 Farah Omran
Farah Omran Mark Christian
Mark Christian- 1Warwick Medical School, University of Warwick, Coventry, United Kingdom
- 2Department of Biosciences, School of Science and Technology, Nottingham Trent University, Nottingham, United Kingdom
Obesity is characterized by a state of chronic inflammation in adipose tissue mediated by the secretion of a range of inflammatory cytokines. In comparison to WAT, relatively little is known about the inflammatory status of brown adipose tissue (BAT) in physiology and pathophysiology. Because BAT and brown/beige adipocytes are specialized in energy expenditure they have protective roles against obesity and associated metabolic diseases. BAT appears to be is less susceptible to developing inflammation than WAT. However, there is increasing evidence that inflammation directly alters the thermogenic activity of brown fat by impairing its capacity for energy expenditure and glucose uptake. The inflammatory microenvironment can be affected by cytokines secreted by immune cells as well as by the brown adipocytes themselves. Therefore, pro-inflammatory signals represent an important component of the thermogenic potential of brown and beige adipocytes and may contribute their dysfunction in obesity.
Introduction
Obesity is generally associated with a systemic low-grade inflammation with adipocytes able to produce and release signaling proteins that contribute to this condition (1–4). Many pathologies are associated with this inflamed state, including cancer, heart disease, type 2 diabetes (T2DM), and neurodegenerative diseases. Additionally, inflammation has been shown to impact the function of BAT with thermogenic activity inhibited by TNFα-induced insulin resistance and proinflammatory cytokines secreted from macrophages (5–8).
Adipose tissue (AT) functions as the body's main organ to maintain energy homeostasis (9). Mammals have two main classes of AT; brown AT (BAT) and white (WAT) that act together to maintain a balance between fat accumulation and energy expenditure (10). AT is characterized by the presence of mature lipid-storing adipocytes and pre-adipocytes (11). However, it is heterogeneous in nature and composed of a wide range of additional cell types including macrophages, neutrophils, lymphocytes, endothelial cells, and nerve endings (12–14). With its secretion of over 100 different adipokines, cytokines, and chemokines, AT is the largest endocrine organ and links metabolism and immunity (15). Undesirable changes in adipokine expression including up-regulation of inflammatory markers and down-regulation of adiponectin are linked to obesity (16).
BAT dissipates energy through the process of non-shivering thermogenesis (10). This is facilitated by a large number of mitochondria, which express high levels of UCP1 (uncoupling protein 1) in the inner membrane (10). In brown adipocytes, the nucleus occupies a central position and triglycerides (TGs) are stored in many small multilocular lipid droplets (LDs) (17, 18). This provides a large LD surface accessible to lipases, which facilitates the rapid lipid consumption for adaptive thermogenesis (18). In contrast, WAT acts as a storage repository with white adipocytes maintaining TG in a single large LD that occupies a central position (16, 19). Its nucleus is located on the periphery and the cell possesses fewer mitochondria than brown adipocytes (16). Adipocytes with brown characteristics located within WAT are known as BRITE (brown-in-white) or beige adipocytes, and are found under conditions such as in response to cold or other stimuli (20–22). Evidence indicates that beige adipocytes are mostly derived from a different cellular lineage to that of classical brown adipocytes and have the capacity to reversibly transition between white and beige adipocytes (23). Due to the widespread prevalence of obesity and its associated diseases, there is considerable research interest in factors that modulate BAT thermogenesis and the beige phenotype to enhance weight loss and reduce morbidity risk. BAT itself has recently being recognized to practice an endocrine role. It can secrete multiple factors which could contribute to the systemic consequences of BAT activity. This also forms an interesting aspect of obesity research as it could lead to the identification of novel brown fat factors to direct drug discovery approaches and ultimately improve metabolic health (24).
The presence of BAT is associated with metabolic health and the amount of BAT is reduced in obesity (25–29). Higher BAT content and activation, such as by BAT transplantation in mice, positively affects glucose and insulin metabolism and body mass and plays a protective role against obesity pathogenesis and associated metabolic disorders such as hyperglycaemia and hyperlipidaemia (30–38). Cold induced thermogenesis, glucose uptake rates and insulin stimulation is severely impaired in BAT in the adiposity state (29, 39–41). Despite the studies showing that reduced BAT in obesity is associated with many negative metabolic consequences, understanding of the underlying mechanisms is limited. Chronic inflammation represents an important mechanism behind the dysfunction of BAT and browning of white adipocytes in obesity.
Inflammatory Cells in Brown Adipose Tissue
Although mainly composed of brown adipocytes and pre-adipocytes BAT also contains a variety of immune cells such as macrophages, neutrophils and lymphocytes (42–44). Inflammation due to infiltration by macrophages and other immune cells is recognized as a key contributor to WAT pathophysiology in adiposity including insulin resistance and other alterations in metabolism (45, 46). Recent studies have identified infiltrated immune cells in BAT and inflammatory processes as contributors to BAT dysfunction in obesity and associated metabolic disorders. Similar to WAT, it is thought that recruitment of immune cells in BAT is a result of lipolysis and the release of fatty acids from stored TG (47). In diet-induced obese mice, after 6 months, BAT presents an increase in immune responses, including genes that indicate broad infiltration of leukocytes, monocytes, M1-macrophages, and cytokine release (48–51). However, BAT appears to be more resistant to macrophage infiltration than WAT in diet induced obese mice as these cells take longer to appear and have a more limited influence on BAT (50, 51). Also, the expression of inflammatory markers is lower in BAT than WAT regardless of diet (52) providing further support that BAT is generally more resistant to inflammation. Ultimately, inflammatory changes and higher expression of inflammation markers (including TNFα and F4/80) are evident in BAT after a persistent high burden of calorie intake (39, 52–54).
Enhanced inflammation is suggested to play a major role in the whitening of BAT that occurs after prolonged exposure to high fat diet at thermoneutrality. This transformation of brown adipocytes to unilocular cells similar to white adipocytes, is a result of a combination of various factors that include triggering macrophage infiltration, brown adipocyte death, and crown-like structure (CLS) formation. Whitened BAT shows CLS formation surrounding adipocytes that contain enlarged endoplasmic reticulum, cholesterol crystals, some degenerating mitochondria, and become surrounded by an increased number of collagen fibrils. BAT gene expression analysis shows that whitened BAT is associated with a strong inflammatory response and activation of nucleotide-binding oligomerization domain-like receptor-3 inflammasome (NLRP3) (72). In addition, the multimodular adaptor protein p62 is involved in multiple functions including inflammation, and it contributes to regulating energy metabolism via control of mitochondrial function in BAT which is another indicator of the importance of inflammation and immune cells pathways in BAT biology (73).
The enhancement in BAT inflammation is considered to be largely a result of the existence and active participation of infiltrated pro-inflammatory immune cells which are listed and reviewed below:
Macrophages
Macrophages are immune cells that serve an important role in the coordination of inflammatory processes (74). Classically activated macrophages (M1) secrete high levels of pro-inflammatory cytokines including TNF-α, MCP-1, IL-1β, and IL-6, whereas alternatively activated macrophages (M2) produce anti-inflammatory cytokines including IL-4 (2). In subcutaneous fat (scWAT), positive roles are reported for M2 macrophages in adaptive thermogenesis. Adipocyte-derived adiponectin signals to activate M2 macrophage proliferation during chronic cold exposure and the depletion of macrophages or adiponectin leads to resistance to cold-induced browning in scWAT (75). M2 macrophage activation also contributes to the beiging effects of adrenomedullin 2 (ADM2) and subsequent increased UCP1 expression in adipocytes (76). ADM2 can be produced by white adipocytes, and its expression is down-regulated in adipose tissues of obese mice (76). M2 macrophage activation is also stimulated by meteorin-like (Metrnl) supporting the link between adaptive thermogenic responses and anti-inflammatory gene programs in fat (77).
In the lean state BAT resident macrophages which are mostly the M2 subtype (78, 79). In obesity, however, BAT is infiltrated with (M1) macrophages which are suggested to play a crucial role in controlling adaptive thermogenesis. Inflammation of BAT caused by infiltrated macrophages reduces thermogenesis and UCP1 activation (39, 43). However, how macrophages affect thermogenesis and BAT biology is controversial (78). Initially, cold-induced thermogenesis was thought to be dependent on the secretion of the cytokines IL-4 and IL-13 by innate lymphoid cells and eosinophils that signal to macrophages as deletion of these cytokines receptors was found to diminish UCP1 expression and heat generation (77, 80). It was also suggested that M2 macrophages participate in this mechanism by secreting catecholamines (77, 80–82). However, this concept was recently challenged (83). It was found that adipose resident macrophages do not express tyrosine hydroxylase (the rate limiting enzyme for the catecholamine synthesis) and chronic treatment of wild type, UCP1−/−, and IL-4 receptor knockout mice with IL-4 failed to increase energy expenditure. In addition, incubation of adipocytes with conditioned medium from IL-4 stimulated macrophages did not induce UCP1 protein expression (83). These data indicate that any role of macrophages in brown fat activation is not through IL-4 stimulated secretion of catecholamines. However, a role of macrophages should not be completely ruled out in thermogenesis.
The main pathway for thermogenesis activation in BAT is via the sympathetic nervous system. It has recently been demonstrated that macrophages play a role in the control of BAT innervation; as selective depletion of the nuclear transcription factor Mecp2 (methyl-CpG- binding protein 2) in macrophages, a murine model of Rett syndrome, leads to spontaneous obesity with compromised homeostatic energy expenditure and thermogenesis of BAT. Specifically, deficiency of Mecp2 in BAT-macrophages causes a reduction of UCP1 gene expression levels that appears to result from impaired sympathetic innervation (43). Moreover, adipose tissue resident macrophages are reported to express a set of genes, or have a subpopulation attached to sympathetic neurons, which regulate norepinephrine levels by controlling its degradation which influences adipose tissue thermogenesis (84, 85).
Mast Cells
Mast cells are immunological classic mediators of allergic reactions and the main secretors of histamine (86). They are present in both WAT and BAT and their number increases in obesity (87, 88). Similar to some macrophages, they are closely associated with the vasculature (88). Brown adipocytes have high levels of histamine contained in mast cells and it is reported to play a role in thermogenesis through the H2-receptor. This action appears to be independent of any effect on noradrenaline stimulated oxygen consumption in isolated brown adipocytes (89). In response to colder temperatures, mast cells secrete histamine, IL-4 and other factors that promote UCP1 expression and the beiging response of WAT (90). Furthermore, it is proposed that acute cold exposure recruits mast cells to the WAT of lean subjects and enhances their degranulation and histamine secretion in both lean and obese subjects. As degranulation positively correlates with UCP1 suggests thermogenesis and beiging enhancement through histamine and secretion of other factors (91). However, these positive associations between mast cells and thermogenesis/beiging of WAT has been challenged. Zhang et al. reported that mast cell deficiency or pharmacological inhibition in mice increases browning of WAT by increasing beige adipocyte differentiation. It has also been demonstrated that mast cell-derived serotonin inhibits WAT browning and systemic energy expenditure (92). The mouse model used for this study has a mutation in c-kit tyrosine kinase and a degree of caution in the interpretation of the outcomes is required. Several alternative (c-kit-independent) genetic models of mast cell depletion have found that there is essentially no effect of mast cells in obesity and related pathologies. That is because diet-induced obese mice with either deficiency or proficiency of mast cells exhibits similar profiles of weight gain, glucose tolerance, insulin sensitivity, metabolic parameters, and AT or liver inflammation (93, 94). Further research is needed to fully understand the role of mast cells in brown and beige adipocytes especially in humans.
T Lymphocytes: Treg and ILC2s Cells
Treg cells are a small subset of T lymphocytes and are considered to be one of the most crucial defense mechanisms in maintaining appropriate immune responses including roles in autoimmunity and inflammation (95). Treg cells appear to be reduced in obesity and also required to maintain a normal adaptive thermogenesis response to cold (96, 97). Depletion of this type of immune cell impairs BAT function which was demonstrated by decreased oxygen consumption and prevention of the activation of thermogenic genes coincident with enhanced inflammation and the invasion of proinflammatory macrophages (96).
ILC2s (IL-33/Group 2 innate lymphoid cells) are a subtype of innate lymphoid cells. ILC2s are activated by epithelial cell-derived cytokines IL-33 and IL-25 as well as thymid stromal lympoiphoidin (TSLP) in response to allergens. In WAT, it has been found that white adipocytes themselves (98) and endothelial cells (99) can express IL-33. ILC2s control eosinophil and pro inflammatory macrophages to initiate type 2 immune responses that prevent helminth infection or promote pathologic allergic inflammation (100). They are found in WAT and their number is decreased in obese mice and humans (101). These cells essentially release IL-5 which maintains macrophage responses and IL-13 which controls eosinophil responses. Both of these cytokines appear to play an indirect role as mediators of beiging of WAT (100). In addition, ILC2s produce an opioid-like peptide, methionine-enkephalin (MetEnk) peptide, which appears to directly upregulate UCP1 in WAT and induces the beiging process (101). The cytokine IL-33 limits the development of spontaneous obesity by increasing numbers of ILC2s and eosinophils. This coincides with beiging and energy expenditure in the WAT of mice by but not BAT. Deletion of IL-33 leads to opposite effects (100, 102, 103).
Impact of Inflammation and Inflammatory Mediators on Brown Adipocyte Function
Obesity mediated upregulation of inflammatory cytokines has been extensively studied in WAT, while relatively little is known about the cytokines involved in the adiposity inflammatory state in BAT and how it affects BAT function and thermogenesis. However, there is an increasing amount of evidence that inflammation directly alters the thermogenic activity of brown fat by impairing its energy expenditure mechanism and glucose uptake. Pro-inflammatory cytokines can affect thermogenesis in BAT (104–106) and also determine the capacity of WAT browning (106, 107). It has been clearly demonstrated that infiltrated macrophages and other immune cells in subcutaneous WAT negatively impact the ability of precursor cells to differentiate into thermogenically active beige adipocytes because of pro-inflammatory cytokine secretion and generating an inflammatory microenvironment (108).
In BAT, the increased expression of inflammatory markers such as TNFα and MCP-1 in obese murine models is accompanied by a decrease in the expression of UCP1 and other markers of thermogenesis as well as lack of fatty acids which are needed as substrate for thermogenesis (49, 109). Also, it is reported that IL-1b reduces the cAMP-mediated induction of UCP1 expression (104), cold-induced thermogenesis in adipocytes in vivo via sirtuin-1 inhibition (SIRT1) (110) and WAT browning (111). Furthermore, Fractalkine, which is an adipocyte-synthesized chemokine, appears to contribute to enhancement of the pro-inflammatory status of BAT and reduced thermogenic gene expression in diet-induced obese mice (59). In contrast, IL-13, which has anti-inflammatory properties, induces GDF15 (growth differentiation factor 15) expression which is found to protect against obesity by inducing thermogenesis, lipolysis, and oxidative metabolism in mice (112, 113), and prevent inflammation through inhibition of M1 macrophage activation (71).
Oncostatin M, a macrophage proinflammatory cytokine impairs BAT thermogenesis and browning capacity of subcutaneous WAT in vivo. Furthermore, it inhibits brown adipocyte differentiation in vitro (114, 115). The pro-inflammatory phenotype induced by Oncostatin M is indicated as a mechanism of downregulating UCP1 expression. The significance of inflammation-driven inhibition of beige adipogenesis in obesity has been highlighted by studies of the interaction between α4-integrin receptor on pro-inflammatory macrophages and VCAM-1 (vascular cell adhesion molecule-1) on adipocytes. This interaction resulted in reduced UCP1 gene expression via the ERK (extracellular signal-regulated kinase) pathway and blockage of α4 integrin led to elevated beige adipogenesis and prevented metabolic dysregulation of the obese AT (113). This mechanism establishes a self-sustained cycle of inflammation driven impairment of the beige phenotype in obesity.
Another inflammatory candidate that might affect BAT biology and thermogenesis is the macrophage secreted factor GDF3 (growth differentiation factor-3) which increases in obesity. It is suggested that GDF3 is responsible for inhibition of β3-adrenoceptors which can lead to reduced lipolysis and consequently the impaired release of fuel for thermogenesis (84). However, although thermogenic gene expression was not restored after deleting activin-like kinase-7 (Alk7) which is the GDF3 receptor (84, 116, 117), deleting Alk7 led to reduced obesity. The metabolic benefit of Alk7 deletion may be attributed to enhanced mitochondrial biogenesis and increased levels of fatty acid oxidation found in this mouse model (116) as browning did not occur.
BAT can respond to immune and inflammatory pathways by the expression of cytokine receptors, Toll like receptors (TLRs), and nucleotide-oligomerization domain-containing proteins (NODs). Activation of these receptors by immune and metabolic signals mediates a negative impact of proinflammatory signaling on BAT thermogenesis (53). In this respect, both LPS and TNFα are found to impair UCP1 in BAT in mice in vivo and in vitro studies (110, 118). In addition, TLR4 activation inhibits β3-adrenergic-induced browning of WAT, whereas TLR4-deletion maintains thermogenic capacity (111). Some inflammatory inducers can lead to greater disruption of WAT browning compared to affecting thermogenesis in BAT. This may be indicative of a greater inflammatory response of WAT compared BAT. For example, depletion of the intestinal microbiota leads to greatly enhanced WAT browning while having only a minor effect on typical BAT (119). Also, LBP (LPS-binding protein) depletion similarly enhances WAT browning (120). This might be explained by a higher basal level of inflammation in subcutaneous WAT compared to BAT (51, 106, 107).
On the molecular level, IKKε (IκB kinase ε) and IRF3 (interferon regulatory factor-3) are among the main inflammation regulators in obesity (121, 122). Deletion of IKKε or IRF3 results in a reduction of inflammatory markers in adipose tissues and enhanced WAT browning with UCP1 expression and energy expenditure increased, while there are only minor effects on BAT (122, 123). The Nod-like receptor 3 (NLRP3) inflammasome multiprotein complex regulates inflammation and macrophage activity by cleaving IL-1b and IL-18 precursors into their active forms. Activation of NLRP3 in macrophages attenuates UCP1 and adaptive thermogenesis induction of white adipocytes and mitochondrial respiration, while NLRP3 deletion prevents UCP1 reduction. The action is through IL-1 as blocking the IL-1 receptor in adipocytes protected thermogenesis activity (111). IEX-1, an immediate early gene, is highly expressed in macrophages in obesity and is responsible for the majority of the obesity associated inflammation in humans and mice and its deletion had profound effects on the browning of WAT. Knockout of IEX-1 prevents HFD-induced inflammation, insulin resistance, and obesity by elevated browning and increasing thermogenic gene expression in WAT. This results from the promotion of M2 macrophages in WAT, but not BAT (124): and further highlights the different immune responses of white and brown adipose tissues.
There is a link between inflammatory stress pathways and the accompanied activation of endoplasmic reticulum (ER) in conditions of disruption of systemic metabolic homeostasis like obesity (125, 126). Brown adipocytes have a relatively small ER content and restricted ER surface area compared to other cell types. Thus, ER adaptation in these cells may require alternative pathways to conventional mechanisms such as chaperone-mediated protein folding and ER expansion (127, 128). In fact, some of the canonical ubiquitin-proteasome system molecules, for example X-box binding protein 1, appear to be dispensable in adipocytes (129). To maintain ER homeostasis and cellular integrity increased proteasomal activity in brown adipocytes is reported to be essential for thermogenic adaptation. This occurs via induction of the induction of the ER-localized transcription factor nuclear factor erythroid-2, like-1 (Nfe2l1, also known as Nrf1) (130). Deletion of Nfe2l1, specifically in brown adipocytes, results in ER stress, inflammation, mitochondrial dysfunction, insulin resistance, and whitening of the BAT (130, 131).
In addition to the direct effects that proinflammatory cytokines may have on brown adipocytes, some of these factors may inhibit activation of adrenergic receptors, stimulation of sympathetic nervous activity and thus local secretion of noradrenaline. As this is the main mechanism of inducing BAT thermogenesis activity and WAT browning, in response to cold and diet, it should be considered when evaluating effects on browning.
Brown Adipocytes Secrete Pro/Anti-Inflammatory Mediators
In addition to the cytokine mediators secreted by infiltrated immune cells, such cytokines may also be secreted by brown adipocytes themselves. Table 1 summarizes what has been studied in BAT.
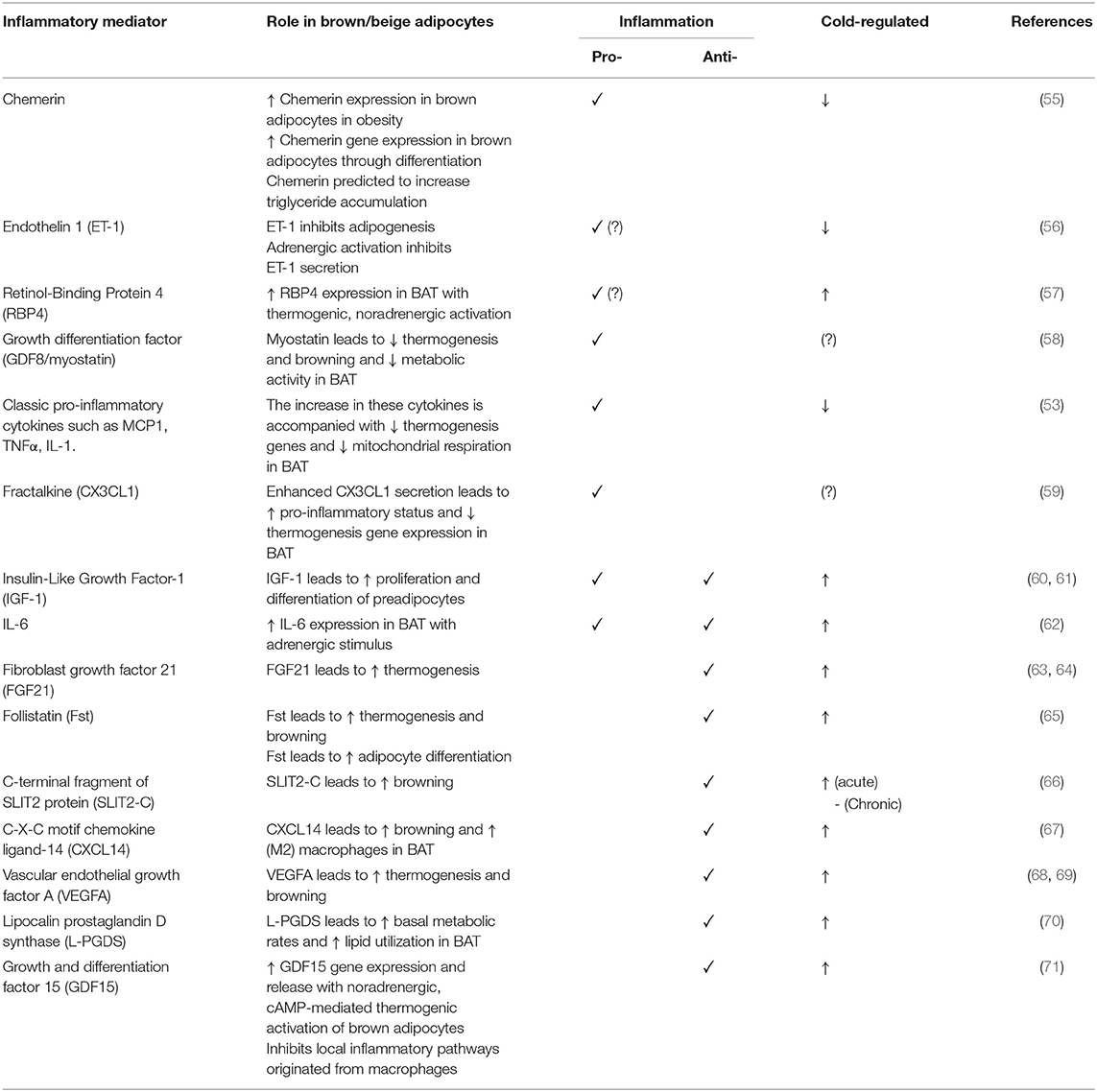
Table 1. Summary of brown adipocyte secreted inflammatory mediators.
Chemerin is an adipokine associated with inflammation markers (e.g., IL-6, TNFα, Leptin) and components of the metabolic syndrome in WAT. It modulates chemotaxis and activation of dendritic cells and macrophages (132–134). Chemerin was found to be secreted by brown adipocytes. Its gene expression levels are increased in obesity and decreased with cold induced thermogenesis and could potentially play a key role as an inflammatory modulator in BAT. However, the lack of correlation between expression levels in BAT and circulating levels make it unclear whether it plays an endocrine role in attracting immune cells (55). At present, it remains to be determined how Chemerin expression is controlled and what is its function in BAT.
Endothelin-1 (ET-1) has pro-inflammatory effects by activating macrophages, resulting in the secretion of pro-inflammatory and chemotactic mediators including TNFα, IL-1, IL-6, and IL-8 (135, 136). ET-1 levels were found to be increased in obesity and enhance lipolysis thereby linking it to insulin resistance in WAT (137). ET-1 is released by brown adipocytes and its secretion is inhibited during adrenergic stimulation (56). Data implicates that it can inhibit thermogenesis via induction of Gq signaling. However, the contribution of ET-1 to inflammation of BAT and the mechanism of thermogenesis repression remains to be fully investigated.
Vascular endothelial growth factor A (VEGFA) is a proangiogenic cytokine. The reported findings concerning VEGFA levels in WAT in obesity are controversial. Deletion of VEGFA in WAT leads to little or no change in the expression of inflammatory markers that contribute to systemic insulin resistance, such as EGF-like module-containing mucin-like hormone receptor-like 1 (Emr1), TNF-α, and MCP-1. Nor were there detectable changes in the expression of mitochondrial genes in WAT (138, 139). In contrast, in BAT VEGFA can induce thermogenic activity and deletion of VEGFA results in reduction of BAT mass, vessel density, and eventually loss of thermogenesis through mitochondrial dysfunction (68, 138). Hence, ablation of VEGFA results in the whitening of BAT. However, the direct effect of VEGFA on inflammatory markers in brown adipocytes is currently unknown.
Retinol-Binding Protein 4 (RBP4) is an adipokine and circulating transporter of vitamin A (retinol) that induces inflammation and promotes the secretion of proinflammatory molecules (140). There has been some controversy regarding the associations and/or causality in the context of obesity and metabolic syndrome. However, in adipocytes, RBP4 appears to have a relevant role in obesity and the development of insulin resistance and diabetes (141). Brown adipocytes release RBP4 when exposed to a thermogenic, noradrenergic activation, but the mechanism associated with this release is unclear. Lipocalin 2 (Lcn2) has been implicated in the release of RBP4 as Lnc2 KO adipose tissue shows increased RBP4 levels while circulating levels are reduced (142). Also, BAT released RBP4 may not be associated with insulin resistance given that cold-induced activation of BAT is associated with insulin sensitization (57).
Fibroblast growth factor 21 (FGF21) is a brown adipokine and a key factor in the regulation of energy homeostasis. In WAT, it induces browning and participates in improving glucose metabolism and weight regulation. Cold induced thermogenesis and adrenergic activation induces FGF21 release from brown adipocytes. In addition to this mechanism, WAT-resident anti-inflammatory invariant natural killer T (iNKT) cells promote the release of FGF-21 by adipocytes and the browning process (143–145). Prevention of hyperglycaemia and hyperlipidaemia is associated with high levels of FGF21 in line with high BAT activity and enhanced energy expenditure (146, 147). Although FGF21 is reported to have anti-inflammatory effects on white adipocytes (148) it remains to be determined if it has a similar action in brown adipocytes.
CXCL14 is a member of the CXC chemokine family and exerts chemoattractive activity for activated macrophages, immature dendritic cells and natural killer cells. In WAT, CXCL14 participates in glucose metabolism (149, 150). It is reported to be secreted by brown adipocytes in response to thermogenic activation. CXCL14 appears to attract M2 macrophages and its deletion leads to impaired BAT thermogenesis activity and low recruitment of macrophages into BAT. CXCL14 enhances the browning of white fat via type 2 cytokine signaling (67).
Fractalkine (CX3CL1) is a chemokine produced by brown adipocytes that plays a role in the recruitment of leukocytes through the fractalkine receptor. Its action seems to be to promote an inflammatory state as deficiency of the fractalkine receptor prevents BAT accumulation of macrophages and leads to reduced expression of pro inflammatory genes (Tnfα, Il1α, and Ccl2) in mice exposed to HFD. Furthermore, the BAT of fractalkine receptor deficient mice shows increased expression of lipolytic enzymes such as adipose triglyceride lipase (Atgl), lipase, hormone-sensitive (Hsl) and monoglyceride lipase (Mgtl) and upregulation of UCP1 and other thermogenesis genes (59). This indicates that fractalkine serves a key role in the local inflammation of BAT tissue and the remodeling on HFD affecting metabolism.
Follistatin (FST) is a soluble glycoprotein that has the capacity to modulate the activities of multiple members of the transforming growth factor (TGF) family, specifically activin A and myostatin (GDF8). TGF-β superfamily cytokines play pivotal roles in regulation of tissue functions including inflammation (151–153). Blockade of TGF-β/Smad3 signaling enhances insulin sensitivity and prevents diet-induced obesity, promotes the browning of WAT with reduced levels of inflammatory cytokines and less inflammatory macrophage infiltration (154–156). FST is upregulated in BAT in response to cold, and is potentially a positive regulator of BAT function by blocking TGF-β signaling pathways, GDF8 actions, and exerting anti-inflammatory effects (65, 157, 158). However, these actions are yet to be explored.
Myostatin (GDF8) is a key member of the transforming growth factor-β (TGF-β) super family and has an essential role in the regulation of overall fat content in mice. Loss of GDF8 leads to a significant increase in lean mass, total energy expenditure, protection against diet-induced obesity, and insulin resistance. GDF8 levels increase in obesity and it is reported to suppresses Irisin leading to activation of inflammatory cytokines and insulin resistance in WAT. GDF8 secretion from brown adipocytes is stimulated by activation of hunger-related neural circuits. It negatively regulates BAT thermogenesis as well as WAT browning, and metabolic activity. Data clearly indicates that GDF8 inhibits brown fat gene expression (157), however, further research is needed to investigate its inflammatory related effects in this tissue (58, 156, 157, 159–161).
Growth and differentiation factor 15 (GDF15) is also known as macrophage inhibiting cytokine-1 and is a member of the TGF-β superfamily (162). GDF15 is suggested to be a reliable predictor of disease progression in certain tumors, inflammatory diseases, cardiovascular disease, and obesity (162). It is reported to decrease food intake, body weight and adiposity, and to improve glucose tolerance under normal and obesogenic diets (163). Furthermore, systemic overexpression of GDF15 was shown to prevent obesity and insulin resistance by increasing the expression of the main thermogenic and lipolytic genes and oxidative metabolism in BAT and WAT (164). It is identified as one of the factors secreted by brown adipocytes through protein kinase A-mediated mechanisms, and highly induced in response to thermogenic activity following stimulus by cold, norepinephrine, and cAMP. GDF15 acts on macrophages in BAT and may mediate inhibition of local inflammatory pathways under conditions of enhanced BAT activity (71).
C-terminal fragment of SLIT2 protein (SLIT2-C) belongs to the Slit family of secreted proteins that play important roles in various physiologic and pathologic activities including inflammatory cell chemotaxis where it exercises an anti-inflammatory role (66, 165). SLIT2-C expression is regulated by PRDM16 and is secreted from beige/brown adipocytes. It induces thermogenesis, WAT browning, and metabolic processes associated with substrate supply to fuel thermogenesis. The pathway for the induction of thermogenesis is independent of β-adrenergic activation, but requires activation of protein kinase A signaling (66). The protease that generates the SLIT2-C fragment as well as the receptor in BAT that binds it are important areas for future investigation.
Lipocalin prostaglandin D synthase (L-PGDS) is expressed in BAT where it has a key role in energy substrate utilization. It is also localized in the central nervous system and it is involved in inflammatory modulations amongst other functions (166). Deletion of L-PGDS leads to inadequate thermogenesis in BAT because of impairment in switching of substrate utilization from glucose to lipids (70, 167). In addition, L-PGDS deficiency induces obesity possibly through the regulation of inflammatory responses (168). However, if that is the case in brown adipocytes, it is yet to be elucidated.
Insulin-Like Growth Factor-1 (IGF-1) appears to play pleiotropic functions and provides signals to macrophages to sustain adipose tissue development and homeostasis. IGF1 signaling integrates immune-metabolic interactions to facilitate macrophage activation status. Cold exposure stimulates elevation of IGF-1 expression in the BAT of rats (169). However, Myeloid-specific ablation of IGF-1 receptor worsens diet induced obesity but not cold induced thermogenesis (170, 171). It is suggested that IGF-1 is released by brown adipocytes and involved in proliferation and differentiation of brown preadipocytes (60, 61, 172). IGF-1 upregulation due to BAT transplantation is proposed to abolish type I diabetes in this experimental model and negatively correlates with glucose, glucagon, and inflammatory cytokines in rodents (31, 173). However, the detailed role of IGF-1 in brown adipocytes inflammation regulation remains an area to be investigated.
Interleukin 6 (IL-6) is secreted by brown adipocytes upon β-adrenergic activation (62). Chronic elevation of IL-6 in the CNS leads to increased UCP1 in BAT, but not in denervated BAT tissue which suggest a central role in IL-6-dependent promotion of thermogenesis (174). Signaling by IL-6 promotes M2 macrophage polarization in BAT (175). Evidence indicates that IL-6 is released from WAT during the differentiation of human beige adipocytes to facilitate the commitment of adipocyte precursors toward beigeing and enhancement of thermogenesis capacity in an autocrine manner (176). The deletion of IL-6 in mice leads to inefficient BAT transplantation with sustained obesity and insulin resistance, and blunted FGF21 increase (33). These data suggest beneficial effects of IL-6 in regulation of BAT metabolism possibly directly or indirectly related to FGF21 actions. However, this contrasts with the action of IL-6 as a potent pro-inflammatory cytokine. This aspect can be demonstrated by plasma IL-6 being elevated in obesity and diabetes, in addition to reduced levels in weight loss (177–179). Furthermore, it is also found to play a major role as a pro-inflammatory cytokine in obese adipose tissue, macrophage polarization, and T cell regulation via STAT3, leading eventually to insulin resistance and worsening diet-induced obesity (180). Moreover, as expected for IL-6 having a typical role as a pro-inflammatory cytokine, its deletion causes reversal of pro-inflammatory signaling in the obese state (179). In terms of browning activation, IL-6 is implicated in inducing inguinal WAT atrophy by accelerating WAT lipolysis and browning (181). In any case, these available contradictory data concerning the role of IL-6 are indicative of both pro- and anti-inflammatory actions. A schematic showing the actions of inflammatory mediators on brown and white adipocytes is presented in Figure 1.
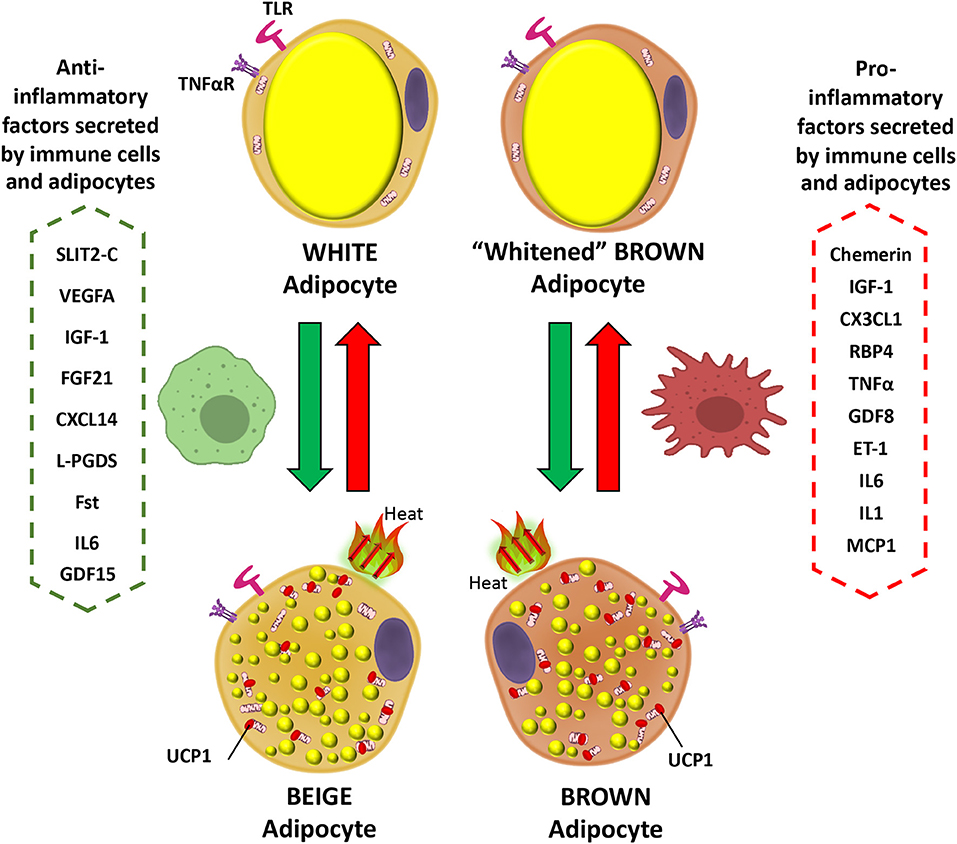
Figure 1. Inflammatory mediator actions on white and brown adipocytes. Pro-inflammatory factors secreted by immune cells and brown/beige adipocytes prevent the expression of brown fat genes in adipocytes, including UCP1, the main thermogenic protein (red arrows). In contrast anti-inflammatory mediators promote the transition of white to beige adipocytes and could prevent expression of the “whitened” brown adipocyte phenotype in brown adipose tissue (green arrows). IGF-1, Insulin-Like Growth Factor-1; CX3CL1, Fractaline; RBP4, Retinol-Binding Protein 4; TNFα, Tumor necrosis factor a; GDF8, Growth differentiation factor 8; ET-1, Endothelin 1; IL6, Interleukin 6; IL1, Interleukin 1; MCP1, Monocyte Chemoattractant Protein-1; SLIT2-C, C-terminal fragment of SLIT2 protein; VEGFA, Vascular endothelial growth factor A; FGF21, Fibroblast growth factor 21; CXCL14, C-X-C motif chemokine ligand-14; L-PGDS, Lipocalin prostaglandin D synthase; Fst, Follistatin; UCP1, Uncoupling Protein 1; GDF15, Growth and differentiation factor 15.
Insulin Sensitivity
Pro-inflammatory signaling can disrupt the insulin signaling cascade and impair the insulin sensitivity of BAT. Although, IL-1, TNF-α, MIF, and IL-6 have consistently been shown to cause insulin resistance in WAT (182, 183), their effects have not been extensively explored in BAT at the molecular level. Elevated inflammatory marker levels in the diet induced obesity state in mice are suggested to be responsible for BAT insulin resistance via AKT (protein kinase B) and ERK pathways (52). TNF-α appears to play an important role in impairing insulin sensitivity of BAT. The mechanism has been discussed in detail and it involves disturbances of both MAP- kinases activation and IRS-2 and AKT (5–7). Mammalian target of rapamycin complex 2 (mTORC2), which activates inflammation, sustains thermogenesis via Akt-induced glucose uptake and glycolysis in BAT. This highlights the significance of glucose metabolism in BAT in thermogenesis and indicates the importance of identifying how inflammation can affect mTORC2-activation in BAT (184, 185). Alleviating the inflammation state in obesity may restore insulin sensitivity as targeting inflammation in diet induced obesity in mice leads to a decrease in adipocyte area, macrophage infiltration, proinflammatory gene expression, along with JNK and NF-κB activation and increased insulin sensitivity via increased AKT phosphorylation (186, 187). In this context, sucrose non-fermenting related kinase (SNRK), a member of the AMPK-related kinase family, is found to suppress inflammation in WAT and is essential for maintaining UCP1 expression for BAT thermogenesis. Dysregulation of this anti-inflammatory kinase leads to induction of insulin resistance in BAT via impairment of the PP2A-Akt pathway (188, 189). As a result, inflammation is a modulator of insulin responses in BAT and is strongly linked to UCP1 expression and thermogenesis. It is important to determine the role of each inflammatory cytokine in insulin resistance and thermogenesis in order to identify therapeutic targets.
Mitochondrial Function Is Affected by Inflammatory Pathways
Inflammation and mitochondrial dysfunction are closely linked with obesity and associated with alteration in mitochondrial function and mass (190, 191). These alterations are further demonstrated by downregulation of mitochondrial biogenesis, oxidative metabolic pathways, and oxidative phosphorylation proteins in WAT in obesity, and a negative correlation with pro-inflammatory cytokines (192). Evidence indicates that proinflammatory cytokines have a significant influence on modulating mitochondrial efficiency leading to effects on energy homeostasis in human white adipocytes. TNF-α most dramatically alters 3T3-L1 adipocyte mitochondrial functions, whereas IL-1β and IL-6 have more modest effects. Moreover, activation of the NLRP3 inflammasome in macrophages attenuates UCP1 induction and mitochondrial respiration in cultures of primary adipocytes possibly via IL-1, while the absence of NLRP3 is protective for UCP1 and adaptive thermogenesis capacity in adipocytes (111, 193, 194).
The activation of pattern recognition receptors in brown adipocytes and subsequent increased inflammation leads to mitochondrial dysfunction and suppression of mitochondrial respiration with reduced UCP1 expression levels and repressed white adipocyte browning capacity in response to adrenergic stimulation. Mechanistically, these effects are likely to involve inhibition of SIRT1 activity (53, 110, 111). Moreover, deletion of TLR4 protected mitochondrial function and thermogenesis in WAT (111). However, there is a suggestion that mitochondrial dysfunction in adipocytes is a primary cause of adipose tissue inflammation, adipocyte enlargement and insulin resistance. According to this hypothesis mitochondrial dysfunction and fatty acid oxidation in adipocytes leads to adipocyte enlargement because of triglyceride accumulation. Furthermore, adipocyte mitochondrial dysfunction leads to pseudo-hypoxia with greater accumulation of hypoxia-inducible factor 1α (HIF-1α), which elevates adipose tissue inflammation and fibrosis (195, 196). Similarly, alteration of mitochondrial capacity in BAT might be functionally associated with defective thermogenesis and energy expenditure in obesity and increased risk to develop obesity-induced insulin resistance.
Using a mouse model of chronic systemic inflammation, which exhibits increased circulating levels of inflammatory cytokines and abnormal regulation of both innate and adaptive immune responses, mitochondrial swelling is detected with severe damage of the cristae, in addition to reduced cold-induced thermogenic capacity and UCP1-dependent mitochondrial respiration (197). Furthermore, low grade inflammation in BAT in obesity is found as a contributor to excess reactive oxygen species (ROS) production and associated oxidative stress, which may cause mitochondrial dysfunction (198–202). Further investigations in BAT confirmed increased inflammation and ROS generation, but this was accompanied by the doubling of mitochondria respiration compared to lean subjects. It is possible that if the obesogenic conditions were maintained for longer, mitochondria would have eventually failed to deal with obesity stress and thermogenic capacity would be ultimately compromised (49). ROS production does not necessarily have negative consequences in BAT. Consistent with beneficial effects of increased ROS, activated BAT thermogenesis in vivo is defined by a substantial increase in mitochondrial ROS levels and pharmacological depletion of mitochondrial ROS leads to hypothermia upon cold exposure, and inhibits UCP1-dependent increases in whole body energy expenditure (203).
Mitochondrial dysfunction resulting from deletion of the mitochondrial transcription factor A (TFAM) leads to adipocyte death coincident with inflammation in WAT and a whitening of BAT with decreased energy expenditure. BAT whitening in these mice is mainly explained by impairment of mitochondrial electron transport chain function, reduced fatty acid oxidation, and increased circulating fatty acids, rather than a conversion of brown to white adipocytes (204). These findings highlight the link between mitochondrial function and inflammation and point to mitochondria dysfunction leading to increased inflammation which could ultimately lead to a vicious cycle.
Anti-Inflammatory Pathways and Bat Function
Strategies that target the inflammatory status may have the potential to reverse adipose tissue dysfunction and prevent progression of metabolic diseases. Suppression of inflammation using pharmacological agents, with reduction of pro-inflammatory cytokines and macrophage infiltration in WAT, improves AKT-phosphorylation in response to insulin along with improved body weight and fat mass (187, 205–207). Cytarabine, which has immunosuppressive actions, is associated with enhanced BAT activity via the AMPK pathway raising the possibility it could be developed for anti-obesity therapy (208). There is also evidence that dietary intervention can have anti-inflammation activity which leads to enhanced insulin sensitivity. Food extracts with a high content of either flavonoids, phenolic compounds, p-coumaric acid, quercetin, or resveratrol have been found to exert systemic anti-inflammatory actions via inhibition of TNF-α-triggered activation of MAPKs and NFκB in human white adipocytes which improve insulin sensitivity (186). Specifically, Curcumin intervention was found to reduce mouse WAT inflammation and increase BAT UCP1 expression via PPAR-dependent and -independent mechanisms. It reduces macrophage infiltration and proinflammation cytokine expression in both macrophages and adipocytes along with increased energy expenditure and body temperature in response to cold (209).
Fatty acids (FA) are another example of dietary constituents that act as inflammation modulators. Importantly, ω3-FAs (n-3 polyunsaturated fatty acids (n-3PUFAs) have anti-inflammatory effects and may significantly impact chronic inflammatory diseases including obesity related disorders (210). An ω3-enriched diet, in non-obesogenic non-inflammatory conditions, leads to synthesis of oxylipins which have an anti-inflammatory response in both WAT and BAT with a macrophage modulation effect, but with no influence on inflammatory cytokine secretion (209). FFAs are active stimulators for members of the rhodopsin-like family of G protein-coupled receptors (GPCRs) including GPR40, GPR41, GPR43, GPR84, and GPR120 (211, 212). GPR120 is highly expressed in both BAT and WAT. and positively impacts metabolic health by stimulating mitochondrial respiration in brown fat via intracellular Ca2+ release which results in mitochondrial depolarization and fragmentation. This occurs along with mitochondrial UCP1 activation, which may act synergistically with mitochondrial fragmentation to increase respiration. GPR120 activation by the agonist TUG-891 upregulates fat combustion in BAT thereby reducing fat mass, while GPR120 deficiency diminishes expression of genes involved in nutrient metabolism (213). Moreover, GPR120 deficiency leads to obesity, glucose intolerance, and hepatic steatosis in mice fed a high-fat diet (214). Importantly, GPR120 mediates the anti-inflammatory and insulin sensitizing effects of ω3-FAs including inhibition of inflammatory pathways and cytokine secretion in adipocytes and macrophages (215, 216). A role for GPR120 in BAT activation and WAT browning in response to cold via FGF21 secretion has also been confirmed (217).
Conclusion
Immune responses pose a significant metabolic challenge for the host due a range of energetically expensive processes including inflammatory mediator production and cell migration and proliferation. There is a trade-off between the energetic demands of immunity and homeothermy that permits a hypometabolic-hypothermic state to favor the immune system. Peripheral insulin resistance provides a mechanism for reallocating metabolic fuels to immune cells due to decreased nutrient storage in fat, muscle, and liver. The precise role of BAT in the hypometabolic-hypothermic state is currently unclear. Although BAT is generally more resistant to inflammatory stimuli than WAT, the repression of thermogenesis by inflammation may be a key energy trade-off to allow sufficient resources for immune responses. Importantly, BAT-mediated thermogenesis reactivation seems to be required for the exit from the hypometabolic-hypothermic state (218).
Many immune and inflammatory cells actively participate in the regulation of BAT thermogenesis, WAT browning and ultimately have the capacity to participate in controlling energy balance, glycemia, and lipidemia. Pro-inflammatory mediators secreted by both immune cells and adipocytes inhibit thermogenesis activation in BAT and browning of WAT in contrast to anti-inflammatory factors that have a positive influence. Additional research is needed to demonstrate the effect of each one of these mediators on brown and beige adipose cells and fully explain the pathways involved at the molecular level that regulate immune cells and brown/beige adipocytes interactions. This could lead to new therapeutic strategies to improve metabolic health and combating obesity and associated metabolic diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by The Graduate School, University of Warwick.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge CARA, The Council for At-Risk Academics, Warwick Medical School.
References
1. McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. (2014) 41:36–48. doi: 10.1016/j.immuni.2014.05.010
2. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
3. Saltiel AR, Olefsky JM. Inflammatory linking obesity and metabolic disease and metabolic disease. J Clin Invest. (2017) 127:1–4. doi: 10.1172/JCI92035
4. Xiao L, Yang X, Lin Y, Li S, Jiang J, Qian S, et al. Large adipocytes function as antigen-presenting cells to activate CD4 + T cells via upregulating MHCII in obesity. Int J Obes. (2016) 40:112–20. doi: 10.1038/ijo.2015.145
5. Valverde AM, Teruel T, Navarro P, Benito M, Lorenzo M. Tumor necrosis factor-α causes insulin receptor substrate-2-mediated insulin resistance and inhibits insulin-induced adipogenesis in fetal brown adipocytes. Endocrinology. (1998) 139:1229–38. doi: 10.1210/endo.139.3.5854
6. Lorenzo M, Alvaro C De, E-e JAS, Ferna S. Insulin resistance induced by tumor necrosis factor-Alpha in myocytes and brown adipocytes. J Anim Sci. (2008) 86:94–104. doi: 10.2527/jas.2007-0462
7. Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem. (2008) 114:183–94. doi: 10.1080/13813450802181047
8. Sakamoto T, Takahashi N, Sawaragi Y, Naknukool S, Yu R, Goto T, et al. Inflammation induced by RAW macrophages suppresses UCP1 mRNA induction via ERK activation in 10T1/2 adipocytes. Am J Physiol Physiol. (2013) 304:C729–38. doi: 10.1152/ajpcell.00312.2012
9. Halberg N, Wernsted I, Scherer P. The adipocyte as an endcrine cell. Endocrinol Metab Clin North Am. (2009) 37:1–15. doi: 10.1016/j.ecl.2008.07.002
10. Lo KA, Sun L. Turning WAT into BAT: a review on regulators controlling the browning of white adipocytes. Biosci Rep. (2013) 33:711–9. doi: 10.1042/BSR20130046
11. Armani A, Mammi C, Marzolla V, Calanchini M, Antelmi A, Rosano GMC, et al. Cellular models for understanding adipogenesis, adipose dysfunction, and obesity. J Cell Biochem. (2010) 110:564–72. doi: 10.1002/jcb.22598
12. Berry DC, Stenesen D, Zeve D, Graff JM. The developmental origins of adipose tissue. Development. (2013) 140:3939–49. doi: 10.1242/dev.080549
13. Martinez-Santibañez, WonCho K, Lumeng CN. Imaging white adipose tissue with confocal microscopy. Methods Enzym. (2014) 537:17–30. doi: 10.1016/B978-0-12-411619-1.00002-1
14. Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. (2006) 55:1537–45. doi: 10.2337/db06-0263
15. MacDougald OA, Burant CF. The rapidly expanding family of adipokines. Cell Metab. (2007) 6:159–61. doi: 10.1016/j.cmet.2007.08.010
16. Rega-Kaun G, Kaun C, Wojta J. More than a simple storage organ: adipose tissue as a source of adipokines involved in cardiovascular disease. Thromb Haemost. (2013) 110:641–50. doi: 10.1160/TH13-03-0212
18. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
19. Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Clin Endocrinol. (2006) 64:355–65. doi: 10.1111/j.1365-2265.2006.02474.x
20. Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. (2010) 285:7153–64. doi: 10.1074/jbc.M109.053942
21. Wu J, Khandekar M, Nuutila P, Schaart G, Huang K, Tu H, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. (2012) 150:366–76. doi: 10.1016/j.cell.2012.05.016
22. Giralt M, Villarroya F. White, brown, beige/brite: different adipose cells for different functions? Endocrinology. (2013) 154:2992–3000. doi: 10.1210/en.2013-1403
23. Rosenwald M, Wolfrum C. The origin and definition of brite versus white and classical brown adipocytes. Adipocyte. (2014) 3:4–9. doi: 10.4161/adip.26232
24. Villarroya F, Cereijo R, Villarroya J, Giralt M. Brown adipose tissue as a secretory organ. Nat Rev Endocrinol. (2017) 13:26–35. doi: 10.1038/nrendo.2016.136
25. Leitner BP, Huang S, Brychta RJ, Duckworth CJ, Baskin AS, McGehee S, et al. Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci USA. (2017) 114:8649–54. doi: 10.1073/pnas.1705287114
26. Vijgen GHEJ, Bouvy ND, Teule GJJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. Brown adipose tissue in morbidly obese subjects. PLoS ONE. (2011) 6:2–7. doi: 10.1371/journal.pone.0017247
27. Matsushita M, Yoneshiro T, Aita S, Kameya T, Sugie H, Saito M. Impact of brown adipose tissue on body fatness and glucose metabolism in healthy humans. Int J Obes. (2014) 38:812–7. doi: 10.1038/ijo.2013.206
28. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. (2009) 360:1500–8. doi: 10.1056/NEJMoa0808718
29. Ferre P, Burnol AF, Leturque A, Terretaz J, Penicaud L, Jeanrenaud B, et al. Glucose utilization in vivo and insulin-sensitivity of rat brown adipose tissue in various physiological and pathological conditions. Biochem J. (1986) 233:249–52. doi: 10.1042/bj2330249
30. Liu X, Wang S, You Y, Meng M, Zheng Z, Liu L, et al. Brown adipose tissue transplantation reverses obesity in Ob/Ob mice. Endocrinology. (2015) 156:2461–9. doi: 10.1210/en.2014-1598
31. Gunawardana SC, Piston DW. Reversal of type 1 diabetes in mice by brown adipose tissue transplant. Diabetes. (2012) 61:674–82. doi: 10.2337/db11-0510
32. Shankar K, Kumar D, Gupta S, Varshney S, Rajan S, Srivastava A, et al. Role of brown adipose tissue in modulating adipose tissue in fl ammation and insulin resistance in high-fat diet fed mice. Eur J Pharmacol. (2019) 854:354–64. doi: 10.1016/j.ejphar.2019.02.044
33. Stanford KI, Middelbeek RJW, Townsend KL, An D, Nygaard EB, Hitchcox KM, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. (2013) 123:215–23. doi: 10.1172/JCI62308
34. Xiaomeng L, Zongji Z, Xiaoming Z, Minghui M, Lan L, Yanyan S, et al. Brown adipose tissue transplantation improves whole-body energy metabolism. Cell Res. (2013) 23:851–4. doi: 10.1038/cr.2013.64
35. Berbeé JFP, Boon MR, Khedoe PPSJ, Bartelt A, Schlein C, Worthmann A, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun. (2015) 6:6356. doi: 10.1038/ncomms7356
36. Koksharova E, Ustyuzhanin D, Philippov Y, Mayorov A, Shestakova M, Shariya M, et al. The relationship between brown adipose tissue content in supraclavicular fat depots and insulin sensitivity in patients with type 2 diabetes mellitus and prediabetes. Diabetes Technol Ther. (2017) 19:96–102. doi: 10.1089/dia.2016.0360
37. Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. (2014) 63:4089–99. doi: 10.2337/db14-0746
38. Wu C, Cheng W, Sun Y, Dang Y, Gong F, Zhu H, et al. Activating brown adipose tissue for weight loss and lowering of blood glucose levels: a microPET study using obese and diabetic model mice. PLoS ONE. (2014) 9:1–14. doi: 10.1371/journal.pone.0113742
39. Sakamoto T, Nitta T, Maruno K, Yeh YS, Kuwata H, Tomita K, et al. Macrophage infiltration into obese adipose tissues suppresses the induction of UCP1 level in mice. Am J Physiol Endocrinol Metab. (2016) 310:E676–87. doi: 10.1152/ajpendo.00028.2015
40. Rissanen A, Orava J, Nuutila P, Noponen T, Parkkola R, Viljanen T, et al. Blunted metabolic responses to cold and insulin stimulation in brown adipose tissue of obese humans. Obes. (2013) 21:2279–87. doi: 10.1002/oby.20456
41. Penicaud L, Ferre P, Terretaz J, Kinebanyan MF, Leturque A, Dore E, et al. Development of obesity in Zucker rats. Early insulin resistance in muscles but normal sensitivity in white adipose tissue. Diabetes. (1987) 36:626–31. doi: 10.2337/diabetes.36.5.626
42. Tran K Van, Gealekman O, Frontini A, Zingaretti MC, Morroni M, Giordano A, et al. The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metab. (2012) 15:222–9. doi: 10.1016/j.cmet.2012.01.008
43. Wolf Y, Boura-Halfon S, Cortese N, Haimon Z, Sar Shalom H, Kuperman Y, et al. Brown-adipose-tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat Immunol. (2017) 18:665–74. doi: 10.1038/ni.3746
44. Kooijman S, van den Heuvel JK, Rensen PCN. Neuronal control of brown fat activity. Trends Endocrinol Metab. (2015) 26:657–68. doi: 10.1016/j.tem.2015.09.008
45. Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. (2008) 4:619–26. doi: 10.1038/ncpendmet0976
46. Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. (2008) 14:222–31. doi: 10.2119/2007-00119.Tilg
47. Chawla A, Nguyen KD, Goh YPS. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. (2011) 11:738–49. doi: 10.1038/nri3071
48. McGregor RA, Kwon EY, Shin SK, Jung UJ, Kim E, Park JHY, et al. Time-course microarrays reveal modulation of developmental, lipid metabolism and immune gene networks in intrascapular brown adipose tissue during the development of diet-induced obesity. Int J Obes. (2013) 37:1524–31. doi: 10.1038/ijo.2013.52
49. Alcalá M, Calderon-Dominguez M, Bustos E, Ramos P, Casals N, Serra D, et al. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci Rep. (2017) 7:1–12. doi: 10.1038/s41598-017-16463-6
50. Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J, Czech MP. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am J Physiol Hear Circ Physiol. (2011) 301:H1425–37. doi: 10.1152/ajpheart.00376.2011
51. Dowal L, Parameswaran P, Phat S, Akella S, Majumdar ID, Ranjan J, et al. Intrinsic properties of brown and white adipocytes have differential effects on macrophage inflammatory responses. Mediators Inflamm. (2017) 2017:9067049. doi: 10.1155/2017/9067049
52. Roberts-Toler C, O'Neill BT, Cypess AM. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity. (2015) 23:1765–70. doi: 10.1002/oby.21134
53. Bae J, Ricciardi CJ, Esposito D, Komarnytsky S, Hu P, Curry BJ, et al. Activation of pattern recognition receptors in brown adipocytes induces inflammation and suppresses uncoupling protein 1 expression and mitochondrial respiration. AJP Cell Physiol. (2014) 306:C918–30. doi: 10.1152/ajpcell.00249.2013
54. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI200319451
55. Hansen IR, Jansson KM, Cannon B, Nedergaard J. Contrasting effects of cold acclimation versus obesogenic diets on chemerin gene expression in brown and brite adipose tissues. Biochim Biophys Acta. (2014) 1841:1691–9. doi: 10.1016/j.bbalip.2014.09.003
56. Klepac K, Kilić A, Gnad T, Brown LM, Herrmann B, Wilderman A, et al. The G q signalling pathway inhibits brown and beige adipose tissue. Nat Commun. (2016) 7:10895. doi: 10.1038/ncomms10895
57. Rosell M, Hondares E, Iwamoto S, Gonzalez FJ, Wabitsch M, Staels B, et al. Peroxisome proliferator-activated receptors-α and -γ and cAMP-mediated pathways, control retinol-binding protein-4 gene expression in brown adipose tissue. Endocrinology. (2012) 153:1162–73. doi: 10.1210/en.2011-1367
58. Steculorum SM, Ruud J, Karakasilioti I, Backes H, Engström Ruud L, Timper K, et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell. (2016) 165:125–38. doi: 10.1016/j.cell.2016.02.044
59. Polyák Á, Winkler Z, Kuti D, Ferenczi S, Kovács KJ. Brown adipose tissue in obesity: fractalkine-receptor dependent immune cell recruitment affects metabolic-related gene expression. Biochim Biophys Acta. (2016) 1861:1614–22. doi: 10.1016/j.bbalip.2016.07.002
60. Yamashita H, Sato Y, Kizaki T, Oh-ishi S, Nagasawa J, Ohno H. Basic fibroblast growth factor (bFGF) contributes to the enlargement of brown adipose tissue during cold acclimation. Pflügers Arch. (1994) 428:352–6. doi: 10.1007/BF00724518
61. Lorenzo M, Valverde AM, Teruel T, Benito M. IGF-I is a mitogen involved in differentiation-related gene expression in fetal rat brown adipocytes. J Cell Biol. (1993) 123:1567–75. doi: 10.1083/jcb.123.6.1567
62. Burysek L, Houstek J. B-Adrenergic stimulation of interleukin-la and interleukin-6 expression in mouse brown adipocytes. FEBS Lett. (1997) 411:83–6. doi: 10.1016/S0014-5793(97)00671-6
63. Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, Papavassiliou AG. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med. (2011) 17:736–40. doi: 10.2119/molmed.2011.00075
64. Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, et al. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J Biol Chem. (2011) 286:12983–90. doi: 10.1074/jbc.M110.215889
65. Braga M, Reddy ST, Vergnes L, Pervin S, Grijalva V, Stout D, et al. Follistatin promotes adipocyte differentiation, browning, and energy metabolism. J Lipid Res. (2014) 55:375–84. doi: 10.1194/jlr.M039719
66. Svensson KJ, Long JZ, Jedrychowski MP, Cohen P, Lo JC, Serag S, et al. A secreted Slit2 fragment regulates adipose tissue thermogenesis and metabolic function. Cell Metab. (2016) 23:454–66. doi: 10.1016/j.cmet.2016.01.008
67. Cereijo R, Gavaldà-Navarro A, Cairó M, Quesada-López T, Villarroya J, Morón-Ros S, et al. CXCL14, a brown adipokine that mediates brown-fat-to-macrophage communication in thermogenic adaptation. Cell Metab. (2018) 28:750–63.e6. doi: 10.1016/j.cmet.2018.07.015
68. Sun K, Kusminski CM, Luby-Phelps K, Spurgin SB, An YA, Wang QA, et al. Brown adipose tissue derived VEGF-A modulates cold tolerance and energy expenditure. Mol Metab. (2014) 3:474–83. doi: 10.1016/j.molmet.2014.03.010
69. Xue Y, Petrovic N, Cao R, Larsson O, Lim S, Chen S, et al. Hypoxia-independent angiogenesis in adipose tissues during cold acclimation. Cell Metab. (2009) 9:99–109. doi: 10.1016/j.cmet.2008.11.009
70. Virtue S, Feldmann H, Christian M, Tan CY, Masoodi M, Dale M, et al. A new role for lipocalin prostaglandin D synthase in the regulation of brown adipose tissue substrate utilization. Diabetes. (2012) 61:3139–47. doi: 10.2337/db12-0015
71. Campderrós L, Moure R, Cairó M, Gavaldà-Navarro A, Quesada-López T, Cereijo R, et al. Brown adipocytes secrete GDF15 in response to thermogenic activation. Obesity. (2019) 27:1606–16. doi: 10.1002/oby.22584
72. Kotzbeck P, Giordano A, Mondini E, Murano I, Severi I, Venema W, et al. Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J Lipid Res. (2018) 59:784–94. doi: 10.1194/jlr.M079665
73. Müller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, et al. p62 Links β-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. (2013) 123:469–78. doi: 10.1172/JCI64209
74. Wynn TA, Chawla A, Pollard JW. Origins and hallmarks of macrophages: development, homeostasis, and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
75. Hui X, Gu P, Zhang J, Nie T, Pan Y, Wu D, et al. Adiponectin enhances cold-induced browning of subcutaneous adipose tissue via promoting M2 macrophage proliferation. Cell Metab. (2015) 22:279–90. doi: 10.1016/j.cmet.2015.06.004
76. Lv Y, Zhang SY, Liang X, Zhang H, Xu Z, Liu B, et al. Adrenomedullin 2 enhances beiging in white adipose tissue directly in an adipocyte-autonomous manner and indirectly through activation of M2 macrophages. J Biol Chem. (2016) 291:23390–402. doi: 10.1074/jbc.M116.735563
77. Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell. (2014) 157:1279–91. doi: 10.1016/j.cell.2014.03.065
78. Reitman ML. How does fat transition from white to beige? Cell Metab. (2017) 26:14–6. doi: 10.1016/j.cmet.2017.06.011
79. Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism. (2017) 72:120–43. doi: 10.1016/j.metabol.2017.04.005
80. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. (2014) 157:1292–308. doi: 10.1016/j.cell.2014.03.066
81. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol. (2017) 18:519–29. doi: 10.1038/ni.3709
82. Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. (2011) 480:104–8. doi: 10.1038/nature10653
83. Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, Van Der Heide V, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med. (2017) 23:623–30. doi: 10.1038/nm.4316
84. Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. (2017) 550:119–23. doi: 10.1038/nature24022
85. Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. (2017) 23:1309–18. doi: 10.1038/nm.4422
86. Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. (2005) 6:135–42. doi: 10.1038/ni1158
87. Chaldakov GN, Tonchev AB, Tuncel N, Atanassova P, Aloe L. Chapter 12: Adipose tissue and mast cells: adipokines as Yin–Yang modulators of inflammation. In: Giamila Fantuzzi TM, editor. Adipose Tissue and Adipokines in Health and Disease (Nutrition and Health). Totowa, NJ: Humana Press (2007). p. 151–9.
88. Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. (2009) 15:940–5. doi: 10.1038/nm.1994
89. Desautels M, Wollin A, Halvorson I, Muralidhara DV, Thornhill J. Role of mast cell histamine in brown adipose tissue thermogenic response to VMH stimulation. Am J Physiol Regul Integr Comp Physiol. (1994) 266:R831–7. doi: 10.1152/ajpregu.1994.266.3.R831
90. Finlin BS, Zhu B, Confides AL, Westgate PM, Harfmann BD, Dupont-Versteegden EE, et al. Mast cells promote seasonal white adipose beiging in humans. Diabetes. (2017) 66:1237–46. doi: 10.2337/db16-1057
91. Finlin BS, Confides AL, Zhu B, Boulanger MC, Memetimin H, Taylor KW, et al. Adipose tissue mast cells promote human adipose beiging in response to cold. Sci Rep. (2019) 9:1–10. doi: 10.1038/s41598-019-45136-9
92. Zhang X, Wang X, Yin H, Zhang L, Feng A, Zhang QX, et al. Functional inactivation of mast cells enhances subcutaneous adipose tissue browning in mice. Cell Rep. (2019) 28:792–803.e4. doi: 10.1016/j.celrep.2019.06.044
93. Chmelar J, Chatzigeorgiou A, Chung KJ, Prucnal M, Voehringer D, Roers A, et al. No role for mast cells in obesity-related metabolic dysregulation. Front Immunol. (2016) 7:1–8. doi: 10.3389/fimmu.2016.00524
94. Gutierrez DA, Muralidhar S, Feyerabend TB, Herzig S, Rodewald HR. Hematopoietic kit deficiency, rather than lack of mast cells, protects mice from obesity and insulin resistance. Cell Metab. (2015) 21:678–91. doi: 10.1016/j.cmet.2015.04.013
95. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
96. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Fat Treg cells: a liaison between the immune and metabolic systems. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
97. Medrikova D, Sijmonsma TP, Sowodniok K, Richards DM, Delacher M, Sticht C, et al. Brown adipose tissue harbors a distinct sub-population of regulatory T cells. PLoS ONE. (2015) 10:e0118534. doi: 10.1371/journal.pone.0118534
98. Wood IS, Wang B, Trayhurn P. IL-33, a recently identified interleukin-1 gene family member, is expressed in human adipocytes. Biochem Biophys Res Commun. (2009) 384:105–9. doi: 10.1016/j.bbrc.2009.04.081
99. Zeyda M, Wernly B, Demyanets S, Kaun C, Hämmerle M, Hantusch B, et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int J Obes. (2013) 37:658–65. doi: 10.1038/ijo.2012.118
100. Brestoff JR, Artis D. Immune regulation of metabolic homeostasis in health and disease. Cell. (2015) 161:146–60. doi: 10.1016/j.cell.2015.02.022
101. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. (2015) 519:242–6. doi: 10.1038/nature14115
102. Lee M, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. (2015) 160:74–87. doi: 10.1016/j.cell.2014.12.011
103. Ding X, Luo Y, Zhang X, Zheng H, Yang X, Yang X, et al. IL-33-driven ILC2/eosinophil axis in fat is induced by sympathetic tone and suppressed by obesity. J Endocrinol. (2016) 231:35–48. doi: 10.1530/JOE-16-0229
104. Goto T, Naknukool S, Yoshitake R, Hanafusa Y, Tokiwa S, Li Y, et al. Proinflammatory cytokine interleukin-1β suppresses cold-induced thermogenesis in adipocytes. Cytokine. (2016) 77:107–14. doi: 10.1016/j.cyto.2015.11.001
105. Rebiger L, Lenzen S, Mehmeti I. Susceptibility of brown adipocytes to pro-inflammatory cytokine toxicity and reactive oxygen species. Biosci Rep. (2016) 36:1–11. doi: 10.1042/BSR20150193
106. García M, del C, Pazos P, Lima L, Diéguez C. Regulation of energy expenditure and brown/beige thermogenic activity by interleukins: new roles for old actors. Int J Mol Sci. (2018) 19:2569. doi: 10.3390/ijms19092569
107. van den Berg SM, van Dam AD, Rensen PCN, de Winther MPJ, Lutgens E. Immune modulation of brown(ing) adipose tissue in obesity. Endocr Rev. (2017) 38:46–68. doi: 10.1210/er.2016-1066
108. Estève D, Boulet N, Volat F, Zakaroff-Girard A, Ledoux S, Coupaye M, et al. Human white and brite adipogenesis is supported by MSCA1 and is impaired by immune cells. Stem Cells. (2015) 33:1277–91. doi: 10.1002/stem.1916
109. Martins FF, Bargut TCL, Aguila MB, Mandarim-de-Lacerda CA. Thermogenesis, fatty acid synthesis with oxidation, and inflammation in the brown adipose tissue of ob/ob (–/–) mice. Ann Anat. (2017) 210:44–51. doi: 10.1016/j.aanat.2016.11.013
110. Nøhr MK, Bobba N, Richelsen B, Lund S, Pedersen SB. Inflammation downregulates UCP1 expression in brown adipocytes potentially via SIRT1 and DBC1 interaction. Int J Mol Sci. (2017) 18:1006. doi: 10.3390/ijms18051006
111. Okla M, Zaher W, Alfayez M, Chung S. Inhibitory effects of Toll-like receptor 4, NLRP3 inflammasome, and interleukin-1β on white adipocyte browning. Inflammation. (2018) 41:626–42. doi: 10.1007/s10753-017-0718-y
112. Lee SE, Kang SG, Choi MJ, Jung SB, Ryu MJ, Chung HK, et al. Growth differentiation factor 15 mediates systemic glucose regulatory action of T-helper type 2 cytokines. Diabetes. (2017) 66:2774–88. doi: 10.2337/db17-0333
113. Chung K-J, Chatzigeorgiou A, Economopoulou M, Garcia-Martin R, Alexaki VI, Mitroulis I, et al. A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat Immunol. (2017) 18:654–64. doi: 10.1038/ni.3728
114. Sanchez-Infantes D, White UA, Elks CM, Morrison RF, Gimble JM, Considine RV, et al. Oncostatin M is produced in adipose tissue and is regulated in conditions of obesity and type 2 diabetes. J Clin Endocrinol Metab. (2014) 99:1–9. doi: 10.1210/jc.2013-3555
115. Sánchez-Infantes D, Cereijo R, Peyrou M, Piquer-Garcia I, Stephens JM, Villarroya F. Oncostatin m impairs brown adipose tissue thermogenic function and the browning of subcutaneous white adipose tissue. Obesity. (2017) 25:85–93. doi: 10.1002/oby.21679
116. Guo T, Marmol P, Moliner A, Björnholm M, Zhang C, Shokat KM, et al. Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. Elife. (2014) 3:e03245. doi: 10.7554/eLife.03245
117. Andersson O, Korach-Andre M, Reissmann E, Ibáñez CF, Bertolino P. Growth/differentiation factor 3 signals through ALK7 and regulates accumulation of adipose tissue and diet-induced obesity. Proc Natl Acad Sci USA. (2008) 105:7252–6. doi: 10.1073/pnas.0800272105
118. Valladares A, Roncero C, Benito M, Porras A. TNF-α inhibits UCP-1 expression in brown adipocytes via ERKs - opposite effect of p38MAPK. FEBS Lett. (2001) 493:6–11. doi: 10.1016/S0014-5793(01)02264-5
119. Suárez-Zamorano N, Fabbiano S, Chevalier C, Stojanović O, Colin DJ, Stevanović A, et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med. (2015) 21:1497–501. doi: 10.1038/nm.3994
120. Gavaldà-Navarro A, Moreno-Navarrete JM, Quesada-López T, Cairó M, Giralt M, Fernández-Real JM, et al. Lipopolysaccharide-binding protein is a negative regulator of adipose tissue browning in mice and humans. Diabetologia. (2016) 59:2208–18. doi: 10.1007/s00125-016-4028-y
121. Olefsky JM. IKKε: a bridge between obesity and inflammation. Cell. (2009) 138:834–6. doi: 10.1016/j.cell.2009.08.018
122. Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest. (2016) 126:2839–54. doi: 10.1172/JCI86080
123. Chiang S, Bazuine M, Lumeng CN, Geletka LM, White NM, Ma J, et al. The protein kinase IKKε regulates energy expenditure, insulin sensitivity and chronic inflammation in obese mice. Cell. (2010) 138:961–75. doi: 10.1016/j.cell.2009.06.046
124. Shahid M, Javed AA, Chandra D, Ramsey HE, Shah D, Khan MF, et al. IEX-1 deficiency induces browning of white adipose tissue and resists diet-induced obesity. Sci Rep. (2016) 6:1–14. doi: 10.1038/srep24135
125. Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, et al. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. (2015) 349:500–6. doi: 10.1126/science.aaa0079
126. Otoda T, Takamura T, Misu H, Ota T, Murata S, Hayashi H, et al. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes. (2013) 62:811–24. doi: 10.2337/db11-1652
127. Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. (2014) 33:418–36. doi: 10.1002/embj.201385014
128. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. (2014) 20:1427–35. doi: 10.1038/nm.3735
129. Gregor MF, Misch ES, Yang L, Hummasti S, Inouye KE, Lee AH, et al. The role of adipocyte XBP1 in metabolic regulation during lactation. Cell Rep. (2013) 3:1430–9. doi: 10.1016/j.celrep.2013.03.042
130. Bartelt A, Widenmaier SB, Schlein C, Johann K, Goncalves RLS, Eguchi K, et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat Med. (2018) 24:292–303. doi: 10.1038/nm.4481
131. Hou Y, Liu Z, Zuo Z, Gao T, Fu J, Wang H, et al. Adipocyte-specific deficiency of Nfe2l1 disrupts plasticity of white adipose tissues and metabolic homeostasis in mice. Biochem Biophys Res Commun. (2018) 503:264–70. doi: 10.1016/j.bbrc.2018.06.013
132. Lehrke M, Becker A, Greif M, Stark R, Laubender RP, Von Ziegler F, et al. Chemerin is associated with markers of inflammation and components of the metabolic syndrome but does not predict coronary atherosclerosis. Eur J Endocrinol. (2009) 161:339–44. doi: 10.1530/EJE-09-0380
133. Rourke JL, Muruganandan S, Dranse HJ, McMullen NM, Sinal CJ. Gpr1 is an active chemerin receptor influencing glucose homeostasis in obese mice. J Endocrinol. (2014) 222:201–15. doi: 10.1530/JOE-14-0069
134. Mattern A, Zellmann T, Beck-Sickinger AG. Processing, signaling, and physiological function of chemerin. IUBMB Life. (2014) 66:19–26. doi: 10.1002/iub.1242
135. Böhm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. (2007) 76:8–18. doi: 10.1016/j.cardiores.2007.06.004
136. Kowalczyk A, Kleniewska P, Kolodziejczyk M, Skibska B, Goraca A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch Immunol Ther Exp. (2015) 63:41–52. doi: 10.1007/s00005-014-0310-1
137. Eriksson AKS, Van Harmelen V, Stenson BM, Åström G, Wåhlén K, Laurencikiene J, et al. Endothelin-1 stimulates human adipocyte lipolysis through the ET A receptor. Int J Obes. (2009) 33:67–74. doi: 10.1038/ijo.2008.212
138. Shimizu I, Maruyama S, Walsh K, Shimizu I, Aprahamian T, Kikuchi R, et al. Vascular rarefaction mediates whitening of brown fat in obesity Find the latest version: vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest. (2014) 124:2099–112. doi: 10.1172/JCI71643
139. Park J, Kim M, Sun K, An YA, Gu X, Scherer PE. VEGF-A - expressing adipose tissue shows rapid beiging and enhanced survival after transplantation and confers IL-4-independent metabolic improvements. Diabetes. (2017) 66:1479–90. doi: 10.2337/db16-1081
140. Farjo KM, Farjo RA, Halsey S, Moiseyev G, Ma J-x. Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor Kappa B-dependent and retinol-independent mechanism. Mol Cell Biol. (2012) 32:5103–15. doi: 10.1128/MCB.00820-12
141. Kotnik P, Fischer-Posovszky P, Wabitsch M. RBP4: a controversial adipokine. Eur J Endocrinol. (2011) 165:703–11. doi: 10.1530/EJE-11-0431
142. Guo H, Foncea R, O'Byrne SM, Jiang H, Zhang Y, Deis JA, et al. Lipocalin 2, a regulator of retinoid homeostasis and retinoid-mediated thermogenic activation in adipose tissue. J Biol Chem. (2016) 291:11216–29. doi: 10.1074/jbc.M115.711556
143. Hondares E, Rosell M, Gonzalez FJ, Giralt M, Iglesias R, Villarroya F. Hepatic FGF21 expression is induced at birth via PPARα in response to milk intake and contributes to thermogenic activation of neonatal brown fat. Cell Metab. (2010) 11:206–12. doi: 10.1016/j.cmet.2010.02.001
144. Fisher FF, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. (2012) 26:271–81. doi: 10.1101/gad.177857.111
145. Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, et al. iNKT cells induce FGF21 for thermogenesis and are required for maximal weight loss in GLP1 therapy. Cell Metab. (2016) 24:510–9. doi: 10.1016/j.cmet.2016.08.003
146. Giralt M, Gavaldà-Navarro A, Villarroya F. Fibroblast growth factor-21, energy balance and obesity. Mol Cell Endocrinol. (2015) 418:66–73. doi: 10.1016/j.mce.2015.09.018
147. Véniant MM, Sivits G, Helmering J, Komorowski R, Lee J, Fan W, et al. Pharmacologic effects of FGF21 are independent of the “browning” of white adipose tissue. Cell Metab. (2015) 21:731–8. doi: 10.1016/j.cmet.2015.04.019
148. Wang N, Zhao T-T, Li S-M, Sun X, Li Z-C, Li Y-H, et al. Fibroblast growth factor 21 exerts its anti-inflammatory effects on multiple cell types of adipose tissue in obesity. Obesity. (2019) 27:399–408. doi: 10.1002/oby.22376
149. Hara T, Tanegashima K. Pleiotropic functions of the CXC-type chemokine CXCL14 in mammals. J Biochem. (2012) 151:469–76. doi: 10.1093/jb/mvs030
150. Hara T, Nakayama Y. Chapter 5 CXCL14 and insulin action. Vitam Horm. (2009) 80:107–23. doi: 10.1016/S0083-6729(08)00605-5
151. Zhang S, Sun WY, Wu JJ, Wei W. TGF-β signaling pathway as a pharmacological target in liver diseases. Pharmacol Res. (2014) 85:15–22. doi: 10.1016/j.phrs.2014.05.005
152. Eckel J. Skeletal muscle: a novel secretory organ. In: The Cellular Secretome and Organ Crosstalk. London: Elsevier (2018). p. 65–90.
153. Choi SC, Han JK. Chapter Five: negativenegative regulation of activin signal transduction. 1st ed. Vol. 85. In: Vitamins and Hormones. London: Elsevier Inc. (2011). p. 79–104.
154. Seong HA, Manoharan R, Ha H. Smad proteins differentially regulate obesity-induced glucose and lipid abnormalities and inflammation via class-specific control of AMPK-related kinase MPK38/MELK activity. Cell Death Dis. (2018) 9:471. doi: 10.1038/s41419-018-0489-x
155. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. (2011) 14:67–79. doi: 10.1016/j.cmet.2011.04.013
156. Koncarevic A, Kajimura S, Cornwall-Brady M, Andreucci A, Pullen A, Sako D, et al. A novel therapeutic approach to treating obesity through modulation of TGFβ signaling. Endocrinology. (2012) 153:3133–46. doi: 10.1210/en.2012-1016
157. Braga M, Pervin S, Norris K, Bhasin S, Singh R. Inhibition of in vitro and in vivo brown fat differentiation program by myostatin. Obesity. (2013) 21:1180–8. doi: 10.1002/oby.20117
158. Pervin S, Singh V, Tucker A, Collazo J, Singh R. Modulation of transforming growth factor-β/follistatin signaling and white adipose browning: therapeutic implications for obesity related disorders. Horm Mol Biol Clin Investig. (2017) 31:1868–91. doi: 10.1515/hmbci-2017-0036
159. Fournier B, Murray B, Gutzwiller S, Marcaletti S, Marcellin D, Bergling S, et al. Blockade of the activin receptor IIB activates functional brown adipogenesis and thermogenesis by inducing mitochondrial oxidative metabolism. Mol Cell Biol. (2012) 32:2871–9. doi: 10.1128/MCB.06575-11
160. Zhang C, McFarlane C, Lokireddy S, Masuda S, Ge X, Gluckman PD, et al. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia. (2012) 55:183–93. doi: 10.1007/s00125-011-2304-4
161. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int J Obes. (2016) 40:434–42. doi: 10.1038/ijo.2015.200
162. Emmerson PJ, Duffin KL, Chintharlapalli S, Wu X. GDF15 and growth control. Front Physiol. (2018) 9:1712. doi: 10.3389/fphys.2018.01712
163. Macia L, Tsai VWW, Nguyen AD, Johnen H, Kuffner T, Shi YC, et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PLoS ONE. (2012) 7:e34868. doi: 10.1371/journal.pone.0034868
164. Chrysovergis K, Wang X, Kosak J, Lee SH, Kim JS, Foley JF, et al. NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int J Obes. (2014) 38:1555–64. doi: 10.1038/ijo.2014.27
165. Tong M, Jun T, Nie Y, Hao J, Fan D. The role of the Slit/Robo signaling pathway. J Cancer. (2019) 10:2694–705. doi: 10.7150/jca.31877
166. Ishii T. Close teamwork between Nrf2 and peroxiredoxins 1 and 6 for the regulation of prostaglandin D2 and E2 production in macrophages in acute inflammation. Free Radic Biol Med. (2015) 88(Part B):189–98. doi: 10.1016/j.freeradbiomed.2015.04.034
167. Lee M-WM, Lee M-WM, Oh K-J. Adipose tissue-derived signatures for obesity and type 2 diabetes: adipokines, batokines and MicroRNAs. J Clin Med. (2019) 8:854. doi: 10.3390/jcm8060854
168. Tanaka R, Miwa Y, Mou K, Tomikawa M, Eguchi N, Urade Y, et al. Knockout of the l-pgds gene aggravates obesity and atherosclerosis in mice. Biochem Biophys Res Commun. (2009) 378:851–6. doi: 10.1016/j.bbrc.2008.11.152
169. Duchamp C, Burton KA, Géloën A, Dauncey MJ. Transient upregulation of IGF-I gene expression in brown adipose tissue of cold-exposed rats. Am J Physiol. (1997) 272:453–60. doi: 10.1152/ajpendo.1997.272.3.E453
170. Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep. (2017) 19:225–34. doi: 10.1016/j.celrep.2017.03.046
171. Chang HR, Kim HJ, Xu X, Ferrante AW. Macrophage and adipocyte IGF1 maintain adipose tissue homeostasis during metabolic stresses. Obesity. (2016) 24:172–83. doi: 10.1002/oby.21354
172. Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia. (2016) 59:1075–88. doi: 10.1007/s00125-016-3933-4
173. Gunawardana SC, Piston DW. Insulin-independent reversal of type 1 diabetes in nonobese diabetic mice with brown adipose tissue transplant. Am J Physiol. (2015) 308:E1043–55. doi: 10.1152/ajpendo.00570.2014
174. Egecioglu E, Anesten F, Schéle E, Palsdottir V. Interleukin-6 is important for regulation of core body temperature during long-term cold exposure in mice. Biomed Rep. (2018) 9:206–12. doi: 10.3892/br.2018.1118
175. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. (2014) 15:423–30. doi: 10.1038/ni.2865
176. Kristóf E, Klusóczki Á, Veress R, Shaw A, Combi ZS, Varga K, et al. Interleukin-6 released from differentiating human beige adipocytes improves browning. Exp Cell Res. (2019) 377:47–55. doi: 10.1016/j.yexcr.2019.02.015
177. DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA. (2013) 110:5133–8. doi: 10.1073/pnas.1215840110
178. Pal M, Febbraio MA, Whitham M. From cytokine to myokine: the emerging role of interleukin-6 in metabolic regulation. Immunol Cell Biol. (2014) 92:331–9. doi: 10.1038/icb.2014.16
179. Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, et al. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. (2000) 85:3338–42. doi: 10.1210/jcem.85.9.6839
180. Priceman SJ, Kujawski M, Shen S, Cherryholmes GA, Lee H, Zhang C, et al. Regulation of adipose tissue T cell subsets by Stat3 is crucial for diet-induced obesity and insulin resistance. Proc Natl Acad Sci USA. (2013) 110:13079–84. doi: 10.1073/pnas.1311557110
181. Han J, Meng Q, Shen L, Wu G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. (2018) 17:1–8. doi: 10.1186/s12944-018-0657-0
182. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol. (2013) 4:52. doi: 10.3389/fendo.2013.00052
183. Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol. (2013) 4:71. doi: 10.3389/fendo.2013.00071
184. Albert V, Svensson K, Shimobayashi M, Colombi M, Muñoz S, Jimenez V, et al. mTORC 2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol Med. (2016) 8:232–46. doi: 10.15252/emmm.201505610
185. Martinez N, Cheng CY, Ketheesan N, Cullen A, Tang Y, Lum J, et al. mTORC2/Akt activation in adipocytes is required for adipose tissue inflammation in tuberculosis. EBioMedicine. (2019) 45:314–27. doi: 10.1016/j.ebiom.2019.06.052
186. Vazquez-Prieto MA, Bettaieb A, Haj FG, Fraga CG, Oteiza PI. (–)-Epicatechin prevents TNFα-induced activation of signaling cascades involved in inflammation and insulin sensitivity in 3T3-L1 adipocytes. Arch Biochem Biophys. (2012) 527:113–8. doi: 10.1016/j.abb.2012.02.019
187. Chen C, Zhang W, Shi H, Zhuo Y, Yang G, Zhang A, et al. A novel benzenediamine derivative FC98 reduces insulin resistance in high fat diet-induced obese mice by suppression of metaflammation. Eur J Pharmacol. (2015) 761:298–308. doi: 10.1016/j.ejphar.2015.06.021
188. Li J, An R, Liu S, Xu H. Deregulation of PP2A-Akt interaction contributes to sucrose non-fermenting related kinase (SNRK) deficiency induced insulin resistance in adipose tissue (P21-071-19). Curr Dev Nutr. (2019) 3(Supplement_1):26–35. doi: 10.1093/cdn/nzz041.P21-071-19
189. Li J, Feng B, Nie Y, Jiao P, Lin X, Huang M, et al. Sucrose nonfermenting-related kinase regulates both adipose inflammation and energy homeostasis in mice and humans. Diabetes. (2018) 67:400–11. doi: 10.2337/db17-0745
190. Putti R, Sica R, Migliaccio V, Lionetti L. Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol. (2015) 6:109. doi: 10.3389/fphys.2015.00109
191. Zorzano A, Liesa M, Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. (2009) 41:1846–54. doi: 10.1016/j.biocel.2009.02.004
192. Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes. (2015) 64:3135–45. doi: 10.2337/db14-1937
193. Keuper M, Sachs S, Walheim E, Berti L, Raedle B, Tews D, et al. Activated macrophages control human adipocyte mitochondrial bioenergetics via secreted factors. Mol Metab. (2017) 6:1226–39. doi: 10.1016/j.molmet.2017.07.008
194. Hahn WS, Kuzmicic J, Burrill JS, Donoghue MA, Foncea R, Jensen MD, et al. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am J Physiol. (2014) 306:1033–45. doi: 10.1152/ajpendo.00422.2013
195. Woo CY, Jang JE, Lee SE, Koh EH, Lee KU. Mitochondrial dysfunction in adipocytes as a primary cause of adipose tissue inflammation. Diabetes Metab J. (2019) 43:247–56. doi: 10.4093/dmj.2018.0221
196. Gurung P, Lukens JR, Kanneganti T-D. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. (2015) 21:193–201. doi: 10.1016/j.molmed.2014.11.008
197. de-Lima-Júnior JC, Souza GF, Moura-Assis A, Gaspar RS, Gaspar JM, Rocha AL, et al. Abnormal brown adipose tissue mitochondrial structure and function in IL10 deficiency. EBioMedicine. (2019) 39:436–47. doi: 10.1016/j.ebiom.2018.11.041
198. Frohnert BI, Bernlohr DA. Protein carbonylation, mitochondrial dysfunction, and insulin resistance. Adv Nutr. (2013) 4:157–63. doi: 10.3945/an.112.003319
199. Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. (2010) 31:364–95. doi: 10.1210/er.2009-0027
200. Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González Á, Esquivel-Chirino C, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci. (2011) 12:3117–32. doi: 10.3390/ijms12053117
201. Bondia-Pons I, Ryan L, Martinez JA. Oxidative stress and inflammation interactions in human obesity. J Physiol Biochem. (2012) 68:701–11. doi: 10.1007/s13105-012-0154-2
202. Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract. (2013) 7:e330–41. doi: 10.1016/j.orcp.2013.05.004
203. Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. (2016) 532:112–6. doi: 10.1038/nature17399
204. Vernochet C, Damilano F, Mourier A, Bezy O, Mori MA, Smyth G, et al. Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J. (2014) 28:4408–19. doi: 10.1096/fj.14-253971
205. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science. (2001) 293:1673–7. doi: 10.1126/science.1061620
206. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. (2007) 356:1517–26. doi: 10.1056/NEJMoa065213
207. Sanchez-Zamora Y, Terrazas LI, Vilches-Flores A, Leal E, Juárez I, Whitacre C, et al. Macrophage migration inhibitory factor is a therapeutic target in treatment of non-insulin-dependent diabetes mellitus. FASEB J. (2010) 24:2583–90. doi: 10.1096/fj.09-147066
208. Brendle C, Stefan N, Stef I, Ripkens S, Soekler M, la Fougère C, et al. Impact of diverse chemotherapeutic agents and external factors on activation of brown adipose tissue in a large patient collective. Sci Rep. (2019) 9:1–8. doi: 10.1038/s41598-018-37924-6
209. Song Z, Revelo X, Shao W, Tian L, Zeng K, Lei H, et al. Dietary curcumin intervention targets mouse white adipose tissue inflammation and brown adipose tissue UCP1 expression. Obesity. (2018) 26:547–58. doi: 10.1002/oby.22110
210. Talukdar S, Olefsky JM, Osborn O. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci. (2011) 32:543–50. doi: 10.1016/j.tips.2011.04.004
211. Hirasawa A, Hara T, Katsuma S, Adachi T, Tsujimoto G. Free fatty acid receptors and drug discovery. Biol Pharm Bull. (2008) 31:1847–51. doi: 10.1248/bpb.31.1847
212. Yonezawa T, Kurata R, Yoshida K, Murayama MA, Cui X, Hasegawa A. Free fatty acids-sensing G protein-coupled receptors in drug targeting and therapeutics. Curr Med Chem. (2013) 20:3855–71. doi: 10.2174/09298673113209990168
213. Schilperoort M, Dam AD, Hoeke G, Shabalina IG, Okolo A, Hanyaloglu AC, et al. The GPR 120 agonist TUG−891 promotes metabolic health by stimulating mitochondrial respiration in brown fat. EMBO Mol Med. (2018) 10:1–18. doi: 10.15252/emmm.201708047
214. Caiazzo R, Van Hul W, Van Gaal L, Horber F, Balkau B, Pigeyre M, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. (2012) 483:350–4. doi: 10.1038/nature10798
215. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
216. Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med. (2014) 20:942–7. doi: 10.1038/nm.3614
217. Quesada-López T, Cereijo R, Turatsinze J, Planavila A, Cairó M, Gavaldà-Navarro A, et al. The lipid sensor GPR120 promotes brown fat activation and FGF21 release from adipocytes. Nat Commun. (2016) 7:13479. doi: 10.1038/ncomms13479
Keywords: inflammation, brown adipose tissue (BAT), cytokine, beige adipocyte, white adipose tissue
Citation: Omran F and Christian M (2020) Inflammatory Signaling and Brown Fat Activity. Front. Endocrinol. 11:156. doi: 10.3389/fendo.2020.00156
Received: 05 February 2020; Accepted: 06 March 2020;
Published: 24 March 2020.
Edited by:
Rosalia Rodriguez-Rodriguez, Universitat Internacional de Catalunya, SpainReviewed by:
Alexander Bartelt, Ludwig Maximilian University of Munich, GermanyMarta Giralt, University of Barcelona, Spain
Copyright © 2020 Omran and Christian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Christian, bWFyay5jaHJpc3RpYW5AbnR1LmFjLnVr