 Luise Fischer1,2
Luise Fischer1,2 Caroline Herkner1Reni Kitte1Sebastian Dohnke1,2Julia Riewaldt2†
Caroline Herkner1Reni Kitte1Sebastian Dohnke1,2Julia Riewaldt2† Karsten Kretschmer2
Karsten Kretschmer2 Annette I. Garbe1*
Annette I. Garbe1*- 1Osteoimmunology, DFG-Center for Regenerative Therapies Dresden (CRTD), Technische Universität Dresden, Dresden, Germany
- 2Molecular and Cellular Immunology/Immune Regulation, DFG-Center for Regenerative Therapies Dresden (CRTD), Technische Universität Dresden, Dresden, Germany
The bone represents surprisingly dynamic structures that are subject to constant remodeling by the concerted action of bone-forming osteoblasts and bone-resorbing osteoclasts - two cell subsets of distinct developmental origin that are key in maintaining skeletal integrity throughout life. In general, abnormal bone remodeling due to dysregulated bone resorption and formation is an early event in the manifestation of various human bone diseases, such as osteopetrosis/osteoporosis and arthritis. But bone remodeling is also closely interrelated with lympho-hematopoietic homeostasis, as the bone marrow niche is formed by solid and trabecular bone structures that provide a framework for the long-term maintenance and differentiation of HSCs (>blood lineage cells and osteoclasts) and MSCs (>osteoblasts). Numerous studies in mice and humans have implicated innate and adaptive immune cells in the dynamic regulation of bone homeostasis, but despite considerable clinical relevance, the exact mechanisms of such immuno-bone interplay have remained incompletely understood. This holds particularly true for CD4+ regulatory T (Treg) cells expressing the lineage specification factor Foxp3: Foxp3+ Treg cells have been shown to play an indispensable role in maintaining immune homeostasis, but may also exert critical non-immune functions, which includes the control of metabolic and regenerative processes, as well as the differentiation of HSCs and function of osteoclasts. Here, we summarize our current knowledge on the T cell/bone interplay, with a particular emphasis on our own efforts to dissect the role of Foxp3+ Treg cells in bone and hematopoietic homeostasis, employing experimental settings of gain- and loss-of-Treg cell function. These data make a strong case that Foxp3+ Treg cells impinge on lympho-hematopoiesis through indirect mechanisms, i.e., by acting on osteoclast development and function, which translates into changes in niche size. Furthermore, we propose that, besides disorders that involve inflammatory bone loss, the modulation of Foxp3+ Treg cell function in vivo may represent a suitable approach to reinstate bone homeostasis in non-autoimmune settings of aberrant bone remodeling.
Introduction
In adult mammals, the bone marrow (BM) is the primary site of hematopoiesis. Hematopoietic stem cells (HSCs) reside within specific niches, which are specialized microenvironments within the BM cavity, providing HSCs with regulatory signals essential for their maintenance, proliferation and differentiation into blood and immune cells. HSCs dynamically regulate their numbers by undergoing asymmetric, self-renewing divisions depending upon cell-intrinsic and cell-extrinsic mechanisms, thereby sustaining hematopoiesis over lifetime (1, 2). Homeostasis of the bone microenvironment critically depends on bone remodeling, a tightly regulated process involving three major cell types: bone-resorbing osteoclasts, bone-forming osteoblasts and osteocytes. Osteocytes originate from mature osteoblasts, residing within the lacuna of the mineralized bone matrix, and act as orchestrators of bone remodeling (3). Under physiological conditions, resorption of damaged bone by osteoclasts is followed by the recruitment of osteoblasts and the formation of new bone, a concerted action critical for the maintenance of skeletal integrity and bone homeostasis. Defects in osteoclastic activity cause osteopetrosis, a disease associated with high bone mass, whereas enhanced osteoclastic bone resorption results in low bone mass and osteoporosis (4). While the direct impact of osteoclasts on maintenance and BM retention of HSCs remains controversial (5–7), there is accumulating evidence that osteoblast lineage cells play a crucial role in the regulation of hematopoiesis (8–10). Since the majority of the cells in the BM are hematopoietic cells, it appears likely that, in addition to stromal niche components, HSCs are regulated by their own progeny. In this context, megakaryocytes have recently been identified as direct regulators of homeostatic HSC quiescence using several mechanisms, including secretion of CXCL4 and TGF-β (11–13), while direct cell-cell interaction of megakaryocytes and bone cells have been described to affect skeletal homeostasis (14–16). Similarly, it has been suggested that the function of osteoblasts in maintaining the HSC niche is controlled by macrophages, whereas “HSC niche macrophages” have been described to govern maintenance of hematopoiesis directly (17–20). However, due to the heterogeneity of macrophages, and since the bone- and BM-residing macrophage populations, including osteoclasts, are primarily defined by their histological location, the precise identity and role of the different macrophage subpopulations in bone remodeling, regeneration, and shaping the HSC niche still need to be defined (21–23).
The impact of cells of the adaptive immune system — B and T lymphocytes — on bone remodeling under physiological conditions is still a matter of debate: while some studies using lymphocyte-deficient mouse models such as Rag−/−, μMT or nude mice described low bone mass phenotypes, others did not (24–27). Given the fact that several populations of mature lymphocytes, such as long-lived memory T and B cells reside in the BM (28, 29) and share common niches with HSCs, it is reasonable to assume that these mature cells impinge on bone and hematopoietic homeostasis. In this context, it has been shown, that after allogeneic BM stem cell transplantation, T cells act as a double-edged sword as they can on the one hand promote graft-versus-host-disease (GvHD), but on the other hand can also provide beneficial effects on the engraftment (30). High-resolution imaging over time in an experimental setup of allogeneic HSC transplantation provided evidence that HSCs preferentially co-localize with CD4+ regulatory T (Treg) cells, expressing the lineage specification factor Foxp3, that accumulate on the endosteal surface of the bones of non-irradiated recipient mice, enabling transplanted stem cells to evade from allogeneic rejection (31, 32). The essential role of Foxp3+ Treg cells in maintaining immune homeostasis is firmly established (33, 34), but additional, non-immunological functions of Treg cells, such as controlling (i) the metabolic function in adipose tissue, (ii) regeneration of muscle cells, (iii) lympho-hematopoiesis, and (iv) osteoclast and osteoblast development and function, become increasingly apparent (35–41).
While much less is known about the interplay between Treg cells and osteoclasts/osteoblasts in bone remodeling and the formation of the HSC niche, a large body of experimental evidence focused on the immuno-bone crosstalk in rheumatoid arthritis (RA), one of the most common human autoimmune diseases. Osteoclasts play a central role in the pathogenesis of RA, where bone destruction is characterized by inflammatory osteolysis, caused by aberrant activation of the immune system, eventually leading to the destruction of surface cartilage and subchondral bone (42, 43). It has been proposed that the onset and pathogenesis of RA critically depends on the balance between two distinct CD4+ T cell subsets: type 17 helper T (TH17) cells, a pathogenic subset of CD4+ T cells that produces IL-17, and thereby promoting osteoclastogenesis, and Treg cells, crucial for the prevention of autoimmune diseases driven by TH17 cells (34, 44). The role of Treg cells in animal models for autoimmune arthritis has been demonstrated by amelioration of the disease following adoptive transfer of Treg cells and induction of an accelerated and more severe form of arthritis after depletion of Treg cells (45–48). Moreover, it has been described that under certain conditions of experimental arthritis a subpopulation of Foxp3+ Treg cells can convert into autoreactive Foxp3− TH17 cells (49).
There is also evidence that Treg cells interact with osteoblasts. For example, studies on intermittent PTH-induced bone anabolism propose that Treg cells are involved in the up-regulation of the expression of the osteogenic factor Wnt10b by CD8+ T cells, which was also suggested to be the main mechanism in the promotion of bone formation by oral supplementation with the probiotic Lactobacillus rhamnosus GG (50–53). On the other hand, Treg cells have been implicated to play a role in bone formation by promoting the differentiation of osteoblasts directly (54).
Although the close relationship between the bone and the immune system has long been recognized (55), the spatial relationship and the interaction between the different cell types within the bone microenvironment as well as the identity of their communication factors, in particular under physiological conditions, is still incompletely understood.
Studies on the interplay between osteoclasts/osteoblasts and Treg cells in the BM microenvironment are hampered by several unresolved issues: (a) osteoclasts are difficult to study due to the lack of reliable methods for their ex vivo purification, owing to their low abundance, large size, and lack of specific surface marker expression. Furthermore, the phenotypic definition of “true” osteoclast precursors and their developmental stages vary considerably; (b) constitutive Treg cell deficiency inevitably results in secondary effects due to systemic autoimmunity and increased systemic levels of inflammatory factors. Mice with constitutive Treg cell deficiency suffer from severe morbidity leading to premature death prior to completion of bone development; (c) due to the unique properties and structure of bone, it is technically challenging to assess and visualize interactions between cells in the BM niche. Thus, it will be essential to develop experimental systems and more advanced imaging that keep these limitations to a minimum.
In this review we discuss the impact of BM-residing Treg cells on the bone microenvironment, central to the development of therapeutic strategies for the treatment of bone diseases and to promote tolerance after stem cell transplantation.
Lympho-Hematopoietic Niche and Foxp3+ Treg Cells
For a long time, HSCs were considered as dormant cells but increasing evidence suggests HSCs as direct targets of inflammatory signals. Earlier studies have identified HSCs as “first responders” during inflammatory responses, e.g., during infections, later it became clear that pro-inflammatory cytokines such as interleukin (IL)-1, IL-6, IL-8, tumor necrosis factor (TNF) and type I and type II interferons (IFNs), G-CSF, and Toll-like receptor (TLR) ligands regulate HSCs not only in response to stress but also under homeostatic conditions. Together with BM niche signals such as CXCL12, basal levels of inflammatory cytokines provided by T cells, NK cells, neutrophils and macrophages control the balance between HSC dormancy and lineage fate decision under homeostatic conditions, while inflammatory conditions promote HSC proliferation and differentiation at the expense of self-renewal, emphasizing the interdependency of the distinct BM niche components in health and disease (56–60). However, increasing evidence is pointing towards regulation of HSC maintenance by distal/systemic factors: in addition to the nervous system (e.g., by oscillation of CXCL12 production) and hormones such as PTH or estrogen that have been described to regulate HSCs from the outside, two recent studies demonstrate that also the liver and the intestine contribute to HSC maintenance under steady-state conditions (61–65). Given that bone remodeling is also highly regulated by systemic factors, further studies are required to dissect direct and indirect contributions of distal organs on hematopoietic and skeletal homeostasis.
In both mouse and man, the T cell compartment in the BM, which constitutes only about 5% of mononuclear BM cells, is characterized by a lower CD4/CD8 T cell ratio and notably, by substantially elevated frequencies of Foxp3+ Treg cells within the CD4+ T cell population compared to peripheral lymphoid organs (66, 67). Like other BM T cells, BM Treg cells exhibit a more activated/memory phenotype. Transcriptional characterization of BM Treg cells revealed a signature distinct from Treg cells in the periphery. The differential expression of cytokine/chemokine receptors such as Il9r, Ccr2, and Cxcr3 and effector molecules such as Il10 and Ctla4 suggests increased suppressive capacity of BM resident Treg cells (68). The transcriptional signature is consistent with the idea that these niche-associated Treg cells contribute to the maintenance of the BM microenvironment as an immune privilege site, crucial for HSC survival (31, 32).
Interestingly, and reminiscent of the above-described observations, our temporal analysis of Treg cells in the BM of wildtype mice revealed that the proportion of Foxp3+ Treg cells among CD4+ T cells further increases with age. While in juvenile mice, Foxp3+ Treg cells account for approximately 20% of the CD4+ T cell population, their frequency has almost doubled in aged mice (Figure 1A). This can be recapitulated by adoptive Treg cell transfers into Treg cell-deficient mice: here we found Foxp3GFP+ Treg cells, which initially comprised about 10–15% of the adoptively transferred splenic bulk CD4+ T cell population, to be selectively enriched in the BM of Foxp3-deficient recipient mice [up to 90% of transferred CD4+ T cells (69)] and in the BM of T and B cell-deficient Rag1−/− recipient mice (up to 40% of transferred CD4+ T cells; Figure 1B), indicating the physiological relevance of the enrichment. Overall, these observations raise the question whether this accumulation of Foxp3+ Treg cells within the BM is due to preferential de novo induction of initially Foxp3-negative CD4+ T cells or preferential recruitment of preformed cells to the bone microenvironment (70, 71).
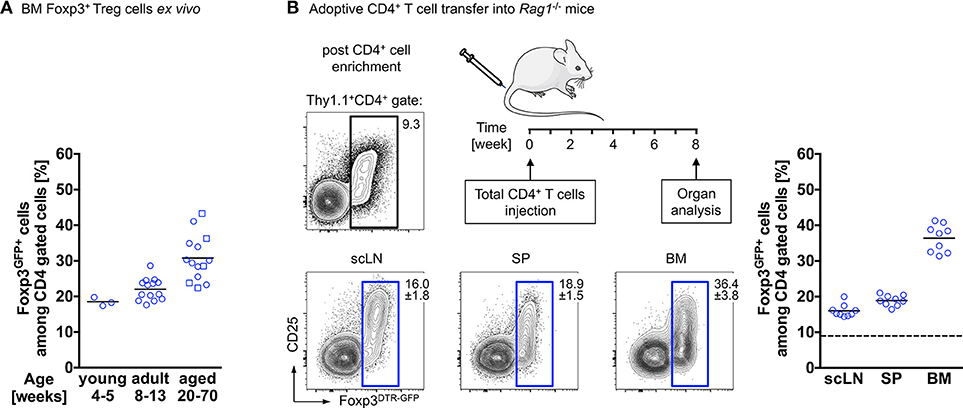
Figure 1. Accumulation of Foxp3+ Treg cells in the BM. (A) Age-related accumulation of BM-resident Foxp3+ Treg cells. Flow cytometry analysis of Foxp3GFP+ cells within the CD4+ T cell population in the BM of wildtype mice of different age groups. Each experiment was performed with at least 3 mice and symbols and lines indicate individual mice and mean values, respectively (young: 4–5 weeks; adult: 8–13 weeks; aged: circles: 20–22 weeks; and squares: 50–70 weeks). (B) Schematic overview of the experimental design. Four-week-old Rag1−/− mice were adoptively transferred with bulk CD4+ T cells (upper plot: Foxp3+ Treg cell proportion among total CD4+ T cells before adoptively transferred). The distributions of gated Foxp3DTR−GFP+ cells in various organs of the recipient mice (scLN, subcutaneous lymph nodes; SP, spleen, and BM, bone marrow) were analyzed by flow cytometry 8 weeks after transfer. Numbers in plots indicate the mean percentages ± SD of gated cells within the respective gate. Graph illustrates the accumulation of Foxp3+ Treg cells in the BM. Data are collected from two independent experiments with 4–5 mice; symbols and lines denote individual mice and mean values, respectively.
Since CD4+ Treg cells were originally identified exclusively by the constitutive expression of the IL-2 receptor alpha chain CD25, employing depleting antibodies directed against CD25 still represents the most widely used loss-of-function approach to characterize Treg cell function in vivo (72, 73). Whether the in vivo administration of anti-CD25 monoclonal antibodies (mAbs) leads to depletion or to functional inactivation of CD25-expressing Treg cells has been controversially discussed (74–77). In mouse models for RA, administration of anti-CD25 mAbs exacerbates the disease (45, 78, 79). However, interpretation of results from studies using this approach to assess the direct impact of Treg cells on bone cells is hampered due to the upregulation of CD25 expression on activated CD4 and CD8 T cells and in addition, due to the existence of CD25-negative Treg cells in particular in peripheral tissues and the BM. Consistently, anti-CD25 treatment spares Foxp3+ Treg cells with a CD25low/− phenotype (80).
Only the identification of Foxp3 as the Treg cell lineage specification factor, necessary and sufficient for their development and function (81–83) allowed specific in vivo targeting of Foxp3+ Treg cells and with this the generation of mouse models with abrogated or enhanced Foxp3+ Treg cell activity.
Lympho-Hematopoiesis in Mice With Constitutive Treg Cell Deficiency
Scurfy mice exhibit a spontaneous loss-of-function mutation in the gene encoding Foxp3, resulting in complete absence of Foxp3 protein and functional CD4+Foxp3+ Treg cells. In addition to systemic autoimmunity targeting several organs such as skin, lung, stomach and liver, severe lympho-hyperproliferation represents another hallmark of the scurfy phenotype, resulting in massively increased secondary lymphoid organs and peripheral immune effector compartments (82, 84).
In initial studies in 3 to 4-week-old scurfy mice, we unexpectedly noticed that the immature B cell compartment in the spleen was essentially completely absent, which appeared to be at odds with the existing mature B cell compartments at the same anatomical site. A more systematic analysis of peripheral B cell compartments revealed even higher numbers of mature B cells in lymph nodes of scurfy mice as compared to wildtype control mice (69). These observations raised several important questions, including the origin of mature B cells in scurfy mice and the possibility that scurfy mice exhibit a heretofore-unappreciated defect in B cell development in the BM, in addition to the well-known hematopoietic bias in scurfy mice towards macrophages (85, 86). When we extended our analysis to lympho-hematopoiesis, it became clear that adolescent scurfy mice exhibit severe B cell developmental defects with near-complete absence of B lymphopoiesis in the BM. Pre-B-II and immature B cell stages were found consistently below the detection level. Several lines of evidence such as the indistinguishability of the BM microenvironment of scurfy and Foxp3-proficient mice within the first week of life with regard to the capacity to support B cell development in vivo argue against a constitutive lympho-hematopoietic defect, but support the assumption of an ontogenetic acquisition of B cell developmental defects in scurfy mice. The apparent inconsistency between previously described significant residual B lymphopoietic activity in the BM of 28-day-old scurfy mice (39) and the almost entire block of B cell development observed in mice of the same age in our study is most likely due to differences in the kinetics of autoimmune manifestations, perhaps due to microbial flora variations between independent colonies of Foxp3-deficient mice.
Overall, our results were further corroborated by other recent studies showing that the constitutive absence of Foxp3+ Treg cells impinges on lympho-hematopoiesis in the BM although significant differences between those studies exist. In conjunction with aberrant B lymphopoiesis and in line with other reports, we detected aberrantly increased proportions and numbers of Lin−Sca-1+c-Kithi (LSK) cells early during ontogeny, suggesting that HSCs might be directly affected by inflammatory cytokines such as type I and type II IFNs. Analysis of in vivo reconstitution capacities of scurfy-derived HSCs in BM irradiation chimeras provided inconsistent results, with either substantially reduced or efficient although delayed reconstitution of lymphoid and myeloid lineages (38, 39, 87–89). Moreover, the underlying mechanism and in particular the direct and/or indirect roles of Treg cells remain a matter of debate. While it has been suggested that an altered cytokine milieu in scurfy mice promotes the differentiation of myeloid lineage cells at the expense of lymphopoiesis (87, 89, 90), indicating that the hematopoietic changes are not directly caused by the lack of Treg cells, additional mechanisms such as significantly increased cell death of developing B cells may affect B lymphopoiesis (88).
Nevertheless, our data indicate that the observed defects may not be exclusively mediated by systemic autoimmunity that negatively feeds back to BM hematopoiesis, as neonatal adoptive Treg cell therapy suppressed exacerbated production of inflammatory cytokines such as IL-17 and IL-6, but not IFN-γ and restored defective thymopoiesis, but was ineffective in recovering defective hematopoiesis and B lymphopoiesis. One possible explanation for this might be the failure to re-establish efficient production of CXCL12 and IL-7, which was consistently decreased in the BM of untreated scurfy mice (69). Thus, it appears likely that the aberrant B lymphopoiesis in scurfy mice is a consequence of the diversion of lymphoid progenitors from the B lymphoid lineage in favor of increased myelopoiesis (85–87, 89, 90) and the corporate action of the enhanced systemic production of helper T cell cytokines and locally reduced expression of B lymphopoiesis-promoting factors in the BM (69).
Overall, our observations indicate that re-establishment of immune homeostasis is not sufficient to reinstate hematopoietic homeostasis. We speculate that the adult BM niche might be irreversibly affected by constitutive Treg cell deficiency, which is in line with the severe bone loss afflicted with pronounced autoimmune pathologies, we and others (87) detected in scurfy mice.
Lympho-Hematopoiesis in Mice With Acute Treg Cell Ablation
In contrast to the above described conventional mouse models and depletion strategies, the diphtheria toxin (DT)-mediated deletion of Foxp3+ Treg cells allows for efficient, selective and temporally controlled depletion of Foxp3+ Treg cells without affecting CD25+ effector T cells (91, 92) and thus has provided essential information on Foxp3+ Treg cell biology in health and disease.
However, striking differences exist with regard to depletion efficiency and severity of clinical symptoms after Treg cell ablation (93, 94). Two independent mouse models with an insertion of a DT receptor (DTR) into either the endogenous Foxp3 locus (91) or a Foxp3 promoter-encoding bacterial artificial chromosome [BAC; these mice were termed “depletion of regulatory T cells” (DEREG) mice, (92)] were generated in parallel. In both models, expression of the DTR under the control of the Foxp3 promoter and administration of DT leads to the induction of apoptotic cell death due to blockage of protein synthesis (95), specifically in Foxp3+ Treg cells. When DT was applied to neonatal mice, both, the BAC-transgenic and the knock-in approach resulted in an autoimmune disease similar in severity to that of Foxp3-deficient scurfy mice (91, 92). However, while DT-induced Treg cell ablation in adult mice did not induce scurfy-like disease in DEREG mice (92), Foxp3DTR knock-in mice succumbed to a severe lympho-hyperproliferative disorder (91), which might be explained by different efficiencies with regard to transgene expression (93). Another study assessed BAC transgenic Foxp3DTR lines (Foxp3.LuciDTR, specifically generated for bioluminescence imaging of Treg cells) for depletion efficiencies and signs of pathology and demonstrated that lines with depletion efficiencies of >95% succumbed to a wasting disease, whereas lines with depleting efficiencies <95% lacked the onset of autoimmunity (96).
To deplete Foxp3+ Treg cells in adult steady-state mice, we therefore took advantage of the well-characterized DEREG mouse model (92–94, 97).
Our results show that upon administration of consecutive doses of DT, Foxp3GFP+ Treg cells in primary (thymus, BM) and secondary (spleen, lymph nodes, peritoneum) lymphoid organs of NOD.Foxp3DTR−GFP mice were highly efficiently depleted with minimal systemic effects due to rapid recovery of the depleted Treg cell compartment. In contrast to DT-treated C57BL/6 and Balb/c mice, NOD control mice did not show any DT side effects. As a read-out for the impact of Treg cell activity on the generation of adaptive immune cells from HSCs, we assessed the developmental B lymphopoietic activity in the absence and presence of Foxp3+ Treg cells. Our comprehensive analysis of the BM niche revealed that acute Foxp3+ Treg cell ablation in Foxp3DTR−GFP mice results in rapid dysregulation of lympho-hematopoiesis, as evidenced by a strongly increased HSC compartment size (Figure 2A) and a strong reduction of BM-residing mature B cells (Figure 2B). Of note, and in contrast to our results obtained in mice with constitutive Treg cell deficiency showing that B cell development was almost completely abrogated (69), we observed that acute Treg cell ablation in adolescent NOD.Foxp3DTR−GFP mice induced only a slight reduction in B cell development. In contrast to scurfy mice, where already the earliest c-Kit+ Pro/Pre-B-I cell precursors were significantly reduced, this compartment remained unaffected by the DT-mediated Treg cell depletion, emphasizing that the effects observed in scurfy mice are not directly caused by Treg cell deficiency.
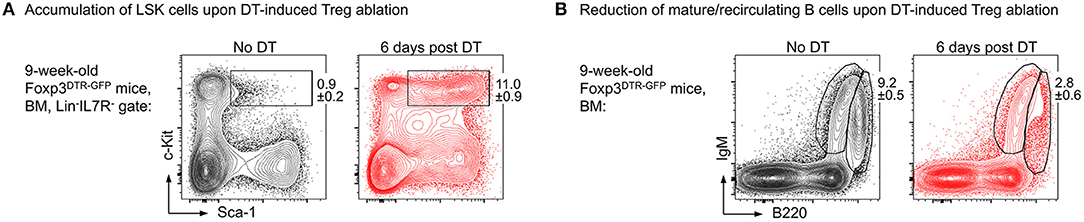
Figure 2. Impact of Foxp3+ Treg cell ablation on the BM niche. Foxp3DTR−GFP mice were i.p. injected with DT on 3 consecutive days and the BM was analyzed at day 6 after the first DT dose by flow cytometry. Representative plots of (A) LSK (Lineage−IL7R−Sca-1+c-Kit+) cells and (B) mature/recirculating B cells (B220highIgM+) in the BM of 9-week-old untreated (left) and DT-treated (right) mice.
The apparent discrepancy between the recently reported almost complete block in B cell development observed in C57BL/6 Foxp3DTR knock-in mice (98) and the only partial block in early B cell development we observed in NOD.Foxp3DTR−GFP mice, could most likely be attributed to scurfy-like lethal autoimmunity and unwanted DT side effects that have been described for the former mouse model but not for the latter (91, 92, 94, 99, 100).
Overall, the prompt alteration of B lymphopoiesis in the BM upon DT-mediated Treg cell ablation in the absence of overt autoimmunity further support a potential direct role of Treg cells in hematopoiesis.
Bone Homeostasis and Foxp3+ Treg Cells
Osteoclast Development and Treg Cells
Osteoclasts, exclusive bone-resorbing cells of hematopoietic origin, are giant multinucleated cells derived from fused precursor cells of the myeloid lineage. The development of mature osteoclasts from early myeloid progenitors is a tightly controlled process involving multiple mediators within the bone microenvironment. Osteoclastogenesis is controlled by ligation of macrophage colony-stimulating factor (c-Fms; M-CSF) and membrane receptors for receptor activator of NF-kB ligand (RANK; RANKL). Under physiological conditions, osteoblast lineage cells, which are of mesenchymal origin, seem to be the main source for M-CSF and RANKL. The central role of M-CSF in survival and proliferation of osteoclast precursors was already revealed decades ago, by studying osteopetrotic op/op mice that bear a natural mutation in the Csf1 gene coding for M-CSF (101, 102). The discovery of the RANK/RANKL/osteoprotegerin (OPG) signaling axis represents another milestone in bone biology: the finding that RANKL-deficient mice develop severe osteoclast-deficient osteopetrosis, whereas overexpression of RANKL or the lack of the decoy receptor for RANKL, OPG, result in osteoporosis, due to excessive osteoclast formation (103–105), emphasize the critical role of these pathways in osteoclastogenesis.
RANKL promotes the differentiation of committed osteoclast precursor cells by inducing the expression of several genes, such as cathepsin K, tartrate-resistant acid phosphatase (TRAP) and calcitonin receptor, as well as transcription factors, such as NF-κB, c-Fos, and the master transcription regulator of osteoclast differentiation NFATc1 (106). Important signals downstream of c-Fms include the PI3K/Akt pathway, crucial for cell survival and the MAPK/ERK pathway essential for survival and proliferation of precursor cells (4).
The discovery that activated T cells express RANKL along with the identification of RANKL as a key differentiation factor for osteoclasts represented one of the first links between the immune and the bone system (107–109). To date, the impact of numerous inflammatory cytokines on the fate of bone cells has been investigated in vitro and in vivo, and it has been shown that for example IL-1, TNF-α, IL-17, and IL-6 promote osteoclastogenesis by either inducing RANKL expression or directly acting on osteoclast precursor cells, whereas IL-10, IL-4, IFN-γ, and GM-CSF have inhibitory effects on osteoclast differentiation and function, illustrating the tight relationship of osteoclast precursor cells with other cells in the bone microenvironment such as innate and adaptive immune cells (9).
Despite extensive research efforts, osteoclast developmental stages as well as the precise identity of bona fide osteoclast precursors have remained poorly defined and thus there is still fundamental lack of reliable methods for ex vivo purification of osteoclasts and their precursors. For this reason, most studies are based on in vitro differentiated osteoclasts derived either from macrophages expanded in vitro in the presence of M-CSF or from unfractionated whole BM cells cultured under osteoclastogenic conditions, i.e., in the presence of M-CSF and RANKL. However, both methods have their drawbacks since interpretation of data obtained from these cultures is hampered by the heterogeneity of the starting population of which only a minor faction represents osteoclast progenitor cells that differ in their growth kinetics and differentiation potential. Moreover, isolation of primary cells from their in vivo microenvironment and subsequent in vitro culture results in altered phenotypic and functional characteristics of these cells.
To characterize and isolate osteoclast precursor cells, we combined complex state-of-the-art experimental approaches such as osteoclast differentiation and functional assessment (110, 111) with high-end flow cytometry and cell sorting of very rare cell populations employing a comprehensive array of hematopoietic cell surface markers suggested in varying combinations in the literature (112–116). Multicolor flow cytometry allowed for identification and prospective isolation of these infrequent cell subsets, which — in some cases — account for <0.1% of total BM cells in wildtype mice. To determine the osteoclastogenic potential of the identified populations, FACS-purified cells were cultured in the presence of M-CSF and RANKL. Differentiated osteoclasts were detected as giant, multinucleated, and TRAP-positive cells.
With regard to CD11b expression on potential progenitor cells earlier studies have yielded contradictory results: while some groups described CD11b+ precursor cells to exhibit high osteoclastogenic potential, others reported on CD11b−/low cells being more potent (114–118). In our hands, B220−CD3−Ter119−CD11b+ BM cells did not have a high osteoclastogenic potential, independent of the expression of Ly6C, whereas the B220−CD3−Ter119−CD11b−/lowLy6Chi cell population contained the majority of osteoclast progenitors (Figure 3, left), which is in accordance with recent findings by Nakamura and colleagues (116). Since that study detected heterogeneous CD115 (c-Fms) and CD117 (c-Kit) expression on B220−CD3−Ter119−CD11b−/lowLy6Chi osteoclast progenitor cells (116) and others identified CD115+CD117+ double positive cells as highly osteoclastogenic (113, 115), we applied in our study a combination of the surface markers used in these studies. Our results show that B220−CD3−Ter119−CD11b−/lowLy6ChiCD115+CD117+ BM cells efficiently differentiate into mature TRAP-expressing osteoclasts in vitro (Figure 3, right). Initial analyses of the BM niche of Foxp3DTR−GFP mice provided first evidence that acute Foxp3+ Treg cell ablation results in substantial changes in osteoclast precursors in the BM.
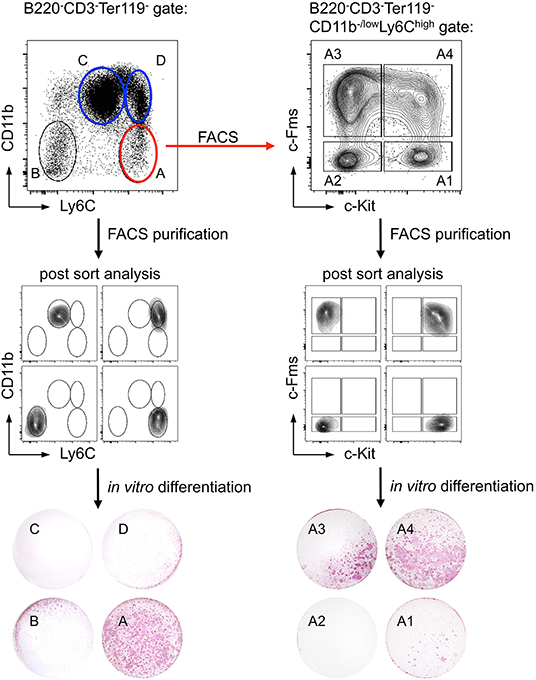
Figure 3. Identification of a cell population with high in vitro osteoclast differentiation potential. Ly6C and CD11b expression of bead-enriched B220−CD3−Ter119− BM cells was detected by flow cytometry. Four distinct populations (A: Ly6ChighCD11b−/low; B: Ly6C−CD11b−; C: Ly6ClowCD11b+; D: Ly6C+CD11b+) were FACS-purified and after post-sort analysis in vitro differentiated under osteoclast-forming conditions (cytokines: M-CSF and RANKL). The differentiated cultures were fixed and a TRAP staining was performed to identify mature osteoclasts. Additionally, population A was further subdivided by the expression of c-Kit and c-Fms (A1: c-Kit+c-Fms−; A2: c-Kit−c-Fms−; A3: c-Kit−c-Fms+; A4: c-Kit+c-Fms+). After FACS-purification and post-sort analysis, the populations were cultured and analyzed for their osteoclastogenic potential.
The sorting of low-abundance prospective primary osteoclast precursors to high purity allow for detailed ex vivo analysis without contamination by unwanted cells and hence represents a powerful method to evaluate how Treg cells are affecting the formation of osteoclast precursors in mouse models with abrogated or enhanced Treg cell activity.
Osteoclast Function and Treg Cells
Foxp3+ Treg cells control adaptive and innate immune responses by the suppression of activation, proliferation and function of various immune cell types, as for example CD4+ helper T cells, CD8+ cytotoxic T cells, B cells, NKT cells, macrophages, and dendritic cells (DCs). Multiple mechanisms of Foxp3+ Treg cell-mediated suppression have been proposed, involving cell contact-dependent and cell contact-independent mechanisms. The basic mechanisms used by Treg cells include (a) secretion of inhibitory cytokines, such as TGF-β, IL-10, and IL-35, (b) induction of cytolysis by for example granzymes, (c) metabolic disruption by e.g., IL-2-deprivation-mediated apoptosis, and (d) functional modification of antigen presenting cells (APCs) such as DCs. Expression of the cytotoxic T-lymphocyte antigen 4 (CTLA-4) enables Treg cells to interact with CD80/CD86 on DCs outcompeting CD28-mediated co-stimulation and thereby indirectly preventing differentiation of effector T cells (119).
In this context, it has been suggested that Treg cells have the ability to suppress osteoclastogenesis in vitro, but the mechanisms of suppression remain incompletely understood and controversial: while some studies identified inhibitory cytokines as key players in Treg cell-mediated suppression of osteoclasts, other reports suggested cell-cell-contact dependent mechanisms (40, 47, 48, 120–122). Most studies addressing the impact of Foxp3+ Treg cells on bone homeostasis in vivo employed mouse models with either constitutive Foxp3 deficiency or constitutive Foxp3 overexpression (48, 87, 123). As mice with constitutive Foxp3 deficiency suffer from a massive autoimmune lymphoproliferative disease and die approximately at 3 weeks of age (84) and overexpression of Foxp3 in CD4+ T cells in vivo results in reduced total numbers and functional impairment of T cells (124, 125) potential direct effects of gain and loss of Foxp3+ Treg cells are difficult to dissect from the impact of systemic immune dysregulation.
To our knowledge, the impact of temporally controlled, transient Foxp3+ Treg cell ablation (91, 92) on skeletal homeostasis under physiological conditions has not been addressed so far.
Osteoclasts and DCs share several features (126, 127). In this context, a recent study proposed that osteoclasts express the co-stimulatory molecules CD80 and CD86 and that Treg cells can regulate osteoclast differentiation via CTLA-4 (128). Moreover, it has been described that BM-residing Treg cells express higher levels of CTLA-4 than peripheral Treg cells (68). Therefore, we hypothesize that this direct cell-cell interaction may indeed play a central role in the crosstalk of osteoclasts and Treg cells.
To investigate the direct interaction of osteoclasts and Treg cells, we performed in vitro co-cultures of Treg cells and BM-derived precursor cells. These experiments revealed that the expression of co-stimulatory molecules on the surface of osteoclasts is down-regulated in the presence of Treg cells which leads to the suppression of osteoclast differentiation and function. To directly study cell-cell interactions, cells harvested from these cultures were subjected to simultaneous analysis by multi-parameter flow cytometry and fluorescence microscopy. Imaging flow cytometry showed the direct cell-cell interaction between individual CD11b+ late osteoclast precursors and Foxp3GFP+ Treg cells which additionally exhibited intracellular red puncta (Figure 4), providing first direct evidence that Treg cells can remove CD80/CD86 from the surface of osteoclast precursors by CTLA-4 mediated trans-endocytosis, potentially leading to reduced co-stimulation by osteoclasts — a mechanism which has been described for the interaction of Treg cells and APCs such as DCs (129).
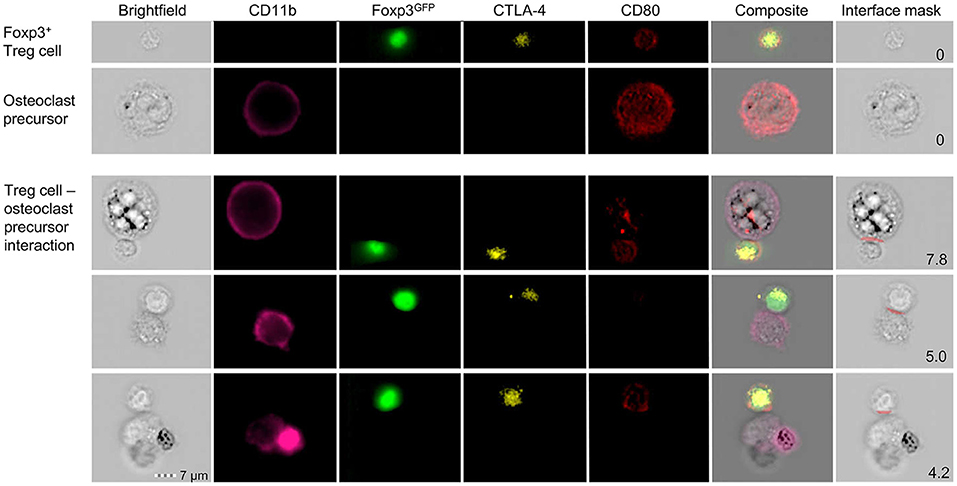
Figure 4. Detailed characterization of in vitro Foxp3+ Treg cell-osteoclast precursor interaction. Representative single cell (upper panels) and cluster (lower panels) analysis by imaging flow cytometry. Stimulated FACS-purified CD4+CD25+Foxp3GFP+ Treg cells and freshly isolated irradiated T-cell-depleted splenocytes were added to the BM culture and cultured under osteoclastogenic conditions. After 48 h, Foxp3+ Treg cells and CD11b+ osteoclast precursors were analyzed for intracellular expression of CD80 and CTLA-4 by imaging flow cytometry. Cell clusters depict physical interaction of CTLA-4+Foxp3GFP+ Treg cells and CD11b+ osteoclast precursors. Co-localization of CD80 in Treg cells indicates trans-endocytosis from CD11b+ osteoclast precursors. Numbers indicate the interface area and illustrate the overlapping fluorescence of Foxp3 and CD11b.
In sum, available data suggest that Treg cells can interfere with bone resorption by affecting osteoclast development and function at multiple stages. However, the mechanism of action is still controversial, emphasizing the complexity of this relationship.
Whether or not T cell regulation of bone and hematopoietic homeostasis requires APCs within the bone microenvironment, is still unknown, but based on the notion that osteoclasts are derived from hematopoietic precursors of the myeloid lineage, express a number of immune receptors and are regulated similarly to macrophages and DCs (130), it is a legitimate question, whether osteoclasts play a role in the active regulation of the immune system, can act e.g., as APCs. Indeed, albeit critically discussed (131), it has been suggested that, in addition to their ability to resorb bone, in vitro differentiated osteoclasts can function as APCs and activate CD4+ and CD8+ cells (132). In this context, it was reported that osteoclasts are capable to cross-present antigens to CD8+ T cells, thereby inducing Foxp3 in these cells, which were characterized as osteoclast-induced regulatory CD8+ T cells (133–135).
In addition, recent data suggest that osteoclasts can on the one hand participate in immunogenic T cell responses under chronic inflammatory diseases (136) and on the other hand exhibit immune suppressive functions in multiple myeloma (137, 138). Whether these proposed immune-modulating functions of osteoclasts play a role under physiological conditions or depend on the pathological conditions still needs to be defined. Data from our own group propose the concomitant up-regulation of c-Fms, RANK, and the ligand for the immune checkpoint molecule programmed death 1 protein (PD-1; PD-L1) on the surface of murine CD11b+ osteoclast precursors cultured under osteoclastogenic conditions, which complements the findings on human osteoclast precursor cells (137).
Overall, these observations open additional new prospects for the role of osteoclast in the intercellular interaction of bone cells and immune cells in health and disease (Figure 5).
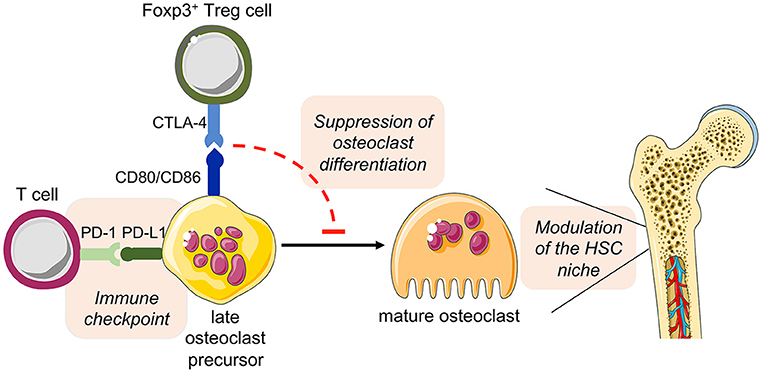
Figure 5. Hypothesized direct cross-talk between osteoclast precursors and T cells and the indirect effect on hematopoiesis. The expression of the costimulatory molecules CD80 and CD86 on osteoclast precursors has been implicated in the interplay with Foxp3+ Treg cells (via the inhibitory molecule CTLA-4), leading to the suppression of the differentiation of osteoclast precursors into mature functional osteoclasts. As a consequence, disturbed bone remodeling results in modulation of the HSC niche. On the other hand, osteoclast precursors are able to upregulate the expression of PD-L1, whose interaction with PD-1 on T cells has immune modulatory functions, thus representing a crucial immune checkpoint. This figure was in part created with modified Servier Medical Art templates, licensed under a Creative Commons Attribution 3.0 Unported license: http://smart.servier.com.
Conclusion
The clinical relevance of the BM niche is reflected by its function as (a) the primary hematopoietic site in the adult, (b) a reservoir for memory cells, and (c) in the support of HSC transplantation. The nature of the HSC niche in the BM is defined by various components of the BM microenvironment. Several independent lines of evidence are pointing towards specialized local functions of BM-residing Treg cells in settings of unwanted (autoimmunity, transplant rejection, GvHD) and insufficient immunity (cancer, chronic infections). Modulation of Treg cell activity in vivo can be achieved by means such as application of IL-2/anti-IL-2 immune complexes to enhance Treg cell function, whereas depletion or other means of interference with Treg cell function promotes immune responses. This raises the question whether comparable strategies may also be suitable to modulate bone remodeling to manipulate lympho-hematopoiesis in the adult BM. Are there niche-associated Treg cell subsets that specifically interact with bone cells such as osteoclasts? Overall, future studies are warranted to further define the molecular pathways involved in the complex intercellular communication between BM-residing Treg cells, hematopoietic stem and progenitor cells, mature immune cells, and the skeletal system. Understanding the immune regulatory mechanisms in the BM microenvironment is central for the development of effective therapeutic strategies for the treatment of bone diseases and to improve current protocols for HSC transplantation and peripheral immune reconstitution.
Author Contributions
LF, CH, RK, SD, and JR designed, performed, and analyzed the experiments. KK and AG conceived the research, guided its design, analysis, and interpretation and wrote the manuscript. All authors contributed to discussions and writing of the manuscript.
Funding
AG was supported by a DFG grant (GA1576/1-2) within the SPP1468 IMMUNOBONE. In addition, AG and KK received funds from the FZT 111 (DFG, Center for Regenerative Therapies Dresden, Cluster of Excellence).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Tim Sparwasser (University Medical Center of the Johannes Gutenberg University, Mainz, Germany) for providing DEREG mice.
References
1. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. (2014) 505:327–34. doi: 10.1038/nature12984
2. Baryawno N, Severe N, Scadden DT. Hematopoiesis: reconciling historic controversies about the Niche. Cell Stem Cell. (2017) 20:590–2. doi: 10.1016/j.stem.2017.03.025
3. Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann N Y Acad Sci. (2007) 1116:281–90. doi: 10.1196/annals.1402.018
4. Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. (2008) 3:457–84. doi: 10.1146/annurev.pathmechdis.3.121806.151431
5. Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. (2006) 12:657–64. doi: 10.1038/nm1417
6. Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SE, Wakkach A, Blin-Wakkach C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med. (2012) 209:537–49. doi: 10.1084/jem.20110994
7. Miyamoto K, Yoshida S, Kawasumi M, Hashimoto K, Kimura T, Sato Y, et al. Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J Exp Med. (2011) 208:2175–81. doi: 10.1084/jem.20101890
8. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. (2003) 425:841–6. doi: 10.1038/nature02040
9. Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev. (2017) 97:1295–349. doi: 10.1152/physrev.00036.2016
10. Walsh MC, Takegahara N, Kim H, Choi Y. Updating osteoimmunology: regulation of bone cells by innate and adaptive immunity. Nat Rev Rheumatol. (2018) 14:146–56. doi: 10.1038/nrrheum.2017.213
11. Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. (2014) 20:1315–20. doi: 10.1038/nm.3707
12. Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. (2014) 20:1321–6. doi: 10.1038/nm.3706
13. Jiang L, Han X, Wang J, Wang C, Sun X, Xie J, et al. SHP-1 regulates hematopoietic stem cell quiescence by coordinating TGF-beta signaling. J Exp Med. (2018) 215:1337–47. doi: 10.1084/jem.20171477
14. Kacena MA, Shivdasani RA, Wilson K, Xi Y, Troiano N, Nazarian A, et al. Megakaryocyte-osteoblast interaction revealed in mice deficient in transcription factors GATA-1 and NF-E2. J Bone Miner Res. (2004) 19:652–60. doi: 10.1359/JBMR.0301254
15. Kacena MA, Gundberg CM, Horowitz MC. A reciprocal regulatory interaction between megakaryocytes, bone cells, and hematopoietic stem cells. Bone. (2006) 39:978–84. doi: 10.1016/j.bone.2006.05.019
16. Kacena MA, Nelson T, Clough ME, Lee SK, Lorenzo JA, Gundberg CM, et al. Megakaryocyte-mediated inhibition of osteoclast development. Bone. (2006) 39:991–9. doi: 10.1016/j.bone.2006.05.004
17. Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. (2010) 116:4815–28. doi: 10.1182/blood-2009-11-253534
18. Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. (2011) 208:261–71. doi: 10.1084/jem.20101688
19. Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med. (2011) 208:251–60. doi: 10.1084/jem.20101700
20. Hur J, Choi JI, Lee H, Nham P, Kim TW, Chae CW, et al. CD82/KAI1 Maintains the dormancy of long-term hematopoietic stem cells through interaction with DARC-expressing macrophages. Cell Stem Cell. (2016) 18:508–21. doi: 10.1016/j.stem.2016.01.013
21. Szade K, Gulati GS, Chan CKF, Kao KS, Miyanishi M, Marjon KD, et al. Where hematopoietic stem cells live: the bone marrow niche. Antioxid Redox Signal. (2018) 29:191–204. doi: 10.1089/ars.2017.7419
22. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. (2019) 20:303–20. doi: 10.1038/s41580-019-0103-9
23. Michalski MN, McCauley LK. Macrophages and skeletal health. Pharmacol Ther. (2017) 174:43–54. doi: 10.1016/j.pharmthera.2017.02.017
24. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. (2007) 109:3839–48. doi: 10.1182/blood-2006-07-037994
25. Lee SK, Kadono Y, Okada F, Jacquin C, Koczon-Jaremko B, Gronowicz G, et al. T lymphocyte-deficient mice lose trabecular bone mass with ovariectomy. J Bone Miner Res. (2006) 21:1704–12. doi: 10.1359/jbmr.060726
26. Horowitz MC, Xi Y, Pflugh DL, Hesslein DG, Schatz DG, Lorenzo JA, et al. Pax5-deficient mice exhibit early onset osteopenia with increased osteoclast progenitors. J Immunol. (2004) 173:6583–91. doi: 10.4049/jimmunol.173.11.6583
27. Sauer AV, Mrak E, Hernandez RJ, Zacchi E, Cavani F, Casiraghi M, et al. ADA-deficient SCID is associated with a specific microenvironment and bone phenotype characterized by RANKL/OPG imbalance and osteoblast insufficiency. Blood. (2009) 114:3216–26. doi: 10.1182/blood-2009-03-209221
28. Di Rosa F. Two Niches in the bone marrow: a hypothesis on life-long T cell memory. Trends Immunol. (2016) 37:503–12. doi: 10.1016/j.it.2016.05.004
29. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. (1997) 388:133–4. doi: 10.1038/40540
30. Geerman S, Nolte MA. Impact of T cells on hematopoietic stem and progenitor cell function: Good guys or bad guys? World J Stem Cells. (2017) 9:37–44. doi: 10.4252/wjsc.v9.i2.37
31. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. (2011) 474:216–9. doi: 10.1038/nature10160
32. Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150(high) bone marrow tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell. (2018) 22:445–53.e5. doi: 10.1016/j.stem.2018.01.017
33. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. (2005) 6:345–52. doi: 10.1038/ni1178
34. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
35. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
36. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
37. Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. (2013) 14:1007–13. doi: 10.1038/ni.2683
38. Chen C, Liu Y, Liu Y, Zheng P. Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J Clin Invest. (2010) 120:4091–101. doi: 10.1172/JCI43873
39. Leonardo SM, Josephson JA, Hartog NL, Gauld SB. Altered B cell development and anergy in the absence of Foxp3. J Immunol. (2010) 185:2147–56. doi: 10.4049/jimmunol.1000136
40. Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, et al. Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. (2007) 56:4104–12. doi: 10.1002/art.23138
41. Wang T, Qiao H, Zhai Z, Zhang J, Tu J, Zheng X, et al. Plumbagin ameliorates collagen-induced arthritis by regulating Treg/Th17 cell imbalances and suppressing osteoclastogenesis. Front Immunol. (2018) 9:3102. doi: 10.3389/fimmu.2018.03102
42. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. (2007) 7:429–42. doi: 10.1038/nri2094
43. Gravallese EM. Bone destruction in arthritis. Ann Rheum Dis. (2002) 61(Suppl. 2):ii84–6. doi: 10.1136/ard.61.suppl_2.ii84
44. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. (2006) 203:2673–82. doi: 10.1084/jem.20061775
45. Morgan ME, Sutmuller RP, Witteveen HJ, van Duivenvoorde LM, Zanelli E, Melief CJ, et al. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. (2003) 48:1452–60. doi: 10.1002/art.11063
46. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. (2005) 52:2212–21. doi: 10.1002/art.21195
47. Kelchtermans H, Geboes L, Mitera T, Huskens D, Leclercq G, Matthys P. Activated CD4+CD25+ regulatory T cells inhibit osteoclastogenesis and collagen-induced arthritis. Ann Rheum Dis. (2009) 68:744–50. doi: 10.1136/ard.2007.086066
48. Zaiss MM, Frey B, Hess A, Zwerina J, Luther J, Nimmerjahn F, et al. Regulatory T cells protect from local and systemic bone destruction in arthritis. J Immunol. (2010) 184:7238–46. doi: 10.4049/jimmunol.0903841
49. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
50. Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. (2009) 10:229–40. doi: 10.1016/j.cmet.2009.07.010
51. Li JY, Walker LD, Tyagi AM, Adams J, Weitzmann MN, Pacifici R. The sclerostin-independent bone anabolic activity of intermittent PTH treatment is mediated by T-cell-produced Wnt10b. J Bone Miner Res. (2014) 29:43–54. doi: 10.1002/jbmr.2044
52. Tyagi AM, Yu M, Darby TM, Vaccaro C, Li JY, Owens JA, et al. The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of WNT10B expression. Immunity. (2018) 49:1116–31.e7. doi: 10.1016/j.immuni.2018.10.013
53. Yu M, D'Amelio P, Tyagi AM, Vaccaro C, Li JY, Hsu E, et al. Regulatory T cells are expanded by Teriparatide treatment in humans and mediate intermittent PTH-induced bone anabolism in mice. EMBO Rep. (2018) 19:156–71. doi: 10.15252/embr.201744421
54. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol. (2015) 6:184. doi: 10.3389/fphar.2015.00184
56. King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol. (2011) 11:685–92. doi: 10.1038/nri3062
57. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. (2017) 130:1693–8. doi: 10.1182/blood-2017-06-780882
58. Christopher MJ, Liu F, Hilton MJ, Long F, Link DC. Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood. (2009) 114:1331–9. doi: 10.1182/blood-2008-10-184754
59. Sato N, Takahashi N, Suda K, Nakamura M, Yamaki M, Ninomiya T, et al. MyD88 but not TRIF is essential for osteoclastogenesis induced by lipopolysaccharide, diacyl lipopeptide, and IL-1alpha. J Exp Med. (2004) 200:601–11. doi: 10.1084/jem.20040689
60. Hayashi S, Yamada T, Tsuneto M, Yamane T, Takahashi M, Shultz LD, et al. Distinct osteoclast precursors in the bone marrow and extramedullary organs characterized by responsiveness to Toll-like receptor ligands and TNF-alpha. J Immunol. (2003) 171:5130–9. doi: 10.4049/jimmunol.171.10.5130
61. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. (2006) 124:407–21. doi: 10.1016/j.cell.2005.10.041
62. Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
63. Lucas D. Leukocyte trafficking and regulation of murine hematopoietic stem cells and their niches. Front Immunol. (2019) 10:387. doi: 10.3389/fimmu.2019.00387
64. Decker M, Leslie J, Liu Q, Ding L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science. (2018) 360:106–10. doi: 10.1126/science.aap8861
65. Casanova-Acebes M, Nicolas-Avila JA, Li JL, Garcia-Silva S, Balachander A, Rubio-Ponce A, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med. (2018) 215:2778–95. doi: 10.1084/jem.20181468
66. Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. (2005) 26:360–6. doi: 10.1016/j.it.2005.04.011
67. Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. (2004) 64:8451–5. doi: 10.1158/0008-5472.CAN-04-1987
68. Glatman Zaretsky A, Konradt C, Depis F, Wing JB, Goenka R, Atria DG, et al. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. (2017) 18:1906–16. doi: 10.1016/j.celrep.2017.01.067
69. Riewaldt J, Duber S, Boernert M, Krey M, Dembinski M, Weiss S, et al. Severe developmental B lymphopoietic defects in foxp3-deficient mice are refractory to adoptive regulatory T cell therapy. Front Immunol. (2012) 3:141. doi: 10.3389/fimmu.2012.00141
70. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. (2005) 6:1219–27. doi: 10.1038/ni1265
71. Kretschmer K, Apostolou I, Jaeckel E, Khazaie K, von Boehmer H. Making regulatory T cells with defined antigen specificity: role in autoimmunity and cancer. Immunol Rev. (2006) 212:163–9. doi: 10.1111/j.0105-2896.2006.00411.x
72. Taguchi O, Takahashi T. Administration of anti-interleukin-2 receptor alpha antibody in vivo induces localized autoimmune disease. Eur J Immunol. (1996) 26:1608–12. doi: 10.1002/eji.1830260730
73. McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. (2002) 168:5979–83. doi: 10.1023/A:1020665614139
74. Kohm AP, McMahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ, et al. Cutting Edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol. (2006) 176:3301–5. doi: 10.4049/jimmunol.176.6.3301
75. Stephens LA, Anderton SM. Comment on “cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J Immunol. (2006) 177:2036; author reply 7–8. doi: 10.4049/jimmunol.177.4.2036
76. Zelenay S, Demengeot J. Comment on “cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J Immunol. (2006) 177:2036–7; author reply 7–8. doi: 10.4049/jimmunol.177.4.2036-a
77. Huss DJ, Pellerin AF, Collette BP, Kannan AK, Peng L, Datta A, et al. Anti-CD25 monoclonal antibody Fc variants differentially impact regulatory T cells and immune homeostasis. Immunology. (2016) 148:276–86. doi: 10.1111/imm.12609
78. Kelchtermans H, De Klerck B, Mitera T, Van Balen M, Bullens D, Billiau A, et al. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther. (2005) 7:R402–15. doi: 10.1186/ar1500
79. Frey O, Petrow PK, Gajda M, Siegmund K, Huehn J, Scheffold A, et al. The role of regulatory T cells in antigen-induced arthritis: aggravation of arthritis after depletion and amelioration after transfer of CD4+CD25+ T cells. Arthritis Res Ther. (2005) 7:R291–301. doi: 10.1186/ar1484
80. Couper KN, Blount DG, de Souza JB, Suffia I, Belkaid Y, Riley EM. Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti-CD25 treatment in malaria-infected mice. J Immunol. (2007) 178:4136–46. doi: 10.4049/jimmunol.178.7.4136
81. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
82. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
83. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. (2003) 4:337–42. doi: 10.1038/ni909
84. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
85. Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. (1999) 162:2546–54.
86. Lee JH, Wang C, Kim CH. FoxP3+ regulatory T cells restrain splenic extramedullary myelopoiesis via suppression of hemopoietic cytokine-producing T cells. J Immunol. (2009) 183:6377–86. doi: 10.4049/jimmunol.0901268
87. Chen TH, Swarnkar G, Mbalaviele G, Abu-Amer Y. Myeloid lineage skewing due to exacerbated NF-kappaB signaling facilitates osteopenia in Scurfy mice. Cell Death Dis. (2015) 6:e1723. doi: 10.1038/cddis.2015.87
88. Chang SE, Guo L, Tian J, Liu Y, Guo Z, Zheng B, et al. Autoimmune bone marrow environment severely inhibits B cell development by inducing extensive cell death and inhibiting proliferation. Autoimmunity. (2012) 45:210–7. doi: 10.3109/08916934.2011.632455
89. Skuljec J, Cabanski M, Surdziel E, Lachmann N, Brennig S, Pul R, et al. Monocyte/macrophage lineage commitment and distribution are affected by the lack of regulatory T cells in scurfy mice. Eur J Immunol. (2016) 46:1656–68. doi: 10.1002/eji.201546200
90. Kim S, Park K, Choi J, Jang E, Paik DJ, Seong RH, et al. Foxp3+ regulatory T cells ensure B lymphopoiesis by inhibiting the granulopoietic activity of effector T cells in mouse bone marrow. Eur J Immunol. (2015) 45:167–79. doi: 10.1002/eji.201444532
91. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. (2007) 8:191–7. doi: 10.1038/ni1428
92. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. (2007) 204:57–63. doi: 10.1084/jem.20061852
93. Lahl K, Sparwasser T. In vivo depletion of FoxP3+ Tregs using the DEREG mouse model. Methods Mol Biol. (2011) 707:157–72. doi: 10.1007/978-1-61737-979-6_10
94. Mayer CT, Lahl K, Milanez-Almeida P, Watts D, Dittmer U, Fyhrquist N, et al. Advantages of Foxp3(+) regulatory T cell depletion using DEREG mice. Immun Inflamm Dis. (2014) 2:162–5. doi: 10.1002/iid3.33
95. Saito M, Iwawaki T, Taya C, Yonekawa H, Noda M, Inui Y, et al. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat Biotechnol. (2001) 19:746–50. doi: 10.1038/90795
96. Suffner J, Hochweller K, Kuhnle MC, Li X, Kroczek RA, Garbi N, et al. Dendritic cells support homeostatic expansion of Foxp3+ regulatory T cells in Foxp3.LuciDTR mice. J Immunol. (2010) 184:1810–20. doi: 10.4049/jimmunol.0902420
97. Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, et al. Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol. (2009) 183:7631–4. doi: 10.4049/jimmunol.0804308
98. Pierini A, Nishikii H, Baker J, Kimura T, Kwon HS, Pan Y, et al. Foxp3(+) regulatory T cells maintain the bone marrow microenvironment for B cell lymphopoiesis. Nat Commun. (2017) 8:15068. doi: 10.1038/ncomms15068
99. Lahl K, Mayer CT, Bopp T, Huehn J, Loddenkemper C, Eberl G, et al. Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J Immunol. (2009) 183:5662–72. doi: 10.4049/jimmunol.0803762
100. Mayer CT, Ghorbani P, Kuhl AA, Stuve P, Hegemann M, Berod L, et al. Few Foxp3(+) regulatory T cells are sufficient to protect adult mice from lethal autoimmunity. Eur J Immunol. (2014) 44:2990–3002. doi: 10.1002/eji.201344315
101. Wiktor-Jedrzejczak WW, Ahmed A, Szczylik C, Skelly RR. Hematological characterization of congenital osteopetrosis in op/op mouse. Possible mechanism for abnormal macrophage differentiation. J Exp Med. (1982) 156:1516–27. doi: 10.1084/jem.156.5.1516
102. Felix R, Cecchini MG, Hofstetter W, Elford PR, Stutzer A, Fleisch H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J Bone Miner Res. (1990) 5:781–9. doi: 10.1002/jbmr.5650050716
103. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. (1999) 402:304–9. doi: 10.1038/35005552
104. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. doi: 10.1038/16852
105. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. (1998) 12:1260–8. doi: 10.1101/gad.12.9.1260
106. Novack DV, Mbalaviele G. Osteoclasts-key players in skeletal health and disease. Microbiol Spectr. (2016) 4:1–19. doi: 10.1128/microbiolspec.MCHD-0011-2015
107. Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. (1997) 272:25190–4. doi: 10.1074/jbc.272.40.25190
108. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. (1998) 93:165–76. doi: 10.1016/S0092-8674(00)81569-X
109. Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. (1998) 95:3597–602. doi: 10.1073/pnas.95.7.3597
110. Lutter AH, Hempel U, Wolf-Brandstetter C, Garbe AI, Goettsch C, Hofbauer LC, et al. A novel resorption assay for osteoclast functionality based on an osteoblast-derived native extracellular matrix. J Cell Biochem. (2010) 109:1025–32. doi: 10.1002/jcb.22485
111. Garbe AI, Roscher A, Schuler C, Lutter AH, Glosmann M, Bernhardt R, et al. Regulation of bone mass and osteoclast function depend on the F-actin modulator SWAP-70. J Bone Miner Res. (2012) 27:2085–96. doi: 10.1002/jbmr.1670
112. Muguruma Y, Lee MY. Isolation and characterization of murine clonogenic osteoclast progenitors by cell surface phenotype analysis. Blood. (1998) 91:1272–9.
113. Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. (1999) 190:1741–54. doi: 10.1084/jem.190.12.1741
114. Jacome-Galarza CE, Lee SK, Lorenzo JA, Aguila HL. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J Bone Miner Res. (2013) 28:1203–13. doi: 10.1002/jbmr.1822
115. Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res. (2006) 21:67–77. doi: 10.1359/JBMR.051007
116. Charles JF, Hsu LY, Niemi EC, Weiss A, Aliprantis AO, Nakamura MC. Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J Clin Invest. (2012) 122:4592–605. doi: 10.1172/JCI60920
117. Li P, Schwarz EM, O'Keefe RJ, Ma L, Looney RJ, Ritchlin CT, et al. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. (2004) 50:265–76. doi: 10.1002/art.11419
118. Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, et al. Tumor necrosis factor-alpha increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem. (2006) 281:11846–55. doi: 10.1074/jbc.M512624200
119. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
120. Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB. Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun. (2007) 357:1046–52. doi: 10.1016/j.bbrc.2007.04.042
121. Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, et al. CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis. (2008) 67:1603–9. doi: 10.1136/ard.2007.080713
122. Luo CY, Wang L, Sun C, Li DJ. Estrogen enhances the functions of CD4(+)CD25(+)Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol. (2011) 8:50–8. doi: 10.1038/cmi.2010.54
123. Zaiss MM, Sarter K, Hess A, Engelke K, Bohm C, Nimmerjahn F, et al. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum. (2010) 62:2328–38. doi: 10.1002/art.27535
124. Khattri R, Kasprowicz D, Cox T, Mortrud M, Appleby MW, Brunkow ME, et al. The amount of scurfin protein determines peripheral T cell number and responsiveness. J Immunol. (2001) 167:6312–20. doi: 10.4049/jimmunol.167.11.6312
125. Kasprowicz DJ, Droin N, Soper DM, Ramsdell F, Green DR, Ziegler SF. Dynamic regulation of FoxP3 expression controls the balance between CD4+ T cell activation and cell death. Eur J Immunol. (2005) 35:3424–32. doi: 10.1002/eji.200526339
126. Xiao Y, Zijl S, Wang L, de Groot DC, van Tol MJ, Lankester AC, et al. Identification of the common origins of osteoclasts, macrophages, and dendritic cells in human hematopoiesis. Stem Cell Reports. (2015) 4:984–94. doi: 10.1016/j.stemcr.2015.04.012
127. Laperine O, Blin-Wakkach C, Guicheux J, Beck-Cormier S, Lesclous P. Dendritic-cell-derived osteoclasts: a new game changer in bone-resorption-associated diseases. Drug Discov Today. (2016) 21:1345–54. doi: 10.1016/j.drudis.2016.04.022
128. Bozec A, Zaiss MM, Kagwiria R, Voll R, Rauh M, Chen Z, et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med. (2014) 6:235ra60. doi: 10.1126/scitranslmed.3007764
129. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
130. Wu Y, Humphrey MB, Nakamura MC. Osteoclasts - the innate immune cells of the bone. Autoimmunity. (2008) 41:183–94. doi: 10.1080/08916930701693180
131. Lorenzo JA. Do osteoclasts have dual roles: bone resorption and antigen presentation? IBMS BoneKEy. (2011) 8:37–40. doi: 10.1138/20110488
132. Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood. (2010) 116:210–7. doi: 10.1182/blood-2009-11-255026
133. Kiesel JR, Buchwald ZS, Aurora R. Cross-presentation by osteoclasts induces FoxP3 in CD8+ T cells. J Immunol. (2009) 182:5477–87. doi: 10.4049/jimmunol.0803897
134. Buchwald ZS, Kiesel JR, DiPaolo R, Pagadala MS, Aurora R. Osteoclast activated FoxP3+ CD8+ T-cells suppress bone resorption in vitro. PLoS ONE. (2012) 7:e38199. doi: 10.1371/journal.pone.0038199
135. Buchwald ZS, Kiesel JR, Yang C, DiPaolo R, Novack DV, Aurora R. Osteoclast-induced Foxp3+ CD8 T-cells limit bone loss in mice. Bone. (2013) 56:163–73. doi: 10.1016/j.bone.2013.05.024
136. Ibanez L, Abou-Ezzi G, Ciucci T, Amiot V, Belaid N, Obino D, et al. Inflammatory osteoclasts prime TNFalpha-producing CD4(+) T cells and express CX3 CR1. J Bone Miner Res. (2016) 31:1899–908. doi: 10.1002/jbmr.2868
137. An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood. (2016) 128:1590–603. doi: 10.1182/blood-2016-03-707547
Keywords: bone disorders, bone microenvironment, lympho-hematopoiesis, osteoclasts, Foxp3+ Treg cells
Citation: Fischer L, Herkner C, Kitte R, Dohnke S, Riewaldt J, Kretschmer K and Garbe AI (2019) Foxp3+ Regulatory T Cells in Bone and Hematopoietic Homeostasis. Front. Endocrinol. 10:578. doi: 10.3389/fendo.2019.00578
Received: 17 May 2019; Accepted: 08 August 2019;
Published: 10 September 2019.
Edited by:
Giacomina Brunetti, University of Bari Aldo Moro, ItalyReviewed by:
Rajeev Aurora, Saint Louis University, United StatesTingyu Wang, Shanghai Jiao-Tong University School of Medicine, China
Copyright © 2019 Fischer, Herkner, Kitte, Dohnke, Riewaldt, Kretschmer and Garbe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annette I. Garbe, YW5uZXR0ZS5nYXJiZUB0dS1kcmVzZGVuLmRl
†Present address: Julia Riewaldt, Cellex Patient Treatment GmbH, Dresden, Germany