 Masahiro Maruyama1
Masahiro Maruyama1 Claire Rhee1Takeshi Utsunomiya1Ning Zhang1Masaya Ueno1
Claire Rhee1Takeshi Utsunomiya1Ning Zhang1Masaya Ueno1 Zhenyu Yao1
Zhenyu Yao1 Stuart B. Goodman1,2*
Stuart B. Goodman1,2*- 1Department of Orthopaedic Surgery, Stanford University, Stanford, CA, United States
- 2Department of Bioengineering, Stanford University, Stanford, CA, United States
The optimal treatment for complex fractures and large bone defects is an important unsolved issue in orthopedics and related specialties. Approximately 5–10% of fractures fail to heal and develop non-unions. Bone healing can be characterized by three partially overlapping phases: the inflammatory phase, the repair phase, and the remodeling phase. Eventual healing is highly dependent on the initial inflammatory phase, which is affected by both the local and systemic responses to the injurious stimulus. Furthermore, immune cells and mesenchymal stromal cells (MSCs) participate in critical inter-cellular communication or crosstalk to modulate bone healing. Deficiencies in this inter-cellular exchange, inhibition of the natural processes of acute inflammation, and its resolution, or chronic inflammation due to a persistent adverse stimulus can lead to impaired fracture healing. Thus, an initial and optimal transient stage of acute inflammation is one of the key factors for successful, robust bone healing. Recent studies demonstrated the therapeutic potential of immunomodulation for bone healing by the preconditioning of MSCs to empower their immunosuppressive properties. Preconditioned MSCs (also known as “primed/ licensed/ activated” MSCs) are cultured first with pro-inflammatory cytokines (e.g., TNFα and IL17A) or exposed to hypoxic conditions to mimic the inflammatory environment prior to their intended application. Another approach of immunomodulation for bone healing is the resolution of inflammation with anti-inflammatory cytokines such as IL4, IL10, and IL13. In this review, we summarize the principles of inflammation and bone healing and provide an update on cellular interactions and immunomodulation for optimal bone healing.
Introduction
The optimal treatment for complex fractures and large bone defects is an important unsolved issue in orthopedics and related specialties. In the United States alone, there are ~8 million bone fractures annually; 5–10% of fractures fail to heal and develop a non-union (1). The average cost for treatment of a bone non-union is estimated to be >US$ 10,000 (2).
Bone healing after injury is a complex biological and biomechanical process. Bone healing can be characterized by three partially overlapping phases: the inflammatory phase, the repair phase, and the remodeling phase (3). Eventual healing is highly dependent on the initial inflammatory phase, which is affected by both the local and systemic responses to the injurious stimulus. Furthermore, immune cells and mesenchymal stromal cells (MSCs) participate in critical inter-cellular communication or crosstalk to modulate bone healing. Thus, understanding and regulating inflammation is one of the key factors for successful, robust bone healing.
This review will summarize the principles of inflammation and bone healing, and provide an update on cellular interactions and immunomodulation for optimal bone healing.
What is Inflammation?
Inflammation is the protective response of tissue to a noxious stimulus, leading to both the removal of harmful stimuli, and the initiation of the healing process (4, 5). Acute inflammation is marked by capillary dilatation and leukocytic migration and infiltration to the local area; this leads to the clinical symptoms of redness, heat, pain, and loss of function (6).
Acute Inflammation
Acute inflammation is initiated by endogenous or exogenous adverse stimuli (4, 6). The acute inflammatory response after injury peaks within the first 24–48 h and is generally complete after 7 days (7, 8). In acute inflammation, tissue-resident cells including tissue macrophages, dendritic cells, lymphocytes, endothelial cells, fibroblasts, and mast cells recognize invading pathogens, or tissue injury byproducts, and release a variety of pro-inflammatory mediators including cytokines, chemokines, and growth factors; this results in the infiltration of polymorphonuclear neutrophils (PMNs), monocytes/macrophages, and lymphocytes into the injured site. PMNs phagocytose and eliminate invading pathogens and tissue debris. Macrophages are polarized to the M1 phenotype by damage-associated molecular patterns (DAMPs, e.g., apoptotic cells and their byproducts), pathogen-associated molecular patterns (PAMPs, e.g., bacterial endotoxin, lipopolysaccharide [LPS]), and pro-inflammatory cytokines (e.g., interferon γ [INFγ], tumor necrosis factor α [TNFα], interleukin 1β [IL1β]) (6, 9). During acute inflammation, M1 macrophages contribute to host defense as well as amplify the inflammatory reaction and recruit additional immune cells (10). M1 macrophages phagocytose and remove micro-organisms, necrotic tissue, and the provisional fibrin matrix (5, 6, 10). In addition, M1 macrophages secrete pro-inflammatory and chemotactic mediators, such as TNFα, IL1β, IL6, and C-C motif chemokine 2 (CCL2) (known as monocyte chemotactic protein 1 [MCP1]) (5, 6). These mediators initiate further recruitment of inflammatory cells and MSCs (5, 6).
The Resolution of Inflammation
The resolution of inflammation was traditionally characterized as a passive process (11, 12). However, more recently, the resolution of inflammation is thought to be an active process, regulated by various mediators and immune cells (13).
The resolution process begins a few hours after the acute inflammatory response is initiated. First, the initial inflammatory stimuli are eliminated; subsequently, pro-inflammatory mediators are suspended, whereas anti-inflammatory (pro-resolving) mediators, such as IL4, IL10, and IL13, and CCL2, are promoted. PMNs cease to infiltrate the injury site and undergo apoptosis and subsequent efferocytosis by macrophages (14). Efferocytosis is the removal of apoptotic cells that are swiftly engulfed and digested by macrophages. Macrophages are polarized from a pro-inflammatory M1 phenotype to an anti-inflammatory tissue-repair M2 phenotype by anti-inflammatory cytokines, resulting in the advancement of the healing processes (9, 15).
Recently it has been proposed that the resolution of acute inflammation may not terminate the local immune response. Subsequent immunological activity after the resolution of the acute inflammatory stage is termed “the post-resolution stage,” to obtain a state of “adapted (or adaptive) homeostasis” at the injury site (13, 16). Adapted homeostasis alters the innate immune environment of tissues after the resolution of inflammation, including modifying the biochemical, phenotypic, and functional aspects of affected cells (13). This new adapted homeostatic environment may lead to new set-points and ranges of immune response, and is important for the maintenance of immune tolerance. This is an area of ongoing active research.
Chronic Inflammation
Chronic inflammation is a state in which acute inflammation, fibrosis, and repair occur simultaneously (17). In chronic inflammation, monocytes/macrophages, lymphocytes, fibroblasts, and other cells are present at the injury site (17, 18). Examples of chronic inflammation include periprosthetic osteolysis due to wear particles after total joint arthroplasty, and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus (SLE) (9, 17). The failure of the resolution stage may lead to chronic inflammation, which may persist for prolonged periods of several weeks, or in some cases, months to years (9, 17). In chronic inflammation, adaptive homeostasis is not achieved. Continued production of pro-inflammatory cytokines such as IFNγ and TNFα continues to polarize macrophages to a pro-inflammatory M1 phenotype rather than an anti-inflammatory tissue-repair M2 phenotype. Therefore, chronic inflammation fails to establish an adaptive homeostatic state (9).
WHAT is Bone Healing?
Bone healing is an intricate regenerative process which can be classified into primary (direct) and secondary (indirect) bone healing (3, 19). Primary bone healing is uncommon in the process of fracture healing and occurs under extremely rigid fixation without any displacement of bony fragments. The fracture site is bridged by Haversian systems (or osteons), similar to the normal bone remodeling. Bone healing by Haversian systems has little or no inflammatory response and is a slow healing process that takes from a few months to a few years to achieve complete healing (3, 19). On the other hand, secondary bone healing is the most common form of fracture healing. Generally, the process of secondary bone healing is characterized by three partially overlapping phases: the inflammatory phase, the repair phase, and the remodeling phase (3) (Figure 1). Eventual healing is highly dependent on the initial inflammatory phase. Furthermore, immune cells and bone marrow-derived MSCs (BM-MSCs) participate in critical inter-cellular communication or crosstalk to modulate bone healing.
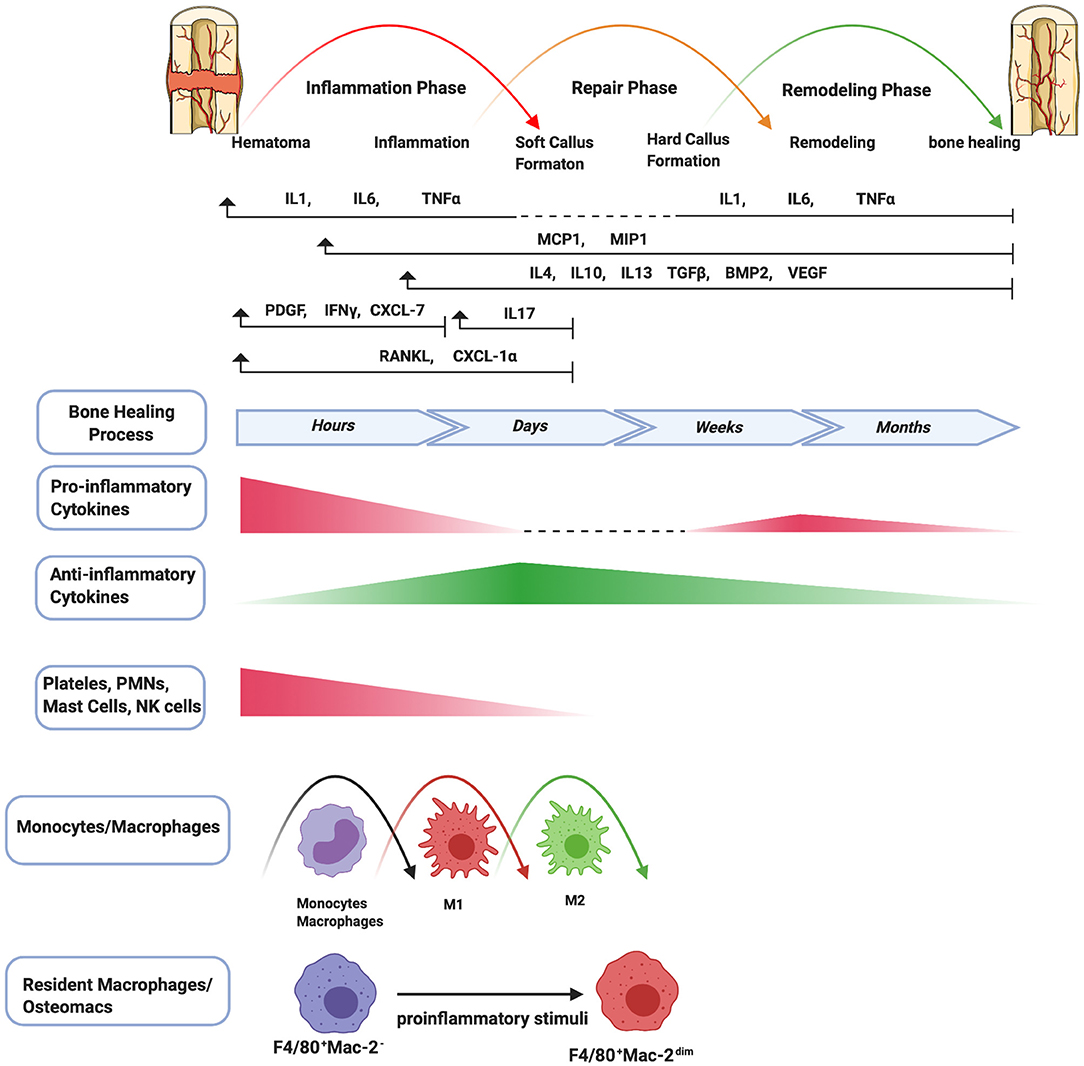
Figure 1. Schematic summary of the stages of bone healing and the temporal pattern of the relative immune cells and cytokines/growth factors expression. Bone healing can be viewed as a three-stage biological phase (inflammation, repair, and remodeling) which can be further divided into six main sub-steps: hematoma, inflammation, soft callus formation, hard callus formation, remodeling, bone healing. After fracture, immune cells including PMNs, NK cells, mast cells, and platelets (platelets are not truly cells as they have no nuclei) are activated in the early stage of the inflammation and the secreted cytokines/chemokines subsequently recruit and activate monocytes/macrophages to further play important roles throughout this process. The pro-inflammatory cytokines including IL1, IL6, TNFα are essential signals during the early stages of bone fracture. In addition, TNFα increases again in the late repair phase, and several pro-inflammatory cytokines (e.g., IL1, IL6, TNFα) are highly expressed in the remodeling phase. The control switch of expression patterns from a pro-inflammatory to an anti-inflammatory response (IL4, IL10, IL13) in the late stages of inflammation is critical to fracture repair.
Inflammatory Phase
An inflammatory response occurs immediately following bone fracture. The trauma leads to blood vessel rupture inside and surrounding the fracture site, resulting in a hematoma. The hematoma micro-environment is initially characterized by local hypoxia, acidity, and lower temperature, and is rich in calcium and lactic acid (20). The hematoma works as a scaffold for recruited inflammatory cells and a variety of cytokines, including IL1, IL6, TNFα, CCL2, and others, to initiate the inflammatory cascade (20). First, PMNs are recruited and then monocytes/macrophages infiltrate into the fracture site (21). Macrophages are polarized to the M1 phenotype. After infiltration of macrophages, the immune response shifts toward adaptive immunity, reflected by the invasion of lymphocytes into the fracture zone. PMNs and macrophages clear the area of dead cells and debris, and the process transforms to the resolution of inflammation, which is a complex and well-regulated activity. In this process, the agents initiating the inflammatory response and the synthesis of pro-inflammatory mediators are reduced, and the immune cells are gradually cleared from the tissue (9). Osteomacs, a special subtype of macrophages residing in bone tissues, are distributed among bone lining cells within both endosteum and periosteum and contribute bone homeostasis. Osteomacs not only sense the original injurious stimulus and initiate the inflammatory cascade, but also provide a source of molecules that begins the essential cellular events for bone healing (22, 23).
During the resolution of acute inflammation, macrophages are polarized from an M1 phenotype to an M2 phenotype by anti-inflammatory cytokines such as IL4, IL10, and IL13. BM-MSCs are attracted locally by cytokines such as TNFα (24) and stromal cell-derived factor 1 (SDF1) (known as chemokine C-X-C motif chemokine ligand 12 [CXCL12]). Recruited inflammatory cells and BM-MSCs participate in critical inter-cellular communication or crosstalk via pro-inflammatory cytokines, anti-inflammatory cytokines, as well as transforming growth factor β (TGFβ), bone morphogenetic proteins (BMPs), and growth factors (e.g., vascular endothelial growth factor [VEGF], platelet-derived growth factor [PDGF] and fibroblast growth factor-2 [FGF-2]) to initiate osteogenesis and angiogenesis (25). This process could also create a reparative granuloma forming a template for the following formation of callus (26). The acute inflammatory response peaks within 24–48 h and disappears at about 1-week post-fracture.
Repair Phase
During the repair phase, callus is formed as vascular buds grow into the area, and the collagen matrix is laid down. Callus formation has two types of processes: intramembranous ossification and endochondral ossification (27). Intramembranous ossification occurs at the periosteum and forms hard callus directly. Periosteal MSCs differentiate into osteoprogenitor cells, which subsequently proliferate and differentiate into osteoblasts that directly form woven bone. Endochondral ossification occurs at the endosteum and bone marrow, and forms soft callus and then hard callus. BM-MSCs differentiate into chondrocytes and secrete a cartilage matrix which forms a cartilaginous template. Chondrocytes subsequently undergo hypertrophic differentiation and mineralize the surrounding matrix to form cartilaginous callus. Finally, hypertrophic chondrocytes undergo apoptosis, resulting in vascular invasion and migration of osteoblasts. Cartilage matrix subsequently coverts to bone matrix (27). Pro-inflammatory cytokines including IL1 and IL6 are absent during this phase (19). TNFα is also diminished in the early repair phase but increases in the late repair phase (24). In the endochondral ossification process, TNFα facilitates chondrocyte apoptosis, resorption of mineralized cartilage, and vascularization (24, 28). Lower levels of TNFα also enhance osteoblast proliferation but high levels inhibit these processes (24). In addition, osteomacs are enriched at sites of bone formation, forming a canopy-like structure over sites of active cuboidal osteoblasts. Osteomacs are associated with maturing of bone tissues in the repair and remodeling phases (22, 23). Alternatively, osteoclasts are highly differentiated multinucleated cells that degrade bone.
Remodeling Phase
At the late stage of bone healing, bone is restored to its original structure, shape, and mechanical properties by remodeling. The balance between osteoblastic and osteoclastic activity which results in lamellar bone deposition and bone resorption plays an important role during the remodeling stage (19, 27). Several pro-inflammatory cytokines (e.g., IL1, IL6, and TNFα) are highly expressed (3, 19, 26).
Is Inflammation Necessary to Obtain Bone Healing?
Inflammation is the crucial first step for bone healing as described above. However, deficiency and inhibition of acute inflammation can lead to impaired bone healing. For example, it is well-known that drugs such as non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, chemotherapeutic agents and others increase the risk for non-union (29, 30). In addition, excess acute inflammation due to severe injuries such as polytrauma, and open fracture also increases the risk for impaired bone healing (31). Furthermore, chronic inflammation is detrimental to bone healing (31).
Deficiency and Inhibition of Acute Inflammation
NOD/SCID-IL2Rγcnull mice are innate and adaptive immunodeficient mice that lack functional monocytes, dendritic cells, natural killer cells, and lymphocytes (32). In addition, there is a defect in the common gamma-chain of the IL2 receptor (γc) such that signaling of IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 is defective (33). The fracture healing process in NOD/SCID-IL2Rγcnull mice demonstrates a callus whose bone content is unaffected during the early healing stage; however, the callus is reduced during the late healing phase and the amount of cartilage is significantly increased, indicating delayed endochondral ossification (34).
The pro-inflammatory cytokine TNFα has a complex role during bone healing, demonstrating biphasic peaks at 72 h and 3 weeks after injury (35). TNFα-receptor-deficient mice showed delayed endochondral and intramembranous bone formation (24, 36, 37). Furthermore, TNFα knockout mice demonstrated poor endochondral bone repair after fracture, but had normal skeletogenesis, implicating TNFα signaling specifically in early fracture repair (38). IL6 has been shown to increase osteoclastogenesis (39). IL6 knockout mice demonstrated delayed fracture healing, with decreased osteoclastogenesis and impaired callus formation, but showed comparable bone healing and strength in the late stage of fracture healing (40, 41). The IL6 signal has two different pathways: the classic signaling pathway via the membrane-bound receptor (mLI-6R) and the trans-signaling pathway via its soluble form (sIL6R) (42). Recent studies using a fracture model in rats demonstrated that the classic signaling pathway regulates the immune response and bone healing, whereas the trans-signaling pathway has a minor effect on the immune response and does not influence bone healing (43); rather it has negative effects on bone healing after severe trauma (44). IL17A, secreted mainly by γδ T cells, has also been characterized as an inflammatory cytokine (45). IL17A expression is induced in the early phase of bone healing and stimulates the proliferation and osteoblastic differentiation of BM-MSCs (45–47). IL17A knockout mice exhibited delayed callus formation and lower bone mineral density due to a decrease in osteoblastic bone formation (45). CCL2 is an important chemokine expressed early in inflammatory conditions. CCL2 deficient mice showed diminished infiltration of macrophages and BM-MSCs, and impaired vascularization, resulting in delayed fracture healing, and less callus formation (48). Thus, pro-inflammatory cytokines and chemokines are critical to bone healing. Conversely, although IL1β promotes the proliferation and differentiation of murine pre-osteoblasts in vitro, IL1β receptor knockout mice had normal bone healing (49).
Prostaglandin E2 (PGE2) plays a key role in bone metabolism including homeostasis, inflammation, and healing (50, 51). Cyclooxygenase 2 (COX-2) is a key enzyme important to PGE2 synthesis. COX-2 knockout mice exhibited reduced osteoblastogenesis and impaired intramembranous and endochondral bone healing (52). NSAIDs have analgesic, antipyretic, and anti-inflammatory effects, and are frequently used to treat musculoskeletal conditions. Continuous use of NSAIDs is associated with delayed bone healing and non-union via inhibition of the COX2-PGE2 pathway (53, 54). In our previous study, co-culture of murine BM-MSCs with undifferentiated M0, pro-inflammatory M1, or anti-inflammatory M2 macrophages showed that celecoxib, a COX-2 selective NSAID, reduced bone mineralization in all co-cultures but most dramatically in the BM-MSC-M1 co-cultures (55). This study re-emphasized the importance of an initial transient acute inflammatory period during fracture healing.
Excess of Inflammation vs. Bone Healing
Excessive acute inflammation can be caused by several stimuli including microbial infection, surgical intervention, and injury due to mechanical, chemical, electrical, or thermal trauma (56), leading to the excessive production of pro-inflammatory cytokines (56, 57). TNFα stimulates osteoclastogenesis and inhibits osteoblast function (58). TNFα overexpression increases NFκB and the mitogen-activated protein kinases (MAPKs), increasing the release of cytokines and chemokines and leading to the activation of osteoclasts (58). In addition, TNFα inhibits BM-MSC differentiation into osteoblasts via the ubiquitin E3 ligase Wwp1 (59, 60). In polytrauma, excessive infiltration of activated neutrophils to the fracture site leads to poor fracture healing (21). Coculture of human BM-MSC with CD4+ T cells promoted increased expression of bone markers and mineralization, however, coculture of human BM-MSC with CD8+ T cells did not (61). Patients with impaired fracture healing had a lower CD4+/CD8+ ratio compared to patients with healed fractures (62). These data indicated that CD4+ T cells promoted osteogenic differentiation of human BM-MSCs. On the other hand, in patients with severe trauma, the CD4+/CD8+ ratio has been reported to be decreased (63). A rat model of tibial fracture with severe overlying muscle injury demonstrated not only excessive and prolonged infiltration of T cells but also a low CD4+/CD8+ ratio at 14 and 28 days, compared to a rat model of tibial fracture only. Furthermore, this rat model of tibial fracture with muscle injury showed persistence of M1 macrophages, resulting in impaired bone healing (64). Thus, excessive and/or prolonged acute inflammation alters the balance of inflammatory cells and inflammatory cytokines, which may lead to poor bone healing.
Chronic Inflammation vs. Bone Healing
Chronic inflammation can be highly detrimental to bone healing. In chronic inflammation, TNFα and the NFκB signaling pathways are continuously upregulated, resulting in the differentiation and activation of osteoclasts (6, 65–68). High and persistent TNFα levels damage tissues and reduce bone volume (58). In the context of high TNFα levels, the eroded bone surface, which is a parameter of bone resorption, was significantly increased histomorphometrically in patients with inflammatory diseases, such as rheumatoid arthritis, Crohn's disease, and bronchial asthma (69). Chronically elevated NFκB activity was associated with the impaired ability of BM-MSCs to form bone (70). Chronic inflammation due to polyethylene particles induced the activation of NFκB pathways, leading to bone resorption in both the femoral intramedullary continuous polyethylene particle infusion model (71) and in the murine calvarial model (72). In addition, prolonged M1-macrophage activation continuously produced cytokines, resulting in bone resorption via increased osteoclast activity and suppression of bone formation by osteoblasts (5). Therefore, failed bone healing in chronic inflammation can be caused by the imbalance of M1/M2 macrophages (5).
What Happens if Bone Healing Does Not Occur?
According to the U.S. Food and Drug Administration (FDA), non-union is defined as a fractured bone that has not healed after a minimum of 9 months since the injury and shows no radiographic progressive bone healing for a minimum of 3 consecutive months (73). Although different definitions have been proposed, non-unions can be radiologically categorized as hypertrophic and atrophic non-unions (74).
Hypertrophic Non-union
On radiographs, a hypertrophic non-union is seen as a large broad callus facing the radiolucent fracture gap and is referred to as a horse-shoe or elephant-foot non-union (75). Hypertrophic non-union is believed to be caused by inadequate immobilization to maintain the necessary vascularity and biologic viability for complete bone healing to occur (76, 77). However, a previous in vitro study (78) demonstrated that cell viability and osteogenic ability of human osteoblasts isolated from hypertrophic non-unions were decreased. Iwakura et al. (79) showed that hypertrophic non-union tissue contained mesenchymal progenitor cells with multilineage capacity, but their proliferative capacity was decreased. Thus, the biological mechanisms and immunoregulatory controls in hypertrophic non-union remain largely unknown.
Atrophic Non-union
Atrophic non-union refers to an inadequate or poorly vascularized mesenchymal tissue with very limited potential for successful healing of the bone ends. Radiographically, there is little callus formation around a fibrous tissue-filled fracture gap (75). An in vitro study demonstrated that the fibroblast-like cells isolated from atrophic non-union tissues in humans had lower cell proliferation and osteogenic ability (80). El-Jawhari et al. (81) showed that BM-MSCs isolated from the iliac crest in patients with a non-union had low proliferative capacity and osteogenic ability compared with those with successful union. In addition, the passage-zero cultured BM-MSCs treated with a mixture of IFNγ, TNFα, IL1, and IL17 showed lower gene expression of indoleamine 2,3-dioxygenase (IDO), prostaglandin E synthetase 2 (PGES2), and TGFβ1, indicating reduced immunosuppressive potential. The immunoregulatory properties of cells in atrophic non-unions have not been fully clarified.
Can we Modulate Inflammation to Facilitate Healing?
As stated previously, an initial and optimal transient stage of acute inflammation is a crucial event during fracture healing. One approach to facilitating bone healing by means of immunomodulation is the preconditioning of MSCs to empower their immunosuppressive properties (82). Preconditioned MSCs (also known as “primed/ licensed/ activated” MSCs) are cultured first with pro-inflammatory cytokines or exposed to hypoxic conditions for a few days to mimic the inflammatory microenvironment prior to their intended application in in vitro and in vivo studies in animals and humans (e.g., differentiation into special cell lineages, loading into scaffolds, and administration to animals and humans). Another approach involves the resolution of inflammation with anti-inflammatory cytokines.
Preconditioned MSC With Pro-Inflammatory Cytokines
Addition of different combinations of inflammatory cytokines to cell culture can dramatically affect the secretory profile and osteogenic ability of MSCs (Figure 2, Table 1). IFNγ-preconditioned MSCs upregulated IDO and the secretion of immunomodulatory molecules, such as PGE2, hepatocyte growth factor (HGF), TGFβ, and CCL2 (82, 89). IFNγ-preconditioned MSCs also suppressed CD4+ and CD 8+ T cell and NK cell proliferation and polarized macrophages to an M2 phenotype (87, 90). However, there are no studies concerning the osteogenic ability of IFNγ-preconditioned MSCs.
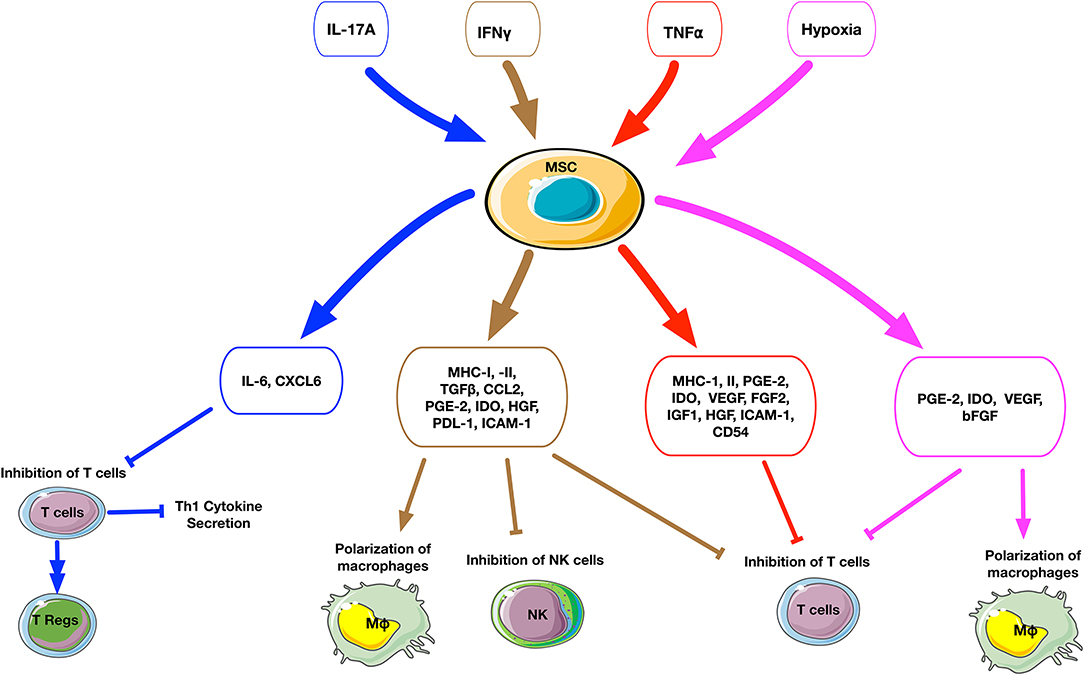
Figure 2. Schematic summary of the cellular and molecular effects after preconditioning of MSCs. The upper small rectangle boxes depict the different stimuli used to precondition MSCs. The middle level boxes indicate the upregulation of cytokines and chemokines by MSCs after stimulation. The effects of preconditioned MSCs on a cellular level are represented on the bottom of the figure. The stimuli factors and their respectively triggered outputs are linked by matched color arrows and boxes. IL17A-preconditioned MSCs increase IL6 secretion and regulatory T cell generation and inhibit Th1 cytokine secretion such as TNFα, IFNγ, IL2, and IL10. IFNγ-preconditioned MSCs promote the secretion of immunomodulatory molecules such as IDO, PGE2, HGF, TGFβ, and CCL2, suppress T cell and NK cell proliferation and polarize macrophages to an M2 phenotype. TNFα-preconditioned MSCs promote the secretion of immunoregulatory mediators such as PGE2, IDO, and HGF, suppress T cell proliferation. Hypoxia-preconditioned MSCs promote the secretion of PGE2, IDO, VEGF, and bFGF, suppress T cell proliferation, and polarize macrophages to an M2 phenotype.
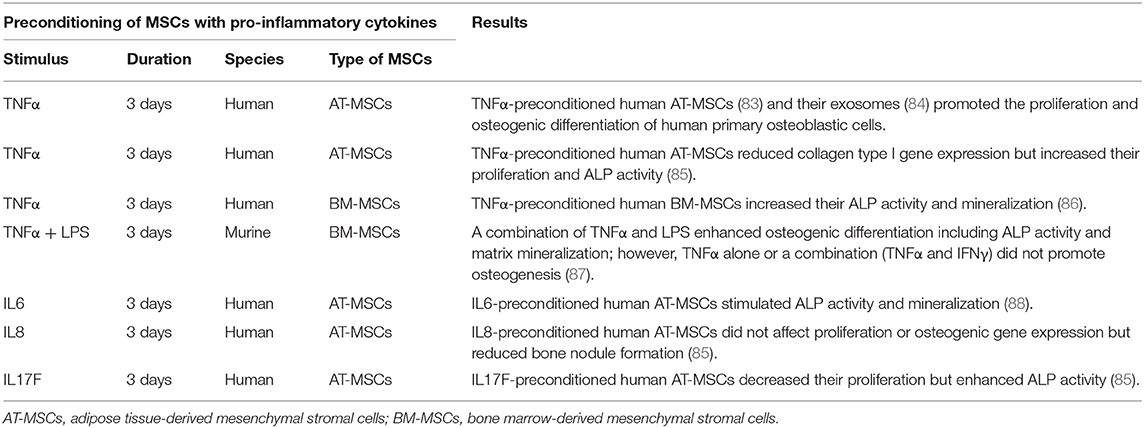
Table 1. Immunomodulation for bone healing by the preconditioning of MSCs with pro-inflammatory cytokines in vitro.
TNFα preconditioning also promotes immunoregulatory mediators such as PGE2, IDO, and HGF, but this is less pronounced compared to IFNγ preconditioning (82, 89). TNFα-preconditioned human adipose tissue-derived MSCs (AT-MSCs) (83) and their exosomes (84) promoted the proliferation and osteogenic differentiation of human primary osteoblastic cells. Another study demonstrated that TNFα-preconditioned human AT-MSCs reduced collagen type I gene expression but increased proliferation and ALP activity (85). Similarly, preconditioned human BM-MSCs exposed to TNFα enhanced their osteogenic capacity (86). However, our previous in vitro study demonstrated that preconditioning of murine BM-MSCs using TNFα alone or a combination TNFα and IFNγ did not promote osteogenesis; however, a combination of TNFα and LPS enhanced osteogenic differentiation including ALP activity and matrix mineralization (87).
IL17A-preconditioned MSCs increased IL6 and regulatory T cell generation and inhibited Th1 cytokine secretion (TNFα, IFNγ, IL2, and IL10) (91). In a rat calvarial defect model, the direct application of IL17A inhibited osteoblast precursor cells and bone regeneration (92). In other studies, IL17A promoted osteoblastic differentiation (46, 93, 94) and inhibited adipogenic differentiation (95) in human BM-MSCs. Ono et al. demonstrated that IL17A accelerated osteoblastogenesis in vitro and in vivo in mice (45). A recent study demonstrated that IL17 stimulated the osteogenic differentiation of the murine BM-MSC niche by using IL6 and IL1β signaling to activate ERK1/2, STAT3, and AKT (47). When these BM-MSCs were exposed to IL17 in 3D coculture with osteocytes, the BM-MSCs showed enhanced osteogenesis. However, studies concerning the osteogenic ability of IL17A-preconditioned MSCs were not reported.
The effects of preconditioning of MSCs by other pro-inflammatory cytokines such as IL6, IL8, or IL17F have been reported. IL6-preconditioned human AT-MSCs demonstrated increased ALP activity and mineralization (85, 88). IL8-preconditioned human AT-MSCs did not show changes in proliferation or osteogenic gene expression, but had reduced bone nodule formation (85). IL17F-preconditioned human AT-MSCs had decreased proliferation but enhanced ALP activity (85).
These in vitro studies suggest that the osteogenic ability of preconditioned MSCs may be influenced not only by pro-inflammatory cytokines but also by the species selected and the tissue of origin. Interestingly, there are few in vivo studies concerning the efficacy of preconditioned MSCs using pro-inflammatory cytokines; further study is needed in this area.
Preconditioned MSC With Hypoxia
The effects of hypoxia-preconditioning on the immunomodulatory and osteogenic capacities of MSCs have become increasingly relevant, particularly as the potential therapeutic conditions of MSCs have an oxygen tension between 1 and 11%, compared to normal ambient oxygen tension (21% O2) (82, 89). Downstream signaling of hypoxia-inducible factors (HIFs) modulates VEGF expression and activation of SDF1 and CXCR4, thus implicating hypoxia as a modifiable element to increase MSC migration and bone healing (96). The HIF1α pathway is tightly linked to skeletal development and bone repair; mice exposed to hypoxic conditions had increased vascularity and bone healing via HIF1α and VEGF mediated pathways (97). MSCs cultured under hypoxic conditions maintain or increase their proliferation rates with increased secretion of growth factors, including VEGF, basic FGF, and PDGF-BB (98–100). In addition, increased lactate production by MSCs under hypoxic conditions could contribute to the polarization of macrophages to an anti-inflammatory M2 phenotype (101).
A recent systematic review paper (102) described that MSCs cultured under severe hypoxic conditions (<2% O2) with long-term exposure increased their proliferation rate and inhibited osteogenic differentiation, whereas MSCs cultured under moderate hypoxic conditions (2–5% O2) with short-term or cyclic exposure had accelerated osteogenic differentiation and inhibited osteoclast function in vitro.
Although in vivo applications of hypoxia-preconditioned MSCs for bone healing are limited (Table 2), Lee et al. studied human BM-MSCs that were expanded under hypoxia with 1% O2 and seeded on hydroxyapatite (HA)/tricalcium phosphate (TCP)-based scaffolds. These hypoxia-preconditioned BM-MSCs/HA/TCP-based scaffolds increased collagen tissue formation in a subcutaneous transplantation model in mice (103). A 21-months-old aged male rat model of hypoxia-preconditioned BM-MSCs with dimethyloxalylglycine (DMOG), a prolyl hydroxylase inhibitor, under 1% O2 conditions improved the repair of a critical-sized mandibular defect (106). The intramuscular injection of hypoxia-preconditioned human BM-MSCs under 1% O2 for 48 h exhibited markedly increased cell survival over 4 weeks in mice (107). A spheroid of hypoxia-preconditioned human BM-MSCs were cultured under 1% O2 for 3 days (105). Hypoxia-preconditioned BM-MSC spheroids had high osteogenic differentiation and VEGF secretion in vitro, and an alginate hydrogel containing hypoxia-preconditioned BM-MSC spheroids improved bone healing of a critical-sized femoral bone defect in male athymic rats at the age of 10–12 weeks. Thus, although the molecular mechanisms of hypoxic conditioning on MSCs remain unclear, these results demonstrate the translatability of biochemical and in vitro studies of MSC hypoxia to in vivo therapies, even under challenging conditions such as critical-sized defects in aged models.
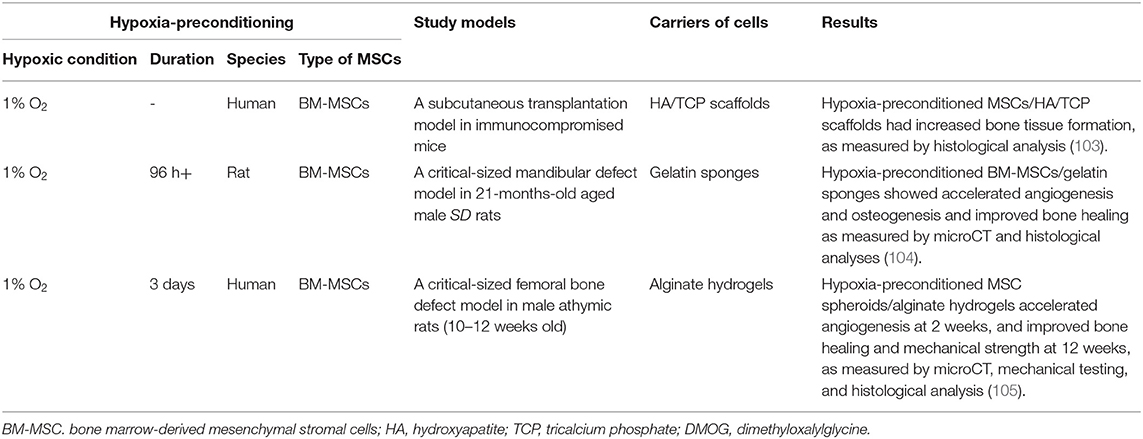
Table 2. Immunomodulation for bone healing by hypoxia-reconditioned MSC in vivo.
Immunomodulation by Accelerating the Resolution of Inflammation via Local Delivery of Anti-inflammatory Cytokines
Direct application of anti-inflammatory cytokines can modify the microenvironment and promote bone healing, particularly when consideration is given to the timing and delivery method (Table 3). Anti-inflammatory cytokines IL4 and IL13 inhibit the proliferation of human osteoblasts (115) but increase collagen secretion, ALP expression, and mineralization (116). Alternatively, IL4 decreased osteogenic differentiation in monoculture of mouse BM-MSCs (108) and human AT-MSCs (88). However, IL4 and IL13 stimulate the polarization of macrophages from the inflammatory M1 phenotype to the anti-inflammatory M2 phenotype (117), and crosstalk between MSCs and macrophages is critical for successful bone healing (5). Therefore, monoculture models may not accurately reflect the full immunomodulatory and osteogenic potential of anti-inflammatory cytokines in vivo. An MSC-macrophage coculture model using either primary murine BM-MSCs (55) or pre-osteoblastic MC3T3 cells (109) with M1 macrophages showed enhanced bone mineralization and ALP activity. Moreover, the addition of IL4 at 72 h to polarize the M1 macrophages to an M2 phenotype further increased calcified matrix formation, compared to introducing IL4 earlier (109, 110). Thus, acute inflammation is necessary to initiate bone healing; the specific timing of the resolution of inflammation is critical for optimal bone formation in vitro.
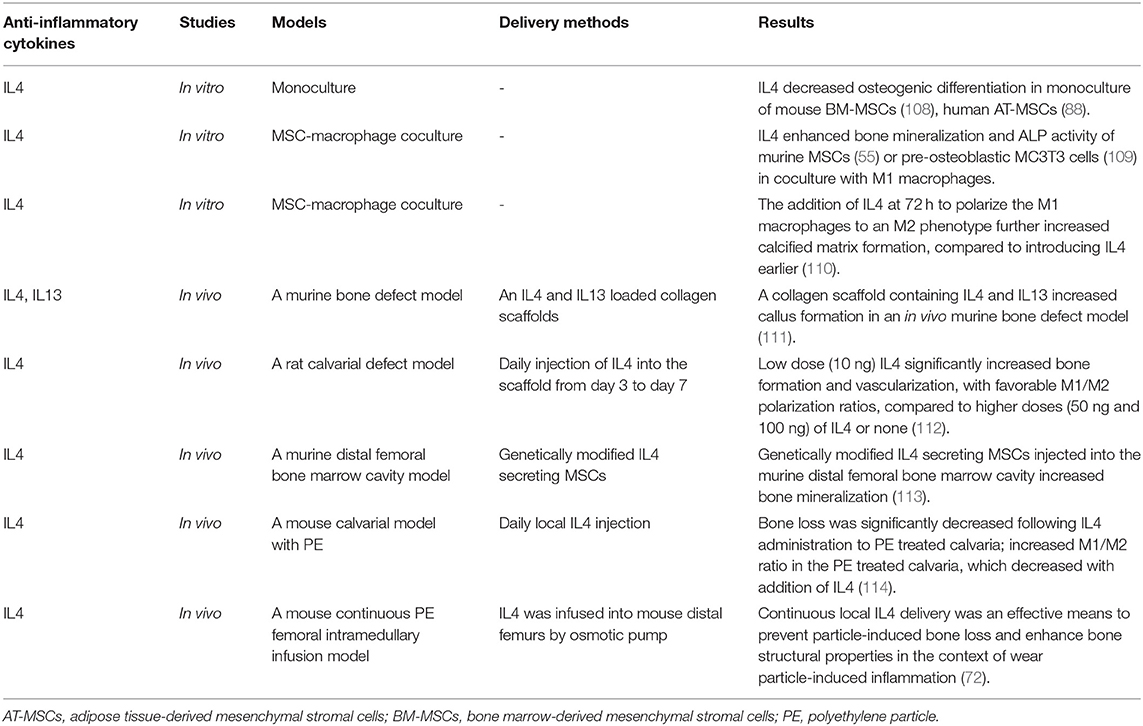
Table 3. Immunomodulation for bone healing by the resolution of inflammation with anti-inflammatory cytokines.
Others have demonstrated that a collagen scaffold containing IL4 and IL13 increased callus formation in an in vivo murine bone defect model (111). Zheng et al. (112) implanted a decellularized bone matrix (DBM) scaffold into a calvarial defect in rats and performed daily injection of different IL4 doses (0, 10, 50, and 100 ng) through the skin directly over the scaffold from day 3 to 7 after surgery. They found that a rat cranial bone defect model with low dose (10 ng) IL4 loaded into decellularized bone matrix significantly increased bone formation and vascularization, with favorable M1/M2 polarization ratios, compared to higher doses (50 and 100 ng) of IL4 or matrix alone. Indeed, as immunomodulation to improve bone healing becomes a therapeutic strategy, immunomodulatory scaffolds are becoming more complex. Other studies have fabricated a functional scaffold with differential release of immunomodulatory molecules, such as a decellularized bone scaffold with sustained release of IL4 via biotin-streptavidin binding (118) and a collagen scaffold with poly(lactic-co-glycolic acid)-multistage silicon particles composite microspheres releasing IL4 (119). A more recent method to provide controlled, direct release of anti-inflammatory cytokines is through cell modification.
Our group (108) has established two types of genetically modified IL-4 secreting BM-MSCs using lentiviral vectors: one is a continuously-IL4-overexpressing BM-MSC driven by the cytomegalovirus (CMV) promoter, and the other is an NFκB-sensing-IL4-overexpressing BM-MSC driven by the NFκB promoter. NFκB-sensing-IL4-overexpressing BM-MSCs produce IL4 when NFκB is activated; thus, these cells secrete IL4 under the conditions of an inflammatory stimulus only. Transplantation of both types of genetically modified IL-4 secreting BM-MSCs into the murine distal femoral bone marrow cavity increased bone mineralization (113).
Although these in vivo studies demonstrated the therapeutic effect of IL4 and IL13 for bone healing, a comprehensive comparison of the optimal dose and delivery timing and methods (e.g., biomaterials and genetically modified MSCs, etc.) of pro-inflammatory cytokines for bone healing is still lacking; further in vivo studies are needed.
IL10 is another potent anti-inflammatory cytokine that can affect bone formation. Mechanistic studies in mice found that IL10 promotes chondrocyte proliferation and differentiation via the bone morphogenetic protein (BMP) pathway, thus influencing endochondral bone formation (120). IL10 deficient mice showed suppressed bone formation and osteoblastogenesis, resulting in osteopenia and increased bone fragility (121, 122). However, various concentrations of IL10 exert different effects on the osteogenesis of human BM-MSCs. Low physiologic concentrations of IL10 (0.01–1.0 ng/ml) activate the p38/MAPK signaling pathway to promote osteogenesis, whereas higher doses of IL10 (10–100 ng/ml) inhibit p38/MAPK signaling by activating NF-kB, inhibiting osteogenesis (123). An in vitro osteoblast-osteoclast coculture model demonstrated that genetically-modified IL10 and TGFβ overexpressing osteoclasts inhibited osteoblast apoptosis and decreased osteoclast formation and bone absorption ability (124). Further translational studies using IL10 and other immunomodulatory molecules are needed.
Immunomodulatory strategies to resolve chronic inflammatory bone disease apply these same principles but require more advanced models and the ability to respond to inflammatory stimuli. For example, in wear particle-induced chronic inflammation, in vivo studies using a mouse calvarial model (114) and continuous polyethylene particle femoral intramedullary infusion model (125) showed that IL4 prevented bone loss and accelerated bone formation by modulating local macrophage polarization to an M2 phenotype. In a recent MSC-macrophage coculture study simulating wear particle-induced inflammation, we demonstrated that NFκB-sensing-IL4-overexpressing BM-MSCs decreased ALP activity and osteocalcin expression (early osteogenic markers) but increased mineralization using Alizarin red staining (late osteogenic marker) (126). These results suggest that NFκB-sensing-IL4-overexpressing BM-MSCs are useful to enhance osteogenesis at a later stage. In combination with preconditioned BM-MSCs, which increased early osteogenic markers, these immunomodulatory strategies may increase bone regeneration at different stages of chronic bone inflammatory disease.
Discussion
This review summarizes current fundamental knowledge underlining the importance of acute inflammation for normal bone healing after injury. Numerous studies have now substantiated that deficiencies in critical cell crosstalk, inhibition of the natural processes of acute inflammation and its resolution, or chronic inflammation due to a persistent adverse stimulus can lead to impaired fracture healing. Thus, an initial and optimal transient stage of acute inflammation is one of the crucial events during fracture healing.
Once the basic principles associated with normal bone healing are clearly understood, then potential strategies for immunomodulation of critical biological events may be exploited (Figure 3). For example, in vitro studies demonstrated that preconditioning of MSCs by pro-inflammatory cytokines or hypoxic conditions enhances their osteogenic potential. However, a comprehensive comparison of the osteogenic ability of MSCs derived from different tissue sources is still limited. In addition, no study has directly and comprehensively compared preconditioning of MSCs using different pro-inflammatory cytokine combinations vs. hypoxic conditions. Furthermore, there are few in vivo studies concerning the efficacy of preconditioned MSCs. Thus, the clinical application of preconditioned MSCs and other novel technologies is not fully known. Further in vivo translational studies are needed in this regard. In vitro studies concerning immunomodulation for the resolution of inflammation using anti-inflammatory cytokines also demonstrated their therapeutic potential to improve bone healing. The clinical application of immunomodulation for bone healing using anti-inflammatory cytokines would not be suitable before 3 days from the onset of acute inflammation. However, the optimal dose, timing, and methods of delivery of pro-inflammatory cytokines have not been fully clarified. Furthermore, similar to preconditioned MSCs, in vivo translational studies concerning the efficacy of immunomodulation for inflammatory resolution for bone healing are limited. Therefore, the therapeutic effects of immunomodulation for bone healing using preconditioned MSCs, anti-inflammatory cytokines to suppress inflammation, or a combination of these strategies should be further evaluated in future in vivo translational studies.
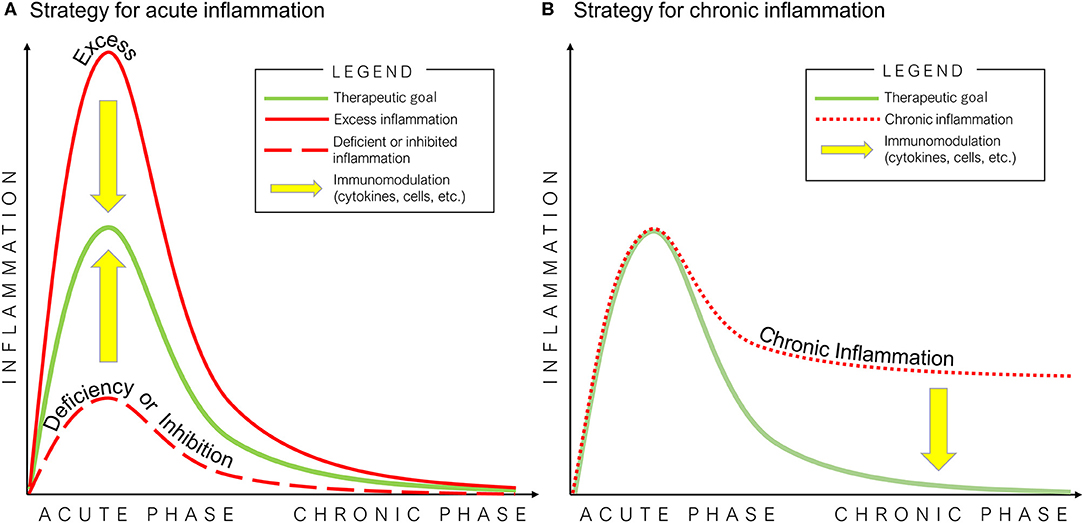
Figure 3. Potential strategies of immunomodulation for bone healing in acute (A) and chronic (B) inflammation. The optimal inflammatory process for bone healing is mediated by an initial and optimal transient stage of acute inflammation, followed by the resolution of inflammation (green line). Excess (solid red line) or deficiency/inhibition of acute inflammation (dashed red line), and chronic inflammation (dotted red line) interfere with the healing of bone defects.
Author Contributions
All authors contributed to the initial concepts and writing of the present manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the generous support of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institute of Health (Grant Nos. R01AR055650, R01AR063717, R01AR073145, and RO1AR72613), the Ellenburg Chair in Surgery, and the Stanford University Medical Scholars Research Grant.
References
2. Hak DJ, Fitzpatrick D, Bishop JA, Marsh JL, Tilp S, Schnettler R, et al. Delayed union and nonunions: epidemiology, clinical issues, and financial aspects. Injury. (2014) 45(Suppl.2):S3–7. doi: 10.1016/j.injury.2014.04.002
3. Claes L, Recknagel S, Ignatius A. Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol. (2012) 8:133–43. doi: 10.1038/nrrheum.2012.1
5. Pajarinen J, Lin T, Gibon E, Kohno Y, Maruyama M, Nathan K, et al. Mesenchymal stem cell-macrophage crosstalk and bone healing. Biomaterials. (2019) 196:80–9. doi: 10.1016/j.biomaterials.2017.12.025
6. Loi F, Cordova LA, Pajarinen J, Lin TH, Yao Z, Goodman SB. Inflammation, fracture and bone repair. Bone. (2016a) 86:119–30. doi: 10.1016/j.bone.2016.02.020
7. Cho TJ, Gerstenfeld LC, Einhorn TA. Differential temporal expression of members of the transforming growth factor beta superfamily during murine fracture healing. J Bone Miner Res. (2002) 17:513–20. doi: 10.1359/jbmr.2002.17.3.513
8. Dimitriou R, Tsiridis E, Giannoudis PV. Current concepts of molecular aspects of bone healing. Injury. (2005) 36:1392–404. doi: 10.1016/j.injury.2005.07.019
9. Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. (2016) 15:551–67. doi: 10.1038/nrd.2016.39
10. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
11. Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. (2012) 13:59–66. doi: 10.1038/nri3362
12. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. (2014) 40:315–27. doi: 10.1016/j.immuni.2014.02.009
13. Feehan KT, Gilroy DW. Is resolution the end of inflammation? Trends Mol Med. (2019) 25:198–214. doi: 10.1016/j.molmed.2019.01.006
14. Reville K, Crean JK, Vivers S, Dransfield I, Godson C. Lipoxin A4 redistributes myosin IIA and Cdc42 in macrophages: implications for phagocytosis of apoptotic leukocytes. J Immunol. (2006) 176:1878–88. doi: 10.4049/jimmunol.176.3.1878
15. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. (2005) 6:1191–7. doi: 10.1038/ni1276
16. Newson J, Stables M, Karra E, Arce-Vargas F, Quezada S, Motwani M, et al. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood. (2014) 124:1748–64. doi: 10.1182/blood-2014-03-562710
17. Lin TH, Tamaki Y, Pajarinen J, Waters HA, Woo DK, Yao Z, et al. Chronic inflammation in biomaterial-induced periprosthetic osteolysis: NF-kappaB as a therapeutic target. Acta Biomater. (2014) 10:1–10. doi: 10.1016/j.actbio.2013.09.034
18. Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. (2008) 181:1232–44. doi: 10.4049/jimmunol.181.2.1232
19. Marsell R, Einhorn TA. The biology of fracture healing. Injury. (2011) 42:551–5. doi: 10.1016/j.injury.2011.03.031
20. Walters G, Pountos I, Giannoudis PV. The cytokines and micro-environment of fracture haematoma: current evidence. J Tissue Eng Regen Med. (2018) 12:e1662–77. doi: 10.1002/term.2593
21. Bastian O, Pillay J, Alblas J, Leenen L, Koenderman L, Blokhuis T. Systemic inflammation and fracture healing. J Leukoc Biol. (2011) 89:669–73. doi: 10.1189/jlb.0810446
22. Miron RJ, Bosshardt DD. OsteoMacs: key players around bone biomaterials. Biomaterials. (2016) 82:1–19. doi: 10.1016/j.biomaterials.2015.12.017
23. Batoon L, Millard SM, Raggatt LJ, Pettit AR. Osteomacs and bone regeneration. Curr Osteoporos Rep. (2017) 15:385–95. doi: 10.1007/s11914-017-0384-x
24. Karnes JM, Daffner SD, Watkins CM. Multiple roles of tumor necrosis factor-alpha in fracture healing. Bone. (2015) 78:87–93. doi: 10.1016/j.bone.2015.05.001
25. Toosi S, Behravan J. Osteogenesis and bone remodeling: a focus on growth factors and bioactive peptides. Biofactors. (2019). doi: 10.1002/biof.1598. [Epub ahead of print].
26. Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. (2003) 88:873–84. doi: 10.1002/jcb.10435
27. Kenkre JS, Bassett J. The bone remodelling cycle. Ann Clin Biochem. (2018) 55:308–27. doi: 10.1177/0004563218759371
28. Lim JC, Ko KI, Mattos M, Fang M, Zhang C, Feinberg D, et al. TNFalpha contributes to diabetes impaired angiogenesis in fracture healing. Bone. (2017) 99:26–38. doi: 10.1016/j.bone.2017.02.014
29. Copuroglu C, Calori GM, Giannoudis PV. Fracture non-union: who is at risk? Injury. (2013) 44:1379–82. doi: 10.1016/j.injury.2013.08.003
30. Foulke BA, Kendal AR, Murray DW, Pandit H. Fracture healing in the elderly: a review. Maturitas. (2016) 92:49–55. doi: 10.1016/j.maturitas.2016.07.014
31. Andrzejowski P, Giannoudis PV. The 'diamond concept' for long bone non-union management. J Orthop Traumatol. (2019) 20:21. doi: 10.1186/s10195-019-0528-0
32. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. (2005) 174:6477–89. doi: 10.4049/jimmunol.174.10.6477
33. Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. (2004) 202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x
34. Rapp AE, Bindl R, Recknagel S, Erbacher A, Muller I, Schrezenmeier H, et al. Fracture healing is delayed in immunodeficient NOD/scidIL2Rgammacnull mice. PLoS ONE. (2016) 11:e0147465. doi: 10.1371/journal.pone.0147465
35. Kon T, Cho TJ, Aizawa T, Yamazaki M, Nooh N, Graves D, et al. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J Bone Miner Res. (2001) 16:1004–14. doi: 10.1359/jbmr.2001.16.6.1004
36. Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Cruceta J, Graves BD, et al. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs. (2001) 169:285–94. doi: 10.1159/000047893
37. Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, et al. Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. (2003) 18:1584–92. doi: 10.1359/jbmr.2003.18.9.1584
38. Mountziaris PM, Mikos AG. Modulation of the inflammatory response for enhanced bone tissue regeneration. Tissue Eng B Rev. (2008) 14:179–86. doi: 10.1089/ten.teb.2008.0038
39. Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin-6. Proc Natl Acad Sci USA. (1993) 90:11924–8. doi: 10.1073/pnas.90.24.11924
40. Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MP. Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone. (2007) 41:928–36. doi: 10.1016/j.bone.2007.07.022
41. Wallace A, Cooney TE, Englund R, Lubahn JD. Effects of interleukin-6 ablation on fracture healing in mice. J Orthop Res. (2011) 29:1437–42. doi: 10.1002/jor.21367
42. Schaper F, Rose-John S. Interleukin-6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. (2015) 26:475–87. doi: 10.1016/j.cytogfr.2015.07.004
43. Prystaz K, Kaiser K, Kovtun A, Haffner-Luntzer M, Fischer V, Rapp AE, et al. Distinct effects of IL-6 classic and trans-signaling in bone fracture healing. Am J Pathol. (2018) 188:474–90. doi: 10.1016/j.ajpath.2017.10.011
44. Kaiser K, Prystaz K, Vikman A, Haffner-Luntzer M, Bergdolt S, Strauss G, et al. Pharmacological inhibition of IL-6 trans-signaling improves compromised fracture healing after severe trauma. Naunyn Schmiedebergs Arch Pharmacol. (2018) 391:523–36. doi: 10.1007/s00210-018-1483-7
45. Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, et al. IL-17-producing gammadelta T cells enhance bone regeneration. Nat Commun. (2016) 7:10928. doi: 10.1038/ncomms10928
46. Croes M, Oner FC, van Neerven D, Sabir E, Kruyt MC, Blokhuis TJ, et al. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone. (2016) 84:262–70. doi: 10.1016/j.bone.2016.01.010
47. Liao C, Zhang C, Jin L, Yang Y. IL-17 alters the mesenchymal stem cell niche towards osteogenesis in cooperation with osteocytes. J Cell Physiol. (2020) 235:4466–80. doi: 10.1002/jcp.29323
48. Ishikawa M, Ito H, Kitaori T, Murata K, Shibuya H, Furu M, et al. MCP/CCR2 signaling is essential for recruitment of mesenchymal progenitor cells during the early phase of fracture healing. PLoS ONE. (2014) 9:e104954. doi: 10.1371/journal.pone.0104954
49. Lange J, Sapozhnikova A, Lu C, Hu D, Li X, Miclau T 3rd, et al. Action of IL-1beta during fracture healing. J Orthop Res. (2010) 28:778–84. doi: 10.1002/jor.21061
50. Raisz LG. Prostaglandins and bone: physiology and pathophysiology. Osteoarthr Cartil. (1999) 7:419–21. doi: 10.1053/joca.1998.0230
51. Lisowska B, Kosson D, Domaracka K. Lights and shadows of NSAIDs in bone healing: the role of prostaglandins in bone metabolism. Drug Des Devel Ther. (2018) 12:1753–8. doi: 10.2147/DDDT.S164562
52. Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O'Keefe RJ. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. (2002) 109:1405–15. doi: 10.1172/jci0215681
53. Marquez-Lara A, Hutchinson ID, Nunez FJr, Smith TL, Miller AN. Nonsteroidal anti-inflammatory drugs and bone-healing: a systematic review of research quality. JBJS Rev. (2016) 4:55. doi: 10.2106/JBJS.RVW.O.00055
54. Lisowska B, Kosson D, Domaracka K. Positives and negatives of nonsteroidal anti-inflammatory drugs in bone healing: the effects of these drugs on bone repair. Drug Des Devel Ther. (2018) 12:1809–14. doi: 10.2147/DDDT.S164565
55. Lu LY, Loi F, Nathan K, Lin TH, Pajarinen J, Gibon E, et al. Pro-inflammatory M1 macrophages promote Osteogenesis by mesenchymal stem cells via the COX-2-prostaglandin E2 pathway. J Orthop Res. (2017) 35:2378–85. doi: 10.1002/jor.23553
56. Valparaiso AP, Vicente DA, Bograd BA, Elster EA, Davis TA. Modeling acute traumatic injury. J Surg Res. (2015) 194:220–32. doi: 10.1016/j.jss.2014.10.025
57. Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. (2007) 38:1336–45. doi: 10.1016/j.injury.2007.10.003
58. Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-alpha on bone homeostasis. Front Immunol. (2014) 5:48. doi: 10.3389/fimmu.2014.00048
59. Zhao L, Huang J, Zhang H, Wang Y, Matesic LE, Takahata M, et al. Tumor necrosis factor inhibits mesenchymal stem cell differentiation into osteoblasts via the ubiquitin E3 ligase Wwp1. Stem Cells. (2011) 29:1601–10. doi: 10.1002/stem.703
60. Adamopoulos IE. Inflammation in bone physiology and pathology. Curr Opin Rheumatol. (2018) 30:59–64. doi: 10.1097/BOR.0000000000000449
61. Grassi F, Cattini L, Gambari L, Manferdini C, Piacentini A, Gabusi E, et al. T cell subsets differently regulate osteogenic differentiation of human mesenchymal stromal cells in vitro. J Tissue Eng Regen Med. (2016) 10:305–14. doi: 10.1002/term.1727
62. Wendler S, Schlundt C, Bucher CH, Birkigt J, Schipp CJ, Volk HD, et al. Immune modulation to enhance bone healing-a new concept to induce bone using prostacyclin to locally modulate immunity. Front Immunol. (2019) 10:713. doi: 10.3389/fimmu.2019.00713
63. Menges T, Engel J, Welters I, Wagner RM, Little S, Ruwoldt R, et al. Changes in blood lymphocyte populations after multiple trauma: association with posttraumatic complications. Crit Care Med. (1999) 27:733–40. doi: 10.1097/00003246-199904000-00026
64. Hurtgen BJ, Ward CL, Garg K, Pollot BE, Goldman SM, McKinley TO, et al. Severe muscle trauma triggers heightened and prolonged local musculoskeletal inflammation and impairs adjacent tibia fracture healing. J Musculoskelet Neuronal Interact. (2016) 16:122–34.
65. Chiu R, Ma T, Smith RL, Goodman SB. Polymethylmethacrylate particles inhibit osteoblastic differentiation of bone marrow osteoprogenitor cells. J Biomed Mater Res A. (2006) 77:850–6. doi: 10.1002/jbm.a.30697
66. Chiu R, Ma T, Smith RL, Goodman SB. Ultrahigh molecular weight polyethylene wear debris inhibits osteoprogenitor proliferation and differentiation in vitro. J Biomed Mater Res A. (2009) 89:242–7. doi: 10.1002/jbm.a.32001
67. Kodaya YRP, Al-Saffar N, Kobayashi A, Scott G, Freeman AR. Bone formation and bone resorption in failed total joint arthroplasties: histomorphometric analysis wit hisochemical and immunohistochemical technique. J Orthop Res. (1996) 14:473–82.
68. Martinez FOSA, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. (2008) 13:453–61. doi: 10.2741/2692
69. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
70. Lin TH, Gibon E, Loi F, Pajarinen J, Cordova LA, Nabeshima A, et al. Decreased osteogenesis in mesenchymal stem cells derived from the aged mouse is associated with enhanced NF-kappaB activity. J Orthop Res. (2017) 35:281–8. doi: 10.1002/jor.23270
71. Lin TH, Pajarinen J, Sato T, Loi F, Fan C, Cordova LA, et al. NF-kappaB decoy oligodeoxynucleotide mitigates wear particle-associated bone loss in the murine continuous infusion model. Acta Biomater. (2016) 41:273–81. doi: 10.1016/j.actbio.2016.05.038
72. Sato T, Pajarinen J, Lin TH, Tamaki Y, Loi F, Egashira K, et al. NF-κB decoy oligodeoxynucleotide inhibits wear particle-induced inflammation in a murine calvarial model. J Biomed Mater Res A. (2015) 103:3872–8. doi: 10.1002/jbm.a.35532
73. Bishop JA, Palanca AA, Bellino MJ, Lowenberg DW. Assessment of compromised fracture healing. J Am Acad Orthop Surg (2012) 20:273–82. doi: 10.5435/JAAOS-20-05-273
74. Toosi S, Behravan N, Behravan J. Nonunion fractures, mesenchymal stem cells and bone tissue engineering. J Biomed Mater Res A. (2018) 106:2552–62. doi: 10.1002/jbm.a.36433
75. Megas P. Classification of non-union. Injury. (2005) 36(Suppl.4):S30–7. doi: 10.1016/j.injury.2005.10.008
76. Gomez-Barrena E, Rosset P, Lozano D, Stanovici J, Ermthaller C, Gerbhard F. Bone fracture healing: cell therapy in delayed unions and nonunions. Bone. (2015) 70:93–101. doi: 10.1016/j.bone.2014.07.033
77. Schlundt C, Bucher CH, Tsitsilonis S, Schell H, Duda GN, Schmidt-Bleek K. Clinical and research approaches to treat non-union fracture. Curr Osteoporos Rep. (2018) 16:155–68. doi: 10.1007/s11914-018-0432-1
78. Hofmann A, Ritz U, Hessmann MH, Schmid C, Tresch A, Rompe JD, et al. Cell viability, osteoblast differentiation, and gene expression are altered in human osteoblasts from hypertrophic fracture non-unions. Bone. (2008) 42:894–906. doi: 10.1016/j.bone.2008.01.013
79. Iwakura T, Miwa M, Sakai Y, Niikura T, Lee SY, Oe K, et al. Human hypertrophic nonunion tissue contains mesenchymal progenitor cells with multilineage capacity in vitro. J Orthop Res. (2009) 27:208–15. doi: 10.1002/jor.20739
80. Bajada S, Marshall MJ, Wright KT, Richardson JB, Johnson WE. Decreased osteogenesis, increased cell senescence and elevated Dickkopf-1 secretion in human fracture non union stromal cells. Bone. (2009) 45:726–35. doi: 10.1016/j.bone.2009.06.015
81. El-Jawhari JJ, Kleftouris G, El-Sherbiny Y, Saleeb H, West RM, Jones E, et al. Defective proliferation and osteogenic potential with altered immunoregulatory phenotype of native bone marrow-multipotential stromal cells in atrophic fracture non-union. Sci Rep. (2019) 9:17340. doi: 10.1038/s41598-019-53927-3
82. Noronha Nc NC, Mizukami A, Caliari-Oliveira C, Cominal JG, Rocha JLM, Covas DT, et al. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res Ther. (2019) 10:131. doi: 10.1186/s13287-019-1224-y
83. Lu Z, Wang G, Dunstan CR, Chen Y, Lu WY, Davies B, et al. Activation and promotion of adipose stem cells by tumour necrosis factor-alpha preconditioning for bone regeneration. J Cell Physiol. (2013) 228:1737–44. doi: 10.1002/jcp.24330
84. Lu Z, Chen Y, Dunstan C, Roohani-Esfahani S, Zreiqat H. Priming adipose stem cells with tumor necrosis factor-alpha preconditioning potentiates their exosome efficacy for bone regeneration. Tissue Eng A. (2017) 23:1212–20. doi: 10.1089/ten.tea.2016.0548
85. Bastidas-Coral AP, Bakker AD, Zandieh-Doulabi B, Kleverlaan CJ, Bravenboer N, Forouzanfar T, et al. Cytokines TNF-alpha, IL-6, IL-17F, and IL-4 differentially affect osteogenic differentiation of human adipose stem cells. Stem Cells Int (2016) 2016:1318256. doi: 10.1155/2016/1318256
86. Croes M, Oner FC, Kruyt MC, Blokhuis TJ, Bastian O, Dhert WJ, et al. Proinflammatory mediators enhance the osteogenesis of human mesenchymal stem cells after lineage commitment. PLoS ONE. (2015) 10:e0132781. doi: 10.1371/journal.pone.0132781
87. Lin T, Pajarinen J, Nabeshima A, Lu L, Nathan K, Jamsen E, et al. Preconditioning of murine mesenchymal stem cells synergistically enhanced immunomodulation and osteogenesis. Stem Cell Res Ther. (2017) 8:277. doi: 10.1186/s13287-017-0730-z
88. Bastidas-Coral AP, Hogervorst JMA, Forouzanfar T, Kleverlaan CJ, Koolwijk P, Klein-Nulend J, et al. IL-6 counteracts the inhibitory effect of IL-4 on osteogenic differentiation of human adipose stem cells. J Cell Physiol. (2019) 234:20520–32. doi: 10.1002/jcp.28652
89. de Witte SF, Franquesa M, Baan CC, Hoogduijn MJ. Toward development of iMesenchymal stem cells for immunomodulatory therapy. Front Immunol. (2015) 6:648. doi: 10.3389/fimmu.2015.00648
90. Philipp D, Suhr L, Wahlers T, Choi YH, Paunel-Gorgulu A. Preconditioning of bone marrow-derived mesenchymal stem cells highly strengthens their potential to promote IL-6-dependent M2b polarization. Stem Cell Res Ther. (2018) 9:286. doi: 10.1186/s13287-018-1039-2
91. Sivanathan KN, Rojas-Canales DM, Hope CM, Krishnan R, Carroll RP, Gronthos S, et al. Interleukin-17A-induced human mesenchymal stem cells are superior modulators of immunological function. Stem Cells. (2015) 33:2850–63. doi: 10.1002/stem.2075
92. Kim Y-G, Park J-W, Lee J-M, Suh J-Y, Lee J-K, Chang B-S, et al. IL-17 inhibits osteoblast differentiation and bone regeneration in rat. Arch Oral Biol. (2014) 59:897–905. doi: 10.1016/j.archoralbio.2014.05.009
93. Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ. (2009) 16:1332–43. doi: 10.1038/cdd.2009.74
94. Noh M. Interleukin-17A increases leptin production in human bone marrow mesenchymal stem cells. Biochem Pharmacol. (2012) 83:661–70. doi: 10.1016/j.bcp.2011.12.010
95. Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem Pharmacol. (2009) 77:1835–44. doi: 10.1016/j.bcp.2009.03.008
96. Lin W, Xu L, Zwingenberger S, Gibon E, Goodman SB, Li G. Mesenchymal stem cells homing to improve bone healing. J Orthop Translat. (2017) 9:19–27. doi: 10.1016/j.jot.2017.03.002
97. Wan C, Gilbert SR, Wang Y, Cao X, Shen X, Ramaswamy G, et al. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc Natl Acad Sci USA. (2008) 105:686–91. doi: 10.1073/pnas.0708474105
98. Crisostomo PR, Wang Y, Markel TA, Wang M, Lahm T, Meldrum DR. Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B- but not JNK-dependent mechanism. Am J Physiol, Cell Physiol. (2008) 294:C675–82. doi: 10.1152/ajpcell.00437.2007
99. Liu L, Gao J, Yuan Y, Chang Q, Liao Y, Lu F. Hypoxia preconditioned human adipose derived mesenchymal stem cells enhance angiogenic potential via secretion of increased VEGF and bFGF. Cell Biol Int. (2013) 37:551–60. doi: 10.1002/cbin.10097
100. Fotia C, Massa A, Boriani F, Baldini N, Granchi D. Prolonged exposure to hypoxic milieu improves the osteogenic potential of adipose derived stem cells. J Cell Biochem. (2015) 116:1442–53. doi: 10.1002/jcb.25106
101. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
102. Camacho-Cardenosa M, Camacho-Cardenosa A, Timon R, Olcina G, Tomas-Carus P, Brazo-Sayavera J. Can hypoxic conditioning improve bone metabolism? A systematic review. Int J Environ Res Public Health. (2019) 16:1799. doi: 10.3390/ijerph16101799
103. Lee JS, Park JC, Kim TW, Jung BJ, Lee Y, Shim EK, et al. Human bone marrow stem cells cultured under hypoxic conditions present altered characteristics and enhanced in vivo tissue regeneration. Bone. (2015) 78:34–45. doi: 10.1016/j.bone.2015.04.044
104. Zhang J, Feng Z, Wei J, Yu Y, Luo J, Zhou J, et al. Repair of critical-sized mandible defects in aged rat using hypoxia preconditioned BMSCs with up-regulation of Hif-1alpha. Int J Biol Sci. (2018) 14:449–60. doi: 10.7150/ijbs.24158
105. Ho SS, Hung BP, Heyrani N, Lee MA, Leach JK. Hypoxic preconditioning of mesenchymal stem cells with subsequent spheroid formation accelerates repair of segmental bone defects. Stem Cells. (2018) 36:1393–403. doi: 10.1002/stem.2853
106. Zhang J, Feng Z, Wei J, Yu Y, Luo J, Zhou J, et al. Repair of critical-sized mandible defects in aged rat using hypoxia preconditioned BMSCs with up-regulation of Hif-1α. Int J Biol Sci. (2018) 14:449–60. doi: 10.7150/ijbs.24158
107. Beegle J, Lakatos K, Kalomoiris S, Stewart H, Isseroff RR, Nolta JA, et al. Hypoxic preconditioning of mesenchymal stromal cells induces metabolic changes, enhances survival, and promotes cell retention in vivo. Stem Cells. (2015) 33:1818–28. doi: 10.1002/stem.1976
108. Lin T, Pajarinen J, Nabeshima A, Lu L, Nathan K, Yao Z, et al. Establishment of NF-kappaB sensing and interleukin-4 secreting mesenchymal stromal cells as an “on-demand” drug delivery system to modulate inflammation. Cytotherapy. (2017) 19:1025–34. doi: 10.1016/j.jcyt.2017.06.008
109. Loi F, Cordova LA, Zhang R, Pajarinen J, Lin TH, Goodman SB, et al. The effects of immunomodulation by macrophage subsets on osteogenesis in vitro. Stem Cell Res Ther. (2016) 7:15. doi: 10.1186/s13287-016-0276-5
110. Nathan K, Lu LY, Lin T, Pajarinen J, Jamsen E, Huang JF, et al. Precise immunomodulation of the M1 to M2 macrophage transition enhances mesenchymal stem cell osteogenesis and differs by sex. Bone Joint Res. (2019) 8:481–8. doi: 10.1302/2046-3758.810.BJR-2018-0231.R2
111. Schlundt C, El Khassawna T, Serra A, Dienelt A, Wendler S, Schell H, et al. Macrophages in bone fracture healing: their essential role in endochondral ossification. Bone. (2018) 106:78–89. doi: 10.1016/j.bone.2015.10.019
112. Zheng ZW, Chen YH, Wu DY, Wang JB, Lv MM, Wang XS, et al. Development of an accurate and proactive immunomodulatory strategy to improve bone substitute material-mediated osteogenesis and angiogenesis. Theranostics. (2018) 8:5482–500. doi: 10.7150/thno.28315
113. Lin T, Pajarinen J, Kohno Y, Maruyama M, Romero-Lopez M, Huang JF, et al. Transplanted interleukin-4–secreting mesenchymal stromal cells show extended survival and increased bone mineral density in the murine femur. Cytotherapy. (2018) 20:1028–36. doi: 10.1016/j.jcyt.2018.06.009
114. Rao AJ, Nich C, Dhulipala LS, Gibon E, Valladares R, Zwingenberger S, et al. Local effect of IL-4 delivery on polyethylene particle induced osteolysis in the murine calvarium. J Biomed Mater Res A. (2013) 101:1926–34. doi: 10.1002/jbm.a.34486
115. Frost A, Jonsson KB, BrändstrÖm H, Ljunghall S, Nilsson O, Ljunggren Ö. Interleukin (IL)-13 and IL-4 inhibit proliferation and stimulate IL-6 formation in human osteoblasts: evidence for involvement of receptor subunits IL-13R, IL-13Rα, and IL-4Rα. Bone. (2001) 28:268–74. doi: 10.1016/s8756-328200449-x
116. Silfversward CJ, Penno H, Frost A, Nilsson O, Ljunggren O. Expression of markers of activity in cultured human osteoblasts: effects of interleukin-4 and interleukin-13. Scand J Clin Lab Invest. (2010) 70:338–42. doi: 10.3109/00365513.2010.488698
117. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
118. Spiller KL, Nassiri S, Witherel CE, Anfang RR, Ng J, Nakazawa KR, et al. Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds. Biomaterials. (2015) 37:194–207. doi: 10.1016/j.biomaterials.2014.10.017
119. Minardi S, Corradetti B, Taraballi F, Byun JH, Cabrera F, Liu X, et al. IL-4 release from a biomimetic scaffold for the temporally controlled modulation of macrophage response. Ann Biomed Eng. (2016) 44:2008–19. doi: 10.1007/s10439-016-1580-z
120. Jung YK, Kim GW, Park HR, Lee EJ, Choi JY, Beier F, et al. Role of interleukin-10 in endochondral bone formation in mice: anabolic effect via the bone morphogenetic protein/Smad pathway. Arthritis Rheum. (2013) 65:3153–64. doi: 10.1002/art.38181
121. Dresner-Pollak R, Gelb N, Rachmilewitz D, Karmeli F, Weinreb M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology. (2004) 127:792–801. doi: 10.1053/j.gastro.2004.06.013
122. Holgersen K, Dobie R, Farquharson C, van't Hof R, Ahmed SF, Hansen AK, et al. Piroxicam treatment augments bone abnormalities in interleukin-10 knockout mice. Inflamm. Bowel Dis. (2015) 21:257–66. doi: 10.1097/MIB.0000000000000269
123. Chen E, Liu G, Zhou X, Zhang W, Wang C, Hu D, et al. Concentration-dependent, dual roles of IL-10 in the osteogenesis of human BMSCs via P38/MAPK and NF-kappaB signaling pathways. FASEB J. (2018) 32:4917–29. doi: 10.1096/fj.201701256RRR
124. Yi L, Li Z, Jiang H, Cao Z, Liu J, Zhang X. Gene modification of transforming growth factor beta (TGF-beta) and interleukin 10 (IL-10) in suppressing Mt sonicate induced osteoclast formation and bone absorption. Med Sci Monit. (2018) 24:5200–7. doi: 10.12659/MSM.909720
125. Sato T, Pajarinen J, Behn A, Jiang X, Lin TH, Loi F, et al. The effect of local IL-4 delivery or CCL2 blockade on implant fixation and bone structural properties in a mouse model of wear particle induced osteolysis. J Biomed Mater Res A. (2016) 104:2255–62. doi: 10.1002/jbm.a.35759
Keywords: bone healing, immunomodulation, inflammation, mesenchymal stromal cell, preconditioning, pro-inflammatory cytokines, anti-inflammatory cytokines
Citation: Maruyama M, Rhee C, Utsunomiya T, Zhang N, Ueno M, Yao Z and Goodman SB (2020) Modulation of the Inflammatory Response and Bone Healing. Front. Endocrinol. 11:386. doi: 10.3389/fendo.2020.00386
Received: 13 March 2020; Accepted: 14 May 2020;
Published: 11 June 2020.
Edited by:
Deborah Veis, Washington University School of Medicine in St. Louis, United StatesReviewed by:
Michaela Tencerova, Institute of Physiology (ASCR), CzechiaPaula H. Stern, Northwestern University, United States
Copyright © 2020 Maruyama, Rhee, Utsunomiya, Zhang, Ueno, Yao and Goodman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart B. Goodman, Z29vZGJvbmVAc3RhbmZvcmQuZWR1