 Bin Zhang
Bin Zhang Virginia M. Miller
Virginia M. Miller Jordan D. Miller
Jordan D. Miller- 1Department of Surgery, Mayo Clinic, Rochester, MN, United States
- 2Robert and Arlene Kogod Center on Aging, Mayo Clinic, Rochester, MN, United States
- 3Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, MN, United States
- 4Department of Cardiovascular Surgery, Mayo Clinic, Rochester, MN, United States
Vascular and cardiac valvular calcification was once considered to be a degenerative and end stage product in aging cardiovascular tissues. Over the past two decades, however, a critical mass of data has shown that cardiovascular calcification can be an active and highly regulated process. While the incidence of calcification in the coronary arteries and cardiac valves is higher in men than in age-matched women, a high index of calcification associates with increased morbidity, and mortality in both sexes. Despite the ubiquitous portending of poor outcomes in both sexes, our understanding of mechanisms of calcification under the dramatically different biological contexts of sex and hormonal milieu remains rudimentary. Understanding how the critical context of these variables inform our understanding of mechanisms of calcification—as well as innovative strategies to target it therapeutically–is essential to advancing the fields of both cardiovascular disease and fundamental mechanisms of aging. This review will explore potential sex and sex-steroid differences in the basic biological pathways associated with vascular and cardiac valvular tissue calcification, and potential strategies of pharmacological therapy to reduce or slow these processes.
Background
Ectopic calcification in cardiovascular tissue was once considered to be a passive consequence of cardiovascular disease processes with increasing age. The association-based clinical observations driving this model painted a remarkably appealing picture, with vascular calcification being evident in roughly 25% of patients at age of 50 years, and soaring to over 60% in patients over the age of 75 years (1). While the incidence of aortic valve calcification was slightly lower, the overall trend for dramatic, age-associated increases was equally robust. From such population-based studies, risk factors for vascular and valvular calcification quickly emerged, include aging, metabolic syndrome, smoking, and male sex (2–5).
The site-specific mechanisms of ectopic calcification within the tissues of the cardiovascular system are incompletely understood. At present, calcification in either the coronary arteries or cardiac valves is often considered an organized, regulated, and active pathological process, with evidence of many molecular pathways paralleling those observed in bone/orthotopic ossification. Despite this apparent conservation of core osteoblastic signaling pathways, mature bone matrix is rarely found in calcifying cardiovascular tissues (6–8). Critically, however, upstream mechanisms regulating the induction and amplification of these signaling pathways are likely to be fundamentally different between cardiovascular and orthotopic tissues, since exposure to oxidative stress amplifies osteogenic signaling in calcifying cardiovascular cells but markedly suppresses calcification in bone-derived osteoblasts (9). Furthermore, the contribution of dystrophic tissue calcification—where amorphous accumulation of calcium occurs in the absence of bone matrix, functional osteogenic signaling, or presence of osteoblast-like cells—remains remarkably elusive in the pathogenesis of aortic valve stenosis.
In recent years, tissue fibrosis emerged as a potential and major contributor to aortic valve dysfunction in experimental animals. In particular, genetically altered mice with a propensity for both hypertension and hyperlipidemia developed hemodynamically-significant aortic valve stenosis associated with structural and molecular changes consistent with activation of fibrogenic signaling, and critically, in the absence of substantive changes in valvular calcification. Consequently, investigation into the role of fibrosis as a clinically-meaningful determinant of the degree of valvular stenosis is an exciting and emerging field.
The Clinical Justification for Exploring the Role of Sex in Cavd
While a number of retrospective studies led many to conclude that “women are protected against aortic valve stenosis by estrogens” (10–12), recent work suggests that the pathobiology underlying the disease process may be fundamentally different. For instance, in aortic valve disease—where calcification was once thought to be the primary and near exclusive driver of valve dysfunction–men had more calcification than women at any given level of valvular stenosis (even after normalizing for body size or aortic root size) (13–15), suggesting that valvular fibrosis may play a greater role in determining cusp movement in women compared to men. Similarly, the site of cardiovascular calcification seems to play an important role in predicting mortality in a sex-dependent manner, with thoracic aortic calcification being a strong predictor of mortality secondary to coronary events predominantly in women (16), whereas thoracic aortic and abdominal aortic calcification are strong predictors of all-cause mortality in men (17).
Collectively, the observation that men have higher prevalence of calcification in atherosclerotic lesions and cardiac valves compared to women at any given decade of life has been an interesting clinical observation, and the biological underpinnings and collective clinical implications of these observations are likely to be of great value in developing sex-specific pharmacological treatments to prevent clinically significant valvular and vascular pathology and dysfunction (15, 18, 19). Herein, we aim to highlight potential cellular mechanisms modulated by sex and sex steroid hormones contributing to key sex differences in cardiovascular calcification, with an overall aim of driving dialogue around critical unanswered questions in the field.
Molecular Signaling involved in Cardiovascular Calcification: aN INITIAL Sex and Sex Hormone Agnostic Perspective
While calcification in the cardiovascular system is often considered an active, regulated process with activation of many fundamental osteogenic signaling cascades being conserved between ectopic cardiovascular calcification and orthotopic bone ossification, upstream mechanisms contributing to the induction, sustained activation, and amplification of these pathways can differ markedly. Interestingly, even during formation of micro-calcific deposits there is expression of proteins which are usually absent (e.g., osteopontin) and/or overexpression of proteins which are usually very low in local tissue (e.g., matrix Gla protein), suggesting that maladaptive processes may be initiated even in the earliest stages of the disease (4, 5, 14, 20, 21). Several of the major molecular signaling pathways involved in regulation of ectopic calcification are ubiquitously active in both males and females (Figure 1).
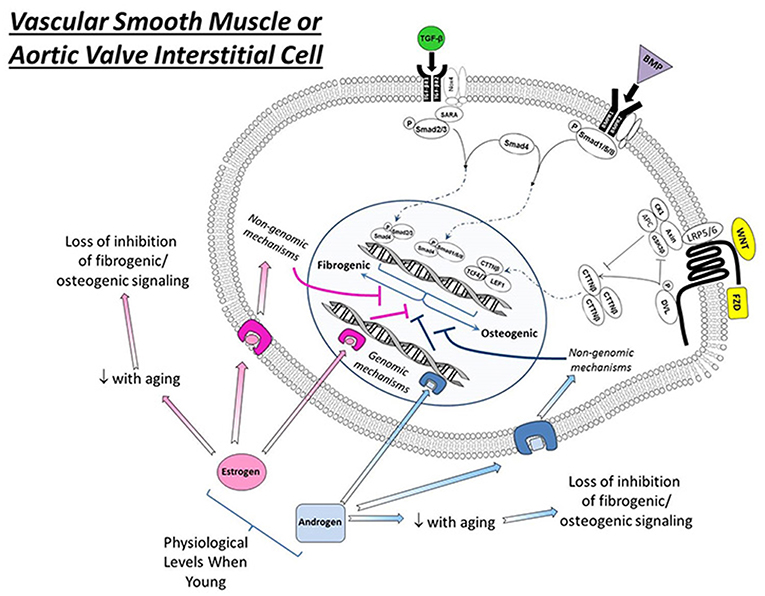
Figure 1. Effects of estrogens and androgen signaling on multiple cellular processes implicated in the regulation of cardiovascular calcification. Note that estrogen can bind to estrogen receptors (ER, resulting in nuclear translation), G-protein coupled estrogen receptors (GPER, eliciting cytosolic signaling), and estrogen binding proteins (EBPs, eliciting cytosolic signaling) to exert a variety of effects—both positive and negative—on molecules influencing ectopic calcification. In general, androgens bind to androgen receptors (AR) and have a smaller number of signal transducing elements compared to estrogens. Abbreviations and impact on calcification: RANKL, receptor activator of nuclear factor κB ligand (promotes calcification); PPAR γ, peroxisome proliferator-activated receptor-γ (prevents calcification); NFκB, nuclear factor κ B (promotes inflammation/calcification); NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase (increases oxidative stress/calcification); SOD, superoxide dismutase (reduces oxidative stress/calcification); O2•−, superoxide anion (increases calcification); p53, tumor protein 53 (promotes inflammation/calcification).
Transforming Growth Factor-β (TGF-β) Signaling
One of the most extensively studied pathways in calcific vascular and valvular disease is TGF-β pathway. While the downstream effects are remarkably context dependent (and in vitro are dependent on substrate stiffness), TGF-β most often induces cell migration, proliferation, and extracellular matrix protein elaboration. Critically, on stiff substrates and matrices, TGFβ robustly induces apoptosis and dystrophic calcific nodule formation in aortic valve interstitial cells from a variety of species, suggesting that the matrix accumulation and sclerosis occurring in early stages of valvular heart disease may shift the phenotypic consequences of increased TGF-β across the spectrum of the disease and contribute to both fibrosis and dystrophic calcification to different extents during the evolution of disease (22, 23).
BMP Signaling
Bone morphogenetic proteins (BMPs), members of the TGF-β superfamily, are significantly increased in ectopic calcification lesions within the cardiovascular system including valvular and vascular tissues (22, 24, 25). Initiated by seminal observations from Demer et al., nearly two decades of work have generated compelling data that BMP signaling plays an integral role in the initiation and progression of cardiovascular calcification. Most paradigms implicating BMP signaling suggest that these morphogens serve as a paracrine signal from nearby resident cells to drive osteogenesis via a BMP-Msx2-Wnt cascade (26–28). Importantly, mechanical stimuli—including non-laminar blood flow patterns exacerbated by multiple disease states–induce both oxidative stress and BMP elaboration from vascular and valvular endothelial cells (29, 30). Furthermore, BMP2- and BMP4-driven osteogenic signaling can be further augmented in conditions where endogenous inhibitors (such as matrix Gla protein) are reduced or absent in a variety of disease states (31).
It is noteworthy that not all bone morphogenetic proteins drive ectopic tissue calcification. For example, BMP-7, which is found in human vascular calcification, slows the progression of arterial calcification in both human and mice with diabetes and hyperlipidemia (32, 33). It is also important to note that while the context dependence of TGF-β signaling has been well-defined by numerous investigators, the role of cell-substrate interactions in the phenotypic consequence of BMP signaling has received much less attention in the literature.
Wnt/β-Catenin Signaling
While increased TGF-β superfamily signaling is a near ubiquitous finding in calcifying cardiovascular tissues, numerous investigators have reported upregulation of other signaling pathways central in bone ossification in diseased vascular and valvular tissues (34). One such pathway is Wnt/β-catenin signaling, where multiple reports have documented increases in Wnt ligand elaboration, low-density lipoprotein receptor-related protein (LRP) receptor components, hyperactivation of canonical β-catenin signaling components, and upregulation of β-catenin transcriptional targets (35, 36).
The Role of Extracellular Vesicles in Cardiovascular Calcification
Recently, numerous studies have implicated extracellular vesicles in the initiation and progression of cardiovascular calcification (37–39). While their precise role remains largely unclear, several studies reported accumulation of nanoparticles that appear to precede (or occur concomitantly) with induction of osteogenic signals in cardiovascular tissue, and aggregation of such vesicles can contribute to formation of larger calcific masses at multiple cardiovascular sites (40–42). Unlike bone, however, where matrix vesicles are derived largely from chondrocytes and osteoblasts, extracellular vesicles accumulating in the cardiovascular system appear to be derived from vascular smooth muscle cells and/or immune cells/macrophages (43, 44). While the composition of each vesicle subset has yet to be comprehensively characterized, it is likely that the cell origin, mechanism/driver of release, vesicle contents, and target tissue in which deposition occurs are all likely determinants of phenotypic/biological outcomes (45, 46).
The Site-Specific Role of Inflammation in Ectopic and Orthotopic Calcification
Tumor necrosis factor-α (TNF-α) is a major cytokine involved in driving both local and systemic inflammation in a variety of cardiovascular pathologies. Thus, it is not surprising that upregulation of TNFα has been shown to augment multiple pathophysiological intracellular signaling cascades involved with vascular and valvular calcification, including interactions with BMP signaling, Msx2-dependent gene transcription, and both canonical and non-canonical Wnt/β-catenin signaling) (35, 47–49). For example, the presence of a TNF-α-Msx2–Wnt/β-catenin cascade acts as a major driver of calcium deposition in aortic valve interstitial cells in vitro (48, 49) and in atherosclerotic lesions in hyperlipidemic mice in vivo (50). While additional mechanistic studies have suggested that antibody-mediated neutralization of TNFα may be effective at slowing initiation or progression of plaque calcification through attenuation of Wnt/β-catenin signaling (3, 50), systemic, long-term suppression of TNFα also puts patients at risk of being immunocompromised, and development of fatal infections.
Impact of Sex Hormones on Processes of Calcification
Although the overall lifetime incidence of atherosclerosis, aortic stenosis, and cumulative death from cardiovascular diseases are remarkably comparable between men and women (51, 52), the primary contributor to the perception of reduced CVD burden in women is the delay in prevalence of atherosclerosis and/or aortic valve stenosis in women compared to men at each decade of life (53, 54). Our understanding of the impact of sex hormones on cardiovascular calcification is compounded by several factors, including marked differences in sex hormone levels over the lifespan of men and women, the effect, type, and timing of hormone replacement on cardiovascular biology in women, and the general paucity of appropriately powered clinical trial data evaluating differential efficacy and effectiveness of drug interventions on men and women. This understanding is complicated further by the fact that many pre-clinical studies have not actively considered the role of sex in the biological pathways being interrogated (via the exclusion of female animals) or have not faithfully recaptured changes in hormones similar to that observed over the lifespan of humans. Given these caveats, we will address the potential roles of both estrogens and androgens on cardiovascular calcification by first providing limited insights from clinical observations, followed by mechanistic insights gleaned largely from pre-clinical animal models.
Role of Estrogens in the Regulation of Cardiovascular Calcification
Clinical Observations
Several seminal studies reported that post-menopausal estrogen treatments may reduce the risk of cardiovascular calcification when administered within the first 5 years of menopause (55, 56). While several recent studies suggest that initiation of estrogen repletion outside of this time period may not confer optimal protection against a myriad of CV complications, the timing, type, and dosing regimens of estrogen that confer vasculo-/valvulo-protection remains a very active field of investigation (57, 58).
Pre-clnical Observations
Given the increased prevalence of subclinical and clinical CVD occurring within first decade following menopause (59, 60), a large amount of effort has been put into understanding the interplay between exposure to either endogenous or exogenous estrogens and several of the abovementioned pathophysiological signaling cascades.
in vitro, estrogen signals through its binding to cytosolic estrogen receptors (such as estrogen receptor α or β), estrogen binding proteins(EBPs), or membrane G-protein-coupled estrogen receptors(Gpr30) (see Figure 2) (61–63). Through a myriad of genomic and non-genomic effects, estrogens can suppress a variety of molecular processes known to drive cardiovascular calcification, including repression of receptor-activator of nuclear factor κB ligand (RANKL) (47, 64) and NFκB signaling, suppression of NADPH oxidase activity in resident cells and inflammatory infiltrates (65, 66) and suppression of p53 (67). Importantly, estrogens do not exert their effects solely by negative regulatory mechanisms, and treatment of cells or animals with exogenous estrogens can drive expression of antioxidant enzymes (in cytosolic, mitochondrial, and lysosomal compartments), and increase nitric oxide synthase activity and expression, both of which have been implicated as key protective mechanisms in cardiovascular calcification (Figure 2) (68–70).
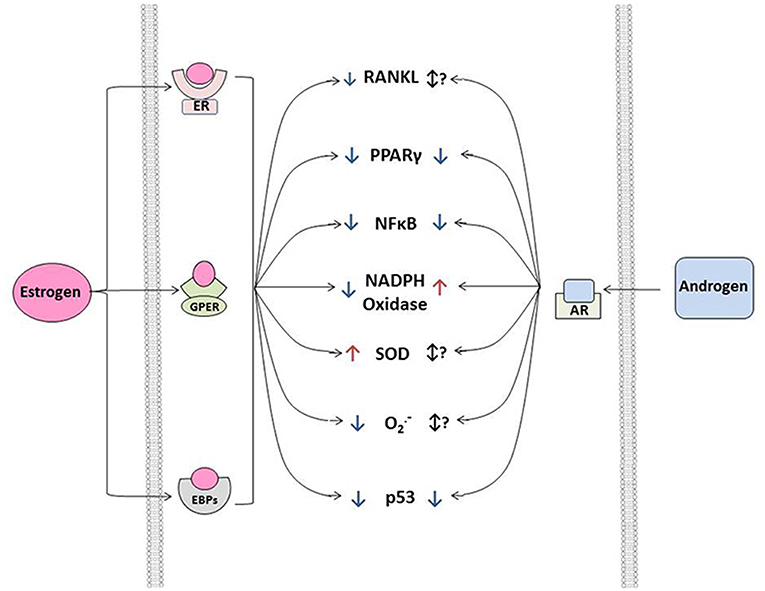
Figure 2. Interactions between estrogen signaling, androgen signaling, and osteogenic signaling in vascular smooth muscle or aortic valve interstitial cells exposed to physiological levels of sex hormones. Note that—in general—both estrogens and androgens suppress osteogenic signaling via both genomic and non-genomic mechanisms in both cell types at physiological levels in relatively early to mid-life stages. Importantly, the therapeutic harnessing of these mechanisms requires substantial research into the context dependence of sex hormone signaling (e.g., timing relative to menopause, level to which hormones should be restored for optimal therapeutic benefit, etc.). TGF-β, transforming growth factor β; BMP, bone morphogenetic protein; Wnt, wingless-related integration site; TGFβR1/2, transforming growth factor beta receptor 1 or 2; BMPR1/2, bone morphogenetic protein receptor 1 or 2; Nox4, NADPH oxidase 4; SARA, smad anchor for receptor activation; Smad, Suppressor of Mothers Against Decapentaplegic; LRP5/6, Low-density lipoprotein receptor-related protein 5 or 6; CK1, Casein kinase 1; DVL, Disheveled protein; Axin, Axin 1 protein; APC, adenomatous polyposis coli protein; GSK3, Glycogen synthase kinase 3; CTTNβ, beta-catenin protein; TCF 4/7, Transcription factor 4 or 7; LEF1, Lymphoid Enhancer Binding Factor 1; FZD, Frizzled receptor.
in vivo, endogenous estrogen levels are critical for protection against cardiovascular calcification at multiple sites, as surgical ovariectomy in young mice results in accelerated development of advanced calcified lesions in both aortic and aortic valve tissue (14). It is likely that a major mechanism whereby estrogen exerts its protective effects is via the suppression of TGF-β-dependent extracellular matrix production and accumulation and downregulation of non-collagenous proteins (71) in cardiovascular lesions, both of which likely serve to prevent increases in micro-environmental stiffness that increase the propensity for apoptosis in response to sustained elevations in TGF-β (72). The interactions between sex hormones and TGF-β signaling are remarkably complex and context dependent, however, and entire reviews have focused on this topic and unanswered questions in the field (73). Furthermore, given previous work suggesting that TGF-β can reciprocally inhibit estrogen receptor signaling via a canonical smad4 interaction (74, 75), the extent to which systemic estrogens can be increased to win over this interplay remains unclear. Given clinical observations of disproportionately augmented TGF-β signaling and fibrosis in women (which is not associated with increases in deposition of dystrophic calcific deposits) (15), it is evident that the net impact of sex hormones on the molecular and phenotypic sequelae of TGF-β signaling will be paramount to the advancement of pharmacotherapies targeting valvular stenosis.
These estrogenic effects are independent of the Y chromosome, as atherosclerosis, vascular calcification, and bone growth were accelerated in a man with estrogen receptor dysfunction (76). Since testosterone is converted to estrogen by aromatase in both females and males, additional insights related to the role of estrogenic signaling in men can be gleaned from studies in which aromatase inhibition was administered. Here, inhibition of aromatase reduced vascular dilatation in men (77, 78), suggesting the net impact of endogenous “testosterone-derived” estrogens is protective in men. Furthermore, we are not aware of additional clinical evidence that short term use of aromatase inhibitors bring benefit for reducing cardiovascular disease incidence in the elderly male patients with low levels of androgen (i.e., attempts to restore testosterone levels through prevention of its degradation) (78, 79). Complementing these data supporting a net protective effect of aromatase-derived estrogens, administration of aromatase inhibitors for 5 years in women (to reduce the recurrence of estrogen-receptor positive breast cancer), also appears to increase incidence of cardiovascular disease (80).
Role of Androgens in the Regulation of Cardiovascular Calcification
Clinical Observations
Numerous studies have shown that, in general, testosterone levels decline relatively linearly after the third decade of life, and can be reduced by more than 50% beyond the sixth decade of life. While the decline in free testosterone is coarsely and inversely related to cardiovascular event rates, causal relationships between changes in testosterone levels and cardiovascular disease remains complicated and highly context dependent (81, 82).
Currently, the vast majority of scientific literature would suggest that supraphysiological levels of testosterone—such as those observed in athletes aiming to improve performance—results in significantly higher levels of coronary artery atherosclerosis compared to non-users of the same age (83–85). Reciprocally, hypogonadal men (testosterone levels <300 ng/dL) have an increased risk of numerous cardiovascular events and complications (86, 87). Of the handful of controlled clinical trials completed to date, most suggest that restoring testosterone levels to mid-normal range does not increase cardiovascular event rates during most follow-up periods (82, 88). Such interventions may, however, increase the volume of non-calcified coronary lesions (89), suggesting that normal levels of testosterone may be pro-atherogenic but not pro-osteogenic/-calcific.
Pre-clinical Observations
Androgens
Numerous studies have probed the interactions between androgen signaling and a variety of pathophysiological signaling cascades in cardiovascular tissues, which have in part been a significant contributor to the controversy surrounding their net impact on cardiovascular diseases. In the context of regulating valvular and vascular disease, several studies have demonstrated a clear role for androgens in the promotion of calcific nodule formation through increasing levels of reactive oxygen species (66, 90–92), repressing PPARγ signaling (93–95), and increasing osteogenic signaling (88, 96, 97) (Figure 2). In line with the aforementioned clinical observations in hypogonadal men, however, physiological levels of androgens may reduce vascular calcification by sustaining eNOS activity (23, 98, 99), reducing TGFβ signaling (100, 101), through the suppression of p53-dependent cellular senescence (67, 102), prevention of cellular apoptosis (91, 103, 104), reducing RANKL signaling (105), suppression of local inflammatory signaling (106–110), and attenuation of pro-thrombotic factor activity (111, 112) (Figure 2).
Protective Androgens vs. Protective Androgen-derived Estrogens
Similar to aging humans, lower serum testosterone is associated with increased risk of cardiovascular calcification in experimental animals and is attenuated by long-term androgen repletion (98, 113). The biological interpretation of this effect is complicated, however, given the fact that both endogenous and exogenous testosterone can be converted to estrogen by aromatase enzymes (114, 115). Seminal studies showing that androgen receptor-dependent signaling has a deleterious impact on CV calcification (via genetic inactivation of the androgen receptor) combined with reports of augmented vascular calcification in men with spontaneous loss-of-function mutations in estrogen receptors also suggests that testosterone-derived aromatases are an underappreciated factor when considering the net impact of androgen signaling on advanced vascular disease (79, 80, 96).
Hormone-Independent Effects of Organismal Sex: The Role of the Chromosomal Complement
Sex hormones aside, the sex chromosomes–the most fundamental and intrinsic determinant of organismal sex–is also likely to be a significant determinant of propensity for cardiovascular calcification. Both X and Y chromosomes have strong linkage associations with cardiovascular disease risk factors such as hypertension, cardiovascular inflammation, immune biology and macrophage function, and organismal metabolism (116–120). Perhaps most critically, cells derived from XX or XY organisms which are treated with identical in vitro conditions show differences in proliferation, fibrosis, and apoptosis in response to various agonists (121, 122). These changes are not restricted solely to vascular tissues, as osteogenic signaling and responses to various agonists also differ in aortic valve interstitial cells from XX and XY animals (123). Thus, while the Y chromosome may be referred to as a “non-recombining desert” in some biological circles (124), its sustained phenotypic impact is of undeniable importance in cardiovascular tissues.
Conceptual Gaps and Controversies in the Field of Cardiovascular Calcification
While tremendous advances have been made in our understanding of cardiovascular calcification over the past several decades, several major gaps remain in our efforts to translate and apply both biological and clinical discoveries to the care of an individual patient.
As appropriate with the scope of this review, one could readily argue that the field's greatest gap relates to our understanding of the impact of biological sex and the sex steroid hormonal milieu on phenotypic and clinical outcomes in diseases where vascular, valvular, or microvascular calcification are of clinical importance. As the role of this critical context becomes clearer with appropriately controlled and sex-balanced pre-clinical and clinical investigation (125–127), we will undoubtedly gain deeper insights into both the pathobiological underpinnings and potential efficacious therapeutic interventions in men and women suffering from calcific cardiovascular diseases.
Perhaps the greatest controversy in the field of cardiovascular calcification—which is not necessarily exclusive from of our understanding of biological sex—is the contexts in which ectopic calcification is driven by non-osteogenic or osteogenic mechanisms. More specifically, while an overwhelming body of evidence suggests that the osteogenic signaling cascades described in this review are present in calcifying tissues from the vast majority of patients with cardiovascular calcification, clinical observations at the time of surgery or autopsy suggest that bone matrix is only evident in a relatively small fraction of this patient population (e.g., 15–25%) (128). Thus, how the cellular decision is made to initiate maladaptive, osteogenic “response to injury” at the earliest stages of microcalcific nodule formation (129) that propagates to true “ectopic bone” or alternatively expands due to progressive and persistent cellular apoptosis to form an amorphous, calcific deposit (130–133) with merely associative increases in osteogenic signaling remains remarkably elusive.
Finally, the role of the biological context of organismal age (and its fundamental biological determinants including changes in sex steroid hormones) in dictating these decisions is only beginning to be understood. While there has been a longstanding association between cellular senescence and tissue calcification [stemming from seminal work by Shanahan et al. (134–137)] more recent work suggests that the pharmacological targeting and/or clearance of senescent cells may also be a viable strategy for slowing progression of vascular calcification and dysfunction (136). The postulate that targeting fundamental biological mechanisms of aging may be a viable strategy to delay onset or prevent progression of cardiovascular calcification is supported by intriguing observation that biomarkers of biological age (e.g., telomere length) are stronger predictors of incidence of valvular heart disease than chronological age (138).
Perspectives on the Future of the Field of Cardiovascular Calcification and Strategies for Addressing the Effects of Sex
While the field of cardiovascular calcification has made tremendous strides in advancing our understanding of osteogenic and non-osteogenic mechanisms contributing to ectopic calcium accrual over the past two decades, it is our opinion that the greatest advances—both scientifically and clinically—will be made in the near future by exploring the context of sex and sex hormones in these phenomena. While many may consider this to be too bold of a statement, it stands on a firm foundation of both clinical and biological reports demonstrating clear sex- and sex hormone-driven differences in the progression of calcific cardiovascular diseases and a glaring lack of success in viable pharmacological strategies to mitigate cardiovascular calcification in elderly persons.
While the National Institutes of Health now mandates consideration of sex as a biological variable in all studies that receive funding (e.g., from pre-clinical animal investigation to human trials), there have not been consistent requirements from publishers and journals requiring reporting of data by sex. Consequently, we would make the following additional suggestions to drive discussion of true “best practices” and accelerate development of critical insights into mechanisms underlying cardiovascular calcification in future studies. First, we suggest that in vitro studies should include independent cell lines derived from each sex of the species being studied, which will allow for characterization of the impact of chromosomal complement and differing epigenetic demarcations in the absence of the sex-steroid and systemic hormonal millieu. With the emergence of evidence that cell line immortalization can drive X and Y chromosomal reconfiguration (139) and the absence of data demonstrating whether such phenomena eliminates sex-dependent molecular responses to exogenous stressors, we also advocate for the use of primary cell lines until comprehensive characterizations of cell phenotype and response are available. Second, we suggest that all sex-disaggregated data should be available within manuscripts and/or online supplements, and that studies should be designed to detect sex differences with appropriate statistical power in an a priori manner. In clinical conditions in which a disease occurs predominantly in one sex vs. the other, we feel a minimum recommendation of having the experimental sample composition be reflective of the sex distribution within the patient population of interest is reasonable. Finally, execution of appropriate hormonal depletion, repletion, or crossover studies would represent a major advance in the field. By appropriate, we refer not only to absolute hormonal levels but also to timing of repletion/depletion (i.e., initiation of the insult later in life, similar to what occurs in humans).
While we believe few investigators would argue that the abovementioned recommendations directly align with the foundational principles of scientific rigor, we are acutely aware that the logistics of implementing such recommendations must be addressed. While doubling the scope and scale of ongoing research projects is neither feasible nor sustainable, we would argue that generation of pre-clinical and clinical datasets that do not inform either the pathobiological underpinnings or clinical care of half of the world's population reduces the relevance and impact of scientific investigation around the globe. At present, we believe there are few viable arguments against the intentional and appropriate inclusion (and subsequent disaggregated presentation) of data from both sexes in studies of cardiovascular calcification, as the number of samples required for demonstrating sex differences should not increase dramatically when truly qualitatively different trends are uncovered. We acknowledge that discovery of such dichotomous sex responses can often spur new lines of investigation that are beyond the scope of the existing project and subsequently require support through additional funding mechanisms. Critically, we commend NIH and several other funding entities for creation of supplemental research awards devoted to supporting more detailed investigation of mechanisms underlying unexpectedly discovered sex differences, as well as creation of recent RFA's that prioritize identification of novel sex differences in cardiovascular calcification and disease.
Ultimately, we feel that appropriately designed and executed clinical studies—including both men and women potentially with independent outcome measures in each sex (e.g., not only valvular function as a primary outcome but predominantly calcification in men and fibrosis in women as secondary outcomes) is an essential step in ensuring that the utility of sex in predicting therapeutic efficacy and effectiveness across the translational spectrum. While beyond the scope of this review [and covered recently in Sritharen et al. (14)], emerging data strongly suggest that incoming risk profiles, outcomes for surgical valve replacement, outcomes for transcatheter valve replacement, and comorbid condition frequency following disease diagnosis differ robustly amongst men and women, and can serve as a critical catalyst for driving impactful investigation in the biological mechanisms underlying such observations.
Summary
The clinical presentation, biological underpinnings, and molecular interactions with sex hormones and biological sex, and ultimate strategies to therapeutically prevent or slow progression of cardiovascular calcification differ dramatically between men and women. While in the United States, the National Institutes of Health (NIH) mandate for inclusion of both sexes in research will undoubtedly serve to advance our understanding of these differences, it is our hope that this review will spur additional genuine interest in understanding critical biological and clinical contexts—including, but not limited to organismal sex—and drive transformative advances in the strategies and tools needed to reduce the growing global burden of calcific vascular and valvular diseases.
Author Contributions
All authors contributed to the drafting and revision of the manuscript and concepts expressed herein.
Funding
This work was supported in part by grants from the NIH U54 AG 44170, R01 AG 053832, R01 HL 141819, and the Mayo Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cannata-Andia JB, Rodriguez-Garcia M, Carrillo-Lopez N, Naves-Diaz M, Diaz-Lopez B. Vascular calcifications: pathogenesis, management, and impact on clinical outcomes. J Am Soc Nephrol. (2006) 17:S267–73. doi: 10.1681/ASN.2006080925
2. Ruiz JL, Hutcheson JD, Aikawa E. Cardiovascular calcification: current controversies and novel concepts. Cardiovasc Pathol. (2015) 24:207–12. doi: 10.1016/j.carpath.2015.03.002
3. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. (2010) 7:528–36. doi: 10.1038/nrcardio.2010.115
4. Shioi A. Molecular mechanisms of vascular calcification. Clin Calcium. (2010) 20:1611–9. doi: 10.1007/s11914-015-0270-3
5. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Mechanisms of medial arterial calcification in diabetes. Curr Pharm Des. (2014) 20: 5870–83. doi: 10.2174/1381612820666140212210451
6. Pillai CLI, Li S, Romay M, Lam L, Lu Y, Huang J, et al. Cardiac fibroblasts adopt osteogenic fates and can be targeted to attenuate pathological heart calcification. Cell Stem Cell. (2017) 20:218–32.e5. doi: 10.1016/j.stem.2016.10.005
7. Nakahara T, Dweck MR, Narula N, Pisapia D, Narula J, Strauss HW. Coronary artery calcification: from mechanism to molecular imaging. JACC Cardiovasc Imaging. (2017) 10:582–93. doi: 10.1016/j.jcmg.2017.03.005
8. Hou M, Song Y, Li Z, Luo C, Ou JS, Yu H, et al. Curcumin attenuates osteogenic differentiation and calcification of rat vascular smooth muscle cells. Mol Cell Biochem. (2016) 420:151–60. doi: 10.1007/s11010-016-2778-y
9. Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med. (2001) 31:509–19. doi: 10.1016/S0891-5849(01)00610-4
10. Nordmeyer J, Eder S, Mahmoodzadeh S, Martus P, Fielitz J, Bass J, et al. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation. (2004) 110:3270–5. doi: 10.1161/01.CIR.0000147610.41984.E8
11. Masjedi S, Ferdous Z. Understanding the role of sex in heart valve and major vascular diseases. Cardiovasc Eng Technol. (2015) 6:209–19. doi: 10.1007/s13239-015-0226-x
12. Thaden JJ, Nkomo VT, Suri RM, Maleszewski JJ, Soderberg DJ, Clavel MA, et al. Sex-related differences in calcific aortic stenosis: correlating clinical and echocardiographic characteristics and computed tomography aortic valve calcium score to excised aortic valve weight. Eur Heart J. (2016) 37:693–9. doi: 10.1093/eurheartj/ehv560
13. Aggarwal SR, Clavel MA, Messika-Zeitoun D, Cueff C, Malouf J, Araoz PA, et al. Sex differences in aortic valve calcification measured by multidetector computed tomography in aortic stenosis. Circ Cardiovasc Imaging. (2013) 6:40–7. doi: 10.1161/CIRCIMAGING.112.980052
14. Sritharen Y, Enriquez-Sarano M, Schaff HV, Casaclang-Verzosa G, Miller JD. Pathophysiology of aortic valve stenosis: is it both fibrocalcific and sex specific? Physiology. (2017) 32:182–96. doi: 10.1152/physiol.00025.2016
15. Simard L, Cote N, Dagenais F, Mathieu P, Couture C, Trahan S, et al. Sex-related discordance between aortic valve calcification and hemodynamic severity of aortic stenosis: is valvular fibrosis the explanation? Circ Res. (2017) 120:681–91. doi: 10.1161/CIRCRESAHA.116.309306
16. Rodondi N, Taylor BC, Bauer DC, Lui LY, Vogt MT, Fink HA, et al. Association between aortic calcification and total and cardiovascular mortality in older women. J Intern Med. (2007) 261:238–44. doi: 10.1111/j.1365-2796.2007.01769.x
17. Estublier C, Chapurlat R, Szulc P. Association of severe disc degeneration with all-cause mortality and abdominal aortic calcification assessed prospectively in older men: findings of a single-center prospective study of osteoporosis in men. Arthritis Rheumatol. (2015) 67:1295–304. doi: 10.1002/art.39055
18. Liyanage L, Lee NJ, Cook T, Herrmann HC, Jagasia D, Litt H, et al. The impact of gender on cardiovascular system calcification in very elderly patients with severe aortic stenosis. Int J Cardiovasc Imaging. (2016) 32:173–9. doi: 10.1007/s10554-015-0752-5
19. Yahagi K, Davis HR, Arbustini E, Virmani R. Sex differences in coronary artery disease: pathological observations. Atherosclerosis. (2015) 239:260–7. doi: 10.1016/j.atherosclerosis.2015.01.017
20. Weiss RM, Miller JD, Heistad DD. Fibrocalcific aortic valve disease: opportunity to understand disease mechanisms using mouse models. Circ Res. (2013) 113:209–22. doi: 10.1161/CIRCRESAHA.113.300153
21. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. (2011) 108:1392–412. doi: 10.1161/CIRCRESAHA.110.234138
22. Dutta P, Lincoln J. Calcific aortic valve disease: a developmental biology perspective. Curr Cardiol Rep. (2018) 20:21. doi: 10.1007/s11886-018-0968-9
23. Liu AC, Gotlieb AI. Transforming growth factor-beta regulates in vitro heart valve repair by activated valve interstitial cells. Am J Pathol. (2008) 173:1275–85. doi: 10.2353/ajpath.2008.080365
24. Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, Conway SJ, Yutzey KE. Bone morphogenetic protein signaling is required for aortic valve calcification. Arterioscler Thromb Vasc Biol. (2016) 36:1398–405. doi: 10.1161/ATVBAHA.116.307526
25. Zhang M, Sara JD, Wang FL, Liu LP, Su LX, Zhe J, et al. Increased plasma BMP-2 levels are associated with atherosclerosis burden and coronary calcification in type 2 diabetic patients. Cardiovasc Diabetol. (2015) 14:64. doi: 10.1186/s12933-015-0214-3
26. Shao JS, Aly ZA, Lai CF, Cheng SL, Cai J, Huang E, et al. Vascular Bmp Msx2 Wnt signaling and oxidative stress in arterial calcification. Ann N Y Acad Sci. (2007) 1117:40–50. doi: 10.1196/annals.1402.075
27. Yung LM, Sanchez-Duffhues G, Ten Dijke P, Yu PB. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res. (2015) 108:278–87. doi: 10.1093/cvr/cvv221
28. Sun L, Rajamannan NM, Sucosky P. Defining the role of fluid shear stress in the expression of early signaling markers for calcific aortic valve disease. PLoS ONE. (2013) 8:e84433. doi: 10.1371/journal.pone.0084433
29. Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med. (2007) 17:48–54. doi: 10.1016/j.tcm.2006.11.005
30. Csiszar A, Lehoux S, Ungvari Z. Hemodynamic forces, vascular oxidative stress, and regulation of BMP-2/4 expression. Antioxid Redox Signal. (2009) 11:1683–97. doi: 10.1089/ars.2008.2401
31. Malhotra R, Burke MF, Martyn T, Shakartzi HR, Thayer TE, O'Rourke C, et al. Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix Gla protein deficiency. PLoS ONE. (2015) 10:e0117098. doi: 10.1371/journal.pone.0117098
32. Davies MR, Lund RJ, Hruska KA. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J Am Soc Nephrol. (2003) 14:1559–67. doi: 10.1097/01.ASN.0000068404.57780.DD
33. Freedman BI, Bowden DW, Ziegler JT, Langefeld CD, Lehtinen AB, Rudock ME, et al. Bone morphogenetic protein 7 (BMP7) gene polymorphisms are associated with inverse relationships between vascular calcification and BMD: the Diabetes Heart Study. J Bone Miner Res. (2009) 24:1719–27. doi: 10.1359/jbmr.090501
34. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc Res. (2008) 77:221–30. doi: 10.1093/cvr/cvm049
35. Al-Aly Z. Arterial calcification: a tumor necrosis factor-alpha mediated vascular Wnt-opathy. Transl Res. (2008) 151:233–9. doi: 10.1016/j.trsl.2007.12.005
36. Gao Y, Huang E, Zhang H, Wang J, Wu N, Chen X, et al. Crosstalk between Wnt/beta-catenin and estrogen receptor signaling synergistically promotes osteogenic differentiation of mesenchymal progenitor cells. PLoS ONE. (2013) 8:e82436. doi: 10.1371/journal.pone.0082436
37. Bakhshian Nik A, Hutcheson JD, Aikawa E. Extracellular vesicles as mediators of cardiovascular calcification. Front Cardiovasc Med. (2017) 4:78. doi: 10.3389/fcvm.2017.00078
38. Aikawa E. Extracellular vesicles in cardiovascular disease: focus on vascular calcification. J Physiol. (2016) 594:2877–80. doi: 10.1113/JP272112
39. Zazzeroni L, Faggioli G, Pasquinelli G. Mechanisms of arterial calcification: the role of matrix vesicles. Eur J Vasc Endovasc Surg. (2018) 55:425–32. doi: 10.1016/j.ejvs.2017.12.009
40. Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. (2004) 15:2857–67. doi: 10.1097/01.ASN.0000141960.01035.28
41. Chen NX, O'Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res. (2008) 23:1798–805. doi: 10.1359/jbmr.080604
42. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. (2015) 116:1312–23. doi: 10.1161/CIRCRESAHA.116.305012
43. Krohn JB, Hutcheson JD, Martinez-Martinez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. (2016) 594:2895–903. doi: 10.1113/JP271338
44. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. (2016) 15:335–43. doi: 10.1038/nmat4519
45. Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol. (2010) 21:103–12. doi: 10.1681/ASN.2009060640
46. Jansen F, Xiang X, Werner N. Role and function of extracellular vesicles in calcific aortic valve disease. Eur Heart J. (2017) 38:2714–6. doi: 10.1093/eurheartj/ehx477
47. Harper E, Forde H, Davenport C, Rochfort KD, Smith D, Cummins PM. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vascul Pharmacol. (2016) 82:30–40. doi: 10.1016/j.vph.2016.02.003
48. Kaden JJ, Kilic R, Sarikoc A, Hagl S, Lang S, Hoffmann U, et al. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med. (2005) 16:869–72. doi: 10.3892/ijmm.16.5.869
49. Yu Z, Seya K, Daitoku K, Motomura S, Fukuda I, Furukawa K. Tumor necrosis factor-alpha accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther. (2011) 337:16–23. doi: 10.1124/jpet.110.177915
50. Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol. (2007) 27:2589–96. doi: 10.1161/ATVBAHA.107.153668
51. Jousilahti P, Vartiainen E, Tuomilehto J, Puska P. Sex, age, cardiovascular risk factors, and coronary heart disease: a prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation. (1999) 99:1165–72. doi: 10.1161/01.CIR.99.9.1165
52. Appelman Y, van Rijn BB, Ten Haaf ME, Boersma E, Peters SA. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis. (2015) 241:211–8. doi: 10.1016/j.atherosclerosis.2015.01.027
53. Mosca L, Barrett-Connor E, Wenger NK. Sex/gender differences in cardiovascular disease prevention: what a difference a decade makes. Circulation. (2011) 124:2145–54. doi: 10.1161/CIRCULATIONAHA.110.968792
54. Pilote L, Dasgupta K, Guru V, Humphries KH, McGrath J, Norris C, et al. A comprehensive view of sex-specific issues related to cardiovascular disease. CMAJ. (2007) 176:S1–44. doi: 10.1503/cmaj.051455
55. Manson JE, Allison MA, Rossouw JE, Carr JJ, Langer RD, Hsia J, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. (2007) 356:2591–602. doi: 10.1056/NEJMoa071513
56. Shahar E, Burke GL, Cushman M, Heckbert SR, Ouyang P, Szklo M. Post menopausal hormones and measures of subclinical atherosclerosis: the multi-ethnic study of atherosclerosis. Prev Med. (2008) 47:38–45. doi: 10.1016/j.ypmed.2007.12.013
57. Miller VM, Harman SM. An update on hormone therapy in postmenopausal women: mini-review for the basic scientist. Am J Physiol Heart Circ Physiol. (2017) 313:H1013–21. doi: 10.1152/ajpheart.00383.2017
58. Subramanya V, Zhao D, Ouyang P, Ying W, Vaidya D, Ndumele CE, et al. Association of endogenous sex hormone levels with coronary artery calcium progression among post-menopausal women in the Multi-Ethnic Study of Atherosclerosis (MESA). J Cardiovasc Comput Tomogr. (2019) 13:41–7. doi: 10.1016/j.jcct.2018.09.010
59. Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. (2007) 297:1465–77. doi: 10.1001/jama.297.13.1465
60. Rosano GM, Vitale C, Marazzi G, Volterrani M. Menopause and cardiovascular disease: the evidence. Climacteric. (2007) 10(Suppl 1):19–24. doi: 10.1080/13697130601114917
61. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. (1997) 138:863–70. doi: 10.1210/endo.138.3.4979
62. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. (2007) 87:905–31. doi: 10.1152/physrev.00026.2006
63. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. (2008) 70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518
64. Osako MK, Nakagami H, Koibuchi N, Shimizu H, Nakagami F, Koriyama H, et al. Estrogen inhibits vascular calcification via vascular RANKL system: common mechanism of osteoporosis and vascular calcification. Circ Res. (2010) 107:466–75. doi: 10.1161/CIRCRESAHA.110.216846
65. Sahoo S, Meijles DN, Pagano PJ. NADPH oxidases: key modulators in aging and age-related cardiovascular diseases? Clin Sci. (2016) 130:317–35. doi: 10.1042/CS20150087
66. Chignalia AZ, Oliveira MA, Debbas V, Dull RO, Laurindo FR, Touyz RM, et al. Testosterone induces leucocyte migration by NADPH oxidase-driven ROS- and COX2-dependent mechanisms. Clin Sci. (2015) 129:39–48. doi: 10.1042/CS20140548
67. Zapata E, Ventura JL, De la Cruz K, Rodriguez E, Damian P, Masso F, et al. Dehydroepiandrosterone inhibits the proliferation of human umbilical vein endothelial cells by enhancing the expression of p53 and p21, restricting the phosphorylation of retinoblastoma protein, and is androgen- and estrogen-receptor independent. FEBS J. (2005) 272:1343–53. doi: 10.1111/j.1742-4658.2005.04563.x
68. Castardo-de-Paula JC, de Campos BH, Amorim DTE, da Silva RV, de Farias CC, Higachi L, et al. Cardiovascular risk and the effect of nitric oxide synthase inhibition in female rats: the role of estrogen. Exp Gerontol. (2017) 97:38–48. doi: 10.1016/j.exger.2017.07.016
69. Nevzati E, Shafighi M, Bakhtian KD, Treiber H, Fandino J, Fathi AR. Estrogen induces nitric oxide production via nitric oxide synthase activation in endothelial cells. Acta Neurochir Suppl. (2015) 120:141–5. doi: 10.1007/978-3-319-04981-6_24
70. Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, et al. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol. (2013) 182:1922–31. doi: 10.1016/j.ajpath.2013.01.037
71. Donley GE, Fitzpatrick LA. Noncollagenous matrix proteins controlling mineralization; possible role in pathologic calcification of vascular tissue. Trends Cardiovasc Med. (1998) 8:199–206. doi: 10.1016/S1050-1738(98)00014-0
72. Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. (2009) 29:936–42. doi: 10.1161/ATVBAHA.108.182394
73. Shah TA, Rogers MB. Unanswered questions regarding sex and BMP/TGF-β signaling. J Dev Biol. (2018) 6:14. doi: 10.20944/preprints201805.0180.v1
74. Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, et al. Smad4 as a transcription corepressor for estrogen receptor alpha. J Biol Chem. (2003) 278:15192–200. doi: 10.1074/jbc.M212332200
75. Giacomini D, Paez-Pereda M, Stalla J, Stalla GK, Arzt E. Molecular interaction of BMP-4, TGF-beta, and estrogens in lactotrophs: impact on the PRL promoter. Mol Endocrinol. (2009) 23:1102–14. doi: 10.1210/me.2008-0425
76. Sinnesael M, Boonen S, Claessens F, Gielen E, Vanderschueren D. Testosterone and the male skeleton: a dual mode of action. J Osteoporos. (2011) 2011:240328. doi: 10.4061/2011/240328
77. Lew R, Komesaroff P, Williams M, Dawood T, Sudhir K. Endogenous estrogens influence endothelial function in young men. Circ Res. (2003) 93:1127–33. doi: 10.1161/01.RES.0000103633.57225.BC
78. Duckles SP, Miller VM. Hormonal modulation of endothelial NO production. Pflugers Arch. (2010) 459:841–51. doi: 10.1007/s00424-010-0797-1
79. Dougherty RH, Rohrer JL, Hayden D, Rubin SD, Leder BZ. Effect of aromatase inhibition on lipids and inflammatory markers of cardiovascular disease in elderly men with low testosterone levels. Clin Endocrinol. (2005) 62:228–35. doi: 10.1111/j.1365-2265.2005.02205.x
80. Lonning PE, Eikesdal HP. Aromatase inhibition 2013: clinical state of the art and questions that remain to be solved. Endocr Relat Cancer. (2013) 20:R183–201. doi: 10.1530/ERC-13-0099
81. Kloner RA, Carson C 3rd, Dobs A, Kopecky S, Mohler ER 3rd. Testosterone and cardiovascular disease. J Am Coll Cardiol. (2016) 67:545–57. doi: 10.1016/j.jacc.2015.12.005
82. Gencer B, Mach F. Testosterone: a hormone preventing cardiovascular disease or a therapy increasing cardiovascular events? Eur Heart J. (2016) 37:3569–75. doi: 10.1093/eurheartj/ehv439
83. Vanberg P, Atar D. Androgenic anabolic steroid abuse and the cardiovascular system. Handb Exp Pharmacol. (2010) 411−57. doi: 10.1007/978-3-540-79088-4_18
84. Sonmez E, Turkdogan KA, Yilmaz C, Kucukbuzcu S, Ozkan A, Sogutt O. Chronic anabolic androgenic steroid usage associated with acute coronary syndrome in bodybuilder. Turk J Emerg Med. (2016) 16:35–7. doi: 10.1016/j.tjem.2014.11.001
85. Major RW, Pierides M, Squire IB, Roberts E. Bodybuilding, exogenous testosterone use and myocardial infarction. QJM. (2015) 108:651–2. doi: 10.1093/qjmed/hcu173
86. Traish AM, Abdou R, Kypreos KE. Androgen deficiency and atherosclerosis: the lipid link. Vascul Pharmacol. (2009) 51:303–13. doi: 10.1016/j.vph.2009.09.003
87. Traish AM, Saad F, Feeley RJ, Guay A. The dark side of testosterone deficiency: III. Cardiovascular disease. J Androl. (2009) 30:477–94. doi: 10.2164/jandrol.108.007245
88. Lopes RA, Neves KB, Carneiro FS, Tostes RC. Testosterone and vascular function in aging. Front Physiol. (2012) 3:89. doi: 10.3389/fphys.2012.00089
89. Budoff MJ, Ellenberg SS, Lewis CE, Mohler ER 3rd, Wenger NK, Bhasin S, et al. Testosterone treatment and coronary artery plaque volume in older men with low testosterone. JAMA. (2017) 317:708–16. doi: 10.1001/jama.2016.21043
90. Chistiakov DA, Myasoedova VA, Melnichenko AA, Grechko AV, Orekhov AN. Role of androgens in cardiovascular pathology. Vasc Health Risk Manag. (2018) 14:283–90. doi: 10.2147/VHRM.S173259
91. Lucas-Herald AK, Alves-Lopes R, Montezano AC, Ahmed SF, Touyz RM. Genomic and non-genomic effects of androgens in the cardiovascular system: clinical implications. Clin Sci. (2017) 131:1405–18. doi: 10.1042/CS20170090
92. Skogastierna C, Hotzen M, Rane A, Ekstrom L. A supraphysiological dose of testosterone induces nitric oxide production and oxidative stress. Eur J Prev Cardiol. (2014) 21:1049–54. doi: 10.1177/2047487313481755
93. Du J, Zhang L, Wang Z. Testosterone inhibits the activity of peroxisome proliferator-activated receptor gamma in a transcriptional transaction assay. Pharmazie. (2009) 64:692–3. doi: 10.1691/ph.2009.9606
94. Sato H, Sugai H, Kurosaki H, Ishikawa M, Funaki A, Kimura Y, et al. The effect of sex hormones on peroxisome proliferator-activated receptor gamma expression and activity in mature adipocytes. Biol Pharm Bull. (2013) 36:564–73. doi: 10.1248/bpb.b12-00868
95. Altman R, Motton DD, Kota RS, Rutledge JC. Inhibition of vascular inflammation by dehydroepiandrosterone sulfate in human aortic endothelial cells: roles of PPARalpha and NF-kappaB. Vascul Pharmacol. (2008) 48:76–84. doi: 10.1016/j.vph.2007.12.002
96. Cho JJ, Cadet P, Salamon E, Mantione K, Stefano GB. The nongenomic protective effects of estrogen on the male cardiovascular system: clinical and therapeutic implications in aging men. Med Sci Monit. (2003) 9:RA63-8.
97. Vasconsuelo A, Pronsato L, Ronda AC, Boland R, Milanesi L. Role of 17beta-estradiol and testosterone in apoptosis. Steroids. (2011) 76:1223–31. doi: 10.1016/j.steroids.2011.08.001
98. Ota H, Akishita M, Akiyoshi T, Kahyo T, Setou M, Ogawa S, et al. Testosterone deficiency accelerates neuronal and vascular aging of SAMP8 mice: protective role of eNOS and SIRT1. PLoS ONE. (2012) 7:e29598. doi: 10.1371/journal.pone.0029598
99. Yu J, Akishita M, Eto M, Koizumi H, Hashimoto R, Ogawa S, et al. Src kinase-mediates androgen receptor-dependent non-genomic activation of signaling cascade leading to endothelial nitric oxide synthase. Biochem Biophys Res Commun. (2012) 424:538–43. doi: 10.1016/j.bbrc.2012.06.151
100. Chung CC, Hsu RC, Kao YH, Liou JP, Lu YY, Chen YJ Androgen attenuates cardiac fibroblasts activations through modulations of transforming growth factor-beta and angiotensin II signaling. Int J Cardiol. (2014) 176:386–93. doi: 10.1016/j.ijcard.2014.07.077
101. Ikeda Y, Aihara K, Sato T, Akaike M, Yoshizumi M, Suzaki Y, et al. Androgen receptor gene knockout male mice exhibit impaired cardiac growth and exacerbation of angiotensin II-induced cardiac fibrosis. J Biol Chem. (2005) 280:29661–6. doi: 10.1074/jbc.M411694200
102. Altieri P, Barisione C, Lazzarini E, Garuti A, Bezante GP, Canepa M, et al. Testosterone antagonizes doxorubicin-induced senescence of cardiomyocytes. J Am Heart Assoc. (2016) 5:e002383. doi: 10.1161/JAHA.115.002383
103. Zhu D, Hadoke PW, Wu J, Vesey AT, Lerman DA, Dweck MR, et al. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci Rep. (2016) 6:24807. doi: 10.1038/srep24807
104. Lopes RA, Neves KB, Pestana CR, Queiroz AL, Zanotto CZ, Chignalia AZ, et al. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am J Physiol Heart Circ Physiol. (2014) 306:H1485–94. doi: 10.1152/ajpheart.00809.2013
105. Martin A, Yu J, Xiong J, Khalid AB, Katzenellenbogen B, Kim SH, et al. Estrogens and androgens inhibit association of RANKL with the pre-osteoblast membrane through post-translational mechanisms. J Cell Physiol. (2017) 232:3798–807. doi: 10.1002/jcp.25862
106. Steffens JP, Herrera BS, Coimbra LS, Stephens DN, Rossa C Jr, Spolidorio LC, et al. Testosterone regulates bone response to inflammation. Horm Metab Res. (2014) 46:193–200. doi: 10.1055/s-0034-1367031
107. Freeman BM, Mountain DJ, Brock TC, Chapman JR, Kirkpatrick SS, Freeman MB, et al. Low testosterone elevates interleukin family cytokines in a rodent model: a possible mechanism for the potentiation of vascular disease in androgen-deficient males. J Surg Res. (2014) 190:319–27. doi: 10.1016/j.jss.2014.03.017
108. Zhang X, Wang LY, Jiang TY, Zhang HP, Dou Y, Zhao JH, et al. Effects of testosterone and 17-beta-estradiol on TNF-alpha-induced E-selectin and VCAM-1 expression in endothelial cells. Analysis of the underlying receptor pathways. Life Sci. (2002) 71:15–29. doi: 10.1016/S0024-3205(02)01567-9
109. Zhang X, Wang L, Dou Y, Zhao J, Jiang T, Qiao Z, et al. Testosterone and estradiol modulate TNF-alpha-induced expression of adhesion molecules in endothelial cells. Methods Find Exp Clin Pharmacol. (2002) 24:125–30. doi: 10.1358/mf.2002.24.3.802295
110. Hatakeyama H, Nishizawa M, Nakagawa A, Nakano S, Kigoshi T, Uchida K. Testosterone inhibits tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in human aortic endothelial cells. FEBS Lett. (2002) 530:129–32. doi: 10.1016/S0014-5793(02)03440-3
111. Glueck CJ, Friedman J, Hafeez A, Hassan A, Wang P Testosterone therapy thrombophilia and hospitalization for deep venous thrombosis-pulmonary embolus an exploratory hypothesis-generating study. Med Hypotheses. (2015) 84:341–3. doi: 10.1016/j.mehy.2015.01.020
112. Jones RD, Nettleship JE, Kapoor D, Jones HT, Channer KS Testosterone and atherosclerosis in aging men: purported association and clinical implications. Am J Cardiovasc Drugs. (2005) 5:141–54. doi: 10.2165/00129784-200505030-00001
113. Li R, Meng X, Zhang Y, Wang T, Yang J, Niu Y, et al. Testosterone improves erectile function through inhibition of reactive oxygen species generation in castrated rats. PeerJ. (2016) 4:e2000. doi: 10.7717/peerj.2000
114. Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA Estrogens in male physiology. Physiol Rev. (2017) 97:995–1043. doi: 10.1152/physrev.00018.2016
115. Patel S, Homaei A, Raju AB, Meher BR. Estrogen: the necessary evil for human health, and ways to tame it. Biomed Pharmacother. (2018) 102:403–11. doi: 10.1016/j.biopha.2018.03.078
116. Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol. (2017) 37:746–56. doi: 10.1161/ATVBAHA.116.307301
117. Ventura-Clapier R, Dworatzek E, Seeland U, Kararigas G, Arnal JF, Brunelleschi S, et al. Sex in basic research: concepts in the cardiovascular field. Cardiovasc Res. (2017) 113:711–24. doi: 10.1093/cvr/cvx066
118. Maan AA, Eales J, Akbarov A, Rowland J, Xu X, Jobling MA, et al. The Y chromosome: a blueprint for men's health? Eur J Hum Genet. (2017) 25:1181–8. doi: 10.1038/ejhg.2017.128
119. Alsiraj Y, Thatcher SE, Blalock E, Fleenor B, Daugherty A, Cassis LA. Sex chromosome complement defines diffuse versus focal angiotensin II-induced aortic pathology. Arterioscler Thromb Vasc Biol. (2018) 38:143–53. doi: 10.1161/ATVBAHA.117.310035
120. Molina E, Clarence EM, Ahmady F, Chew GS, Charchar FJ. Coronary artery disease: why we should consider the Y chromosome. Heart Lung Circ. (2016) 25:791–801. doi: 10.1016/j.hlc.2015.12.100
121. Bacakova L, Baudysova M. Vascular smooth muscle cells of male and female rats in culture, migration, proliferation and chromosome number. Physiol Bohemoslov. (1990) 39:449–58.
122. Bacakova L, Pellicciari C, Bottone MG, Lisa V, Mares V. A sex-related difference in the hypertrophic versus hyperplastic response of vascular smooth muscle cells to repeated passaging in culture. Histol Histopathol. (2001) 16:675–84. doi: 10.14670/HH-16.675
123. McCoy CM, Nicholas DQ, Masters KS. Sex-related differences in gene expression by porcine aortic valvular interstitial cells. PLoS ONE. (2012) 7:e39980. doi: 10.1371/journal.pone.0039980
124. Quintana-Murci L, Fellous M. The human Y chromosome: the biological role of a “functional wasteland.” J Biomed Biotechnol. (2001) 1:18–24. doi: 10.1155/S1110724301000080
125. Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from ATVB council. Arterioscler Thromb Vasc Biol. (2018) 38:292–303. doi: 10.1161/ATVBAHA.117.309524
126. Ouyang P, Wenger NK, Taylor D, Rich-Edwards JW, Steiner M, Shaw LJ, et al. Strategies and methods to study female-specific cardiovascular health and disease: a guide for clinical scientists. Biol Sex Differ. (2016) 7:19. doi: 10.1186/s13293-016-0073-y
127. Raz L, Miller VM. Considerations of sex and gender differences in preclinical and clinical trials. Handb Exp Pharmacol. (2012) 127–47. doi: 10.1007/978-3-642-30726-3_7
128. Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. (2001) 103:1522–8. doi: 10.1161/01.CIR.103.11.1522
129. Bertazzo S, Gentleman E, Cloyd KL, Chester AH, Yacoub MH, Stevens MM. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat Mater. (2013) 12:576–83. doi: 10.1038/nmat3627
130. Shioi A, Ikari Y Plaque calcification during atherosclerosis progression and regression. J Atheroscler Thromb. (2018) 25:294–303. doi: 10.5551/jat.RV17020
131. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. (2014) 34:715–23. doi: 10.1161/ATVBAHA.113.302070
132. Fang K, Chen Z, Liu M, Peng J, Wu P. Apoptosis and calcification of vascular endothelial cell under hyperhomocysteinemia. Med Oncol. (2015) 32:403. doi: 10.1007/s12032-014-0403-z
133. Jeremy RW. Calcific aortic valve disease: insights into the genetics of vascular ageing. Circ Cardiovasc Genet. (2017) 10:e002012. doi: 10.1161/CIRCGENETICS.117.002012
134. Xia ZY, Hu Y, Xie PL, Tang SY, Luo XH, Liao EY, et al. Runx2/miR-3960/miR-2861 positive feedback loop is responsible for osteogenic transdifferentiation of vascular smooth muscle cells. Biomed Res Int. (2015) 2015:624037. doi: 10.1155/2015/624037
135. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
136. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. (2016) 15:973–7. doi: 10.1111/acel.12458
137. Bielak-Zmijewska A, Wnuk M, Przybylska D, Grabowska W, Lewinska A, Alster O, et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology. (2014) 15:47–64. doi: 10.1007/s10522-013-9477-9
138. Kurz DJ, Kloeckener-Gruissem B, Akhmedov A, Eberli FR, Buhler I, Berger W, et al. Degenerative aortic valve stenosis, but not coronary disease, is associated with shorter telomere length in the elderly. Arterioscler Thromb Vasc Biol. (2006) 26:e114–7. doi: 10.1161/01.ATV.0000222961.24912.69
Keywords: age-related cardiovascular parameters, aging, aortic valve stenosis, cardiovascular calcification, epidemiology, estrogen, hemodynamics, testosterone
Citation: Zhang B, Miller VM and Miller JD (2019) Influences of Sex and Estrogen in Arterial and Valvular Calcification. Front. Endocrinol. 10:622. doi: 10.3389/fendo.2019.00622
Received: 29 May 2019; Accepted: 27 August 2019;
Published: 20 September 2019.
Edited by:
Dawn A. Lowe, University of Minnesota Twin Cities, United StatesReviewed by:
Kristyn Simcha Masters, University of Wisconsin-Madison, United StatesElena Aikawa, Brigham and Women's Hospital and Harvard Medical School, United States
Copyright © 2019 Zhang, Miller and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordan D. Miller, bWlsbGVyLmpvcmRhbkBtYXlvLmVkdQ==