 Lian Duan1†
Lian Duan1† Jie Liu
Jie Liu Lijia Cui
Lijia Cui Lin Lu
Lin Lu Hui Pan
Hui Pan Yong Yao
Yong Yao- 1Key Laboratory of Endocrinology of National Health Commission, Department of Endocrinology, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 2Department of Neurosurgery, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 3Department of Hematology, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 4Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
Primary pituitary lymphoma (PPL) represents an extremely rare entity. Here, we have reported two recently identified cases of immunocompetent PPL having diffuse large B-cell lymphoma by surgical biopsy. Both patients had hypopituitarism, with one patient developing right ptosis. In both patients, MRI and FDG-PET/CT depicted sellar mass that extended into the cavernous sinus with the right sphenoid also present in one of the patients. No systemic disease was found in these two patients. Surprisingly, we found that both patients had infiltrative lesions in sphenoid sinus mucosa pathologically, but the sphenoid bones that composed the sellar base were visually intact during the biopsy procedure. Chemotherapy was administered to both patients, where one patient achieved remission at the recent follow-up, whereas the other one did not respond to the treatment. The diagnosis of PPL is usually difficult if solely dependent on history, clinical presentation, biochemical indexes, and radiographic findings. We have also updated and reviewed the epidemiologic features, clinical presentations, pathological characteristics, potential mechanisms, therapeutic orientation, and prognostic advances of PPL. A total of 40 cases (including ours and four pediatric patients), histologically diagnosed, were analyzed in terms of clinical presentation, endocrine abnormality, radiological features, pathology, treatment, and follow-up. Hypopituitarism and headache were the most common presentation of PPL, while diabetes insipidus was reported in 13 patients (43.3%). B cell lymphoma was the most common type of pathology, followed by T-cell and NK/T cell. PPL was more invasive in nature at the suprasellar region (72.5%), cavernous sinus (52.5%), and sphenoidal sinus (27.5%) in 29, 21, and 11 patients, respectively. Pediatric patients with PPL seem to be different compared to their adult counterparts in terms of pathogenesis, clinical presentation, and radiological features. The management of PPL usually follows the treatment protocols for PCNSL but has a poor prognosis compared to the pituitary involvement of systemic lymphoma.
Introduction
Primary pituitary lymphoma (PPL) is an extremely rare clinical entity with only 40 immunocompetent cases identified and reported to date. It is commonly considered to be a diffuse lymphoma that is limited to the sellar or parasellar regions without any evidence of systemic involvement. The disease shows a slight but not significant female prevalence with an onset at around the age of 60 years (1–3). Regarding the histology, a large majority are of B cell origin compared to T cell or NK/T cell origin, as in the case for primary CNS lymphoma (3, 4). The clinical presentations are always atypical, including headache, hypopituitarism, diplopia, hemianopia, and fever (5). The PPL prognosis is usually poor, which emphasizes the importance of earlier detection and management to improve the outcomes (6). Here, we have reported two recently diagnosed cases of PPL with a comprehensive review of our current understanding of PPL. We also concluded that PPL tends to spread to the sphenoidal area through vascular and bony approaches, which might be a distinct entity from primary CNS lymphoma.
Methods
We conducted a literature review using the PubMed database. Keywords of “pituitary lymphoma”, “pituitary tumor”, and “lymphoma” were used to select adequate papers till May 2020 where the diagnosis of PPL was confirmed histologically. PPL caused by systemic lymphoma or the ones that occurred in immunosuppressed patients were excluded. Only studies in English were considered. Also, we have reviewed references of relevant studies. Some detailed information that was not provided by the original papers was again referred through the latest update (2), acquired through the e-mail contact with the corresponding authors of each article. A total of 36 adult cases (Table 1) with four pediatric cases (Table 2) were reviewed in terms of clinical presentation, endocrine abnormality, radiological features, pathology, treatment, and follow-up (Figure 1). Firstly, we present our two cases.
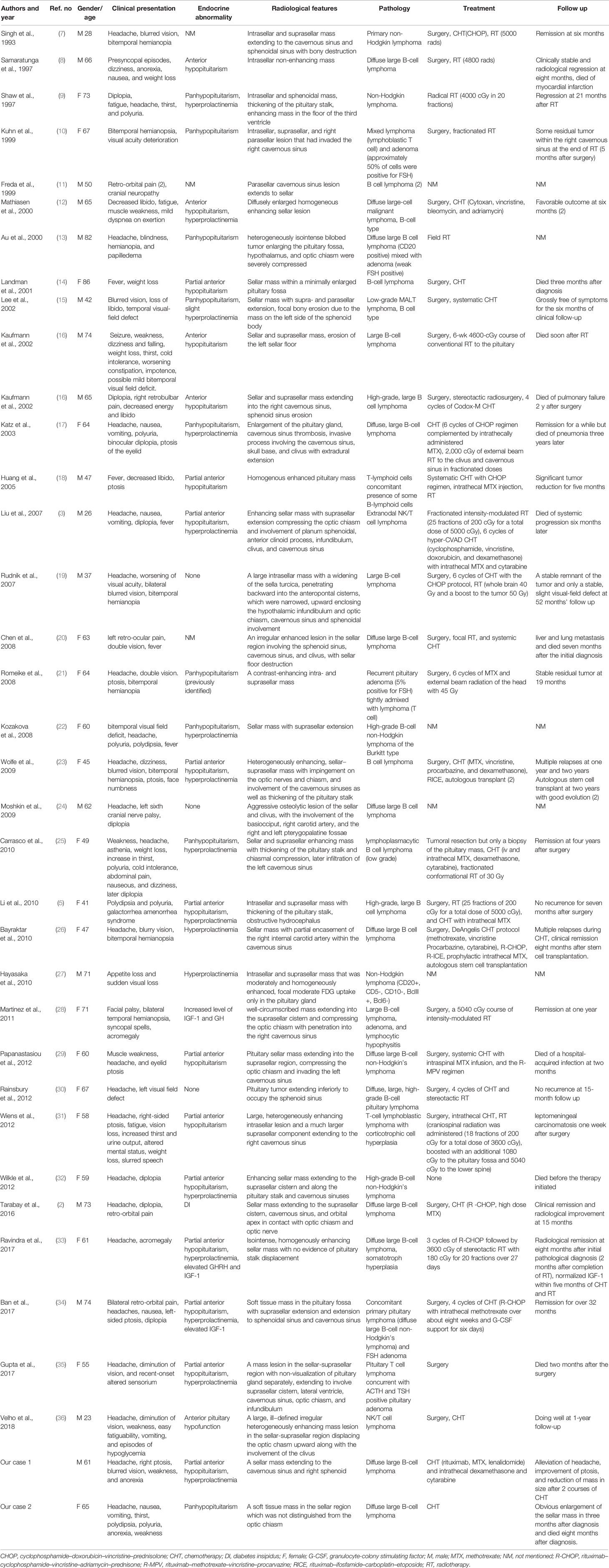
Table 1 The clinical summary of 36 immunocompetent adult PPL patients.
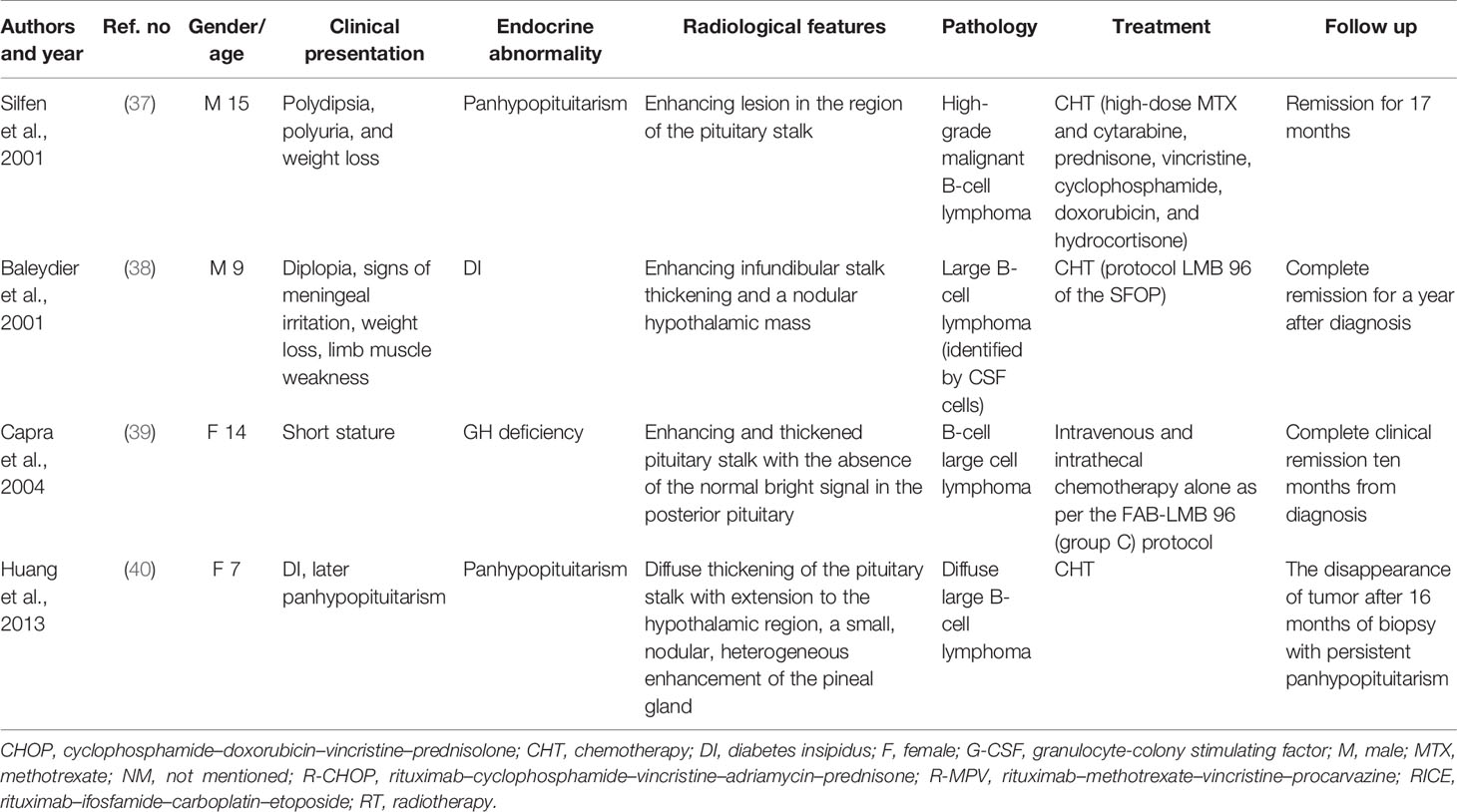
Table 2 The clinical summary of 4 pediatric immunocompetent PPL patients.
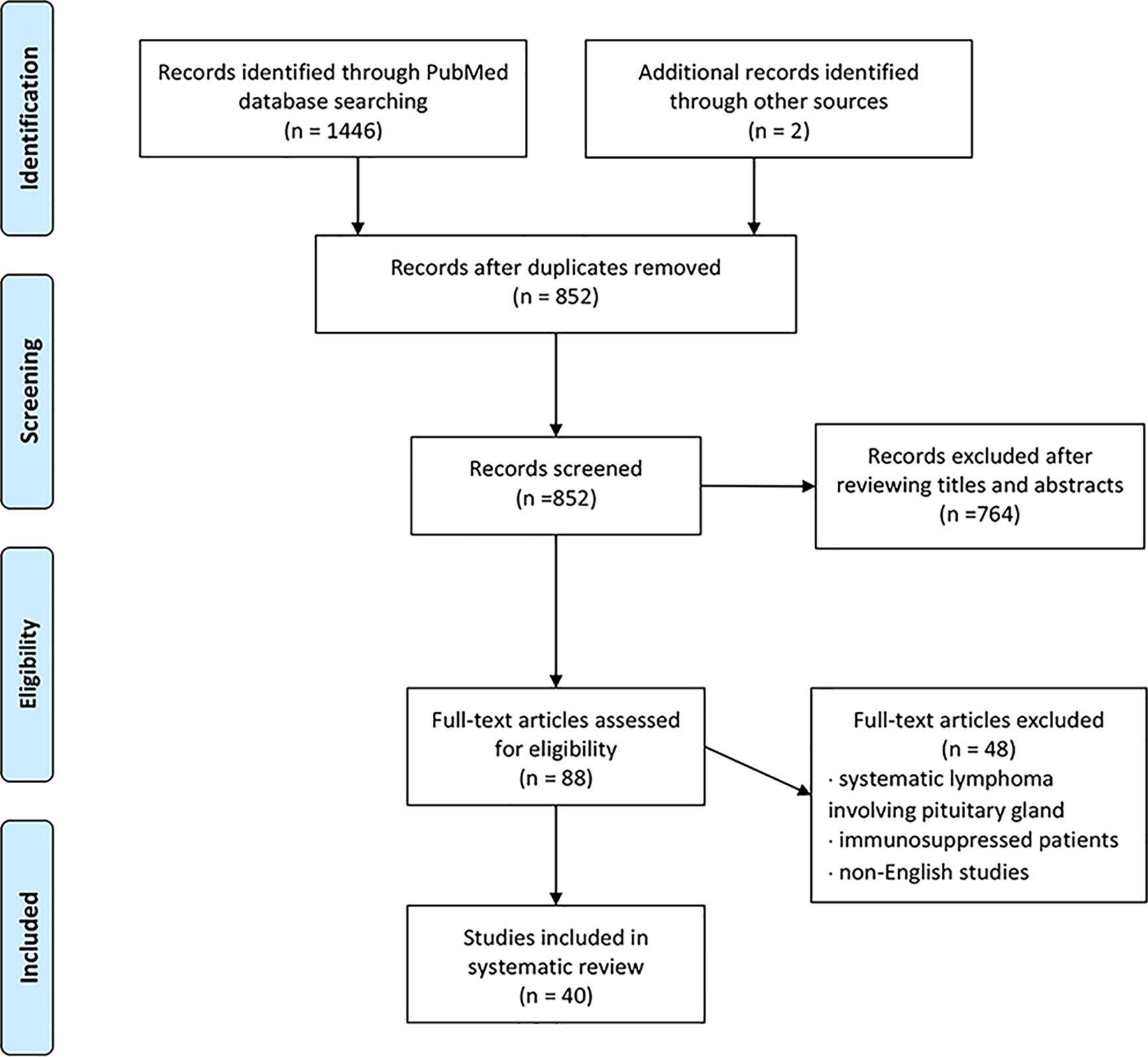
Figure 1 Flowchart of study selection using the PRISMA guidelines.
Case Presentation
Case 1
A 61-year-old male was presented to our clinic reporting episodes of headache and hypophyseal mass for nine months and right eyelid ptosis for four months. In January 2019, he started with an intermittent headache, blurred vision, weakness, and anorexia. He was then admitted to a local hospital for further examination. His serum sodium level was 127 mmol/L with normal blood pressure. The biochemical indexes indicated that he suffered from hypopituitarism. The symptoms of diabetes insipidus were not observed. Pituitary MRI showed a suspicious low signal in the right-wing of the pituitary. Further, he had a history of partial thyroidectomy ten years ago due to a thyroid nodule, with unknown postoperative pathological results. Based on the above evidence, an initial diagnosis of autoimmune hypophysitis was made. The patient then started the replacement therapy using 30 mg hydrocortisone per day and Euthyrox at 50 µg qd, which alleviated headache and improved the right eye acuity.
Five months later, the patient developed sudden right eyelid ptosis. A new MRI disclosed that the pituitary mass had invaded the right cavernous sinus. Glucocorticoid impact therapy with an initial dose of 500 mg qd was initiated, which improved his right eyelid ptosis on the second day of therapy. Nevertheless, a relapse of ptosis occurred three months later and another MRI revealed a 1.3 × 0.9 cm sellar mass with extension to the right cavernous sinus and internal carotid artery. Immunology tests, including ANA, ANCA, serum IgG4, and ACL were reported negative. Lumbar puncture and thoracoabdominal CT scan did not show any remarkable findings. The diagnosis of autoimmune hypophysitis could not be excluded and the cortisol treatment was continued (prednisone 50 mg qd) in addition to 75 mg azathioprine per day.
In October 2019, he was referred to our clinic with several tests, including an electrolyte, serum and urine osmolality, sex hormone (FSH, LH, E, P, T, PRL), IGF-1, thyroid (TSH, FT3, FT4), immunology (IgG4, ANCA, sACE, ESR) and tumor markers tests (CEA, AFP, βhCG), which were all within the normal range. The MRI indicated a possible sellar macroadenoma (12.7 × 6.7 × 11.1 mm) with Knosp IV; the progression of the sellar lesion is shown in Figure 2. Physical examination revealed truncal obesity and diabetes mellitus due to glucocorticoid treatment. His right eye showed ptosis with loss of pupil’s direct/indirect light reflex and ocular motility. FDG-PET/CT revealed a hypermetabolic lesion in the right sellar with infiltration to the right sphenoid sinus, suggesting invasive pituitary adenoma. The systemic disease was, therefore, excluded.
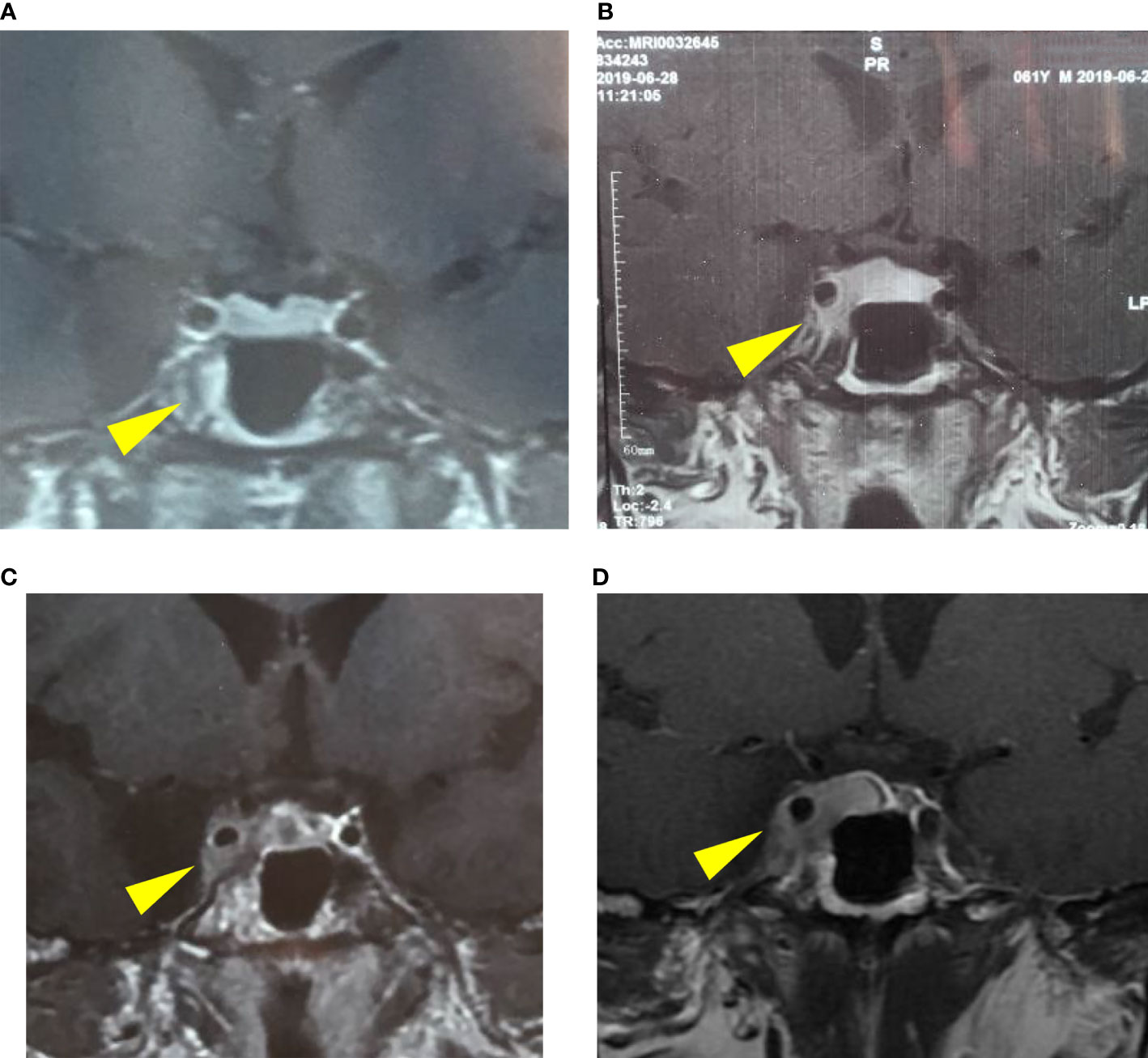
Figure 2 The progression of the pituitary lesion in MRI: (A) a suspicious low signal in the right-wing of the pituitary (January 15, 2019), (B) enlargement of the pituitary lesion involving right cavernous sinus (June 28, 2019), (C) involvement of the right cavernous sinus and internal carotid artery (September 24, 2019), (D) a possible macroadenoma, Knosp IV (October 17, 2019).
The patient underwent a sellar biopsy using an endonasal transsphenoidal approach and the mass on the right side of the sellar displayed a grayish-white hard tissue that looked similar to fish-meat with insufficient blood supply on tissue modality. Pathological analysis established the diagnosis of diffuse large B-cell lymphoma. Cell immunohistochemistry was positive for p53, Bcl-2, Bcl-6, C-MYC, CD20, and CD5, whereas the index for pituitary hormones and epithelial cells tested negative. Cell proliferation index Ki-67 was found to be 70%. Fluorescent in situ hybridization (FISH) of EBER ISH was negative, and no abnormality was found in CSF and bone marrow. The diagnosis of primary pituitary lymphoma was eventually established, and the patient received chemotherapy (rituximab 800 mg iv d1, MTX 7 g iv 4 h d2, lenalidomide 25 mg po d1–14) with 5 mg intrathecal dexamethasone and 50 mg cytarabine. After two courses of chemotherapy (R2-MTX), the patient achieved remission from prolonged headache and ptosis. MRI confirmed a significant reduction in the size of the sellar mass (Figure 3).
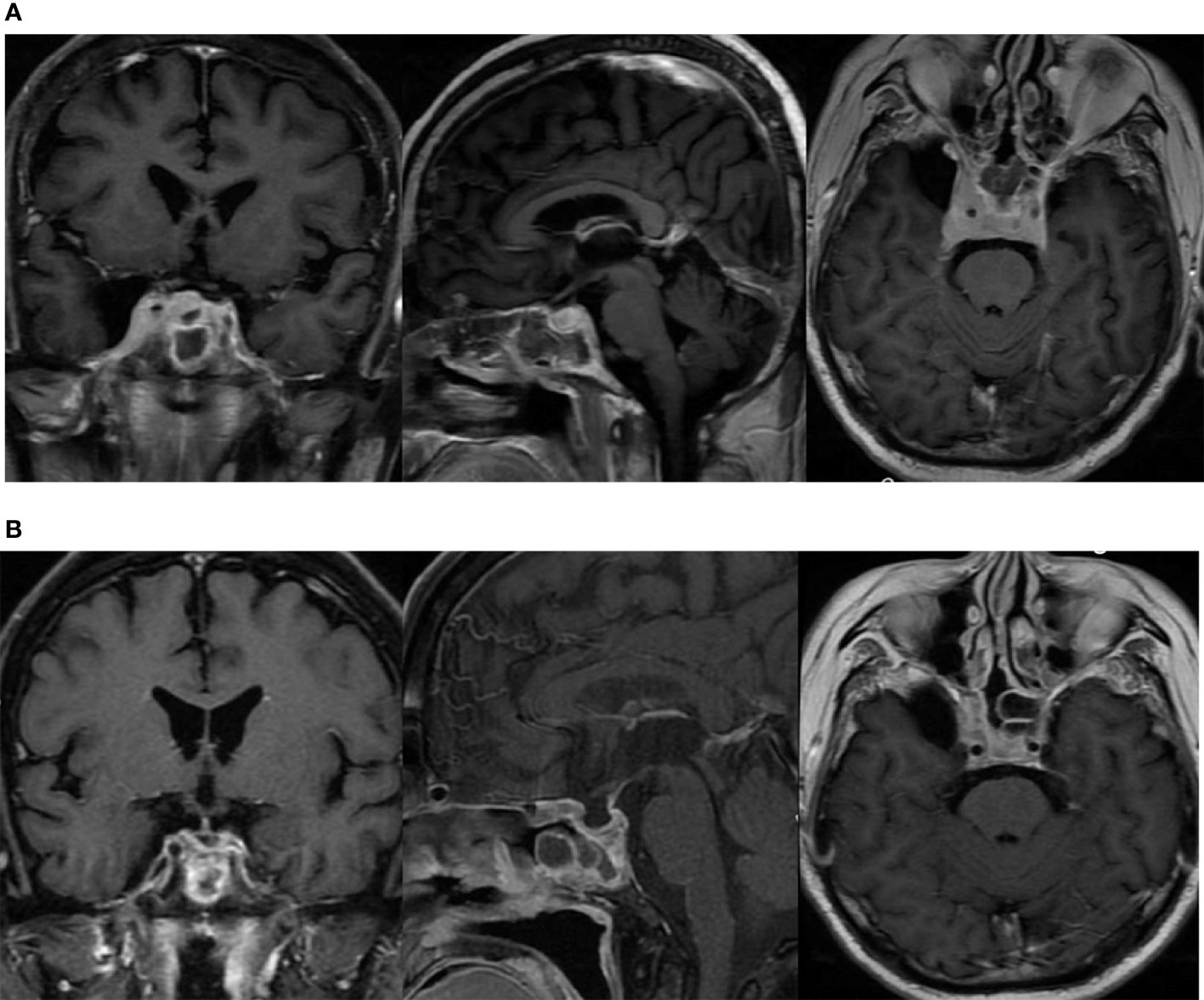
Figure 3 The lesion in the sellar region involving bilateral cavernous sinus as shown in MRI. (A) before chemotherapy; (B) after two courses of chemotherapy (R2-MTX).
Case 2
A 65-year-old woman was presented to our clinic with complaints of headache, nausea, and vomiting. Two months earlier, she had developed symptoms of intermittent headache and nausea with no obvious precipitating factors. Then she gradually had thirst, polydipsia, polyuria, anorexia, and weakness. The biochemical indexes measurement showed values indicative for anterior hypopituitarism, with FT4 of 8 pmol/L, TSH of 0.11 µIU/ml (normal range is 0.380–4.340 µIU/ml), LH< 0.01 mIU/ml (normal range is 1.24–8.62 mIU/ml), FSH of 1.16 mIU/ml (normal range is 1.27–19.26 mIU/ml), ACTH (8 AM) of 1.91 pmol/L (normal range is 2.2–17.6 pmol/L), serum cortisol (8 AM) of 0.536 µg/dl (normal range is 4.0–22.3 µg/dl), and IGF-1 of 150 ng/ml (normal range is 94–252 ng/ml). She had hypernatremia (serum sodium of 150 mmol/L), increased plasma osmolality (304 Osm/kgH2O), and hyposthenuria (98 Osm/kgH2O). The examination of the visual field and visual acuity were found normal, although the MRI revealed a 1.5 × 1.3 × 2.2 cm soft tissue mass in the sellar region, which had no defects in the optic chiasm. The PET/CT identified a pituitary mass with a high intake of FDG in the sellar region and had no other remarkable findings. Replacement therapy with desmopressin, prednisone acetate, and Euthyrox was then administered, which provided the remission from thirst, but headache continued with intermittent nausea and vomiting. In December 2019, the patient underwent a sellar biopsy using an endoscopic transsphenoidal approach. Pathological analysis revealed diffuse large B-cell lymphoma. Immunohistology studies showed CD3, CD5, CD20, Bcl-2, Bcl-6, C-MYC, PAX-5, Mum-1, and p53 to be positive. Cell proliferation index Ki-67 was 90%. Fluorescent in situ hybridization (FISH) for EBER ISH was negative. Therefore, the patient was diagnosed with primary pituitary diffuse large B-cell lymphoma. The patient then received chemotherapy in her local hospital. However, in February 2020, a recent MRI revealed an obvious enlargement of the sellar mass compared to the former one, which may indicate the patient’s insensitivity to chemotherapy. Our latest follow-up revealed that the patient died in July 2020, which is eight months after the diagnosis.
Results
Age and Sex
Out of the 36 adult cases reviewed here, 19 patients (52.8%) were female and 17 patients (47.2%) were male (F: M was 1.12:1) and the mean age was found to be 58 years (mean age for males was 56 years and 61 years for females). Out of the four pediatric patients, two were of each sex (Figure 4).
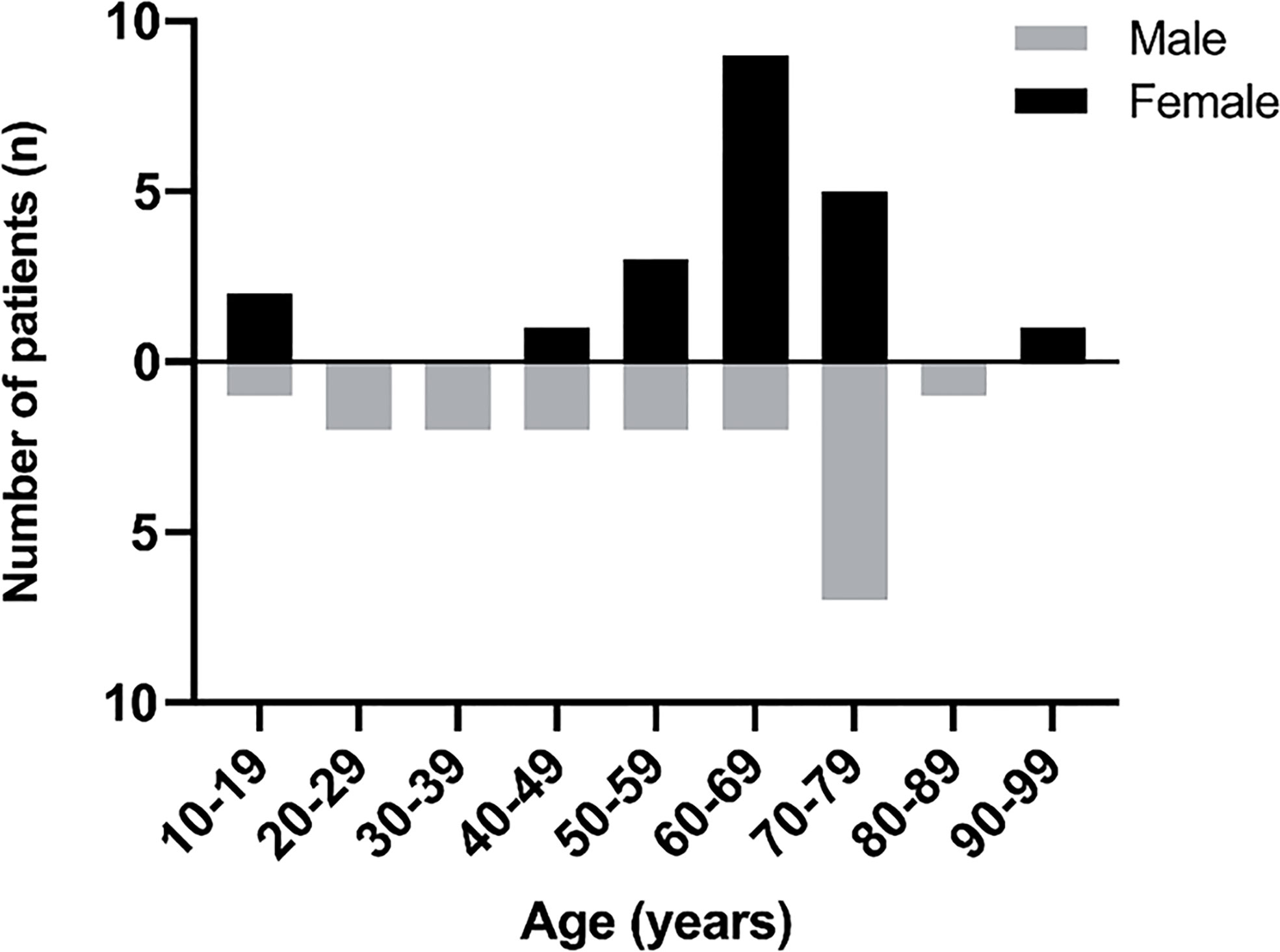
Figure 4 The age and sex of PPL patients.
Tumor Location and Pathology Type
B cell lymphoma was the most common pathology type in PPL patients (in 32 patients, 80%), followed by T cell (in five patients, 12.5%) and NK/T cell (in two patients, 5%). Out of the five cases of T cell type, four had concomitant pituitary adenomas or B cell lymphoma, showing the tendency of T cell type PPL. Tumor location was identified through radiological findings of MRI or FDG-PET/CT, or both of them, in which 37 patients had a tumor in the intrasellar region (92.5%), 29 patients in the suprasellar region (72.5%), 21 patients in cavernous sinus (52.5%), and 11 patients in the sphenoidal sinus (27.5%) (Figure 5).
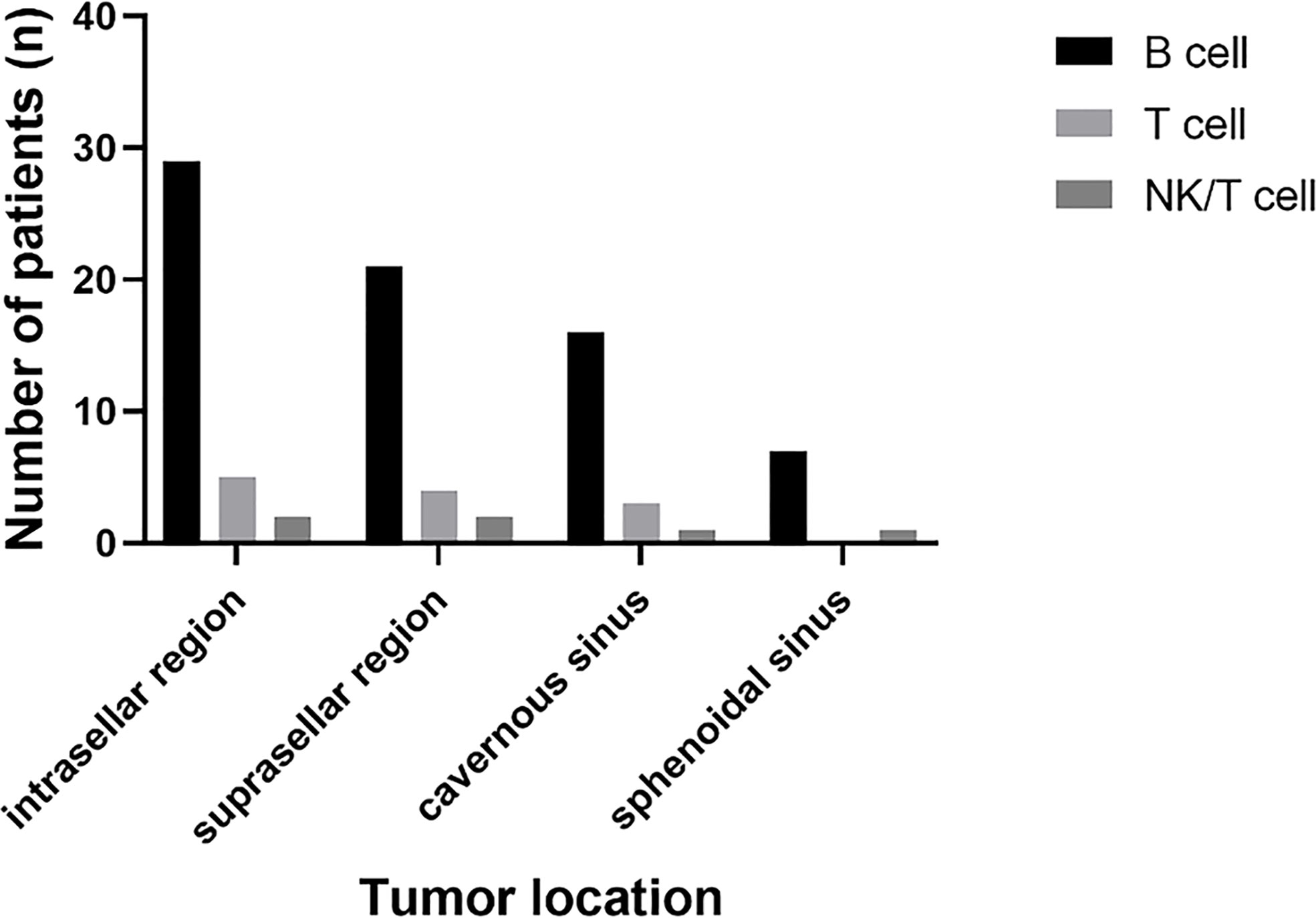
Figure 5 The summary of the tumor location and pathology type in PPL patients.
Clinical Presentation
Hypopituitarism (in 30 patients, 75%) and headache (in 23 patients, 57.5%) were the most common presentations of PPL, followed by hemianopia (in 12 patients, 30%) and diplopia (in 12 patients, 30%). Other symptoms included fatigue, weight loss, fever, retro-orbital pain, nausea, and vomiting (Figure 6).
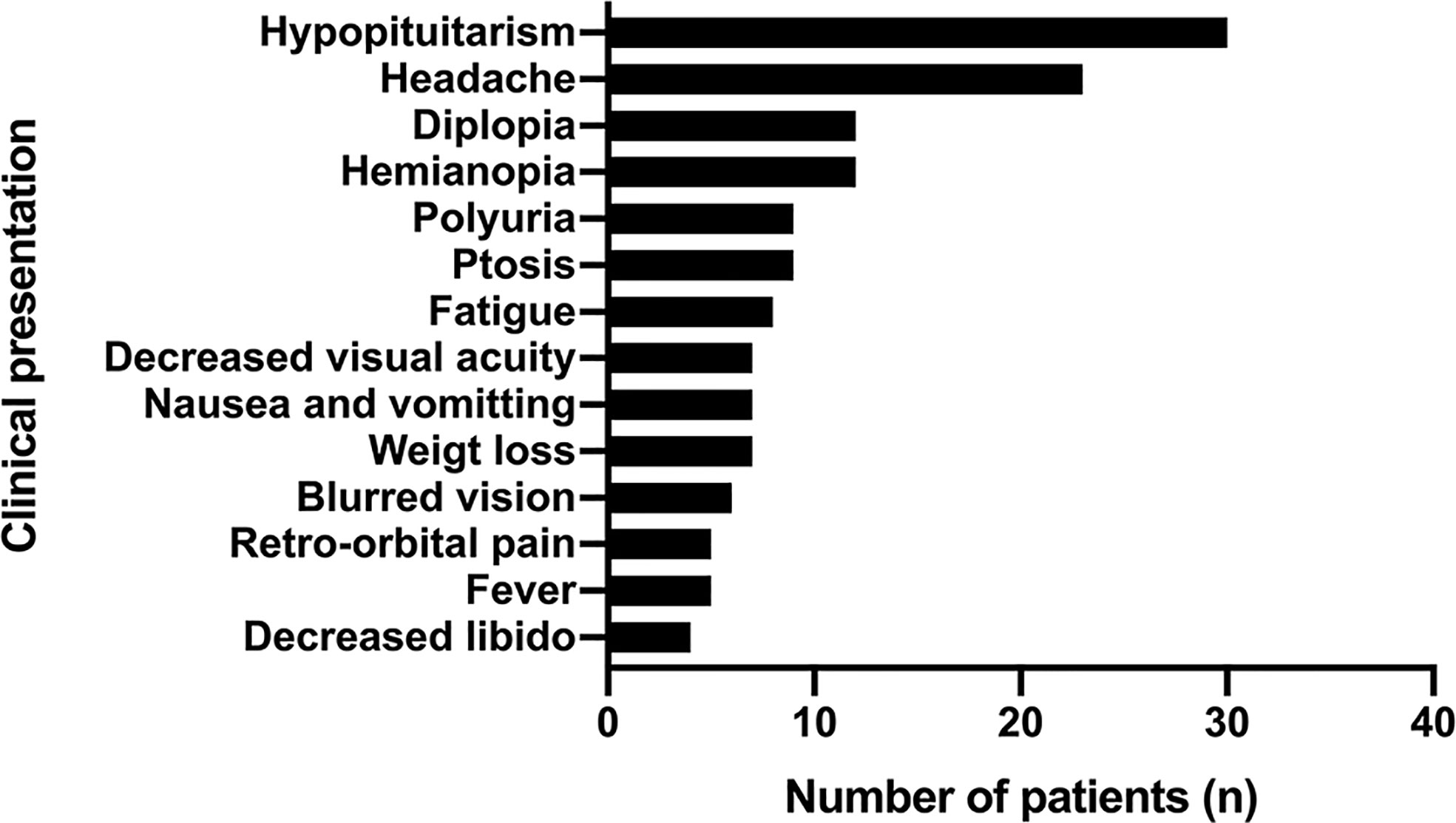
Figure 6 The common clinical presentations of PPL patients.
Among the patients with hypopituitarism (30 patients, 75%), panhypopituitarism was present in 11 patients (36.7%). Notably, diabetes insipidus was reported in 13 patients (43.3%). Apart from the patients concomitant with panhypopituitarism, only five patients showed complete anterior hypopituitarism (16.7%), while 12 patients showed partial anterior hypopituitarism (40%) (Figure 7).

Figure 7 The hypopituitarism in PPL patients.
Treatment Strategies and Outcome
The therapeutic regimen for PPL is usually a combination of surgery, chemotherapy, and/or radiotherapy. Since the case reports included in our study had an unbalanced follow-up duration, it might have resulted in inevitable statistical bias and the possibility that the tumor recurrence and metastasis were neglected. Thus, we attempted to evaluate the response to treatment as “responding” or “non-responding”, where “responding” indicated that the patient had achieved remission or had stable remnant tumor without progression after the therapies were initiated whereas, the “non-responding” implied that the patient had tumor progression, death, metastasis, or relapses after the treatment was adopted.
In this review, treatment strategies were reported in 35 patients, out of which 10 patients (28.6%) received a single treatment modality while 25 patients (71.4%) received combined ones. Only one patient underwent just a resection but died soon after the surgery. Two patients received radiotherapy, but only one of them was sensitive to it, and the other showed no response to the treatment. Notably, seven patients received chemotherapy, and six of them (85.7%) turned out to be effective. In the combined treatment group, four out of five patients (80%) responded to chemotherapy and radiotherapy, six out of nine patients (66.7%) responded to resection and chemotherapy, whereas two out of four patients (50%) responded to resection and radiotherapy while five out of seven patients (71.4%) responded to resection, chemotherapy, and radiotherapy.
Mortality
Among 40 patients of PPL, 12 of them (30%) died, where deaths were caused by the tumor (3), myocardial infarction (1), pulmonary failure (1), pneumonia (1), infection (2), and metastasis (2) while one patient died soon after the treatment, whereas the other one died before the treatment. The median mortality time was 8.7 months (P25:2 months, P75:8 months).
Differential Diagnosis
The diagnosis of PPL was not suspected in 27 patients, and before biopsy could confirm PPL, other diagnoses were made. Pituitary adenoma was the most common in the differential diagnosis (18 cases, 66.7%), followed by lymphocytic hypophysitis (four cases, 14.8%), Langerhans cell histiocytosis (three cases, 11.1%), metastases (three cases, 11.1%), and lymphoma (three cases, 11.1%). Meningioma and germinoma were found in two cases (7.4%), while other diagnoses included sphenoid sinusitis, granulomatous disease, schwannoma, intrasellar solitary plasmacytoma, sellar/suprasellar hemangiopericytoma, and rhabdomyosarcoma of the sphenoid sinus.
Discussion
Primary CNS lymphoma (PCNSL) in immunocompetent patients is rare and represents about 2–4% of all intracranial neoplasms and 4–6% of all extranodal lymphomas (41–43). However, in recent years, a rising incidence has been recognized, particularly in patients older than 60 years (43). Although PPL can develop in immunosuppressed individuals, it seems to be irrelevant to the immune status of PCNSL patients. Therefore, further studies are required to investigate the underlying mechanism of this pathological damage. Previous studies tend to categorize PPL as a sub-population of PCNSL, but we recommend it to be considered as a distinct entity from PCNSL. The origins of anatomic regions involved in the PPL are embryologically different from that of the cerebral parenchyma, as these anatomic structures are outside the blood–brain barrier (2).
Surprisingly, we found that both patients had infiltrative lesions in sphenoid sinus mucosa pathologically, but the sphenoid bones that composed the sellar base were visually intact during the biopsy procedure. In this review, we identified 13 patients (32.5%) with sphenoidal involvement, where seven patients (58.3%) showed no evidence of bony erosion. Notably, two patients, including the one in our case had no radiographic evidence of sphenoidal involvement but were confirmed histologically (14). Because most cases reviewed here did not undergo pathological analysis of sphenoidal mucosa, it is highly likely that there were more patients with sphenoidal involvement of PPL. Therefore, we speculated that PPL tends to spread to the sphenoidal area through vascular and bony approaches. The vascular approach was considered because a large proportion of PPL cases demonstrated sphenoidal involvement without bony erosion. Bony erosion happens in 8% of Hodgkin’s Lymphoma (44) cases and 15% in diffuse large B-cell lymphoma, which is less common at the early stage of the lymphoma system (45) but is still a viable way by which the tumor spreads to the sphenoidal area with or without visual changes in the sphenoidal bones. To the best of our knowledge, it is the first time that the characteristic of sphenoidal involvement is concluded for PPL, which might be different from other lymphomatous pathologies. Hence, we hypothesized that the biopsy of the mucosal sphenoid sinus could be an alternative to sellar biopsy, therefore, reducing the surgical risk while conducting diagnosis.
PPL is more invasive in nature when compared to other pituitary pathologies like adenomas or lymphocytic hypophysitis. It is often aggressive and may extend to the sphenoid sinus, cavernous sinus, or the floor of the third ventricle (26). PPL is usually located in the sellar or parasellar region. But we have also found the involvement of clivus and pituitary stalk (2). The correlation between tumor location and pathology type was not identified, which means that a radiological finding cannot indicate the specific pathology of lymphoma (Figure 5). We noticed that four patients developed metastasis after the diagnosis of PPL. The pathology of these patients did not follow the prevalence of B cell lymphoma, where three of them showed symptoms of fever. Reports of leptomeningeal dissemination, metastasis lesions in the bone marrow, liver, lung, adrenal gland, retroperitoneal lymph nodes raised the possibility that PPL might be the initial lesion location for the systemic disease (3, 18, 20, 31). Further studies are required to test and confirm this hypothesis.
PPL usually has atypical characteristics that make it difficult to distinguish from other pituitary tumors. Endocrine abnormalities and neurological deficits are found to assist tumor localization and explain some specific clinical presentations. Hypopituitarism (30 patients, 75%) and headache (23 patients, 57.5%) were the most common presentations of PPL, followed by hemianopia in 12 patients (30%) and diplopia in 12 patients (30%). Other symptoms included fatigue, weight loss, fever, retro-orbital pain, nausea, and vomiting. About 80% of the patients represented a cranial nerve deficit (II and III CN were most frequently involved), which means that it extended to the cavernous sinus (2). Hypopituitarism in PPL usually presents with various symptoms, including fatigue, muscle weakness, loss of libido, amenorrhea, thirst, and polyuria (3). Anterior hypopituitarism was present in about 70% of the cases, and more than one-third had diabetes insipidus associated with poor prognosis, which was also seen in our review (1, 2). Moreover, the brain tissues taken at the post-mortem of the PCNSL patients identified pituitary gland involvement in about 25% of cases, particularly in the posterior lobe and not the anterior one (46). Such a frequent involvement of the posterior pituitary should be paid more attention. Distinct radiological features were not identified in the sellar and suprasellar lymphomas (29). Therefore, the pathological analysis was required to confirm the diagnosis and adjust the treatment.
As for diagnosis, it seems that pituitary biopsy is currently a gold standard for confirmation. Medical history, physical exam and radiological criteria is highly unlikely to reach the correct diagnosis. Surprisingly, some biomarkers were developed for early detection of PCNSL. Royer-Perron et al. reviewed several biomarkers of PCNSL for diagnostic purpose, such as different microRNAs (miR-21, miR-19, miR-92a, etc.), which could be tested in CSF and vitreous fluid, and IL-10 in the CSF (47). Other biomarkers tested in CSF include Circulating U2 small nuclear RNA fragments, Tumor cell-free DNA, Neopterin, Osteopontin, sTACI, sBCMA, APRIL,BAFF, VSIG4, GPNMB4, and APOC2 (47–50). Based on the studies of the markers above, we believe the specific markers for PPL would be developed soon. However, these markers could not replace the value of biopsy so far and need to be evaluated within a larger patient cohort.
The specific pathogenesis of PPL is still controversial. PPL may either derive from neoplastic transformation of normal lymphocytes that enter the CNS due to inflammatory processes or from the transformation of normal resident lymphoid tissue in the CNS (2, 28). Also, some multipotent cells, i.e., chromophobes, marginal zone cells, follicular cells, folliculo stellate cells, and colony-forming units, might activate and proliferate to develop pituitary lymphoma (29). Risk factors, including AIDS and other immunodeficiency states, along with lymphocytic hypophysitis and pituitary adenomas increase the susceptibility to PPL (1). PPL may share similar pathogenesis with primary thyroid lymphomas, noted more commonly in patients with coexisting Hashimoto thyroiditis (HT) (26). The patient of case 1 had undergone partial thyroidectomy due to a thyroid nodule, but we could not confirm whether it was associated with HT and if it was possibly a potential pathogenic factor. EBV infection is another precipitating factor that induces oncogenic protein expression with subsequent loss of apoptosis and increased proliferation of lymphocytes, but both our patients were found to be EBER-ISH negative (51).
Pediatric patients with PPL seem to be different from their adult counterparts in terms of pathogenesis, clinical presentation, and radiological features. A data suggested that PPL in pediatric patients may be a different disease from that found in adults (40). They tend to have a smaller tumor size, mainly located in the pituitary stalk, which is easily misdiagnosed as germ cell tumors or Langerhans cell histiocytosis that occurs more often in children (39, 40). More importantly, these children were not likely to develop a symptom of headache. No distinct difference was observed in gender and age, and a good prognosis was achieved under chemotherapy. However, more studies are required for further investigation.
The management of PPL usually follows the protocols of treatment for PCNSL because of the lack of studies conducted on this extremely rare disease (2). The therapeutic regimen for PPL is usually a combination of surgery, chemotherapy, and/or radiotherapy (including gamma knife surgery and common radiotherapy) (5). The surgical treatment using the transsphenoidal approach may be a viable option considering the specific location and bulk mass of the PPL lesion. However, the surgical intervention suggests no obvious benefits to the outcome of PCNSL, and the main purpose of it remains to establish a diagnosis via biopsy (42, 52).
Chemotherapy was proven effective in the treatment of PCNSL. The therapeutic regimen consisted of high-dose chemotherapy based on methotrexate (HD-MTX) combined with rituximab and other cytostatic drugs that can penetrate the blood–brain barrier and are highly recommended if the patient’s general condition permits (53). Both our patients received chemotherapy as the first-line of treatment but with varied clinical outcomes. Case 1 patient was sensitive to chemotherapy, which was confirmed by MRI that revealed a significant reduction in the size of the sellar mass, while the Case 2 patient showed a larger sellar mass compared to the previous radiological findings. Pathologically, both our patients showed a Bcl-2, Bcl-6, and C-MYC positive pattern indicating a poor prognosis. The index of p53 was positive in both patients and was more strongly expressed in the Case 2 patient, which might have contributed to the distinction of response against chemotherapy. Notably, the Case 2 patient exhibited aberrantly high positivity of Bcl-6 (95%+) and C-MYC (80%+), also the Ki-67 index had increased up to 90%, which indicated a much poorer prognosis. Given the dominant use of methotrexate in the treatment of PPL, we speculate that the insensitivity of chemotherapy in the Case 2 patient might have resulted from drug resistance or ineffectiveness. Several alternative drugs such as pemetrexed and ibrutinib and salvage chemoimmunotherapy with rituximab, ifosfamide, and etoposide (R-IE regimen) were found to be clinically effective to some degree in relapsed or refractory PCNSL (54–56). Proper antitumor agents based on mRNA expression of drug-resistance genes in tumor tissues, including MDR-1, MRP-1, MRP-2, MXR-1, MGMT, GST-pei, and topoisomerase II alpha can also be selected accordingly (57).
Moreover, high-dose chemotherapy with autologous stem cell transplantation (HD-AST), which was first shown as an efficient therapeutic approach to recurrence, was proposed as a valuable alternative to the whole brain radiotherapy (WBRT) to consolidate the first-line of treatment (58). Two of the PPL patients reviewed in our study had received HD-AST after the occurrence of multiple relapses, but only one patient showed a good evolution (23, 26). Molecular studies are targeting PCNSL, which may be a novel therapeutic option in the future. Immune-mediated therapies and targeted therapies are under investigation and will achieve a more precise treatment with less damaging clinical outcomes (59).
PPL has a much poorer prognosis when compared to the pituitary involvement of systemic lymphoma (6). The overall mean survival rate of immunocompetent PPL patients was 14.4 months. There was no significant difference in terms of survival rates according to the adjuvant treatment strategies, although a combination of radiotherapy and chemotherapy provided a little longer mean survival rate than radiotherapy or chemotherapy alone (2). A newly developed prognostic score for PCNSL (Taipei Score) may offer disease risk stratification and facilitate clinical decision-making. Here, an age ≥80, deep brain lesions, and ECOG ≥2 were considered independent risk factors for PFS where each factor was assigned one point and there were four distinct risk groups (0–3 points) (60). Currently, the prediction of prognosis and treatment modalities of PPL follow the protocols of PCNSL due to its extreme rarity and also the scarcity of adequate research materials. Although PPL might have a different nature from PCNSL, we should also notice the similarity between these two pathologies. We believe that an exclusive prognostic score for PPL will be developed one day with more studies and cases being reported. Fortunately, with the advancing therapeutic strategies nowadays, the treatment outcomes are improving, and also PCNSL is now considered curable in some cases (59).
Conclusion
PPL is an emerging rare clinical entity with a poor prognosis, where advanced therapeutic strategies with a more precise and less damaging effect are under investigation. We are less likely to reach a confirmative diagnosis without the pathological analysis since distinguishing PPL from other sellar tumors is difficult just through the clinical presentations or radiographic findings. Molecular studies are required in the future to investigate the pathological mechanism and establish the biomarkers for early detection and therapeutic orientation. The prognostic score for PCNSL might be a useful tool to predict the clinical outcome of PPL. Since only 40 cases are reported to date, further investigations about PPL are expected in the future, and more details about the distinction between PPL and PCNSL are yet to be studied.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding authors.
Author Contributions
LD and JL drafted the manuscript and conducted the literature review. LC and XZ performed clinical management for patients. YZ guided the hematological treatment. BP conducted the pathological analysis. LL and HP edited the manuscript and verified the literature review. YY and HZ conceived the research idea and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Key Research and Development Program of China (no. 2016YFC0901501) and CAMS Innovation Fund for Medical Science (CAMS-2016-I2M-1-002)
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Giustina A, Gola M, Doga M, Rosei EA. Clinical review 136: Primary lymphoma of the pituitary: an emerging clinical entity. J Clin Endocrinol Metab (2001) 86(10):4567–75. doi: 10.1210/jcem.86.10.7909
2. Tarabay A, Cossu G, Berhouma M, Levivier M, Daniel RT, Messerer M. Primary pituitary lymphoma: an update of the literature. J Neurooncol (2016) 130(3):383–95. doi: 10.1007/s11060-016-2249-z
3. Liu JK, Sayama C, Chin SS, Couldwell WT. Extranodal NK/T-cell lymphoma presenting as a pituitary mass. Case report and review of the literature. J Neurosurg (2007) 107(3):660–5. doi: 10.3171/JNS-07/09/0660
4. Bataille B, Delwail V, Menet E, Vandermarcq P, Ingrand P, Wager M, et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg (2000) 92(2):261–6. doi: 10.3171/jns.2000.92.2.0261
5. Li Y, Zhang Y, Xu J, Chen N. Primary pituitary lymphoma in an immunocompetent patient: a rare clinical entity. J Neurol (2012) 259(2):297–305. doi: 10.1007/s00415-011-6179-6
6. Koiso T, Akutsu H, Takano S, Yamamoto T, Ishikawa E, Okoshi Y, et al. Malignant lymphoma in the parasellar region. Case Rep Med (2014) 2014:747280–. doi: 10.1155/2014/747280
7. Singh VP, Mahapatra AK, Dinde AK. Sellar-suprasellar primary malignant lymphoma: case report. Indian J Cancer (1993) 30(2):88–91.
8. Samaratunga H, Perry-Keene D, Apel RL. Primary Lymphoma of Pituitary Gland: A Neoplasm of Acquired Malt? Endocr Pathol (1997) 8(4):335–41. doi: 10.1007/BF02739936
9. Shaw JA, Strachan FM, Sawers HA, Bevan JS. Non-Hodgkin lymphoma with panhypopituitarism, hyperprolactinaemia and sixth nerve palsy. J R Soc Med (1997) 90(5):274–5. doi: 10.1177/014107689709000512
10. Kuhn D, Buchfelder M, Brabletz T, Paulus W. Intrasellar malignant lymphoma developing within pituitary adenoma. Acta Neuropathol (1999) 97(3):311–6. doi: 10.1007/s004010050990
11. Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am (1999) 28(1):81–117, vi. doi: 10.1016/S0889-8529(05)70058-X
12. Mathiasen RA, Jarrahy R, Cha ST, Kovacs K, Herman VS, Ginsberg E, et al. Pituitary lymphoma: a case report and literature review. Pituitary (2000) 2(4):283–7. doi: 10.1023/A:1009969417380
13. Au WY, Kwong YL, Shek TW, Leung G, Ooi C. Diffuse large-cell B-cell lymphoma in a pituitary adenoma: an unusual cause of pituitary apoplexy. Am J Hematol (2000) 63(4):231–2. doi: 10.1002/(SICI)1096-8652(200004)63:4<231::AID-AJH14>3.0.CO;2-Z
14. Landman RE, Wardlaw SL, McConnell RJ, Khandji AG, Bruce JN, Freda PU. Pituitary lymphoma presenting as fever of unknown origin. J Clin Endocrinol Metab (2001) 86(4):1470–6. doi: 10.1210/jcem.86.4.7389
15. Lee JH, Lee HK, Choi CT, Huh J. Mucosa-associated lymphoid tissue lymphoma of the pituitary gland: MR imaging features. AJNR Am J Neuroradiol (2002) 23(5):838–40.
16. Kaufmann TJ, Lopes MB, Laws ER Jr., Lipper MH. Primary sellar lymphoma: radiologic and pathologic findings in two patients. AJNR Am J Neuroradiol (2002) 23(3):364–7.
17. Katz BJ, Jones RE, Digre KB, Warner JE, Moore KR. Panhypopituitarism as an initial manifestation of primary central nervous system non-Hodgkin’s lymphoma. Endocr Pract (2003) 9(4):296–300. doi: 10.4158/EP.9.4.296
18. Huang YY, Lin SF, Dunn P, Wai YY, Hsueh C, Tsai JS. Primary pituitary lymphoma presenting as hypophysitis. Endocr J (2005) 52(5):543–9. doi: 10.1507/endocrj.52.543
19. Rudnik A, Larysz D, Blamek S, Larysz P, Bierzynska-Macyszyn G, Wlaszczuk P, et al. Primary pituitary lymphoma. Folia Neuropathol (2007) 45(3):144–8.
20. Chen SM, Chang CN, Wei KC, Jung SM, Chuang CC. Sellar lymphoma mimicking sphenoid infection presenting with cavernous sinus syndrome. J Clin Neurosci (2008) 15(10):1148–51. doi: 10.1016/j.jocn.2007.08.021
21. Romeike BF, Joellenbeck B, Stein H, Loddenkemper C, Hummel M, Firsching R, et al. Precursor T-lymphoblastic lymphoma within a recurrent pituitary adenoma. Acta Neurochir (Wien) (2008) 150(8):833–6. doi: 10.1007/s00701-008-1585-y
22. Kozakova D, Machalekova K, Brtko P, Szepe P, Vanuga P, Pura M. Primary B-cell pituitary lymphoma of the Burkitt type: case report of the rare clinic entity with typical clinical presentation. Cas Lek Cesk (2008) 147(11):569–73.
23. Quintero Wolfe S, Hood B, Barker J, Benveniste RJ. Primary central nervous system lymphoma mimicking pituitary apoplexy: case report. Pituitary (2009) 12(1):76–9. doi: 10.1007/s11102-008-0084-8
24. Moshkin O, Muller P, Scheithauer BW, Juco J, Horvath E, Patterson BJ, et al. Primary pituitary lymphoma: a histological, immunohistochemical, and ultrastructural study with literature review. Endocr Pathol (2009) 20(1):46–9. doi: 10.1007/s12022-009-9062-6
25. Carrasco CA, Rojas ZD, Chiorino R, Gonzalez G. Primary pituitary lymphoma in immunocompetent patient: diagnostic problems and prolonged follow-up. Pituitary (2012) 15(1):93–6. doi: 10.1007/s11102-010-0219-6
26. Bayraktar S, Bassini W, Goodman M. Primary pituitary lymphoma: idiopathic anasarca with relapse in bone marrow only. Acta Haematol (2010) 123(2):121–5. doi: 10.1159/000272920
27. Hayasaka K, Koyama M, Yamashita T. Primary pituitary lymphoma diagnosis by FDG-PET/CT. Clin Nucl Med (2010) 35(3):205. doi: 10.1097/RLU.0b013e3181cc64a2
28. Martinez JH, Davila Martinez M, Mercado de Gorgola M, Montalvo LF, Tome JE. The coexistence of an intrasellar adenoma, lymphocytic hypophysitis, and primary pituitary lymphoma in a patient with acromegaly. Case Rep Endocrinol (2011) 2011:941738–. doi: 10.1155/2011/941738
29. Papanastasiou L, Pappa T, Dasou A, Kyrodimou E, Kontogeorgos G, Samara C, et al. Case report: Primary pituitary non-Hodgkin’s lymphoma developed following surgery and radiation of a pituitary macroadenoma. Hormones (Athens) (2012) 11(4):488–94. doi: 10.14310/horm.2002.1382
30. Rainsbury P, Mitchell-Innes A, Clifton N, Khalil H. Primary lymphoma of the pituitary gland: an unusual cause of hemianopia in an immunocompetent patient. JRSM Short Rep (2012) 3(8):55. doi: 10.1258/shorts.2012.012067
31. Wiens AL, Hagen MC, Bonnin JM, Rizzo KA. T-cell lymphoblastic lymphoma/leukemia presenting as a pituitary mass lesion: a case report and review of the literature. Neuropathology (2012) 32(6):668–74. doi: 10.1111/j.1440-1789.2012.01314.x
32. Wilkie MD, Al-Mahfoudh R, Javadpour M. A rare case of primary sellar lymphoma presenting a diagnostic challenge. Br J Neurosurg (2012) 26(5):782–3. doi: 10.3109/02688697.2012.674578
33. Ravindra VM, Raheja A, Corn H, Driscoll M, Welt C, Simmons DL, et al. Primary pituitary diffuse large B-cell lymphoma with somatotroph hyperplasia and acromegaly: case report. J Neurosurg (2017) 126(5):1725–30. doi: 10.3171/2016.5.JNS16828
34. Ban VS, Chaudhary BR, Allinson K, Santarius T, Kirollos RW. Concomitant Primary CNS Lymphoma and FSH-Pituitary Adenoma Arising Within the Sella. Entirely Coincidental? Neurosurgery (2017) 80(1):E170–e5. doi: 10.1093/neuros/nyw003
35. Gupta RK, Saran RK, Srivastava AK, Jagetia A, Garg L, Sharma MC. T cell lymphoblastic lymphoma/leukemia within an adrenocorticotropic hormone and thyroid stimulating hormone positive pituitary adenoma: A cytohistological correlation emphasizing importance of intra-operative squash smear. Neuropathology (2017) 37(4):358–64. doi: 10.1111/neup.12375
36. Velho V, Guha A, Naik H, Bhople L, Jain N. Unravelling Hitherto Unreported Masses Camouflaged as Pituitary Macro Adenomas. Asian J Neurosurg (2018) 13(4):1005–7. doi: 10.4103/ajns.AJNS_17_17
37. Silfen ME, Garvin JH Jr., Hays AP, Starkman HS, Aranoff GS, Levine LS, et al. Primary central nervous system lymphoma in childhood presenting as progressive panhypopituitarism. J Pediatr Hematol Oncol (2001) 23(2):130–3. doi: 10.1097/00043426-200102000-00013
38. Baleydier F, Galambrun C, Manel AM, Guibaud L, Nicolino M, Bertrand Y. Primary lymphoma of the pituitary stalk in an immunocompetent 9-year-old child. Med Pediatr Oncol (2001) 36(3):392–5. doi: 10.1002/mpo.1094
39. Capra M, Wherrett D, Weitzman S, Dirks P, Hawkins C, Bouffet E. Pituitary stalk thickening and primary central nervous system lymphoma. J Neurooncol (2004) 67(1–2):227–31. doi: 10.1023/B:NEON.0000021863.06620.48
40. Huang AP, Wu MZ, Kuo MF. Pediatric primary lymphoma of the pituitary stalk: a different disease entity from the adult form? J Formos Med Assoc (2013) 112(3):171–2. doi: 10.1016/j.jfma.2012.02.005
41. Villano JL, Koshy M, Shaikh H, Dolecek TA, McCarthy BJ. Age, gender, and racial differences in incidence and survival in primary CNS lymphoma. Br J Cancer (2011) 105(9):1414–8. doi: 10.1038/bjc.2011.357
42. Batchelor TT. Primary central nervous system lymphoma: A curable disease. Hematol Oncol (2019) 37 Suppl 1:15–8. doi: 10.1002/hon.2598
43. Grommes C, DeAngelis LM. Primary CNS Lymphoma. J Clin Oncol (2017) 35(21):2410–8. doi: 10.1200/JCO.2017.72.7602
44. Gaudio F, Pedote P, Niccoli Asabella A, Ingravallo G, Sindaco P, Alberotanza V, et al. Bone Involvement in Hodgkin’s Lymphoma: Clinical Features and Outcome. Acta Haematol (2018) 140(3):178–82. doi: 10.1159/000490489
45. Stacy RC, Jakobiec FA, Herwig MC, Schoenfield L, Singh A, Grossniklaus HE. Diffuse large B-cell lymphoma of the orbit: clinicopathologic, immunohistochemical, and prognostic features of 20 cases. Am J Ophthalmol (2012) 154(1):87–98.e1. doi: 10.1016/j.ajo.2012.01.021
46. Onda K, Wakabayashi K, Tanaka R, Takahashi H. Intracranial malignant lymphomas: clinicopathological study of 26 autopsy cases. Brain Tumor Pathol (1999) 16(1):29–35. doi: 10.1007/BF02478899
47. Royer-Perron L, Hoang-Xuan K, Alentorn A. Primary central nervous system lymphoma: time for diagnostic biomarkers and biotherapies? Curr Opin Neurol (2017) 30(6):669–76. doi: 10.1097/WCO.0000000000000492
48. Mulazzani M, Huber M, Borchard S, Langer S, Angele B, Schuh E, et al. APRIL and BAFF: novel biomarkers for central nervous system lymphoma. J Hematol Oncol (2019) 12(1):102. doi: 10.1186/s13045-019-0796-4
49. Rimelen V, Ahle G, Pencreach E, Zinniger N, Debliquis A, Zalmaï L, et al. Tumor cell-free DNA detection in CSF for primary CNS lymphoma diagnosis. Acta Neuropathol Commun (2019) 7(1):43. doi: 10.1186/s40478-019-0692-8
50. Waldera-Lupa DM, Poschmann G, Kirchgaessler N, Etemad-Parishanzadeh O, Baberg F, Brocksieper M, et al. A Multiplex Assay for the Stratification of Patients with Primary Central Nervous System Lymphoma Using Targeted Mass Spectrometry. Cancers (Basel) (2020) 12(7):1732. doi: 10.3390/cancers12071732
51. Brandsma D, Bromberg JEC. Primary CNS lymphoma in HIV infection. Handb Clin Neurol (2018) 152:177–86. doi: 10.1016/B978-0-444-63849-6.00014-1
52. Hoang-Xuan K, Bessell E, Bromberg J, Hottinger AF, Preusser M, Rudà R, et al. Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: guidelines from the European Association for Neuro-Oncology. Lancet Oncol (2015) 16(7):e322–e32. doi: 10.1016/S1470-2045(15)00076-5
53. von Baumgarten L, Illerhaus G, Korfel A, Schlegel U, Deckert M, Dreyling M. The Diagnosis and Treatment of Primary CNS Lymphoma. Dtsch Arztebl Int (2018) 115(25):419–26. doi: 10.3238/arztebl.2018.0419
54. Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov (2017) 7(9):1018–29. doi: 10.1158/2159-8290.CD-17-0613
55. Mappa S, Marturano E, Licata G, Frezzato M, Frungillo N, Ilariucci F, et al. Salvage chemoimmunotherapy with rituximab, ifosfamide and etoposide (R-IE regimen) in patients with primary CNS lymphoma relapsed or refractory to high-dose methotrexate-based chemotherapy. Hematol Oncol (2013) 31(3):143–50. doi: 10.1002/hon.2037
56. Raizer JJ, Rademaker A, Evens AM, Rice L, Schwartz M, Chandler JP, et al. Pemetrexed in the treatment of relapsed/refractory primary central nervous system lymphoma. Cancer (2012) 118(15):3743–8. doi: 10.1002/cncr.26709
57. Kunishio K, Okada M, Matsumoto Y, Nagao S. Preliminary individual adjuvant chemotherapy for primary central nervous system lymphomas based on the expression of drug-resistance genes. Brain Tumor Pathol (2004) 21(2):57–61. doi: 10.1007/BF02484511
58. Houillier C, Soussain C, Ghesquieres H, Soubeyran P, Chinot O, Taillandier L, et al. Management and outcome of primary CNS lymphoma in the modern era: An LOC network study. Neurology (2020) 94(10):e1027–39. doi: 10.1212/WNL.0000000000008900
59. Graham MS, DeAngelis LM. Improving outcomes in primary CNS lymphoma. Best Pract Res Clin Haematol (2018) 31(3):262–9. doi: 10.1016/j.beha.2018.07.006
Keywords: primary pituitary lymphoma, large B-cell lymphoma, sellar mass, primary CNS lymphoma, radiotherapy, chemotherapy
Citation: Duan L, Liu J, Zhang Y, Cui L, Zhai X, Pan B, Lu L, Pan H, Yao Y and Zhu H (2021) Primary Pituitary Lymphoma in Immunocompetent Patients: A Report on Two Case Studies and the Review of Literature. Front. Endocrinol. 11:562850. doi: 10.3389/fendo.2020.562850
Received: 16 May 2020; Accepted: 21 December 2020;
Published: 04 February 2021.
Edited by:
Monica Livia Gheorghiu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Giulia Cossu, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandMirtha Adriana Guitelman, Durand Hospital, Argentina
Copyright © 2021 Duan, Liu, Zhang, Cui, Zhai, Pan, Lu, Pan, Yao and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Yao, ZnJlZXRpZ2VyeWFvQDE2My5jb20=; Huijuan Zhu, c2hlbmd4aW4yMDA0QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship