 Chiara Mele1,2
Chiara Mele1,2 Monica Mencarelli3
Monica Mencarelli3 Marina Caputo4,5
Marina Caputo4,5 Stefania Mai6
Stefania Mai6 Loredana Pagano7
Loredana Pagano7 Gianluca Aimaretti1,5
Gianluca Aimaretti1,5 Massimo Scacchi2
Massimo Scacchi2 Alberto Falchetti8,9
Alberto Falchetti8,9 Paolo Marzullo1,2*
Paolo Marzullo1,2*- 1Department of Translational Medicine, University of Piemonte Orientale, Novara, Italy
- 2Istituto Auxologico Italiano, IRCCS, Division of General Medicine, S. Giuseppe Hospital, Piancavallo, Italy
- 3Istituto Auxologico Italiano, IRCCS, Laboratory of Molecular Biology, S. Giuseppe Hospital, Piancavallo, Italy
- 4Department of Health Sciences, University of Piemonte Orientale, Novara, Italy
- 5Division of Endocrinology, University Hospital “Maggiore della Carità”, Novara, Italy
- 6Istituto Auxologico Italiano, IRCCS, Laboratory of Metabolic Research, S. Giuseppe Hospital, Piancavallo, Italy
- 7Division of Endocrinology, Diabetology and Metabolism, Department of Medical Sciences, University of Turin, Turin, Italy
- 8Istituto Auxologico Italiano, IRCCS, Rehabilitation Unit, S. Giuseppe Hospital, Unit for Bone Metabolism Diseases, Verbania, Italy
- 9Diabetes & Lab of Endocrine and Metabolic Research, Dept. of Clinical Sciences & Community Health, University of Milan, Milan, Italy
Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal dominant inherited tumor syndrome, associated with parathyroid, pituitary, and gastro-entero-pancreatic (GEP) neuroendocrine tumors (NETs). MEN1 is usually consequent to different germline and somatic mutations of the MEN1 tumor suppressor gene, although phenocopies have also been reported. This review analyzed main biomedical databases searching for reports on MEN1 gene mutations and focused on aggressive and aberrant clinical manifestations to investigate the potential genotype-phenotype correlation. Despite efforts made by several groups, this link remains elusive to date and evidence that aggressive or aberrant clinical phenotypes may be related to specific mutations has been provided by case reports and small groups of MEN1 patients or families. In such context, a higher risk of aggressive tumor phenotypes has been described in relation to frameshift and non-sense mutations, and predominantly associated with aggressive GEP NETs, particularly pancreatic NETs. In our experience a novel heterozygous missense mutation at c.836C>A in exon 6 was noticed in a MEN1 patient operated for macro-prolactinoma, who progressively developed recurrent parathyroid adenomas, expanding gastrinomas and, long after the first MEN1 manifestation, a neuroendocrine uterine carcinoma. In conclusion, proof of genotype-phenotype correlation is limited but current evidence hints at the need for long-term interdisciplinary surveillance in patients with aggressive phenotypes and genetically confirmed MEN1.
Introduction
Multiple endocrine neoplasia type 1 (MEN1) syndrome (OMIM#131100) is a rare autosomal dominant inherited tumor syndrome with high penetrance, that is typically associated with parathyroid, pituitary, and gastro-entero-pancreatic (GEP) neuroendocrine tumors (NETs), either functioning or nonfunctioning (1, 2). Adding to the intrinsic burden of MEN1, a different combination of endocrine and non-endocrine tumors may develop, i.e. carcinoids (thymic, bronchial), adrenocortical tumors, facial angiofibromas, lipomas, and collagenomas (3–5).
The clinical behavior of MEN1 largely depends on tumor spread and histological features, as well as type and degree of hormone hypersecretion, risk of tumor recurrence, and duration of surveillance (6). The MEN1 gene (NM_130799.2) is a tumor-suppressor gene located on chromosome 11q13 (7, 8). This gene spans approximatively 9,000 bp of genomic DNA containing 10 exons and is transcribed into a 2.8 kb mRNA with the translational start codon (ATG) in exon-2 and the stop-codon in exon-10. The protein product of the MEN1 gene is called MENIN, of which different isoforms have been reported: long isoform 1, 615 amino acids, chosen as the canonical sequence, short isoform 2, 610 amino acids length, and isoform 3 consisting of 575 amino acids (9). MENIN nuclear localization sequences (NLSs) are located in its C-terminal region and directly interact with DNA in a sequence-independent manner to serve as a scaffold protein that controls gene expression and cell signaling (10). The majority of patients with the inherited form of the disease carry germline mutations of the MEN1 gene (11). Germline heterozygous inactivating mutations were found in the coding region of the MEN1 gene in index cases and affected family members, together with loss of heterozygosity (LOH) for markers at the MEN1 gene locus in their tumors as expected for a causative tumor suppressor gene (12–14). To date, many germline or somatic mutations have been described both in MEN1 families and sporadic cases (15–18).
MEN1 gene mutations are distributed throughout the coding region without particular hot spot regions. Since the original description of the MEN1 gene, different germline MEN1 mutations have been identified across its coding sequences (6, 15). Approximatively 10% of the MEN1 mutations arise de novo and are later transmitted to following generations (19). More than 10% of mutations are nonsense mutations, 40% frameshift insertions and deletions, 25% missense mutations while 11% are splice site defects (20). Generally, the MEN1-related tumors occurrence requires inheritance of a germline mutation of MEN1 gene together with a somatic mutation in the DNA of tumor, leading to LOH, according to the two hits model by Knudsons’ (13, 15). However, in multifocal MEN1-gastrinomas, with Zollinger–Ellison syndrome, it has been described that LOH at 11q13 region can be found in less than 50% of patients, and within the same patient different tumoral foci frequently exhibited different patterns of LOH, ranging from LOH limited only to 11q13 to loss of the whole chromosome or no LOH, suggesting that each focus of gastrinoma may arise by an independent second hit (21). It has been also hypothesized that preneoplastic G-cells hyperplastic lesions, and similarly to what described in somatostatin‐secreting tumors from the same patients, may retain both MEN1 alleles and that LOH and/or MEN1 gene mutation, reported to be present in 54% of tumors lacking evidence of LOH, may account for the initiation step of neoplastic lesions (21). Lack of mutations have been reported in 5–25% of patients with a clinical diagnosis of MEN1, constituting the so-called “phenocopies” (15, 22, 23). Of note, a recent study on 189 patients with typical MEN1 phenotype described a higher prevalence (74%) of mutation-negative cases than previously reported (24).
Interest has increasingly focused on the genetic mutations associated with MEN1 hinting at potential genotype-phenotype correlations. To date, genotype-phenotype correlations in MEN1 have been difficult to assess, and the nature of mutations appears to play a null role in clinical MEN1 features, i.e. age of onset, multicentricity, recurrence or markers of aggressiveness (25, 26). Opposed to MEN1 syndrome, the existence of a strong genotype-phenotype correlation has been demonstrated in genetic studies of MEN2 syndrome (1). In the present narrative review, we sought to summarize evidences from the literature on peculiar MEN1 gene mutations with a focus on aggressive and/or aberrant clinical presentations. The search for relevant original publications and case reports written in English was performed on PubMed, Embase, Scopus, and Google Scholar using the NCI Dictionary of Cancer Terms: multiple endocrine neoplasia type 1 OR MEN1 AND phenotype OR genotype, multiple endocrine neoplasia type 1 OR MEN1 AND genotype-phenotype correlation, multiple endocrine neoplasia type 1 OR MEN1 AND aggressive, multiple endocrine neoplasia type 1 OR MEN1 AND aberrant. As an index case, we describe a MEN1 patient harboring a novel sporadic germline mutation and late aggressive clinical features, so as to further discuss the potential clues underlying a genotype–phenotype correlation in MEN1.
Gene Mutations Involved in the Pathogenesis of MEN1
Since the MEN1 gene has been identified, 1,698 mutations have been described (17). Of these, approximately 85% are germline and 15% are somatic mutations (15, 27). Germline MEN1 mutations consist of about 460 different mutations, distributed throughout the 1,830 bp coding region and splice sites of the MEN1 gene (27).
Different types of MEN1 gene mutations and their frequencies have been summarized in Table 1. These mutations are dispersed throughout the coding region of the gene rather than being clustered (27). Approximately 75% of MEN1 mutations are inactivating, as it often happens for tumor suppressor genes (17). A few mutations repeatedly described in unrelated families, refer to 9 sites in the MEN1 gene, and account for over 20% of all germline mutations (27). These mutations are recurrent and possibly represent hot spots. Deletion or insertion of these hot spots elements has also been reported in association with DNA sequence repeats, DNA stretches of long strips of either single nucleotides or shorter repeat elements (28).

Table 1 Different types of gene mutations associated with MEN1 and their frequencies.
Approximatively 5–10% of MEN1 patients may show no evident germline mutations in the MEN1 gene coding region (12, 14, 28–31) but they may harbor large mutations or deletions in the promoter or untranslated regions, which have not been described to date (27). Large deletions are hard to detect by conventional Sanger sequencing, and multiplex ligation-dependent probe amplification (MLPA) (32) or next-generation sequencing (NGS) technology, specifically whole genome testing, or long-read sequencing, the latter enabling also detection of intronic or promoter mutations, are predicted to more accurately extrapolate information on such large gene rearrangements (33).
Additional genetic mechanisms have been recently investigated for their possible involvement in MEN1 phenotype (27). Particular interest has focused on the p27Kip1 protein, which is encoded by the CDKN1B gene and is located downstream of MEN1-driven tumorigenesis (34). The V109G variant of p27 has been reported to influence the clinical manifestation of MEN1 subjects who carry truncating MEN1 gene mutations (34). Carriers of both genetic variants, i.e. truncating MEN1 mutation and V109G variant, show a more aggressive clinical behavior with a worse prognosis of the syndrome (35). These mechanisms will subsequently be detailed.
The Classical Clinical Spectrum of MEN1
Clinical manifestations of MEN1 are predominantly associated with classical endocrine tumors and their relative secretion products (28). Typically, MEN1 is characterized by the presence of several endocrine tumors in the parathyroids, the pituitary gland and the GEP tract. Possible concomitant bronchial, thymic, type II gastric entero-chromaffin-Like (ECL) NETs, and adrenocortical tumors have also been reported. Likewise, a variable number of other endocrine and non-endocrine tumors have been described in the context of MEN1 phenotype, such as central nervous system (CNS) and cutaneous tumors, and will be subsequently summarized (36). MEN1 can affect all age groups, from 5 to 82 years (37, 38), although clinical and/or biochemical manifestations onset in nearly 95% of patients by the fifth decade (39). MEN1 affects both genders equally, and a recent series of 734 cases described a 57.8% female predominance (40).
Parathyroid Tumors
Parathyroid tumors, resulting in primary hyperparathyroidism (PHPT), affect up to 95% of MEN1 patients and represent the first manifestation of the syndrome, with more than 85% of cases between ages 20 and 25 years (1, 39, 41, 42). PHPT manifests with hypercalcemia in 100% of affected patients by age 50 years (43). Compared to sporadic PHPT, bone disease and urolithiasis in MEN1-related PHPT reportedly show an early onset and higher severity (44–46). Interestingly, Kanazawa and colleagues demonstrated, in isolated Men1 knock-out osteoblasts model, that menin may play a key role in bone development, remodeling, and maintenance of bone mass in vivo (47), although in patients with germline MEN1 mutations a specific genotype-phenotype correlation has never been described in this regard.
GEP-NETs Tumors
GEP tumors occur in 70–80% of patients and mainly consist of gastrinomas (the most frequent functioning pancreatic tumor), glucagonomas, insulinomas, vaso-active intestinal peptidomas (VIPomas), and non-functioning tumors (28, 39, 42).
More than 80% of MEN1-associated gastrinomas exhibit, at pathology, multiple microgastrinomas within the first and second duodenum portion (48). Gastrinomas are usually associated with hypergastrinemia, increased gastric acid secretion and peptic ulcers, a clinical combination usually referred to as the Zollinger-Ellison syndrome (ZES) (22). Approximatively 50% of MEN1-associated duodenal microgastrinomas harbor LOH at the MEN1 locus (49). MEN1-associated gastrinomas exhibit a malignant course and metastasize to local lymph nodes and liver in about 50% of cases, even before diagnosis (48, 50). Liver metastases negatively affect prognosis and survival, whereas lymph node metastases do not seem to affect the prognosis (51).
Interestingly, considering that most patients with ZES are treated for long periods with proton pump inhibitors (PPI), a debate has recently emerged on the potential sensitivity to PPI-related secondary hypergastrinemia, leading to a potential increased risk of gastric neuroendocrine tumors, as well as other tumors (52).
In the context of MEN1, insulinomas are the second most prevalent functioning pancreatic tumor. They are characterized by hypoglycemia and the typical Whipple’s triad, which is the first clinical manifestation of MEN1 syndrome in about 10% of subjects (36, 53). MEN1 insulinomas usually manifest as single benign lesions (54), often sized >1 cm, in the setting of multiple islet macroadenomas (55).
Glucagonomas and VIPomas are infrequent and often present as large lesions (>3 cm) with predominant benign behavior. Glucagonomas occur in fewer than 3% of subjects with MEN1 (56). Symptoms are often vague and the typical signs of skin rash, anemia, weight loss, and stomatitis can be absent, while mass effects can be present (22). In asymptomatic MEN1 patients, the presence of the tumor can be suspected upon pancreatic imaging in the presence of glucose intolerance and hyperglucagonemia (22). VIPomas are clinically characterized by watery diarrhea with achlorhydria and hypokalemia (57).
Non-functioning GEP NETs are the most frequent tumor types. They occur in approximately 55% of MEN1 subjects and are often multiple, asymptomatic or cause compressive symptoms (58). An accurate identification of non-functioning GEP NETs is clinically important for three main reasons: 1) these tumors could have a malignant course, which represent the most frequent cause of death in MEN1 patients; 2) several studies demonstrated that non-functioning GEP NETs are associated with a worse prognosis compared to other functioning tumors; 3) the absence of a clinical syndrome and specific biomarkers can result in delayed diagnosis, which increase the mortality rate (22, 59).
Anterior Pituitary Tumors
Anterior pituitary tumors occur in about 30% of subjects and represent the first phenotypic manifestation of MEN1 in up to 42% of sporadic cases (28, 41, 42, 60). In 65–85% of MEN1 patients, pituitary tumors are represented by macroadenomas, a proportion that exceeds that recorded in sporadic tumors (55, 60). In about 30% of cases, pituitary tumors are locally invasive (60). Prolactinomas are the most prevalent pituitary adenomas in the context of MEN1 (65%), followed by somatotropinomas, ACTH-secreting tumors, and non-functioning tumors, the frequency of which is often overlooked (61). Clinical manifestations of pituitary adenomas in patients with MEN1 parallel those of sporadic tumors, hence they depend on hormone hypersecretion, tumor size, pathological features, and pituitary reserve. Depending on specific pituitary hormone hypersecretion, patients could manifest symptoms of hyperprolactinemia (e.g. galactorrhea, amenorrhea, and infertility in women, impotence and infertility in men) or develop somatic and metabolic alterations associated with Cushing’s disease or acromegaly. Large and/or invasive pituitary tumors could compress adjacent structures including the normal pituitary tissue and the optic chiasm, leading to hypopituitarism and/or visual disturbance (22).
Other Endocrine Tumor Types
Adrenal cortical tumors are not rare in patients with MEN1, occurring in approximately one-fourth of patients with genetically confirmed MEN1 (5). Less than 10% of subjects with adrenal tumors show hormonal hypersecretion (62–64), mainly consisting of primary hyperaldosteronism and/or hypercortisolism (65). In 2002, Langer et al. conducted a clinical study with the aim of monitoring 66 patients with confirmed MEN1 germline mutations in a screening program that included evaluation of the adrenal glands. They observed that patients with mutations in exons 2 and 10 of the MEN1 gene develop adrenal lesions more often than subjects with other mutations of the MEN1 gene. The malignant potential of MEN-1-related adrenal neoplasia is of clinical relevance (5).
Approximately 10% of MEN1 patients can develop thymic, bronchial, or type II gastric ECL carcinoids. Thymic NETs are aggressive malignant tumors that preferentially occur in male smokers (66). Their detection is largely dependent on imaging studies. In women, carcinoids are primarily multicentric and metasynchronous bronchial NETs and their course is generally indolent (66, 67). However, Lecomte et al. described cases of poorly differentiated and aggressive bronchial NETs, which are associated with an increased mortality (68).
Non-endocrine Tumors
MEN1 patients can also develop lipomas, collagenomas, facial angiofibromas, CNS tumors including meningiomas and ependymomas, and smooth-muscle tumors, including leiomyomas (1, 4, 30, 39, 41, 51).
Skin tumors tend to be multiple and their diagnosis often precedes the onset of clinical hormone-dependent manifestations, thus contributing to early diagnosis of MEN1 (69, 70). Subcutaneous, pleural, visceral, or retroperitoneal lipomas (34%), facial angiofibromas (88%), and collagenomas (72%) may occur frequently in patients with MEN1 (1, 71). CNS tumors include asymptomatic meningiomas in 8% of MEN1 patients (70), while ependymomas and schwannomas affect about 1% of cases (72). Recently, the case of a MEN1-related mature teratoma and yolk sac testis tumor has been described (73).
Aggressive and Aberrant Phenotype of MEN1
Cases of MEN1 phenotypes featured by aggressive or aberrant presentation, malignant evolution, and unfavorable clinical course have been reported in the literature and will be followingly summarized.
Parathyroid Carcinoma
The presence of parathyroid carcinoma in association with MEN1 has been so far described in 16 cases (74–84). The clinical characteristics of these cases are reported in Table 2. In 2016, a single tertiary care center study conducted from 1997 to 2013 in a cohort of 348 patients with MEN1 syndrome, reported only one case of parathyroid carcinoma with a prevalence of 0.28% (80). In the same year, Christakis et al. collected 291 cases with a genetic and/or clinical diagnosis of MEN1 from the MD Anderson patients’ database (81). Hyperparathyroidism was diagnosed in 242 of these patients (83.2%) with two of them receiving a histopathologic diagnosis of parathyroid carcinoma which accounted for an overall prevalence of 0.8%, while 1 patient (0.4%) received a diagnosis of atypical parathyroid neoplasm. Then, progression to malignancy does not seem to be a prerogative of MEN1 parathyroid tumors. Interestingly, MEN1 gene an inactivating mutation and a splicing mutation, previously identified in subjects who developed malignant lesions, has been reported, thus suggesting a possible genotype−phenotype association (89, 90).
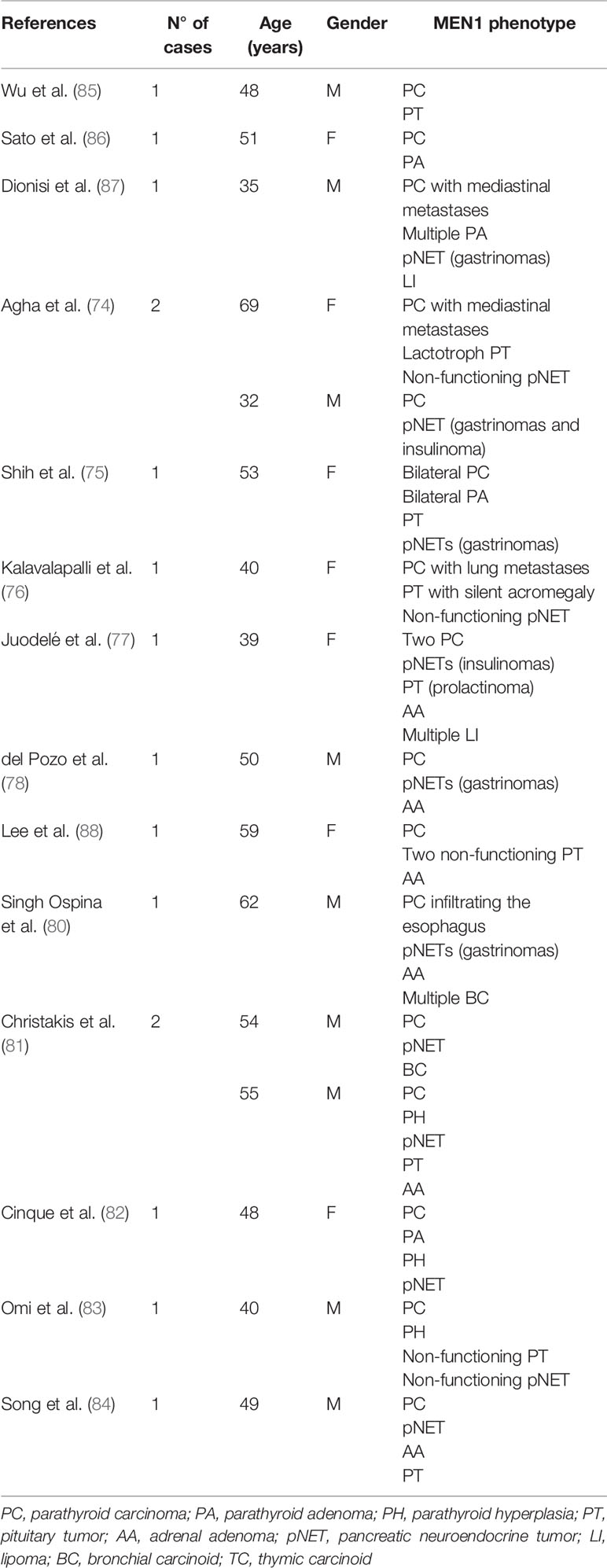
Table 2 Summary of clinical features of 16 patients with parathyroid carcinoma and MEN1.
Malignant Insulinoma and Glucagonoma
Malignant insulinomas are rare, hence scant data exist on their prevalence and clinical presentation in MEN1 patients. In 2011, Hasani-Ranjbar et al. described a large family encompassing several members from three generations who were evaluated for MEN1. Genetic analysis was performed in all family members using PCR amplification of coding regions followed by direct sequencing. In three brothers presenting with hypoglycemia, the presence of insulinomas was confirmed and in two cases it was malignant, according to the surgery and pathology report. Two of these presented with hyperparathyroidism as well. Mutation screening revealed the presence of a two nucleotides deletion in the exon 2 resulting in a non-functional gene product (91). Recently, Novruzov et al. reported another case of malignant insulinoma with multiple metastatic lesions in the right lung, liver, and pancreas, in a 54-year-old man, who had a previous history parathyroid surgery and left thyroid lobectomy (92).
Only one case of malignant glucagonoma with cervical metastases has been reported to date in MEN1 (88).
Pituitary Carcinoma
Only four cases of pituitary carcinoma have been described in association with MEN1 (79–82). In 2005, Benito and co-workers described the first case of a woman with a MEN1 associated gonadotroph carcinoma, who developed a temporal lobe metastasis (93). One year later, Gordon et al. presented the case of a 47-year-old male patient with MEN1 who was affected by parathyroid adenomas, non-functioning pancreatic tumors and a metastatic prolactinoma presenting as a cervical spinal cord lesion (94). Another case of malignant prolactinoma was described by Philippon and co-workers (95). Finally, Scheithauer et al. described the case of a 19-year-old man with a peculiar MEN1 phenotype, characterized by a parathyroid adenoma, pancreatic islet cell tumors in association with two enlarged hepatic hilar lymph nodes that were not biopsied, and a non-functioning pituitary mass with supra- and parasellar invasion, harboring craniospinal and systemic metastases (96).
Adrenocortical Carcinoma
The incidence of adrenocortical carcinoma in patients with MEN1 has been reported as ranging between 1.4 and 6% (65, 97). The prevalence of adrenocortical carcinoma is reportedly 10 times higher in patients with adrenal tumors and MEN1 as compared to those with adrenal incidentalomas without MEN1 (64). Adrenocortical carcinoma can exhibit familial aggregation in MEN1 patients. In reviewing literature, we could document 22 cases of adrenocortical carcinoma associated with MEN1 (5, 63, 65, 97–105). The most peculiar and aggressive MEN1 phenotypes associated with adrenocortical carcinoma were recently described (104, 105). Wang et al. described the case of a 51-year-old man with MEN1-associated bilateral parathyroid adenoma, multiple pNETs, and left adrenocortical carcinoma, which metastasized to supraclavicular and mediastinal lymph nodes, bilateral lung, and uncinate process of pancreas (104). In the same year, Harada et al. reported the case of a 68-year-old woman with a complex MEN1 phenotype characterized by pancreatic insulinoma, breast cancer, non-functioning pituitary tumor, parathyroid adenoma, and a myxoid variant of adrenocortical carcinoma without metastases (105).
Other Neoplasms
Ovarian NETs are rare and comprise 0.1% of all ovarian tumors. To date, few cases of primary ovarian NETs in women with MEN1 syndrome have been described in association with clinical manifestations of MEN1, but without genetic testing (106–109). More recently, clinical cases of ovarian NETs have been reported in genetically tested MEN1 cases. Jhawar et al. reported on a genetically confirmed case of MEN1 associated with an ovarian NET in a 33-year-old woman (109). Also, the case of an atypical ovarian carcinoid has been described as the first manifestation of an otherwise occult MEN1 syndrome in a 30-year old woman, who later developed a contralateral lesion two years after initial diagnosis (108). In this case, subsequent work-up allowed identification of simultaneous multifocal endocrine tumors involving parathyroids, thymus, adrenal glands, and pancreas, along with metastatic lesions in lymph nodes, liver, and bones.
Non-endocrine Malignancies
Recent studies suggest a general role of menin in carcinogenesis that may affect the risk and clinical course of developing common non-endocrine neoplasms (22). The most frequent non-endocrine neoplasm in MEN1 is breast cancer, which is in the MEN1 setting characterized by earlier onset as compared to non-MEN1 patients (110). The calculated relative risk for breast cancer in MEN1 women is 2.83 (111), which advises to categorize the MEN1 gene as a moderate risk factor for breast cancer (112). Interestingly, it has been also described a patient harboring both MEN1 and BRCA1 germline mutations in whom the severity of the MEN1-related biochemical and clinical findings did not differ from those for other affected family members lacking the BRCA1 mutation, but she did not develop any BRCA1-related malignancies (113). Other cancers that have been anecdotally described in association with MEN1 include hepatocellular carcinoma (114), melanoma (115–118), lung adenocarcinoma (103), renal cell carcinoma (119, 120), papillary thyroid cancer (120–122), and prostate cancer (120, 123).
The Natural History of MEN1 Patients
MEN1 patients have an increased risk of premature death. Earlier studies reported an average life-span of 50 years in affected patients (124, 125). The leading cause of death is complications related to hypergastrinemia and hyperparathyroidism. Although the improvement of medical and surgical management has remarkably decreased the risk of premature deaths for such causes, survival curves in MEN1 patients remain significantly affected when compared to the general population (126). Main negative prognostic factors in MEN1 patients include clinical features, i.e. disease duration, presence of non-ZES functional syndromes, number of parathyroidectomies, occurrence of thymic carcinoid, family history and, in case of ZES, a previous acid-reducing surgery. Also, entity of hypergastrinemia and tumor features, such as pNETs size, liver metastases, distant metastases, number of lesions at imaging, and tumor growth, are also reported to play a role (126).
To date, there are only few prospective studies evaluating the long-term course and causes of death of MEN1 patients. In 2013, Ito et al. conducted a prospective study with the aim of describing the current course of MEN1 patients late in the disease history and the causes of death at present (126). Opposed to previous reports, this and other large MEN1 series reported that patients with MEN1 rarely die for causes related to hormone excess per se (126–129), while the likelihood of death increases in the presence of pNET tumors with a malignant behavior. Among these, gastrinomas account for more than one half of pNET-related deaths (130). Intriguingly, causes of death in one third of MEN1 patients do not involve MEN1-related causes, such as cardiovascular disease and neoplasms arising from other sites like colorectal, renal, lung, breast, and oral cancers (126–129). It remains unclear whether these are directly related to MEN1 and the role of menin in regulating growth-related processes has been hypothesized to play a role (4, 131, 132). Among the cardiovascular diseases, conditions at particular risk of complications include hyperparathyroidism and glucose intolerance/diabetes, these latter being reported to occur with a higher frequency in MEN1 patients (133–135).
Diagnosis and Management of MEN1
Beyond work-up strategies for identification of MEN1-associated tumors, the simultaneous presence of at least two of the three characteristic tumors (parathyroids, pituitary, or pancreatic islets) is considered pathognomonic for MEN1 (23). The current clinical practice guidelines recommend three criteria for MEN1 diagnosis (22): 1) the presence of one of the MEN1-associated tumors in a first-degree relative of a patient with MEN1 syndrome; 2) the identification of a germline MEN1 mutation in a subject, who could be asymptomatic and has not yet developed radiological or biochemical signs of tumor onset; 3) the presence of two or more primary MEN1-associated endocrine tumors (pituitary, parathyroid, or pancreatic islets).
However, genetic testing in MEN1 patients meeting the clinical criteria could be negative (23, 89). Some studies demonstrated that negative testing for mutations is frequent in clinical MEN1-like presentations including a combination of pituitary and parathyroid tumors (136, 137), while GEP NET appeared more frequently and earlier in MEN1-positive probands, and its development under 30 years seems to be a predictor of a positive genetic test (24).
Biochemical screening for the MEN1 tumors onset in asymptomatic members of families with MEN1 syndrome is useful, since early diagnosis and treatment help reducing morbidity and mortality from these tumors (138). Attempts to screen for MEN1 tumors in asymptomatic relatives of affected individuals largely rely on the measurement of calcium, prolactin, IGF-I, and gastrointestinal hormones (22, 139). However, the beneficial effect of routine screening and the timing for genetic testing in pre-symptomatic individuals remain questionable (140) and psychologically stressful (141).
Medical imaging plays a key role in detection, staging, presurgical planning, and postsurgical surveillance (142). As summarized in Table 3, different imaging techniques are available to detect MEN1-associated tumors based on clinical practice guidelines (22). Imaging is particularly important for patients harboring a clinical diagnosis of MEN1, but showing no mutation, so as to aid distinguishing sporadic coincidental cases from true MEN1 (143). In this context, clinical observations showed that mutation-negative patients often have a more favorable clinical course (143) and may receive a less intensive follow-up to reduce radiation exposure, healthcare costs and anxiety (23). With regard to MEN1-PHPT, existing a genetic underlying predisposition to multiglandular disease, parathyroid imaging may probably result more useful to localize recurrent and/or persistent parathyroid disease after a sub-total/total parathyroidectomy (90).
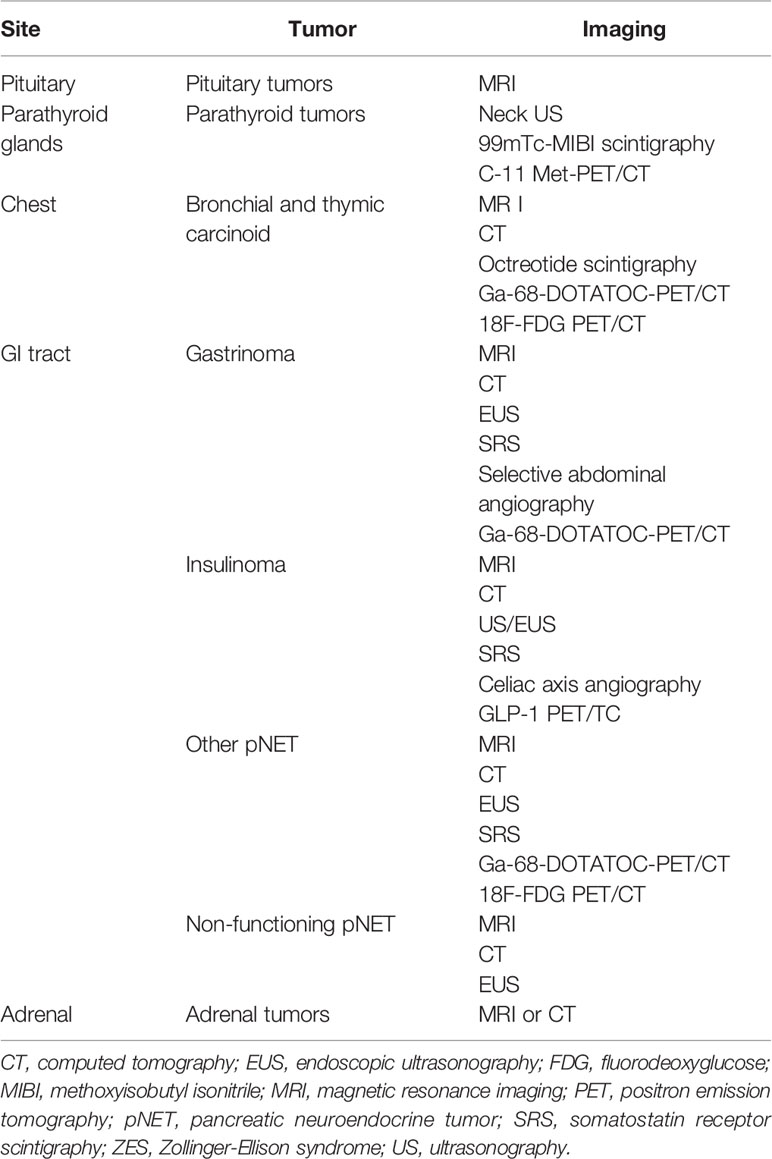
Table 3 Imaging techniques used to detect the main MEN1-associated tumors.
Surgical management remains the cornerstone of MEN1 treatment, with medical therapy being used to control hormone hypersecretion and disease symptoms depending on the tumor extension and histotype, although the antiproliferative effect of somatostatin analogs and everolimus have been repetitively shown in sporadic tumors (139, 144). In the case of multiple MEN1-associated tumors, surgical success is less frequent (22).
In MEN1-PHPT, surgery by means of subtotal or total parathyroidectomy is the treatment of choice, the latter followed by intramuscular reimplantation (brachioradialis muscle of the non-dominant forearm or sternocleidomastoid muscle) of parathyroid tissue fragments generally from the gland which shows the smallest dimensions at the neck surgery (22, 90). The optimal timing of surgery is debated and should be evaluated individually. Early surgery could be difficult because glands are minimally enlarged, which might predispose the patient to recurrence and reoperation; at the same time, longstanding hyperparathyroidism predisposes patient to more severe bone disease (145). Clinical practice guidelines recommend open bilateral neck exploration over the minimally invasive parathyroidectomy, because all the parathyroids gland are usually affected in MEN1 patients (22). If surgery is not possible because of patient’s refusal, inoperability, or negative imaging, the calcimimetic agent can be used, even if only small series showed that cinacalcet is effective in reducing serum calcium levels in MEN1 patients (43, 146, 147).
Treatments of MEN1 associated pituitary, adrenals, thymic, and bronchopulmonary tumors are similar to that for non-MEN1 tumors. However, it is important to remember that MEN1 thymic carcinoids, with a near-total prevalence in MEN1 smoker males, are associated with a very high lethality and, therefore, prophylactic thymectomy should be considered at the moment of neck surgery for MEN1-PHPT in male patients (148).
The primary treatment for GH-, ACTH-, TSH-secreting and symptomatic non-functioning pituitary tumors should involve selective transsphenoidal surgical resection if clinically feasible, with the curative intent of eradicating the tumor, or debulking the tumor mass if compressive symptoms occur (149). Treatment outcomes of these tumors in MEN1 syndrome are less successful than sporadic tumors (60). In the case of 1) patient’s inoperability, 2) post-surgical tumor residual or 3) tumor relapse, 4) unfeasible reoperation, and 5) before contemplating the use of pituitary radiotherapy, clinicians generally employ medical or third-line therapies depending on tumor type, clinical burden, individual responsiveness, local therapy availabilities, and center experience. First- or second-generation somatostatin analogs (SSAs), GH receptor antagonist and dopamine agonists are used in case of GH-secreting adenomas; while second-generation SSA, adrenolytic medications, glucocorticoid receptor-antagonist, or adrenalectomy are used for persistent Cushing’s disease. In case of TSH-omas, first-generation SSAs, dopamine agonists, or anti-thyroid medications can be used. Conversely, prolactinomas without neurological involvement are primarily treated with long-acting dopamine agonists. In the case of hypopituitarism, replacement therapy should be initiated as per individual needs.
Treatment of adrenal tumors consists of surgery for functioning tumors and non-functioning tumors with atypical characteristics, tumor size >4 cm, or significant tumor growth over a 6-months period (22, 110). In case of thymic and bronchopulmonary carcinoids, surgery is the treatment of choice. Where disease is advanced, additional therapies such as radiotherapy and chemotherapy or adrenolytic drugs should be considered (22, 110).
Surgery represents the treatment of choice also in case of functional GEP NETs. However, treatment outcomes of these tumors in MEN1 syndrome are less successful than sporadic tumors for different reasons (150):
1) MEN1 subjects often develop multiple gastrinomas, thus reducing the probability of surgical cure rates compared to similar sporadic solitary tumors. In fact, only 15% of MEN1 patients are free of disease immediately after surgery as compared to approximately 45% of non-MEN1 patients (22, 151, 152).
2) Occult metastatic disease is more frequent in MEN1 patients with NETs than in patients with sporadic endocrine tumors. For instance, a metastatic disease is present in up to 50% of subjects with MEN1-related insulinomas, whereas less than 10% of sporadic insulinomas are malignant (57).
3) MEN1-associated tumors are often larger, more aggressive, and resistant to treatment than sporadic ones. In particular, about 85% of pituitary tumors in MEN1 patients are macroadenomas, as opposed to 64% in non-MEN1 subjects, they more frequently infiltrate surrounding tissues, and show persistent hormone hypersecretion after treatment in more than 45% of cases (60, 153).
The average life expectancy in MEN1 patients with GEP NETs is reported to be shorter than in MEN1 patients without (59). However, tumor size <20 mm shows a poor tendency to grow and/or metastasize over a long monitoring period (154), irrespective of the underlying MEN1 genotype (155). In line with this evidence, other studies investigated the role of surgery vs surveillance on survival and liver metastatization in nonfunctioning pancreatic NETs (pNETs) (156–158) after stratification by size (≤20 vs. >20 mm) as well as proliferation indices, i.e. mitotic count and Ki67 (159). These authors demonstrated that MEN1 patients with small nonfunctioning pNETs (≤20 mm) can be managed by watchful waiting, hereby avoiding major surgery without loss of oncological safety (156–158).
With regards to medical therapy, a number of observational, longitudinal, and randomized placebo-controlled studies have been conducted in sporadic NETs using somatostatin analogs, peptide receptor radionuclide therapy (PRRT), (mTOR) signaling inhibitors, and receptor tyrosine kinase (RTK) inhibitors, all collectively showing a statistically significant effect on disease progression (160). Particularly PNETs are difficult to treat medically in MEN1 due to differences in growth potential, concomitant development of other tumors, and relative insensitivity to treatment, such that medical treatments for MEN1-related tumors have not been properly evaluated, but rather have been employed based on recognized effects in patients without MEN1 (161).
Genotype-Phenotype Correlations in MEN1
A MEN1 gene mutation can be detrimental to gene function or result in a protein product retaining residual functions. An aberrant menin protein becomes impaired in its functions through pathological interaction with transcription factors such as Smad3, JunD, and NFκB as well as nuclear receptors, or with proteins implicated in the apoptotic cascade such as caspase-3, p53, or p21 (162). Lips et al. hypothesized that MEN1 germline mutations can selectively affect menin binding to its targets and lead to distinctly aggressive clinical phenotypes (162).
Moreover, it is known that a clustering of mutations is observed in some regions of exon-2 and exon-10 which have been attributed to the nature of the underlying repetitive nucleotides prone to DNA polymerase errors (30, 56). Also, another observation of clustering of mutations at specific nucleotides in apparently unrelated families has been attributed to founder effects (29, 163–168). However, highlighting rare tumors among family members that are not seen among other individuals with the same mutation may suggest other mechanisms besides the mutation that can account for these phenotypes such as epigenetics, changes in other parts of the genome, environmental influences, immunogenicity, etc. Specifically, it is known that dysregulation of some miRNAs could account for parathyroid tumorigenesis (169–171), and this could also happen in MEN1 GEP/NETs carcinogenesis (172). Finally, epigenetic alterations could be hardly involved also in MEN1 GEP-NET tumorigenesis (173).
Although an effort has been made to characterize the potential genotype-phenotype correlation in MEN1, this link remains debated to date and no definitive evidence has been recognized (15, 25, 174). However, some authors described a heavier or lighter clinical burden in association with some specific mutations, and they will be subsequently described.
MEN1 missense mutations have been described in association with familiar isolated hyperparathyroidism (FIHP), an autosomal dominant disease that potentially represents an early stage or a milder presentation of MEN1 attributable to an allelic variant of the MEN1 gene (175–180). Peculiarly, most of MEN1 gene germline mutations identified in FIHP are seemingly in-frame deletions or mild missense mutations (180).
On the other hand, studies performed in four kindreds from Newfoundland demonstrated that a single nonsense mutation in the MEN1 gene (R460X) was predominantly associated with prolactinomas, carcinoids and parathyroid tumors (167, 181–183), though the same mutation has been described in other MEN1 cases with milder MEN1 clinical features (26, 30, 137, 164, 184–188). Therefore, there is currently no evidence of a genotype-phenotype correlation for this mutation.
In 2011, Raef and colleagues described a MEN1 family showing an aggressive tumor behavior associated with a monoallelic 5 kb deletion of genomic DNA, involving the MEN1 promoter and exons 1 and 2 (189). LOH analysis identified a somatic deletion within the MEN1 locus 11q13 and the 11p15 imprinting control region (ICR) of the maternal chromosome 11. Following methylation analysis of ICR, ICR1 hypermethylation and ICR2 hypomethylation were demonstrated in tumor specimens. These genetic alterations were found in association with the development of multiple malignant pNETs. Likewise, Ishida et al. described the case of a MEN1 patient with a relapsing macroprolactinoma co-stained for FSH showing histological features of malignancy and associated with a metastasizing non-functioning pNET. This phenotype was associated with menin and p27Kip1 down-regulation (190).
In 2014, Bartsch et al. retrospectively analyzed a cohort of 71 genetically confirmed MEN1 patients with the aim of evaluating the relationship between MEN1 mutations in different interacting domains of menin and the pNETs phenotype. The authors demonstrated that patients with MEN1 mutations leading to loss of interaction with checkpoint kinase 1 (CHES1-LOI) displayed a higher risk of malignant pNETs with an aggressive course of the disease and disease-related death (191). Longuini et al. analyzed a cohort of one hundred Brazilian MEN1 germline mutant carriers, genotyping them for the coding p27 c.326T>G (V109G) variant. They suggest that the p27 tumor suppressor gene could represent a disease modifier gene in MEN1 syndrome cases associated with MEN1 germline mutations (34). Subsequently, Circelli and colleagues confirmed, in a smaller sample of 55 Italian MEN1 patients, the possible impact of this CDKN1B polymorphism on the clinical course of the disease, since the MEN1-related aggressive tumors, or other malignancies, were more frequent in those patients with the CDKN1B V109G genetic variant (35). However, currently it is still not possible to clearly state whether this p27 variant behaves as a real modifying gene or not.
In 2016, Skalniak and collegues described a three generations family with MEN1 caused by a previously undescribed in-frame deletion c.1231_1233delGCC (Ala411del) in the MEN1 gene. Even if the family members differed for their phenotypic features, all NETs showed an aggressive behavior and a high mortality rate (192). Lastly, a clinical survey by Palermo et al. found a strong genotype-phenotype correlation with aggressive MEN1-related GEP NETs. In particular, the authors described three MEN1 patients carrying a novel heterozygous germline mutation in exon 10 of the MEN1 gene, c.1561_1571 delACTGTCGCTGG corresponding to T521 frame-shit effect, which was associated with malignant GEP NETs lesions and a higher rate of malignancy (193). In this regard, it is correct to point out that patients with symptomatic MEN1 gastrinomas, a long-time treatment by H2-blockers or PPIs, may stimulate gastric neuroendocrine cells proliferation contributing to the clinical outcome and severity of GEP-NETs (194).
At odds with previous studies, a retrospective-prospective study on a large Italian cohort by Marini et al. analyzed MEN1 mutation sites and features in relation to the affected menin functional domains and clinical presentations. The authors observed a wide variability in the age of disease onset and clinical severity even in the presence of the same mutation, implying a lack of direct genotype-phenotype correlations. The authors speculated that other genetic or epigenetic factors may intervene in individual MEN1 tumorigenesis (20). Nevertheless, in this large cohort a stronger association was documented between aggressive phenotypes and non-sense or frameshift mutations, than with missense mutations.
It is worth to point out that the described correlations were generally not tested in family members with the identical genotype to determine whether all of the family members had the aggressive phenotypes. Therefore, it is difficult to state whether there is a genotype-phenotype correlation between aggressive MEN1 phenotype with any type of mutation.
A Novel MEN1 Gene Mutation Related to Aggressive Phenotype
In the context of the potential genotype-phenotype correlation, we report on the case of a 69-year-old woman with a previously unrecognized long history of MEN1, who was referred to our Unit in 2017 for complicated obesity. As common practice, patient signed an informed consent to collect clinical, biochemical, and genetic data. The investigation was approved by the local ethics committee, functioning according to the fourth edition of the Guidelines on the Practice of Ethics Committees in Medical Research With Human Participants. Unfortunately, because of the suffering for her long clinical history, not adequately managed from the beginning, and for the diagnosis that came only on the occasion of her hospitalization at our hospital unit, understandably exhausted, she did not give consent to carry out molecular studies on any of her surgically removed tissues. Her MEN1-related clinical record was noticeable for 1) a pituitary macroprolactinoma cured after trans-sphenoidal adenomectomy and external conventional radiotherapy in 1980, 2) PHPT surgically treated by excision of a single upper left parathyroid adenoma in 1981 at the age of 35 years, 3) two duodenal gastrinomas diagnosed in 2005 and left untreated according to the patient’s choice. While the diagnosis of pituitary and parathyroid disease was nearly synchronous, her osteoporotic bone involvement was severe due to dual dorsal vertebral fractures causing an early exaggerated thoracic kyphosis and compromising her biomechanical performance. Reportedly, no genetic analysis had been performed on the patient or her relatives at that stage. Upon admission to our unit, the clinical and imaging work-up showed: a normal (unstimulated) pituitary function; hypercalcemia with 3.5× elevation of PTH levels due to an apparently single left parathyroid adenoma identified at ultrasound and MIBG-scintigraphy; hypergastrinemia and high chromogranin A levels associated with endoscopic, scintigraphic (octroscan) and MRI evidence of three separate lesions located in the anterior gastric wall, in the duodenal loop, and in the pancreas head. A CT scan excluded systemic metastases. Because of associated ZES, she started proton pump-inhibitors. For her osteoporosis, she started antiresorptive treatment with oral alendronate as she refused i.v. administration with amino-bisphosphonates available by this route. After surgical and anesthesiological consultation, she underwent total parathyroidectomy and auto-transplantation of three parathyroid fragments within the brachioradialis muscle. Abdominal surgery was contraindicated for her poor clinical conditions, and she was started on octreotide LAR (30 mg/28d), leading to clinical control and near-normalization of gastrin and CgA levels. She was subsequently followed up at 3 months intervals. Due to new onset metrorrhagia, the patient was re-admitted to our unit in 2018 to undergo a diagnostic workup. Transvaginal ultrasound and pelvic MRI showed endometrial thickening, and hysteroscopic endomyometrial biopsy revealed a high-grade neuroendocrine endometrial carcinoma (G3) staining negative for gastrin and showing immunopositivity for p16, p53, CKpan, chromogranin A, vimentin, estrogen, and progesterone. Ki-67 was 50%. The patient’s conditions contraindicated gynecological surgery, and she refused palliative antineoplastic treatment.
Genetic analysis by sequencing of the coding sequence of the MEN1 gene (NM_130799.2) and CDKN1B (NM_004064.3) resulted in the identification of the missense variant c.836C>A in exon 6 resulting in the amino acid change p.A279D in heterozygous state. This variant was not found in genomic variation databases (1000Genomes, ExAC, ESP, dbSNP, Alamut, HGMD Professional) and represented a non-conservative change leading to the switch from a hydrophobic to a basic amino acid (BLOSUM62=-2; Grantham Distance =126), located in a very conserved position in the protein domain involved in the interaction of menin protein with FANCD2. Previous in silico prediction tools of variation effects, such as SIFT, Mutation Tester, Poly Phen, Provean, and A-GVGD, suggested a probable pathogenic effect of this substitution on the aberrant patient’s phenotype. Deletions in MEN1 gene were excluded through MLPA analysis (MRC Holland, P017-D1 probemix). Potential overlap with other published gene mutations sharing the same amino acid sequence or functional domain effects was also excluded. We subsequently evaluated the co-segregation of the afore-mentioned MEN1 variant in the proband family (Figure 1). A total of 16 relatives, including her old parents, were screened but none was found to carry the index mutation or any other variant, likewise none was presenting clinical manifestations suggestive of MEN1. Further, the possible contribution to the proband phenotype of BRCA1/2 mutations was excluded through NGS (Illumina, NY, USA) and MLPA analysis (MRC Holland, P002-D1 BRCA1 and P090-B1 BRCA2 probe mix).
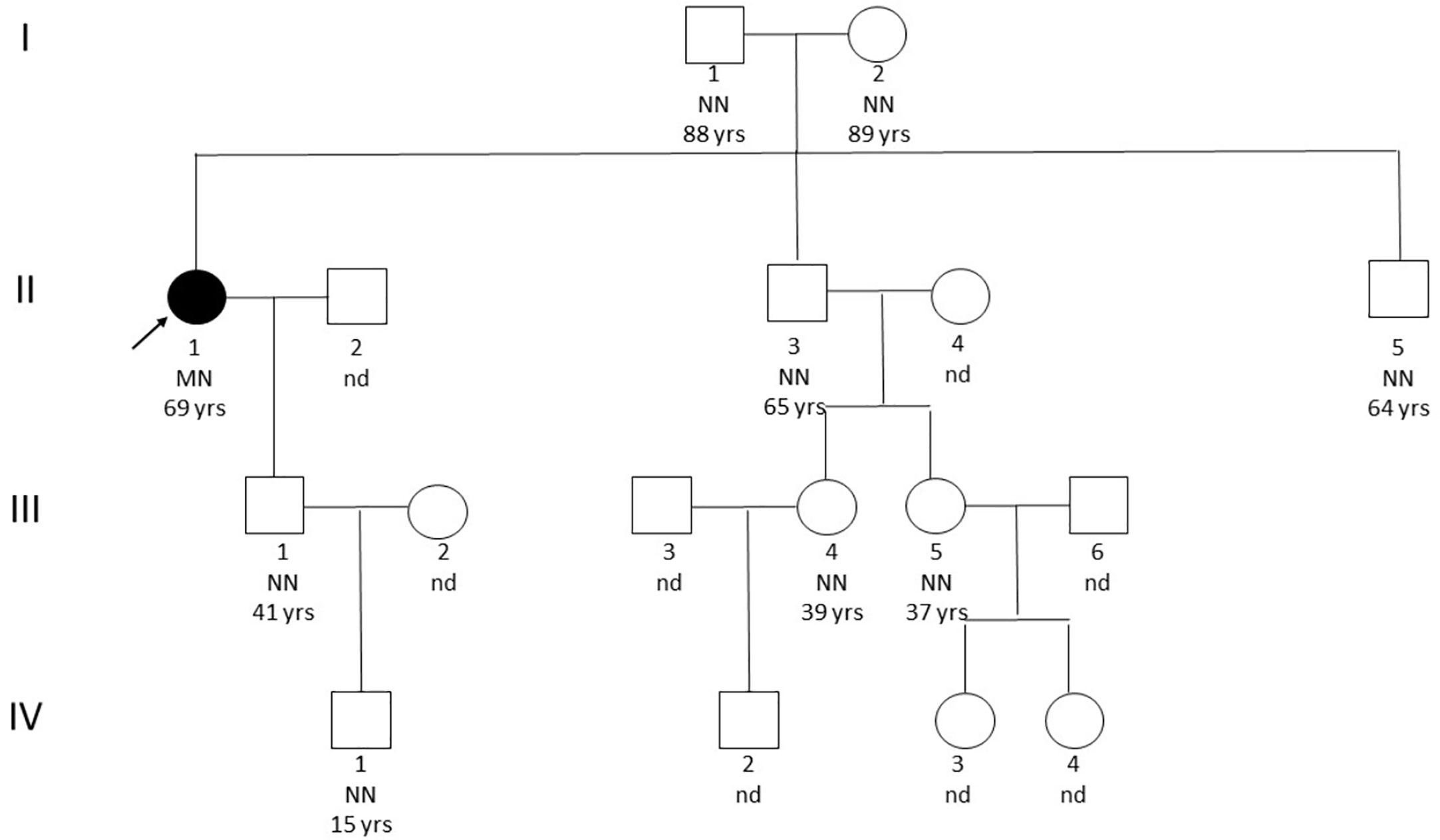
Figure 1 Pedigree. The black arrow indicates the affected patient. NN, homozygote without mutation; MN, heterozygous with mutation; nd, not screened.
Discussion
Clinical guidelines focus on early detection of MEN1-related tumors, namely parathyroid adenomas, GEP NETs, and pituitary adenomas (22, 139, 144). An aggressive or aberrant behavior of endocrine tumors has been occasionally described in patients with MEN1 syndrome, raising the following questions: do some MEN1 patients with specific mutations carry an increased risk of aberrant clinical progression requiring in-depth diagnostic and therapeutic assessment? Also, are tumor-related manifestations or the disease course dependent on the type of mutation? Earlier studies failed to show strong genotype-phenotype associations, while more novel studies seem to dispute this viewpoint. It is suggestive to speculate that the type of MEN1 gene mutation could influence the clinical manifestations of MEN1. In MEN1 syndrome exhibiting a non-aggressive phenotype, frameshift or nonsense leading to a truncated and consequently inactivated protein have been identified in most cases (120). Likewise, FIHP is characterized by the onset of primary hyperparathyroidism alone and is related to specific mutations of MEN1 gene that often include missense mutations and only occasionally nonsense or frameshift mutations (22, 89). Conversely, some specific mutations seem to be associated with a less favorable prognosis. For example, subjects with MEN1 mutations leading to a loss of interaction with the checkpoint kinase 1-interacting domain have a higher risk of malignant pNETs with aggressive phenotype and higher prevalence of disease-related death (192). Peculiarly, patients with mutations that affect the JunD-interacting domain have a higher risk of death for a typical MEN1 tumor, requiring a more aggressive therapeutic approach (195). In keeping with these indications, the novel missense variant herein reported c.836C>A resulting in the amino acid change p.A279D in heterozygous state, leads to a change from alanine to aspartic acid with potential aggressive behavior. At odds with studies minimizing the clinical impact of missense mutations compared to frameshift or non-sense mutations (20, 24), the missense variant described in our index case stands out for its aberrant and aggressive clinical manifestations developing long after the first clinical manifestation of MEN1.
Conclusions
Current clinical practice guidelines for MEN1 recommend a screening program for MEN1 patients and their families with the aim of reducing morbidity and mortality and achieving an early detection of MEN1-related tumors (22, 139). Although genotype-phenotype correlations are difficult to demonstrate, our index case and other reports suggest that patients with suggestive genotype-phenotype correlations should undergo a closer follow-up and surveillance with an interdisciplinary approach. In fact, our team was involved and intervened only at a relatively advanced stage in the clinical history of this case, when the patient was understandably exhausted and did not give consent to carry out molecular studies on her surgically removed tissues. Consequently, we lack molecular data on menin expression and function, LOH studies at the tumor tissue level, as also on possible involvement of specific miRNAs. This aspect further confirms the importance that subjects affected by rare and complex pathologies such this are taken in charge from the beginning by expert multidisciplinary teams, also capable of managing the psychological aspects and implications linked to genetic and repeatedly clinical management complexity.
Author Contributions
Conceptualization and methodology, CM, MM, PM. Original draft preparation, CM, MM, MC, AF, SM. Review and editing, CM, LP, AF, PM. Supervision, GA, MS, AF. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab (2001) 86:5658–71. doi: 10.1210/jcem.86.12.8070
2. Falchetti A, Marini F, Tonelli F, Brandi ML. Lessons from genes mutated in multiple endocrine neoplasia (MEN) syndromes. Ann Endocrinol (Paris) (2005) 66:195–205. doi: 10.1016/s0003-4266(05)81751-2
3. Agarwal SK. Multiple endocrine neoplasia type 1. Front Horm Res (2013) 41:1–15. doi: 10.1159/000345666
4. Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med (1998) 129:484–94. doi: 10.7326/0003-4819-129-6-199809150-00011
5. Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg (2002) 26:891–6. doi: 10.1007/s00268-002-6492-4
6. Concolino P, Costella A, Capoluongo E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet (2016) 209:36–41. doi: 10.1016/j.cancergen.2015.12.002
7. Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjöld M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature (1988) 332:85–7. doi: 10.1038/332085a0
8. Thakker RV, Bouloux P, Wooding C, Chotai K, Broad PM, Spurr NK, et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. N Engl J Med (1989) 321:218–24. doi: 10.1056/NEJM198907273210403
9. UniProtKB - O00255 (MEN1_HUMAN). Available at: https://www.uniprot.org/uniprot/O00255 (Accessed June 24, 2020).
10. Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci (2013) 38:394–402. doi: 10.1016/j.tibs.2013.05.005
11. Romei C, Pardi E, Cetani F, Elisei R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J Oncol (2012) 2012:705036. doi: 10.1155/2012/705036
12. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science (1997) 276:404–7. doi: 10.1126/science.276.5311.404
13. Knudson AG. Antioncogenes and human cancer. Proc Natl Acad Sci U S A (1993) 90:10914–21. doi: 10.1073/pnas.90.23.10914
14. Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Eur Consortium MEN1 Hum Mol Genet (1997) 6:1177–83. doi: 10.1093/hmg/6.7.1177
15. Lemos MC, Thakker RV. Multiple endocrine neoplasia type1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat (2008) 29:22–32. doi: 10.1002/humu.20605
16. The Human Gene Mutation Database. Available at: http://www.hgmd.cf.ac.uk/ (Accessed January 11, 2020).
17. The UMD - MEN1 mutations database. Available at: http://www.umd.be/MEN1/ (Accessed April 02, 2020).
18. LOVD v.3.0 - Leiden Open Variation Database. Online gene-centered collection and display of DNA variants. Available at: http://www.lovd.nl/ (Accessed March 22, 2020).
19. Marini F, Falchetti A, Del Monte F, Carbonell Sala S, Gozzini A, Luzi E, et al. Multiple endocrine neoplasia type 1. Orphanet J Rare Dis (2006) 1:38. doi: 10.1186/1750-1172-1-38
20. Marini F, Giusti F, Fossi C, Cioppi F, Cianferotti L, Masi L, et al. Multiple endocrine neoplasia type 1: analysis of germline MEN1 mutations in the Italian multicenter MEN1 patient database. Endocrine (2018) 62:215–33. doi: 10.1007/s12020-018-1566-8
21. Anlauf M, Perren A, Henopp T, Rudolf T, Garbrecht N, Schmitt A, et al. Allelic deletion of the MEN1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut (2007) 56:637–44. doi: 10.1136/gut.2006.108910
22. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230
23. de Laat JM, van Leeuwaarde RS, Valk GD. The Importance of an Early and Accurate MEN1 Diagnosis. Front Endocrinol (Lausanne) (2018) 9:533. doi: 10.3389/fendo.2018.00533
24. Kövesdi A, Tóth M, Butz H, Szücs N, Sármán B, Pusztai P, et al. True MEN1 or phenocopy? Evidence for geno-phenotypic correlations in MEN1 syndrome. Endocrine (2019) 65:451–9. doi: 10.1007/s12020-019-01932-x
25. Wautot V, Vercherat C, Lespinasse J, Chambe B, Lenoir GM, Zhang CX, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat (2002) 20:35–47. doi: 10.1002/humu.10092
26. Kouvaraki MA, Lee JE, Shapiro SE, Gagel RF, Sherman SI, Sellin RV, et al. Genotype–phenotype analysis in multiple endocrine neoplasia type 1. Arch Surg (2002) 137:641–7. doi: 10.1001/archsurg.137.6.641
27. Falchetti A. Genetics of multiple endocrine neoplasia type 1 syndrome: what’s new and what’s old. F1000Res (2017) 24:6. doi: 10.12688/f1000research.7230.1
28. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol (2014) 386:2–15. doi: 10.1016/j.mce.2013.08.002
29. Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR, Skarulis MC, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet (1997) 6:1169–75. doi: 10.1093/hmg/6.7.1169
30. Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet (1998) 62:232–44. doi: 10.1086/301729
31. Laitman Y, Jaffe A, Schayek H, Friedman E. De novo mutation in MEN1 is not associated with parental somatic mosaicism. Endoc Relat Cancer (2017) 24:L1–3. doi: 10.1530/ERC-16-0446
32. Schouten JP, Mc Elgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res (2002) 30:e57. doi: 10.1093/nar/gnf056
33. De Sousa SM, Hardy TS, Scott HS, Torpy DJ. Genetic Testing in Endocrinology. Clin Biochem Rev (2018) 39:17–28.
34. Longuini VC, Lourenço DM Jr, Sekiya T, Meirelles O, Goncalves TD, Coutinho FL, et al. Association between the p27 rs2066827 variant and tumor multiplicity in patients harboring MEN1 germline mutations. Eur J Endocrinol (2014) 171:335–42. doi: 10.1530/EJE-14-0130
35. Circelli L, Ramundo V, Marotta V, Sciammarella C, Marciello F, Del Prete M, et al. Multidisciplinary Group for NeuroEndocrine Tumours of Naples. Prognostic role of the CDNK1B V109G polymorphism in multiple endocrine neoplasia type 1. J Cell Mol Med (2015) 19:1735–41. doi: 10.1111/jcmm.12552
36. Al-Salameh A, Baudry C, Cohen R. Update on multiple endocrine neoplasia Type 1 and 2. Presse Med (2018) 47:722–31. doi: 10.1016/j.lpm.2018.03.005
37. Stratakis CA, Schussheim DH, Freedman SM, Keil MF, Pack SD, Agarwal SK, et al. Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2000) 85:4776–80. doi: 10.1210/jcem.85.12.7064
38. Giusti F, Cianferotti L, Boaretto F, Cetani F, Cioppi F, Colao A, et al. Multiple endocrine neoplasia syndrome type 1: institution, management, and data analysis of a nationwide multicenter patient database. Endocrine (2017) 58:349–59. doi: 10.1007/s12020-017-1234-4
39. Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM (1996) 89:653–69. doi: 10.1093/qjmed/89.9.653
40. Goudet P, Bonithon-Kopp C, Murat A, Ruszniewski P, Niccoli P, Ménégaux F, et al. Gender-related differences in MEN1 lesion occurrence and diagnosis: a cohort study of 734 cases from the Groupe d’etude des Tumeurs Endocrines. Eur J Endocrinol (2011) 165:97–105. doi: 10.1530/EJE-10-0950
41. Calender A, Giraud S, Cougard P, Chanson P, Lenoir G, Murat A, et al. Multiple endocrine neoplasia type 1 in France: clinical and genetic studies. J Intern Med (1995) 238:263–8. doi: 10.1111/j.1365-2796.1995.tb00933.x
42. Benson L, Ljunghall S, Akerström G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type. Am J Med (1987) 82:731–7. doi: 10.1016/0002-9343(87)90008-8
43. Falchetti A, Marini F, Luzi E, Tonelli F, Brandi ML. Multiple endocrine neoplasms. Best Pract Res Clin Rheumatol (2008) 22:149–63. doi: 10.1016/j.berh.2007.11.010
44. Eller-Vainicher C, Chiodini I, Battista C, Viti R, Mascia ML, Massironi S, et al. Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity. J Bone Miner Res (2009) 24:1404–10. doi: 10.1359/jbmr.090304
45. Christopoulos C, Antoniou N, Thempeyioti A, Calender A, Economopoulos P. Familial multiple endocrine neoplasia type I: the urologist is first on the scene. BJU Int (2005) 96:884–7. doi: 10.1111/j.1464-410X.2005.05731.x
46. Lourenço DM Jr, Coutinho FL, Toledo RA, Montenegro FL, Correia-Deur JE, Toledo SP. Early-onset, Progressive, Frequent, Extensive, and Severe Bone Mineral and Renal Complications in Multiple Endocrine Neoplasia Type 1-associated Primary Hyperparathyroidism. J Bone Miner Res (2010) 25:2382–91. doi: 10.1002/jbmr.125
47. Kanazawa I, Canaff L, Abi Rafeh J, Angrula A, Li J, Riddle RC, et al. Osteoblast menin regulates bone mass in vivo. J Biol Chem (2015) 290:3910–24. doi: 10.1074/jbc.M114.629899
48. Hoffmann KM, Furukawa M, Jensen RT. Duodenal neuroendocrine tumors: classification, functional syndromes, diagnosis and medical treatment. Best Pract Res Clin Gastroenterol (2005) 19:675–97. doi: 10.1016/j.bpg.2005.05.009
49. Anlauf M, Perren A, Klöppel G. Endocrine precursor lesions and microadenomas of the duodenum and pancreas with and without MEN1: criteria, molecular concepts and clinical significance. Pathobiology (2007) 74:279–84. doi: 10.1159/000105810
50. Fendrich V, Langer P, Waldmann J, Bartsch DK, Rothmund M. Management of sporadic and multiple endocrine neoplasia type 1 gastrinomas. Br J Surg (2007) 94:1331–41. doi: 10.1002/bjs.5987
51. Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, et al. Multiple endocrine neoplasia type 1 (MEN1): Not only inherited endocrine tumors. Genet Med (2009) 11:825–35. doi: 10.1097/GIM.0b013e3181be5c97
52. Lee L, Ramos-Alvarez I, Ito T, Jensen RT. Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong PPI Treatment in Man Based on Studies of Patients with Zollinger-Ellison Syndrome. Int J Mol Sci (2019) 20:5128. doi: 10.3390/ijms20205128
53. Mele C, Brunani A, Damascelli B, Tichà V, Castello L, Aimaretti G, et al. Non-surgical ablative therapies for inoperable benign insulinoma. J Endocrinol Invest (2018) 41:153–62. doi: 10.1007/s40618-017-0738-3
54. Mignon M, Ruszniewski P, Podevin P, Sabbagh L, Cadiot G, Rigaud D, et al. Current approach to the management of gastrinoma and insulinoma in adults with multiple endocrine neoplasia type I. World J Surg (1993) 17:489–97. doi: 10.1007/BF01655108
55. Brandi ML, Bordi C, Tonelli F, Falchetti A, Marx SJ. Multiple endocrine neoplasia type 1. In: Bilezikian JP, Raisz GA, Rodan LG, editors. Principles of bone biology, 3rd ed. San Diego, CA: Academic Press Co (2008). p. 1345–74.
56. Thakker RV. Multiple endocrine neoplasia type 1. In: DeGroot L, Jameson JL, editors. Endocrinology, 6th ed. Philadelphia: Elsevier (2010). p. 2719–41.
57. Akerström G, Hellman P. Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab (2007) 21:87–109. doi: 10.1016/j.beem.2006.12.004
58. Thomas-Marques L, Murat A, Delemer B, Penfornis A, Cardot-Bauters C, Baudin E, et al. Groupe des Tumeurs Endocrines (GTE). Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol (2006) 101:266–73. doi: 10.1111/j.1572-0241.2006.00367.x
59. Triponez F, Dosseh D, Goudet P, Cougard P, Bauters C, Murat A, et al. Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg (2006) 243:265–72. doi: 10.1097/01.sla.0000197715.96762.68
60. Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab (2002) 87:457–65. doi: 10.1210/jcem.87.2.8145
61. Syro LV, Scheithauer BW, Kovacs K, Toledo RA, Londoño FJ, Ortiz LD, et al. Pituitary tumors in patients with MEN1 syndrome. Clinics (Sao Paulo) (2012) 67:43–8. doi: 10.6061/clinics/2012(sup01)09
62. Beckers A, Abs R, Willems PJ, van der Auwera B, Kovacs K, Reznik M, et al. Aldosterone-secreting adrenal adenoma as part of multiple endocrine neoplasia type 1 (MEN1): loss of heterozygosity for polymorphic chromosome 11 deoxyribonucleic acid markers, including the MEN1 locus. J Clin Endocrinol Metab (1992) 75:564–70. doi: 10.1210/jcem.75.2.1639957
63. Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Theodorsson E, et al. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (1992) 75:76–81. doi: 10.1210/jcem.75.1.1352309
64. Honda M, Tsukada T, Horiuchi T, Tanaka R, Yamaguchi K, Obara T, et al. Primary hyperparathyroidism associated with aldosterone-producing adrenocortical adenoma and breast cancer: relation to MEN1 gene. Intern Med (2004) 43:310–4. doi: 10.2169/internalmedicine.43.310
65. Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’etude des Tumeurs Endocrines database. Eur J Endocrinol (2012) 166:269–79. doi: 10.1530/EJE-11-0679
66. Teh BT, McArdle J, Chan SP, Menon J, Hartley L, Pullan P, et al. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Med (Baltimore) (1997) 76:21–9. doi: 10.1097/00005792-199701000-00002
67. Gibril F, Chen YJ, Schrump DS, Vortmeyer A, Zhuang Z, Lubensky IA, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2003) 88:1066 –81. doi: 10.1210/jc.2002-021314
68. Lecomte P, Binquet C, Le Bras M, Tabarin A, Cardot-Bauters C, Borson-Chazot F, et al. Histologically Proven Bronchial Neuroendocrine Tumors in MEN1: A GTE 51-Case Cohort Study. World J Surg (2018) 42:143–52. doi: 10.1007/s00268-017-4135-z
69. Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol (1997) 133:853– 7. doi: 10.1001/archderm.1997.03890430067009
70. Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab (2004) 89:5328–36. doi: 10.1210/jc.2004-0218
71. Vidal A, Iglesias MJ, Fernandez B, Fonseca E, Cordido F. Cutaneous lesions associated to multiple endocrine neoplasia syndrome type 1. J Eur Acad Dermatol Venereol (2008) 22:835–8. doi: 10.1111/j.1468-3083.2008.02578.x
72. Kato H, Uchimura I, Morohoshi M, Fujisawa K, Kobayashi Y, Numano F, et al. Multiple endocrine neoplasia type 1 associated with spinal ependymoma. Intern Med (1996) 35:285–9. doi: 10.2169/internalmedicine.35.285
73. Chiloiro S, Capoluongo ED, Schinzari G, Concolino P, Rossi E, Martini M, et al. First Case of Mature Teratoma and Yolk Sac Testis Tumor Associated to Inherited MEN-1 Syndrome. Front Endocrinol (Lausanne) (2019) 10:365. doi: 10.3389/fendo.2019.00365
74. Agha A, Carpenter R, Bhattacharya S, Edmonson SJ, Carlsen E, Monson JP. Parathyroid carcinoma in multiple endocrine neoplasia type 1 (MEN1) syndrome: two case reports of an unrecognised entity. J Endocrinol Invest (2007) 30:145–9. doi: 10.1007/BF03347413
75. Shih RY, Fackler S, Maturo S, True MW, Brennan J, Wells D. Parathyroid carcinoma in multiple endocrine neoplasia type 1 with a classic germline mutation. Endocr Pract (2009) 15:567–72. doi: 10.4158/EP09045.CRR1
76. Kalavalapalli S, Talapatra I, O’ Connell IPM. A complex case of Multiple Endocrine Neoplasia type 1 with Metastatic Parathyroid Carcinoma. Cent Eur J Med (2010) 5:53–8. doi: 10.2478/s11536-009-0116-4
77. Juodelė L, Serapinas D, Sabaliauskas G, Krasauskienė A, Krasauskas V, Verkauskienė R, et al. Carcinoma of two parathyroid glands caused by a novel MEN1 gene mutation - a rare feature of the MEN 1 syndrome. Medicina (Kaunas) (2011) 47:635–9. doi: 10.3390/medicina47110092
78. del Pozo C, García-Pascual L, Balsells M, Barahona MJ, Veloso E, González C, et al. Parathyroid carcinoma in multiple endocrine neoplasia type 1. Case report and review of the literature. Hormones (Athens) (2011) 10:326–31. doi: 10.14310/horm.2002.1325
79. Lee KM, Kim EJ, Choi WS, Park WS, Kim SW. Intrathyroidal parathyroid carcinoma mimicking a thyroid nodule in a MEN type 1 patient. J Clin Ultrasound (2014) 42:212–4. doi: 10.1002/jcu.22090
80. Singh Ospina N, Sebo TJ, Thompson GB, Clarke BL, Young WF Jr. Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 - case report and review of the literature. Clin Endocrinol (Oxf) (2016) 84:244–9. doi: 10.1111/cen.12714
81. Christakis I, Busaidy NL, Cote GJ, Williams MD, Hyde SM, Silva Figueroa AM, et al. Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature. Int J Surg (2016) 31:10–6. doi: 10.1016/j.ijsu.2016.05.035
82. Cinque L, Sparaneo A, Cetani F, Coco M, Clemente C, Chetta M, et al. Novel association of MEN1 gene mutations with parathyroid carcinoma. Oncol Lett (2017) 14:23–30. doi: 10.3892/ol.2017.6162
83. Omi Y, Horiuchi K, Haniu K, Tokura M, Nagai E, Isozaki O, et al. Parathyroid carcinoma occurred in two glands in multiple endocrine neoplasia 1: a report on a rare case. Endocr J (2018) 65:245–52. doi: 10.1507/endocrj.EJ17-0409
84. Song A, Yang Y, Liu S, Nie M, Jiang Y, Li M, et al. Prevalence of Parathyroid Carcinoma and Atypical Parathyroid Neoplasms in 153 Patients With Multiple Endocrine Neoplasia Type 1: Case Series and Literature Review. Front Endocrinol (2020) 11:557050. doi: 10.3389/fendo.2020.557050
85. Wu CW, Huang CI, Tsai ST, Chiang H, Lui WY, P’eng FK. Parathyroid carcinoma in a patient with non-secretory pituitary tumor: a variant of multiple endocrine neoplasia type-I? Eur J Surg Oncol (1992) 18:517–20.
86. Sato M, Miyauchi A, Namihira H, Bhuiyan MM, Imachi H, Murao K, et al. A newly recognized germline mutation of MEN1 gene identified in a patient with parathyroid adenoma and carcinoma. Endocrine (2000) 12:223–6. doi: 10.1385/ENDO:12:3:223
87. Dionisi S, Minisola S, Pepe J, De Geronimo S, Paglia F, Memeo L, et al. Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: presentation as hypercalcemic crisis. Mayo Clin Proc (2002) 77:866–9. doi: 10.4065/77.8.866
88. Butte JM, Montero PH, Solar A, Torres J, Olmos PR, Goñi I, et al. Cervical metastases of glucagonoma in a patient with multiple endocrine neoplasia type 1: report of a case. Surg Today (2008) 38:1137–43. doi: 10.1007/s00595-008-3763-1
89. Falchetti A. Genetics of parathyroids disorders: Overview. Best Pract Res Clin Endocrinol Metab (2018) 32:781–90. doi: 10.1016/j.beem.2018.09.011
90. Eller Vainicher C, Falchetti A. Management of Familial Hyperparathyroidism Syndromes: MEN1, MEN2, MEN4, HPT-Jaw Tumour, Familial Isolated Hyperparathyroidism, FHH, and Neonatal Severe Hyperparathyroidism. Best Pract Res Clin Endocrinol Metab (2018) 32:861–75. doi: 10.1016/j.beem.2018.09.010
91. Hasani-Ranjbar S, Amoli MM, Ebrahim-Habibi A, Gozashti MH, Khalili N, Sayyahpour FA, et al. A new frameshift MEN1 gene mutation associated with familial malignant insulinomas. Fam Cancer (2011) 10:343–8. doi: 10.1007/s10689-010-9412-z
92. Novruzov F, Mehmetbeyli L, Aliyev JA, Abbasov B, Mehdi E. Metastatic Insulinoma Controlled by Targeted Radionuclide Therapy With 177Lu-DOTATATE in a Patient With Solitary Kidney and MEN-1 Syndrome. Clin Nucl Med (2019) 44:e415–7. doi: 10.1097/RLU.0000000000002500
93. Benito M, Asa SL, Livolsi VA, West VA, Snyder PJ. Gonadotroph tumor associated with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2005) 90:570–4. doi: 10.1210/jc.2004-1373
94. Gordon MV, Varma D, McLean CA, Bittar RG, Burgess JR, Topliss DJ. Metastatic prolactinoma presenting as a cervical spinal cord tumour in multiple endocrine neoplasia type one (MEN-1). Clin Endocrinol (Oxf) (2007) 66:150–2. doi: 10.1111/j.1365-2265.2006.02697.x
95. Philippon M, Morange I, Barrie M, Barlier A, Taieb D, Dufour H, et al. Long-term control of a MEN1 prolactin secreting pituitary carcinoma after temozolomide treatment. Ann Endocrinol (Paris) (2012) 73:225–9. doi: 10.1016/j.ando.2012.03.001
96. Scheithauer BW, Kovacs K, Nose V, Lombardero M, Osamura YR, Lloyd RV, et al. Multiple endocrine neoplasia type 1-associated thyrotropin-producing pituitary carcinoma: report of a probable de novo example. Hum Pathol (2009) 40:270–8. doi: 10.1016/j.humpath.2008.06.013
97. Griniatsos JE, Dimitriou N, Zilos A, Sakellariou S, Evangelou K, Kamakari S, et al. Bilateral adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1 (MEN1) and a novel mutation in the MEN1 gene. World J Surg Oncol (2011) 9:6. doi: 10.1186/1477-7819-9-6
98. Skogseid B, Rastad J, Gobl A, Larsson C, Backlin K, Juhlin C, et al. Adrenal lesion in multiple endocrine neoplasia type 1. Surgery (1995) 118:1077–82. doi: 10.1016/s0039-6060(05)80117-5
99. Waldmann J, Bartsch DK, Kann PH, Fendrich V, Rothmund M, Langer P. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg (2007) 392:437–43. doi: 10.1007/s00423-006-0124-7
100. Haase M, Anlauf M, Schott M, Schinner S, Kaminsky E, Scherbaum WA, et al. A new mutation in the menin gene causes the multiple endocrine neoplasia type 1 syndrome with adrenocortical carcinoma. Endocrine (2011) 39:153–9. doi: 10.1007/s12020-010-9424-3
101. Kharb S, Pandit A, Gundgurthi A, Garg MK, Brar KS, Kannan N, et al. Hidden diagnosis of multiple endocrine neoplasia-1 unraveled during workup of virilization caused by adrenocortical carcinoma. Indian J Endocrinol Metab (2013) 17:514–8. doi: 10.4103/2230-8210.111672
102. Goudet P, Dalac A, Le Bras M, Cardot-Bauters C, Niccoli P, Lévy-Bohbot N, et al. MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d’étude des Tumeurs Endocrines. J Clin Endocrinol Metab (2015) 100:1568–77. doi: 10.1210/jc.2014-3659
103. Ohara N, Kaneko M, Ikeda M, Ishizaki F, Suzuki K, Maruyama R, et al. Lung adenocarcinoma and adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1. Respir Med Case Rep (2016) 20:77–81. doi: 10.1016/j.rmcr.2016.12.002
104. Wang W, Han R, Ye L, Xie J, Tao B, Sun F, et al. Adrenocortical carcinoma in patients with MEN1: a kindred report and review of the literature. Endocr Connect (2019) 8:230–8. doi: 10.1530/EC-18-0526
105. Harada K, Yasuda M, Hasegawa K, Yamazaki Y, Sasano H, Otsuka F. A novel case of myxoid variant of adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1. Endocr J (2019) 66:739–44. doi: 10.1507/endocrj.EJ19-0067
106. Duh QY, Hybarger CP, Geist R, Gamsu G, Goodman PC, Gooding GA, et al. Carcinoids associated with multiple endocrine neoplasia syndromes. Am J Surg (1987) 154:142–8. doi: 10.1016/0002-9610(87)90305-9
107. Spaulding R, Alatassi H, Stewart Metzinger D, Moghadamfalahi M. Ependymoma and carcinoid tumor associated with ovarian mature cystic teratoma in a patient with multiple endocrine neoplasia I. Case Rep Obstet Gynecol (2014) 2014:712657. doi: 10.1155/2014/712657
108. Lou L, Zhou L, Wang W, Li H, Li Y. Atypical ovarian carcinoid tumor with widespread skeletal metastases: a case report of multiple endocrine neoplasia type 1 in a young woman. BMC Cancer (2019) 19:1107. doi: 10.1186/s12885-019-6332-7
109. Jhawar S, Lakhotia R, Suzuki M, Welch J, Agarwal SK, Sharretts J, et al. Clinical presentation and management of primary ovarian neuroendocrine tumor in multiple endocrine neoplasia type 1. Endocrinol Diabetes Metab Case Rep (2019) 2019:19–0040. doi: 10.1530/EDM-19-0040
110. van Leeuwaarde RS, Dreijerink KM, Ausems MG, Beijers HJ, Dekkers OM, de Herder WW, et al. MEN1-Dependent Breast Cancer: Indication for Early Screening? Results From Dutch MEN1 Study Group J Clin Endocrinol Metab (2017) 102:2083–90. doi: 10.1210/jc.2016-3690
111. Dreijerink KM, Goudet P, Burgess JR, Valk GD. International Breast Cancer in MEN1 Study Group. Breast-cancer predisposition in multiple endocrine neoplasia type 1. N Engl J Med (2014) 371:583–4. doi: 10.1056/NEJMc1406028
112. National Collaborating Centre for Cancer. NICE Clinical Guidelines, No. 164. Familial breast cancer: classification and care of people at risk at familial breast cancer and related risks in people with a family history of breast cancer. Cardiff, UK: National Collaborating Centre for Cancer (2013).
113. Papi L, Palli D, Masi L, Putignano AL, Congregati C, Zanna I, et al. Germline mutations in MEN1 and BRCA1 genes in a woman with familial multiple endocrine neoplasia type 1 and inherited breast-ovarian cancer syndromes: a case report. Cancer Genet Cytogenet (2009) 195:75–9. doi: 10.1016/j.cancergencyto.2009.06.019
114. Xu B, Li SH, Zheng R, Gao SB, Ding LH, Yin ZY, et al. Menin promotes hepatocellular carcinogenesis and epigenetically up-regulates Yap1 transcription. Proc Natl Acad Sci U S A (2013) 110:17480–5. doi: 10.1073/pnas.1312022110
115. Böni R, Vortmeyer AO, Huang S, Burg G, Hofbauer G, Zhuang Z. Mutation analysis of the MEN1 tumour suppressor gene in malignant melanoma. Melanoma Res (1999) 9:249–52. doi: 10.1097/00008390-199906000-00006
116. Nord B, Platz A, Smoczynski K, Kytölä S, Robertson G, Calender A, et al. Malignant melanoma in patients with multiple endocrine neoplasia type 1 and involvement of the MEN1 gene in sporadic melanoma. Int J Cancer (2000) 87:463–7. doi: 10.1002/1097-0215(20000815)87:4<463::aid-ijc1>3.0.co;2-8
117. Baldauf C, Vortmeyer AO, Koch CA, Sticherling M. Combination of multiple skin malignancies with multiple endocrine neoplasia type 1: coincidental or pathogenetically related? Dermatology (2009) 219:365–7. doi: 10.1159/000193058
118. Brown GT, Cowen EW, Lee CC. Malignant melanoma masquerading as an angiofibromas in a patient with MEN-1. JAMA Dermatol (2015) 151:105–6. doi: 10.1001/jamadermatol.2014.2186
119. Mallek R, Mostbeck G, Walter RM, Herold CH, Imhof H, Tscholakoff D. Contrast MRI in multiple endocrine neoplasia type 1 (MEN) associated with renal cell carcinoma. Eur J Radiol (1990) 10:105–8. doi: 10.1016/0720-048x(90)90116-s
120. Perakakis N, Flohr F, Kayser G, Thomusch O, Parsons L, Billmann F, et al. Multiple endocrine neoplasia type 1 associated with a new germline Men1 mutation in a family with atypical tumor phenotype. Hormones (Athens) (2016) 15:113–7. doi: 10.14310/horm.2002.1626
121. Kim HJ, Park JS, Kim CS, Kang ES, Cha BS, Lim SK, et al. Ahn CW. A case of multiple endocrine neoplasia type 1 combined with papillary thyroid carcinoma. Yonsei Med J (2008) 49:503–6. doi: 10.3349/ymj.2008.49.3.503
122. Hill KA, Yip L, Carty SE, McCoy KL. Concomitant Thyroid Cancer in Patients with Multiple Endocrine Neoplasia Type 1 Undergoing Surgery for Primary Hyperparathyroidism. Thyroid (2019) 29:252–7. doi: 10.1089/thy.2017.0675
123. Paris PL, Sridharan S, Hittelman AB, Kobayashi Y, Perner S, Huang G, et al. An oncogenic role for the multiple endocrine neoplasia type 1 gene in prostate cancer. Prostate Cancer Prostatic Dis (2009) 12:184–91. doi: 10.1038/pcan.2008.45
124. Majewski JT, Wilson SD. The MEA-I syndrome: an all or none phenomenon? Surgery (1979) 86:475–84. doi: 10.5555/uri:pii:0039606079900412
125. Wilkinson S, Teh BT, Davey KR, McArdle JP, Young M, Shepherd JJ. Cause of death in multiple endocrine neoplasia type 1. Arch Surg (1993) 128:683–90. doi: 10.1001/archsurg.1993.01420180085016
126. Ito T, Igarashi H, Uehara H, Berna MJ, Jensen RT. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Med (Baltimore) (2013) 92:135–81. doi: 10.1097/MD.0b013e3182954af1
127. Geerdink EA, Van der Luijt RB, Lips CJ. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol (2003) 149:577–82. doi: 10.1530/eje.0.1490577
128. Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg (2010) 34:249–55. doi: 10.1007/s00268-009-0290-1
129. Kouvaraki MA, Shapiro SE, Cote GJ, Lee JE, Yao JC, Waguespack SG, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg (2006) 30:643–53. doi: 10.1007/s00268-006-0360-y
130. Gibril F, Venzon DJ, Ojeaburu JV, Bashir S, Jensen RT. Prospective study of the natural history of gastrinoma in patients with MEN1: definition of an aggressive and a nonaggressive form. J Clin Endocrinol Metab (2001) 86:5282–93. doi: 10.1210/jcem.86.11.8011
131. Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol (2007) 265-266:34–41. doi: 10.1016/j.mce.2006.12.032
132. Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer (2008) 113:1807–43. doi: 10.1002/cncr.23648
133. Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care (2005) 28:1769–78. doi: 10.2337/diacare.28.7.1769
134. McCallum RW, Parameswaran V, Burgess JR. Multiple endocrine neoplasia type 1 (MEN 1) is associated with an increased prevalence of diabetes mellitus and impaired fasting glucose. Clin Endocrinol (Oxf) (2006) 65:163–8. doi: 10.1111/j.1365-2265.2006.02563.x
135. van Wijk JP, Dreijerink KM, Pieterman CR, Lips CJ, Zelissen PM, Valk GD. Increased prevalence of impaired fasting glucose in MEN1 gene mutation carriers. Clin Endocrinol (Oxf) (2012) 76:67–71. doi: 10.1111/j.1365-2265.2011.04166.x
136. Hai N, Aokis N, Shlmatsu A, Mod T, Kosugi S. Clinical features of multiple endocrine neoplasia type 1 (MENI) phenocopy without germline MEN7 gene mutations: analysis of 20 Japanese sporadic cases with MEN1. Clin Endocrinol (Oxf) (2000) 52:509–18. doi: 10.1046/j.1365-2265.2000.00966.x
137. Klein RD, Salih S, Bessoni J, Bale AE. Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med (2005) 7:131–8. doi: 10.1097/01.gim.0000153663.62300.f8
138. O’Toole D, Grossman A, Gross D, Delle Fave G, Barkmanova J, O’Connor J, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: biochemical markers. Neuroendocrinology (2009) 90:194–202. doi: 10.1159/000225948
139. Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, Caplin M, et al. Vienna Consensus Conference participants. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology (2016) 103:153–71. doi: 10.1159/000443171
140. Manoharan J, Raue F, Lopez CL, Albers MB, Bollmann C, Fendrich V, et al. Is Routine Screening of Young Asymptomatic MEN1 Patients Necessary? World J Surg (2017) 41:2026–32. doi: 10.1007/s00268-017-3992-9
141. van Leeuwaarde RS, Pieterman CRC, Bleiker EMA, Dekkers OM, van der Horst-Schrivers AN, Hermus AR, et al. High Fear of Disease Occurrence Is Associated With Low Quality of Life in Patients With Multiple Endocrine Neoplasia Type 1: Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab (2018) 103:2354–61. doi: 10.1210/jc.2018-00259
142. Manoharan J, Albers MB, Bartsch DK. The future: diagnostic and imaging advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer (2017) 24:T209–25. doi: 10.1530/ERC-17-0231
143. de Laat JM, van der Luijt RB, Pieterman CR, Oostveen MP, Hermus AR, Dekkers OM, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med (2016) 14:182. doi: 10.1186/s12916-016-0708-1
144. Delle Fave G, O’Toole D, Sundin A, Taal B, Ferolla P, Ramage JK, et al. Vienna Consensus Conference participants. ENETS Consensus Guidelines Update for Gastroduodenal Neuroendocrine Neoplasms. Neuroendocrinology (2016) 103:119–24. doi: 10.1159/000443168
145. Giusti F, Tonelli F, Brandi ML. Primary hyperparathyroidism in multiple endocrine neoplasia type 1: when to perform surgery? Clinics (Sao Paulo) (2012) 67:141–4. doi: 10.6061/clinics/2012(sup01)23
146. Moyes VJ, Monson JP, Chew SL, Akker SA. Clinical Use of Cinacalcet in MEN1 Hyperparathyroidism. Int J Endocrinol (2010) 2010:906163. doi: 10.1155/2010/906163
147. Falchetti A, Cilotti A, Vaggelli L, Masi L, Amedei A, Cioppi F, et al. A Patient With MEN1-associated Hyperparathyroidism, Responsive to Cinacalcet. Nat Clin Pract Endocrinol Metab (2008) 4:351–7. doi: 10.1038/ncpendmet0816
148. Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, Avenia N, et al. Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series. J Clin Endocrinol Metab (2005) 90:2603–9. doi: 10.1210/jc.2004-1155
149. Molitch ME. Diagnosis and Treatment of Pituitary Adenomas: A Review. JAMA (2017) 317:516–24. doi: 10.1001/jama.2016.19699
150. Kamilaris CDC, Stratakis CA. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front Endocrinol (Lausanne) (2019) 10:339. doi: 10.3389/fendo.2019.00339
151. Imamura M, Komoto I, Ota S, Hiratsuka T, Kosugi S, Doi R, et al. Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol (2011) 17:1343–53. doi: 10.3748/wjg.v17.i10.1343
152. Norton JA, Jensen RT. Role of surgery in Zollinger-Ellison syndrome. J Am Coll Surg (2007) 205:S34–S3724. doi: 10.1016/j.jamcollsurg.2007.06.320
153. Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, et al. Groupe d’études des Tumeurs Endocrines. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1patients. Am J Surg Pathol (2008) 32:534–54312. doi: 10.1097/PAS.0b013e31815ade45
154. Sakurai A, Katai M, Yamashita K, Mori J, Fukushima Y, Hashizume K. Long-term follow-up of patients with multiple endocrine neoplasia type 1. Endocr J (2007) 54:295–302. doi: 10.1507/endocrj.k06-147
155. Pieterman CRC, de Laat JM, Twisk JWR, van Leeuwaarde RS, de Herder WW, Dreijerink KMA, et al. Long-Term Natural Course of Small Nonfunctional Pancreatic Neuroendocrine Tumors in MEN1-Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab (2017) 102:3795–805. doi: 10.1210/jc.2017-00372
156. Partelli S, Cirocchi R, Crippa S, Cardinali L, Fendrich V, Bartsch DK, et al. Systematic review of active surveillance versus surgical management of asymptomatic small non-functioning pancreatic neuroendocrine neoplasms. Br J Surg (2017) 104:34–41. doi: 10.1002/bjs.10312
157. Nell S, Verkooijen HM, Pieterman CRC, de Herder WW, Hermus AR, Dekkers OM, et al. Management of MEN1 Related Nonfunctioning Pancreatic NETs: A Shifting Paradigm: Results From the Dutch MEN1 Study Group. Ann Surg (2018) 267:1155–60. doi: 10.1097/SLA.0000000000002183
158. Triponez F, Sadowski SM, Pattou F, Cardot-Bauters C, Mirallié E, Le Bras M, et al. Long-term Follow-up of MEN1 Patients Who Do Not Have Initial Surgery for Small ≤2 cm Non functioning Pancreatic Neuroendocrine Tumors, an AFCE and GTE Study: Association Francophone de Chirurgie Endocrinienne & Groupe d’Etude des Tumeurs Endocrines. Ann Surg (2018) 268:158–64. doi: 10.1097/SLA.0000000000002191
159. Klöppel G, Klimstra DS, Hruban RH, Adsay V, Capella C, Couvelard A, et al. Pancreatic neuroendocrine tumors: Update on the new World Health Organization classification. AJSP: Rev Rep (2017) 22:233–9. doi: 10.1097/PCR.0000000000000211
160. Stueven AK, Kayser A, Wetz C, Amthauer H, Wree A, Tacke F, et al. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int J Mol Sci (2019) 20:3049. doi: 10.3390/ijms20123049
161. Frost M, Lines KE, Thakker RV. Current and emerging therapies for PNETs in patients with or without MEN1. Nat Rev Endocrinol (2018) 14:216–27. doi: 10.1038/nrendo.2018.3
162. Lips CJ, Dreijerink KM, Höppener JW. Variable clinical expression in patients with a germline MEN1 disease gene mutation: clues to a genotype-phenotype correlation. Clinics (Sao Paulo) (2012) 67:49–56. doi: 10.6061/clinics/2012(sup01)10
163. Burgess JR, Nord B, David R, Greenaway TM, Parameswaran V, Larsson C, et al. Phenotype and phenocopy: the relationship between genotype and clinical phenotype in a single large family with multiple endocrine neoplasia type 1 (MEN 1). Clin Endocrinol (Oxf) (2000) 53:205–21. doi: 10.1046/j.1365-2265.2000.01032.x
164. Cardinal JW, Bergman L, Hayward N, Sweet A, Warner J, Marks L, et al. A report of a national mutation testing service for the MEN1 gene: clinical presentations and implications for mutation testing. J Med Genet (2005) 42:69–74. doi: 10.1136/jmg.2003.017319
165. Kytölä S, Villablanca A, Ebeling T, Nord B, Larsson C, Höög A, et al. Founder effect in multiple endocrine neoplasia type 1 (MEN 1) in Finland. J Med Genet (2001) 38:185–9. doi: 10.1136/jmg.38.3.185
166. Lourenço DM Jr, Toledo RA, Mackowiak II, Coutinho FL, Cavalcanti MG, Correia-Deur JE, et al. Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. Eur J Endocrinol (2008) 159:259–74. doi: 10.1530/EJE-08-0153
167. Olufemi SE, Green JS, Manickam P, Guru SC, Agarwal SK, Kester MB, et al. Common ancestral mutation in the MEN1 gene is likely responsible for the prolactinoma variant of MEN1 (MEN1Burin) in four kindreds from Newfoundland. Hum Mutat (1998) 11:264–9. doi: 10.1002/(SICI)1098-1004(1998)11:4<264::AID-HUMU2>3.0.CO;2-V
168. Vierimaa O, Ebeling TM, Kytölä S, Bloigu R, Eloranta E, Salmi J, et al. Multiple endocrine neoplasia type 1 in Northern Finland; clinical features and genotype phenotype correlation. Eur J Endocrinol (2007) 157:285–94. doi: 10.1530/EJE-07-0195
169. Luzi E, Brandi ML. Are microRNAs involved in the endocrine-specific pattern of tumorigenesis in multiple endocrine neoplasia type 1? Endocr Pract (2011) 17:58–63. doi: 10.4158/EP11062.RA
170. Luzi E, Marini F, Tognarini I, Carbonell Sala S, Galli G, Falchetti A, et al. Ribozyme-mediated compensatory induction of menin-oncosuppressor function in primary fibroblasts from MEN1 patients. Cancer Gene Ther (2010) 17:814–25. doi: 10.1038/cgt.2010.39
171. Luzi E, Ciuffi S, Marini F, Mavilia C, Galli G, Brandi ML. Analysis of differentially expressed microRNAs in MEN1 parathyroid adenomas. Am J Transl Res (2017) 9:1743–53.
172. Gurung B, Katona BW, Hua X. Menin-mediated regulation of miRNA biogenesis uncovers the IRS2 pathway as a target for regulating pancreatic beta cells. Oncoscience (2014) 1:562–6. doi: 10.18632/oncoscience.79
173. Lin W, Watanabe H, Peng S, Francis JM, Kaplan N, Pedamallu CS, et al. Dynamic epigenetic regulation by menin during pancreatic islet tumor formation. Mol Cancer Res (2015) 13:689–98. doi: 10.1158/1541-7786.MCR-14-0457
174. Turner JJ, Leotlela PD, Pannett AA, Forbes SA, Bassett JH, Harding B, et al. Frequent occurrence of an intron 4 mutation in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2002) 87:2688–93. doi: 10.1210/jcem.87.6.8607
175. Kassem M, Kruse TA, Wong FK, Larsson C, The BT. Familial isolated hyperparathyroidism as a variant of multiple endocrine neoplasia type 1 in a large Danish pedigree. J Clin Endocrinol Metab (2000) 85:165–7. doi: 10.1210/jcem.85.1.6299
176. Miedlich S, Lohmann T, Schneyer U, Lamesch P, Paschke R. Familial isolated primary hyperparathyroidism–a multiple endocrine neoplasia type 1 variant? Eur J Endocrinol (2001) 145:155–60. doi: 10.1530/eje.0.1450155
177. Villablanca A, Wassif WS, Smith T, Höög A, Vierimaa O, Kassem M, et al. Involvement of the MEN1 gene locus in familial isolated hyperparathyroidism. Eur J Endocrinol (2002) 147:313–22. doi: 10.1530/eje.0.1470313
178. Pannett AA, Kennedy AM, Turner JJ, Forbes SA, Cavaco BM, Bassett JH, et al. Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol (Oxf) (2003) 58:639–46. doi: 10.1046/j.1365-2265.2003.01765.x
179. Hannan FM, Nesbit MA, Christie PT, Fratter C, Dudley NE, Sadler GP, et al. Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab (2008) 4:53–8. doi: 10.1038/ncpendmet0718
180. Miyauchi A, Sato M, Matsubara S, Ohye H, Kihara M, Matsusaka K, et al. A family of MEN1 with a novel germline missense mutation and benign polymorphisms. Endocr J (1998) 45:753–9. doi: 10.1507/endocrj.45.753
181. Kong C, Ellard S, Johnston C, Farid NR. Multiple endocrine neoplasia type 1Burin from Mauritius: a novel MEN1 mutation. J Endocrinol Invest (2001) 24:806–10. doi: 10.1007/BF03343931
182. Hao W, Skarulis MC, Simonds WF, Weinstein LS, Agarwal SK, Mateo C, et al. Multiple endocrine neoplasia type 1 variant with frequent prolactinoma and rare gastrinoma. J Clin Endocrinol Metab (2004) 89:3776–84. doi: 10.1210/jc.2003-031511
183. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab (2009) 94:1826–34. doi: 10.1210/jc.2008-2083
184. Giraud S, Zhang CX, Serova-Sinilnikova O, Wautot V, Salandre J, Buisson N, et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet (1998) 63:455–67. doi: 10.1086/301953
185. Sato K, Yamazaki K, Zhu H, Kanbe M, Iihara M, Wada Y, et al. Somatic mutations of the multiple endocrine neoplasia type 1 (MEN1) gene in patients with sporadic, nonfamilial primary hyperparathyroidism. Surgery (2000) 127:337–41. doi: 10.1067/msy.2000.104165
186. Bergman L, Boothroyd C, Palmer J, Grimmond S, Walters M, The B, et al. Identification of somatic mutations of the MEN1 gene in sporadic endocrine tumours. Br J Cancer (2000) 83:1003–8. doi: 10.1054/bjoc.2000.1385
187. Matsuzaki LN, Canto-Costa MH, Hauache OM. Cushing’s disease as the first clinical manifestation of multiple endocrine neoplasia type 1 (MEN1) associated with an R460X mutation of the MEN1 gene. Clin Endocrinol (Oxf) (2004) 60:142–3. doi: 10.1111/j.1365-2265.2004.01943.x
188. Ellard S, Hattersley AT, Brewer CM, Vaidya B. Detection of an MEN1 gene mutation depends on clinical features and supports current referral criteria for diagnostic molecular genetic testing. Clin Endocrinol (Oxf) (2005) 62:169–75. doi: 10.1111/j.1365-2265.2005.02190.x
189. Raef H, Zou M, Baitei EY, Al-Rijjal RA, Kaya N, Al-Hamed M, et al. A novel deletion of the MEN1 gene in a large family of multiple endocrine neoplasia type 1 (MEN1) with aggressive phenotype. Clin Endocrinol (Oxf) (2011) 75:791–800. doi: 10.1111/j.1365-2265.2011.04134.x
190. Ishida E, Yamada M, Horiguchi K, Taguchi R, Ozawa A, Shibusawa N, et al. Attenuated expression of menin and p27 (Kip1) in an aggressive case of multiple endocrine neoplasia type 1 (MEN1) associated with an atypical prolactinoma and a malignant pancreatic endocrine tumor. Endocr J (2011) 58:287–96. doi: 10.1507/endocrj.k10e-158
191. Bartsch DK, Slater EP, Albers M, Knoop R, Chaloupka B, Lopez CL, et al. Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab (2014) 99:E2387–91. doi: 10.1210/jc.2013-4432
192. Skalniak A, Sokołowski G, Jabrocka-Hybel A, Piątkowski J, Białas M, Gilis-Januszewska A, et al. A novel in-frame deletion in MEN1 (p.Ala416del) causes familial multiple endocrine neoplasia type 1 with an aggressive phenotype and unexpected inheritance pattern. Mol Med Rep (2016) 14:2061–6. doi: 10.3892/mmr.2016.5462
193. Palermo A, Capoluongo E, Del Toro R, Manfrini S, Pozzilli P, Maggi D, et al. A novel germline mutation at exon 10 of MEN1 gene: a clinical survey and positive genotype-phenotype analysis of a MEN1 Italian family, including monozygotic twins. Hormones (Athens) (2018) 17:427–35. doi: 10.1007/s42000-018-0044-2
194. Marx SJ, Simonds WF. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev (2005) 26:615–66. doi: 10.1210/er.2003-0037
Keywords: MEN1, genotype, phenotype, mutations, tumors
Citation: Mele C, Mencarelli M, Caputo M, Mai S, Pagano L, Aimaretti G, Scacchi M, Falchetti A and Marzullo P (2020) Phenotypes Associated With MEN1 Syndrome: A Focus on Genotype-Phenotype Correlations. Front. Endocrinol. 11:591501. doi: 10.3389/fendo.2020.591501
Received: 04 August 2020; Accepted: 16 October 2020;
Published: 18 November 2020.
Edited by:
Wen Zhou, Case Western Reserve University, United StatesReviewed by:
Jean-Yves Scoazec, Institut Gustave Roussy, FranceFrancesco Giudici, University of Florence, Italy
Copyright © 2020 Mele, Mencarelli, Caputo, Mai, Pagano, Aimaretti, Scacchi, Falchetti and Marzullo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Marzullo, cGFvbG8ubWFyenVsbG9AbWVkLnVuaXVwby5pdA==