 Stefanie Parisien-La Salle
Stefanie Parisien-La Salle Nadine Dumas
Nadine Dumas Geneviève Rondeau1
Geneviève Rondeau1 Isabelle Bourdeau
Isabelle Bourdeau- 1Division of Endocrinology, Department of Medicine, Research Center, Centre Hospitalier de l'Université de Montréal (CHUM), Montreal, QC, Canada
- 2Division of Genetics, Department of Medicine, Research Center, Centre Hospitalier de l'Université de Montréal (CHUM), Montreal, QC, Canada
- 3Department of Pathology and Cellular Biology, Centre Hospitalier de l'Université de Montréal (CHUM), Montreal, QC, Canada
Pheochromocytomas (PHEOs) are a rare cause of endocrine hypertension that requires genetic counseling since at least 30% of PHEOs are associated with a germline mutation in a susceptibility gene. Neurofibromatosis type 1, NF1 is amongst the 16 known causing genes for pheochromocytomas/paragangliomas. We report a case of a 73-year-old man with PHEO in whom genetic testing revealed a large pathogenic heterozygous deletion of 1.14 Mb encompassing the entire coding sequence of the NF1 gene while the patient showed no signs of clinical NF1.This case illustrates that the diagnosis of NF1 should not be excluded in patients with PHEO in the absence of clinical diagnosis of the disease and support that older patients with PHEO should also be offered genetic counseling.
Introduction
Pheochromocytomas (PHEOs) are rare catecholamine-secreting tumors that can cause endocrine hypertension with a prevalence of 0.4–2% in hypertensive patients (1). Germline mutations can be identified in at least 30% of patients with PHEOs and paragangliomas (2). In light of this, genetic testing is now recommended for all patients diagnosed with PHEO (3, 4). Some of these mutations can cause hereditary syndromes such as neurofibromatosis type 1 (NF1), von Hippel-Lindau (VHL), and multiple endocrine neoplasia type 2 (MEN2) (2). NF1 is a neurocutaneous disease caused by a mutation in the NF1 gene that encodes for the neurofibromin protein (5). The NF1 gene is located on the chromosome 17q11.2 (2). Two of the following criteria are needed to establish the diagnosis of NF1: inguinal or axillary freckling, two or more neurofibromas, six or more café-au-lait macules, two or more Lisch nodules, optic gliomas, distinctive bone lesions, or a first degree relative with NF1 (6). We report here the case of a man with pheochromocytoma that was diagnosed at 73 years-old carrying an unsuspected NF1 gene deletion but with no clinical manifestations of NF1.
Case Report
Following coronary artery bypass grafting, a 73-year-old man presented with symptoms of palpitation, headache, sweating, and blurred vision. His initial systolic blood pressure was over 240 mmHg. The patient's medical history included type 2 diabetes, dyslipidemia, coronary heart disease, prostate cancer and long-standing hypertension that used to be well-controlled with 2 medications; Metoprolol 25 mg twice daily and Valsartan 80 mg daily. However, his hypertension had been harder to control for the past 2 years. His family history was unremarkable except for cases of sudden death for his maternal uncle and aunts. No familial syndromes were noted. Patient was worked up for severe postoperative hypertension with a computed tomography (CT) scan which revealed a small right adrenal mass measured at 31 mm (Figure 1). Biochemical testing included measurements of urinary fractionated metanephrines which revealed high normetanephrines with a value of 880 nmol/d (N < 240). However, metanephrines (148 nmol/d: N < 275), dopamine (936 nmol/d: N <2570), epinephrines (undetectable: N < 110), and norepinephrines (456 nmol/d: N < 440) were within the normal limits. Plasma free normetanephrines were 2.10 nmol/L (N <1.20) and free metanephrines 0.4 nmol/L (N < 0.48). The patient had normal calcitonin levels. A meta-iodobenzylguanidine (MIBG) scan showed intense and moderate uptake of a 2 ×2 ×3 cm mass of the right adrenal mass with no distant metastases. The patient underwent adrenalectomy and the diagnosis of PHEO was confirmed by histology post-operatively (Figure 2). PHEO of the Adrenal gland Scaled Score (PASS) was 4, and the ki67 proliferation index was less than one percent.
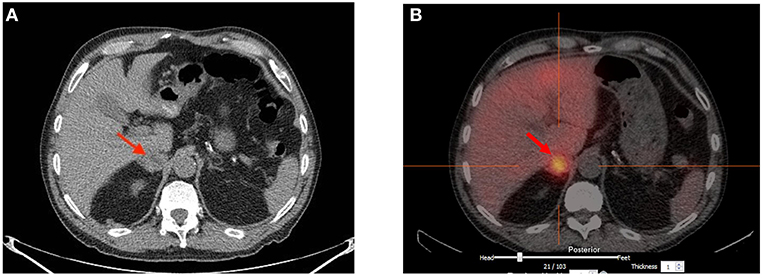
Figure 1. (A) Computed Tomography scan showing a 3 cm heterogeneous right adrenal mass of undetermined significance. (B) Strong Meta-iodobenzylguanidine (MIBG) uptake of the 2 × 2 × 3 cm right adrenal mass with no evidence of distant metastasis.
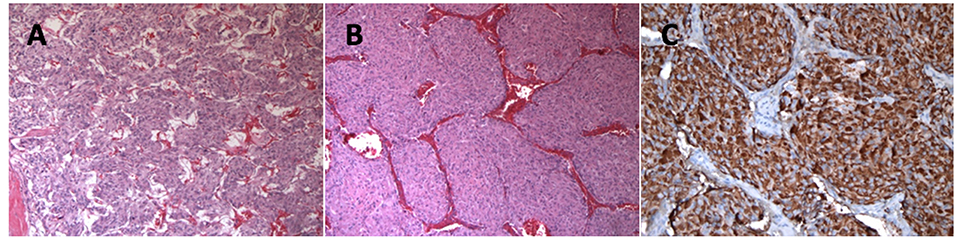
Figure 2. (A) More typical Zellballen's architecture with small tumor nests and very focal tumor cell spindling (Hematoxylin and eosin staining, 100 x). (B) Large tumor nests were also present (Hematoxylin and eosin staining, 100 x). (C) Chromogranine A immunostaining showing strong granular cytoplasmic positivity in the tumor cells (Chromogranine A staining, 200 x).
Methods
After genetic counseling, the patient gave written consent for genetic analysis. Leucocyte DNA was obtained. A genetic panel of 14 susceptibility genes for PHEOs and paragangliomas was performed. The following genes were evaluated for sequence changes and exonic deletions/duplications (VHL, RET, NF1, SDHD, SDHB, SDHC, SDHA/SDHAF2, TMEM127, MAX, FH, EGLN1, KIF1B, MEN1) (Invitae, San Francisco, CA). Following the identification of a gross deletion, a comparative genomic hybridization (CGH) was performed using Agilent-CGXTM-HD, 4 ×180K oligonucleotide array (CHU Ste Justine, Unité Biologie Médicale: Génétique, Montréal, Quebec) to identify the boundaries of this genetic deletion. A written informed consent was obtained from the patient for the publication of this case report.
Results
Panel genetic testing revealed a gross heterozygous germline deletion of the genomic region encompassing the full-coding sequence of the NF1 gene (NM_000267.3). In order to better identify the boundaries and the extends of this event CGH array was performed. The analysis showed a deletion of 15 oligonucleotides at 17q11.2 (position 29, 116, 494-30, 260, 501) (Genome Browser UCSC 2009hg19 assembly). The size of this large pathogenic heterozygous deletion is estimated at 1.14 Mb. In addition to the NF1 gene, this deletion includes 10 OMIM genes: CRLF3, ATAD5, TEFM, ADAP2 RNF135, OMG, EVI2A, RAB11FIP4, MIR193A and 7 other genes: DPRXP4, MIR4733, MIR4724, MIR4725, MIR365B, COPRS, UTP6 (Figure 3). Patient was re-examined in light of these findings but showed no signs of clinical neurofibromatosis type 1 except for a pectum excavatum that may be associated with NF1.
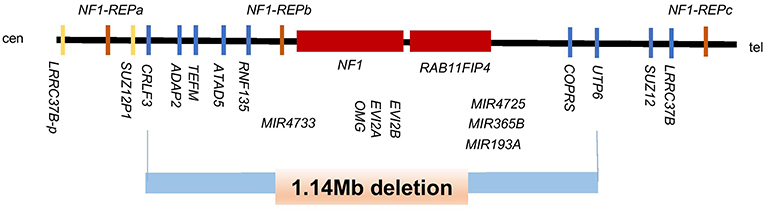
Figure 3. Schematic representation of the genomic region at 17q11.2 harboring the NF1 gene and its flanking genes. The deletion of 1.14-Mb is shown with the blue bar and includes 11 OMIM genes (CRLF3, ATAD5, TEFM, ADAP2, RNF135, OMG, EVI2B, EVI2A, NF1, RAB11FIP4, MIRI193A) and, 7 other genes (DPRXP4, MIR4733, MIR4724, MIR4725, MIR365B, CORPS, UTP6). Modified from Kehrer-Sawatzki et al. (7).
Discussion
To our knowledge, we report the oldest patient carrying a large deletion encompassing the entire NF1 gene in the context of an apparently sporadic pheochromocytoma. Very recently, a case series described three patients with PHEOs carrying unsuspected NF1 mutations (2). In contrast to our case, these patients were younger (22, 39, and 56 years) and carried only point mutations in the NF1 gene rather than the loss of the entire gene. In this report, only one of the three patients had discreet signs of NF1 on a repeat examination (2) (Table 1). This shows that a clinical examination is insufficient to exclude a NF1 diagnosis in a case of PHEO.
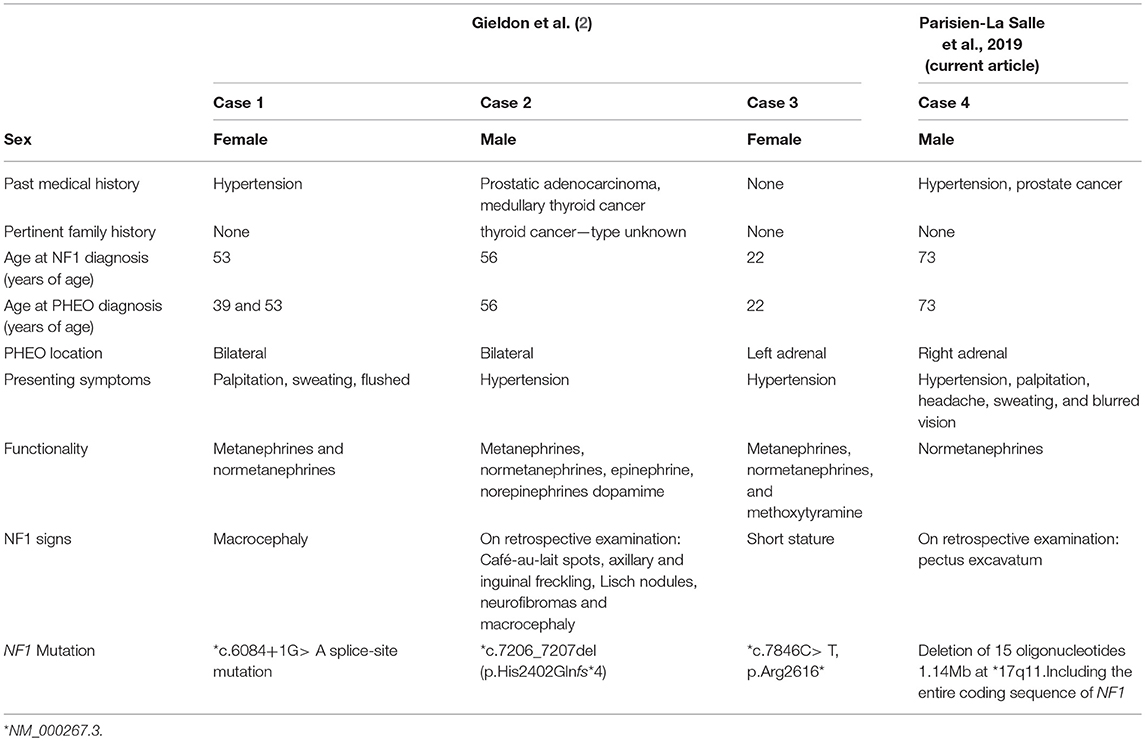
Table 1. Clinical and biochemical descriptions of patients with PHEO and NF1 without clinical NF1 diagnosis described previously and including the current report.
In our case, the tumor secreted normetanephrines. NF1 cases of PHEO have been shown to secrete metanephrines or normetanephrines (2). Gross deletions of the NF1 genes have been described previously in individuals with neurofibromatosis type 1 (8, 9). Large deletions of the NF1 gene and its flanking regions are frequently associated with a more severe clinical phenotype of NF1 (dysmorphic features, overgrowth, hypotonia, and cognitive impairment) that was not present in this patient (7).
NF1 is a heterogeneous disorder affecting ~1/3,000 births (10). This disease causes multi-organ involvement, which can include PHEOs in 0.1–7% (2, 10). The incidence might be underestimated due to the lack of formal recommendations to universally screen biochemically NF1 patients for PHEOs, as there are in other genetic syndromes such as Von-Hippel-Lindau or MEN2 (10, 11). Recent data showed that 31% of PHEOs in NF1 are found incidentally and only 7.3% were diagnosed following biochemical testing (12). Moreover, PHEOs tend to be diagnosed at an older age in patients with NF1 when compared to other genetic mutations (48 vs. 30 years old) (10).
Diagnosis of NF1 is important as it is associated with an elevated risk of associated malignancies, including malignant peripheral nerve sheath tumors, breast cancer, gastrointestinal stromal tumors, brain, and central nervous system tumors (13). This entails these patients to a more rigorous screening and active surveillance. Furthermore, recognition of NF1 is important for familial screening. In addition to de novo mutations NF1 may be inherited (14). The patient reported here did not have a family history of NF1. However, NF1 being an autosomal dominant disorder, appropriate genetic counseling for family members is essential (15). Overall, patients with NF1 need a medical follow-up by a multidisciplinary team to assess and manage complications, counsel on genetic screening (6).
Our findings support the recommendation of more recent guidelines that all patients, independently of age and the clinical presentation of PHEO, should be offered genetic counseling (3, 4). This is illustrated in this 73 year-old man carrying an unsuspected major NF1 deletion. Moreover, genetic testing should not only include evaluation for gene sequence changes but also gene deletion/duplication. Finally, the diagnosis of NF1 should not be excluded in patients with PHEO in the absence of clinical diagnosis of the disease.
Data Availability
This manuscript contains previously unpublished data. The name of the repository and accession number(s) are not available.
Author Contributions
SP-L collected the data and wrote the manuscript with the support of IB. ND genetic counselor that met the patient. GR endocrinologist that was involved in patients' care. IB supervised the project and wrote the manuscript with the support of SP-L. ML pathologist that analyzed the specimen and provided the pictures of the tumors' histology. All authors discussed the results and contributed to the final manuscript.
Funding
This study was supported by a salary grant to IB from Fonds de Recherche du Québec-Santé.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patient that kindly accepted that we report his case.
Abbreviations
PHEO, Pheochromocytoma; NF1, Neurofibromatosis type 1.
References
1. Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Practice. (2000) 6:249–52. doi: 10.4158/EP.6.3.249
2. Gieldon L, Masjkur JR, Richter S, Darr R, Lahera M, Aust D, et al. Next-generation panel sequencing identifies NF1 germline mutations in three patients with pheochromocytoma but no clinical diagnosis of neurofibromatosis type 1. Eur J Endocrinol. (2018) 178:K1–9. doi: 10.1530/EJE-17-0714
3. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. (2017) 13:233–47. doi: 10.1038/nrendo.2016.185
4. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
5. Cimino PJ, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol. (2018) 148:799–811. doi: 10.1016/B978-0-444-64076-5.00051-X
6. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. (2007) 44:81–8. doi: 10.1136/jmg.2006.045906
7. Kehrer-Sawatzki H, Mautner VF, Cooper DN. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet. (2017) 136:349–76. doi: 10.1007/s00439-017-1766-y
8. Rasmussen SA, Colman SD, Ho VT, Abernathy CR, Arn PH, Weiss L, et al. Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet. (1998) 35:468–71. doi: 10.1136/jmg.35.6.468
9. Kayes LM, Burke W, Riccardi VM, Bennett R, Ehrlich P, Rubenstein A, et al. Deletions spanning the neurofibromatosis 1 gene: identification and phenotype of five patients. Am J Hum Genet. (1994) 54:424–36.
10. Moramarco J, El Ghorayeb N, Dumas N, Nolet S, Boulanger L, Burnichon N, et al. Pheochromocytomas are diagnosed incidentally and at older age in neurofibromatosis type 1. Clin Endocrinol. (2017) 86:332–9. doi: 10.1111/cen.13265
11. Petr EJ, Else T. Pheochromocytoma and Paraganglioma in Neurofibromatosis type 1: frequent surgeries and cardiovascular crises indicate the need for screening. Clin Diabetes Endocrinol. (2018) 4:15. doi: 10.1186/s40842-018-0065-4
12. Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF Jr, Bancos I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol. (2017) 86:141–9. doi: 10.1111/cen.13163
13. Stewart DR, Korf BR, Nathanson KL, Stevenson DA, Yohay K. Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2018) 20:671–82. doi: 10.1038/gim.2018.28
14. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet Part A. (2010) 152a:327–32. doi: 10.1002/ajmg.a.33139
Keywords: neurofibromatosis, deletion, phenotype, pheochromocytoma, hypertension
Citation: Parisien-La Salle S, Dumas N, Rondeau G, Latour M and Bourdeau I (2019) Isolated Pheochromocytoma in a 73-Year-Old Man With No Clinical Manifestations of Type 1 Neurofibromatosis Carrying an Unsuspected Deletion of the Entire NF1 Gene. Front. Endocrinol. 10:546. doi: 10.3389/fendo.2019.00546
Received: 11 June 2019; Accepted: 22 July 2019;
Published: 20 August 2019.
Edited by:
Krzysztof Reiss, LSU Health Sciences Center New Orleans, Louisiana State University, United StatesReviewed by:
Luis Del Valle, LSU Health Sciences Center New Orleans, Louisiana State University, United StatesSalah A. Mohamed, Department of Cardiac and Thoracic Vascular Surgery, University Medical Center Schleswig-Holstein, Germany
Copyright © 2019 Parisien-La Salle, Dumas, Rondeau, Latour and Bourdeau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Bourdeau, aXNhYmVsbGUuYm91cmRlYXVAdW1vbnRyZWFsLmNh