 Sara Della Torre
Sara Della Torre- Department of Pharmaceutical Sciences, University of Milan, Milan, Italy
There is extensive evidence supporting the interplay between metabolism and immune response, that have evolved in close relationship, sharing regulatory molecules and signaling systems, to support biological functions. Nowadays, the disruption of this interaction in the context of obesity and overnutrition underlies the increasing incidence of many inflammatory-based metabolic diseases, even in a sex-specific fashion. During evolution, the interplay between metabolism and reproduction has reached a degree of complexity particularly high in female mammals, likely to ensure reproduction only under favorable conditions. Several factors may account for differences in the incidence and progression of inflammatory-based metabolic diseases between females and males, thus contributing to age-related disease development and difference in life expectancy between the two sexes. Among these factors, estrogens, acting mainly through Estrogen Receptors (ERs), have been reported to regulate several metabolic pathways and inflammatory processes particularly in the liver, the metabolic organ showing the highest degree of sexual dimorphism. This review aims to investigate on the interaction between metabolism and inflammation in the liver, focusing on the relevance of estrogen signaling in counteracting the development and progression of non-alcoholic fatty liver disease (NAFLD), a canonical example of metabolic inflammatory-based liver disease showing a sex-specific prevalence. Understanding the role of estrogens/ERs in the regulation of hepatic metabolism and inflammation may provide the basis for the development of sex-specific therapeutic strategies for the management of such an inflammatory-based metabolic disease and its cardio-metabolic consequences.
Introduction
Liver Metabolism and Inflammation: Two Sides of the Same Coin
The liver is one of the most complex organs in the body, performing a multitude of functions, including the macronutrient metabolism, glucose, lipid and cholesterol homeostasis, protein and amino acid metabolism, detoxification and drug metabolism (1). The liver is also an immunological organ, being responsible for the production of acute phase proteins, complement components, cytokines and chemokines, and contains large, diverse populations of resident immune cells (2). Under physiological conditions, the liver is constantly exposed to dietary and gut-derived bacterial products with inflammatory potential and is engaged in tissue remodeling, all process requiring a tight regulation of the inflammatory response to maintain tissue and organ homeostasis and to redistribute the energy resources during the rising of an inflammatory response (3–5).
A close and coordinated regulation of metabolic and immune responses has been conserved through evolution, with lower and higher organisms sharing common ancestral structures and common key regulatory molecules and signaling systems (6). However, the integration between metabolic and inflammatory pathways have been set up in the context of nutrient limitations and have not evolved and adapted to the current habits and lifestyles, where overnutrition and the reduced physical activity lead to chronic disturbance of metabolic homeostasis and to aberrant immune responses (4, 7). The metabolic overload and the lack of metabolic homeostasis typical of obesity and obesity-associated metabolic diseases trigger a sustained and chronic inflammatory response, that, by converse, can disrupt systemic metabolic functions, thus fostering a vicious cycle that favors the progression of metabolic diseases (4). In the liver, the inability to resolve inflammation may lead to chronic pathological inflammation and to a disrupted tissue homeostasis, which can promote hepatic steatosis, fibrosis, cirrhosis, and liver failure (3, 5, 8–10).
Although the higher prevalence of obesity among female population, women result to be somewhat protected from the obesity-associated cardio-metabolic consequences, such as non-alcoholic fatty liver disease (NAFLD), at least until menopause (11). The reason of that likely relies on the tight regulation of metabolic and inflammatory processes that may have reached its maximum degree of complexity in the liver of female mammals, where the regulation of hepatic metabolism is under the control of sexual hormones, estrogens in particular, and is subjugated to the reproductive needs (12–15). In view of the tight link between energy homeostasis and reproduction, liver diseases show a sex-specific prevalence (16, 17) and are associated with reproductive dysfunctions in women (14, 18). Nowadays, changes in dietary and lifestyle habits as well as the increased lifespan of women, that spend more than 1/3 of their lives in post-menopause, can explain the increased incidence in female population of cardio-metabolic diseases, which are previously considered male-prevalent (16, 19, 20). In this view, research programs aimed to unravel the role of estrogen signaling in the regulation of metabolic and inflammatory processes may have a significant impact on the design of new therapies that can counteract the development of NAFLD and the associated cardio-metabolic consequences in a sex-specific fashion.
NAFLD, a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence
With respect to young, fertile women, men and post-menopausal women show an increased incidence of metabolic and inflammatory-based liver diseases (14, 18, 21). Among them, a canonical example is NAFLD, a syndrome characterized by excessive triglyceride (TG) accumulation within hepatocytes (22), that has reached epidemic proportions and represents an increasing public health issue due to its emerging association with several extra-hepatic diseases (23, 24), cardiovascular diseases (CVDs) in particular (25, 26). Indeed, cardiovascular mortality represents the commonest cause (45%) of death in NAFLD patients, followed by cancer (36%) and then liver-related mortality (7%) (27).
NAFLD is closely linked with peripheral insulin resistance and hepatic insulin resistance (28–30), a condition where insulin fails to suppress hepatic glucose production (HPG, which accounts for 90% of endogenous glucose production) but promotes lipid synthesis leading to hyperglycemia, hypertriglyceridemia and hepatic steatosis (31). Therefore, there is a significant correlation between HPG and the extent of liver fat in NAFLD patients (32) as well as between NAFLD and other metabolic insulin-resistant disorders such as type 2 diabetes mellitus (T2DM) (33, 34) and sarcopenia (35). Notably, women show an improved glycemic control, a greater peripheral and hepatic insulin sensitivity and a reduced HPG with respect to men (36–38), likely a consequence of a sex-dimorphic regulation of glucose homeostasis (39), to which the hepatic signaling of sexual hormones strongly contributes (40, 41), thus leading to a different susceptibility to NAFLD between the two sexes.
In the liver of NAFLD patients, TG accumulation it is due to increased de novo lipogenesis (DNL) (42, 43), increased delivery of fatty acids (FAs) to the liver (42, 44), and decreased lipid clearance consequent to impaired FA oxidation and lower lipid secretion (45, 46). Hepatocellular damage and fat-derived factors mediate the local activation of a pro-inflammatory response by hepatocytes and non-parenchymal cells, including Kupffer cells (KCs) and hepatic stellate cells (HSCs) (4, 47–49), that promote the recruitment of other immune cells, including neutrophils, T-lymphocytes and, mainly, macrophages (50).
The impaired mitochondrial oxidation (42, 51) and the up-regulation of both peroxisomal β-oxidation (52) and microsomal ω-oxidation (53) of FAs lead to chronic oxidative stress and result in the generation of reactive oxidative species (ROS) within the hepatocytes (42, 45). In addition to mitochondria—that are considered the most relevant source of ROS—and to peroxisomes and microsomes, the endoplasmic reticulum stress and enzymes as NADPH oxidase (NOX), cytochrome P450 2E1 (CYP2E1), cyclooxygenases, and lipoxygenases also produce ROS (54). According to the most valuable theories (22), the production of lipotoxic lipid intermediates and the excessive production of ROS further trigger a pro-inflammatory response that contributes to the progression of NAFLD to non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis and hepatocellular carcinoma (HCC) (46, 55, 56). Pro-inflammatory cytokines released by immune cells intensify the inflammatory process, that hinders the liver to orchestrate a proper tissue regeneration by replacing the hepatocytes subjected to cell death or apoptosis, as occurs under physiological conditions (57). Possibly as an unsuccessful effort against liver injury and tissue regeneration, HSCs become activated and differentiate into myofibroblasts, that, in turn, express actin and diverse types of collagen, leading to extracellular matrix deposition and fibrosis and liver degeneration (58–60).
In the presence of increased flux of free fatty acids (FFAs) and of chronic, low-grade inflammation, the liver acts as both a target of and a contributor to systemic chronic inflammation, triggering or boosting the progression of NAFLD and several extra-hepatic diseases (23), including atherosclerosis (61–63), cardiovascular diseases (62–64), chronic kidney disease (65), osteoporosis (66), and inflammatory bowel disease (67).
From Metabolism to Liver Injury: Role of Obesity and Nutrients in NAFLD Development
Although several factors might contribute to hepatic steatosis, including genetic and epigenetic factors (68, 69), obesity represents the main trigger of NAFLD development and progression. However, independently of energy intake, also the macronutrient composition of the diet can be associated with NAFLD/NASH development (70). Different epidemiological studies have, therefore, demonstrated that dietary habits may directly promote NAFLD/NASH, by modulating hepatic TG accumulation and antioxidant activity and, indirectly, by affecting insulin sensitivity and the postprandial TG metabolism (70). Several studies have identified the overconsumption of fats (saturated fats and trans-fats, in particular) and sugars (fructose, in particular) as the main nutritional mediators of NAFLD development (71–73), while the role of proteins and amino acids in NAFLD etiology has been less investigated and still raises controversies (70).
Obesity
The rising trends in obesity has been linked with the increase in the incidence and severity of NAFLD, with an estimated global prevalence of 25–30% worldwide, rising up to 90% in morbidly obese patients (26, 74). In obese NAFLD patients, ~60% of hepatic FAs are derived from FFAs released by the adipose tissue as a consequence of an enhanced lipolysis and taken up by the liver via the increased uptake mediated by CD36 (cluster of differentiation 36) (75–77). To a less extent, hepatic lipid deposits derive from dietary FAs (~15%) and from increased synthesis of new lipids (~25%) from ingested carbohydrates that reach to a greater extent the liver due to the insulin resistance of the muscle (43, 75, 78). The exposure of hepatocytes to high lipid and carbohydrate levels promotes lipotoxicity and glucotoxicity, that, in turn, lead to mitochondrial defects, endoplasmic reticulum stress and oxidative stress (45, 79). The ectopic accumulation of lipid toxic intermediates triggers the activation of inflammatory pathways, cellular dysfunction, and lipoapoptosis, all features favoring NAFLD progression and liver injury (22, 80, 81).
Obesity also affects the liver through the unbalanced secretion of adipokines, exerting different effects on insulin resistance, hepatic steatosis, inflammation and fibrosis (82). For example, the obesity-associated reduction of adiponectin levels promotes insulin resistance and hepatic steatosis, while the increased levels of leptin foster hepatic inflammation (82). In the adipose tissue of obese people, the infiltration and activation of immune cells (macrophages, B-lymphocytes, T-lymphocytes and neutrophils) that produce pro-inflammatory cytokines (e.g., interleukin 1β, IL-1β; interleukin 6, IL-6; tumor necrosis factor-alpha, TNF-α) impair the dynamic antagonism between adipokines and cytokines and facilitate the progression of steatosis, inflammation and fibrosis (82).
Under obesogenic-like conditions, in addition to adipose tissue, the impaired regulation of metabolic process and signaling pathways in other tissues showing a strong interplay with the liver, including the skeletal muscle (83–85) and the gut-microbiota (86, 87), can further negatively affect the hepatic metabolic homeostasis and boost the progression of NAFLD.
In addition to genetic factors (88, 89), estrogen signaling strongly contributes to sex differences in obesity and associated cardio-metabolic consequences such as NAFLD (20, 21, 39, 90–93). With respect to pre-menopausal women, lean and obese men tend to accrue more visceral fat, that, having a greater lipolytic potential than subcutaneous adipose tissue, strongly contributes to increased FFA flux to the liver, where FFAs mediate insulin resistance and NAFLD pathogenesis (94, 95). After menopause, there is a redistribution of fat toward visceral depots and a lower inhibition of adipose lipolysis, all changes that fuel the FFA flux to the liver and increase the risk of developing NAFLD in post-menopausal women (92, 94).
Sex-specific and estrogen-mediated differences in obesity-induced NAFLD are ascribable also to impaired regulation of metabolic process in extrahepatic tissues showing a cross-talk with the liver, such as the adipose tissue and the skeletal muscle, that under obesogenic conditions display increased insulin resistance and increased inflammation that might further aggravate the hepatic dysmetabolism (96–106).
Dietary Sugars
Over the past century, the increased intake of added sugars, fructose in particular, is associated with increased incidence of hepatic steatosis and liver inflammation (107–109). Unlike glucose, ingested fructose by-passes the rate-limiting step of glycolysis and is preferentially metabolized by the liver, where it stimulates hepatic DNL acting mainly through SREBP1c (sterol regulatory element-binding protein 1c) and ChREBP (carbohydrate responsive element-binding protein), inhibits the mitochondrial β-oxidation of long-chain FAs, induces endoplasmic reticulum stress, and promotes TG formation and hepatic steatosis (73, 110, 111). Owing to the molecular instability of its five-membered furanose ring, fructose promotes protein fructosylation and formation of ROS, yielding to hepatocellular damage and to the development of a pro-inflammatory response (73).
Even after a single meal, fructose strongly up-regulates an inflammatory cascade through increased hepatic JNK (c-Jun N-terminal kinase) activity and induces hepatic insulin resistance, all effects occurring specifically in hepatocytes (112). Recent reports suggest that fructose can also induce liver inflammation by acting directly on inflammatory cells, where it drives the production of pro-inflammatory cytokines (IL-6 and IL-1β) that further promotes an aberrant lipid metabolism (107, 109, 112, 113). The high intake of fructose can also lead to gut microbiota dysbiosis and contribute to inflammation, insulin resistance and NAFLD progression (114).
The consequences of extended fructose consumption on liver health are different between the two sexes, with males being more responsive to fructose and showing higher hepatic postprandial DNL and higher prevalence of NAFLD compared to females (115–117). These differences can likely be a direct consequence of the sex-specific modulation of glucose metabolism (39, 118) and of the specific relevance of estrogen signaling in regulating hepatic glucose metabolism and in promoting insulin sensitivity (119–121), acting also through FGF21 (fibroblast growth factor 21) signaling (122, 123). Accordingly, high fructose intake exacerbates the progression of NAFLD in ovariectomized (OVX) female mice by enhancing liver cell destruction, macrophage accumulation, and progression of fibrosis, all negative effects that can be reverted by 17β-estradiol supplementation (121).
Dietary Fatty Acids
The increased intake of dietary FAs is strongly associated with obesity and the development of obesity-associated metabolic diseases, such as NAFLD (124–127). Dietary regimens enriched in fats contribute to increase the hepatic pool of FAs, where they promote DNL and the generation of lipotoxicity through a sustained oxidation (128). Dietary FAs influence NAFLD pathogenesis also by modulating the gene transcription of specific enzymes and regulating various metabolic pathways involved in lipid metabolism (129, 130). Modern western diets are particularly enriched in saturated and trans FAs that are particularly detrimental for hepatic health, because they induce insulin resistance and fatty liver and promote liver injury by altering the composition of plasma cell membrane, thus impairing cellular homeostasis and amplifying the already sustained inflammatory signaling, that, in turn, boosts insulin resistance and apoptosis (127, 128, 131, 132). Conversely, diets enriched in ω3 polyunsaturated FAs (ω3 PUFAs), such as the Mediterranean diet (133), may be particularly effective in counteracting the early stages of NAFLD (134), limiting insulin resistance, oxidative stress, DNL and TG deposition in the liver (135, 136) and preventing the development of liver-associated cardio-metabolic diseases (137). ω3 PUFAs exert anti-inflammatory actions by preventing the alteration of cell membrane phospholipid composition and the disruption of lipid rafts, by inhibiting the activation of NF-κB (nuclear factor-kappa B), by reducing expression of inflammatory genes and by activating PPARγ (peroxisome proliferator-activated receptor γ) (138).
With respect to the female counterparts, men and male rodents show a higher propensity of developing hepatic steatosis/NAFLD that derives from the combination of increased FA import, DNL, and storage of lipids within the liver and lower dietary FA oxidation and secretion (91, 139). By comparing control and LERKO (liver-specific Estrogen Receptor alpha KO) mice, a recent study demonstrates that the liver ability of females to cope with the excess of dietary lipids strongly relies on the activity of hepatic ERα, that confers to females a higher metabolic flexibility (91).
Different dietary fatty acid regimens can also change the composition and the ratio of FAs in liver plasma cell membrane in gender-specific manner, another mechanism that can further explain the sex-specific incidence of NAFLD (140).
Furthermore, maternal high-fat diet can promote and even program hepatic steatosis/NAFLD and liver inflammation of offspring in a sexually dimorphic manner by altering gut microbiota (141) that has been shown relevant for the achievement of hepatic sexual dimorphism (142).
Dietary Amino Acids
While the hazardous effects of high-carbohydrate and high-fat diets upon hepatic structure/function are well-recognized, the potential effects of dietary regimens enriched in proteins and amino acids (AAs) on hepatic health are partly clarified and still raise controversies. Indeed, while several studies show a beneficial role exerted by high-protein diets in reducing body weight and in reverting hepatic steatosis, other studies suggest that high-protein diets can instead promote the development of NAFLD (143). The reasons of these contradictory effects on liver health can be ascribable to differences in dietary regimens (e.g., diet composition and protein source) and on the functional status of the liver (143).
Among AAs, branched chain amino acids (BCAAs: leucine, isoleucine, and valine), that account for 20% of total protein intake (144), exert beneficial effects on hepatic health as they alleviate hepatic steatosis and liver injury and prevent hepatic fibrosis and the development of HCC in NASH mouse models (145, 146). By contrast, elevated circulating BCAAs are strongly associated with several metabolic disorders, including obesity and insulin-resistant metabolic diseases (147, 148). NAFLD patients show a low hepatic content of BCAAs, that changes with the progression of the pathology, likely as a consequence of impaired expression of hepatic BCAA-degrading enzymes (149, 150). Furthermore, a recent study demonstrates that plasma BCAA levels display sex-dimorphic changes with increasing severity of NAFLD, independently of BMI, insulin resistance and age (151), suggesting a sex-specific regulation of BCAA metabolism and/or a sex-specific role of BCAAs in NAFLD development, as supported by pre-clinical studies (91). Indeed, although their causative or associative role has not yet clarified, among AAs and several other metabolites, BCAAs result the pathway most affected in the liver of a mouse model of diet-induced obesity (91). Notably, the decrease in AAs and, especially, in BCAAs correlates with increased lipid deposition in the liver of male, but not female mice; in fact, when exposed to an excess of dietary lipids, female mice, contrary to males, preserve the hepatic AA homeostasis, an effect associated with the ability to counteract liver lipid deposition (91), suggesting that the metabolism of BCAAs might have a key role in driving hepatic steatosis in a sex-specific fashion. The female-specific ability to preserve BCAA homeostasis and counteract liver lipid deposition is dependent on hepatic ERα, as it is lost in LERKO female mice (91), and it is likely a consequence of an higher metabolic flexibility conferred by hepatic ERα, that, in the female liver, adapts the hepatic metabolism to hormonal status and to nutrient availability, amino acids in particular (13, 15, 152, 153).
Sex Differences in NAFLD Onset, Development and Progression
NAFLD is more common in men, in whom it has a 2.0–3.5-fold higher prevalence than in fertile women; however, after menopause the incidence of NAFLD increases significantly to reach the levels seen in men, owing to the putative protective effect of estrogens (14, 17, 154). Indeed, gender-specific prevalence of NAFLD is related to age: while men commonly display an increasing prevalence of NAFLD during adulthood from young to middle-age, the prevalence of NAFLD in women occurs ~10 years later than in men, rising after the age of 50 years, peaking at 60–69 years, and declining after 70 years (16). This last trend indicates that the increased incidence of NAFLD in aging women relies more on the lack of estrogens than on aging per se, even though aging may exacerbate the progression of NAFLD by negatively impacting on metabolic (155) and inflammatory processes (156). According to this view, young oophorectomized women (157) as well as young women suffering of other reproductive dysfunctions characterized by altered estrogen levels (such as Polycystic ovary syndrome, PCOS) (14, 158) show increased incidence of NAFLD with respect to young fertile women.
Even if the exact etiology of NAFLD in post-menopausal women is still unclear, the association of NAFLD with the cessation of ovarian activity and with other ovarian dysfunctions such as PCOS (159) suggests that estrogens protect against its development and progression. Notably, with respect to their control counterparts, pre-menopausal, post-menopausal, and PCOS women with NAFLD exhibit a significantly lower concentration of serum 17β-estradiol, which is the principal active estrogen (158). Accordingly, hormone replacement therapy (HRT) reduces the risk of developing NAFLD for post-menopausal women (16, 160).
In pediatric populations, NAFLD prevalence is higher in boys than in girls (161), even though sex differences are less relevant with respect to adult population, suggesting that the achievement of complete sexual differentiation is required to accomplish the sex-specific prevalence and features of such a pathology. Such a hypothesis is sustained by several studies showing a strict association between puberty and features of NAFLD (162) and between earlier age at menarche and the prevalence of NAFLD later in life (163–165).
Although the prevalence of NAFLD is undoubtfully higher in men than women, less clear is the sex-specific incidence of liver injury associated with NAFLD progression to NASH and fibrosis. In fact, some studies suggest that women have a lower risk of developing NASH and fibrosis (166–169), while others do not find differences between the two sexes (170, 171) or, even, indicate that women are more susceptible than men to an inflammatory-driven degeneration of NAFLD toward more harmful conditions (172–176). Most of these studies, however, has several limitations and important potential bias, as they do not differentiate between pre- and post-menopausal women or do not often consider the timing/duration of menopause, which may give confounding and contradictory results (12). By converse, consistent with the hypothesis that estrogens exert beneficial effects on liver health, menopause, premature menopause and prolonged estrogen deficiency have been independently associated with significant fibrosis in women with NAFLD (177, 178).
NAFLD incidence is increased in obese people suffering of other obesity-associated cardio-metabolic diseases; nevertheless, several mechanistic and longitudinal studies have indicated that NAFLD is an independent risk factor for atherogenesis (179–181) and CVDs (23, 63, 182–184) apart from other metabolic disorders. Although still debated, the causal relationship independent of other metabolic risk factors seems to rely on the systemic inflammatory milieu initiated in part by liver-secreted cytokines and molecules (23, 63). In addition to enhanced inflammation, a growing body of evidence indicates that, along with NAFLD progression, the alteration of cholesterol and lipoprotein metabolism (185–187) and the excessive generation of ROS may lead to the accumulation of oxidized low-density lipoprotein (ox-LDL) in the liver (188–190) and to macrophages transformation into foam cells, which is a hallmark of atherosclerosis.
Given such a correlation between NAFLD and CVDs, it is not surprising that, while in the general population women are less prone to CVDs under the age of 50 years, after menopause, women lose this protection and show a higher risk of developing NAFLD and cardio-metabolic associated consequences (191–193).
Sex Differences in the Regulation of Metabolism and Inflammation in the Liver
Sex-specific prevalence, progression and outcomes of hepatic diseases and their associated co-morbidities might be considered the resultant of sex differences typifying the male and female liver phenotype.
The liver is the major metabolic organ in mammals with the highest degree of sexual dimorphism (194, 195). Most of the sex differences in liver gene expression are dictated by the temporal pattern of circulating growth hormone (GH), which is sex dependent (highly pulsatile in males and more continuous in females) (196, 197) and under gonadal control (198–200). GH regulates the sexually dimorphic patterns of a large number of liver-expressed genes, including various plasma and urinary proteins, cytochromes P450 (CYPs, which contribute to sex differences in sex steroid hormone metabolism), enzymes devoted to steroid and foreign compound metabolism, and various receptors and signaling molecules involved in a broad range of physiological processes (194, 197, 201, 202). GH pattern carries out its sexual differentiating action of liver functions through multiple intracellular signaling pathways, including the transcription factor signal transducer and activator of transcription 5b (STAT5b) (203–205), hepatocyte nuclear factors 3β, 4α and 6 (HNF3β, HNF4α, HNF6) (206, 207) as well as their signaling cross-talk (208–210). GH dimorphic action on hepatic gene expression is also dependent on sex-specific regulation of DNA methylation and chromatin structure (197, 205, 211–215), resulting in major changes in sex-based liver functions. The hepatic responsiveness to GH dimorphic action changes during development (216–218) and remains dynamic during adult life (205, 217), charging the liver of the possibility to adapt its functions to the needs of the organism throughout life.
GH and its signaling pathway, acting mainly through insulin-like growth factor-I (IGF-I), regulate lipid metabolism in the liver (219) and play an important role in antagonizing NAFLD, by directly reducing DNL in the hepatocytes and by inactivating HSCs, therefore limiting fibrosis (220). According to this, GH deficiency in adults and in obese children is associated with increased prevalence of NAFLD and NASH, while GH replacement therapy improves these conditions (220, 221). In mice, the liver-specific ablation of the GH receptor (GHR) increases lipid uptake and DNL, resulting in hepatic steatosis that cannot be reverted by IGF-1 treatment (219).
GH and its signaling may have a key role also in the liver disease progression, by regulating excessive inflammation and allowing liver regeneration (222). By converse, during inflammation, the liver can become resistant to GH actions, through mechanisms involving proinflammatory cytokines such as IL-6, TNF-α, and IL-1β (223–226), thus worsening metabolic alterations.
In addition to the well-known dimorphic activity of GH, hepatic sexual dimorphism depends on several other factors, including genetic (213, 215) and epigenetic (227, 228) factors, diet (141, 229, 230), circadian rhythm (231, 232), gut microbiota (142) and sexual hormones (12, 152, 233).
In spite of the fact that our knowledge of the entity of hepatic sexual dimorphism under physio-pathological conditions remains very limited (12), several evidences, including the sex-specific prevalence, incidence, progression and outcomes of hepatic diseases such as NAFLD (17, 234, 235), indicate that, among the factors contributing to hepatic sexual dimorphism, estrogens and their receptors recover a key role.
Estrogens can regulate sex differences in the liver through direct and indirect mechanisms, that are both affected by and able to prevail over sex-based genetic background and sexual hormone-dependent regulatory activities. Estrogen activity can contribute to the sexual dimorphism of the liver directly (21, 91, 152, 236, 237) and indirectly, by regulating GH action, both in the central nervous system and locally (198, 233, 238–240). Several experimental models with impaired/lost estrogen signaling support the involvement of estrogen dependent pathways in the regulation of hepatic metabolism, also in a sexually dimorphic fashion (14, 91, 152, 153, 241).
Estrogen-mediated contribution of hepatic sexual dimorphism likely arises from different metabolic costs of reproduction and from higher metabolic flexibility acquired and perfected through evolution by the female liver of mammals to adapt the hepatic metabolism to nutrient availability to sustain the energy needs of reproductive function (12, 13, 152, 242). In view of these evidences, although androgens and androgen receptor (AR) contribute to the sex-based hepatic phenotype in a direct or indirect fashion, by acting on GH dependent pathways (200, 215, 233, 243) and by regulating the accessibility of DNA to several transcription factors through chromatin remodeling (244, 245), this review will focus in particular on the role of estrogen signaling in the regulation of metabolic-driven inflammatory process at the basis of NAFLD development and progression.
NAFLD and Liver Inflammation
In NAFLD, the increased flux of FFAs, the generation of lipotoxicity and oxidative stress and insulin resistance concur in activating JNK and NF-κB signaling pathways, resulting in the increased production of pro-inflammatory cytokines, including IL-6 and TNF-α (4, 48, 49, 246). JNK is a member of mitogen activated protein kinases, which activation in fatty liver is associated with insulin resistance, activation of apoptosis and development of NASH (247–249). JNK pathway is differentially regulated between males and females during liver injury (250, 251), likely through an estrogen- and ERα-mediated inhibition of lipotoxicity-induced hepatic mitochondrial oxidative stress and, in turn, of JNK signaling pathway, thus avoiding the over-regulation of pro-inflammatory and pro-apoptotic process (252).
NF-κB is a transcription factor involved in innate and adaptive immune responses playing an essential role in the regulation of inflammatory signaling pathways in the liver. Under normal conditions, NF-κB is sequestered in the cytoplasm by the binding with IκB proteins; in response to stimulation by pathogenic stimuli, the degradation of the NF-κB inhibitor α (IκBα) allows the translocation of NF-κB to the nucleus, where it induces the expression of target genes encoding inflammatory mediators, such as TNF-α and IL-6 (4, 253). Persistent activation of the NF-κB pathway in the liver leads to a chronic inflammatory state and to insulin resistance, that further promote the development of NAFLD and NASH (81, 254). NF-κB and its downstream signaling pathway are under the inhibitory control of estrogen signaling (255–257), a regulation that accounts for sex- and menopause-associated over-regulation of hepatic inflammatory process and for the progression of NAFLD toward more harmful conditions such as NASH, fibrosis and HCC (16, 49, 230, 258, 259).
Homeostatic inflammation is tightly regulated by mechanisms acting to resolve inflammation in order to avoid excessive inflammation and pathological consequences. In the liver, the propagation or the resolution of inflammation mostly relies on the polarization abilities of KCs (the resident macrophages) and of the recruited macrophages (260, 261). Once activated by exogenous or endogenous danger signals, macrophages undergo pro-inflammatory or anti-inflammatory and reparative phenotype, respectively promoting or attenuating hepatic steatosis and liver injury in NAFLD (50, 258, 260, 261). As occurs in other physio-pathoplogical contexts (262, 263), estrogens might promote the skewing of pro-inflammatory macrophages toward anti-inflammatory macrophages and accelerate the resolution of inflammation and the tissue repair in the liver, thus contributing to limit NAFLD progression in pre-menopausal women with respect to men and post-menopausal women (258). Accordingly, a longer duration of estrogen deficiency increases the risk of developing fibrosis among post-menopausal women with NAFLD (177) as well as in OVX female mice fed with HFD (264).
Although the FA-induced activation of NOD-like receptor (NLR) NLRP3 inflammasome, which promotes IL-1β production, has been implicated in the progression of NAFLD to NASH (265–267), the potential role of estrogens in directly modulating NLRP3 inflammasome in the progression of NAFLD to NASH has been very poorly investigated (268) and remains unclear. By converse, estrogens suppress HCC through the ERβ-mediated upregulation of the NLRP3 inflammasome (269), likely contributing to the sex differences in HCC prevalence (270).
Liver Regeneration and Inflammation
Inflammation triggers many chronic and degenerative diseases, but it also aims to eliminate damaged cells and initiate tissue repair and regeneration, through highly conserved mechanisms (271). Tissue repair and regeneration is particularly important for the liver, especially in response to injury, an ability essential for the maintenance of the hepatic metabolic functions (57).
The process of liver repair and regeneration relies on the proliferative capacity of existing mature hepatocytes in response to environmental cues and can be divided in two phases: a “priming phase,” where inflammatory mediators (e.g., IL-6, TNFα) trigger the inflammation-induced regeneration, and a “proliferation phase,” where mitogens (including hepatocyte growth factor, HGF; transforming growth factor-α, TGF-α; epidermal growth factor, EGF) and auxiliary mitogens (including bile acids; endothelial growth factor, VEGF; insulin-like growth factor system, IGF system; estrogens) carry out the proliferation of hepatocytes, also through the interaction with the liver-resident immune cells (272–275).
Among inflammatory mediators, IL-6 plays a key role in the liver regeneration, as it is responsible for activating ~40% of the genes that are immediately activated by transcription factors following partial hepatectomy (276, 277). According to that, mice lacking IL-6 show reduced hepatocyte proliferation, that can be restored with IL-6 administration (275). In addition to IL-6, also TNF-α is involved in the priming phase of liver regeneration, which requires the expression of inducible nitric oxide synthase (eNOS) to block the potential pro-apoptotic effect of TNFα signaling and trigger liver regeneration (5, 275). IL-6 and TNFα are released mainly by KCs, thus promoting hepatocyte proliferation. The KCs activation is mediated through the NF-κB signaling pathway triggered either by lipopolysaccharide (LPS)/Toll-like receptor4 (TLR4) signaling or by the components of the complement system like C3a and C5a (274, 275). While KC depletion is associated with impaired liver regeneration, the depletion of other liver-resident immune cells such as NK (natural killer) cells enhances liver regeneration due to reduced production of TNFα and IFNγ (interferon-γ), a negative feedback mechanism aimed at regulating the process of liver regeneration (5).
Males and females differ for their ability to regenerate the hepatic tissue in response to injury, with male animals showing a time-delay in the recovery process associated with a higher recruitment of monocytes (278), a difference that depends on both, estrogen and androgen signaling pathways (279–281). In regenerating livers, estrogens act mainly through ERα (281, 282), but also through ERβ (279), with ERα and ERβ orchestrating cell proliferation and differentiation, respectively. The relation between estrogens and IL-6 could be particularly complex, being IL-6 able to influence estrogen levels and, therefore, estrogen-dependent modulation of liver regeneration process (275).
A recent study demonstrated that estrogen and ERα might play an important role also in the accumulation of fats in the liver by modulating CD36 during the early phase of liver regeneration, when fatty acids, triglycerides and cholesterol are required for the proliferation of hepatocyte and for the formation of new cell membrane (283).
The Lack of Estrogen Signaling Impairs the Regulation of Hepatic Metabolism and Inflammation: Lessons From Estrogen Deficient and Knockout Mice
Estrogen Deficiency in Females
The relevance of estrogen signaling in the regulation of female hepatic metabolism and inflammation has been investigated in several pre-clinical studies recapitulating the effects of estrogen deficiency observed in post-menopausal women (14, 18, 169, 284, 285).
In the liver of ovariectomized (OVX) female mice, the lack of estrogens leads to hepatic insulin resistance, to enhanced DNL and FA import, and to reduced FA oxidation and secretion, resulting in increased body weight and fat mass and in fatty liver (14, 21, 153, 286, 287). In OVX females, the administration of estrogens improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor FOXO1 (Forkhead Box O1) (288), prevents hepatic fat deposition by inhibiting DNL (153, 289), facilitates the VLDL (very low density lipoprotein)-mediated export of lipids from the liver by increasing hepatic VLDL-TG production and expression of microsomal triglyceride transfer protein (21, 290, 291) and sustains the β-oxidation of FAs by inducing expression of PPAR-α (peroxisome proliferator-activated receptor α) and FGF21 (123, 289).
Although estrogen replacement has been shown effective in reducing hepatic steatosis (123, 153, 287, 289, 291), however, the administration of constant amount of estrogens or SERMs (selective estrogen receptor modulators) partially restores a proper regulation of hepatic metabolism (123, 292). The reason for that likely resides on the fact that the administration of constant amount of estrogens does not reproduce the physiological oscillation of estrogen levels typical of the reproductive cycle and, therefore, fails to reproduce the cyclic activation of hepatic ERα, which is responsible for a tuned modulation of hepatic metabolism in females (15, 153, 292).
Moreover, estrogens may have a significant impact on hepatic metabolism depending on their route of delivery. For example, while transdermal estradiol reduces plasma TGs by increasing the rate of VLDL-TG clearance without affecting VLDL-TG production (293, 294), oral delivery of estradiol increases VLDL production and plasma TGs, indicating the liver the most responsible of estrogen's effects on increasing VLDL-TGs (21).
The lack of estrogens is associated with increases in lipotoxicity, pro-inflammatory cytokines (e.g., TNFα, IL-1β, and IL-6) and oxidative stress and with decreases in anti-inflammatory cytokines (e.g., IL-10, interleukin 10) and antioxidant defense, all changes that can be reverted or, at least, mitigated by HRT (295–297). When exposed to high intake of dietary lipids, the liver of OVX female mice displays increased expression of Mcp-1 (monocyte chemoattractant protein-1) and Ccr2 (monocyte chemokine receptor 2) that trigger the recuitment of macrophages and promote hepatic fibrosis, endoplasmic reticulum stress and apoptosis, all changes that are improved by estradiol treatment (264).
Estrogen Deficiency in Males
Even in the liver of males, estrogen action is relevant for the regulation of glucose homeostasis, insulin sensitivity, lipid metabolism, and in the prevention of hepatic steatosis (298, 299). Estrogen deficiency in men with mutations in the gene codifying for aromatase (CYP19A1, the enzyme converting testosterone in estrogen) show impaired glucose and lipid liver metabolism (300, 301). Aromatase KO (ArKO) mice display increased adiposity, glucose intolerance and insulin resistance in both sexes (302); in male ArKO mice, increased insulin resistance is primarily due to increased hepatic gluconeogenesis through the induction of G6Pase (glucose 6-phosphatase) and Pepck (phosphoenolpyruvate carboxykinase) expression (299). By contrast, only ArKO males, but not females, show impaired lipid and lipoprotein metabolism and develop hepatic steatosis (302, 303). The administration of estrogens reverses the hepatic steatosis, by reducing the expression of genes involved in DNL (e.g., Fasn, fatty acid synthase; Acaca, acetyl-CoA carboxylase α; Scd-1, stearoyl-CoA desaturase-1) and fatty acid uptake (e.g., Adrp, adipocyte differentiated regulatory protein) (302, 304) and by restoring the expression of enzymes involved in FA oxidation (e.g., Cat, catalase; Mcad, medium-chain acyl-CoA dehydrogenase) (305). Although the precise mechanism of estrogen action in the liver of males have not been fully elucidated, studies performed in KO mice suggest that estradiol mediats PPARα signaling in protecting against hepatic steatosis (306, 307).
Estrogen deficiency in ArKO males is also responsible for increased hepatic mitochondrial apoptosis and altered permeability of the mitochondrial membranes, that are restored by supplementation of 17β-estradiol (308).
ERα in Females
Estrogens can mediate their biologic effects in the female liver acting mainly through the estrogen receptor α (ERα, the isoform most expressed at the hepatic level) (15, 152) through a number of mechanisms, including the regulation of gene transcription by the direct binding to specific estrogen responsive elements (ERE) or by the tethering with other DNA-binding factors and by non-genomic action through membrane-associated ERα (15, 153, 309–313).
While the lack of ERβ does not affect hepatic phenotype (314), the role of ERα in the regulation of hepatic metabolism and inflammation has been highlighted by several studies performed with total body (ERαKO) and liver-specific (LERKO) ERα knockout mice (311). ERαKO mice mostly recapitulate the metabolic phenotype of OVX animals, with increased body weight, visceral adiposity, glucose production, insulin resistance, and hepatic steatosis associated with increased hepatic inflammatory signaling (21, 314–316). Differently from control mice, ERαKO mice are not able to antagonize the induction of cytokines in consequence to a pro-inflammatory stimulus, indicating that ERα protects the liver against liver inflammation (317).
The LERKO mouse represents a useful tool to elucidate the specific relevance of ERα in the liver, especially in the hepatocytes, as this mouse model has been obtained by crossing mice expressing floxed ERα with mice expressing Cre-recombinase under the control of albumin promoter, that it is specifically expressed in the hepatocyte cells (13). Although improperly, LERKO can be considered as liver-specific ERα KO mice, being hepatocytes the most abundant cell type in the liver (57) and being ERα the receptor for estrogens most expressed in the hepatocytes (152, 241).
Compared to control counterparts, LERKO females show an impaired regulation of genes relevant in the regulation of hepatic lipid and lipoprotein metabolism during estrous cycle progression (15), with aging and after ovariectomy (153). As a consequence, LERKO females show increased deposition of lipids in the liver and an impaired regulation of lipoprotein synthesis, leading to a reduced cholesterol efflux to the liver, impaired hepatic cholesterol clearance, high circulating cholesterol levels and increased susceptibility to atherosclerosis (15, 237).
Additional studies have confirmed the role of ERα in preventing hepatic steatosis by showing that liver-specific knockdown of ERα is sufficient to induce hepatic steatosis through a mechanism that seems to involve the regulation of small heterodimer partner (SHP), a transcription factor important in the regulation of hepatic metabolic processes and in the protection against hepatic inflammation (318, 319).
The livers of LERKO mice exhibit a greater expression of genes involved in the inflammatory process (e.g., Tnfα; Il-1β; interleukin-12 beta, Il-12β; Ccr2) and collagen deposition (sequestosome1, Sqstm1; vimentin, Vim; serpine1, Serpine); according to that, LERKO females display portal infiltration of mononuclear leukocytes and portal or centrilobular collagen deposition in the liver (15).
The action of the hepatic ERα is particularly relevant when mice are subjected to excess of dietary lipids: with the lack of hepatic ERα, LERKO females result no more protected against the excess of dietary lipids and accumulate lipids in the liver (91), a condition resembling what happens in OVX mice and post-menopausal women (14). However, differently from control OVX females, estrogen treatment fails to prevent lipid deposition in the liver of LERKO females, further stressing the specific relevance of hepatic ERα in the regulation of female hepatic metabolism (287, 320).
Also transgenic mice in which the expression of ERα is limited to the cytoplasm develop hepatic steatosis (312), suggesting that the protective effects of estrogens on liver health can be mediated by both, classical and non-nuclear mechanisms (321, 322).
ERα in Males
Similar to females, ERαKO male mice develop insulin resistance, impaired glucose tolerance, increased adiposity and marked hepatic steatosis (314, 315, 323). Despite its reduced expression compared to females (15, 152), also in males the liver-specific disruption of ERα signaling leads to altered expression of genes involved in carbohydrate and lipid metabolism (241, 324–326). Hepatic ERα plays a key role in the maintenance of hepatic metabolism, by suppressing hepatic gluconeogenesis and by decreasing DNL through its direct binding to the promoters of genes involved in gluconeogenesis (e.g., Pepck, G6Pase) and lipid metabolism (e.g., Fasn, Acaca) (241) and through the modulation of FOXO1 phosphorylation (326). As a consequence of the lack of ERα-dependent regulatory activity, LERKO males display elevated hepatic glucose production (HGP), liver insulin resistance, increased hepatic lipogenesis and liver lipid deposition (241, 326).
Recent studies suggest that hepatic ERα is required for the estrogen-mediated programming of the hepatic metabolism of males, contributing to hepatic sexual dimorphism (152) and accounting for the sex-specific metabolic response to diets enriched in lipids (91). In the liver of males, ERα is required also for optimal immune-metabolic function, as its lack causes increased expression of several inflammatory genes (327).
GPER
In addition to membrane localized ERs, estrogens can signal through G-protein coupled Estrogen Receptor (GPER, also called Gpr30), a cell surface receptor which role in the regulation of liver metabolism has recently emerged (328–330). After the binding with estrogens, GPER activate multiple non-genomic pathways, as well as the transcriptional programs through the regulation of target genes (331–334) in diverse cell types and tissues, including the liver (335).
GPER has been functionally implicated in several physiological and pathological process (335) and, in particular, in the regulation of metabolism (328, 332, 336–338) and immune response (339–341). In the liver, GPER plays a role in modulating lipid metabolism, in lowering circulating lipid levels and in reducing inflammation (20), as confirmed by several pre-clinical and clinical studies. Individuals carrying a hypofunctional genetic variant of GPER show increased plasma LDL cholesterol; according to that, the activation of GPER induces the expression of the LDL receptor (LDLR) in HepG2 liver cells (342). A recent study demonstrates that GPER mediates the estrogen-dependent reduction of LDLR degradation by preventing the internalization of PCSK9 (proprotein convertase subtilisin/kexin type 9), thus resulting in a higher LDL uptake by liver cells and, consequently, to lower circulating LDL cholesterol (343).
In OVX female mice, the activation of GPER lowers the levels of circulating lipids, reduces the expression of lipogenic and pro-inflammatory genes, and increases the expression of genes involved in lipid oxidation in the liver (329). In a KO mouse model, the lack of GPER leads to increased lipid accumulation in the liver and decreased circulating HDL levels in females, but not males (344), highlighting a sex-specific role of GPER in the metabolic homeostasis (329).
GPER signaling is associated with the immune and anti-inflammatory response, as revealed by its role in counteracting a variety of pathological conditions, including diabetes and obesity (330, 338, 345), atherosclerosis (346, 347), asthma (348), neuroinflammation (349, 350), and cancer (97, 351). In the liver, the lack of GPER enhances immune cell infiltration, fibrosis, and the production of inflammatory factors, such as IL-6, IL-1β, and TNFα in a mouse model of HCC (351). The activation of GPER signaling is effective in reducing the expression of IL-6, but not the viability and proliferation of hepatoma cells, suggesting that GPER action against hepatic tumorigenesis occurs through the regulation of inflammatory response rather than the direct modulation of tumor growth and invasion (351).
Although these studies suggest a direct involvement of GPER in the regulation of metabolism and inflammation in the liver, especially in females, it cannot be excluded that the hepatic effects due to the lack of its signaling are the results of a more complex interaction among metabolic tissues. Indeed, mice lacking GPER show increased adiposity, decreased insulin sensitivity, defective glucose/lipid homeostasis, and inflammation (337, 338, 352), all features that might indirectly affect the hepatic metabolism, pointing to the need of liver-specific GPER models to clarify the specific role of GPER in the hepatic tissue.
Estrogens and Key Cell Types in Liver Metabolism and Inflammation
The liver is composed of several cell types, each of them having unique functions in the regulation of metabolism and immune response and showing interactions with the other cell type, thus cooperating at multiple levels in the regulation of the hepatic function. The major cell types contributing to the main liver functions are hepatocyctes, Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells, and cholangiocytes.
Hepatocyctes
Hepatocyctes represent the most abundant cell type in the liver (accounting for 80% of liver mass) and are involved in several functions, including lipid and carbohydrate metabolism (353), protein synthesis (354), detoxification and drug metabolism (355, 356), and the secretion of coagulation and complement factors (353, 354, 357). In the hepatocytes, estrogens, mainly acting through ERα, limit gluconeogenesis (241, 288) preventing increased HGP and insulin resistance (288), limit the uptake of FFAs, inhibit DNL (153) and promotes FA oxidation (289) and export (287), thus preventing lipid deposition in the liver and the generation of lipotoxicity and ROS (252) that trigger a pro-inflammatory response acting as the driver of NAFLD progression and liver degeneration (81, 247). Estrogen signaling facilitates the resolution of inflammation by inhibiting the production of pro-inflammatory cytokines (264), regulates apoptotic process (358, 359), and promotes liver cell regeneration (279–282), thus limiting or preventing liver injury. As recapitulated by studies performed in OVX and LERKO females, the lack of the regulatory activity of estrogens in the hepatocytes favors the development and progression of NAFLD and, likely, of the associated cardio-metabolic diseases (e.g., atherogenesis) (16).
Kupffer Cells (KCs)
Kupffer cells (KCs) represent one-third of the non-parenchymal cells in the liver and account for 80–90% of tissue macrophages present in the body, acting as immune sentinels (360). KCs are important members of the innate and adaptive immune systems, serving as a first line of defense against bacteria, microbial debris and endotoxins derived from the gastrointestinal tract. Once activated, KCs trigger an inflammatory response by producing a panel of pro-inflammatory cytokines, including TNF-α, IL-1β, and IFN-γ, and provide to the clearance of phagocytosable particles (361). Given their role in the regulation of inflammatory and innate responses, KCs are considered as potential targets for the treatment of liver diseases, including NAFLD (360, 362, 363). Male and female KCs are different from a morphological (364) and functional point of view (258), contributing to sex differences in liver inflammation and regeneration (280) and in the prevalence and progression of NAFLD (17, 258), ALD (alcoholic liver disease) (365) and HCC (259, 366). Estrogens result involved in the sensitization of KCs to toxic stimuli (367) and in driving the pro/anti-inflammatory polarization of KCs, that exerts a key role in the resolution or progression of inflammation, thus counteracting or promoting the development of liver diseases (50, 368, 369). The estrogen-dependent regulation of cytokine production by KCs is predominantly mediated via ERα (370, 371), resulting the isoform most expressed in these cell types (372).
Hepatic Stellate Cells (HSCs)
Although comprising only 5% of the liver cells, hepatic stellate cells (HSCs) play a central role in liver metabolism, especially in retinol metabolism and lipid storage (373). In healthy liver, HSCs are quiescent and store 80% of total liver retinol, that is released depending on its extracellular status. In injured liver, HSCs become activated and transform into myofibroblasts; activated HSCs lose their retinols and produce a considerable amount of extracellular matrix, thus leading to liver fibrosis (59). Although sex differences in the morphological expression of male/female HSCs has not been observed (364), several studies have demonstrated that estrogen inhibits the activation of HSCs and reduces liver fibrosis (374, 375), suggesting that estrogen signaling might account for the sex-specific prevalence of hepatic fibrosis. Although the molecular mechanism has not been fully clarified, estrogens seem to act through ERβ (376) and GPER (377), given that ERα is not expressed in these cells (378).
Liver Sinusoidal Endothelial Cells (LSECs)
Liver sinusoidal endothelial cells (LSECs), which comprise ~50% of liver non-parenchymal cells, are highly specialized endothelial cells containing many small pores or fenestrations, which provide open channels that facilitate the transfer of substrates between the blood and the liver parenchyma and regulate lipoprotein traffic to and from the hepatocytes (379, 380). Their unique morphology gives to LSECs a high endocytic capacity, enabling them to act as effective scavengers and promote the clearance of lipids and macromolecules and small particulates from the blood. The impairment of their function is associated with the development of extra-hepatic pathologies, including atherosclerosis (380). LSECs exert a key role in the innate and adaptive immunity, promoting the presentation of antigens and favoring the removal and clearance of circulating antigens and viruses (381). In addition to their roles as pathogen recognition and antigen-presenting cells, LSECs also have a critical role in the recruitment of leukocytes into liver tissue, thus influencing the composition of hepatic immune population. The balance between tolerance and effector immune responses driven by LSECs might promote the resolution or the progression of the immune response, eventually leading to several chronic liver diseases, including NAFLD, cirrhosis, fibrosis, liver failure and HCC (381, 382).
In LSECs, estrogens, even by modulating the levels and the nuclear occupancy of ER (372), enhance the production of nitric oxide (NO) and regulate the hepatic sinusoidal microcirculation (383, 384), likely explaining the higher incidence of liver cirrhosis with portal hypertension in men and post-menopausal women than pre-menopausal women (385).
Cholangiocytes
Cholangiocytes are the epithelial cells lining the intrahepatic and extrahepatic bile ducts; these cells participate in bile production and secretion and, although to a less extent than hepatocytes, have a role in the liver development, regeneration and repair (386, 387). Cholangiocytes can be activated by a variety of insults, including infections, cholestasis, and xenobiotics (386), leading to increased proliferation and to pro-fibrotic and pro-inflammatory secreted factors (388), that can favor the development of cholangiopathies and cholangiocarcinoma (389–392).
Cholangiocytes are targets of estrogen action: by acting through both ERα and ERβ and by activating either genomic or non-genomic pathways, estrogens play a key role in the regulation of proliferative and secretory activities of cholangiocytes (393, 394). The lack of estrogens in OVX females decreases the expression of ERs (2.5-fold for ERα and 35-fold for ERβ), leading to reduced cholangiocyte proliferation and bile duct mass; conversely, the administration of 17β-estradiol during bile duct ligation in OVX rats induced a normalization of bile duct mass, cholangiocyte proliferation, and apoptosis (395). Also in males, estrogens exerts a major role in stimulating cholangiocyte proliferation by preventing the increase of cholangiocyte apoptosis and loss of cholangiocyte proliferation (396). Notably, the altered expression and/or activation of ERα and ERβ is often associated with a high risk of primary biliary diseases (397–399).
Impaired bile flow leads to cholestasis, a pathology characterized by elevated levels of bile acid in the liver and serum followed by hepatocyte and biliary injury, that shows an increased incidence in women receiving estrogen for contraception or hormone replacement therapy, or in susceptible women during pregnancy. Although the molecular mechanisms involved in cholestasis remain controversial, recent findings suggest that estrogens may influence its course by directly modulating the patho-physiology of cholangiocytes, which are the primary target of damage in this disease (393).
Although each cell type plays a specific role in the liver and expresses a unique gene (400) and proteomic profile (401), only the cooperation among different cell types enables the liver to achieve its functions (402), a consideration that should be taken into account when performing in vitro studies, in which the cross-talk among liver cell types is lost or partially reproduced (403, 404). In this view, although challenging, the recent advances in the generation of human liver organoids might represent a potential, more reliable tool for the in vitro analysis of liver-specific biological processes and for disease modeling and drug screening at near-physiological conditions (405).
Conclusion
The data summarized in this review outline the role of estrogens and their receptors in antagonizing the metabolic and inflammatory alterations that trigger and boost NAFLD development, thus determining its sex-dependent prevalence and its lower incidence in fertile females (Figure 1, Table 1).
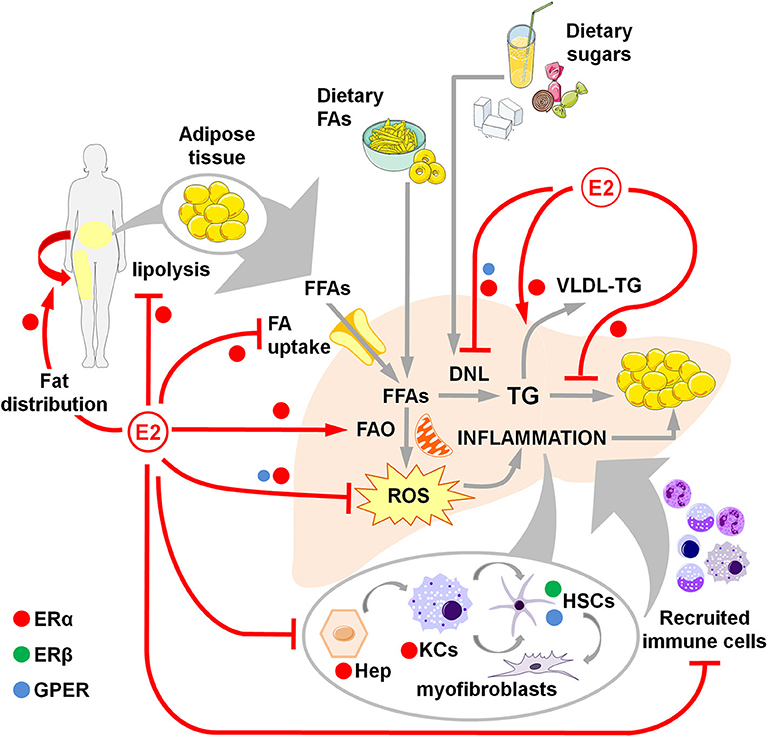
Figure 1. Overview of estrogen action through ERα, ERβ and GPER in counteracting NAFLD development and progression in women. Estrogens favor fat distributionr to subcutaneous deposits, inhibit adipose tissue lipolysis and reduce the uptake of FFAs, thus limiting the flux of FFAs to the liver. Estrogens limit dietary-induced DNL and facilitate the export of lipids as VLDL-TG. Estrogens promote the FA β-oxidation and prevent the activation of a sustained alternative FA oxidation that triggers lipotoxicity and the generation of ROS that, in turn, activate a pro-inflammatory response. Hepatocellular damage and fat-derived factors mediate the local activation of a pro-inflammatory response by hepatocytes, KCs and HSCs, that promote the degeneration of hepatic tissue and the recruitment of extra-hepatic immune cells that boost the inflammatory response and worsen the metabolic alterations. DNL, de novo lipogenesis; E2, estrogens (mainly 17β-estradiol); FAs, fatty acids; FFAs, free fatty acids; FAO, fatty acid oxidation; Hep, hepatocytes; HSCs, hepatic stellate cells; KCs, Kupffer cells; ROS, reactive oxygen species; TG, triglycerides; VLDL, very-low density lipoprotein.
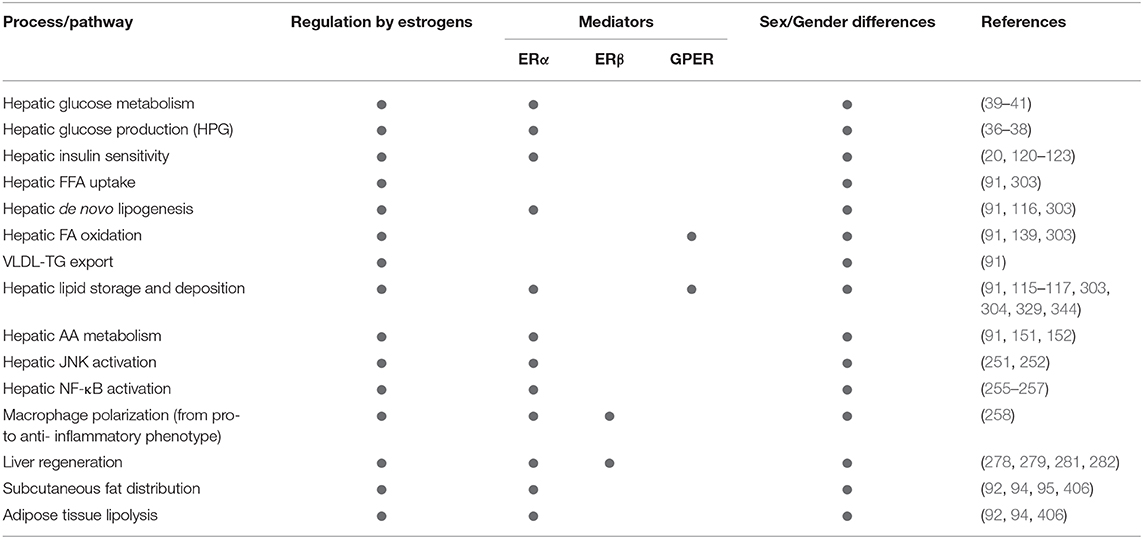
Table 1. Summarizing the relevance of estrogen signaling, ERα, ERβ, and GPER in the sex-specific regulation of metabolic and inflammatory pathways relevant in NAFLD development and progression.
Estrogen-mediated effects likely arise from higher metabolic flexibility gained and perfected through evolution by the female liver to adapt the hepatic metabolism to the reproductive function (12, 13, 152, 242). Playing the liver the most relevant role in the accomplishment of energy requirements, during evolution the hepatic metabolism has been sharpened, in a sex-specific fashion, to reach an accurate interconnection of regulatory mechanisms aimed to sustain the energy needs of reproductive functions that are greatly different between the two sexes. The dynamic regulation of hepatic metabolism should have acquired a maximum degree of complexity in the liver of females that, compared to males, have to be more flexible in adapting their hepatic metabolism to the different, more variable, reproductive stages (reproductive cycle progression, pregnancy, lactation) that entail different energy requirements. The female liver had to develop and mold mechanisms able to sense and modulate efficiently the hepatic metabolism accordingly to the hormonal rhythm of estrogen fluctuations during the reproductive cycle and in other reproductive stages (pregnancy, lactation). In this view, in the liver of female mammals, estrogen signaling has therefore acquired a tight control on the hepatic metabolism through a sequence of well-tuned and intertwined events that have been perfectly tuned to secure reproduction only in favorable energy conditions and to support the energy needs of the different reproductive stages (14, 153, 235).
The high metabolic dynamicity conferred by estrogens to female liver contributes to prevent and limit the surge and progression of metabolic and inflammatory alterations in the liver, a mechanism underlying the increased incidence of NAFLD associated with the decline in liver metabolic flexibility after menopause.
The effects of estrogens and their receptors on the regulation of liver metabolism and inflammation may be direct or indirect, acting—for example—through other transcription factors and nuclear receptors (NRs) (407) with relevant and sex-specific activities in the liver (408) and in the NAFLD pathogenesis (409–411). Such an interplay might be particularly complex and regulated in the female liver: in view of its action in the regulation of reproductive process, ERα might have acquired in the female liver a regulatory role over these signaling pathways to adapt liver metabolism and inflammation to hormonal and nutritional status to accomplish the metabolic adaptations required to support the energy needs of reproduction. According with this idea, the lack of estrogens impairs the regulation of some NR signaling, including PPARα (289, 412) and glucocorticoid receptor (GR) (413), exerting pivotal roles in the regulation of hepatic metabolism and inflammation (414–416), thus favoring NAFLD development.
Despite the extensive, although probably underestimated, awareness on hepatic sex differences, the molecular mechanisms determining the sex-specific incidence of liver pathologies such as NAFLD are far to be unraveled. This knowledge has been prevented and affected by several limitations that stem from: (a) the paucity of available data on both sexes coming from pre- and clinical studies in which females are often underrepresented (12, 417); (b) the inability to enroll females in clinical studies (418–420); (c) the limited and, in some cases, misleading conclusions reached by experimental designs that did not take into account the relative contribution of genetic and hormonal backgrounds and exclude the sexual hormones as potential confounding factors (12, 417); (d) the lack of proper research tools helpful in investigating the genetic and/or hormonal factors relevant for the hepatic sexual dimorphism or the inability to use the available research tools in the best way (421, 422); (e) the fragmentary and still incomplete view coming from several studies that often do not share common protocols or lack of significance for the low number of the samples analyzed (422, 423); (f) the low, still insufficient commitment dedicated to dissemination of the results obtained from sex/gender research, such as educational programs addressed to health professionals (researchers, clinicians, scientific training programs, health institutions, etc.) and to society in general (419, 423–425); g) the still limited policies aimed at promoting sex/gender research programs (426–428). All these aspects contribute to our limited understanding of the nature and relevance of hepatic sexual dimorphism, thus preventing, so far, the development of more efficacious, sex-specific therapies against liver pathologies such as NAFLD, which incidence is greatly increasing in EU and accounts for €35 billions only in Germany, France, Italy, and United Kingdom (234, 429). Furthermore, the partial unawareness of the relevance of hepatic sexual dimorphism in the liver physio-pathology is a contributing cause to the development of associated cardio-metabolic diseases, such as atherogenesis and CVDs (430, 431). Similarly, the lack of sex-specific pharmacological treatments (that should differ in terms of molecules, dose, timing and risk of adverse drug reaction between the two sexes) leads often to drug-induced hepatotoxicity, representing the main cause of withdrawal of drugs from the market and the main reason of liver transplants (17).
In this view, a deeper understanding of the mechanisms underlining the sex-specific incidence of NAFLD and the role of estrogen signaling pathways will likely yield the basis for the design of more personalized hepatic therapies that would significantly improve the quality of life of a large section of our society as well as of men and women which experience impaired/lost hormonal signaling (i.e., due to gonadal failure, aging, exposure to endocrine disrupting chemicals).
Author Contributions
SDT: figure preparation, writing, review, and editing.
Funding
In our lab, the work on women's health was supported by the European Union's grant ERC-2012 (ERC-Advanced Grant Ways, 322977). SDT acknowledges support from EMBO (award ASTF 184 - 2016).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Trefts E, Gannon M, Wasserman DH: The liver. Curr Biol. (2017) 27:R1147–51. doi: 10.1016/j.cub.2017.09.019
2. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43:S54–62. doi: 10.1002/hep.21060
3. Heymann F, Tacke F. Immunology in the liver — from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
4. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
5. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. (2016) 13:267–76. doi: 10.1038/cmi.2016.3
6. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity. (2017) 47:406–20. doi: 10.1016/j.immuni.2017.08.009
7. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. (2005) 115:1111–9. doi: 10.1172/JCI25102
8. Czaja AJ. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J Gastroenterol. (2014) 20:2515. doi: 10.3748/wjg.v20.i10.2515
9. Hijona E, Hijona L, Arenas JI, Bujanda L. Inflammatory mediators of hepatic steatosis. Mediators Inflamm. (2010) 2010:837419. doi: 10.1155/2010/837419
10. Martínez-Esparza M. Inflammatory status in human hepatic cirrhosis. World J Gastroenterol. (2015) 21:11522. doi: 10.3748/wjg.v21.i41.11522
11. Kautzky-Willer A, Handisurya A. Metabolic diseases and associated complications: sex and gender matter! Eur J Clin Invest. (2009) 39:631–48. doi: 10.1111/j.1365-2362.2009.02161.x
12. Della Torre S, Maggi A. Sex differences: a resultant of an evolutionary pressure? Cell Metab. (2017) 25:499–505. doi: 10.1016/j.cmet.2017.01.006
13. Della Torre S, Rando G, Meda C, Stell A, Chambon P, Krust A, et al. Amino acid-dependent activation of liver estrogen receptor alpha integrates metabolic and reproductive functions via IGF-1. Cell Metab. (2011) 13:205–14. doi: 10.1016/j.cmet.2011.01.002
14. Della Torre S, Benedusi V, Fontana R, Maggi A. Energy metabolism and fertility—a balance preserved for female health. Nat Rev Endocrinol. (2014) 10:13–23. doi: 10.1038/nrendo.2013.203
15. Della Torre S, Mitro N, Fontana R, Gomaraschi M, Favari E, Recordati C, et al. An essential role for liver ERα in coupling hepatic metabolism to the reproductive cycle. Cell Rep. (2016) 15:360–71. doi: 10.1016/j.celrep.2016.03.019
16. Ballestri S, Nascimbeni F, Baldelli E, Marrazzo A, Romagnoli D, Lonardo A. NAFLD as a sexual dimorphic disease: role of gender and reproductive status in the development and progression of nonalcoholic fatty liver disease and inherent cardiovascular risk. Adv Ther. (2017) 34:1291–326. doi: 10.1007/s12325-017-0556-1
17. Buzzetti E, Parikh PM, Gerussi A, Tsochatzis E. Gender differences in liver disease and the drug-dose gender gap. Pharmacol Res. (2017) 120:97–108. doi: 10.1016/j.phrs.2017.03.014
18. Brady CW. Liver disease in menopause. World J Gastroenterol. (2015) 21:7613. doi: 10.3748/wjg.v21.i25.7613
19. Gerdts E, Regitz-Zagrosek V. Sex differences in cardiometabolic disorders. Nat Med. (2019) 25:1657–1666. doi: 10.1038/s41591-019-0643-8
20. Mauvais-Jarvis F. Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity. (2018). Available online at: http://www.vlebooks.com/vleweb/product/openreader?id=none&isbn=9783319701783 (accessed May 21, 2020)
21. Palmisano BT, Zhu L, Stafford JM. Role of estrogens in the regulation of liver lipid metabolism. In: Mauvais-Jarvis F, editor. Sex Gender Factors Affecting Metabolic Homeostasis, Diabetes Obesity Advances in Experimental Medicine Biology. Cham: Springer International Publishing (2017). p. 227–56. doi: 10.1007/978-3-319-70178-3_12
22. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
23. Adams LA, Anstee QM, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. (2017) 66:1138–53. doi: 10.1136/gutjnl-2017-313884
24. Mikolasevic I, Milic S, Turk Wensveen T, Grgic I, Jakopcic I, Stimac D, et al. Nonalcoholic fatty liver disease - a multisystem disease? World J Gastroenterol. (2016) 22:9488. doi: 10.3748/wjg.v22.i43.9488
25. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease: estes. Hepatology. (2018) 67:123–33. doi: 10.1002/hep.29466
26. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
27. Bhatia LS, Curzen NP, Calder PC, Byrne CD. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur Heart J. (2012) 33:1190–200. doi: 10.1093/eurheartj/ehr453
28. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. (2017) 13:572–87. doi: 10.1038/nrendo.2017.80
29. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. (2016) 126:12–22. doi: 10.1172/JCI77812
30. Watt MJ, Miotto PM, de Nardo W, Montgomery MK. The liver as an endocrine organ—linking NAFLD and insulin resistance. Endocr Rev. (2019) 40:1367–93. doi: 10.1210/er.2019-00034
31. Santoleri D, Titchenell PM. Resolving the paradox of hepatic insulin resistance. Cell Mol Gastroenterol Hepatol. (2019) 7:447–56. doi: 10.1016/j.jcmgh.2018.10.016
32. Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. (2011) 14:804–10. doi: 10.1016/j.cmet.2011.11.004
33. Seko Y, Sumida Y, Tanaka S, Mori K, Taketani H, Ishiba H, et al. Insulin resistance increases the risk of incident type 2 diabetes mellitus in patients with non-alcoholic fatty liver disease: HOMA-IR predicts development T2DM in NAFLD. Hepatol Res. (2018) 48:E42–51. doi: 10.1111/hepr.12925
34. Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol. (2017) 14:32–42. doi: 10.1038/nrgastro.2016.147
35. de Bandt J-P, Jegatheesan P, Tennoune-El-Hafaia N. Muscle loss in chronic liver diseases: the example of nonalcoholic liver disease. Nutrients. (2018) 10:1195. doi: 10.3390/nu10091195
36. Couchepin C, Le K-A, Bortolotti M, da Encarnacao JA, Oboni J-B, Tran C, et al. Markedly blunted metabolic effects of fructose in healthy young female subjects compared with male subjects. Diabetes Care. (2008) 31:1254–6. doi: 10.2337/dc07-2001
37. ter Horst KW, Gilijamse PW, de Weijer BA, Kilicarslan M, Ackermans MT, Nederveen AJ, et al. Sexual dimorphism in hepatic, adipose tissue, and peripheral tissue insulin sensitivity in obese humans. Front Endocrinol. (2015) 6:182. doi: 10.3389/fendo.2015.00182
38. Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal J-F, Montagner A, et al. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia. (2020) 63:453–61. doi: 10.1007/s00125-019-05040-3
39. Mauvais-Jarvis F. Gender differences in glucose homeostasis and diabetes. Physiol Behav. (2018) 187:20–3. doi: 10.1016/j.physbeh.2017.08.016
40. Grossmann M, Wierman ME, Angus P, Handelsman DJ. Reproductive endocrinology of nonalcoholic fatty liver disease. Endocr Rev. (2019) 40:417–46. doi: 10.1210/er.2018-00158
41. Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol. (2015) 2015:294278. doi: 10.1155/2015/294278
42. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. (2018) 75:3313–27. doi: 10.1007/s00018-018-2860-6
43. Lambert JE, Ramos–Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. (2014) 146:726–35. doi: 10.1053/j.gastro.2013.11.049
44. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. (2013) 48:434–41. doi: 10.1007/s00535-013-0758-5
45. Engin A. Non-alcoholic fatty liver disease. In: Engin AB, Engin A, editors. Obesity Lipotoxicity Advances in Experimental Medicine Biology. Cham: Springer International Publishing (2017). p. 443–67. doi: 10.1007/978-3-319-48382-5_19
46. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
47. Day CP. From fat to inflammation. Gastroenterology. (2006) 130:207–10. doi: 10.1053/j.gastro.2005.11.017
48. Luedde T, Schwabe RF. NF-κB in the liver—linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2011) 8:108–18. doi: 10.1038/nrgastro.2010.213
49. Sharma M, Mitnala S, Vishnubhotla RK, Mukherjee R, Reddy DN, Rao PN. The riddle of nonalcoholic fatty liver disease: progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. J Clin Exp Hepatol. (2015) 5:147–58. doi: 10.1016/j.jceh.2015.02.002
50. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
51. Sunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab. (2017) 28:250–60. doi: 10.1016/j.tem.2016.11.006
52. Moreno-Fernandez ME, Giles DA, Stankiewicz TE, Sheridan R, Karns R, Cappelletti M, et al. Peroxisomal β-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight. (2018) 3:1–19. doi: 10.1172/jci.insight.93626
53. Reddy JK, Sambasiva Rao M. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol-Gastrointest Liver Physiol. (2006) 290:G852–8. doi: 10.1152/ajpgi.00521.2005
54. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. (2020) 152:116–41. doi: 10.1016/j.freeradbiomed.2020.02.025
55. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. (2019) 16:411–28. doi: 10.1038/s41575-019-0145-7
56. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis—new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. (2013) 10:627–36. doi: 10.1038/nrgastro.2013.149
57. Stanger BZ. Cellular homeostasis and repair in the mammalian liver. Annu Rev Physiol. (2015) 77:179–200. doi: 10.1146/annurev-physiol-021113-170255
58. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
59. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
60. Zhang C-Y, Yuan W-G, He P, Lei J-H, Wang C-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol. (2016) 22:10512. doi: 10.3748/wjg.v22.i48.10512
61. Stols-Gonçalves D, Hovingh GK, Nieuwdorp M, Holleboom AG. NAFLD and atherosclerosis: two sides of the same dysmetabolic coin? Trends Endocrinol Metab. (2019) 30:891–902. doi: 10.1016/j.tem.2019.08.008
62. Stahl EP, Dhindsa DS, Lee SK, Sandesara PB, Chalasani NP, Sperling LS. Nonalcoholic fatty liver disease and the heart. J Am Coll Cardiol. (2019) 73:948–63. doi: 10.1016/j.jacc.2018.11.050
63. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. (2020) 69:1691–705. doi: 10.1136/gutjnl-2020-320622
64. Motamed N, Rabiee B, Poustchi H, Dehestani B, Hemasi GR, Khonsari MR, et al. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin Res Hepatol Gastroenterol. (2017) 41:31–8. doi: 10.1016/j.clinre.2016.07.005
65. Mantovani A, Zaza G, Byrne CD, Lonardo A, Zoppini G, Bonora E, et al. Nonalcoholic fatty liver disease increases risk of incident chronic kidney disease: a systematic review and meta-analysis. Metabolism. (2018) 79:64–76. doi: 10.1016/j.metabol.2017.11.003
66. Upala S, Jaruvongvanich V, Wijarnpreecha K, Sanguankeo A. Nonalcoholic fatty liver disease and osteoporosis: a systematic review and meta-analysis. J Bone Miner Metab. (2017) 35:685–93. doi: 10.1007/s00774-016-0807-2
67. Principi M, Iannone A, Losurdo G, Mangia M, Shahini E, Albano F, et al. Nonalcoholic fatty liver disease in inflammatory bowel disease: prevalence and risk factors. Inflamm Bowel Dis. (2018) 24:1589–96. doi: 10.1093/ibd/izy051
68. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. (2018) 68:268–79. doi: 10.1016/j.jhep.2017.09.003
69. Kovalic AJ, Banerjee P, Tran QT, Singal AK, Satapathy SK. Genetic and epigenetic culprits in the pathogenesis of nonalcoholic fatty liver disease. J Clin Exp Hepatol. (2018) 8:390–402. doi: 10.1016/j.jceh.2018.04.001
70. Perdomo CM, Frühbeck G, Escalada J. Impact of nutritional changes on nonalcoholic fatty liver disease. Nutrients. (2019) 11:677. doi: 10.3390/nu11030677
71. Eslamparast T, Tandon P, Raman M. Dietary composition independent of weight loss in the management of non-alcoholic fatty liver disease. Nutrients. (2017) 9:800. doi: 10.3390/nu9080800
72. ter Horst K, Serlie M. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients. (2017) 9:981. doi: 10.3390/nu9090981
73. Lim JS, Mietus-Snyder M, Valente A, Schwarz J-M, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. (2010) 7:251–64. doi: 10.1038/nrgastro.2010.41
74. Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism. (2016) 65:1017–25. doi: 10.1016/j.metabol.2016.01.012
75. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. (2005) 115:1343–51. doi: 10.1172/JCI23621
76. Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci USA. (2009) 106:15430–5. doi: 10.1073/pnas.0904944106
77. Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology. (2016) 157:570–85. doi: 10.1210/en.2015-1866
78. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. (2007) 104:12587–94. doi: 10.1073/pnas.0705408104
79. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. (2016) 65:1049–61. doi: 10.1016/j.metabol.2016.02.014
80. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: how metabolic complications of overnutrition favour lipotoxicity pro-inflammatory fatty liver disease. In: Yu J, editor. Obesity, Fatty Liver Liver Cancer Advances in Experimental Medicine Biology., Vol. 1061. Singapore: Springer Singapore, p. 19–44. doi: 10.1007/978-981-10-8684-7_3
81. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
82. Polyzos SA, Kountouras J, Mantzoros CS. Leptin in nonalcoholic fatty liver disease: a narrative review. Metabolism. (2015) 64:60–78. doi: 10.1016/j.metabol.2014.10.012
83. Jornayvaz FR, Samuel VT, Shulman GI. The role of muscle insulin resistance in the pathogenesis of atherogenic dyslipidemia and nonalcoholic fatty liver disease associated with the metabolic syndrome. Annu Rev Nutr. (2010) 30:273–90. doi: 10.1146/annurev.nutr.012809.104726
84. Nachit M, Leclercq IA. Emerging awareness on the importance of skeletal muscle in liver diseases: time to dig deeper into mechanisms! Clin Sci. (2019) 133:465–81. doi: 10.1042/CS20180421
85. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. (2012) 148:852–71. doi: 10.1016/j.cell.2012.02.017
86. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
87. Safari Z, Gérard P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci. (2019) 76:1541–58. doi: 10.1007/s00018-019-03011-w
88. Link JC, Reue K. Genetic basis for sex differences in obesity and lipid metabolism. Annu Rev Nutr. (2017) 37:225–45. doi: 10.1146/annurev-nutr-071816-064827
89. Zore T, Palafox M, Reue K. Sex differences in obesity, lipid metabolism, and inflammation—A role for the sex chromosomes? Mol Metab. (2018) 15:35–44. doi: 10.1016/j.molmet.2018.04.003
90. Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K. Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol. (2017) 37:746–56. doi: 10.1161/ATVBAHA.116.307301
91. Meda C, Barone M, Mitro N, Lolli F, Pedretti S, Caruso D, et al. Hepatic ERα accounts for sex differences in the ability to cope with an excess of dietary lipids. Mol Metab. (2020) 32:97–108. doi: 10.1016/j.molmet.2019.12.009
92. Palmer BF, Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. (2015) 402:113–9. doi: 10.1016/j.mce.2014.11.029
93. Palmisano BT, Zhu L, Eckel RH, Stafford JM. Sex differences in lipid and lipoprotein metabolism. Mol Metab. (2018) 15:45–55. doi: 10.1016/j.molmet.2018.05.008
94. Pedersen SB, Kristensen K, Hermann PA, Katzenellenbogen JA, Richelsen B. Estrogen controls lipolysis by up-regulating α2A-adrenergic receptors directly in human adipose tissue through the estrogen receptor α. implications for the female fat distribution. J Clin Endocrinol Metab. (2004) 89:1869–78. doi: 10.1210/jc.2003-031327
95. Verrijken AN, Francque S, van Gaal L. The role of visceral adipose tissue in the pathogenesis of non-alcoholic fatty liver disease. Eur Endocrinol. (2010) 7:96–103. doi: 10.17925/EE.2011.07.02.96
96. Azzu V, Vacca M, Virtue S, Allison M, Vidal-Puig A. Adipose tissue-liver cross talk in the control of whole-body metabolism: implications in nonalcoholic fatty liver disease. Gastroenterology. (2020) 158:1899–912. doi: 10.1053/j.gastro.2019.12.054
97. Cortes E, Sarper M, Robinson B, Lachowski D, Chronopoulos A, Thorpe SD, et al. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. (2019) 20:e46556. doi: 10.15252/embr.201846556
98. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. (2017) 23:804–14. doi: 10.1038/nm.4350
99. Gancheva S, Jelenik T, Álvarez-Hernández E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev. (2018) 98:1371–415. doi: 10.1152/physrev.00015.2017
100. Gao X, Salomon C, Freeman DJ. Extracellular vesicles from adipose tissue—a potential role in obesity and type 2 diabetes? Front Endocrinol. (2017) 8:202. doi: 10.3389/fendo.2017.00202
101. Gerner RR, Wieser V, Moschen AR, Tilg H. Metabolic inflammation: role of cytokines in the crosstalk between adipose tissue and liver. Can J Physiol Pharmacol. (2013) 91:867–872. doi: 10.1139/cjpp-2013-0050
102. Kurt Z, Barrere-Cain R, LaGuardia J, Mehrabian M, Pan C, Hui ST, et al. Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol Sex Differ. (2018) 9:46. doi: 10.1186/s13293-018-0205-7
103. Meyer MR, Clegg DJ, Prossnitz ER, Barton M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors: obesity, insulin resistance and diabetes: role of oestrogen. Acta Physiol. (2011) 203:259–69. doi: 10.1111/j.1748-1716.2010.02237.x
104. Varghese M, Griffin C, McKernan K, Eter L, Lanzetta N, Agarwal D, et al. Sex differences in inflammatory responses to adipose tissue lipolysis in diet-induced obesity. Endocrinology. (2019) 160:293–312. doi: 10.1210/en.2018-00797
105. Vasanthakumar A, Chisanga D, Blume J, Gloury R, Britt K, Henstridge DC, et al. Sex-specific adipose tissue imprinting of regulatory T cells. Nature. (2020) 579:581–5. doi: 10.1038/s41586-020-2040-3
106. Zhang Y, Lu D, Wang R, Fu W, Zhang S. Relationship between muscle mass/strength and hepatic fat content in post-menopausal women. Medicina. (2019) 55:629. doi: 10.3390/medicina55100629
107. Basaranoglu M. Fructose as a key player in the development of fatty liver disease. World J Gastroenterol. (2013) 19:1166. doi: 10.3748/wjg.v19.i8.1166
108. Jegatheesan P, de Bandt J. Fructose and NAFLD: the multifaceted aspects of fructose metabolism. Nutrients. (2017) 9:230. doi: 10.3390/nu9030230
109. Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. (2018) 68:1063–75. doi: 10.1016/j.jhep.2018.01.019
110. Softic S, Stanhope KL, Boucher J, Divanovic S, Lanaspa MA, Johnson RJ, et al. Fructose and hepatic insulin resistance. Crit Rev Clin Lab Sci. (2020) 57:308–22. doi: 10.1080/10408363.2019.1711360
111. Tappy L, Lê K-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. (2010) 90:23–46. doi: 10.1152/physrev.00019.2009
112. Wei Y, Wang D, Pagliassotti MJ. Fructose selectively modulates c-jun N-terminal kinase activity and insulin signaling in rat primary hepatocytes. J Nutr. (2005) 135:1642–6. doi: 10.1093/jn/135.7.1642
113. Alwahsh SM, Gebhardt R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch Toxicol. (2017) 91:1545–63. doi: 10.1007/s00204-016-1892-7
114. Lambertz J, Weiskirchen S, Landert S, Weiskirchen R. Fructose: a dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front Immunol. (2017) 8:1159. doi: 10.3389/fimmu.2017.01159
115. Hyer MM, Dyer SK, Kloster A, Adrees A, Taetzsch T, Feaster J, et al. Sex modifies the consequences of extended fructose consumption on liver health, motor function, and physiological damage in rats. Am J Physiol Regul Integr Comp Physiol. (2019) 317:R903–11. doi: 10.1152/ajpregu.00046.2019
116. Low W, Cornfield T, Charlton C, Tomlinson J, Hodson L. Sex differences in hepatic de novo lipogenesis with acute fructose feeding. Nutrients. (2018) 10:1263. doi: 10.3390/nu10091263
117. Morrell A, Tripet BP, Eilers BJ, Tegman M, Thompson D, Copié V, et al. Copper modulates sex-specific fructose hepatoxicity in nonalcoholic fatty liver disease (NALFD) Wistar rat models. J Nutr Biochem. (2020) 78:108316. doi: 10.1016/j.jnutbio.2019.108316
118. Comitato R, Saba A, Turrini A, Arganini C, Virgili F. Sex hormones and macronutrient metabolism. Crit Rev Food Sci Nutr. (2015) 55:227–41. doi: 10.1080/10408398.2011.651177
119. Mauvais-Jarvis F. Menopause, estrogens, glucose homeostasis in women. In: Mauvais-Jarvis F, editor. Sex Gender Factors Affecting Metabolic Homeostasis, Diabetes Obesity Advances in Experimental Medicine Biology. Cham: Springer International Publishing (2017). p. 217–25. doi: 10.1007/978-3-319-70178-3_11
120. Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. (2013) 34:309–38. doi: 10.1210/er.2012-1055
121. Ohashi T, Kato M, Yamasaki A, Kuwano A, Suzuki H, Kohjima M, et al. Effects of high fructose intake on liver injury progression in high fat diet induced fatty liver disease in ovariectomized female mice. Food Chem Toxicol. (2018) 118:190–7. doi: 10.1016/j.fct.2018.05.006
122. Chukijrungroat N, Khamphaya T, Weerachayaphorn J, Songserm T, Saengsirisuwan V. Hepatic FGF21 mediates sex differences in high-fat high-fructose diet-induced fatty liver. Am J Physiol-Endocrinol Metab. (2017) 313:E203–12. doi: 10.1152/ajpendo.00076.2017
123. Kim JH, Meyers MS, Khuder SS, Abdallah SL, Muturi HT, Russo L, et al. Tissue-selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol Metab. (2014) 3:177–90. doi: 10.1016/j.molmet.2013.12.009
124. Ferramosca A. Modulation of hepatic steatosis by dietary fatty acids. World J Gastroenterol. (2014) 20:1746. doi: 10.3748/wjg.v20.i7.1746
125. Parry SA, Hodson L. Influence of dietary macronutrients on liver fat accumulation and metabolism. J Investig Med. (2017) 65:1102–15. doi: 10.1136/jim-2017-000524
126. Silva Figueiredo P, Carla Inada A, Marcelino G, Maiara Lopes Cardozo C, de Cássia Freitas K, de Cássia Avellaneda Guimarães R, et al. Fatty acids consumption: the role metabolic aspects involved in obesity and its associated disorders. Nutrients. (2017) 9:1158. doi: 10.3390/nu9101158
127. Yki-Järvinen H. Nutritional modulation of non-alcoholic fatty liver disease and insulin resistance. Nutrients. (2015) 7:9127–38. doi: 10.3390/nu7115454
128. Zámbó V, Simon-Szabó L, Szelényi P, Kereszturi É, Bánhegyi G, Csala M. Lipotoxicity in the liver. World J Hepatol. (2013) 5:550. doi: 10.4254/wjh.v5.i10.550
129. Jump DB. Fatty acid regulation of hepatic lipid metabolism. Curr Opin Clin Nutr Metab Care. (2011) 14:115–20. doi: 10.1097/MCO.0b013e328342991c
130. Leamy AK, Egnatchik RA, Young JD. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog Lipid Res. (2013) 52:165–74. doi: 10.1016/j.plipres.2012.10.004
131. Dhibi M, Brahmi F, Mnari A, Houas Z, Chargui I, Bchir L, et al. The intake of high fat diet with different trans fatty acid levels differentially induces oxidative stress and non alcoholic fatty liver disease (NAFLD) in rats. Nutr Metab. (2011) 8:65. doi: 10.1186/1743-7075-8-65
132. da Silva-Santi L, Antunes M, Caparroz-Assef S, Carbonera F, Masi L, Curi R, et al. Liver fatty acid composition and inflammation in mice fed with high-carbohydrate diet or high-fat diet. Nutrients. (2016) 8:682. doi: 10.3390/nu8110682
133. Anania C, Perla FM, Olivero F, Pacifico L, Chiesa C. Mediterranean diet and nonalcoholic fatty liver disease. World J Gastroenterol. (2018) 24:2083–94. doi: 10.3748/wjg.v24.i19.2083
134. Yang J, Fernández-Galilea M, Martínez-Fernández L, González-Muniesa P, Pérez-Chávez A, Martínez JA, et al. Oxidative stress and non-alcoholic fatty liver disease: effects of omega-3 fatty acid supplementation. Nutrients. (2019) 11:872. doi: 10.3390/nu11040872
135. Baratta F, Pastori D, Polimeni L, Bucci T, Ceci F, Calabrese C, et al. Adherence to mediterranean diet and non-alcoholic fatty liver disease: effect on insulin resistance: Am J Gastroenterol. (2017) 112:1832–9. doi: 10.1038/ajg.2017.371
136. Gepner Y, Shelef I, Komy O, Cohen N, Schwarzfuchs D, Bril N, et al. The beneficial effects of Mediterranean diet over low-fat diet may be mediated by decreasing hepatic fat content. J Hepatol. (2019) 71:379–388. doi: 10.1016/j.jhep.2019.04.013
137. Abenavoli L, Boccuto L, Federico A, Dallio M, Loguercio C, Di Renzo L, et al. Diet and non-alcoholic fatty liver disease: the mediterranean way. Int J Environ Res Public Health. (2019) 16:3011. doi: 10.3390/ijerph16173011
138. Calder PC. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans. (2017) 45:1105–15. doi: 10.1042/BST20160474
139. Pramfalk C, Pavlides M, Banerjee R, McNeil CA, Neubauer S, Karpe F, et al. Sex-specific differences in hepatic fat oxidation and synthesis may explain the higher propensity for NAFLD in men. J Clin Endocrinol Metab. (2015) 100:4425–33. doi: 10.1210/jc.2015-2649
140. Ranković S, Popović T, Martačić JD, Petrović S, Tomić M, Ignjatović Ð, et al. Liver phospholipids fatty acids composition in response to different types of diets in rats of both sexes. Lipids Health Dis. (2017) 16:94. doi: 10.1186/s12944-017-0483-9
141. Wankhade UD, Zhong Y, Kang P, Alfaro M, Chintapalli SV, Piccolo BD, et al. Maternal high-fat diet programs offspring liver steatosis in a sexually dimorphic manner in association with changes in gut microbial ecology in mice. Sci Rep. (2018) 8:16502. doi: 10.1038/s41598-018-34453-0
142. Weger BD, Gobet C, Yeung J, Martin E, Jimenez S, Betrisey B, et al. The mouse microbiome is required for sex-specific diurnal rhythms of gene expression and metabolism. Cell Metab. (2019) 29:362–82.e8. doi: 10.1016/j.cmet.2018.09.023
143. de Chiara F, Ureta Checcllo C, Ramón Azcón J. High protein diet and metabolic plasticity in non-alcoholic fatty liver disease: myths and truths. Nutrients. (2019) 11:2985. doi: 10.3390/nu11122985
144. Harris RA, Joshi M, Jeoung NH, Obayashi M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr. (2005) 135:1527S−30. doi: 10.1093/jn/135.6.1527S
145. Honda T, Ishigami M, Luo F, Lingyun M, Ishizu Y, Kuzuya T, et al. Branched-chain amino acids alleviate hepatic steatosis and liver injury in choline-deficient high-fat diet induced NASH mice. Metabolism. (2017) 69:177–87. doi: 10.1016/j.metabol.2016.12.013
146. Takegoshi K, Honda M, Okada H, Takabatake R, Matsuzawa-Nagata N, Campbell JS, et al. Branched-chain amino acids prevent hepatic fibrosis and development of hepatocellular carcinoma in a non-alcoholic steatohepatitis mouse model. Oncotarget. (2017) 8:18191–205. doi: 10.18632/oncotarget.15304
147. Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. (2014) 10:723–36. doi: 10.1038/nrendo.2014.171
148. Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. (2012) 15:606–14. doi: 10.1016/j.cmet.2012.01.024
149. Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. (2018) 67:145–58. doi: 10.1002/hep.29465
150. Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, et al. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids. (2015) 47:603–15. doi: 10.1007/s00726-014-1894-9
151. Grzych G, Vonghia L, Bout M-A, Weyler J, Verrijken A, Dirinck E, et al. Plasma BCAA changes in patients with NAFLD are sex dependent. J Clin Endocrinol Metab. (2020) 105:dgaa175. doi: 10.1210/clinem/dgaa175
152. Della Torre S, Mitro N, Meda C, Lolli F, Pedretti S, Barcella M, et al. Short-term fasting reveals amino acid metabolism as a major sex-discriminating factor in the liver. Cell Metab. (2018) 28:256–67.e5. doi: 10.1016/j.cmet.2018.05.021
153. Villa A, Della Torre S, Stell A, Cook J, Brown M, Maggi A. Tetradian oscillation of estrogen receptor is necessary to prevent liver lipid deposition. Proc Natl Acad Sci USA. (2012) 109:11806–11. doi: 10.1073/pnas.1205797109
154. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology. (2019) 70:1457–69. doi: 10.1002/hep.30626
155. Gong Z, Tas E, Yakar S, Muzumdar R. Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging. Mol Cell Endocrinol. (2017) 455:115–30. doi: 10.1016/j.mce.2016.12.022
156. Stahl EC, Haschak MJ, Popovic B, Brown BN. Macrophages in the aging liver and age-related liver disease. Front Immunol. (2018) 9:2795. doi: 10.3389/fimmu.2018.02795
157. Matsuo K, Gualtieri MR, Cahoon SS, Jung CE, Paulson RJ, Shoupe D, et al. Surgical menopause and increased risk of nonalcoholic fatty liver disease in endometrial cancer. Menopause. (2016) 23:189–96. doi: 10.1097/GME.0000000000000500
158. Gutierrez-Grobe Y, Ponciano-Rodríguez G, Ramos MH, Uribe M, Méndez-Sánchez N. Prevalence of non alcoholic fatty liver disease in premenopausal, posmenopausal and polycystic ovary syndrome women. The role of estrogens. Ann Hepatol. (2010) 9:402–9. doi: 10.1016/S1665-2681(19)31616-3
159. Lonardo A, Mantovani A, Lugari S, Targher G. NAFLD in some common endocrine diseases: prevalence, pathophysiology, and principles of diagnosis and management. Int J Mol Sci. (2019) 20:2841. doi: 10.3390/ijms20112841
160. Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. (2002) 122:1649–57. doi: 10.1053/gast.2002.33573
161. Anderson EL, Howe LD, Jones HE, Higgins JPT, Lawlor DA, Fraser A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: a systematic review and meta-analysis. PLoS ONE. (2015) 10:e0140908. doi: 10.1371/journal.pone.0140908
162. Suzuki A, Abdelmalek MF, Schwimmer JB, Lavine JE, Scheimann AO, Unalp–Arida A, et al. Association between puberty and features of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2012) 10:786–94. doi: 10.1016/j.cgh.2012.01.020
163. Lu J, Zhang J, Du R, Wang T, Xu M, Xu Y, et al. Age at menarche is associated with the prevalence of non-alcoholic fatty liver disease later in life: 初潮年龄与成年后非酒精性脂肪性肝病患病风险相关 J Diabetes. (2017) 9:53–60. doi: 10.1111/1753-0407.12379
164. Mueller NT, Pereira MA, Demerath EW, Dreyfus JG, MacLehose RF, Carr JJ, et al. Earlier menarche is associated with fatty liver and abdominal ectopic fat in midlife, independent of young adult BMI: the CARDIA study: menarcheal timing, abdominal fat, and NAFLD. Obesity. (2015) 23:468–74. doi: 10.1002/oby.20950
165. Yi KH, Hwang JS, Lim SW, Lee JA, Kim DH, Lim JS. Early menarche is associated with non-alcoholic fatty liver disease in adulthood. Pediatr Int. (2017) 59:1270–5. doi: 10.1111/ped.13422
166. Al-hamoudi W, El-Sabbah M, Ali S, Altuwaijri M, Bedewi M, Adam M, et al. Epidemiological, clinical, and biochemical characteristics of Saudi patients with nonalcoholic fatty liver disease: a hospital-based study. Ann Saudi Med. (2012) 32:288–92. doi: 10.5144/0256-4947.2012.288
167. Hossain N, Afendy A, Stepanova M, Nader F, Srishord M, Rafiq N, et al. Independent predictors of fibrosis in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2009) 7:1224–9.e2. doi: 10.1016/j.cgh.2009.06.007
168. Loomis AK, Kabadi S, Preiss D, Hyde C, Bonato V, St. Louis M, et al. Body mass index and risk of nonalcoholic fatty liver disease: two electronic health record prospective studies. J Clin Endocrinol Metab. (2016) 101:945–52. doi: 10.1210/jc.2015-3444
169. Yang JD, Abdelmalek MF, Pang H, Guy CD, Smith AD, Diehl AM, et al. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology. (2014) 59:1406–14. doi: 10.1002/hep.26761
170. Argo CK, Northup PG, Al-Osaimi AMS, Caldwell SH. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol. (2009) 51:371–9. doi: 10.1016/j.jhep.2009.03.019
171. Calzadilla Bertot L, Adams L. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci. (2016) 17:774. doi: 10.3390/ijms17050774
172. Bambha K, Belt P, Abraham M, Wilson LA, Pabst M, Ferrell L, et al. Ethnicity and nonalcoholic fatty liver disease. Hepatology. (2012) 55:769–80. doi: 10.1002/hep.24726
173. Singh DK, Sakhuja P, Malhotra V, Gondal R, Sarin SK. Independent predictors of steatohepatitis and fibrosis in asian indian patients with non-alcoholic steatohepatitis. Dig Dis Sci. (2008) 53:1967–76. doi: 10.1007/s10620-007-0074-0
174. Tapper EB, Krajewski K, Lai M, Challies T, Kane R, Afdhal N, et al. Simple non-invasive biomarkers of advanced fibrosis in the evaluation of non-alcoholic fatty liver disease. Gastroenterol Rep. (2014) 2:276–80. doi: 10.1093/gastro/gou034
175. Wang Z, Xu M, Hu Z, Hultström M, Lai E. Sex-specific prevalence of fatty liver disease and associated metabolic factors in Wuhan, south central China. Eur J Gastroenterol Hepatol. (2014) 26:1015–21. doi: 10.1097/MEG.0000000000000151
176. Yang JD, Abdelmalek MF, Guy CD, Gill RM, Lavine JE, Yates K, et al. Patient sex, reproductive status, and synthetic hormone use associate with histologic severity of nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. (2017) 15:127–31.e2. doi: 10.1016/j.cgh.2016.07.034
177. Klair JS, Yang JD, Abdelmalek MF, Guy CD, Gill RM, Yates K, et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease: steatohepatitis/metabolic liver disease. Hepatology. (2016) 64:85–91. doi: 10.1002/hep.28514
178. Yoneda M, Thomas E, Sumida Y, Eguchi Y, Schiff ER. The influence of menopause on the development of hepatic fibrosis in nonobese women with nonalcoholic fatty liver disease. Hepatology. (2014) 60:1792. doi: 10.1002/hep.27097
179. Gummesson A, Strömberg U, Schmidt C, Kullberg J, Angerås O, Lindgren S, et al. Non-alcoholic fatty liver disease is a strong predictor of coronary artery calcification in metabolically healthy subjects: a cross-sectional, population-based study in middle-aged subjects. PLoS ONE. (2018) 13:e0202666. doi: 10.1371/journal.pone.0202666
180. Lee SB, Park G-M, Lee J-Y, Lee BU, Park JH, Kim BG, et al. Association between non-alcoholic fatty liver disease and subclinical coronary atherosclerosis: an observational cohort study. J Hepatol. (2018) 68:1018–24. doi: 10.1016/j.jhep.2017.12.012
181. Ozturk K, Uygun A, Guler AK, Demirci H, Ozdemir C, Cakir M, et al. Nonalcoholic fatty liver disease is an independent risk factor for atherosclerosis in young adult men. Atherosclerosis. (2015) 240:380–6. doi: 10.1016/j.atherosclerosis.2015.04.009
182. Cai J, Zhang X-J, Ji Y-X, Zhang P, She Z-G, Li H. Nonalcoholic fatty liver disease pandemic fuels the upsurge in cardiovascular diseases. Circ Res. (2020) 126:679–704. doi: 10.1161/CIRCRESAHA.119.316337
183. Lu H, Liu H, Hu F, Zou L, Luo S, Sun L. Independent association between nonalcoholic fatty liver disease and cardiovascular disease: a systematic review and meta-analysis. Int J Endocrinol. (2013) 2013:124958. doi: 10.1155/2013/124958
184. Tana C, Ballestri S, Ricci F, Di Vincenzo A, Ticinesi A, Gallina S, et al. Cardiovascular risk in non-alcoholic fatty liver disease: mechanisms and therapeutic implications. Int J Environ Res Public Health. (2019) 16:3104. doi: 10.3390/ijerph16173104
185. van den Berg EH, Gruppen EG, Ebtehaj S, Bakker SJL, Tietge UJF, Dullaart RPF. Cholesterol efflux capacity is impaired in subjects with an elevated fatty liver index, a proxy of non-alcoholic fatty liver disease. Atherosclerosis. (2018) 277:21–7. doi: 10.1016/j.atherosclerosis.2018.07.028
186. Cohen D, Fisher E. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis. (2013) 33:380–8. doi: 10.1055/s-0033-1358519
187. Xu X, Lu L, Dong Q, Li X, Zhang N, Xin Y, et al. Research advances in the relationship between nonalcoholic fatty liver disease and atherosclerosis. Lipids Health Dis. (2015) 14:158. doi: 10.1186/s12944-015-0141-z
188. Ho C-M, Ho S-L, Jeng Y-M, Lai Y-S, Chen Y-H, Lu S-C, et al. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J Inflamm. (2019) 16:1–8. doi: 10.1186/s12950-019-0211-5
189. Houben T, Brandsma E, Walenbergh SMA, Hofker MH, Shiri-Sverdlov R. Oxidized LDL at the crossroads of immunity in non-alcoholic steatohepatitis. Biochim Biophys Acta BBA. (2017) 1862:416–29. doi: 10.1016/j.bbalip.2016.07.008
190. Spahis S, Delvin E, Borys J-M, Levy E. Oxidative stress as a critical factor in nonalcoholic fatty liver disease pathogenesis. Antioxid Redox Signal. (2017) 26:519–41. doi: 10.1089/ars.2016.6776
191. Allen AM, Therneau TM, Mara KC, Larson JJ, Watt KD, Hayes SN, et al. Women with nonalcoholic fatty liver disease lose protection against cardiovascular disease: a longitudinal cohort study. Am J Gastroenterol. (2019) 114:1764–71. doi: 10.14309/ajg.0000000000000401
192. Kim MK, Ahn CW, Nam JS, Md SK, Park JS, Kim KR. Association between nonalcoholic fatty liver disease and coronary artery calcification in postmenopausal women. Menopause. (2015) 22:1323–7. doi: 10.1097/GME.0000000000000503
193. Wells LG. Cardiovascular risk factors: does sex matter? Curr Vasc Pharmacol. (2016) 14:452–7. doi: 10.2174/1570161114666160722113116
194. Roy AK, Chatterjee B. Sexual dimorphism in the liver. Annu Rev Physiol. (1983) 45:37–50. doi: 10.1146/annurev.ph.45.030183.000345
195. Yang X. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. (2006) 16:995–1004. doi: 10.1101/gr.5217506
196. Lichanska AM, Waters MJ. How growth hormone controls growth, obesity and sexual dimorphism. Trends Genet. (2008) 24:41–7. doi: 10.1016/j.tig.2007.10.006
197. Waxman DJ, O'Connor C. Growth hormone regulation of sex-dependent liver gene expression. Mol Endocrinol. (2006) 20:2613–29. doi: 10.1210/me.2006-0007
198. Fernández-Pérez L, Guerra B, Díaz-Chico JC, Flores-Morales A. Estrogens regulate the hepatic effects of growth hormone, a hormonal interplay with multiple fates. Front Endocrinol. (2013) 4:66. doi: 10.3389/fendo.2013.00066
199. Leung K-C, Johannsson G, Leong GM, Ho KKY. Estrogen regulation of growth hormone action. Endocr Rev. (2004) 25:693–721. doi: 10.1210/er.2003-0035
200. Meinhardt UJ, Ho KKY. Modulation of growth hormone action by sex steroids. Clin Endocrinol. (2006) 65:413–22. doi: 10.1111/j.1365-2265.2006.02676.x
201. Mode A, Gustafsson J-Å. Sex and the liver – a journey through five decades. Drug Metab Rev. (2006) 38:197–207. doi: 10.1080/03602530600570057
202. Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol. (2009) 76:215–28. doi: 10.1124/mol.109.056705
203. Clodfelter KH, Holloway MG, Hodor P, Park S-H, Ray WJ, Waxman DJ. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol. (2006) 20:1333–51. doi: 10.1210/me.2005-0489
204. Connerney J, Lau-Corona D, Rampersaud A, Waxman DJ. Activation of male liver chromatin accessibility and STAT5-dependent gene transcription by plasma growth hormone pulses. Endocrinology. (2017) 158:1386–405. doi: 10.1210/en.2017-00060
205. Zhang Y, Laz EV, Waxman DJ. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol. (2012) 32:880–96. doi: 10.1128/MCB.06312-11
206. Holloway MG, Miles GD, Dombkowski AA, Waxman DJ. Liver-specific hepatocyte nuclear factor-4α deficiency: greater impact on gene expression in male than in female mouse liver. Mol Endocrinol. (2008) 22:1274–86. doi: 10.1210/me.2007-0564
207. Wiwi CA, Gupte M, Waxman DJ. Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4α-deficient mice. Mol Endocrinol. (2004) 18:1975–87. doi: 10.1210/me.2004-0129
208. Conforto TL, Steinhardt GF, Waxman DJ. Cross talk between GH-regulated transcription factors HNF6 and CUX2 in adult mouse liver. Mol Endocrinol. (2015) 29:1286–302. doi: 10.1210/me.2015-1028
209. Holloway MG, Laz EV, Waxman DJ. Codependence of growth hormone-responsive, sexually dimorphic hepatic gene expression on signal transducer and activator of transcription 5b and hepatic nuclear factor 4α. Mol Endocrinol. (2006) 20:647–60. doi: 10.1210/me.2005-0328
210. Park S-H, Wiwi CA, Waxman DJ. Signalling cross-talk between hepatocyte nuclear factor 4α and growth-hormone-activated STAT5b. Biochem J. (2006) 397:159–68. doi: 10.1042/BJ20060332
211. Lau-Corona D, Suvorov A, Waxman DJ. Feminization of male mouse liver by persistent growth hormone stimulation: activation of sex-biased transcriptional networks and dynamic changes in chromatin states. Mol Cell Biol. (2017) 37:e00301-17. doi: 10.1128/MCB.00301-17
212. Laz EV, Sugathan A, Waxman DJ. Dynamic in vivo binding of STAT5 to growth hormone-regulated genes in intact rat liver. Sex-specific binding at low- but not high-affinity stat5 sites. Mol Endocrinol. (2009) 23:1242–54. doi: 10.1210/me.2008-0449
213. Ling G, Sugathan A, Mazor T, Fraenkel E, Waxman DJ. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol Cell Biol. (2010) 30:5531–44. doi: 10.1128/MCB.00601-10
214. Meyer RD, Laz EV, Su T, Waxman DJ. Male-Specific hepatic Bcl6: growth hormone-induced block of transcription elongation in females and binding to target genes inversely coordinated with STAT5. Mol Endocrinol. (2009) 23:1914–26. doi: 10.1210/me.2009-0242
215. Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol. (2013) 33:3594–610. doi: 10.1128/MCB.00280-13
216. Conforto TL, Waxman DJ. Sex-specific mouse liver gene expression: genome-wide analysis of developmental changes from pre-pubertal period to young adulthood. Biol Sex Differ. (2012) 3:9. doi: 10.1186/2042-6410-3-9
217. Lowe R, Gemma C, Rakyan VK, Holland ML. Sexually dimorphic gene expression emerges with embryonic genome activation and is dynamic throughout development. BMC Genomics. (2015) 16:295. doi: 10.1186/s12864-015-1506-4
218. Wauthier V, Sugathan A, Meyer RD, Dombkowski AA, Waxman DJ. Intrinsic sex differences in the early growth hormone responsiveness of sex-specific genes in mouse liver. Mol Endocrinol. (2010) 24:667–78. doi: 10.1210/me.2009-0454
219. Liu Z, Cordoba-Chacon J, Kineman RD, Cronstein BN, Muzumdar R, Gong Z, et al. Growth hormone control of hepatic lipid metabolism. Diabetes. (2016) 65:3598–609. doi: 10.2337/db16-0649
220. Takahashi Y. The role of growth hormone and insulin-like growth factor-I in the liver. Int J Mol Sci. (2017) 18:1447. doi: 10.3390/ijms18071447
221. Liang S, Yu Z, Song X, Wang Y, Li M, Xue J. Reduced growth hormone secretion is associated with nonalcoholic fatty liver disease in obese children. Horm Metab Res. (2018) 50:250–6. doi: 10.1055/s-0043-124970
222. Ishikawa M, Brooks AJ, Fernández-Rojo MA, Medina J, Chhabra Y, Minami S, et al. Growth hormone stops excessive inflammation after partial hepatectomy allowing liver regeneration and survival via induction of H2-Bl/HLA-G. Hepatology. (2020). doi: 10.1002/hep.31297. [Epub a head of print].
223. Ahmed TA, Buzzelli MD, Lang CH, Capen JB, Shumate ML, Navaratnarajah M, et al. Interleukin-6 inhibits growth hormone-mediated gene expression in hepatocytes. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G1793–803. doi: 10.1152/ajpgi.00547.2006
224. Buzzelli MD, Nagarajan M, Radtka JF, Shumate ML, Navaratnarajah M, Lang CH, et al. Nuclear actor-κB mediates the inhibitory effects of tumor necrosis factor-α on growth hormone-inducible gene expression in liver. Endocrinology. (2008) 149:6378–88. doi: 10.1210/en.2007-1574
225. Thissen J-P, Verniers J. Inhibition by interleukin-1β and tumor necrosis factor-α of the insulin-like growth factor i messenger ribonucleic acid response to growth hormone in rat hepatocyte primary culture 1. Endocrinology. (1997) 138:1078–84. doi: 10.1210/endo.138.3.4966
226. Zhao Y, Xiao X, Frank SJ, Lin HY, Xia Y. Distinct mechanisms of induction of hepatic growth hormone resistance by endogenous IL-6, TNF-α, and IL-1β. Am J Physiol-Endocrinol Metab. (2014) 307:E186–98. doi: 10.1152/ajpendo.00652.2013
227. Nohara K, Baba T, Murai H, Kobayashi Y, Suzuki T, Tateishi Y, et al. Global DNA methylation in the mouse liver is affected by methyl deficiency and arsenic in a sex-dependent manner. Arch Toxicol. (2011) 85:653–61. doi: 10.1007/s00204-010-0611-z
228. Takasugi M, Hayakawa K, Arai D, Shiota K. Age- and sex-dependent DNA hypomethylation controlled by growth hormone in mouse liver. Mech Ageing Dev. (2013) 134:331–7. doi: 10.1016/j.mad.2013.05.003
229. Lomas-Soria C, Reyes-Castro LA, Rodríguez-González GL, Ibáñez CA, Bautista CJ, Cox LA, et al. Maternal obesity has sex-dependent effects on insulin, glucose and lipid metabolism and the liver transcriptome in young adult rat offspring: maternal obesity programs liver transcriptome changes in rat offspring. J Physiol. (2018) 596:4611–28. doi: 10.1113/JP276372
230. Stanimirovic J, Obradovic M, Jovanovic A, Sudar-Milovanovic E, Zafirovic S, Pitt SJ, et al. A high fat diet induces sex-specific differences in hepatic lipid metabolism and nitrite/nitrate in rats. Nitric Oxide. (2016) 54:51–9. doi: 10.1016/j.niox.2016.02.007
231. Bur IM, Cohen-Solal AM, Carmignac D, Abecassis P-Y, Chauvet N, Martin AO, et al. The circadian clock components CRY1 and CRY2 are necessary to sustain sex dimorphism in mouse liver metabolism. J Biol Chem. (2009) 284:9066–73. doi: 10.1074/jbc.M808360200
232. Lu Y-F, Jin T, Xu Y, Zhang D, Wu Q, Zhang Y-KJ, et al. Sex differences in the circadian variation of cytochrome P450 genes and corresponding nuclear receptors in mouse liver. Chronobiol Int. (2013) 30:1135–43. doi: 10.3109/07420528.2013.805762
233. Zheng D, Wang X, Antonson P, Gustafsson J-Å, Li Z. Genomics of sex hormone receptor signaling in hepatic sexual dimorphism. Mol Cell Endocrinol. (2018) 471:33–41. doi: 10.1016/j.mce.2017.05.025
234. Blachier M, Leleu H, Peck-Radosavljevic M, Valla D-C, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. (2013) 58:593–608. doi: 10.1016/j.jhep.2012.12.005
235. Maggi A, Della Torre S. Sex, metabolism and health. Mol Metab. (2018) 15:3–7. doi: 10.1016/j.molmet.2018.02.012
236. Della Torre S, Lolli F, Ciana P, Maggi A. Sexual dimorphism estrogen action in mouse liver. In: F. Mauvais-Jarvis F, editor. Sex Gender Factors Affecting Metabolic Homeostasis, Diabetes Obesity Advances in Experimental Medicine Biology. Cham: Springer International Publishing (2017). p. 141–51. doi: 10.1007/978-3-319-70178-3_8
237. Zhu L, Shi J, Luu TN, Neuman JC, Trefts E, Yu S, et al. Hepatocyte estrogen receptor alpha mediates estrogen action to promote reverse cholesterol transport during Western-type diet feeding. Mol Metab. (2018) 8:106–16. doi: 10.1016/j.molmet.2017.12.012
238. Addison ML, Rissman EF. Sexual dimorphism of growth hormone in the hypothalamus: regulation by estradiol. Endocrinology. (2012) 153:1898–907. doi: 10.1210/en.2011-1982
239. Fernández-Pérez L, de Mirecki-Garrido M, Guerra B, Díaz M, Díaz-Chico JC. Sex steroids and growth hormone interactions. Endocrinol Nutr. (2016) 63:171–80. doi: 10.1016/j.endoen.2016.04.006
240. Ramirez MC, Bourguignon NS, Bonaventura MM, Lux-Lantos V, Libertun C, Becu-Villalobos D. Neonatal xenoestrogen exposure alters growth hormone-dependent liver proteins and genes in adult female rats. Toxicol Lett. (2012) 213:325–31. doi: 10.1016/j.toxlet.2012.07.015
241. Qiu S, Vazquez JT, Boulger E, Liu H, Xue P, Hussain MA, et al. Hepatic estrogen receptor α is critical for regulation of gluconeogenesis and lipid metabolism in males. Sci Rep. (2017) 7:1661. doi: 10.1038/s41598-017-01937-4
242. Fontana R, Torre S. The deep correlation between energy metabolism and reproduction: a view on the effects of nutrition for women fertility. Nutrients. (2016) 8:87. doi: 10.3390/nu8020087
243. van Nas A, GuhaThakurta D, Wang SS, Yehya N, Horvath S, Zhang B, et al. Elucidating the role of gonadal hormones in sexually dimorphic gene coexpression networks. Endocrinology. (2009) 150:1235–49. doi: 10.1210/en.2008-0563
244. Biddie SC. Chromatin architecture and the regulation of nuclear receptor inducible transcription: nuclear receptors and the epigenetic regulation of chromatin. J Neuroendocrinol. (2011) 23:94–106. doi: 10.1111/j.1365-2826.2010.02079.x
245. He HH, Meyer CA, Chen MW, Jordan VC, Brown M, Liu XS. Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res. (2012) 22:1015–25. doi: 10.1101/gr.133280.111
246. Papa S, Bubici C, Zazzeroni F, Franzoso G. Mechanisms of liver disease: cross-talk between the NF-κB and JNK pathways. Biol Chem. (2009) 390:965–76. doi: 10.1515/BC.2009.111
247. Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H, et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. (2018) 24:2661–72. doi: 10.3748/wjg.v24.i25.2661
248. Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. (2017) 6:174–84. doi: 10.1016/j.molmet.2016.12.001
249. Yan H, Gao Y, Zhang Y. Inhibition of JNK suppresses autophagy and attenuates insulin resistance in a rat model of nonalcoholic fatty liver disease. Mol Med Rep. (2017) 15:180–6. doi: 10.3892/mmr.2016.5966
250. Du K, Williams CD, McGill MR, Jaeschke H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol Appl Pharmacol. (2014) 281:58–66. doi: 10.1016/j.taap.2014.09.002
251. Win S, Min RWM, Chen CQ, Zhang J, Chen Y, Li M, et al. Expression of mitochondrial membrane–linked SAB determines severity of sex-dependent acute liver injury. J Clin Invest. (2019) 129:5278–93. doi: 10.1172/JCI128289
252. Galmés-Pascual BM, Martínez-Cignoni MR, Morán-Costoya A, Bauza-Thorbrügge M, Sbert-Roig M, Valle A, et al. 17β-estradiol ameliorates lipotoxicity-induced hepatic mitochondrial oxidative stress and insulin resistance. Free Radic Biol Med. (2020) 150:148–60. doi: 10.1016/j.freeradbiomed.2020.02.016
253. Rahman MM, McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol. (2011) 9:291–306. doi: 10.1038/nrmicro2539
254. Chen Z, Yu R, Xiong Y, Du F, Zhu S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. (2017) 16:203. doi: 10.1186/s12944-017-0572-9
255. Evans MJ, Eckert A, Lai K, Adelman SJ, Harnish DC. Reciprocal antagonism between estrogen receptor and NF-κB activity in vivo. Circ Res. (2001) 89:823–30. doi: 10.1161/hh2101.098543
256. Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-κB. Trends Endocrinol Metab. (2005) 16:46–52. doi: 10.1016/j.tem.2005.01.004
257. Sun B, Karin M. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene. (2008) 27:6228–44. doi: 10.1038/onc.2008.300
258. Ministrini S, Montecucco F, Sahebkar A, Carbone F. Macrophages in the pathophysiology of NAFLD: the role of sex differences. Eur J Clin Invest. (2020) 50:1–9. doi: 10.1111/eci.13236
259. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. (2007) 317:121–4. doi: 10.1126/science.1140485
260. Li C, Xu MM, Wang K, Adler AJ, Vella AT, Zhou B. Macrophage polarization and meta-inflammation. Transl Res. (2018) 191:29–44. doi: 10.1016/j.trsl.2017.10.004
261. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. (2014) 59:2034–42. doi: 10.1002/hep.26754
262. Toniolo A, Fadini GP, Tedesco S, Cappellari R, Vegeto E, Maggi A, et al. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J Clin Endocrinol Metab. (2015) 100:E50–8. doi: 10.1210/jc.2014-2751
263. Villa A, Rizzi N, Vegeto E, Ciana P, Maggi A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci Rep. (2015) 5:15224. doi: 10.1038/srep15224
264. Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, et al. Estrogen deficiency worsens steatohepatitis in mice fed high-fat and high-cholesterol diet. Am J Physiol-Gastrointest Liver Physiol. (2011) 301:G1031–43. doi: 10.1152/ajpgi.00211.2011
265. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
266. Wan X, Xu C, Yu C, Li Y. Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can J Gastroenterol Hepatol. (2016) 2016:6489012. doi: 10.1155/2016/6489012
267. Zhang N-P, Liu X-J, Xie L, Shen X-Z, Wu J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest. (2019) 99:749–63. doi: 10.1038/s41374-018-0177-6
268. Farruggio S, Cocomazzi G, Marotta P, Romito R, Surico D, Calamita G, et al. Genistein and 17β-estradiol protect hepatocytes from fatty degeneration by mechanisms involving mitochondria, inflammasome and kinases activation. Cell Physiol Biochem. (2020) 54:401–16. doi: 10.33594/000000227
269. Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai W, et al. Estrogen suppresses hepatocellular carcinoma cells through ERβ-mediated upregulation of the NLRP3 inflammasome. Lab Invest. (2015) 95:804–16. doi: 10.1038/labinvest.2015.63
270. Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. (2004) 127:S5–16. doi: 10.1053/j.gastro.2004.09.011
271. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. (2016) 529:307–15. doi: 10.1038/nature17039
272. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. (2006) 43:S45–53. doi: 10.1002/hep.20969
273. Mao SA, Glorioso JM, Nyberg SL. Liver regeneration. Transl Res. (2014) 163:352–62. doi: 10.1016/j.trsl.2014.01.005
274. Michalopoulos GK. Advances in liver regeneration. Expert Rev Gastroenterol Hepatol. (2014) 8:897–907. doi: 10.1586/17474124.2014.934358
275. Tao Y, Wang M, Chen E, Tang H. Liver regeneration: analysis of the main relevant signaling molecules. Mediators Inflamm. (2017) 2017:4256352. doi: 10.1155/2017/4256352
276. Arai M, Yokosuka O, Chiba T, Imazeki F, Kato M, Hashida J, et al. Gene expression profiling reveals the mechanism and pathophysiology of mouse liver regeneration. J Biol Chem. (2003) 278:29813–8. doi: 10.1074/jbc.M212648200
277. Li W. Global changes in interleukin-6–dependent gene expression patterns in mouse livers after partial hepatectomy. Hepatology. (2001) 33:1377–86. doi: 10.1053/jhep.2001.24431
278. Bizzaro D, Crescenzi M, Di Liddo R, Arcidiacono D, Cappon A, Bertalot T, et al. Sex-dependent differences in inflammatory responses during liver regeneration in a murine model of acute liver injury. Clin Sci. (2018) 132:255–72. doi: 10.1042/CS20171260
279. Kao T-L, Kuan Y-P, Cheng W-C, Chang W-C, Jeng L-B, Yeh S, et al. Estrogen receptors orchestrate cell growth and differentiation to facilitate liver regeneration. Theranostics. (2018) 8:2672–82. doi: 10.7150/thno.23624
280. Sutti S, Tacke F. Liver inflammation and regeneration in drug-induced liver injury: sex matters! Clin Sci. (2018) 132:609–13. doi: 10.1042/CS20171313
281. Tsugawa Y, Natori M, Handa H, Imai T. Estradiol accelerates liver regeneration through estrogen receptor α. Clin Exp Gastroenterol. (2019) 12:331–6. doi: 10.2147/CEG.S214196
282. Uebi T, Umeda M, Imai T. Estrogen induces estrogen receptor alpha expression and hepatocyte proliferation in the livers of male mice. Genes Cells. (2015) 20:217–23. doi: 10.1111/gtc.12214
283. Srisowanna N, Choijookhuu N, Yano K, Batmunkh B, Ikenoue M, Nhat Huynh Mai N, et al. The effect of estrogen on hepatic fat accumulation during early phase of liver regeneration after partial hepatectomy in rats. ACTA Histochem Cytochem. (2019) 52:67–75. doi: 10.1267/ahc.19018
284. Hassan MM, Botrus G, Abdel-Wahab R, Wolff RA, Li D, Tweardy D, et al. Estrogen replacement reduces risk and increases survival times of women with hepatocellular carcinoma. Clin Gastroenterol Hepatol. (2017) 15:1791–9. doi: 10.1016/j.cgh.2017.05.036
285. Louet J-F, LeMay C, Mauvais-Jarvis F. Antidiabetic actions of estrogen: insight from human and genetic mouse models. Curr Atheroscler Rep. (2004) 6:180–5. doi: 10.1007/s11883-004-0030-9
286. D'Eon TM, Souza SC, Aronovitz M, Obin MS, Fried SK, Greenberg AS. Estrogen regulation of adiposity and fuel partitioning: evidence of genomic and non-genomic regulation of lipogenic and oxidative pathways. J Biol Chem. (2005) 280:35983–91. doi: 10.1074/jbc.M507339200
287. Zhu L, Brown WC, Cai Q, Krust A, Chambon P, McGuinness OP, et al. Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway-selective insulin resistance. Diabetes. (2013) 62:424–34. doi: 10.2337/db11-1718
288. Yan H, Yang W, Zhou F, Li X, Pan Q, Shen Z, et al. Estrogen improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor Foxo1. Diabetes. (2019) 68:291–304. doi: 10.2337/db18-0638
289. Paquette A, Wang D, Jankowski M, Gutkowska J, Lavoie J-M. Effects of ovariectomy on PPARα, SREBP-1c, and SCD-1 gene expression in the rat liver. Menopause. (2008) 15:1169–75. doi: 10.1097/gme.0b013e31817b8159
290. Barsalani R, Chapados N, Lavoie J-M. Hepatic VLDL-TG production and MTP gene expression are decreased in ovariectomized rats: effects of exercise training. Horm Metab Res. (2010) 42:860–7. doi: 10.1055/s-0030-1267173
291. Palmisano BT, Le TD, Zhu L, Lee YK, Stafford JM. Cholesteryl ester transfer protein alters liver and plasma triglyceride metabolism through two liver networks in female mice. J Lipid Res. (2016) 57:1541–51. doi: 10.1194/jlr.M069013
292. Della Torre S, Biserni A, Rando G, Monteleone G, Ciana P, Komm B, et al. The conundrum of estrogen receptor oscillatory activity in the search for an appropriate hormone replacement therapy. Endocrinology. (2011) 152:2256–65. doi: 10.1210/en.2011-0173
293. Smith GI, Reeds DN, Okunade AL, Patterson BW, Mittendorfer B. Systemic delivery of estradiol, but not testosterone or progesterone, alters very low density lipoprotein-triglyceride kinetics in postmenopausal women. J Clin Endocrinol Metab. (2014) 99:E1306–10. doi: 10.1210/jc.2013-4470
294. Zegura B, Guzic-Salobir B, Sebestjen M, Keber I. The effect of various menopausal hormone therapies on markers of inflammation, coagulation, fibrinolysis, lipids, and lipoproteins in healthy postmenopausal women. Menopause. (2006) 13:643–50. doi: 10.1097/01.gme.0000198485.70703.7a
295. Benedusi V, Martini E, Kallikourdis M, Villa A, Meda C, Maggi A. Ovariectomy shortens the life span of female mice. Oncotarget. (2015) 6:10801–11. doi: 10.18632/oncotarget.2984
296. Kireev RA, Tresguerres ACF, Garcia C, Borras C, Ariznavarreta C, Vara E, et al. Hormonal regulation of pro-inflammatory and lipid peroxidation processes in liver of old ovariectomized female rats. Biogerontology. (2010) 11:229–43. doi: 10.1007/s10522-009-9242-2
297. Rogers NH, Perfield JW, Strissel KJ, Obin MS, Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology. (2009) 150:2161–8. doi: 10.1210/en.2008-1405
298. Toda K, Toda A, Ono M, Saibara T. Lack of 17β-estradiol reduces sensitivity to insulin in the liver and muscle of male mice. Heliyon. (2018) 4:e00772. doi: 10.1016/j.heliyon.2018.e00772
299. van Sinderen ML, Steinberg GR, Jørgensen SB, To SQ, Knower KC, Clyne CD, et al. Hepatic glucose intolerance precedes hepatic steatosis in the male aromatase knockout (ArKO) mouse. PLoS ONE. (2014) 9:e87230. doi: 10.1371/journal.pone.0087230
300. Chen Z, Wang O, Nie M, Elison K, Zhou D, Li M, et al. Aromatase deficiency in a Chinese adult man caused by novel compound heterozygous CYP19A1 mutations: effects of estrogen replacement therapy on the bone, lipid, liver and glucose metabolism. Mol Cell Endocrinol. (2015) 399:32–42. doi: 10.1016/j.mce.2014.09.016
301. Maffei L, Rochira V, Zirilli L, Antunez P, Aranda C, Fabre B, et al. A novel compound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol. (2007) 67:218–24. doi: 10.1111/j.1365-2265.2007.02864.x
302. Jones MEE, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA. (2000) 97:12735–40. doi: 10.1073/pnas.97.23.12735
303. Amano A, Kondo Y, Noda Y, Ohta M, Kawanishi N, Machida S, et al. Abnormal lipid/lipoprotein metabolism and high plasma testosterone levels in male but not female aromatase-knockout mice. Arch Biochem Biophys. (2017) 622:47–58. doi: 10.1016/j.abb.2017.03.007
304. Hewitt KN, Pratis K, Jones MEE, Simpson ER. Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology. (2004) 145:1842–8. doi: 10.1210/en.2003-1369
305. Nemoto Y, Toda K, Ono M, Fujikawa-Adachi K, Saibara T, Onishi S, et al. Altered expression of fatty acid–metabolizing enzymes in aromatase-deficient mice. J Clin Invest. (2000) 105:1819–25. doi: 10.1172/JCI9575
306. Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, et al. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator- activated receptor alpha- deficient mice. J Clin Invest. (1998) 102:1083–91. doi: 10.1172/JCI3949
307. Yoshikawa T. Aromatase-deficient (ArKO) mice are retrieved from severe hepatic steatosis by peroxisome proliferator administration. Hepatol Res. (2002) 22:278–87. doi: 10.1016/S1386-6346(01)00145-0
308. Moro L, Arbini AA, Hsieh J-T, Ford J, Simpson ER, Hajibeigi A, et al. Aromatase deficiency inhibits the permeability transition in mouse liver mitochondria. Endocrinology. (2010) 151:1643–52. doi: 10.1210/en.2009-1450
309. Ahlbory-Dieker DL, Stride BD, Leder G, Schkoldow J, Trölenberg S, Seidel H, et al. DNA binding by estrogen receptor-α is essential for the transcriptional response to estrogen in the liver and the uterus. Mol Endocrinol. (2009) 23:1544–55. doi: 10.1210/me.2009-0045
310. Gao H, Fält S, Sandelin A, Gustafsson J-Å, Dahlman-Wright K. Genome-wide identification of estrogen receptor α-binding sites in mouse liver. Mol Endocrinol. (2008) 22:10–22. doi: 10.1210/me.2007-0121
311. Hart-Unger S, Arao Y, Hamilton KJ, Lierz SL, Malarkey DE, Hewitt SC, et al. Hormone signaling and fatty liver in females: analysis of estrogen receptor α mutant mice. Int J Obes. (2017) 41:945–54. doi: 10.1038/ijo.2017.50
312. Pedram A, Razandi M, O'Mahony F, Harvey H, Harvey BJ, Levin ER. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci Signal. (2013) 6:ra36. doi: 10.1126/scisignal.2004013
313. Yasrebi A, Rivera JA, Krumm EA, Yang JA, Roepke TA. Activation of estrogen response element–independent erα signaling protects female mice from diet-induced obesity. Endocrinology. (2017) 158:319–34. doi: 10.1210/en.2016-1535
314. Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, et al. Evidence that oestrogen receptor-α plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. (2006) 49:588–97. doi: 10.1007/s00125-005-0105-3
315. Barros RPA, Gustafsson J-Å. Estrogen receptors and the metabolic network. Cell Metab. (2011) 14:289–99. doi: 10.1016/j.cmet.2011.08.005
316. Ribas V, Nguyen MTA, Henstridge DC, Nguyen A-K, Beaven SW, Watt MJ, et al. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am J Physiol-Endocrinol Metab. (2010) 298:E304–19. doi: 10.1152/ajpendo.00504.2009
317. Evans MJ, Lai K, Shaw LJ, Harnish DC, Chadwick CC. Estrogen receptor α inhibits il-1β induction of gene expression in the mouse liver. Endocrinology. (2002) 143:2559–70. doi: 10.1210/endo.143.7.8919
318. Noh J-R, Kim Y-H, Kim D-K, Hwang JH, Kim K-S, Choi D-H, et al. Small heterodimer partner deficiency increases inflammatory liver injury through C-X-C motif chemokine ligand 2-driven neutrophil recruitment in mice. Toxicol Sci. (2018) 163:254–64. doi: 10.1093/toxsci/kfy030
319. Wang X, Lu Y, Wang E, Zhang Z, Xiong X, Zhang H, et al. Hepatic estrogen receptor α improves hepatosteatosis through upregulation of small heterodimer partner. J Hepatol. (2015) 63:183–90. doi: 10.1016/j.jhep.2015.02.029
320. Guillaume M, Riant E, Fabre A, Raymond-Letron I, Buscato M, Davezac M, et al. Selective liver estrogen receptor α modulation prevents steatosis, diabetes, and obesity through the anorectic growth differentiation factor 15 hepatokine in mice. Hepatol Commun. (2019) 3:908–24. doi: 10.1002/hep4.1363
321. Camporez JPG, Jornayvaz FR, Lee H-Y, Kanda S, Guigni BA, Kahn M, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. (2013) 154:1021–8. doi: 10.1210/en.2012-1989
322. Chambliss KL, Barrera J, Umetani M, Umetani J, Kim SH, Madak-Erdogan Z, et al. Nonnuclear estrogen receptor activation improves hepatic steatosis in female mice. Endocrinology. (2016) 157:3731–41. doi: 10.1210/en.2015-1629
323. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA. (2000) 97:12729–34. doi: 10.1073/pnas.97.23.12729
324. Khristi V, Ratri A, Ghosh S, Pathak D, Borosha S, Dai E, et al. Disruption of ESR1 alters the expression of genes regulating hepatic lipid and carbohydrate metabolism in male rats. Mol Cell Endocrinol. (2019) 490:47–56. doi: 10.1016/j.mce.2019.04.005
325. Khristi V, Ratri A, Ghosh S, Borosha S, Dai E, Chakravarthi VP, et al. Liver transcriptome data of Esr1 knockout male rats reveals altered expression of genes involved in carbohydrate and lipid metabolism. Data Brief. (2019) 22:771–80. doi: 10.1016/j.dib.2018.12.089
326. Zhu L, Martinez MN, Emfinger CH, Palmisano BT, Stafford JM. Estrogen signaling prevents diet-induced hepatic insulin resistance in male mice with obesity. Am J Physiol-Endocrinol Metab. (2014) 306:E1188–97. doi: 10.1152/ajpendo.00579.2013
327. Winn NC, Jurrissen TJ, Grunewald ZI, Cunningham RP, Woodford ML, Kanaley JA, et al. Estrogen receptor-α signaling maintains immunometabolic function in males and is obligatory for exercise-induced amelioration of nonalcoholic fatty liver. Am J Physiol-Endocrinol Metab. (2019) 316:E156–67. doi: 10.1152/ajpendo.00259.2018
328. Sharma G, Prossnitz ER. GPER/GPR30 knockout mice: effects of GPER on metabolism. In: Eyster KM, editor. Estrogen Receptors Methods in Molecular Biology. New York, NY: Springer New York (2016). p. 489–502. doi: 10.1007/978-1-4939-3127-9_38
329. Sharma G, Prossnitz ER. G-protein-coupled estrogen receptor (GPER) sex-specific metabolic homeostasis. In: Mauvais-Jarvis F, editor. Sex Gender Factors Affecting Metabolic Homeostasis, Diabetes Obesity. Advances in Experimental Medicine Biology. Cham: Springer International Publishing (2017). p. 427–53. doi: 10.1007/978-3-319-70178-3_20
330. Sharma G, Mauvais-Jarvis F, Prossnitz ER. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J Steroid Biochem Mol Biol. (2018) 176:31–7. doi: 10.1016/j.jsbmb.2017.02.012
331. Barton M, Filardo EJ, Lolait SJ, Thomas P, Maggiolini M, Prossnitz ER. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J Steroid Biochem Mol Biol. (2018) 176:4–15. doi: 10.1016/j.jsbmb.2017.03.021
332. Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol. (2014) 389:71–83. doi: 10.1016/j.mce.2014.02.002
333. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein–coupled receptor GPR30. Annu Rev Physiol. (2008) 70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518
334. Revankar CM. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
335. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. (2011) 7:715–26. doi: 10.1038/nrendo.2011.122
336. Barton M, Prossnitz ER. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol Metab. (2015) 26:185–92. doi: 10.1016/j.tem.2015.02.003
337. Mårtensson UEA, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. (2009) 150:687–98. doi: 10.1210/en.2008-0623
338. Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. (2013) 154:4136–45. doi: 10.1210/en.2013-1357
339. Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol. (2009) 214:67–77. doi: 10.1016/j.jneuroim.2009.06.023
340. Brunsing RL, Prossnitz ER. Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled estrogen receptor (GPER) agonist G-1: IL-10 induction by GPER agonist G-1. Immunology. (2011) 134:93–106. doi: 10.1111/j.1365-2567.2011.03471.x
341. Brunsing RL, Owens KS, Prossnitz ER. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing Conditions. J Immunother. (2013) 36:190–6. doi: 10.1097/CJI.0b013e31828d8e3b
342. Hussain Y, Ding Q, Connelly PW, Brunt JH, Ban MR, McIntyre AD, et al. G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism: cellular and population genetic studies. Arterioscler Thromb Vasc Biol. (2015) 35:213–21. doi: 10.1161/ATVBAHA.114.304326
343. Fu W, Gao X-P, Zhang S, Dai Y-P, Zou W-J, Yue L-M. 17β-estradiol inhibits PCSK9-mediated LDLR degradation through GPER/PLC activation in HepG2 cells. Front Endocrinol. (2020) 10:930. doi: 10.3389/fendo.2019.00930
344. Meoli L, Isensee J, Zazzu V, Nabzdyk CS, Soewarto D, Witt H, et al. Sex- and age-dependent effects of Gpr30 genetic deletion on the metabolic and cardiovascular profiles of diet-induced obese mice. Gene. (2014) 540:210–6. doi: 10.1016/j.gene.2014.02.036
345. Sharma G, Hu C, Staquicini DI, Brigman JL, Liu M, Mauvais-Jarvis F, et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci Transl Med. (2020) 12:eaau5956. doi: 10.1126/scitranslmed.aau5956
346. Meyer MR, Fredette NC, Barton M, Prossnitz ER. G protein-coupled estrogen receptor inhibits vascular prostanoid production and activity. J Endocrinol. (2015) 227:61–9. doi: 10.1530/JOE-15-0257
347. Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci Rep. (2015) 4:7564. doi: 10.1038/srep07564
348. Itoga M, Konno Y, Moritoki Y, Saito Y, Ito W, Tamaki M, et al. G-protein-coupled estrogen receptor agonist suppresses airway inflammation in a mouse model of asthma through IL-10. PLoS ONE. (2015) 10:e0123210. doi: 10.1371/journal.pone.0123210
349. Correa J, Ronchetti S, Labombarda F, De Nicola AF, Pietranera L. Activation of the G protein-coupled estrogen receptor (GPER) increases neurogenesis and ameliorates neuroinflammation in the hippocampus of male spontaneously hypertensive rats. Cell Mol Neurobiol. (2019) 40:711–23. doi: 10.1007/s10571-019-00766-5
350. Mendes-Oliveira J, Lopes Campos F, Videira RA, Baltazar G. GPER activation is effective in protecting against inflammation-induced nigral dopaminergic loss and motor function impairment. Brain Behav Immun. (2017) 64:296–307. doi: 10.1016/j.bbi.2017.04.016
351. Wei T, Chen W, Wen L, Zhang J, Zhang Q, Yang J, et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. (2016) 382:195–202. doi: 10.1016/j.canlet.2016.08.012
352. Prossnitz ER, Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol. (2015) 153:114–26. doi: 10.1016/j.jsbmb.2015.06.014
353. Nagarajan SR, Paul-Heng M, Krycer JR, Fazakerley DJ, Sharland AF, Hoy AJ. Lipid and glucose metabolism in hepatocyte cell lines and primary mouse hepatocytes: a comprehensive resource for in vitro studies of hepatic metabolism. Am J Physiol-Endocrinol Metab. (2019) 316:E578–89. doi: 10.1152/ajpendo.00365.2018
354. Fabris G, Dumortier O, Pisani DF, Gautier N, Van Obberghen E. Amino acid-induced regulation of hepatocyte growth: possible role of Drosha. Cell Death Dis. (2019) 10:566. doi: 10.1038/s41419-019-1779-7
355. Grant DM. Detoxification pathways in the liver. J Inherit Metab Dis. (1991) 14:421–30. doi: 10.1007/BF01797915
356. Almazroo OA, Miah MK, Venkataramanan R. Drug metabolism in the liver. Clin Liver Dis. (2017) 21:1–20. doi: 10.1016/j.cld.2016.08.001
357. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. (2013) 14:996–1006. doi: 10.1038/ni.2691
358. Fukui M, Senmaru T, Hasegawa G, Yamazaki M, Asano M, Kagami Y, et al. 17β-Estradiol attenuates saturated fatty acid diet-induced liver injury in ovariectomized mice by up-regulating hepatic senescence marker protein-30. Biochem Biophys Res Commun. (2011) 415:252–7. doi: 10.1016/j.bbrc.2011.10.025
359. Miyashita T, Toyoda Y, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Hepatoprotective effect of tamoxifen on steatosis and non-alcoholic steatohepatitis in mouse models. J Toxicol Sci. (2012) 37:931–42. doi: 10.2131/jts.37.931
360. Li P, He K, Li J, Liu Z, Gong J. The role of Kupffer cells in hepatic diseases. Mol Immunol. (2017) 85:222–9. doi: 10.1016/j.molimm.2017.02.018
361. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. (2006) 26:1175–86. doi: 10.1111/j.1478-3231.2006.01342.x
362. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
363. Vonghia L, Van Herck MA, Weyler J, Francque S. Targeting myeloid-derived cells: new frontiers in the treatment of non-alcoholic and alcoholic liver disease. Front Immunol. (2019) 10:563. doi: 10.3389/fimmu.2019.00563
364. Marcos R, Lopes C, Malhão F, Correia-Gomes C, Fonseca S, Lima M, et al. Stereological assessment of sexual dimorphism in the rat liver reveals differences in hepatocytes and Kupffer cells but not hepatic stellate cells. J Anat. (2016) 228:996–1005. doi: 10.1111/joa.12448
365. Wagnerberger S, Fiederlein L, Kanuri G, Stahl C, Millonig G, Mueller S, et al. Sex-specific differences in the development of acute alcohol-induced liver steatosis in mice. Alcohol Alcohol. (2013) 48:648–56. doi: 10.1093/alcalc/agt138
366. Wands J. Hepatocellular carcinoma and sex. N Engl J Med. (2007) 357:1974–6. doi: 10.1056/NEJMcibr075652
367. Ikejima K, Enomoto N, Iimuro Y, Ikejima A, Fang D, Xu J, et al. Estrogen increases sensitivity of hepatic Kupffer cells to endotoxin. Am J Physiol Gastrointest Liver Physiol. (1998) 274:G669–76. doi: 10.1152/ajpgi.1998.274.4.G669
368. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
369. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. (2013) 3:785–97. doi: 10.1002/cphy.c120026
370. Shimizu T, Suzuki T, Yu H-P, Yokoyama Y, Choudhry MA, Bland KI, et al. The role of estrogen receptor subtypes on hepatic neutrophil accumulation following trauma-hemorrhage: direct modulation of CINC-1 production by Kupffer cells. Cytokine. (2008) 43:88–92. doi: 10.1016/j.cyto.2008.04.001
371. Suzuki T, Shimizu T, Yu H-P, Hsieh Y-C, Choudhry MA, Bland KI, et al. 17β-estradiol administration following trauma-hemorrhage prevents the increase in Kupffer cell cytokine production and MAPK activation predominately via estrogen receptor-α. Surgery. (2006) 140:141–8. doi: 10.1016/j.surg.2006.02.014
372. Vickers AEM, Lucier GW. Estrogen receptor levels and occupancy in hepatic sinusoidal endothelial and Kupffer cells are enhanced by initiation with diethylnitrosamine and promotion with 17α-ethinylestradiol in rats. Carcinogenesis. (1996) 17:1235–42. doi: 10.1093/carcin/17.6.1235
373. Shmarakov IO, Jiang H, Liu J, Fernandez EJ, Blaner WS. Hepatic stellate cell activation: A source for bioactive lipids. Biochim Biophys Acta BBA - Mol Cell Biol Lipids. (2019) 1864:629–42. doi: 10.1016/j.bbalip.2019.02.004
374. Shimizu I, Mizobuchi Y, Yasuda M, Shiba M, Ma Y-R, Horie T, et al. Inhibitory effect of oestradiol on activation of rat hepatic stellate cells in vivo and in vitro. Gut. (1999) 44:127–36. doi: 10.1136/gut.44.1.127
375. Xu J-W. Estrogen reduces CCL4- induced liver fibrosis in rats. World J Gastroenterol. (2002) 8:883. doi: 10.3748/wjg.v8.i5.883
376. Zhang B, Zhang C-G, Ji L-H, Zhao G, Wu Z-Y. Estrogen receptor β selective agonist ameliorates liver cirrhosis in rats by inhibiting the activation and proliferation of hepatic stellate cells: ERβ agonist and liver cirrhosis. J Gastroenterol Hepatol. (2018) 33:747–55. doi: 10.1111/jgh.13976
377. Cortes E, Lachowski D, Rice A, Thorpe SD, Robinson B, Yeldag G, et al. Tamoxifen mechanically deactivates hepatic stellate cells via the G protein-coupled estrogen receptor. Oncogene. (2019) 38:2910–22. doi: 10.1038/s41388-018-0631-3
378. Zhou Y, Shimizu I, Lu G, Itonaga M, Okamura Y, Shono M, et al. Hepatic stellate cells contain the functional estrogen receptor β but not the estrogen receptor α in male and female rats. Biochem Biophys Res Commun. (2001) 286:1059–65. doi: 10.1006/bbrc.2001.5479
379. Li R, Oteiza A, Sørensen KK, McCourt P, Olsen R, Smedsrød B, et al. Role of liver sinusoidal endothelial cells and stabilins in elimination of oxidized low-density lipoproteins. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G71–81. doi: 10.1152/ajpgi.00215.2010
380. Sørensen KK, Simon-Santamaria J, McCuskey RS, Smedsrød B. Liver sinusoidal endothelial cells. In: Terjung R, editor. Comprehensive Physiology, Hoboken, NJ: John Wiley & Sons, Inc (2015). p. 1751–74. doi: 10.1002/cphy.c140078
381. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells — gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. (2018) 15:555–67. doi: 10.1038/s41575-018-0020-y
382. DeLeve L, Maretti-Mira A. Liver sinusoidal endothelial cell: an update. Semin Liver Dis. (2017) 37:377–87. doi: 10.1055/s-0037-1617455
383. Sakamoto M, Ueno T, Nakamura T, Hashimoto O, Sakata R, Kin M, et al. Estrogen upregulates nitric oxide synthase expression in cultured rat hepatic sinusoidal endothelial cells. J Hepatol. (2001) 34:858–64. doi: 10.1016/S0168-8278(01)00023-X
384. Sakamoto M, Ueno T, Nakamura T, Sakata R, Hasimoto O, Torimura T, et al. Improvement of portal hypertension and hepatic blood flow in cirrhotic rats by oestrogen. Eur J Clin Invest. (2005) 35:220–5. doi: 10.1111/j.1365-2362.2005.01476.x
385. Zhang B, Wu Z-Y. Estrogen derivatives: novel therapeutic agents for liver cirrhosis and portal hypertension. Eur J Gastroenterol Hepatol. (2013) 25:263–70. doi: 10.1097/MEG.0b013e32835ab5dc
386. Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol. (2019) 16:269–81. doi: 10.1038/s41575-019-0125-y
387. Raven A, Lu W-Y, Man TY, Ferreira-Gonzalez S, O'Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. (2017) 547:350–4. doi: 10.1038/nature23015
388. Pinto C, Giordano DM, Maroni L, Marzioni M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim Biophys Acta BBA Mol Basis Dis. (2018) 1864:1270–8. doi: 10.1016/j.bbadis.2017.07.024
389. Fabris L, Spirli C, Cadamuro M, Fiorotto R, Strazzabosco M. Emerging concepts in biliary repair and fibrosis. Am J Physiol Gastrointest Liver Physiol. (2017) 313:G102–16. doi: 10.1152/ajpgi.00452.2016
390. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. (2014) 383:2168–79. doi: 10.1016/S0140-6736(13)61903-0
391. Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma — evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. (2018) 15:95–111. doi: 10.1038/nrclinonc.2017.157
392. Tam PKH, Yiu RS, Lendahl U, Andersson ER. Cholangiopathies – towards a molecular understanding. EBioMedicine. (2018) 35:381–93. doi: 10.1016/j.ebiom.2018.08.024
393. Alvaro D. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. (2006) 12:3537. doi: 10.3748/wjg.v12.i22.3537
394. Alvaro D, Alpini G, Onori P, Perego L, Baroni GS, Franchitto A, et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology. (2000) 119:1681–91. doi: 10.1053/gast.2000.20184
395. Alvaro D, Gigliozzi A, Attili AF, Onori P, Franchitto A, Vetuschi A, et al. Effect of ovariectomy on the proliferative capacity of intrahepatic rat cholangiocytes. Gastroenterology. (2002) 123:336–44. doi: 10.1053/gast.2002.34169
396. Svegliati-Baroni G, Ghiselli R, Marzioni M, Alvaro D, Mocchegiani F, Saccomanno S, et al. Estrogens maintain bile duct mass and reduce apoptosis after biliodigestive anastomosis in bile duct ligated rats. J. Hepatol. (2006) 44:1158–66. doi: 10.1016/j.jhep.2005.10.032
397. Alvaro D, Invernizzi P, Onori P, Franchitto A, De Santis A, Crosignani A, et al. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J Hepatol. (2004) 41:905–12. doi: 10.1016/j.jhep.2004.08.022
398. Cao H, Zhu B, Qu Y, Zhang W. Abnormal expression of ERα in cholangiocytes of patients with primary biliary cholangitis mediated intrahepatic bile duct inflammation. Front Immunol. (2019) 10:2815. doi: 10.3389/fimmu.2019.02815
399. Meyer SK, Probert PME, Lakey AK, Leitch AC, Blake LI, Jowsey PA, et al. Environmental xenoestrogens super-activate a variant murine ER beta in cholangiocytes. Toxicol Sci. (2016) 156:54–71. doi: 10.1093/toxsci/kfw234
400. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
401. Ding C, Li Y, Guo F, Jiang Y, Ying W, Li D, et al. A cell-type-resolved liver proteome. Mol Cell Proteomics. (2016) 15:3190–202. doi: 10.1074/mcp.M116.060145
402. Kmieć Z. Cooperation of Liver Cells in Health and Disease. Berlin, Heidelberg: Springer Berlin Heidelberg. (2001).
403. Cassim S, Raymond V-A, Lapierre P, Bilodeau M. From in vivo to in vitro: major metabolic alterations take place in hepatocytes during and following isolation. PLoS ONE. (2017) 12:e0190366. doi: 10.1371/journal.pone.0190366
404. Suurmond CE, Lasli S, Dolder FW, Ung A, Kim H, Bandaru P, et al. In vitro human liver model of nonalcoholic steatohepatitis by coculturing hepatocytes, endothelial cells, and kupffer cells. Adv Healthc Mater. (2019) 8:1901379. doi: 10.1002/adhm.201901379
405. Akbari S, Arslan N, Senturk S, Erdal E. Next-generation liver medicine using organoid models. Front Cell Dev Biol. (2019) 7:345. doi: 10.3389/fcell.2019.00345
406. Tchernof A, Després J-P. Pathophysiology of human visceral obesity: an update. Physiol Rev. (2013) 93:359–404. doi: 10.1152/physrev.00033.2011
407. de Bosscher K, Desmet SJ, Clarisse D, Estébanez-Perpiña E, Brunsveld L. Nuclear receptor crosstalk — defining the mechanisms for therapeutic innovation. Nat Rev Endocrinol. (2020) 16:363–77. doi: 10.1038/s41574-020-0349-5
408. Rando G, Wahli W. Sex differences in nuclear receptor-regulated liver metabolic pathways. Biochim Biophys Acta BBA Mol Basis Dis. (2011) 1812:964–73. doi: 10.1016/j.bbadis.2010.12.023
409. Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta BBA Gene Regul Mech. (2016) 1859:1083–99. doi: 10.1016/j.bbagrm.2016.03.002
410. Cvitanović Tomaš T, Urlep Ž, Moškon M, Mraz M, Rozman D. Liversex computational model: sexual aspects in hepatic metabolism and abnormalities. Front Physiol. (2018) 9:360. doi: 10.3389/fphys.2018.00360
411. Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther. (2017) 179:142–57. doi: 10.1016/j.pharmthera.2017.05.011
412. Jalouli M, Carlsson L, Améen C, Lindén D, Ljungberg A, Michalik L, et al. Sex difference in hepatic peroxisome proliferator-activated receptor α expression: influence of pituitary and gonadal hormones. Endocrinology. (2003) 144:101–9. doi: 10.1210/en.2002-220630
413. Quinn MA, Xu X, Ronfani M, Cidlowski JA. Estrogen deficiency promotes hepatic steatosis via a glucocorticoid receptor-dependent mechanism in mice. Cell Rep. (2018) 22:2690–701. doi: 10.1016/j.celrep.2018.02.041
414. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr Rev. (2018) 39:760–802. doi: 10.1210/er.2018-00064
415. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. (2015) 62:720–33. doi: 10.1016/j.jhep.2014.10.039
416. Rizzo G, Fiorucci S. PPARs and other nuclear receptors in inflammation. Curr Opin Pharmacol. (2006) 6:421–7. doi: 10.1016/j.coph.2006.03.012
417. Freeman A, Stanko P, Berkowitz LN, Parnell N, Zuppe A, Bale TL, et al. Inclusion of sex and gender in biomedical research: survey of clinical research proposed at the University of Pennsylvania. Biol Sex Differ. (2017) 8:22. doi: 10.1186/s13293-017-0139-5
418. Harris DJ, Douglas PS. Enrollment of women in cardiovascular clinical trials funded by the national heart, lung, and blood institute. N Engl J Med. (2000) 343:475–80. doi: 10.1056/NEJM200008173430706
419. Holdcroft A. Gender bias in research: how does it affect evidence based medicine? J R Soc Med. (2007) 100:2–3. doi: 10.1177/014107680710000102
420. Liu KA, DiPietro Mager NA. Women's involvement in clinical trials: historical perspective and future implications. Pharm Pract. (2016) 14:708. doi: 10.18549/PharmPract.2016.01.708
421. Mauvais-Jarvis F, Arnold AP, Reue K. A guide for the design of pre-clinical studies on sex differences in metabolism. Cell Metab. (2017) 25:1216–30. doi: 10.1016/j.cmet.2017.04.033
422. McGregor AJ, Hasnain M, Sandberg K, Morrison MF, Berlin M, Trott J. How to study the impact of sex and gender in medical research: a review of resources. Biol Sex Differ. (2016) 7:46. doi: 10.1186/s13293-016-0099-1
423. Regitz-Zagrosek V. Sex and gender differences in health: science & society series on sex and science. EMBO Rep. (2012) 13:596–603. doi: 10.1038/embor.2012.87
424. Day S, Mason R, Lagosky S, Rochon PA. Integrating and evaluating sex and gender in health research. Health Res Policy Syst. (2016) 14:75. doi: 10.1186/s12961-016-0147-7
425. Gahagan J, Gray K, Whynacht A. Sex and gender matter in health research: addressing health inequities in health research reporting. Int J Equity Health. (2015) 14:12. doi: 10.1186/s12939-015-0144-4
426. Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature. (2014) 509:282–3. doi: 10.1038/509282a
427. Doyal L. Sex, gender, and health: the need for a new approach. BMJ. (2001) 323:1061–3. doi: 10.1136/bmj.323.7320.1061
428. Johnson J, Sharman Z, Vissandjée B, Stewart DE. Does a change in health research funding policy related to the integration of sex and gender have an impact? PLoS ONE. (2014) 9:e99900. doi: 10.1371/journal.pone.0099900
429. Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. (2016) 64:1577–86. doi: 10.1002/hep.28785
430. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol. (2016) 65:589–600. doi: 10.1016/j.jhep.2016.05.013
Keywords: NAFLD (non-alcoholic fatty liver disease), estrogens, estrogen receptors, liver, sex differences
Citation: Della Torre S (2020) Non-alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 11:572490. doi: 10.3389/fendo.2020.572490
Received: 14 June 2020; Accepted: 20 August 2020;
Published: 18 September 2020.
Edited by:
Nicolas Venteclef, Sorbonne Universités, FranceReviewed by:
Benoit Pourcet, Université de Lille, FranceMagdalene K. Montgomery, The University of Melbourne, Australia
Copyright © 2020 Della Torre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Della Torre, c2FyYS5kZWxsYXRvcnJlQHVuaW1pLml0