by environmental factors. Cells Tissues Organs. (2011) 194:274–8. doi: 10.1159/000324647
70. Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. (2013) 288:17432–40. doi: 10.1074/jbc.M112.444190
71. Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R, et al. Glucocorticoid dose determines osteocyte cell fate. FASEB J. (2011) 25:3366–76. doi: 10.1096/fj.11-182519
72. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. (2003) 423:356–61. doi: 10.1038/nature01661
73. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. (2008) 105:18460–5. doi: 10.1073/pnas.0809850105
74. Kimura A, Naka T, Kishimoto T. IL-6-dependent and-independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci USA. (2007) 104:12099–104. doi: 10.1073/pnas.0705268104
75. Abu-Amer Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-κB. J Clin Invest. (2001) 107:1375–85. doi: 10.1172/JCI10530
76. Moreno JL, Kaczmarek M, Keegan AD, Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: irreversible inhibition of the differentiation program activated by RANKL. Blood. (2003) 102:1078–86. doi: 10.1182/blood-2002-11-3437
77. Park-Min K-H, Ji J-D, Antoniv T, Reid AC, Silver RB, Humphrey MB, et al. IL-10 suppresses calcium-mediated costimulation of receptor activator NF-κB signaling during human osteoclast differentiation by inhibiting TREM-2 expression. J Immunol. (2009) 183:2444–55. doi: 10.4049/jimmunol.0804165
78. Zaiss MM, Axmann R, Zwerina J, Polzer K, Gückel E, Skapenko A, et al. Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. (2007) 56:4104–12. doi: 10.1002/art.23138
79. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
80. Quinn JM, Itoh K, Udagawa N, Häusler K, Yasuda H, Shima N, et al. Transforming growth factor β affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res. (2001) 16:1787–94. doi: 10.1359/jbmr.2001.16.10.1787
81. Galvin RJS, Gatlin CL, Horn JW, Fuson TR. TGF-β enhances osteoclast differentiation in hematopoietic cell cultures stimulated with RANKL and M-CSF. Biochem Biophys Res Commun. (1999) 265:233–9. doi: 10.1006/bbrc.1999.1632
82. Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. (2000) 15:1477–88. doi: 10.1359/jbmr.2000.15.8.1477
83. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
84. Horwood NJ. Macrophage polarization and bone formation: a review. Clin Rev Allergy Immunol. (2016) 51:79–86. doi: 10.1007/s12016-015-8519-2
85. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
86. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
87. Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. (2005) 115:282–90. doi: 10.1172/JCI23394
88. Zwerina J, Redlich K, Polzer K, Joosten L, Krönke G, Distler J, et al. TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. (2007) 104:11742–7. doi: 10.1073/pnas.0610812104
89. Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou N. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. (2003) 32:1–7. doi: 10.1016/S8756-3282(02)00915-8
90. Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-α and IL-17. Rheumatology. (2008) 47:1635–40. doi: 10.1093/rheumatology/ken363
91. Kurihara N, Bertolini D, Suda T, Akiyama Y, Roodman GD. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol. (1990) 144:4226–30.
92. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. (2000) 106:1481–8. doi: 10.1172/JCI11176
93. Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL–RANK interaction. J Exp Med. (2000) 191:275–86. doi: 10.1084/jem.191.2.275
94. Kitaura H, Sands MS, Aya K, Zhou P, Hirayama T, Uthgenannt B, et al. Marrow stromal cells and osteoclast precursors differentially contribute to TNF-α-induced osteoclastogenesis in vivo. J Immunol. (2004) 173:4838–46. doi: 10.4049/jimmunol.173.8.4838
95. Zou W, Hakim I, Tschoep K, Endres S, Bar-Shavit Z. Tumor necrosis factor-α mediates RANK ligand stimulation of osteoclast differentiation by an autocrine mechanism. J Cell Biochem. (2001) 83:70–83. doi: 10.1002/jcb.1202
96. Ho VW, Sly LM. Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol Biol. (2009) 531:173–85. doi: 10.1007/978-1-59745-396-7_12
97. Tjiu J-W, Chen J-S, Shun C-T, Lin S-J, Liao Y-H, Chu C-Y, et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J Invest Dermatol. (2009) 129:1016–25. doi: 10.1038/jid.2008.310
98. Sonomoto K, Yamaoka K, Oshita K, Fukuyo S, Zhang X, Nakano K, et al. Interleukin-1β induces differentiation of human mesenchymal stem cells into osteoblasts via the Wnt-5a/receptor tyrosine kinase-like orphan receptor 2 pathway. Arthritis Rheum. (2012) 64:3355–63. doi: 10.1002/art.34555
99. Ma T, Miyanishi K, Trindade MC, Genovese M, Regula D, Smith RL, et al. Interleukin 1 receptor antagonist inhibits localized bone formation in vivo. J Rheum. (2003) 30:2547–52. Available online at: http://www.jrheum.org/content/30/12/2547
100. Nanes MS. Tumor necrosis factor-α: molecular and cellular mechanisms in skeletal pathology. Gene. (2003) 321: 1–15. doi: 10.1016/S0378-1119(03)00841-2
101. Perrien DS, Brown EC, Fletcher TW, Irby DJ, Aronson J, Gao GG, et al. Interleukin-1 and tumor necrosis factor antagonists attenuate ethanol-induced inhibition of bone formation in a rat model of distraction osteogenesis. J Pharmacol Exp Ther. (2002) 303:904–8. doi: 10.1124/jpet.102.039636
102. Huang H, Kim H, Chang E, Lee Z, Hwang S, Kim H, et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Diff. (2009) 16:1332–43. doi: 10.1038/cdd.2009.74
103. Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, et al. IL-17-producing [gamma][delta] T cells enhance bone regeneration. Nat Commun. (2016) 7:10928. doi: 10.1038/ncomms10928
104. Kim Y-G, Park J-W, Lee J-M, Suh J-Y, Lee J-K, Chang B-S, et al. IL-17 inhibits osteoblast differentiation and bone regeneration in rat. Arch Oral Biol. (2014) 59:897–905. doi: 10.1016/j.archoralbio.2014.05.009
105. Hess K, Ushmorov A, Fiedler J, Brenner RE, Wirth T. TNFα promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-κB signaling pathway. Bone. (2009) 45:367–76. doi: 10.1016/j.bone.2009.04.252
106. Ding J, Ghali O, Lencel P, Broux O, Chauveau C, Devedjian J, et al. TNF-α and IL-1β inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci. (2009) 84:499–504. doi: 10.1016/j.lfs.2009.01.013
107. Duque G, Huang DC, Macoritto M, Rivas D, Yang XF, Ste-Marie LG, et al. Autocrine regulation of interferon γ in mesenchymal stem cells plays a role in early osteoblastogenesis. Stem Cells. (2009) 27:550–8. doi: 10.1634/stemcells.2008-0886
108. Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, et al. Mesenchymal stem cell–based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat Med. (2011) 17:1594. doi: 10.1038/nm.2542
109. Cho T-J, Kim J, Chung C, Yoo W, Gerstenfeld L, Einhorn T, et al. Expression and role of interleukin-6 in distraction osteogenesis. Calcif Tissue Int. (2007) 80:192–200. doi: 10.1007/s00223-006-0240-y
110. Sammons J, Ahmed N, El-Sheemy M, Hassan H. The role of BMP-6, IL-6, and BMP-4 in mesenchymal stem cell-dependent bone development: effects on osteoblastic differentiation induced by parathyroid hormone and vitamin D3. Stem Cells Develop. (2004) 13:273–80. doi: 10.1089/154732804323099208
111. Blanchard F, Duplomb L, Baud'huin M, Brounais B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. (2009) 20:19–28. doi: 10.1016/j.cytogfr.2008.11.004
112. Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, et al. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone. (2006) 39:505–12. doi: 10.1016/j.bone.2006.02.074
113. Bellido T, Borba VZ, Roberson P, Manolagas SC. Activation of the Janus Kinase/STAT (signal transducer and activator of transcription) signal transduction pathway by interleukin-6-type cytokines promotes osteoblast differentiation 1. Endocrinology. (1997) 138:3666–76. doi: 10.1210/en.138.9.3666
114. Song HY, Jeon ES, Kim JI, Jung JS, Kim JH. Oncostatin M promotes osteogenesis and suppresses adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells. J Cell Biochem. (2007) 101:1238–51. doi: 10.1002/jcb.21245
115. Guihard P, Danger Y, Brounais B, David E, Brion R, Delecrin J, et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells. (2012) 30:762–72. doi: 10.1002/stem.1040
116. Schlundt C, El Khassawna T, Serra A, Dienelt A, Wendler S, Schell H, et al. Macrophages in bone fracture healing: their essential role in endochondral ossification. Bone. (2018) 106: 78–89. doi: 10.1016/j.bone.2015.10.019
117. Loi F, Córdova LA, Zhang R, Pajarinen J, Lin T-H, Goodman SB, et al. The effects of immunomodulation by macrophage subsets on osteogenesis in vitro. Stem Cell Res Ther. (2016) 7:1–11. doi: 10.1186/s13287-016-0276-5
118. Bonewald L, Dallas S. Role of active and latent transforming growth factor β in bone formation. J Cell Biochem. (1994) 55:350–7. doi: 10.1002/jcb.240550312
119. Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. (2006) 38:1424. doi: 10.1038/ng1916
120. Sil P, Muse G, Martinez J. A ravenous defense: canonical and non-canonical autophagy in immunity. Curr Opin Immunol. (2018) 50:21–31. doi: 10.1016/j.coi.2017.10.004
121. Pua HH, Guo J, Komatsu M, He Y-W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. (2009) 182:4046–55. doi: 10.4049/jimmunol.0801143
122. Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. (2009) 5:625–35. doi: 10.4161/auto.5.5.8133
123. Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. (2008) 455:396. doi: 10.1038/nature07208
124. Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol. (2012) 22:R540–5. doi: 10.1016/j.cub.2012.06.001
125. Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. (2011) 13:1319–27. doi: 10.1111/j.1462-5822.2011.01632.x
126. Mostowy S, Cossart P. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. (2012) 22:283–91. doi: 10.1016/j.tcb.2012.03.006
127. Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. (2015) 16:1014. doi: 10.1038/ni.3273
128. Sanjuan MA, Dillon CP, Tait SWG, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. (2007) 450:1253–7. doi: 10.1038/nature06421
129. Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy. (2014) 10:192–200. doi: 10.4161/auto.26927
130. Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, et al. Autophagy is required for CSF-1–induced macrophagic differentiation and acquisition of phagocytic functions. Blood. (2012) 119:4527–31. doi: 10.1182/blood-2011-11-392167
131. Nakahira K, Haspel JA, Rathinam VA, Lee S.-J., Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222. doi: 10.1038/ni.1980
132. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221. doi: 10.1038/nature09663
133. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP 3 inflammasome activation and assembly. Immunol Rev. (2015) 265:35–52. doi: 10.1111/imr.12286
134. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. (2016) 164:896–10. doi: 10.1016/j.cell.2015.12.057
135. Shi J, Wong J, Piesik P, Fung G, Zhang J, Jagdeo J, et al. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. (2013) 9:1591–03. doi: 10.4161/auto.26059
136. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. (2016) 166:288–98. doi: 10.1016/j.cell.2016.05.051
137. Chen Z, Bachhuka A, Han S, Wei F, Lu S, Visalakshan RM, et al. Tuning chemistry and topography of nanoengineered surfaces to manipulate immune response for bone regeneration applications. ACS Nano. (2017) 11:4494–06. doi: 10.1021/acsnano.6b07808
138. Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. (2000) 289:1508–14. doi: 10.1126/science.289.5484.1508
139. Taubman MA, Valverde P, Han X, Kawai T. Immune response: the key to bone resorption in periodontal disease. J Periodontol. (2005) 76:2033–41. doi: 10.1902/jop.2005.76.11-S.2033
140. Wang C-Y, Stashenko P. Characterization of bone-resorbing activity in human periapical lesions. J Endod. (1993) 19:107–11. doi: 10.1016/S0099-2399(06)80503-0
141. Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. (2007) 26:79–92. doi: 10.1016/j.immuni.2006.10.018
142. Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology. (2010) 139:1630–41. doi: 10.1053/j.gastro.2010.07.006
143. Wildenberg ME, Vos ACW, Wolfkamp SC, Duijvestein M, Verhaar AP, Te Velde AA, et al. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology. (2012) 142:1493–03. doi: 10.1053/j.gastro.2012.02.034
144. Tomić S, Janjetović K, Mihajlović D, Milenković M, Kravić-Stevović T, Marković Z, et al. Graphene quantum dots suppress proinflammatory T cell responses via autophagy-dependent induction of tolerogenic dendritic cells. Biomaterials. (2017) 146:13–28. doi: 10.1016/j.biomaterials.2017.08.040
145. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. (2013) 15:406. doi: 10.1038/ncb2708
146. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889. doi: 10.1038/ni.1937
147. Heusinkveld M, van Steenwijk PJDV, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, et al. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. (2011) 187:1157–65. doi: 10.4049/jimmunol.1100889
148. Vomero M, Barbati C, Colasanti T, Perricone C, Novelli L, Ceccarelli F, et al. Autophagy and rheumatoid arthritis: current knowledges and future perspectives. Front Immunol. (2018) 9:1577. doi: 10.3389/fimmu.2018.01577
149. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
150. Sorice M, Iannuccelli C, Manganelli V, Capozzi A, Alessandri C, Lococo E, et al. Autophagy generates citrullinated peptides in human synoviocytes: a possible trigger for anti-citrullinated peptide antibodies. Rheumatology. (2016) 55:1374–85. doi: 10.1093/rheumatology/kew178
151. Lin N-Y, Beyer C, Gießl A, Kireva T, Scholtysek C, Uderhardt S, et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann Rheum Dis. (2013) 72:761–8. doi: 10.1136/annrheumdis-2012-201671
152. Shin Y-J, Han S-H, Kim D-S, Lee G-H, Yoo W-H, Kang Y-M, et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res Ther. (2010) 12:R19. doi: 10.1186/ar2921
153. Xu K, Xu P, Yao J-F, Zhang Y-G, Hou W-K, Lu S-M. Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm Res. (2013) 62:229–37. doi: 10.1007/s00011-012-0572-1
154. Kato M, Ospelt C, Gay RE, Gay S, Klein K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. (2014) 66:40–8. doi: 10.1002/art.38190
155. Li S, Chen J-W, Xie X, Tian J, Deng C, Wang J, et al. Autophagy inhibitor regulates apoptosis and proliferation of synovial fibroblasts through the inhibition of PI3K/AKT pathway in collagen-induced arthritis rat model. Am J Trans Res. (2017) 9:2065–76. Available online at: http://www.ajtr.org/files/ajtr0046568.pdf
156. van Loosdregt J, Rossetti M, Spreafico R, Moshref M, Olmer M, Williams GW, et al. Increased autophagy in CD4+ T cells of rheumatoid arthritis patients results in T-cell hyperactivation and apoptosis resistance. Eur J Immunol. (2016) 46:2862–70. doi: 10.1002/eji.201646375
157. Barbati C, Vomero M, Colasanti T, Ceccarelli F, Marcosano M, Miranda F, et al. Microparticles and autophagy: a new frontier in the understanding of atherosclerosis in rheumatoid arthritis. Immunol Res. (2018) 66:655–62. doi: 10.1007/s12026-018-9053-0
158. Farid SS, Azizi G, Mirshafiey A. Anti-citrullinated protein antibodies and their clinical utility in rheumatoid arthritis. Int J Rheum Dis. (2013) 16:379–86. doi: 10.1111/1756-185X.12129
159. Wahab AA, Mohammad M, Rahman MM, Said MSM. Anti-cyclic citrullinated peptide antibody is a good indicator for the diagnosis of rheumatoid arthritis. Pak J Med Sci. (2013) 29:773. doi: 10.12669/pjms.293.2924
160. Krishnamurthy A, Joshua V, Hensvold AH, Jin T, Sun M, Vivar N, et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. (2016) 75:721–9. doi: 10.1136/annrheumdis-2015-208093
161. Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med. (2011) 208:2625–32. doi: 10.1084/jem.20110640
162. Colasanti T, Fiorito S, Alessandri C, Serafino A, Andreola F, Barbati C, et al. Diesel exhaust particles induce autophagy and citrullination in Normal Human Bronchial Epithelial cells. Cell Death Dis. (2018) 9:1073. doi: 10.1038/s41419-018-1111-y
163. Dai Y, Hu S. Recent insights into the role of autophagy in the pathogenesis of rheumatoid arthritis. Rheumatology. (2015) 55:403–10. doi: 10.1093/rheumatology/kev337
164. Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. (2005) 65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640
165. Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. (2009) 164:541–51. doi: 10.1016/j.neuroscience.2009.08.014
166. Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, et al. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum. (2010) 62:2294–302. doi: 10.1002/art.27504
167. Sanchez CP, He Y-Z. Bone growth during rapamycin therapy in young rats. BMC Pediatr. (2009) 9:3. doi: 10.1186/1471-2431-9-3
168. Westenfeld R, Schlieper G, Wöltje M, Gawlik A, Brandenburg V, Rutkowski P, et al. Impact of sirolimus, tacrolimus and mycophenolate mofetil on osteoclastogenesis—implications for post-transplantation bone disease. Nephrol Dial Trans. (2011) 26:4115–23. doi: 10.1093/ndt/gfr214
169. Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. (2005) 35:1482–90. doi: 10.1002/eji.200425405
170. Habib S, El Andaloussi A, Elmasry K, Handoussa A, Azab M, Elsawey A, et al. PDL-1 blockade prevents T cell exhaustion, inhibits autophagy, and promotes clearance of Leishmania donovani. Infect Immun. (2018) 86:e00019–18. doi: 10.1128/IAI.00019-18
171. Chang KH, Sengupta A, Nayak RC, Duran A, Lee SJ, Pratt RG, et al. p62 is required for stem cell/progenitor retention through inhibition of IKK/NF-κB/Ccl4 signaling at the bone marrow macrophage-osteoblast niche. Cell Rep. (2014) 9:2084–97. doi: 10.1016/j.celrep.2014.11.031
172. Zhou Y, Huang R, Fan W, Prasadam I, Crawford R, Xiao Y Mesenchymal stromal cells regulate the cell mobility and the immune response during osteogenesis through secretion of vascular endothelial growth factor A. J Tissue Eng Regen Med. (2018) 12:e566–78. doi: 10.1002/term.2327
173. Hashimoto I, Koizumi K, Tatematsu M, Minami T, Cho S, Takeno N, et al. Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells. Eur J Cancer. (2008) 44:1022–9. doi: 10.1016/j.ejca.2008.02.043
174. Braun M, Qorraj M, Büttner M, Klein F, Saul D, Aigner M, et al. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia. (2016) 30:1788. doi: 10.1038/leu.2016.58
175. Chen Z, Teo AE, McCarty N. ROS-induced CXCR4 signaling regulates mantle cell lymphoma (MCL) cell survival and drug resistance in the bone marrow microenvironment via autophagy. Clin Cancer Res. (2016) 22:187–99. doi: 10.1158/1078-0432.CCR-15-0987
176. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. (2014)111:3526–31. doi: 10.1073/pnas.1322831111
177. Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. (2006) 84:448–54. doi: 10.1111/j.1440-1711.2006.01454.x
178. Keller CW, Fokken C, Turville SG, Lünemann A, Schmidt J, Münz C, et al. TNF-α induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J Biol Chem. (2011) 286:3970–80. doi: 10.1074/jbc.M110.159392
179. Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Souquère S, et al. NF-κB activation represses tumor necrosis factor-α-induced autophagy. J Biol Chem. (2006) 281:30373–82. doi: 10.1074/jbc.M602097200
180. Park H-J, Lee SJ, Kim S-H, Han J, Bae J, Kim SJ, et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol. (2011) 48:720–7. doi: 10.1016/j.molimm.2010.10.020
181. Zhao J, Sun Y, Shi P, Dong J-N, Zuo L-G, Wang H-G, et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int Immunopharmacol. (2015) 26:221–8. doi: 10.1016/j.intimp.2015.03.033
182. Ip WE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. (2017) 356:513–9. doi: 10.1126/science.aal3535
183. Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, et al. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. (2009) 69:8844–52. doi: 10.1158/0008-5472.CAN-08-4401
Keywords: osteoimmunology, bone remodeling, autophagy, immunomodulation, stem cell
Citation: Xiao L and Xiao Y (2019) The Autophagy in Osteoimmonology: Self-Eating, Maintenance, and Beyond. Front. Endocrinol. 10:490. doi: 10.3389/fendo.2019.00490
Received: 17 December 2018; Accepted: 08 July 2019;
Published: 23 July 2019.
Edited by:
Giacomina Brunetti, University of Bari Aldo Moro, ItalyReviewed by:
Carlo Perricone, Sapienza Università di Roma, ItalyFayez Safadi, Northeast Ohio Medical University, United States
Copyright © 2019 Xiao and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yin Xiao, eWluLnhpYW9AcXV0LmVkdS5hdQ==