 Jesús Argente
Jesús Argente Katrina Tatton-Brown2
Katrina Tatton-Brown2 Dagmar Lehwalder
Dagmar Lehwalder- 1Hospital Infantil Universitario Niño Jesús, Universidad Autónoma de Madrid, CIBER de Fisiopatología de la Obesidad y Nutrición, Instituto de Salud Carlos III and IMDEA Institute, Madrid, Spain
- 2Institute of Cancer Research, St George's University Hospital NHS Foundation Trust, London and St George's University of London, London, United Kingdom
- 3Global Medical Affairs, Merck Healthcare KGaA, Darmstadt, Germany
- 4Department of Pediatrics, University of Leipzig, Leipzig, Germany
The second 360° European Meeting on Growth Hormone Disorders, held in Barcelona, Spain, in June 2017, included a session entitled Pragmatism vs. Curiosity in Genetic Diagnosis of Growth Disorders, which examined current concepts of genetics and growth in the clinical setting, in terms of both growth failure and overgrowth. For patients with short stature, multiple genes have been identified that result in GH deficiency, which may be isolated or associated with additional pituitary hormone deficiencies, or in growth hormone resistance, primary insulin-like growth factor (IGF) acid-labile subunit deficiency, IGF-I deficiency, IGF-II deficiency, IGF-I resistance, and primary PAPP-A2 deficiency. While genetic causes of short stature were previously thought to primarily be associated with the GH–IGF-I axis, it is now established that multiple genetic anomalies not associated with the GH–IGF-I axis can result in short stature. A number of genetic anomalies have also been shown to be associated with overgrowth, some of which involve the GH–IGF-I axis. In patients with overgrowth in combination with an intellectual disability, two predominant gene families, the epigenetic regulator genes, and PI3K/AKT pathway genes, have now been identified. Specific processes should be followed for decisions on which patients require genetic testing and which genes should be examined for anomalies. The decision to carry out genetic testing should be directed by the clinical process, not merely for research purposes. The intention of genetic testing should be to direct the clinical options for management of the growth disorder.
Introduction
Human linear growth continues from the embryonic stage through to adolescence and early adulthood. During the fetal stage it is mainly controlled by insulin and growth factors and is affected by maternal health, nutrition and placental function; growth failure at this stage results in a baby being born small for gestational age. During childhood, the hypothalamic-pituitary control of the synthesis and release of growth hormone (GH), and the effects of the GH–insulin-like growth factor (IGF)-I axis on tissues throughout the body, becomes increasingly important in determining height growth. The synthesis and release of gonadotropins by the pituitary, and the increases in sex steroid, GH and IGF-I concentrations, become significant throughout puberty, and the pubertal growth spurt continues until near-adult height is reached. However, normal skeletal growth and development depends on multiple factors and can be affected by numerous genetic anomalies, including many that are not associated with the GH–IGF-I axis. If abnormal growth in either direction occurs during the pediatric period, children frequently require referral to an endocrine specialist for assessment and diagnosis.
While there are clearly environmental influences on height, which may relate to the worldwide increase in average height through the twentieth century, genetic differences have a major influence on height variation (1, 2). Height, which is one of the most heritable phenotypes in humans, shows a binomial distribution, with abnormalities involving both growth failure and overgrowth. It is a polygenic trait, and studies in multiple cohorts of twins have identified ~700 common variants that captured ~60% of heritability (1–5). For 83 identified height-associated coding variants with minor allele frequencies, effects of up to 2 cm per allele were observed, which was more than 10-times the average effect of common variants (4). Results from such genome-wide association studies have indicated several new genetic candidates, although the loci of the variants involved in height are not randomly distributed; examination of genes enriched for these loci are implicated in novel pathways involved in growth, some of which contribute to various syndromes of abnormal skeletal growth (3, 6).
While such studies indicate genes with biological relevance, genetic testing of children with abnormal growth should not just involve research to identify gene anomalies associated with growth defects, but should also provide pragmatic aspects to determine the diagnostic causes. The following report is based upon presentations from a meeting, funded by Merck Healthcare Global Medical Affairs, that aimed to examine the various genetic causes of growth disorders and the treatment options. A report from the session on adherence and personalized GH treatment in the management of growth disorders will be published separately.
Genetic Testing for Factors Associated With Short Stature
Multiple genetic anomalies have been shown to be associated with growth failure and short stature. The short stature may be either proportionate or disproportionate, severe or mild, and have pre-natal and/or post-natal origin (7, 8). While GH treatment has been approved for use in a number of different conditions, growth response varies greatly, both between conditions and within conditions, and it is important to understand the genetic background to the condition in order to help predict the response to treatments.
Genetic testing may, therefore, be carried out for practical reasons to determine the diagnosis and treatment options in patients with growth failure; however, research is also necessary to identify genetic abnormalities that are associated with specific phenotypes linked with short stature. Development of both pragmatic and research aspects have been carried out over time, and new genetic associations are being found as the technologies become more sensitive, cheaper and more widely applied.
The GH–IGF-I Axis
Growth can be affected at various different developmental stages and at different points and facets of the GH–IGF-I axis. The genetic alterations initially identified as being associated with growth disorders are shown in Figure 1. Genetic disorders may cause defects in pituitary development during the embryonic stage, which most frequently results in multiple pituitary hormone deficiencies. There may also be abnormalities within the pituitary gland or in hypothalamic signaling, which affect GH synthesis and release by the pituitary. Finally, there may be defects in the actions of GH and IGF-I through abnormalities of receptors in target tissues, causing resistance to the effects of GH or IGF-I. These defects are usually associated with intrauterine growth retardation.
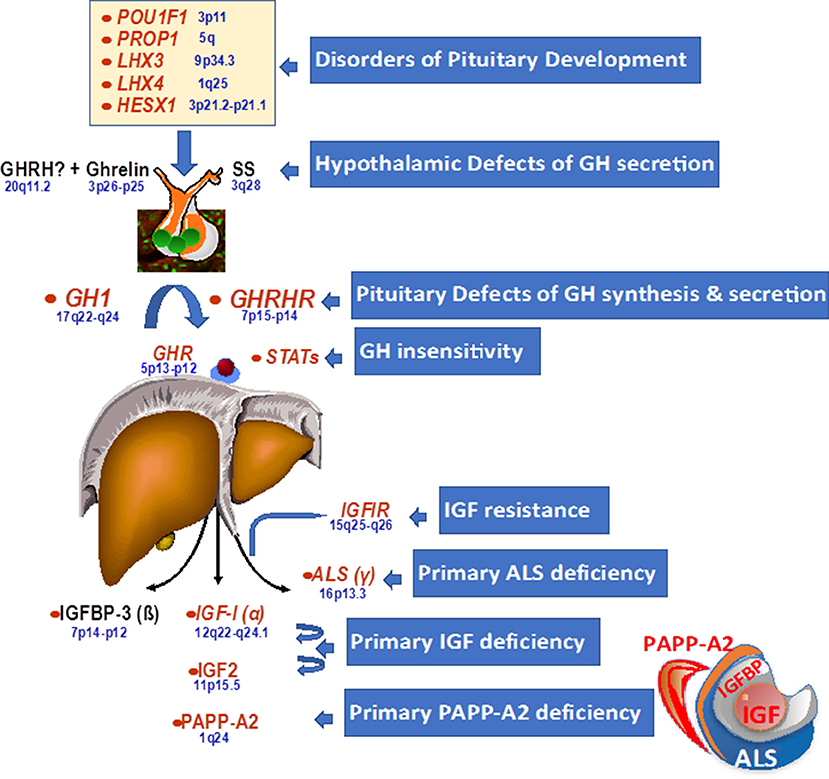
Figure 1. Genetic defects within the hypothalamo-pituitary axis.
GH Deficiency
Isolated GH deficiency or inactivity is a major cause of growth failure, and multiple genetic abnormalities have been shown over time using whole genome sequencing approaches (Table 1). In many familial cases, there are defects in, or deletions of, the gene coding for GH (GH1) or the GH-releasing hormone receptor (GHRHR), which lead to either classical GH deficiency or GH that is biologically inactive (9, 10); in cases where GH is inactive, as in Kowarski syndrome, the apparent serum concentration of GH may be normal or even slightly elevated (11). The condition may also be associated with other syndromic effects, such as ciliopathies including Alström syndrome, where genetic abnormalities are associated with functional GH deficiency (12). Ciliopathies are often associated with skeletal problems, short stature and nephronophthisis, as seen with IFT172 mutations that cause short-rib thoracic dysplasia (13). While there are few data on GH status with ciliopathies, a case of functional GH deficiency, with low IGF-I, that responded to GH treatment has been described in a patient with an IFT172 mutation (14).
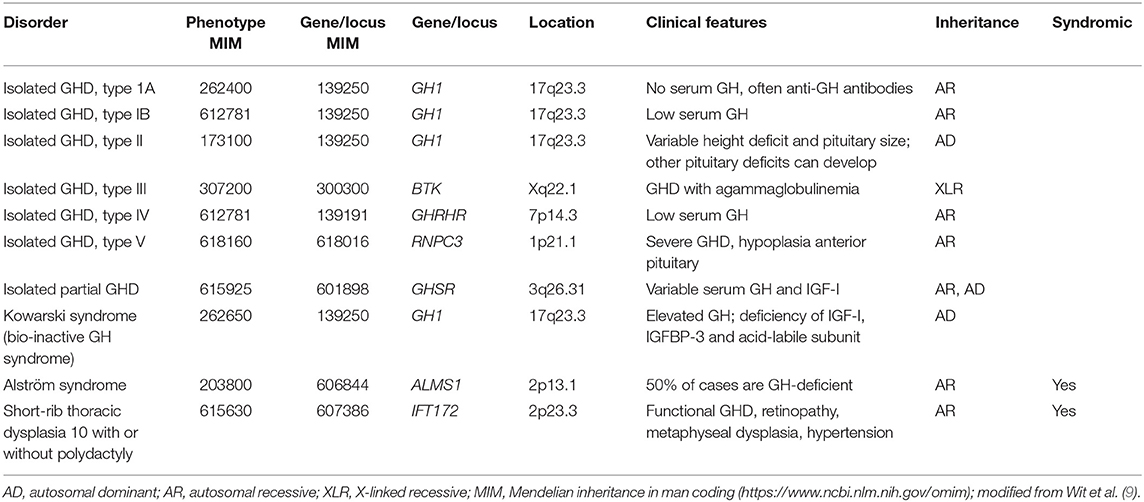
Table 1. Genetic abnormalities associated with isolated GH deficiency (GHD) or bio-inactivity.
Biallelic mutations in RNPC3 have also been associated with familial isolated GH deficiency with hypoplasia of the anterior pituitary (15). The gene encodes a structural component of the nuclear ribonucleoprotein complex that acts as a minor spliceosome in the removal of specific introns for a number of genes. The mutations were identified as being in the RNA recognition motif of the protein, which binds to U12 and acts as a bridge between U12 small nuclear RNA and U11 small nuclear RNA of the intron recognition complex. The growth failure was post-natal and proportionate, with only mild microcephaly, anterior pituitary hypoplasia, and normal psychomotor development. GH treatment of these patients was effective, despite severe short stature and late initiation of treatment (16).
Patients with isolated GH deficiency are reported to develop combined pituitary hormone deficiency in about 5–45% of cases (17, 18). For cases of combined pituitary hormone deficiencies, the genetics appear to be more complicated and the genetic etiology is frequently unidentified (Table 2). Genetic defects may occur sporadically, with no family history, or may be familial; they may be dominant where a defect in only one allele is associated with the condition, recessive where abnormalities of both alleles produce the effect, or X-linked where the defect comes from an unaffected mother and generally affects only males (19). Reported frequencies of genetic mutations in patients with combined pituitary hormone deficiencies varies greatly between countries and ethnic groups (20). For PROP1, POU1F1, and HESX1, mutation frequencies are <10% in patients with sporadic combined pituitary hormone deficiency in Western Europe, although much higher in Eastern Europe. Despite the low prevalence in Western Europe, testing for mutations of these genes is routinely carried out as part of the work-up for such patients (20).
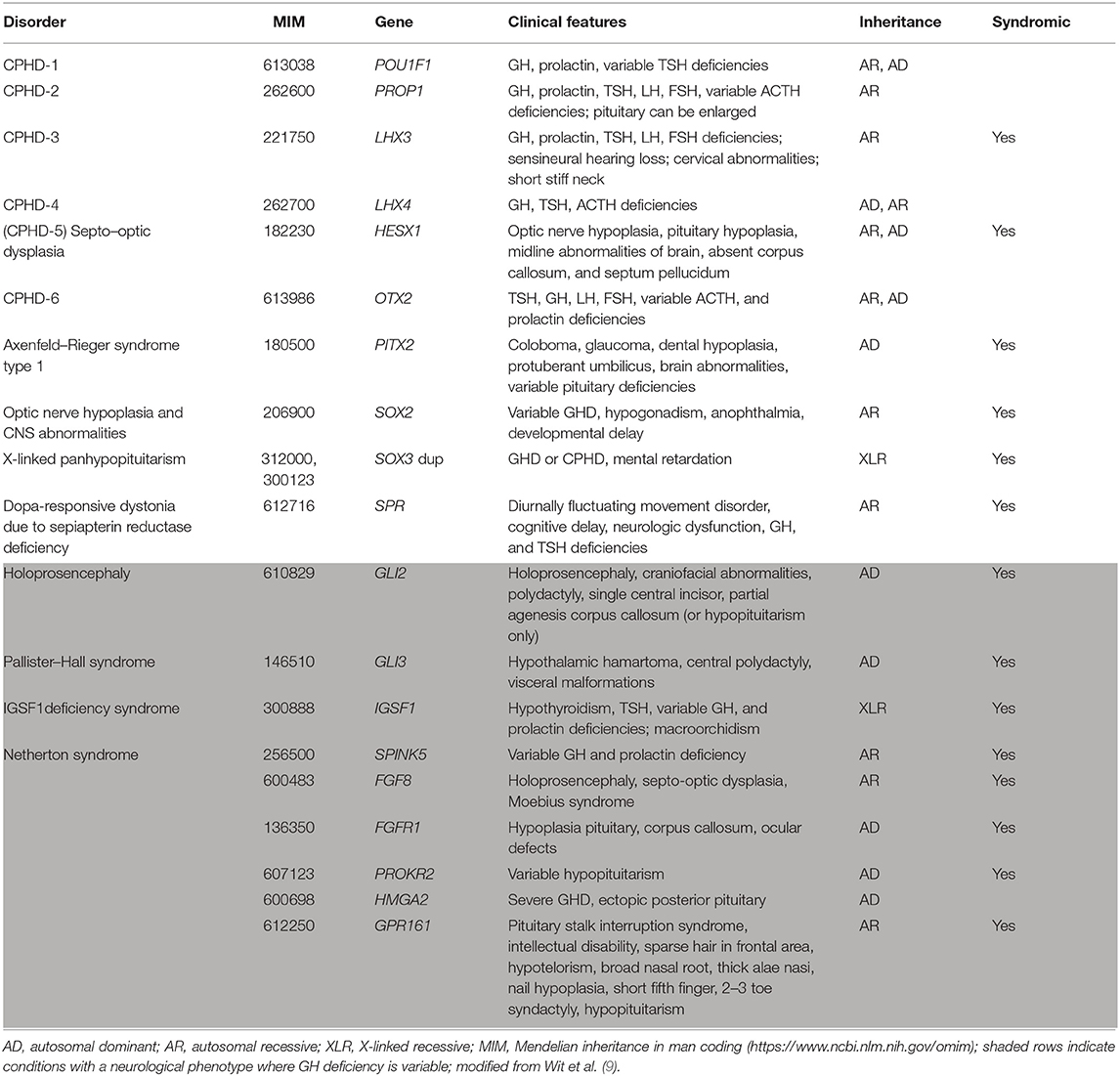
Table 2. Genetic abnormalities associated with combined pituitary hormone deficiencies (CPHD).
Anomalies of the classic transcription factor genes cause very specific profiles of hormone deficiencies due to abnormal pituitary development; the most prevalent defects in patients with growth failure are in PROP1, which was reported in a literature analysis to occur in 6.7% of worldwide sporadic cases of combined pituitary hormone deficiency and 11.2% when familial cases were included (21). Mutations of PROP1 cause the significant clinical feature of pituitary enlargement in some cases, particularly in childhood and adolescence (22), that sometimes helps in the diagnosis. Defects in genes for other transcription factors involved in pituitary development are associated with other syndromic features. For defects in PITX, SOX2, SOX3, and SPR, short stature is the predominant phenotype, while for others (shown in the shaded area of Table 2) there is a more severe neurological phenotype, and GH deficiency may or may not be present as one part of the syndrome.
Using a candidate-gene approach to identify mutations in more than 900 patients with short stature due to GH deficiency, gene defects could be identified in only about 10% of cases (23). Mutations were identified more frequently in patients with combined pituitary hormone deficiencies (14.7%) than in those with isolated GH deficiency (6.5%). In patients with combined pituitary hormone deficiencies, the most frequently identified mutations were in PROP1 (11.6% of patients), followed by LHX3 (1.2%), whereas the gene mutations in patients with isolated GH deficiency were most frequently found in GH1 (4.8%), and GHRHR (1.1%). However, the proportion of patients with identified defects is increasing over time as more genetic variants are investigated.
Genetic abnormalities also had an effect on clinical outcomes following GH treatment (23). For 24 patients with an identified mutation and who reached near-adult height, the height standard deviation score (SDS) at start of GH treatment was −4.1, compared with a baseline height SDS of −2.9 in 191 patients without an identified mutation. The mean near-adult height SDS was −0.7 in those with a mutation vs. −0.9 in those without, and the respective mean gain in height SDS at near-adult height was 3.4 vs. 2.0, which was significantly different (p < 0.001). Thus, those with an identified mutation are likely to be shorter at the start of GH therapy and have a better response to GH treatment. For specific gene mutations, patients with a defect in GH1 (n = 4) had an initial height SDS of −4.2 and a height SDS gain of 3.4 to reach a near-adult height SDS of −0.8. On average, these patients remained slightly shorter than normal height. For patients with a PROP1 mutation, initial height SDS was −3.6, with a gain of 3.6 to reach height SDS 0.0 at near-adult height, and thus were normal height.
GH Resistance and IGF-I Deficiency
The genetic abnormalities causing GH resistance or IGF-I deficiency/insensitivity (Table 3) are mainly associated with intrauterine growth retardation and being born small for gestational age. The concept of GH resistance and IGF-I deficiency is becoming more complex and cannot now be considered as a single clinical entity, but is a continuum of genetic, phenotypic, and biochemical abnormalities (24). Genetic variants influence both total IGF-I concentrations and, through changes in binding proteins, free IGF-I concentrations; therefore, it is important to determine which methodologies should be used in the diagnosis. It is often not easy to identify from the phenotype which genes should be examined, because serum levels of GH and IGFs may be decreased, normal or increased in patients with the same genetic defect. Classic examples of GH resistance are due to mutations in the GH receptor (GHR) gene, and a large number of missense and splice mutations and deletions in GHR have been identified in patients with short stature (24). Mutations in GHR are most generally associated with Laron syndrome, in which patients also have very low IGF-I levels (9, 11, 25). Mutations have also been reported in the signal transducer and activator of transcription gene, STAT5B, which is part of the signaling cascade of the GH receptor; in addition to growth failure, clinical features of these patients frequently involve immunodeficiency and pulmonary fibrosis (24, 26), although some missense mutations of STAT5B have recently been reported to not cause severe immune and pulmonary problems (27).
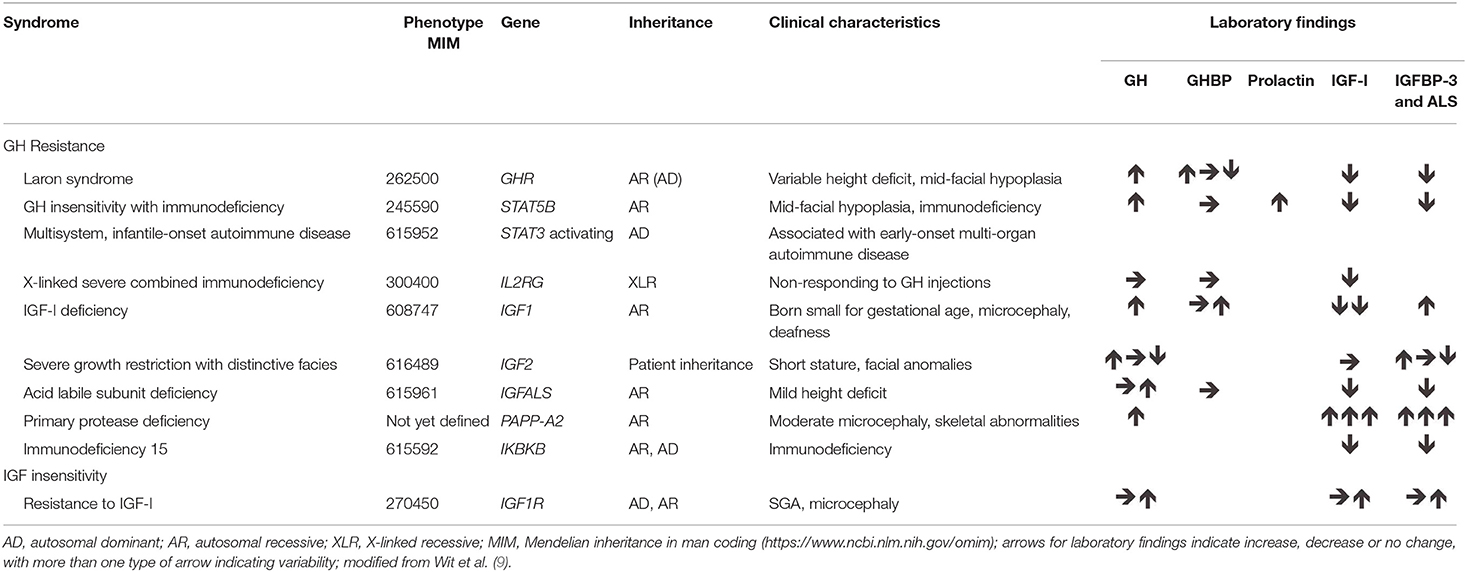
Table 3. Genetic abnormalities associated with GH resistance and IGF-I insensitivity.
More recently, defects in PAPP-A2 have been identified in children with progressive post-natal growth failure from two unrelated families (28, 29). The gene encodes pregnancy-associated plasma protein (PAPP)-A2, which is a metalloproteinase that cleaves insulin-like growth factor binding protein (IGFBP)-3 and IGFBP-5. Serum concentrations of total IGF-I, IGF-II, IGFBP-3, acid-labile subunit, and insulin were all increased in these patients; however, free IGF-I concentrations were very low, resulting in lack of negative feedback on GH synthesis and release. X-radiography of the fibulae, tibiae, and femurs showed that the long bones were very thin; bone density was low, with abnormal trabecular structure, and mild to moderate microcephaly was observed. After 1 year of treatment with recombinant human IGF-I, two Spanish patients showed a good response of growth velocity, height gain, and trabecular bone score (30, 31). A report has recently been published on the history and clinical implications of defects in PAPP-A2 in human growth (32). The specific degradation of IGFBP-3 and IGFBP-5 by the active metalloproteinase results in release of free IGF-I, and it has been demonstrated that the identified mutations in the PAPP-A2 gene, producing inactive PAPP-A2, cause increased binding of IGF-I and low bioactivity, with consequent reduced skeletal growth (33).
Identified defects resulting in GH resistance also include mutations in the IGF1 and IGF2 genes. Abnormalities of IGF1 are rare in humans, with only about 9 cases reported (34, 35), although both homozygous and heterozygous mutations, and heterozygous deletions have been identified in patients with short stature (9, 36, 37); however, homozygous carriers generally have more severe growth failure than those with heterozygous defects. Homozygous IGF1 defects that result in complete loss of functional IGF-I are associated with extreme growth failure, both pre-natal and post-natal, together with sensorineural deafness, microcephaly and mental retardation (34–38).
More recently, IGF2 abnormalities have been detected in a number of patients; the clinical data indicated that IGF-II affects both pre-natal and post-natal growth and the genetic defects are associated with clinical features of Silver–Russell syndrome (39). While IGF2 defects have only recently been identified, it is possible that they may be more common than initially suspected. Imprinting disorders, with hypomethylation of genetic control regions, may result in down-regulation of IGF2 expression by the paternal allele and are involved in short stature due to both pre-natal and post-natal growth failure in Silver-Russell syndrome and Temple syndrome (40). A clinical scoring system has been derived for Silver-Russell syndrome, and a consensus guideline stated that first line molecular testing should include analysis of methylation of the H19/IGF2 domain of the 11p15 region in patients with a high likelihood of the syndrome (41). The guideline also concluded that GH treatment improved body composition and linear growth in these patients.
Defects in IGF1R that result in resistance to the effects of IGF-I have been described in a number of patients who had intrauterine growth retardation and failure of catch-up growth (9, 42, 43). The defects can occur in the external domain, the internal domain or in the transmembrane domain of the receptor. The condition is associated with microcephaly and psychomotor retardation in some, but not all, cases and there may be other clinical pathologies, such as impaired glucose tolerance, dysmorphic features, and cardiac abnormalities, but the pattern is inconsistent (42, 44); patients with homozygous mutations may show stronger syndromic features compared with heterozygous carriers (45). Recently, a case was described with both a deletion of IGF1R and a duplication of the paternal IGF2 region, resulting in appropriate pre-natal growth but retardation of post-natal growth, indicating the key role of IGF-II in fetal growth and the more important role of the GH–IGF-I axis in post-natal growth (46). In children with IGF1R defects, IGF-I concentration is generally high and above normal during GH treatment, although this may be considered acceptable owing to the reduced sensitivity.
As a result of these studies of patients with GH resistance, an algorithm was designed to try to predict which patients should have genetic screening for mutations (Figure 2). This is difficult because of the inconsistent patterns of clinical and laboratory features (Table 3). For patients with intrauterine growth retardation, possibly with microcephaly, and who have sporadic occurrence of psychomotor retardation and deafness, it may be worthwhile to look for mutations in IGF1; however, genotyping is somewhat ambiguous in these patients. Patients with familial occurrence of short stature related to being born small for gestational age who have high IGF-I should be checked for abnormalities of IGF1R. A recent study has suggested a clinical score for selecting patients for genetic testing of IGF1R defects, and indicated that testing should be carried out for children born small for gestational age, with low birth weight and/or length, and who remained short, had microcephaly and had IGF-I SDS >0 at presentation (47). Patients with low serum IGF-II concentration and clinical features of Silver–Russell syndrome, although not necessarily full Silver–Russell syndrome, should be examined for defects in IGF2 (48).
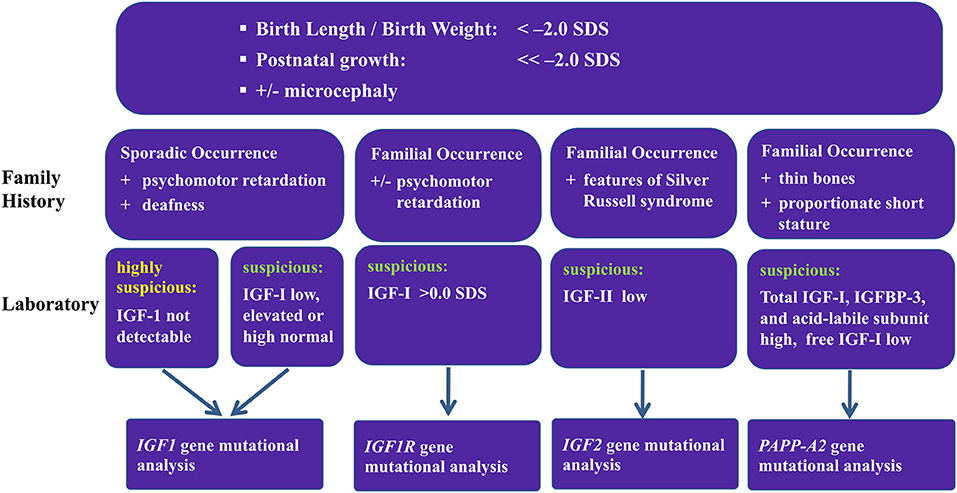
Figure 2. Proposed algorithm for screening patients for IGF-related genetic abnormalities.
In patients shown to have severe primary IGF-I deficiency, recombinant human IGF-I appears to be effective and has been approved for use (7, 49). IGF-I has also been tested in combination with GH in patients with reduced IGF-I concentrations and GH sufficiency (50). Combined GH and IGF-I treatment has also been suggested for patients with GH insufficiency plus IGF-I deficiency due to heterozygous dominant-negative GHR variants (51). However, IGF-I treatment has not been approved for use in such cases, and the specific clinical conditions in which GH, IGF-I, or the combination of these, may be effective need to be identified; genetic testing should help to determine which treatment approach may be useful to improve linear growth (7, 52).
For 51 patients born small for gestational age and with failure of catch-up growth, treatment with GH resulted in a median gain in height SDS of 0.64 in the first year; for nine patients born small for gestational age and without catch-up growth due to genetic defects that resulted in dysfunctional IGF-I receptors, the median first year height SDS response was 0.24 (Pfäffle R, unpublished data). The difference between the overall cohort and the patients with abnormal IGF1R was significant (p = 0.013). However, the GH dose given to the patients with dysfunctional IGF-I receptors was kept slightly lower than the dose for the patients born small for gestational age because of concerns of excessive serum IGF-I concentration, which could have partially accounted for the difference. A variable response to GH treatment of children with IGF1R deletions or mutations was seen in other studies, with GH in most, though not all, having a beneficial effect (44). The response in children with an IGF1R defect was suggested to be less than the response seen in short children born SGA due to other causes, although adult height gain was similar and may be related to duration of treatment (47). While GH treatment for children born small for gestational age and without spontaneous catch-up growth is indicated to be initiated only at age 4 years or above, starting GH treatment at a very young age (<4 years) in such children has been described to be more effective than in those who were older at GH initiation (53), at least during 3 years of therapy.
Genetic Anomalies Not Associated With the GH–IGF-I Axis
Genome wide association studies and next-generation sequencing have indicated a large number of new genes associated with short stature, although in many cases the exact mechanism remains unknown (54). Until relatively recently, it was thought that linear growth in childhood was primarily influenced by the GH–IGF-I axis and that abnormal growth due to genetic causes were related to defects in that system. However, it has recently been suggested that rather than focusing on the GH–IGF-I axis, genetic studies should be more concerned with the biology of the growth plate (55). Cases reported as idiopathic short stature may be due to genetic disorders previously only considered to be associated with skeletal dysplasias.
Hypochondroplasia and achondroplasia are the most common skeletal dysplasias that cause short stature and are associated with abnormalities of the FGFR3 gene (56, 57). Fibroblast growth factor receptor 3 negatively regulates chondrogenesis and mutations in the gene result in increased activity, which disrupts normal functioning of the proliferation zone of the growth plates and impairs bone elongation. While heritability is autosomal dominant, the majority of cases arise from spontaneous mutations and ~80% of cases are born to parents of average height. Diagnosis is generally at birth or in early infancy. No formal diagnostic criteria have been published although there are well-defined clinical and radiological characteristics, including rhizomelic short stature with short limbs and short fingers, limited elbow extension, small chest, macrocephaly, and flattened mid-facial features (56). GH treatment is not an approved treatment, except in Japan, but increases growth velocity over the first few years. Very few studies have followed patients to adult height, but the long-term effect appears to be limited (58). The Indian hedgehog paracrine system also controls chondrocyte proliferation and differentiation and defects can result in short stature (57, 59). Autosomal dominant mutations in the IHH gene result in mild skeletal dysplasias with brachydactyly, but patients may be diagnosed as born small for gestational age or as idiopathic short stature with defects only identified through whole exome sequencing, and the growth failure appeared to be improved by GH in the short-term (59).
Aggrecan is a key component of the cartilage extracellular matrix of the growth plate and articular surfaces. Abnormalities in the ACAN gene, which encodes the proteoglycan, results in proportionate or mildly disproportionate short stature, generally with advanced bone aging and premature growth cessation (60, 61). ACAN defects are a common cause of idiopathic short stature and born small for gestational age; homozygous abnormalities result in severe skeletal dysplasia while heterozygous defects cause milder skeletal dysplasia (61, 62). A clinical scoring system has been proposed to identify children likely to have an ACAN mutation, although there is a variable spectrum of phenotypes (63). GH treatment has been reported to be effective, but very few studies have been carried out (62, 63).
Patients with anomalies of the short stature homeobox-containing (SHOX) gene also show a wide spectrum of phenotypes, from Leri-Weill dischondrosteosis, with mesomelia and Madelung deformity, to non-specific short stature (64). SHOX deficiency is one of the most common genetic defects associated with short stature in humans. In patients with idiopathic short stature, the prevalence of anomalies in the SHOX gene or its regulatory elements, including deletions, duplications and insertions, has been reported to range from about 6 to 22%, with increasing prevalence reported as genetic tests have become more accurate (65). The severity of the condition does not appear to relate to specific gene anomalies, and individuals within a family could have the same defect but different phenotype (65, 66); however, SHOX deletions or mutations are more associated with Leri-Weill syndrome whereas enhancer region defects are more likely to be associated with less severe features of idiopathic short stature (67). SHOX protein expressed by the gene is a transcription factor that regulates the expression of various other genes in the growth plate, including ACAN and FGFR3; while the mechanism has not been completely elucidated, SHOX protein influences chondrocyte proliferation, hypertrophy, maturation, and apoptosis, thus regulating long bone growth (65, 68). The SHOX gene is located in the pseudoautosomal region and normal physiology requires two copies of the gene, with haploinsufficiency occurring when only one functioning copy of the gene is present. Short stature due to SHOX deficiency is, therefore, a frequent occurrence in children with Turner syndrome, with all or part of one X-chromosome missing. A need for screening of SHOX gene defects may be predicted from abnormal limb-to-trunk ratios, x-radiography and family history, and predictive algorithms have been proposed (64, 65). Where clinical features are severe, deletions are seen in about 80 to 90% and genetic evaluations should include chromosomal microarray analysis or SNP array analysis; for patients with less severe characteristics, exome sequencing is used more frequently (64, 65). GH treatment of children with SHOX deficiency is an approved indication and is effective in improving adult height by 7 to 10 cm (65, 69).
SHOX gene anomalies are found in ~70% of children with Leri-Weill dischondrosteosis (68). However, similar phenotypic features, including short stature but without Madelung deformity, occur with defects in the natriuretic peptide receptor-B gene, NPR2. NPR-B is the receptor for C-type natriuretic peptide, which is a regulator of chondrocyte differentiation and hypertrophy in the growth plates, and may be one of the genes activated by SHOX proteins (65). NPR2 defects have been identified in about 2 to 6% of cases of idiopathic short stature (70, 71). Biallelic loss of function mutations cause Maroteaux-type acromesomelic dysplasia, while heterozygous mutations result in less severe short stature (70, 71). Therefore, patients may have severe skeletal dysplasia or non-specific skeletal abnormalities, but phenotype characteristics of patients with NPR2 gene defects are currently not well established (72), and patients with short stature due to NPR2 defects may only be detected where a relative has acromesomelic dysplasia (71). Very few patients with NPR2 defects have currently been treated off-label with GH, but increased growth appeared to be modest and further studies would be required to demonstrate long-term efficacy (70, 72).
Genetic Factors Associated With Overgrowth
Overgrowth With No Associated Intellectual Problems
By contrast to gene defects that cause short stature, alternative defects in the same genes can result in overgrowth. Gain-of-function mutations in NPR2 have been identified in a number of patients with skeletal overgrowth, with associated features of scoliosis and macrodactyly, with markedly large big toes (71, 73, 74). The ligand for the receptor is C-type natriuretic peptide (CNP) and overproduction due to translocation of the NPPC gene also caused tall stature with macrodactyly and skeletal dysplasia (75, 76). The mechanism of excess skeletal growth due to increased activation of the CNP/NRPB system was reported to be through overproduction of cyclic guanosine monophosphate within the growth plate chondrocytes (73, 74, 76).
Beckwith-Wiedemann syndrome is one of the most common overgrowth conditions and is the converse of Silver-Russell syndrome, due to molecular anomalies in the chromosome 11p15 imprinting region (47, 77, 78). Duplications of the imprinting region is associated with over-expression of the IGF2 gene and patients are reported to have both pre-natal and post-natal overgrowth, with associated anomalies including macroglossia, organomegaly, abdominal wall defects, neonatal hypoglycemia, and predisposition to embryonic tumors (77). Because of the increased risk of tumors associated with the condition, a consensus meeting devised a scoring system based on both cardinal and suggestive clinical features of the syndrome. This allowed identification of patients requiring molecular testing and provided a flowchart for analysis of gain of methylation within the 11p15 genetic region and copy number variation (78). The guidelines also provided recommendations for management of various complications, including overgrowth that continues beyond childhood, although that occurs in only 43 to 65% of patients.
Overgrowth, with either gigantism occurring before epiphyseal growth plate fusion or acromegaly due to excessive skeletal growth at any time, can be caused by a number of genetic defects that result in benign adenomas, with excess GH and IGF-I synthesis and release. Gigantism and acromegaly are rare in children, although pediatric cases have aggressive features; identified genetic conditions include, amongst others, familial isolated pituitary adenoma and the more recently described X-linked acrogigantism (X-LAG), which have been reviewed recently and an algorithm for genetic testing and counseling proposed (79, 80). Because such diagnoses are generally made at a young age and there is frequently resistance to medical therapy, analysis of the genetic basis is needed to predict therapeutic options. The most common condition is familial isolated pituitary adenoma, which is usually diagnosed in childhood or adolescence, requires at least two cases of pituitary adenomas in the family and the tumor is large and invasive (81). Inactivating mutations or deletions of the aryl hydrocarbon receptor-interacting protein gene, AIP, predispose patients to the condition and are the most frequently identified genetic cause of pituitary gigantism (82). Prevalence of AIP mutations in cases of sporadic pituitary adenomas is reported to be ~4%, but may be up to 20% in children and adolescents with gigantism and/or macroadenomas (83). Although no genetic defects are found in a majority of patients with familial isolated pituitary adenoma (84), a recent study found novel variants of the MEN1 gene in some cases where no defects in AIP were observed (85). Patients with sporadic pituitary adenomas and identified AIP mutations have been mainly young males, with median age of emergence at 17.5 years, the tumor tends to be larger than in patients without AIP mutations and tumors were more often resistant to somatostatin analogs and required surgical intervention (80, 82, 83).
X-LAG syndrome is very rare, but is the most frequently identified cause of gigantism in pre-pubertal patients, with diagnosis generally before the age of 3 years (80, 86). The condition is characterized by increased linear growth and weight that can be seen before 12 months of age, although gestational size may be normal; patients often have increased appetite and hunger and most identified cases also had hyperprolactinemia (82, 86). Because of the very early onset of overgrowth, adult height may be greatly increased at >4.5 SDS (87, 88). Both GH and IGF-I are markedly elevated, but GH releasing hormone is also elevated and the adenomas remain sensitive suggesting that therapeutic antagonists of the GH releasing hormone receptor could reduce the excess GH secretion (87, 89). The condition is linked to a germline micro-duplication of chromosome Xq26.3 and is associated with the GPR101 gene (86–88). A number of mutations in GPR101 have also been identified in patients with sporadic acromegaly (86, 90). GPR101 encodes a G-protein coupled receptor, the function of which has not currently been fully elucidated, that is predominantly expressed in the hypothalamus and fetal pituitary glands, and it may have a role in pituitary ontogenesis (86, 87, 90). The disorder is X-linked, arising spontaneously in most cases; because index cases are generally female and more than 70% of cases are female, it has been suggested that affected hemizygous male embryos may be less viable, although familial cases from mother to son have been reported (79, 82, 87). X-LAG syndrome does not have any specific association with intellectual disability. However, a chromosome Xq26 microduplication was found in a 4-year old boy referred for examination for X-LAG (91). The genetic duplications are detected using array comparative genome hybridization (82, 87); this is normally clinical grade analysis, where duplications may be smaller than the assay resolution, and when assayed by high definition molecular cytogenetics the duplication did not include the GPR101 gene (91). The patient did not show the phenotype of gigantism or GH excess and the study showed the importance of the correct genetic assay and provided supporting evidence for GPR101 being the gene involved in X-LAG.
Overgrowth With Intellectual Disability
Overgrowth-intellectual disability (OGID) syndromes are a family of genetic disorders characterized by excessive growth (height and/or head circumference ≥2 SD above the mean compared with the age-related peer group) in combination with an intellectual disability (92). Over the last decade, since the introduction of next-generation sequencing technologies that permit the examination of thousands of genes in a single experiment, the genetic cause of many OGID syndromes has been determined. A gene discovery study of patients with OGID syndromes, reported in 2017, included 710 participants and identified 14 different genes among 50% of affected individuals (93). Children with overgrowth phenotypes without intellectual disability were excluded from the study. Two key OGID gene families were identified: the epigenetic gene regulator family (six genes, 44% of the overall patients) and the PI3K/mTOR pathway gene family (five genes, 6% of the overall patients); in addition, variants in three genes involved in, as yet, undetermined pathways were identified (Table 4).
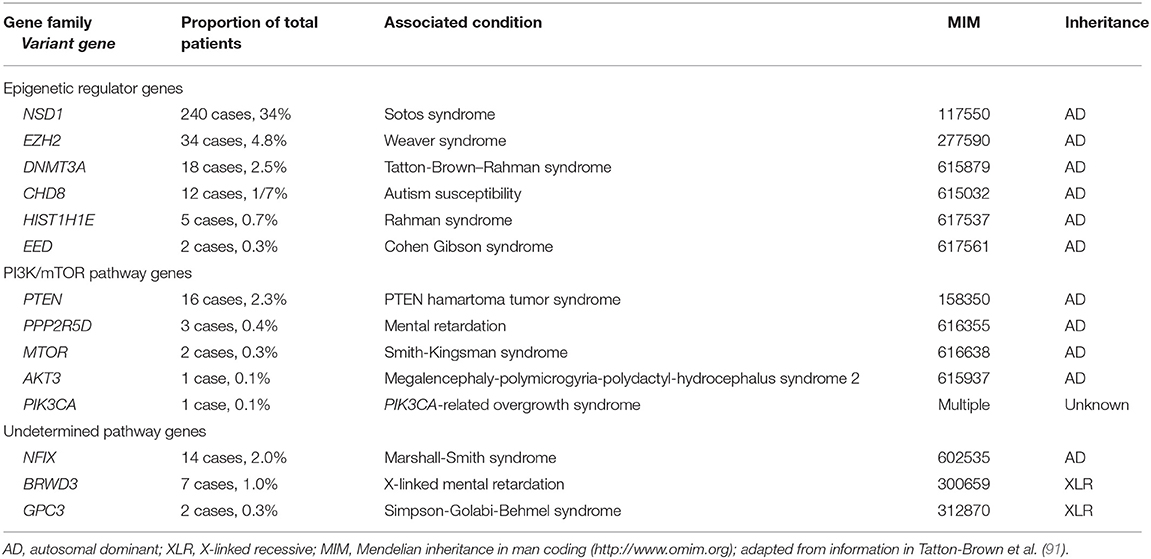
Table 4. Genetic abnormalities identified among 710 patients with OGID syndromes.
The proteins encoded by genes involved in epigenetic regulation were (1) histone methyltransferases targeting specific lysine residues of histone tails (NSD1, EED, EZH2), (2) a DNA methyltransferase (DNMT3A), and (3) genes involved in chromatin remodeling (CHD8) and stabilization of higher-order chromatin structures (HIST1H1E).
Of the OGID genes identified in the study, NSD1 was the most frequently mutated (240 children, 34% overall). Nuclear receptor binding SET domain protein 1 (NSD1) catalyzes the methylation of lysine residue 36 of histone H3 (H3K36) and, to a lesser degree, lysine residue 20 of histone H4 (H4K20), a methylation mark primarily associated with transcriptional activation (94). NSD1 variants cause Sotos syndrome (MIM 117550), associated with increased growth, an intellectual disability and a characteristic facial appearance with a broad forehead, down-slanting palpebral fissures, long thin face, tall chin, malar flushing and sparse fronto-temporal hair (95, 96).
EZH2 and EED encode components of the polycomb repressor complex 2 (PRC2), which catalyzes the methylation of histone 3 at lysine 27 (H3K27) associated with transcriptional repression (97). Pathogenic variants within EZH2 and EED cause very similar OGID phenotypes; respectively, Weaver syndrome (MIM 277590) and Cohen Gibson syndrome (MIM 617561), characterized by tall stature, variable intellectual disability and characteristic facial appearance of “stuck-on” chin, horizontal skin crease, and hypertelorism (96, 98, 99).
DNMT3A pathogenic variants cause Tatton-Brown–Rahman overgrowth syndrome (TBRS; MIM 615879) (100). Children with TBRS characteristically have variable intellectual disability, autism spectrum disorders and increased growth (93, 100–102). A range of different medical issues are additionally associated with TBRS, including hypermobility of joints, obesity, kyphoscoliosis, and congenital cardiac anomalies (100).
HIST1H1E encodes the linker histone H1.4, and recently five unrelated probands were identified with de novo heterozygous frameshift variants (protein truncated variants, PTVs) that were clustered in a 12 base-pair region of the carboxy-terminal domain (93). Histone H1.4 is one of the 11 histones that mediate formation of chromatin structures that regulate gene expression. It is hypothesized that the clustered PTVs all generate the same open-reading frame, with the shorter mutant protein associated with a reduced net positive charge compared to the wild type, thereby disrupting normal binding to negatively-charged DNA (93, 103). Patients with HIST1H1E pathogenic variants have a variable intellectual disability and a characteristic appearance of high hairline and frontal bossing.
With regard to the PI3K/mTOR pathway genes, PTEN pathogenic variants have been an established cause of PTEN hamartoma tumor syndrome (PHTS; MIM 158350), also known for many years as Bannayan-Riley-Ruvalcaba syndrome in childhood (92, 96). The encoded PTEN protein is a negative regulator of the PI3K/mTOR pathway, and affected children are macrocephalic, and may have an intellectual disability and autistic spectrum disorder. In addition, they have a predisposition to develop cancers in adulthood (Cowden syndrome) (104). PIK3CA pathogenic variants are primarily associated with macrocephaly and asymmetric growth, which may be progressive (92, 96). Treatment with drugs that block the PI3K/mTOR pathway, such as sirolimus and everolimus that are already in use as anti-cancer agents, have been shown in vitro and in animal models to be effective in reducing the progressive overgrowth and may potentially improve cognition (93, 105, 106). Such treatments in humans have shown varying success in reducing the regional overgrowth, although the side-effect profile, most notably the increased infection rate, is significant [(106, 107), and ClinicalTrials.gov: NCT02991807 and NCT00971789].
Conclusions
Human height follows a binomial distribution; however, individuals that lie at the extremes of the distribution may potentially have problems that require investigation. Height is a highly heritable classic polygenic trait and, therefore, part of any investigation frequently involves genetic analysis. Genome-wide studies have identified ~700 common associated variants with effects on height and 83 minor allele variants that had effects on growth of up to 2 cm per allele. Many genes have effects on the GH–IGF-I axis, and new genes are constantly being added to the list. Research is fundamental to increase knowledge of the genes and mechanisms involved in the pathophysiology of different diseases. However, in the clinical setting, genetic studies are required to progress from phenotype, auxology, and hormone measurements to more precise genetic diagnoses and treatments.
New technologies, such as whole exome sequencing and whole genome sequencing, are increasing the speed, precision and availability of genetic diagnoses, with decreasing costs (108–110). Some areas of the genome have limited capture by whole exome sequencing, making it difficult to go from a diagnosis based on phenotype and medical background to proof from genotype. Therefore, next-generation sequencing techniques, such as targeting exome sequencing, are being developed, to target specific collections of candidate genes with the use of chromosomal microarrays to detect copy number variations (16, 111). Such techniques can be directed to make a specific diagnosis, while whole exome and whole genome sequencing may be more useful for research purposes to identify new genetic variations associated with specific conditions: identifying the genes involved in combined pituitary hormone deficiency may be more useful as a scientific interest to provide new insights into pathogenesis and novel treatment strategies; identifying the genes involved in Turner syndrome will be more useful from a diagnostic point of view, when appropriate genetic counseling and treatment monitoring can be carried out; and detecting the genes involved in GH resistance could be useful for both scientific research and for identifying potential clinical consequences.
While short stature is a frequent cause of referral to a pediatric endocrinologist, screening for tall stature is generally considered a lower priority. However, it is important to identify pathological causes of gigantism and overgrowth, particularly because certain genetic conditions may be resistant to medical therapy or predispose to tumors. With regard to the OGID syndromes, while there is considerable phenotypic overlap between many of the individual conditions, patients with epigenetic regulator gene variants more frequently have tall stature in association with macrocephaly, whereas those with PI3K/mTOR pathway gene variants more usually have isolated macrocephaly. Research into the genetic basis of these syndromes provides a background understanding of the normal regulation and development of growth. On a practical basis, knowledge of the genotype in patients with OGID syndromes and in patients with growth failure is likely to be increasingly useful for pharmacogenomics and gene-directed therapies.
Author Contributions
All authors critically revised the current work for important intellectual content and gave final approval of the version of the publication to be published. All authors have agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of it are appropriately investigated and resolved.
Funding
The 2nd 360° European Meeting on Growth Hormone Disorders was funded by Merck Healthcare KGaA, Darmstadt, Germany. No payment has been offered or will be offered to the authors for authoring this publication and no compensation will be provided for the time they spend on the publication development.
Disclaimer
Not all indications described in this publication are approved for all growth hormone preparations.
Conflict of Interest Statement
DL is an employee of Merck Healthcare KGaA, Darmstadt, Germany. RP reports lecture fees from Merck, Novo Nordisk, Pfizer, and Sandoz.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Medical writing assistance was provided by Peter Bates, Cambridge Medical Writing Services, UK, funded by Merck Healthcare KGaA, Darmstadt, Germany.
References
1. Silventoinen K, Sammalisto S, Perola M, Boomsma DI, Cornes BK, Davis C, et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. (2003) 6:399–408. doi: 10.1375/twin.6.5.399
2. Jelenkovic A, Hur YM, Sund R, Yokoyama Y, Siribaddana SH, Hotopf M, et al. Genetic and environmental influences on adult human height across birth cohorts from 1886 to 1994. Elife. (2016) 5:e20320. doi: 10.7554/eLife.20320
3. Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. (2014) 46:1173–86. doi: 10.1038/ng.3097
4. Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. (2017) 542:186–90. doi: 10.1038/nature21039
5. Jelenkovic A, Yokoyama Y, Sund R, Hur YM, Harris JR, Brandt I, et al. Associations between birth size and later height from infancy through adulthood: an individual based pooled analysis of 28 twin cohorts participating in the CODATwins project. Early Hum Dev. (2018) 120:53–60. doi: 10.1016/j.earlhumdev.2018.04.004
6. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. (2010) 467:832–8. doi: 10.1038/nature09410
7. Argente J. Challenges in the management of short stature. Horm Res Paediatr. (2016) 85:2–10. doi: 10.1159/000442350
8. Argente J, Pérez-Jurado LA. Genetic causes of proportionate short stature. Best Pract Res Clin Endocrinol Metab. (2018) 32:499–522. doi: 10.1016/j.beem.2018.05.012
9. Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. (2016) 174:R145–73. doi: 10.1530/EJE-15-0937
10. Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, et al. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. (2009) 94:3191–9. doi: 10.1210/jc.2008-2783
11. Mullis PE. Genetics of GHRH, GHRH-receptor, GH and GH-receptor: its impact on pharmacogenetics. Best Pract Res Clin Endocrinol Metab. (2011) 25:25–41. doi: 10.1016/j.beem.2010.06.006
12. Romano S, Maffei P, Bettini V, Milan G, Favaretto F, Gardiman M, et al. Alström syndrome is associated with short stature and reduced GH reserve. Clin Endocrinol (Oxf). (2013) 79:529–36. doi: 10.1111/cen.12180
13. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. (2013) 93:915–25. doi: 10.1016/j.ajhg.2013.09.012
14. Lucas-Herald AK, Kinning E, Iida A, Wang Z, Miyake N, Ikegawa S, et al. A case of functional growth hormone deficiency and early growth retardation in a child with IFT172 mutations. J Clin Endocrinol Metab. (2015) 100:1221–4. doi: 10.1210/jc.2014-3852
15. Argente J, Flores R, Gutiérrez-Arumí A, Verma B, Martos-Moreno GÁ, Cuscó I, et al. Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Mol Med. (2014) 6:299–306. doi: 10.1002/emmm.201303573
16. Martos-Moreno GÁ, Travieso-Suárez L, Pozo-Román J, Muñoz-Calvo MT, Chowen JA, Frilander MJ, et al. Response to growth hormone in patients with RNPC3 mutations. EMBO Mol Med. (2018) 10:e9143. doi: 10.15252/emmm.201809143
17. Blum WF, Deal C, Zimmermann AG, Shavrikova EP, Child CJ, Quigley CA, et al. Development of additional pituitary hormone deficiencies in pediatric patients originally diagnosed with idiopathic isolated GH deficiency. Eur J Endocrinol. (2014) 170:13–21. doi: 10.1530/EJE-13-0643
18. Otto AP, França MM, Correa FA, Costalonga EF, Leite CC, Mendonca BB, et al. Frequent development of combined pituitary hormone deficiency in patients initially diagnosed as isolated growth hormone deficiency: a long term follow-up of patients from a single center. Pituitary. (2015) 18:561–7. doi: 10.1007/s11102-014-0610-9
19. Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, et al. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev. (2016) 37:636–75. doi: 10.1210/er.2016-1101
20. Elizabeth M, Hokken-Koelega ACS, Schuilwerve J, Peeters RP, Visser TJ, de Graaff LCG. Genetic screening of regulatory regions of pituitary transcription factors in patients with idiopathic pituitary hormone deficiencies. Pituitary. (2018) 21:76–83. doi: 10.1007/s11102-017-0850-6
21. De Rienzo F, Mellone S, Bellone S, Babu D, Fusco I, Prodam F, et al. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol. (2015) 83:849–60. doi: 10.1111/cen.12849
22. Obermannova B, Pfaeffle R, Zygmunt-Gorska A, Starzyk J, Verkauskiene R, Smetanina N, et al. Mutations and pituitary morphology in a series of 82 patients with PROP1 gene defects. Horm Res Paediatr. (2011) 76:348–54. doi: 10.1159/000332693
23. Pfäffle RW, Klammt J, Pfäffle HM, Amselem S, Legendre M, Sobrier ML, et al. Genetic screening of patients with congenital GH deficiency in the GeNeSIS observational program: mutation frequency, phenotype and growth outcomes. Horm Res Paediatr. (2017) 88(suppl. 1):FC2. doi: 10.1159/000481424
24. David A, Hwa V, Metherell LA, Netchine I, Camacho-Hübner C, Clark AJ, et al. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. (2011) 32:472–97. doi: 10.1210/er.2010-0023
25. Rosenbloom AL. A half-century of studies of growth hormone insensitivity/ Laron syndrome: a historical perspective. Growth Horm IGF Res. (2016) 28:46–50. doi: 10.1016/j.ghir.2015.08.001
26. Hwa V. STAT5B deficiency: impacts on human growth and immunity. Growth Horm IGF Res. (2016) 28:16–20. doi: 10.1016/j.ghir.2015.12.006
27. Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. (2018) 9:2105. doi: 10.1038/s41467-018-04521-0
28. Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. (2016) 8:363–74. doi: 10.15252/emmm.201506106
29. Muñoz-Calvo MT, Barrios V, Pozo J, Martos-Moreno GA, Hawkins FG, Domene HM, et al. A new syndrome of short stature, mild microcephaly, skeletal abnormalities and high circulating IGF1, IGFBP3 and ALS associated with a homozygous mutation in the gene for pregnancy-associated plasma protein A2 (PAPP-A2, pappalysin2). Endocr Rev. (2015) 36:OR03–1. doi: 10.1210/endo-meetings.2015.PE.1.OR03-1
30. Hawkins-Carranza FG, Muñoz-Calvo MT, Martos-Moreno GÁ, Allo-Miguel G, Del Río L, Pozo J, Chowen JA, et al. rhIGF-1 treatment increases bone mineral density and trabecular bone structure in children with PAPP-A2 deficiency. Horm Res Paediatr. (2018) 89:200–4. doi: 10.1159/000486336
31. Muñoz-Calvo MT, Barrios V, Pozo J, Chowen JA, Martos-Moreno GA, Hawkins F, et al. Treatment with recombinant human insulin-like growth factor-1 improves growth in patients with PAPP-A2 deficiency. J Clin Endocrinol Metab. (2016) 101:3879–83. doi: 10.1210/jc.2016-2751
32. Argente J, Pérez-Jurado LA. History and clinical implications of PAPP-A2 in human growth: when reflecting on idiopathic short stature leads to a specific and new diagnosis: understanding the concept of “low IGF-I availability”. Growth Hormone IGF Res. (2018) 40:17–9. doi: 10.1016/j.ghir.2018.04.001
33. Argente J, Chowen JA, Pérez-Jurado LA, Frystyk J, Oxvig C. One level up: abnormal proteolytic regulation of IGF activity plays a role in human pathophysiology. EMBO Mol Med. (2017) 9:1338–45. doi: 10.15252/emmm.201707950
34. Netchine I, Azzi S, Le Bouc Y, Savage MO. IGF1 molecular anomalies demonstrate its critical role in fetal, postnatal growth and brain development. Best Pract Res Clin Endocrinol Metab. (2011) 25:181–90. doi: 10.1016/j.beem.2010.08.005.
35. Domené HM, Fierro-Carrión G. Genetic disorders of GH action pathway. Growth Horm IGF Res. (2018) 38:19–23. doi: 10.1016/j.ghir.2017.12.004
36. Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, Chen JW, et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. (2005) 90:2855–64. doi: 10.1210/jc.2004-1254
37. Batey L, Moon JE, Yu Y, Wu B, Hirschhorn JN, Shen Y, et al. A novel deletion of IGF1 in a patient with idiopathic short stature provides insight into IGF1 haploinsufficiency. J Clin Endocrinol Metab. (2014) 99:E153–9. doi: 10.1210/jc.2013-3106
38. Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. (1996) 335:1363–7. doi: 10.1056/NEJM199610313351805
39. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. (2015) 373:349–56. doi: 10.1056/NEJMoa1415227
40. Abi Habib W, Brioude F, Azzi S, Rossignol S, Linglart A, Sobrier ML, et al. Transcriptional profiling at the DLK1/MEG3 domain explains clinical overlap between imprinting disorders. Sci Adv. (2019) 5:eaau9425. doi: 10.1126/sciadv.aau9425
41. Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. (2017) 13:105–24. doi: 10.1038/nrendo.2016.138
42. Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. (2003) 349:2211–22. doi: 10.1056/NEJMoa010107
43. Klammt J, Kiess W, Pfäffle R. IGF1R mutations as cause of SGA. Best Pract Res Clin Endocrinol Metab. (2011) 25:191–206. doi: 10.1016/j.beem.2010.09.012
44. Ester WA, van Duyvenvoorde HA, de Wit CC, Broekman AJ, Ruivenkamp CA, Govaerts LC, et al. Two short children born small for gestational age with insulin-like growth factor 1 receptor haploinsufficiency illustrate the heterogeneity of its phenotype. J Clin Endocrinol Metab. (2009) 94:4717–27. doi: 10.1210/jc.2008-1502
45. Prontera P, Micale L, Verrotti A, Napolioni V, Stangoni G, Merla G. A new homozygous IGF1R variant defines a clinically recognizable incomplete dominant form of SHORT syndrome. Hum Mutat. (2015) 36:1043–7. doi: 10.1002/humu.22853
46. Giabicani E, Chantot-Bastaraud S, Bonnard A, Rachid M, Whalen S, Netchine I, Brioude F. Roles of type 1 insulin-like growth factor (IGF) receptor and IGF-II in growth regulation: evidence from a patient carrying both an 11p paternal duplication and 15q deletion. Front Endocrinol (Lausanne). (2019) 10:263. doi: 10.3389/fendo.2019.00263
47. Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, et al. Phenotypic features and response to growth hormone treatment of patients with a molecular defect of the IGF-1 receptor. J Clin Endocrinol Metab. (2019) 104:3157–71. doi: 10.1210/jc.2018-02065
48. Tümer Z, López-Hernández JA, Netchine I, Elbracht M, Grønskov K, Gede LB, et al. Structural and sequence variants in patients with Silver-Russell syndrome or similar features-curation of a disease database. Hum Mutat. (2018) 39:345–64. doi: 10.1002/humu.23382
49. Bang P, Polak M, Woelfle J, Houchard A; EU IGFD Registry Study Group. Effectiveness and safety of rhIGF-1 therapy in children: the European Increlex® Growth Forum Database experience. Horm Res Paediatr. (2015) 83:345–57. doi: 10.1159/000371798
50. Backeljauw PF, Miller BS, Dutailly P, Houchard A, Lawson E, Hale DE, et al. Recombinant human growth hormone plus recombinant human insulin-like growth factor-1 coadministration therapy in short children with low insulin-like growth factor-1 and growth hormone sufficiency: results from a randomized, multicenter, open-label, parallel-group, active treatment-controlled trial. Horm Res Paediatr. (2015) 83:268–79. doi: 10.1159/000371799
51. Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, et al. Novel dominant-negative GH receptor mutations expands the spectrum of GHI and IGF-I deficiency. J Endocr Soc. (2017) 1:345–58. doi: 10.1210/js.2016-1119.
52. Rosenfeld RG. How aggressively should we treat short stature? Horm Res Paediatr. (2015) 83:280–1. doi: 10.1159/000374114
53. Argente J, Gracia R, Ibáñez L, Oliver A, Borrajo E, Vela A, et al. Improvement in growth after two years of growth hormone therapy in very young children born small for gestational age and without spontaneous catch-up growth: results of a multicenter, controlled, randomized, open clinical trial. J Clin Endocrinol Metab. (2007) 92:3095–101. doi: 10.1210/jc.2007-0078
54. Dauber A. IGF-I deficiency in the era of genomics: lessons learned. Horm Res Paediatr. (2017) 88:418–20. doi: 10.1159/000481285
55. Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. (2015) 11:735–46. doi: 10.1038/nrendo.2015.165
56. Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet J Rare Dis. (2019) 14:1. doi: 10.1186/s13023-018-0972-6
57. Ornitz DM, Legeai-Mallet L. Achondroplasia: development, pathogenesis, and therapy. Dev Dyn. (2017) 246:291–309. doi: 10.1002/dvdy.24479
58. Miccoli M, Bertelloni S, Massart F. Height outcome of recombinant human growth hormone treatment in achondroplasia children: a meta-analysis. Horm Res Paediatr. (2016) 86:27–34. doi: 10.1159/000446958
59. Vasques GA, Funari MFA, Ferreira FM, Aza-Carmona M, Sentchordi-Montané L, Barraza-García J, et al. IHH gene mutations causing short stature with nonspecific skeletal abnormalities and response to growth hormone therapy. J Clin Endocrinol Metab. (2018) 103:604–14. doi: 10.1210/jc.2017-02026
60. Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab. (2014) 99:E1510–8. doi: 10.1210/jc.2014-1332
61. Hauer NN, Sticht H, Boppudi S, Büttner C, Kraus C, Trautmann U, et al. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci Rep. (2017) 7:12225. doi: 10.1038/s41598-017-12465-6
62. Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J Clin Endocrinol Metab. (2017) 102:460–9. doi: 10.1210/jc.2016-3313
63. van der Steen M, Pfundt R, Maas SJWH, Bakker-van Waarde WM, Odink RJ, Hokken-Koelega ACS. ACAN gene mutations in short children born SGA and response to growth hormone treatment. J Clin Endocrinol Metab. (2017) 102:1458–67. doi: 10.1210/jc.2016-2941
64. Binder G, Rappold GA. SHOX deficiency disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. GeneReviews®. Seattle, WA: University of Washington (2018).
65. Marchini A, Ogata T, Rappold GA. A track record on SHOX: from basic research to complex models and therapy. Endocr Rev. (2016) 37:417–48. doi: 10.1210/er.2016-1036
66. Huber C, Rosilio M, Munnich A, Cormier-Daire V. French SHOX GeNeSIS Module. High incidence of SHOX anomalies in individuals with short stature. J Med Genet. (2006) 43:735–9. doi: 10.1136/jmg.2006.040998
67. Rosilio M, Huber-Lequesne C, Sapin H, Carel JC, Blum WF, Cormier-Daire V. Genotypes and phenotypes of children with SHOX deficiency in France. J Clin Endocrinol Metab. (2012) 97:E1257–65. doi: 10.1210/jc.2011–3460
68. Fukami M, Seki A, Ogata T. SHOX haploinsufficiency as a cause of syndromic and nonsyndromic short stature. Mol Syndromol. (2016) 7:3–11. doi: 10.1159/000444596
69. Benabbad I, Rosilio M, Child CJ, Carel JC, Ross JL, Deal CL, et al. Safety outcomes and near-adult height gain of growth hormone-treated children with SHOX deficiency: data from an observational study and a clinical trial. Horm Res Paediatr. (2017) 87:42–50. doi: 10.1159/000452973
70. Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, Belinchón A, Barreda-Bonis AC, Vasques GA, et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri-Weill dyschondrosteosis. J Clin Endocrinol Metab. (2015) 100:E1133–42. doi: 10.1210/jc.2015-1612
71. Wang SR, Jacobsen CM, Carmichael H, Edmund AB, Robinson JW, Olney RC, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat. (2015) 36:474–81. doi: 10.1002/humu.22773
72. Jacob M, Menon S, Botti C, Marshall I. Heterozygous NPR2 mutation in two family members with short stature and skeletal dysplasia. Case Rep Endocrinol. (2018) 2018:7658496. doi: 10.1155/2018/7658496
73. Miura K, Kim OH, Lee HR, Namba N, Michigami T, Yoo WJ, et al. Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am J Med Genet A. (2014) 164A:156–63. doi: 10.1002/ajmg.a.36218
74. Hannema SE, van Duyvenvoorde HA, Premsler T, Yang RB, Mueller TD, Gassner B, et al. An activating mutation in the kinase homology domain of the natriuretic peptide receptor-2 causes extremely tall stature without skeletal deformities. J Clin Endocrinol Metab. (2013) 98:E1988–98. doi: 10.1210/jc.2013-2358
75. Ko JM, Bae JS, Choi JS, Miura K, Lee HR, Kim OH, et al. Skeletal overgrowth syndrome caused by overexpression of C-type natriuretic peptide in a girl with balanced chromosomal translocation, t(1;2)(q41;q37.1). Am J Med Genet A. (2015) 167A:1033–8. doi: 10.1002/ajmg.a.36884
76. Bocciardi R, Ravazzolo R. C-type natriuretic peptide and overgrowth. Endocr Dev. (2009) 14:61–6. doi: 10.1159/000207477
77. Heide S, Chantot-Bastaraud S, Keren B, Harbison MD, Azzi S, Rossignol S, et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J Med Genet. (2018) 55:205–13. doi: 10.1136/jmedgenet-2017-104919.
78. Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. (2018) 14:229–49. doi: 10.1038/nrendo.2017.166
79. Hannah-Shmouni F, Trivellin G, Stratakis CA. Genetics of gigantism and acromegaly. Growth Horm IGF Res. (2016) 30–31:37–41. doi: 10.1016/j.ghir.2016.08.002
80. Hannah-Shmouni F, Stratakis CA. An update on the genetics of benign pituitary adenomas in children and adolescents. Curr Opin Endocr Metab Res. (2018) 1:19–24. doi: 10.1016/j.coemr.2018.04.002
81. Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab. (2010) 95:E373–83. doi: 10.1210/jc.2009-2556
82. Beckers A, Petrossians P, Hanson J, Daly AF. The causes and consequences of pituitary gigantism. Nat Rev Endocrinol. (2018) 14:705–20. doi: 10.1038/s41574-018-0114-1
83. Lecoq AL, Kamenický P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas–what to screen for? Nat Rev Endocrinol. (2015) 11:43–54. doi: 10.1038/nrendo.2014.181
84. Daly A, Cano DA, Venegas E, Petrossians P, Dios E, Castermans E, et al. AIP and MEN1 mutations and AIP immunohistochemistry in pituitary adenomas in a tertiary referral center. Endocr Connect. (2019) EC-19-0027. doi: 10.1530/EC-19-0027
85. Yarman S, Tuncer FN, Serbest E. Three novel MEN1 variants in AIP-negative familial isolated pituitary adenoma patients. Pathobiology. (2019) 86:128–134. doi: 10.1159/000495252
86. Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. (2014) 371:2363–74. doi: 10.1056/NEJMoa1408028
87. Beckers A, Rostomyan L, Potorac I, Beckers P, Daly AF. X-LAG: How did they grow so tall? Ann Endocrinol (Paris). (2017) 78:131–6. doi: 10.1016/j.ando.2017.04.013
88. Rostomyan L, Daly AF, Petrossians P, Nachev E, Lila AR, Lecoq AL, et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr Relat Cancer. (2015) 22:745–57. doi: 10.1530/ERC-15-0320
89. Daly AF, Lysy PA, Desfilles C, Rostomyan L, Mohamed A, Caberg JH, et al. GHRH excess and blockade in X-LAG syndrome. Endocr Relat Cancer. (2016) 23:161–70. doi: 10.1530/ERC-15-0478
90. Hou ZS, Tao YX. Mutations in GPR101 as a potential cause of X-linked acrogigantism and acromegaly. Prog Mol Biol Transl Sci. (2019) 161:47–67. doi: 10.1016/bs.pmbts.2018.10.003
91. Trivellin G, Sharwood E, Hijazi H, Carvalho CMB, Yuan B, Tatton-Brown K, et al. Xq26.3 duplication in a boy with motor delay and low muscle tone refines the X-linked acrogigantism genetic locus. J Endocr Soc. (2018) 2:1100–8. doi: 10.1210/js.2018-00156
92. Tatton-Brown K, Weksberg R. Molecular mechanisms of childhood overgrowth. Am J Med Genet C Semin Med Genet. (2013) 163C:71–5. doi: 10.1002/ajmg.c.31362
93. Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet. (2017) 100:725–36. doi: 10.1016/j.ajhg.2017.03.010
94. Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem. (2011) 286:8361–8. doi: 10.1074/jbc.M110.204115
95. Tatton-Brown K, Cole TRP, Rahman N. Sotos syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2015).
96. Edmondson AC, Kalish JM. Overgrowth syndromes. J Pediatr Genet. (2015) 4:136–43. doi: 10.1055/s-0035-1564440
97. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. (2002) 298:1039–43. doi: 10.1126/science.1076997
98. Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, et al. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A. (2013) 161A:2972–80. doi: 10.1002/ajmg.a.36229
99. Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. (2014) 5:3127. doi: 10.1038/ncomms4127
100. Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, et al. The Tatton-Brown-Rahman Syndrome: a clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. (2018) 3:46. doi: 10.12688/wellcomeopenres.14430.1
101. Xin B, Cruz Marino T, Szekely J, Leblanc J, Cechner K, Sency V, et al. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin Genet. (2017) 91:623–8. doi: 10.1111/cge.12878
102. Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. (2014) 46:385–8. doi: 10.1038/ng.2917
103. Harshman SW, Young NL, Parthun MR, Freitas MA. H1 histones: current perspectives and challenges. Nucleic Acids Res. (2013) 41:9593–609. doi: 10.1093/nar/gkt700
104. Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. (2013) 163C:114–21. doi: 10.1002/ajmg.c.31364
105. di Blasio L, Puliafito A, Gagliardi PA, Comunanza V, Somale D, Chiaverina G, et al. PI3K/mTOR inhibition promotes the regression of experimental vascular malformations driven by PIK3CA-activating mutations. Cell Death Dis. (2018) 9:45. doi: 10.1038/s41419-017-0064-x
106. Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. (2018) 558:540–6. doi: 10.1038/s41586-018-0217–9
107. Parker VER, Keppler-Noreuil KM, Faivre L, Luu M, Oden NL, De Silva L, et al. Safety and efficacy of low-dose sirolimus in the PIK3CA-related overgrowth spectrum. Genet Med. (2019) 21(5):1189–98. doi: 10.1038/s41436-018-0297-9
108. Shapiro L, Chatterjee S, Ramadan DG, Davies KM, Savage MO, Metherell LA, et al. Whole-exome sequencing gives additional benefits compared to candidate gene sequencing in the molecular diagnosis of children with growth hormone or IGF-1 insensitivity. Eur J Endocrinol. (2017) 177:485–501. doi: 10.1530/EJE-17-0453
109. Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. (2015) 97:199–215. doi: 10.1016/j.ajhg.2015.06.009
110. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. (2011) 12:745–55. doi: 10.1038/nrg3031
Keywords: growth hormone, genetics, short stature, overgrowth, diagnosis
Citation: Argente J, Tatton-Brown K, Lehwalder D and Pfäffle R (2019) Genetics of Growth Disorders—Which Patients Require Genetic Testing? Front. Endocrinol. 10:602. doi: 10.3389/fendo.2019.00602
Received: 05 February 2019; Accepted: 19 August 2019;
Published: 06 September 2019.
Edited by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanReviewed by:
Lorenzo Iughetti, University of Modena and Reggio Emilia, ItalyYoun Hee Jee, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), United States
Copyright © 2019 Argente, Tatton-Brown, Lehwalder and Pfäffle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Argente, amVzdXMuYXJnZW50ZUBmdW5kYWNpb25lbmRvLm9yZw==; Roland Pfäffle, Um9sYW5kLlBmYWVmZmxlQG1lZGl6aW4udW5pLWxlaXB6aWcuZGU=