 Pavlos Fanis1,2
Pavlos Fanis1,2 Nicos Skordis1,3
Nicos Skordis1,3 Meropi Toumba1,4
Meropi Toumba1,4 Nikoletta Papaioannou1,2
Nikoletta Papaioannou1,2 Anestis Makris1
Anestis Makris1 Andreas Kyriakou5
Andreas Kyriakou5 Vassos Neocleous1,2
Vassos Neocleous1,2 Leonidas A. Phylactou1,2*
Leonidas A. Phylactou1,2*- 1Department of Molecular Genetics, Function and Therapy, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
- 2Cyprus School of Molecular Medicine, Nicosia, Cyprus
- 3Pediatric Endocrine Clinic, Paedi Center for Specialized Pediatrics, Nicosia, Cyprus
- 4Department of Pediatrics, Iasis Hospital, Paphos, Cyprus
- 5Developmental Endocrinology Research Group, School of Medicine, University of Glasgow, Glasgow, United Kingdom
Background: Central Precocious Puberty (CPP) is clinically defined by the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys. To date, mutations in the coding region of KISS1, KISS1R, PROKR2, DLK1, and MKRN3 genes have been reported as causative for CPP. This study investigated the presence of causative mutations in both the promoter and the 5′-UTR regions of the MKRN3 gene.
Methods: Sanger DNA sequencing was used for screening the proximal promoter and 5′-UTR region of the MKRN3 gene in a group of 73 index girls with CPP. Mutations identified were cloned in luciferase reporter gene vectors and transiently transfected in GN11 cells in order to check for changes in the activity of the MKRN3 promoter. GN11 cells were previously checked for Mkrn3 expression using lentivirus mediated knock-down. In silico analysis was implemented for the detection of changes in the mRNA secondary structure of the mutated MKRN3 5′-UTR.
Results: Three novel heterozygous mutations (−166, −865, −886 nt upstream to the transcription start site) located in the proximal promoter region of the MKRN3 gene were identified in six non-related girls with CPP. Four of these girls shared the −865 mutation, one the −166, and another one the −886. A 5′-UTR (+13 nt downstream to the transcription start site) novel mutation was also identified in a girl with similar clinical phenotype. Gene reporter assay evaluated the identified promoter mutations and demonstrated a significant reduction of MKRN3 promoter activity in transfected GN11 cells. In silico analysis for the mutated 5′-UTR predicted a significant change of the mRNA secondary structure. The minimum free energy (MFE) of the mutated 5′-UTR was higher when compared to the corresponding wild-type indicating less stable RNA secondary structure.
Conclusion: Our findings demonstrated novel genetic alterations in the promoter and 5′-UTR regulatory regions of the MKRN3 gene. These changes add to another region to check for the etiology of CPP.
Introduction
Central precocious puberty (CPP) is characterized by the premature activation of the hypothalamic-pituitary-gonadal axis due to the early activation of pulsatile Gonadotropin Releasing hormone (GnRH) secretion. Central precocious puberty is clinically defined by the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys and is associated with a range of clinical and biological implications (1–3). The complex procedure of pubertal timing and progress are influenced by interactions of nutritional, environmental, socioeconomic, and genetic factors (4).
Strong evidence of the association of genetic factors on pubertal timing has been shown by population studies (5, 6). Using Genome Wide Association studies (GWAs) several genes have been associated with an increased growth and development, the regulation of the age at menarche, influence of energy homeostasis, and hormone regulation (7). The role of genetic determinants has been also illustrated by the similar age at menarche in mothers and daughters and among members of an ethnic group (8). Analysis among CPP patients has shown that 27.5% of cases are familial, thus suggesting an autosomal mode of inheritance (9). Although, the evidence suggests that age at the onset of puberty development is determined by genetic factors, the genetic etiology of CPP is largely unknown.
Several studies have used a candidate gene approach in an effort to identify genes associated with pubertal disorders. Currently, there is a steady increase in the number of genes associated with the development of hypogonadotropic hypogonadism and the Kallmann syndrome (10, 11). On the contrary only limited and rare molecular defects have been identified in individuals with CPP (12).
The genes that were discovered to be related with CPP and early GnRH secretion were the KISS1, its G protein-coupled receptor, GPR54 (KISS1R), PROKR2, DLK1, and MKRN3 (13–16). More precisely, the autosomal dominant GPR54 mutation (p.Arg386Pro) was the first identified mutant that was proved to lead to prolonged activation of GnRH secretion through its ligand kisseptin (KISS1) (13). Another study that followed identified the p.Pro74Ser in the KISS1 gene which is a defect that leads to the degradation resistance of kisspeptin and to the elevated availability of the protein (14). Therefore, these two gain-of-function KISS1/KISS1R mutations were the only causative mutations identified in CPP patients and that resulted to upregulation of the KISS1/KISS1R system leading to GnRH secretion and HPG activation (17). Similarly, a gain-of-function heterozygous mutation in the PROKR2 (p.Cys242fsTer305) gene led to CPP by increasing the activity of the coexisting wild-type proteins (18). DLK1 was the most recent gene in which genetic alterations were identified as a causal factor for CPP and in a recent report has also been associated with the age at menarche (8, 19). DLK1 is maternally imprinted and its mutated allele follows the paternal mode of inheritance, such a case was a recent report with a large deletion of exon 1 in the DLK1 gene (16). A second report followed and identified in female CPP patients three different frameshift mutations in the DLK1, also consistent with the maternal imprinting of the gene (20).
Nowadays, the most common genetic causes of CPP are the reported loss-of-function mutations in the MKRN3 gene. Since the first ground breaking discovery of MKRN3 gene, one of the major genes known to be causing CPP (15), various studies by other research groups identified a variety of other MKNR3 loss-of-function mutations (21–27). MKRN3 gene is located in the Prader-Willi syndrome (PWS)-related region (15q11-q13) on chromosome 15. The maternal allele of the gene is imprinted therefore MKRN3 gene is expressed only from the paternal allele. All affected patients reported with familial CPP inherited the MKRN3 mutations from their fathers (28).
MKRN3 contains five zing finger domains: three C3H1 motifs, one C3HC4 RING motif, and one MKRN-specific Cys-His domain (29). C3H1 zinc-finger motifs are responsible for RNA binding while the RING motif is detected in E3 ubiquitin ligases and hence, it is assumed that has an ubiquitin-ligase activity (30). The MKRN3 protein is highly expressed in the developing brain including arcuate nucleus (15). It is believed that, MKRN3 is involved in protein degradation, therefore it might have an inhibitory effect in the pulsatile GnRH secretion (30). The exact mechanism that accomplish this effect or by which mechanism MKRN3 deficiency results in early reactivation of GnRH secretion is still unknown.
Although most of the reported studies describe loss-of-function mutations in the coding region of MKRN3, defects in the regulatory regions of the gene were described in two recent studies (19, 31). In the first study a four nucleotide deletion (c.-150_-147delTCAG) in the proximal promoter region of the MKRN3 gene was found to be responsible for causing CPP (19). The second study describe a single nucleotide substitution at position 19 (MKRN3:g.+19C>T) from the transcription start site (TSS) in the 5′-UTR region of the MKRN3 gene that is associated with CPP (31).
In this study, we report four novel heterozygous mutations located in the proximal promoter and 5′-UTR regions of the MKRN3 gene in seven non-related girls with CPP. Functional analyses of the identified novel promoter/5′-UTR mutations indicated a robust correlation to the development of the disease.
Materials and Methods
Patients Under Study
The promoter/5′-UTR of the MKRN3 gene was screened for variations in 73 index CPP girls referred to the Department of Molecular Genetics, Function and Therapy at the Cyprus Institute of Neurology and Genetics. Central precocious puberty in these girls was clinically evaluated by the development of secondary sexual characteristics before the age of 8 years, the basal and LHRH stimulated LH levels, advanced bone age, ovarian function on the pelvic ultrasound and normal central nervous system MRI. Mutations in the coding sequence of the MKRN3, KISS1, KISS1R, and DLK1 genes were previously excluded. Written informed consent was obtained from parents of all patients under the age of 16 that participated in the study. The project was approved by the Cyprus National Ethics Committee and all methods were performed in accordance with the relevant guidelines and regulations.
DNA Preparation
Genomic DNA was isolated from peripheral blood using the Gentra Puregene Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. The DNA concentration and purity were measured using the Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
PCR Amplification and Sequencing
Variation analysis of the MKRN3 proximal promoter and 5′-UTR region (−984 to +99 nucleotides (nt) from the TSS) was performed by PCR amplification followed by conventional Sanger DNA sequence analysis. The PCR reaction mixture was performed at a final volume of 20 μl and contained 2 μl AmpliTaq Gold PCR buffer I (10x), 2 μl dNTPs (2 mM), 0.5 μl of each primer (5 μM), 0.2 μl AmpliTaq Gold (0.5 U/μl) and 100 ng of genomic DNA. Amplification was performed with an initial denaturing temperature at 95°C for 10 min, followed by 35 cycles of denaturation (95°C, 30 s), annealing (58°C, 60 s), extension (72°C, 90 s), with a final extension at 72°C for 7 min. The following primers were used for PCR amplification; MKRN3_prom_F: 5′-TAACAGAATTCAGTATGCAGTCA−3′, MKRN3_prom_R: 5′-CGTGGGCTTCTGAGGGA−3′. The PCR products were cleaned-up with ExoSAP-IT reagent (Applied Biosystems, Foster City, CA) and sequenced using the same primers as for the PCR and the following internal primers; MKRN3_prom_int_F1: 5′-AGGGGACAGTGTCTTATTAG−3′, MKRN3_prom_int_F2: 5′-GAAGAGATTAAAGTAAAACC−3′ on a 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA).
Vector Construction and Lentivirus Mediated Knock-Down
The wild-type and mutant proximal promoter/5′-UTR MKRN3 sequences (−984_+99 nt relative to TSS) were amplified from human genomic DNA by PCR using the Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA), cloned to pMIR_REPORT firefly luciferase reporter gene vector using KpnI and BamHI restriction sites (excision and replacement of the existing CMV promoter) and verified by sequencing.
For mouse Mkrn3, a clone from the TRC Mission (short hairpin) shRNA library (32) (Sigma Aldrich, St. Louis, MO, USA) was used for lentivirus mediated knockdown experiments in GN11 cells, including a scramble shRNA control clone (SHC002). GN11 cells were transduced with lentivirus and harvested 6 days later. Lentivirus was produced by transient transfection of 293T cells as described before (33, 34). The following clone was used from the TRC shRNA library: TRCN0000418150 (shMkrn3).
Cell Culture and Transient Transfection
For the transfection studies we used the GnRH mouse neuronal GN11 cell line. GN11 cell line express very low levels of Gnrh1 mRNA and serve as cell culture model to study the rare population of hypothalamic GnRH neurons (35, 36). The GN11 cell line was kindly provided by Prof. Davide Lovisolo and Susanna Antoniotti (Department of Life Sciences and Systems Biology, University of Torino). GN11 cells were grown in Dulbecco's Modified Eagle Medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and 1% Penicillin/Streptomycin (Invitrogen, Carlsbad, CA, USA) in a humid environment at 37°C with 5% CO2. Twenty four hours before transfection cells were plated in a 6-well plate at a density of 4 × 105. The next day GN11 cells were transiently transfected with 500 ng/well of the pMIR_REPORT firefly luciferase vector together with 50 ng/well of the pGL4.74 Renilla luciferase vector (internal control) using the Lipofectamine 3000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA). Twenty four hours later, total RNA from the GN11 transfected cells was isolated, cDNA was prepared and qPCR was performed (see RNA extraction, Reverse Transcription (RT) and qPCR section).
RNA Extraction, Reverse Transcription (RT), and qPCR
Total RNA was isolated from GN11 cells with Trizol reagent (Invitrogen, Carlsbad, CA, USA). Before cDNA preparation the RNA was treated with DNaseI (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using the One Taq RT-PCR Kit (New England Biolabs, Ipswich, MA, USA) and amplified with the ABI 7900HT Fast Real-time PCR System (Applied Biosystems, New Jersey, USA). Each sample was amplified in triplicate. KAPA SYBR® FAST qPCR Master Mix (2X) Kit (Kapa Biosystems, Wilmington, MA, USA) was used to quantify the amplified products.
Relative expression analysis was performed as described before (37). Cycle threshold levels were calculated for each gene and normalized to values obtained for the endogenous house-keeping gene. For Mkrn3 knock-down experiments the 18S rRNA gene was used as a housekeeping gene and for the gene reporter assay the Renilla luciferase gene was used. The following primers were used for the Mkrn3 knock-down experiments; 18S_RT_F: 5′-TGGTTGATCCTGCCAGTAG-3′, 18S_RT_R: 5′-CGACCAAAGGAACCATAACT-3′, Mkrn3_RT_F: 5′-TCCTGGACAGCCTTACCG-3′, Mkrn3_RT_R: 5′-TATGCACACCTGTCCCCAC-3′. For gene reporter assay the Fluc_RT_F: 5′-CTCACTGAGACTACATCAGC−3′ and Fluc_RT_R: 5′-TCCAGATCCACAACCTTCGC−3′ were used for the detection of firefly luciferase and the Rluc_RT_F: 5′-GAAGAGGGCGAGAAAATGGT−3′ and Rluc_RT_R: 5′-CCCTTCTCCTTGAATGGCTC−3′ were used for the detection of Renilla luciferase by qPCR.
Western Blot Analysis
After protein transfer, the nitrocellulose membranes were blocked in 1% bovine serum albumin (BSA), incubated with the appropriate primary antibodies and developed using enhanced chemiluminescence. The membranes were probed with the following primary antibodies: Mkrn3 (ab177203), obtained from Abcam (Abcam, Cambridge, MA) and β-Actin (monoclonal AC-15, A1978) from Sigma (Sigma, St. Louis, MO, USA). HRP-conjugated IgG Goat Anti-mouse and Goat Anti-rabbit (Jackson ImmunoResearch, PA USA) secondary antibodies were used. The anti-MKRN3 antibody was used in 1:1,000 dilution and the anti-ACTIN antibody was used in 1:10,000 dilution in a solution of 1% non-fat dried milk in TBS/Tween. Secondary antibodies were used in a 1:20,000 dilution in non-fat dried milk in TBS/Tween. The levels of Mkrn3 protein were normalized with the corresponding β-Actin loading control within the same immunoblot. Visualization of the immunoblots and acquisition of the images was carried out the UVP BioSpectrum Imaging System (UVP, LLC, Upland, CA, USA). Dynamic Total Time Exposure settings were used, with binning set at 12.1 MP, 149% and 1 × 1 interpolation. Exposure times varied depending on the primary antibody used.
RNA Secondary Structure Analysis
The 99 nt 5′-UTR mRNA of MKRN3, was submitted to the RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) with default parameters to predict the potential secondary structure. Secondary structures were predicted for the wild-type and the MKRN3:g.+13C>T mutant mRNA. Potential minimum free energy (MFE) structures, centroid structures, and positional entropies were obtained (38).
Statistical Analysis
Statistical analysis was performed with the GraphPad Prism 8 software using two tailed unpaired t-test with confidence level of 95%. P < 0.05 were considered significant.
Results
Identification of Novel MKRN3 Promoter/5′-UTR Mutations
Sequencing analysis of the promoter and 5′-UTR regions of the MKRN3 gene in the cohort of 73 index girls with CPP revealed three novel heterozygous mutations (−166, −865, −886 nt upstream to the TSS) located in the proximal promoter region and one novel heterozygous mutation located in the 5′-UTR (+13 nt downstream to the TSS) region (Figure 1). Three of these girls shared the promoter MKRN3 mutations: g.-865G>A, g.-166G>A and g.-886C>T. A fourth 7.6 years old girl at the time of diagnosis was identified with the 5′-UTR MKRN3:g.+13C>T mutation. All identified mutations were traced in dbSNP with the following reference SNP ID numbers: rs184950120 (MKRN3:g.+13C>T), rs188505875 (MKRN3:g.-166G>A), rs139233681 (MKRN3:g.-865G>A), and rs74005577 (MKRN3:g.-886C>T) but they have never been linked with CPP. The allele frequencies of the identified mutations in different population studies are described in Table 1. In addition the four mutations were absent in samples from 50 individuals that were randomly selected from the general Cypriot population. Investigation by DNA sequencing of both parents for two of the CPP girls bearing the MKRN3:g.-865G>A mutation and one bearing the MKRN3:g.+13C>T revealed the presence of the novel mutations only in the fathers. DNA from the parents of the remaining three CPP girls was not available. Since the MKRN3 gene is maternally imprinted with only the paternal allele expressed, these findings were in agreement with the paternal mode of inheritance for the MKRN3 mutations (15).
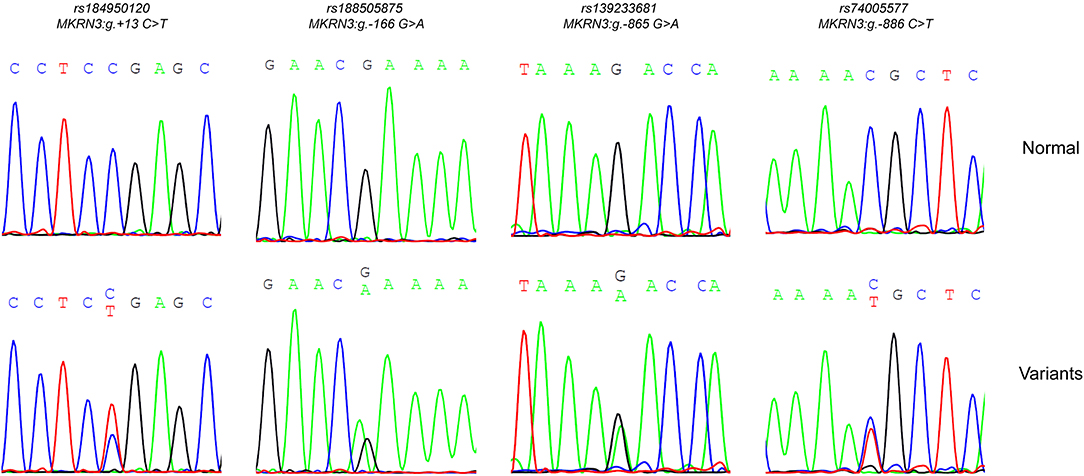
Figure 1. DNA sequencing analysis. Part of the sequencing electropherograms of the MKRN3 proximal promoter showing the novel heterozygous mutations identified; rs184950120 (MKRN3:g.+13C>T), rs188505875 (MKRN3:g.-166G>A), rs139233681 (MKRN3:g.-865G>A), and rs74005577 (MKRN3:g.-886C>T). For each mutation the corresponding normal sequencing electropherograms is showed.
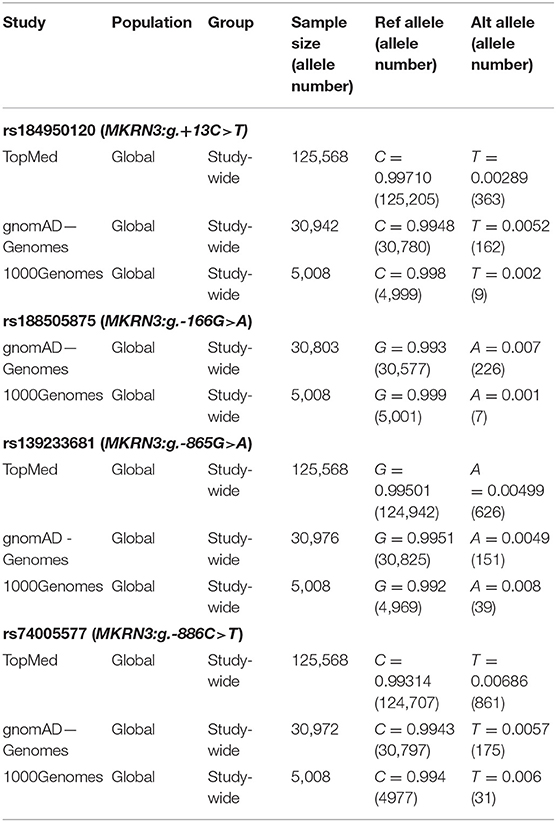
Table 1. Allele frequencies of the identified mutations in different population studies.
Evaluation of the Identified Novel Mutations in the Promoter/5′-UTR Region of the MKRN3 Gene
Luciferase reporter assay was employed to investigate the hypothesis that the identified novel MKRN3 mutations were the functional cause of the CPP. The MKRN3 promoter/5′-UTR containing either the wild-type or the different mutations was isolated and cloned into a luciferase gene reporter vector. Promoter/5′-UTR constructs containing the wild-type, the MKRN3:g.+13C>T, the MKRN3:g.-166G>A, the MKRN3:g.-865G>A, or the MKRN3:g.-886C>T mutations were designed and tested for activity in GN11 cells. GN11 cells express low levels of Gnrh1 mRNA and maintain many of the phenotypic characteristics of immature GnRH neurons in vitro therefore, serve as cell culture model to study the rare population of hypothalamic GnRH neurons (35, 39).
GN11 cells were first tested if they are a suitable model to perform the gene reporter assay. Thus, protein extracts were isolated, loaded on an SDS-PAGE gel and tested for endogenous Mkrn3 expression by staining with an anti-Mkrn3 antibody. Since there was lack of information for the specificity of the antibody we perform knock-down experiment of the endogenous Mkrn3 using shRNA. We used a shRNA against Mkrn3 (shMkrn3) that reduced the mRNA and protein levels of Mkrn3 indicating the presence of Mkrn3 expression in GN11 cells (Figures 2A,B).
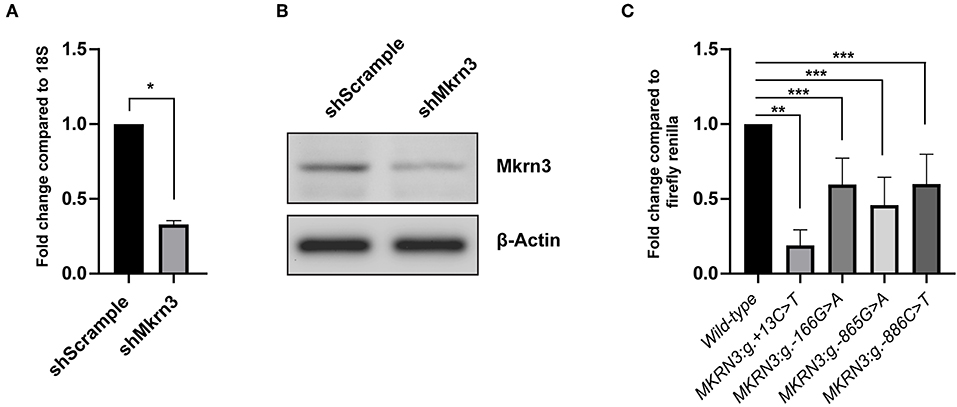
Figure 2. (A,B) shRNA knockdown efficiency on (A) RNA and (B) protein Mkrn3 levels. GnRH expressing GN11 cells were treated with the indicated shRNA. (A) Total mRNA analyzed by RT-qPCR. Relative expression of Mkrn3 was analyzed with respect to the 18S gene. (B) Whole cell lysates were analyzed by western blotting with Mkrn3 antibody. The β-Actin stained membrane serves as loading control. The shMkrn3 reduces the mRNA and protein levels of Mkrn3. As a negative control a shRNA containing a scrambled sequence was used (shScrample). (C) The novel MKRN3 promoter/5′-UTR mutations reduce the promoter activity in GN11 cells. The MKRN3 promoter reporter gene constructs (spanning nucleotides −984_+99 relative to TSS) containing the MKRN3:g.+13C>T, MKRN3:g.-166G>A, MKRN3:g.-865G>A, and MKRN3:g.-886C>T mutations were transiently transfected in GN11 cells. Luciferase activities were calculated relative to the wild-type MKRN3 promoter reporter construct. Results are the average of three independent experiments with each sample assayed in triplicate. *P < 0.0001; **P < 0.001; ***P < 0.05.
The novel promoter MKRN3:g.-166G>A, MKRN3:g.-865G>A, and MKRN3:g.-886C>T mutations were found to significantly decrease the transcription of the luciferase gene in GN11 cells by ~0.4 fold relative to the wild-type (Figure 2C). The strongest effect with ~0.8 fold decrease in the transcription of the luciferase gene was observed with the novel MKRN3:g.+13C>T mutation which is located in the 5′-UTR region of the MKRN3 gene (Figure 2C, second column bar).
Complete clinical characteristics were available for six out of seven of these cases (Table 2). The median age at puberty onset of these patients was 6.55 years and the median age at referral was 8.05 years. These patients displayed the typical characteristics of precocious puberty (Table 2).
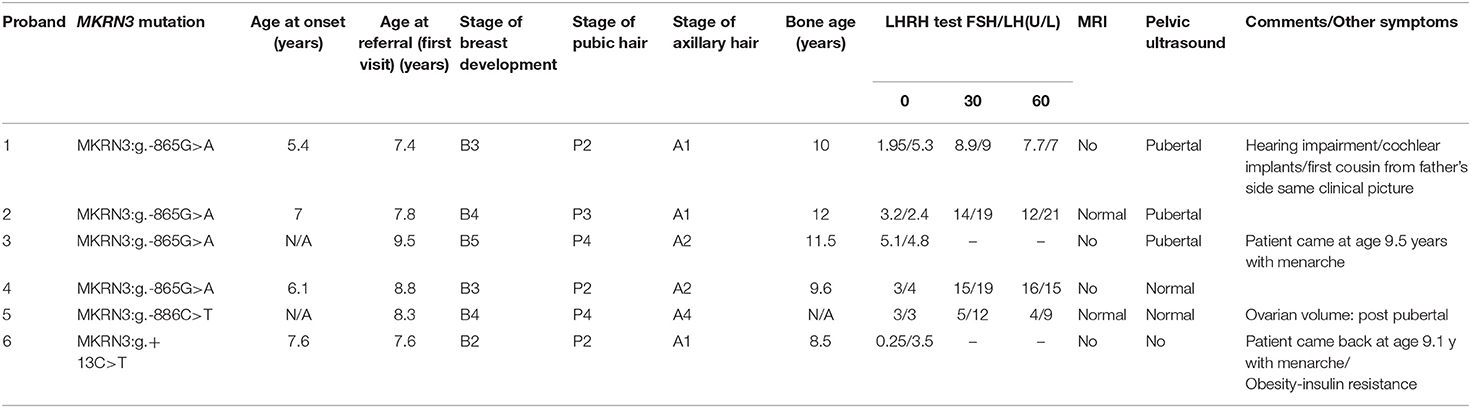
Table 2. Clinical and laboratory characteristics for six girls with MKRN3 mutations.
The affected girl bearing the novel MKRN3:g.+13C>T mutation was referred due to premature sexual maturation at the age of 7.5 years. She was at Tanner stage 2 of puberty with elevated gonadotrophins, bone age advancement for 2 years compared to chronological age while a pelvic ultrasound showed ovarian function. She was treated with GnRH analogs to suppress her pubertal progression with the aim to increase her final height potential and to alleviate any psychological issues. Additionally she was obese with body mass index (BMI) more than +2SDS for her age and gender and of course her height was taller than her midparental target height. During her 2 years of follow up with CPP treatment her weight increased further and she showed signs of insulin resistance for which she was commenced on treatment with metformin.
These results demonstrate that the MKRN3 promoter and especially the 5′-UTR novel mutations have a negative effect on gene regulation.
In silico Analyses of the Novel MKRN3:g.+13C>T 5′-UTR Mutation
Since the luciferase reporter assay of the novel 5′-UTR mutation indicated a strong effect in the reduction of the transcriptional activity of the gene we further investigated this by performing an in silico mRNA secondary MFE structure prediction. Secondary MFE structure of the wild-type MKRN3 5′-UTR mRNA was compared to the secondary MFE structure of the MKRN3:g.+13C>T 5′-UTR mutant mRNA. Figures 3A,B shows the predicted MFE and centroid MFE secondary structures of the MKRN3 5′-UTR mRNA. The effect of the mutated nucleotide is visible as it changes the MFE secondary structures of the MKRN3:g.+13C>T mutant compared to the wild-type 5′-UTR mRNA. Moreover, the centroid secondary structure MFE values of the predicted mRNA secondary structures were also found to be higher in the MKRN3:g.+13C>T mutant when compared to the wild-type form (Table 3). This finding could be attributed to a probable reduced stability of the secondary mRNA structure.
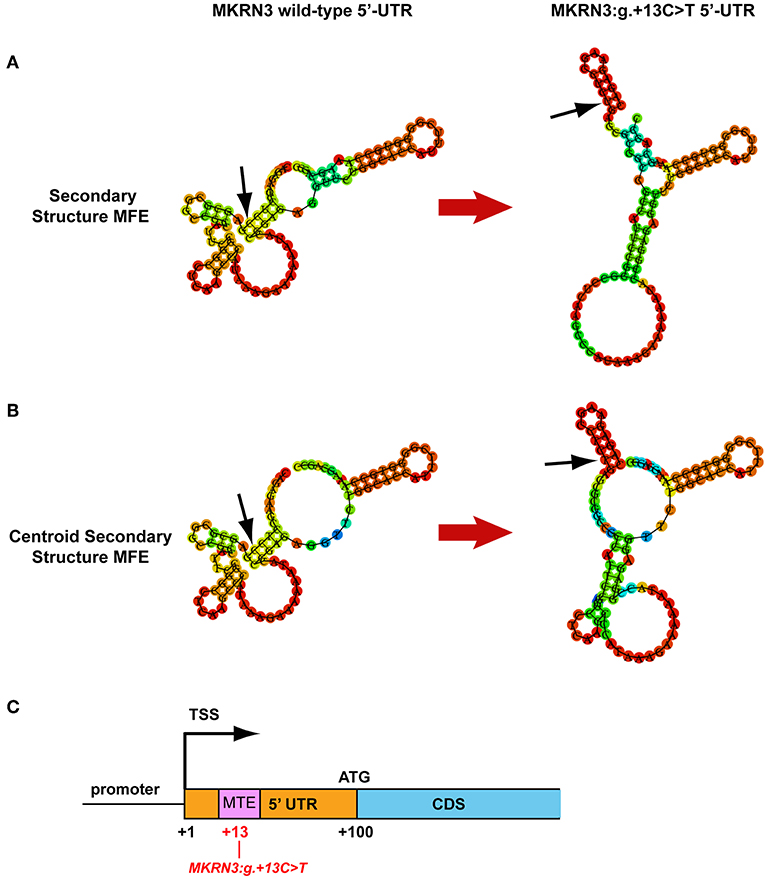
Figure 3. MKRN3 5′-UTR in silico predictions. (A,B) Predicted MKRN3 5′-UTR mRNA secondary structures showing the MFE and centroid MFE secondary structure, respectively, for the wild- type and MKRN3:g.+13C>T mutant. (C) Position of the Motif Ten Element (MTE) in relation with the MKRN3:g.+13C>T mutation. Black arrows indicate the position of the MKRN3:g.+13 nucleotide.
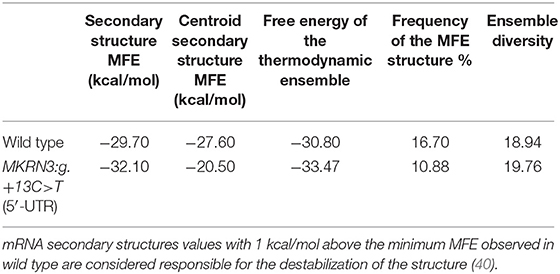
Table 3. Minimum free energy (MFE) and centroid secondary structure MFE values.
Furthermore, using the MatInspector tool of Genomatix software suit we performed in silico analysis of the 5′-UTR genomic region for alterations in binding sites downstream of the TSS. Comparison between the wild-type and mutated sequence predicted that the novel 5′UTR MKRN3:g.+13C>T mutation would lead to the loss of a putative Motif Ten Element (MTE) binding site that can promote transcription by RNA polymerase II (41) (Figure 3C).
Discussion
Mutations in the intronless MKRN3 gene can cause CPP, a gonadotropin-dependent precocious puberty due to the early activation of the hypothalamic-pituitary-gonadal axis (2). To date, ~40 different mutations in the MKRN3 gene have been reported and associated with CPP (21–25, 42). MKRN3 gene is an imprinted gene expressed only from the paternal allele; therefore affected patients with familial CPP inherited the MKRN3 mutations from their fathers. Most of the published studies describe causative mutations in the coding region of MKRN3 gene with only two recent studies report defects in the regulatory regions of the gene (19, 31). In the first study a CPP causative small deletion (c.-150_-147delTCAG) in the promoter region of the MKRN3 gene was identified (19). The second study described a CPP causative mutation (MKRN3:g.+19C>T) located in the 5′-UTR region of the MKRN3 gene (31).
In this study we identified three novel heterozygous mutations located in the proximal promoter and one in the 5′-UTR region of the MKRN3 gene in a total number of seven non-related girls with CPP. Four of these girls shared the MKRN3:g.-865G>A mutation, one the MKRN3:g.-166G>A and another one the MKRN3:g.-886C>T mutation, all located in the proximal promoter of the gene. Interestingly, a 7.6 years old girl with CPP at the time of diagnosis was identified with the novel MKRN3:g.+13C>T mutation in the 5′-UTR region. A large study involving ~370,000 women also identified the MKRN3:g.+13C>T 5′-UTR mutation as a putative marker of puberty timing when paternally inherited (43).
In silico analysis using the MatInspector tool for the identification of possible changes in transcription factor (TF) binding sites in the proximal promoter of wild-type and mutated MKRN3 gene predicted various modifications (Figure 4). Specifically, when the MKRN3:g.-166G>A is present, a putative binding site for the SOX4 is created (Figure 4B). SOX4 is a transcription factor involved in the regulation of embryonic development and in the determination of the cell fate. Additionally, this same transcription factor is mainly expressed in neural cells that have already been dedicated to neuronal differentiation (44, 45) and is highly expressed in the majority of hypothalamic GnRH neurons in adult mice (46). We postulate that with the creation of its binding site, SOX4 is recruited to the promoter of MKRN3 and act as a repressor that would lead to decrease of the expression of MKRN3. The MKRN3:g.-865G>A mutation lead to loss of the PRDM14 transcription factor binding site (Figure 4C). MKRN3 promoter is predicted to have two binding sites for the PRDM14 at positions −240 and −865 relative to TSS (Figure 4). PRDM14 recruits PRMT5 to mediate histone arginine methylation and control neural stem cell differentiation (47). In this study we hypothesize that loss of one of the two PRDM14 binding sites in the MKRN3 promoter might lead to reduction in MKRN3 expression. Similarly, MKRN3:g.-886C>T mutation lead to loss of one out of two HMX2 transcription factor binding sites in the MKRN3 promoter (Figure 4D); therefore reduction in MKRN3 expression. HMX2 is a transcription factor which is expressed in the developing hypothalamus and especially in the growth hormone-releasing hormone (GHRH) neurons of the arcuate nucleus (48). Gene reporter assay studies in GN11 cells with vectors containing the different mutated promoters' revealed significant reduction in the MKRN3 promoter activity, suggesting that the mutations identified in the promoter region had a negative effect on MKRN3 transcription.
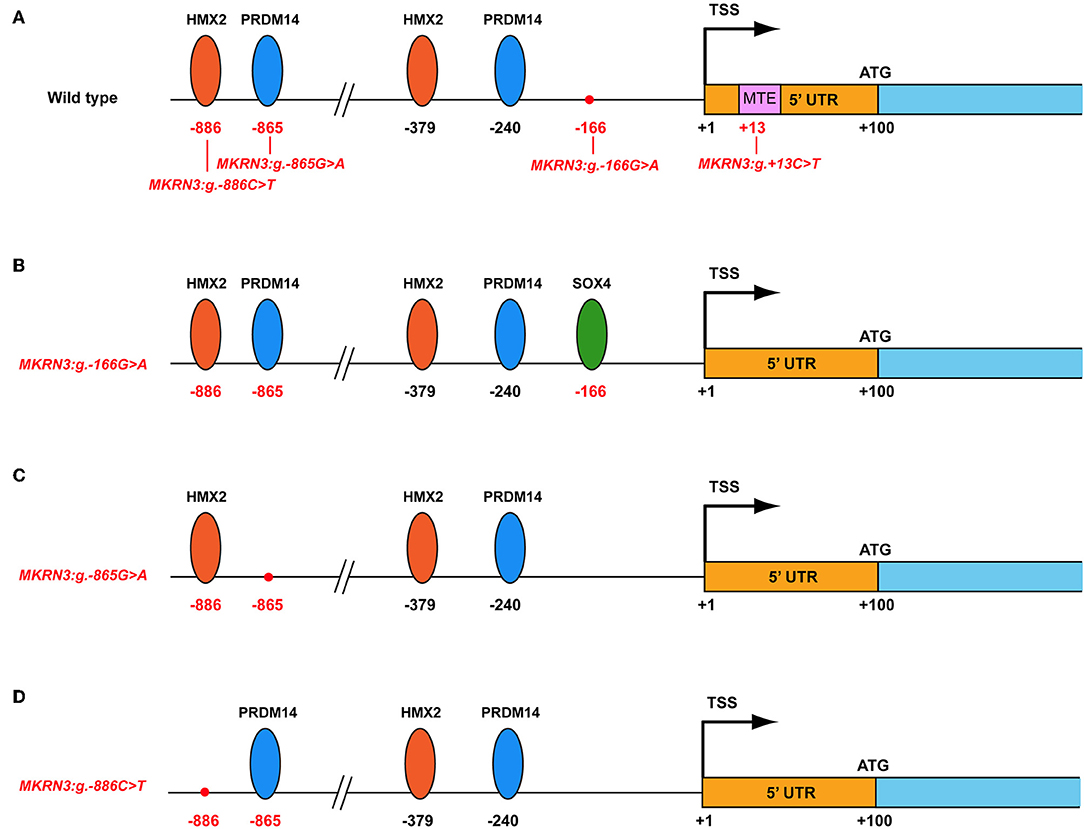
Figure 4. Schematic representation of the MKRN3 gene with various predicted transcription factors. The various transcription factors are indicated with oval shapes. The positions of the mutations identified are indicated in relation with TSS. (A) Wild-type MKRN3 promoter region. (B) MKRN3 promoter region with the novel MKRN3:g.-166G>A mutation. (C) MKRN3 promoter region with the novel MKRN3:g.-865G>A mutation and (D) MKRN3 promoter region with the novel MKRN3:g.-886C>T mutation. TSS: transcription start site; HMX2: H6 Family Homeobox 2; PRDM14: PR/SET Domain 14; SOX4: SRY-Box 4.
The novel MKRN3:g.+13C>T mutation located in the 5′-UTR had the strongest effect with the gene reporter assay. The fact that secondary structures often occur within 5′-UTR with followed impact in the regulation of translation (49), lead us to perform mRNA secondary MFE structure prediction. Comparison of the MKRN3 wild-type and the MKRN3:g.+13C>T mutated 5′-UTR mRNA secondary structures showed distinct differences. The higher centroid secondary structure MFE values of the mutant mRNA indicate less stable mRNA secondary structure. Moreover, using the MatInspector prediction tool the core promoter transcription factor binding site MTE located in 5′-UTR region is lost in the presence of the MKRN3:g.+13C>T mutation indicating a negative impact on MKRN3 transcription.
Although genetic analysis of both parents for only two of the CPP girls bearing the MKRN3:g.-865G>A and the MKRN3:g.+13C>T mutations confirmed the paternal mode of inheritance for the MKRN3 mutations, it is hoped in future genetic analyses will be performed also for the parents of the rest of CPP girls described in this study. Finally, as mutations in the promoter and 5′-UTR region of the MKRN3 gene are present and cause CPP, we cannot exclude the possibility of the presence of mutations in the promoter region of other CPP related genes for these (and other) CPP patients.
In summary, our current findings suggest that novel mutations in the MKRN3 proximal promoter and especially in the 5′-UTR region resulted in MKRN3 deficiency therefore contributing to the clinical manifestation of CPP phenotype.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the A.G. Leventis Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Davide Lovisolo and Susanna Antoniotti (Department of Life Sciences and Systems Biology, University of Torino) for providing the GN11 cells.
References
1. Plant TM. 60 years of neuroendocrinology: the hypothalamo-pituitary-gonadal axis. J Endocrinol. (2015) 226:T41–54. doi: 10.1530/JOE-15-0113
2. Carel JC, Leger J. Clinical practice. Precocious puberty. N Engl J Med. (2008) 358:2366–77. doi: 10.1056/NEJMcp0800459
3. Leka-Emiri S, Chrousos GP, Kanaka-Gantenbein C. The mystery of puberty initiation: genetics and epigenetics of idiopathic central precocious puberty (ICPP). J Endocrinol Invest. (2017) 40:789–802. doi: 10.1007/s40618-017-0627-9
4. Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. (2003) 24:668–93. doi: 10.1210/er.2002-0019
5. Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Mol Genet Metab. (2003) 80:1–10. doi: 10.1016/S1096-7192(03)00107-0
6. Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. (2001) 86:2364–8. doi: 10.1210/jcem.86.6.7603
7. Phillip M, Lazar L. Precocious puberty: growth and genetics. Horm Res. (2005) 64 (Suppl. 2):56–61. doi: 10.1159/000087760
8. Perry JR, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. (2014) 514:92–97. doi: 10.1038/nature13545
9. de Vries L, Kauschansky A, Shohat M, Phillip M. Familial central precocious puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab. (2004) 89:1794–800. doi: 10.1210/jc.2003-030361
10. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2009) 5:569–76. doi: 10.1038/nrendo.2009.177
11. Semple RK, Topaloglu AK. The recent genetics of hypogonadotrophic hypogonadism - novel insights and new questions. Clin Endocrinol. (2010) 72:427–35. doi: 10.1111/j.1365-2265.2009.03687.x
12. Shin YL. An update on the genetic causes of central precocious puberty. Ann Pediatr Endocrinol Metab. (2016) 21:66–9. doi: 10.6065/apem.2016.21.2.66
13. Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. (2008) 358:709–15. doi: 10.1056/NEJMoa073443
14. Silveira LG, Noel SD, Silveira-Neto AP, Abreu AP, Brito VN, Santos MG, et al. Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab. (2010) 95:2276–80. doi: 10.1210/jc.2009-2421
15. Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. (2013) 368:2467–75. doi: 10.1056/NEJMoa1302160
16. Dauber A, Cunha-Silva M, Macedo DB, Brito VN, Abreu AP, Roberts SA, et al. Paternally inherited DLK1 deletion associated with familial central precocious puberty. J Clin Endocrinol Metab. (2017) 102:1557–67. doi: 10.1210/jc.2016-3677
17. Bulcao Macedo D, Nahime Brito V, Latronico AC. New causes of central precocious puberty: the role of genetic factors. Neuroendocrinology. (2014) 100:1–8. doi: 10.1159/000366282
18. Fukami M, Suzuki E, Izumi Y, Torii T, Narumi S, Igarashi M, et al. Paradoxical gain-of-function mutant of the G-protein-coupled receptor PROKR2 promotes early puberty. J Cell Mol Med. (2017) 21:2623–6. doi: 10.1111/jcmm.13146
19. Macedo DB, Franca MM, Montenegro LR, Cunha-Silva M, Best DS, Abreu AP, et al. Central precocious puberty caused by a heterozygous deletion in the MKRN3 promoter region. Neuroendocrinology. (2018) 107:127–32. doi: 10.1159/000490059
20. Gomes LG, Cunha-Silva M, Crespo RP, Ramos CO, Montenegro LR, Canton A, et al. DLK1 is a novel link between reproduction and metabolism. J Clin Endocrinol Metab. (2018) 104:2112–20. doi: 10.1210/jc.2018-02010
21. Settas N, Dacou-Voutetakis C, Karantza M, Kanaka-Gantenbein C, Chrousos GP, Voutetakis A. Central precocious puberty in a girl and early puberty in her brother caused by a novel mutation in the MKRN3 gene. J Clin Endocrinol Metab. (2014) 99:E647–51. doi: 10.1210/jc.2013-4084
22. Macedo DB, Abreu AP, Reis AC, Montenegro LR, Dauber A, Beneduzzi D, et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J Clin Endocrinol Metab. (2014) 99:E1097–103. doi: 10.1210/jc.2013-3126
23. Schreiner F, Gohlke B, Hamm M, Korsch E, Woelfle J. MKRN3 mutations in familial central precocious puberty. Horm Res Paediatr. (2014) 82:122–6. doi: 10.1159/000362815
24. de Vries L, Gat-Yablonski G, Dror N, Singer A, Phillip M. A novel MKRN3 missense mutation causing familial precocious puberty. Hum Reprod. (2014) 29:2838–43. doi: 10.1093/humrep/deu256
25. Neocleous V, Shammas C, Phelan MM, Nicolaou S, Phylactou LA, Skordis N. In silico analysis of a novel MKRN3 missense mutation in familial central precocious puberty. Clin Endocrinol. (2016) 84:80–4. doi: 10.1111/cen.12854
26. Christoforidis A, Skordis N, Fanis P, Dimitriadou M, Sevastidou M, Phelan MM, et al. A novel MKRN3 nonsense mutation causing familial central precocious puberty. Endocrine. (2017) 56:446–9. doi: 10.1007/s12020-017-1232-6
27. Grandone A, Capristo C, Cirillo G, Sasso M, Umano GR, Mariani M, et al. Molecular screening of MKRN3, DLK1, and KCNK9 genes in girls with idiopathic central precocious puberty. Horm Res Paediatr. (2017) 88:194–200. doi: 10.1159/000477441
28. Jong MT, Gray TA, Ji Y, Glenn CC, Saitoh S, Driscoll DJ, et al. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet. (1999) 8:783–93. doi: 10.1093/hmg/8.5.783
29. Lin WD, Wang CH, Tsai FJ. Genetic screening of the makorin ring finger 3 gene in girls with idiopathic central precocious puberty. Clin Chem Lab Med. (2016) 54:e93–6. doi: 10.1515/cclm-2015-0408
30. Liu H, Kong X, Chen F. MKRN3 functions as a novel ubiquitin E3 ligase to inhibit Nptx1 during puberty initiation. Oncotarget. (2017) 8:85102–9. doi: 10.18632/oncotarget.19347
31. Lu W, Wang J, Li C, Sun M, Hu R, Wang W. A novel mutation in 5'-UTR of Makorin ring finger 3 gene associated with the familial precocious puberty. Acta Biochim Biophys Sin. (2018) 50:1291–3. doi: 10.1093/abbs/gmy124
32. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. (2006) 124:1283–98. doi: 10.1016/j.cell.2006.01.040
33. Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. (1997) 15:871–5. doi: 10.1038/nbt0997-871
34. Makrides N, Panayiotou E, Fanis P, Karaiskos C, Lapathitis G, Malas S. Sequential role of SOXB2 factors in GABAergic neuron specification of the dorsal midbrain. Front Mol Neurosci. (2018) 11:152. doi: 10.3389/fnmol.2018.00152
35. Maggi R, Pimpinelli F, Molteni L, Milani M, Martini L, Piva F. Immortalized luteinizing hormone-releasing hormone neurons show a different migratory activity in vitro. Endocrinology. (2000) 141:2105–12. doi: 10.1210/en.141.6.2105
36. Zhen S, Dunn IC, Wray S, Liu Y, Chappell PE, Levine JE, et al. An alternative gonadotropin-releasing hormone (GnRH) RNA splicing product found in cultured GnRH neurons and mouse hypothalamus. J Biol Chem. (1997) 272:12620–5. doi: 10.1074/jbc.272.19.12620
37. Fanis P, Gillemans N, Aghajanirefah A, Pourfarzad F, Demmers J, Esteghamat F, et al. Five friends of methylated chromatin target of protein-arginine-methyltransferase[prmt]-1 (chtop), a complex linking arginine methylation to desumoylation. Mol Cell Proteomics. (2012) 11:1263–73. doi: 10.1074/mcp.M112.017194
38. Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The vienna RNA websuite. Nucleic Acids Res. (2008) 36:W70–4. doi: 10.1093/nar/gkn188
39. Radovick S, Wray S, Lee E, Nicols DK, Nakayama Y, Weintraub BD, et al. Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proc Natl Acad Sci USA. (1991) 88:3402–6. doi: 10.1073/pnas.88.8.3402
40. Shu W, Bo X, Liu R, Zhao D, Zheng Z, Wang S. RDMAS: a web server for RNA deleterious mutation analysis. BMC Bioinformatics. (2006) 7:404. doi: 10.1186/1471-2105-7-404
41. Lim CY, Santoso B, Boulay T, Dong E, Ohler U, Kadonaga JT. The MTE, a new core promoter element for transcription by RNA polymerase II. Genes Dev. (2004) 18:1606–17. doi: 10.1101/gad.1193404
42. Ortiz-Cabrera NV, Riveiro-Alvarez R, Lopez-Martinez MA, Perez-Segura P, Aragon-Gomez I, Trujillo-Tiebas MJ, et al. Clinical exome sequencing reveals MKRN3 pathogenic variants in familial and non-familial idiopathic central precocious puberty. Horm Res Paediatr. (2017) 87:88–94. doi: 10.1159/000453262
43. Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al., Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. (2017) 49:834–41. doi: 10.1038/ng.3841
44. Uwanogho D, Rex M, Cartwright EJ, Pearl G, Healy C, Scotting PJ, et al. Embryonic expression of the chicken Sox2, Sox3 and Sox11 genes suggests an interactive role in neuronal development. Mech Dev. (1995) 49:23–36. doi: 10.1016/0925-4773(94)00299-3
45. Cheung M, Abu-Elmagd M, Clevers H, Scotting PJ. Roles of Sox4 in central nervous system development. Brain Res Mol Brain Res. (2000) 79:180–91. doi: 10.1016/S0169-328X(00)00109-1
46. Kim HD, Choe HK, Chung S, Kim M, Seong JY, Son GH, et al. Class-C SOX transcription factors control GnRH gene expression via the intronic transcriptional enhancer. Mol Endocrinol. (2011) 25:1184–96. doi: 10.1210/me.2010-0332
47. Chittka A, Nitarska J, Grazini U, Richardson WD. Transcription factor positive regulatory domain 4 (PRDM4) recruits protein arginine methyltransferase 5 (PRMT5) to mediate histone arginine methylation and control neural stem cell proliferation and differentiation. J Biol Chem. (2012) 287:42995–3006. doi: 10.1074/jbc.M112.392746
48. Wang W, Grimmer JF, Van De Water TR, Lufkin T. Hmx2 and Hmx3 homeobox genes direct development of the murine inner ear and hypothalamus and can be functionally replaced by Drosophila Hmx. Dev Cell. (2004) 7:439–53. doi: 10.1016/j.devcel.2004.06.016
Keywords: MKRN3, central precocious puberty (CPP), MKRN3 promoter region, MKRN3 5′-UTR, gene mutations
Citation: Fanis P, Skordis N, Toumba M, Papaioannou N, Makris A, Kyriakou A, Neocleous V and Phylactou LA (2019) Central Precocious Puberty Caused by Novel Mutations in the Promoter and 5′-UTR Region of the Imprinted MKRN3 Gene. Front. Endocrinol. 10:677. doi: 10.3389/fendo.2019.00677
Received: 19 March 2019; Accepted: 18 September 2019;
Published: 04 October 2019.
Edited by:
Eli Hershkovitz, Soroka Medical Center, IsraelReviewed by:
Anna Grandone, University of Campania Luigi Vanvitelli, ItalySuzy D. C. Bianco, University of Miami Miller School of Medicine, United States
Copyright © 2019 Fanis, Skordis, Toumba, Papaioannou, Makris, Kyriakou, Neocleous and Phylactou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonidas A. Phylactou, bGFwaHlsYWNAY2luZy5hYy5jeQ==