 Kinning Poon
Kinning Poon- Department of Biological Sciences, SUNY College at Old Westbury, Old Westbury, NY, United States
Excessive dietary fat intake has extensive impacts on several physiological systems and can lead to metabolic and nonmetabolic disease. In animal models of ingestion, exposure to a high fat diet during pregnancy predisposes offspring to increase intake of dietary fat and causes increase in weight gain that can lead to obesity, and without intervention, these physiological and behavioral consequences can persist for several generations. The hypothalamus is a region of the brain that responds to physiological hunger and fullness and contains orexigenic neuropeptide systems that have long been associated with dietary fat intake. The past fifteen years of research show that prenatal exposure to a high fat diet increases neurogenesis of these neuropeptide systems in offspring brain and are correlated to behavioral changes that induce a pro-consummatory and obesogenic phenotype. Current research has uncovered several potential molecular mechanisms by which excessive dietary fat alters the hypothalamus and involve dietary fatty acids, the immune system, gut microbiota, and transcriptional and epigenetic changes. This review will examine the current knowledge of dietary fat-associated changes in the hypothalamus and the potential pathways involved in modifying the development of orexigenic peptide neurons that lead to changes in ingestive behavior, with a special emphasis on inflammation by chemokines.
Introduction
The latest National Health and Nutrition Examination Survey reports 39.8% of adults and 18.5% of youth in the United States to be obese (1), with the ingestion of a diet rich in saturated fats or high in omega-6 fatty acids to be a risk factor in developing obesity (2–5). The ingestion of a High-fat diet (HFD) in humans and in adult animal models have harmful effects to physiological organ systems by inducing a state of systemic inflammation that is related to the increased risk for developing disease including cardiovascular disease, diabetes, and cancer (6–8), and also has cognitive effects in humans (9, 10). These disease states have been correlated to HFD-induced cellular changes in all organ systems, such as adipose tissue, liver, heart, kidneys, and the gut itself (11, 12). The effects of a HFD are not exhaustive to peripheral organ systems and can also directly impact the central nervous system (13, 14), with the central effects governing ingestive behavior. Ingestion of a HFD and obesity prior to mating, during pregnancy, and postnatally also induces similar physiological and central phenotypic outcome in offspring. Animal studies have revealed a transgenerational effect of HFD intake or obesity from both paternal and maternal sides prior to and during pregnancy on over-consummatory behavior in offspring (15, 16), with similar findings in humans from the 1958 British birth cohort study (17). Recently, the fields of developmental biology, metabolomics, nutrition, and ingestive behavior have linked the systemic and central nervous system effects of a fat-rich diet to the activation of the immune system, with inflammatory mediators playing a potential role in mediating HFD changes on the periphery, the brain and on developmental processes (18, 19). The connectivity of these disciplines on the outcome of HFD intake and obesity bring to light the complexity between inflammation and a HFD in adult animal models and in offspring that are prenatally-exposed to this diet, particularly on specific inflammatory mediators such as the chemokines. This article will begin with a brief historical perspective on ingestive behavior and examine in more detail the intersection of other fields in relation to inflammation by chemokines to uncover cellular mechanisms of HFD effects.
Dietary Fat on Stimulating Neurochemical Systems in the Hypothalamus
Several decades of research have well-established in adult animal models that the ingestion of a fat-rich diet consisting of high levels of saturated fats (20) alters brain neurochemistry, particularly in the hypothalamus (21–23) where there exists populations of neurons that express several neurochemicals involved in controlling dietary fat intake (24–26). While the ingestion of a HFD can impact the levels of orexigenic and anorexigenic neuropeptides, the larger effect primarily occurs on the former (27, 28). Several orexigenic neuropeptides, with some of the main players to include enkephalin, galanin, orexin, and melanin-concentrating hormone, have been positively linked to HFD intake that increases expression and peptide levels in the brain. The injection of these neuropeptides or analogs of these neuropeptides directly into the hypothalamus can in turn stimulate excessive HFD intake (20, 29–33). The circuitry maintaining these peptide neurons are complex, with hormones released from the gut, such as ghrelin and cholecystokinin (34, 35), to directly impact hypothalamic peptide neuron excitation and inhibition, respectively, and stimulate or inhibit feeding behavior (Figure 1).
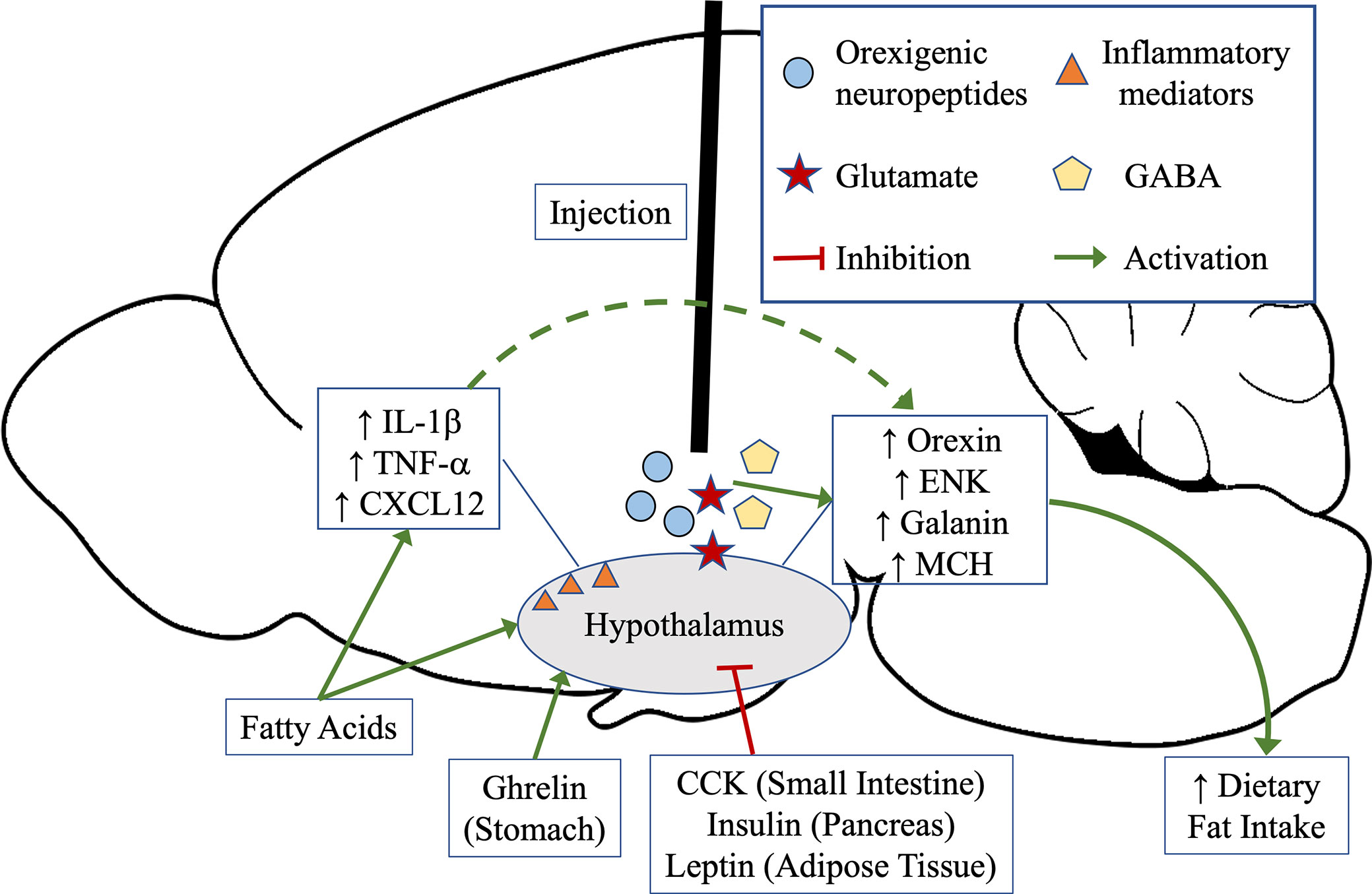
Figure 1 Hunger and satiety can stimulate the release of gut neuropeptides that directly signal the hypothalamus. Ghrelin, released from the stomach during hunger, activates the orexigenic peptide neurons in the hypothalamus to signal food intake, whereas cholecystokinin (CCK) from the intestine, insulin from the pancreas, or leptin from adipose tissue, inhibits orexigenic neuropeptide signaling in the hypothalamus to signal satiety and high levels of macronutrients. Diets rich in fats can directly impact activation of the orexigenic neuropeptide signaling in the hypothalamus, leading to increased ingestive behavior, and additionally induce a state of inflammation. Inflammatory mediators have also been shown to affect orexigenic neuropeptides in the hypothalamus. Other neurotransmitters, such as glutamate and GABA, have also been shown to stimulate orexigenic neuropeptide release and can lead to increase in dietary fat intake.
These early neuropeptide studies expanded to include local hypothalamic circuitry involved in the control of neuropeptide release. Two of the main classical inhibitory and excitatory neurotransmitters, γ-Aminobutyric acid (GABA) and glutamate, have been examined and have been shown to contribute to dietary-fat sensitive neuropeptide function. Glutamatergic innervation of the hypothalamus has been closely linked to ingestive behavior, with a glutamate analog injected into the hypothalamus or the surrounding intracerebroventricular region to increase food intake (36). To date, glutamatergic neuronal contacts with peptide neurons include neuropeptide Y (37), leptin (38), melanin-concentrating hormone (39), and orexin (40). Glutamatergic neurons that may be involved in stimulating the release of peptides from hypothalamic neurons during hunger to initiate food intake can also be depressed during short-term HFD intake and may act as a control mechanism to both initiate feeding and prevent over-ingestion of dietary fat. (41, 42). Linehan et al, found that excitatory signaling on orexin neurons could be depressed during short-term HFD intake but had reduced sensitivity to this signaling during long-term HFD regimes, suggesting extended glutamate signaling can exacerbate orexigenic neuropeptide signaling that leads to increased dietary fat intake (42). Long-term HFD exposure, in female mice in particular, can increase the number of glutamatergic neurons in the hypothalamus (43) and may over time increase neuronal signaling of peptide neurons to stimulate behavioral changes. Likewise, GABA agonists can exert similar effects as glutamate signaling in stimulating feeding (44). Both glutamatergic and GABAergic signaling in the brain can be impacted by overactivity and cellular stress. Prolonged exposure to a HFD can cause changes to the hypothalamic proteome involved in neuronal health and viability, and suggests these effects can lead to increased cellular stress, mitochondrial dysfunction, and altered synaptic plasticity (45), factors that impact glutamatergic and GABAergic signaling in the brain. These HFD-induced brain neurochemistry changes in adults leading to behavioral and physiological changes could potentially impact reproduction and offspring development.
The extensive documented effects of a HFD in adult animals on neuropeptides and neuronal connectivity is observed in offspring born from paternal or maternal obesity. The 1958 British birth cohort study show that both paternal and maternal obesity predisposes offspring to becoming obese, with maternal factors having a more pronounced phenotypic change in offspring compared to paternal obesity, and the combination of obesity in both parents have additive effects in offspring (17). In animal models, in utero exposure to a diet rich in fats with or without prior maternal obesity, predisposes offspring to have increased body weight (46–48), increases the risk of developing obesity (46), increases tendencies to consume other substances of abuse such as nicotine and ethanol (47, 49, 50), and is associated with an increase in the incidence of psychiatric disorders (Figure 2) (51–53). Ingestion of a HFD during pregnancy increases lipid accumulation in the placenta (54), while in offspring decreases the circulation of essential fatty acids (54), and increases the levels of triglycerides and adipocyte size (55, 56). Prenatal HFD paired with a postnatal HFD regime can cause several effects in offspring that is exerted on a macro and microscale level, including a reduction in the variation of DNA methylation in peripheral organs and changes the expression of genes involved in inflammation and RNA processing (48). The paternal inheritance of HFD-induced obesity paired with non-obese maternal pregnancy has similar physiological effects in offspring. Paternal obesity induces in male offspring increase in body weight, impairs glucose and insulin sensitivity and increases leptin levels while female offspring also has increased adiposity. Further breeding of these offspring reveals persistence of these physiological changes in F2 females but not males, suggesting HFD-induced obesity on the paternal side without maternal obesity contributes to female susceptibility to metabolic disorders (57, 58). Studies also show paternal gene imprinting of alleles related to body fat accumulation, Igf2 and Peg3, to be decreased in obesity-resistance mice compared to obesity-prone mice, suggesting a likelihood of paternal gene transmission in offspring resulting in diet-induced obesity (59). Although HFD-induced paternal obesity has been shown to effect physiological outcomes in offspring, the field has primarily focused on maternal contributions due to the large effects evoked in offspring brain neurochemistry.
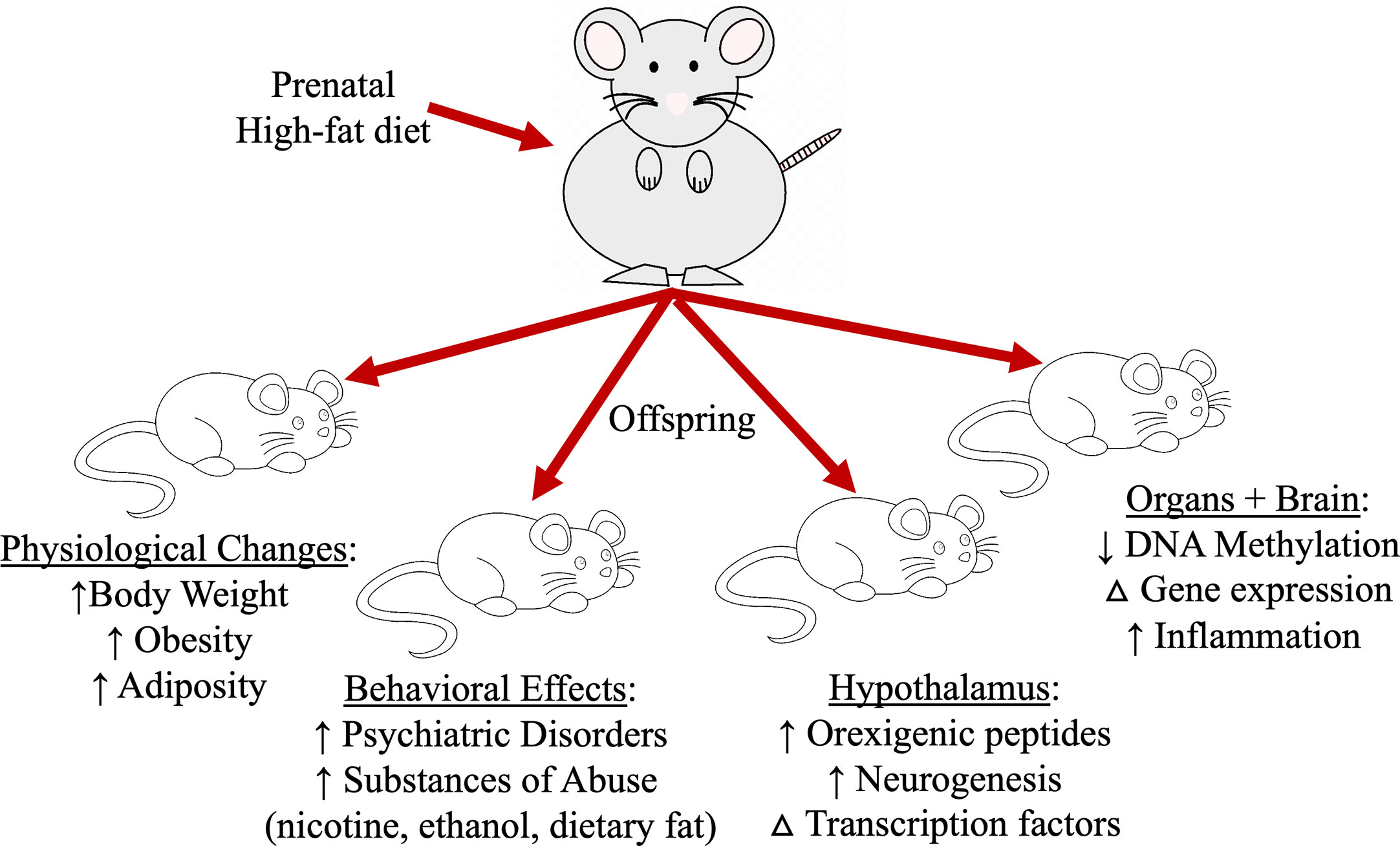
Figure 2 Prenatal high-fat diet exposure impacts several aspects of offspring physiology and behavior that leads to excessive dietary fat intake and increasing the risk for obesity.
In maternal models of HFD-intake, brain neurochemistry is changed such that offspring excessively consume dietary fat when presented with a high-fat diet choice (46, 60). This exposure during gestation increases in the hypothalamus of offspring the mRNA expression and peptide levels of the very same orexigenic neuropeptides that are altered during adult HFD intake (46). This increase is also accompanied by a pronounced rise in the neurogenesis of these peptide neurons (46, 61). While the relationship between dietary fat and neuropeptides in the hypothalamus is well established, the factors involved in causing neurochemical and neurogenesis changes in the brain of offspring are still under speculation, although many studies have provided some insight regarding specific pathways that may be involved. Prenatal HFD exposure can impact DNA methylation of several genes related to dietary fat intake (62, 63) as well as the levels and activity of several transcription factors that have been associated with brain development (64), such as transcriptional enhancer factor-1 (TEF-1), yes-associated protein-1 (YAP-1), and the family of peroxisome proliferator-activated receptors (PPARs), that in turn effect the expression of some of the orexigenic neuropeptides (65–68). The transcription factors TEF-1 and YAP-1 has been linked to prenatal HFD effects and plays a large role in organ formation and brain development during embryogenesis (69–72), and activation of these transcription factors have been found to control neuronal proliferation and differentiation (73–75). Both TEF-1 and YAP-1 were inactivated or decreased during in utero HFD exposure and may promote increased neurogenesis events (65). The PPAR family of transcription factors are fat sensitive and control lipid and carbohydrate metabolism and inflammation (76). While PPAR-δ is colocalized with hypothalamic ENK neurons, reduction of transcriptional regulator results in increased ENK expression and levels, suggesting a regulatory mechanism that is protective of HFD-exposure on neurons (66). A commonality of these factors is their relation to the immune system and the activation of inflammatory mediators. An abundance of research has directed the field toward inflammatory processes and inflammation, which are now thought to play a large role in both the physiological and central effects induced by the intake of a HFD and in obesity.
Dietary Fat Intake Induces Global Systemic Inflammation
The ingestion of a HFD and the obese state produces an increase in systemic inflammation and has been reported to occur in almost every peripheral tissue and physiological system examined to date (77–80). This low-grade inflammation is characterized by an elevation of cytokines and chemokines (81, 82) that is atypical from the disease state and is accompanied by the activation of cellular inflammatory pathways (77, 83) and immune cells. This inflammation occurs whether HFD intake is acute or chronic and has been linked to diseases produced by metabolic syndrome. This inflammation has been linked to excessive levels of saturated fatty acids and omega-6 fatty acids in human western diets (84–86) and in HFD used in animal model studies (87–89).Central nervous system inflammation and activation of immune cells have also been linked to psychiatric disorders such as depression and bipolar disorder (90, 91), implicating the extensive effects HFD-induced inflammation has on the brain and behavior.
In the hypothalamus and in brain regions involved in the emotional component of ingestive behavior such as the amygdala and hippocampus (88, 89, 92, 93), there is an increase in many classical cytokines and activation of inflammatory pathways (94–96). These changes in inflammatory mediators can alter orexigenic neuropeptide levels in the hypothalamus (97–100), stimulate weight gain and increase HFD intake in animal models (101), while inhibition of hypothalamic inflammation with antibodies targeted for cytokines results in a reduction of food intake in obese animals (88, 96, 102). Neuroglial cells in the brain, including microglia, astrocytes, and oligodendrocytes, in conjunction with neurons, may contribute to this inflammation by releasing inflammatory mediators during exposure to excessive dietary fatty acids. Evidence suggests that the immediate inflammatory effect caused by a HFD in the brain can be due to several factors, including infiltration of peripheral macrophages (103), stimulation of resident microglia (94, 104), and direct astrocyte activation, which can increase inflammatory signaling and positively contribute to HFD intake and the onset of obesity (105, 106). The crosstalk between neuroglia, cytokine release, and dietary fat seem to directly impact neuronal signaling.
The exact pathway and the timing of the original inflammatory trigger across physiological systems and its subsequent effects in the brain is not known; is dietary fat itself the inflammatory trigger in the entire body or does the initial inflammatory trigger in the intestines where dietary fat is absorbed lead to additional inflammation across all physiological systems? For instance, long-term dietary fat intake can stimulate inflammation in the gut and alter intestinal microbiota leading to a leaky environment that is more susceptible to absorbing digested fats into the bloodstream (107, 108), with the increase in absorbed fatty acids to enter the brain and themselves induce central inflammatory effects. In contrast, reduction or alteration of gut microbiota results in a reduction of dietary fat intake (109, 110) and may prevent the central inflammatory effects by dietary fat. Based on this observation, it is possible that gut inflammation may precede brain inflammation (111) with the initial localized inflammation of the intestines producing central inflammatory effects. However, a single day of HFD intake itself can increase hypothalamic inflammation (77), suggesting that the increased load of absorbed fatty acids from the HFD itself may be directly causing the low-grade systemic inflammation in the body and brain, with longer term HFD ingestion to additionally contribute to gut inflammation and increase dietary fatty acid absorption that exacerbates the effects of dietary fat on the brain. This also suggests that the pathway of dietary fat intake and induction of inflammation conforms to a perpetual positive circuit and if continued over the course of reproductive age, can also lead to a generational effect.
Maternal Inflammation Is Similar to Excessive Dietary Fat Intake
The impact of a HFD on inflammation is evidenced in maternal humans and in animal models that have widely established significant metabolic effects in offspring. In humans, increase of a marker of inflammation, C-reactive protein, induced by obesity has been correlated with increased incidence of obesity in offspring (112, 113) and suggests this inflammation may be involved in the prenatal programming of ingestive behavioral changes that leads to obesity. During pregnancy, several inflammatory mediators play a role in placental formation and in organogenesis events, and must maintain a delicate balance of immune function and immune suppression to ensure fetal survival (114–116). During pregnancy in obese humans, increased secretion of inflammatory mediators contributes to lowered blood flow into the placenta and potential dysfunction (113). It is possible that HFD-induction of a low-grade systemic inflammation (18) may disrupt the normal placental environment and impact the developing embryo. Prenatal exposure to a HFD and maternal obesity increases macrophage accumulation (117) and cytokine levels in the placenta (118, 119) that creates a more leaky environment to increase dietary fatty acids permeability through the placenta and into the embryo. Prenatal HFD also increases inflammation in several fetal organ systems that is found to be sustained in postnatal offspring, with this hyperinflammation to be further triggered during exposure to foreign pathogenic molecules (56, 119–121). The most notable physiological change in inflammatory profile is found in adipose tissue and closely mirrors the effect of a HFD in adult animals, revealing larger diameter adipocytes, upregulation of genes associated with adipogenesis, and increased association of these adipocytes with macrophages (56, 122, 123). One possibility for the increase in prenatal HFD associated inflammation may be due to the changes in maternal gut microbiota that result in increased intestinal inflammation and fatty acid absorption. The excess fatty acids and inflammatory mediators in the circulatory system of the mother in conjunction with the change in immune cell recruitment can allow these biological agents to enter the placenta and the fetus (119, 124, 125). These studies further show these microbiota changes in the mother to alter offspring microbiota and inflammatory profiles that persist postnatally and into adulthood (126–128). The combination of a HFD increasing inflammation in the mother and in the embryo may alter the course of brain development during gestation.
Prenatal HFD exposure increases in the hypothalamus of offspring the expression of cytokines and chemokines (104, 129, 130), compromises the blood brain barrier (131), the number of astrocytes (132), macrophage infiltration into the brain (103), activation of microglia (133), and immunoglobulin levels in microglia (94). These effects of a HFD in offspring on inflammation, immune cells, and inflammatory mediators can last well into adulthood (134–136) and has been linked to a number of disorders and diseases (137). This interplay between the immune system and HFD induced changes in fetal development is further solidified with prenatal inflammation challenges. Exposure to inflammatory mediators or challenges to the immune system during pregnancy results in increased body weight, increased risk of the development of obesity, and increased ingestive behavior in offspring (138, 139). In contrast, treatment during pregnancy with a natural anti-inflammatory compound, resveratrol, prevented brain changes and physiological changes associated with prenatal high-fat diet exposure (140, 141). The consequences of this heightened immune response in fetal brain may be rewiring of neuronal architecture that leads to postnatal behavioral and physiological consequences in offspring. These aspects of a HFD on maternal immune function, placental formation, and the fetus itself suggest that inflammatory mediators and immune cells play a unique and diverse role during pregnancy and embryogenesis.
Modulation and Development of Hypothalamic Peptide Neurons by Chemokines
A unique class of inflammatory mediators that have recently been shown to play a role in the HFD effects on brain neurochemistry in offspring are the chemokines. Chemokines are a large class of small chemotactic cytokines that induce chemotactic activity in the immune system and has additional non-immunogenic function in the brain (142, 143). Generally, chemokines have been shown to regulate the development, migration and function of cells in several peripheral organs in addition to the brain, with genetic deletion of specific chemokines, such as C-C motif ligand 2 (CCL2) or C-X-C motif chemokine 12 (CXCL12), to modify embryonic development or in certain cases be lethal (144–146). For example, conditional genetic knockout of CXCL12 or its receptor CXCR4 in organ systems has been shown to result in defective blood vessel development in kidneys (147) and thicken alveolar tissue reducing oxygen exchange in lungs of mice (148). In an early 1990’s study conducted by Plata-Salaman CR, et al, they infused several chemokines into the intracerebroventricular zone in rats, measured feeding outcomes, and found that several chemokines reduced acute food intake, providing the first evidence showing the involvement of chemokines in the neuronal regulation of feeding (149). Since then, several chemokines have been linked to metabolism and brain neurochemicals, and given the nature of chemokines, may also be involved in the neurogenesis effects of a HFD on hypothalamic peptide neurons. The chemokine CXCL12 has been shown to be involved in neurogenesis events by regulating neuronal migration and proliferation in several brain regions (150–153) and controlling neuropositioning of newly-born neurons (154). Knockdown of CXCL12 causes impaired proliferation, migration, and differentiation of neurons (145, 155) suggesting modifications to this system can effect peptide neuron formation during pregnancy.
In adult rat models of HFD intake, the chemokine CXCL12 is increased in circulation and the brain while its receptors, C-X-C chemokine receptor 4 (CXCR4) and C-X-C chemokine receptor 7 (CXCR7), are increased in the hypothalamus and third ventricular injection of CXCL12 into the brain stimulates the expression of ENK in the paraventricular nucleus of the hypothalamus (87). Likewise, prenatal HFD exposure during the window of hypothalamus development significantly elevates maternal circulating levels of CXCL12 and also in offspring stimulates ENK expression in conjunction with CXCL12 and its receptors, CXCR4 and CXCR7, in the paraventricular nucleus of the hypothalamus (Figure 3). Maternal injection of CXCL12 during this same hypothalamic developmental time period in pregnancy to achieve chemokine levels similar to a prenatal HFD paradigm can increase the genesis of ENK neurons in the paraventricular nucleus of the hypothalamus in offspring (156) and produce similar effects in increasing anxiolytic and ingestive behavior in offspring. These studies suggest that one pathway of HFD in its physiological and behavioral effects may be mediated through the CXCL12 chemokine pathway, with this chemokine to directly effect ENK in the paraventricular nucleus of the hypothalamus.
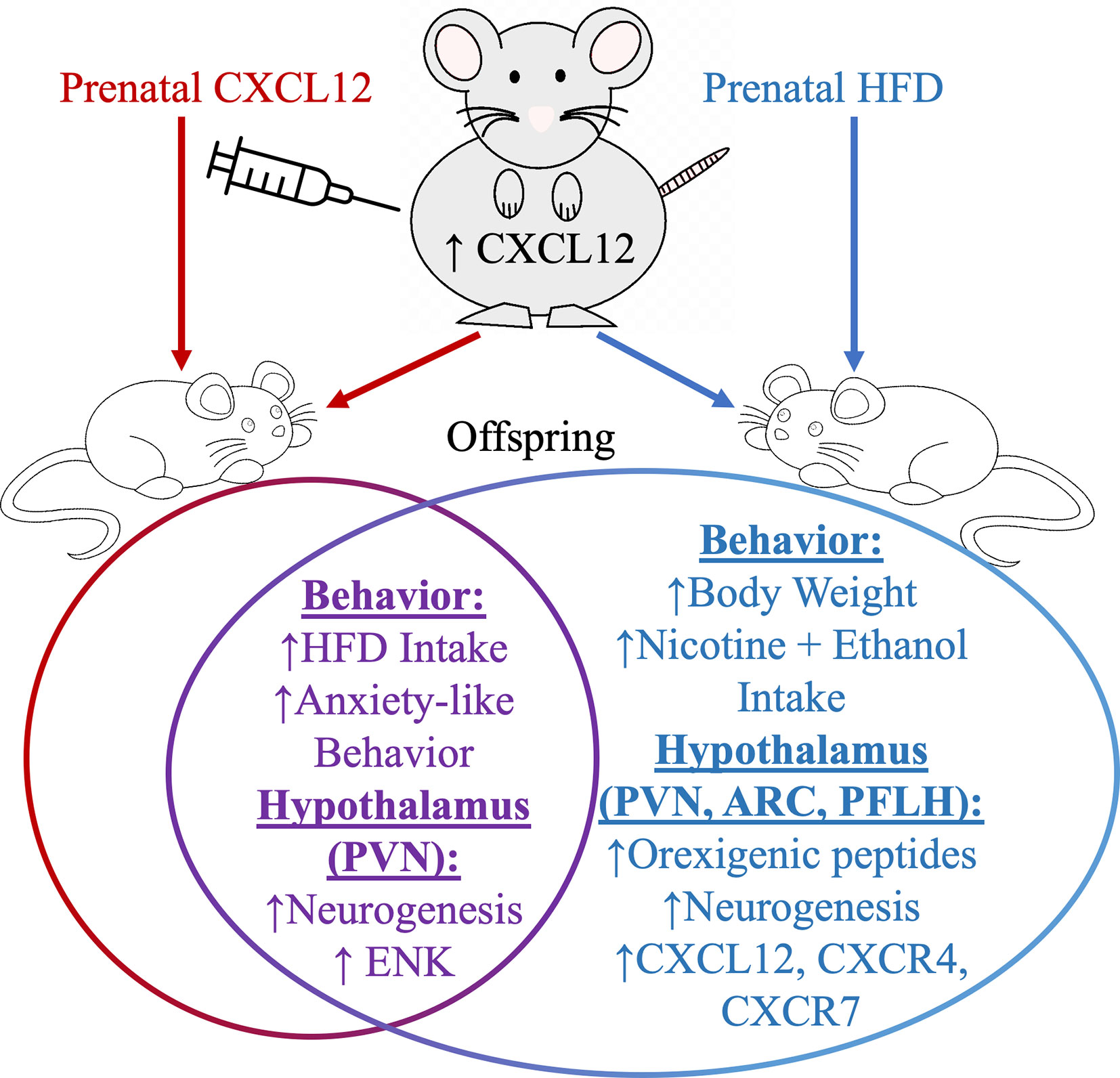
Figure 3 Prenatal high-fat diet ingestion can increase circulating levels of CXCL12 in pregnant rats and also induces in offspring several changes in both behavior and in orexigenic peptide neurons in the arcuate nucleus (ARC), perifornical lateral hypothalamus (PFLH), and in the paraventricular nucleus of the hypothalamus (PVN). Prenatal HFD and prenatal CXCL12 overlap in their effects in offspring on increasing ENK levels in the PVN, and in increasing high-fat diet intake and anxiety-like behaviors.
Another chemokine that is transformed by and may be involved in the physiological and behavioral effects of a HFD is the chemokine, CCL2, also known as monocyte-chemoattractant protein-1 (MCP-1). This chemokine is known to be highly expressed in the obese state and is closely associated with the intake of a HFD (157), with genetic deletion or pharmacological blockade of its receptor, C-C chemokine receptor 2 (CCR2), to reverse or eliminate HFD-induced effect (158). This chemokine has much broader range in the brain due to its diverse co-expression with neuropeptides in several brain areas, including the hypothalamus and thalamus (159–161), and in developmental studies with other substances of abuse reveal close association with migratory radial glial cells (161). If the CCR2 receptor is centrally blocked by an antagonist, weight-loss induction by triggering foreign molecule inflammation through lipopolysaccharide injection is blocked (162), suggesting reduced brain activity of the CCR2 receptor may contribute to increased dietary fat intake and subsequent weight gain. In examining hypothalamic neurons extracted from embryos exposed to low-fat diet conditions, stimulation of hypothalamic neurons by CCL2 increases migration of hypothalamic neurons and the expression of the orexigenic peptides, enkephalin and galanin (159). In contrast, prenatal HFD exposure inhibited CCL2’s ability to produce these same effects (163), suggesting this diet severely disrupts the normal functioning of the CCL2/CCR2 signaling pathway in offspring brain to reduce over-ingestion of dietary fat. Other chemokines that have been implicated in the neuronal effects of a HFD include C-X3-C motif ligand 1 (CX3CL1) and C-C motif ligand 5 (CCL5) and their receptors (164, 165). The CX3CL1 is one of the first chemokines to be expressed during early hypothalamic inflammation (164), has been shown to be a marker for developing metabolic syndrome (166), its presence associated with increased propensity to develop obesity and increased recruitment of white blood cells into adipose tissue (167), and found to be highly expressed in the hypothalamus in HFD-induced obesity in mice (164). Reduction of CX3CL1 expression in the hypothalamus of mice provide mild reduction in brain inflammation and glucose tolerance, but not adiposity measures and body mass, suggesting this chemokine has a specific role in controlling HFD-induced obesity-related glucose metabolism (164). The chemokine CCL5 has been found to be co-expressed in the hypothalamus with insulin receptors and found to also be involved in insulin resistance derived from HFD-induced obesity (168). Intracerebroventricular injection of CCL5 stimulated the mRNA expression of orexin and melanin-concentrating hormone in the hypothalamus (169), suggesting this chemokine is another target of HFD effects that explicitly functions to activate these two fat-associated peptide neurons. A recent study by Fioravante et al, revealed that a chemokine decoy receptor, Ackr2, to be involved in the physiological and neurological effects of a HFD. This receptor is found in the hypothalamus and co-expressed in NPY and POMC neurons. After the intake of an acute HFD regimen, mice that were obesity-prone compared to obesity-resistant had much lower levels of the Akr2 receptor in the hypothalamus (170), suggesting that the increased presence of this decoy chemokine receptor reduces chemokine inflammation that occurs with HFD intake. With over 50 chemokines to date and a few studies showing individual chemokines to be involved in specific aspects of HFD-intake and obesity, future studies are needed to further clarify the role of individual chemokines in mediating HFD effects on brain development and ingestive behavior.
Future Directions
While the extensive research in the field of ingestive behavior has uncovered a large body of knowledge that details how a HFD causes changes in the developing body and the brain, many questions still need to be addressed. While research has primarily focused on the maternal effects of HFD ingestion due to the larger effect on offspring, evidence shows that the paternal side can additively contribute to offspring phenotype and singularly effect female offspring more than male offspring. This suggests that future studies should focus on male contributions in the transmission of HFD-induced obesity proneness to offspring. There is a lack of brain studies in particular and examining the paternal effects on offspring brain neurochemistry and should be explored. While the extensive inflammatory effects induced by a HFD on the entire body is also well-documented, there are many players involved and, particularly with chemokines, shown to have specific function in mediating certain aspects of a HFD on development, cellular and neuronal profiles, and on behavior. It is therefore imperative to continue individualized studies on each inflammatory mediator to explore the function and role that each chemokine may play during HFD intake, particularly during the developmental period. Lastly, the penetration of maternal immune cells, inflammatory mediators, and fatty acids into the embryos during development is not known. Exploration of this topic could elucidate whether it is maternal HFD itself or maternal inflammation that is directly impacting developmental outcomes.
Conclusions
Ingestion of a HFD and prenatal exposure to this diet has similar effects on activating inflammatory pathways in adult, embryonic, and postnatal animal models. The system-wide effects of dietary fat and the abundance of immune factors and ubiquitous expression of inflammatory mediators in peripheral organ systems and in the cells of the central nervous system suggest several regulatory pathways are involved in distinct aspects of ingestive behavior. While a few studies show positive results with the use of anti-inflammatory compounds to prevent prenatal changes in offspring, research on the long-term ramifications in offspring of non-selectively decreasing inflammation during pregnancy still needs to be performed. Similar to the individual chemokine studies such as with CXCL12, future studies focusing on relating each inflammatory mediator with the specific neuronal outcome and behavior may reveal the degree and the type of anti-inflammatory supplements to safely ingest during pregnancy as a preventative measure.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Dr. Manya Mascareno for reviewing the manuscript.
References
1. Hales CM, Carroll MD, Fryar CD, Ogden CL. “Prevalence of Obesity Among Adults and Youth: United States, 2015-2016”. In: NCHS Data Brief. Hyattsville MD: National Center for Health Statistics (2017). p. 1–8.
2. Engin A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv Exp Med Biol (2017) 960:1–17. doi: 10.1007/978-3-319-48382-5_1
3. Oscai LB. Dietary-induced severe obesity: a rat model. Am J Physiol (1982) 242:R212–5. doi: 10.1152/ajpregu.1982.242.3.R212
4. Dooley C, Ryan AS. Role of Dietary Macronutrients and Fatty Acids in Obesity and Metabolic Risk in Older Adults. Int J Obes Nutr Sci (2019) 1:6–10. doi: 10.18689/ijons-1000102
5. Torres-Castillo N, Silva-Gomez JA, Campos-Perez W, Barron-Cabrera E, Hernandez-Canaveral I, Garcia-Cazarin M, et al. High Dietary omega-6:omega-3 PUFA Ratio Is Positively Associated with Excessive Adiposity and Waist Circumference. Obes Facts (2018) 11:344–53. doi: 10.1159/000492116
6. Snell-Bergeon JK, Chartier-Logan C, Maahs DM, Ogden LG, Hokanson JE, Kinney GL, et al. Adults with type 1 diabetes eat a high-fat atherogenic diet that is associated with coronary artery calcium. Diabetologia (2009) 52:801–9. doi: 10.1007/s00125-009-1280-4
7. Matias I, Dias S, Carvalho T. Modulating the Metabolic Phenotype of Cancer Microenvironment. Adv Exp Med Biol (2020) 1219:403–11. doi: 10.1007/978-3-030-34025-4_21
8. Paniagua JA, Perez-Martinez P, Gjelstad IM, Tierney AC, Delgado-Lista J, Defoort C, et al. A low-fat high-carbohydrate diet supplemented with long-chain n-3 PUFA reduces the risk of the metabolic syndrome. Atherosclerosis (2011) 218:443–50. doi: 10.1016/j.atherosclerosis.2011.07.003
9. Jena PK, Sheng L, Di Lucente J, Jin LW, Maezawa I, Wan YY. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J (2018) 32:2866–77. doi: 10.1096/fj.201700984RR
10. Holloway CJ, Cochlin LE, Emmanuel Y, Murray A, Codreanu I, Edwards LM, et al. A high-fat diet impairs cardiac high-energy phosphate metabolism and cognitive function in healthy human subjects. Am J Clin Nutr (2011) 93:748–55. doi: 10.3945/ajcn.110.002758
11. Zhukova NV, Novgorodtseva TP, Denisenko YK. Effect of the prolonged high-fat diet on the fatty acid metabolism in rat blood and liver. Lipids Health Dis (2014) 13:49. doi: 10.1186/1476-511X-13-49
12. Wali JA, Jarzebska N, Raubenheimer D, Simpson SJ, Rodionov RN, O’Sullivan JF. Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms-A Narrative Review. Nutrients (2020) 12:1505. doi: 10.3390/nu12051505
13. Tamashiro KL. Developmental and environmental influences on physiology and behavior–2014 Alan N. Epstein Research Award. Physiol Behav (2015) 152:508–15. doi: 10.1016/j.physbeh.2015.08.019
14. Ryan KK, Woods SC, Seeley RJ. Central nervous system mechanisms linking the consumption of palatable high-fat diets to the defense of greater adiposity. Cell Metab (2012) 15:137–49. doi: 10.1016/j.cmet.2011.12.013
15. Sarker G, Berrens R, von Arx J, Pelczar P, Reik W, Wolfrum C, et al. Transgenerational transmission of hedonic behaviors and metabolic phenotypes induced by maternal overnutrition. Transl Psychiatry (2018) 8:195. doi: 10.1038/s41398-018-0243-2
16. Chang E, Hafner H, Varghese M, Griffin C, Clemente J, Islam M, et al. Programming effects of maternal and gestational obesity on offspring metabolism and metabolic inflammation. Sci Rep (2019) 9:16027. doi: 10.1038/s41598-019-52583-x
17. Cooper R, Hypponen E, Berry D, Power C. Associations between parental and offspring adiposity up to midlife: the contribution of adult lifestyle factors in the 1958 British Birth Cohort Study. Am J Clin Nutr (2010) 92:946–53. doi: 10.3945/ajcn.2010.29477
18. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
19. Chen M, Lu B, Li Y, Wang Y, Zheng H, Zhong D, et al. Metabolomics insights into the modulatory effects of long-term compound polysaccharide intake in high-fat diet-induced obese rats. Nutr Metab (Lond) (2018) 15:8. doi: 10.1186/s12986-018-0246-2
20. Barson JR, Karatayev O, Gaysinskaya V, Chang GQ, Leibowitz SF. Effect of dietary fatty acid composition on food intake, triglycerides, and hypothalamic peptides. Regul Peptides (2012) 173:13–20. doi: 10.1016/j.regpep.2011.08.012
21. Bellinger LL, Bernardis LL. The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol Behav (2002) 76:431–42. doi: 10.1016/S0031-9384(02)00756-4
22. Morris MJ, Beilharz JE, Maniam J, Reichelt AC, Westbrook RF. Why is obesity such a problem in the 21st century? The intersection of palatable food, cues and reward pathways, stress, and cognition. Neurosci Biobehav Rev (2015) 58:36–45. doi: 10.1016/j.neubiorev.2014.12.002
23. Barson JR, Morganstern I, Leibowitz SF. Similarities in hypothalamic and mesocorticolimbic circuits regulating the overconsumption of food and alcohol. Physiol Behav (2011) 104:128–37. doi: 10.1016/j.physbeh.2011.04.054
24. Barson JR, Morganstern I, Leibowitz SF. Complementary roles of orexin and melanin-concentrating hormone in feeding behavior. Int J Endocrinol (2013) 2013:983964. doi: 10.1155/2013/983964
25. Myers MG Jr., Olson DP. Central nervous system control of metabolism. Nature (2012) 491:357–63. doi: 10.1038/nature11705
26. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature (2006) 443:289–95. doi: 10.1038/nature05026
27. Beck B, Jhanwar-Uniyal M, Burlet A, Chapleur-Chateau M, Leibowitz SF, Burlet C. Rapid and localized alterations of neuropeptide Y in discrete hypothalamic nuclei with feeding status. Brain Res (1990) 528:245–9. doi: 10.1016/0006-8993(90)91664-3
28. Sanchez J, Cladera MM, Llopis M, Palou A, Pico C. The different satiating capacity of CHO and fats can be mediated by different effects on leptin and ghrelin systems. Behav Brain Res (2010) 213:183–8. doi: 10.1016/j.bbr.2010.04.051
29. Linehan V, Fang LZ, Parsons MP, Hirasawa M. High-fat diet induces time-dependent synaptic plasticity of the lateral hypothalamus. Mol Metab (2020) 36:100977. doi: 10.1016/j.molmet.2020.100977
30. Chang GQ, Karatayev O, Barson JR, Chang SY, Leibowitz SF. Increased enkephalin in brain of rats prone to overconsuming a fat-rich diet. Physiol Behav (2010) 101:360–9. doi: 10.1016/j.physbeh.2010.06.005
31. Chang GQ, Karatayev O, Davydova Z, Leibowitz SF. Circulating triglycerides impact on orexigenic peptides and neuronal activity in hypothalamus. Endocrinology (2004) 145:3904–12. doi: 10.1210/en.2003-1582
32. Kyrkouli SE, Stanley BG, Seirafi RD, Leibowitz SF. Stimulation of feeding by galanin: anatomical localization and behavioral specificity of this peptide’s effects in the brain. Peptides (1990) 11:995–1001. doi: 10.1016/0196-9781(90)90023-X
33. Robert JJ, Orosco M, Rouch C, Jacquot C, Cohen Y. Effects of opiate agonists and an antagonist on food intake and brain neurotransmitters in normophagic and obese “cafeteria” rats. Pharmacol Biochem Behav (1989) 34:577–83. doi: 10.1016/0091-3057(89)90561-3
34. Deng PY, Xiao Z, Jha A, Ramonet D, Matsui T, Leitges M, et al. Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability. J Neurosci (2010) 30:5136–48. doi: 10.1523/JNEUROSCI.5711-09.2010
35. Serrenho D, Santos SD, Carvalho AL. The Role of Ghrelin in Regulating Synaptic Function and Plasticity of Feeding-Associated Circuits. Front Cell Neurosci (2019) 13:205. doi: 10.3389/fncel.2019.00205
36. Stanley BG, Ha LH, Spears LC, Dee MG. 2nd, Lateral hypothalamic injections of glutamate, kainic acid, D,L-alpha-amino-3-hydroxy-5-methyl-isoxazole propionic acid or N-methyl-D-aspartic acid rapidly elicit intense transient eating in rats. Brain Res (1993) 613:88–95. doi: 10.1016/0006-8993(93)90458-Y
37. Stanley BG, Urstadt KR, Charles JR, Kee T. Glutamate and GABA in lateral hypothalamic mechanisms controlling food intake. Physiol Behav (2011) 104:40–6. doi: 10.1016/j.physbeh.2011.04.046
38. Zheng H, Liu X, Li Y, Patel KP. A Hypothalamic Leptin-Glutamate Interaction in the Regulation of Sympathetic Nerve Activity. Neural Plast (2017) 2017:2361675. doi: 10.1155/2017/2361675
39. Shah BP, Vong L, Olson DP, Koda S, Krashes MJ, Ye C, et al. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proc Natl Acad Sci U S A (2014) 111:13193–8. doi: 10.1073/pnas.1407843111
40. Meister B. Neurotransmitters in key neurons of the hypothalamus that regulate feeding behavior and body weight. Physiol Behav (2007) 92:263–71. doi: 10.1016/j.physbeh.2007.05.021
41. Alpar A, Harkany T. Orexin neurons use endocannabinoids to break obesity-induced inhibition. Proc Natl Acad Sci U S A (2013) 110:9625–6. doi: 10.1073/pnas.1307389110
42. Linehan V, Fang LZ, Hirasawa M. Short-term high-fat diet primes excitatory synapses for long-term depression in orexin neurons. J Physiol (2018) 596:305–16. doi: 10.1113/JP275177
43. Glendining KA, Fisher LC, Jasoni CL. Maternal Obesity Modulates Expression of Satb2 in Hypothalamic VMN of Female Offspring. Life (Basel) (2020) 10(4):48. doi: 10.3390/life10040048
44. Delgado TC. Glutamate and GABA in Appetite Regulation. Front Endocrinol (Lausanne) (2013) 4:103. doi: 10.3389/fendo.2013.00103
45. McLean FH, Campbell FM, Langston RF, Sergi D, Resch C, Grant C, et al. A high-fat diet induces rapid changes in the mouse hypothalamic proteome. Nutr Metab (Lond) (2019) 16:26. doi: 10.1186/s12986-019-0352-9
46. Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci (2008) 28:12107–19. doi: 10.1523/JNEUROSCI.2642-08.2008
47. Bocarsly ME, Barson JR, Hauca JM, Hoebel BG, Leibowitz SF, Avena NM. Effects of perinatal exposure to palatable diets on body weight and sensitivity to drugs of abuse in rats. Physiol Behav (2012) 107:568–75. doi: 10.1016/j.physbeh.2012.04.024
48. Keleher MR, Zaidi R, Shah S, Oakley ME, Pavlatos C, El Idrissi S, et al. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS One (2018) 13:e0192606. doi: 10.1371/journal.pone.0192606
49. Morganstern I, Lukatskaya O, Moon SH, Guo WR, Shaji J, Karatayev O, et al. Stimulation of nicotine reward and central cholinergic activity in Sprague-Dawley rats exposed perinatally to a fat-rich diet. Psychopharmacol (Berl) (2013) 230:509–24. doi: 10.1007/s00213-013-3178-6
50. Karatayev O, Baylan J, Leibowitz SF. Increased intake of ethanol and dietary fat in galanin overexpressing mice. Alcohol (2009) 43:571–80. doi: 10.1016/j.alcohol.2009.09.025
51. Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, et al. Trends in the prevalence of developmental disabilities in US children, 1997-2008. Pediatrics (2011) 127:1034–42. doi: 10.1542/peds.2010-2989
52. Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcin C, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res (2012) 5:160–79. doi: 10.1002/aur.239
53. Schaefer CA, Brown AS, Wyatt RJ, Kline J, Begg MD, Bresnahan MA, et al. Maternal prepregnant body mass and risk of schizophrenia in adult offspring. Schizophr Bull (2000) 26:275–86. doi: 10.1093/oxfordjournals.schbul.a033452
54. Louwagie EJ, Larsen TD, Wachal AL, Baack ML. Placental lipid processing in response to a maternal high-fat diet and diabetes in rats. Pediatr Res (2018) 83:712–22. doi: 10.1038/pr.2017.288
55. Snajder D, Peric Kacarevic Z, Grgic A, Bijelic N, Fenrich M, Belovari T, et al. Effect of different combination of maternal and postnatal diet on adipose tissue morphology in male rat offspring. J Matern Fetal Neonatal Med (2019) 32:1838–46. doi: 10.1080/14767058.2017.1419181
56. Umekawa T, Sugiyama T, Du Q, Murabayashi N, Zhang L, Kamimoto Y, et al. A maternal mouse diet with moderately high-fat levels does not lead to maternal obesity but causes mesenteric adipose tissue dysfunction in male offspring. J Nutr Biochem (2015) 26:259–66. doi: 10.1016/j.jnutbio.2014.10.012
57. Consitt LA, Saxena G, Slyvka Y, Clark BC, Friedlander M, Zhang Y, et al. Paternal high-fat diet enhances offspring whole-body insulin sensitivity and skeletal muscle insulin signaling early in life. Physiol Rep (2018) 6(5):e13583. doi: 10.14814/phy2.13583
58. Fullston T, Ohlsson Teague EM, Palmer NO, DeBlasio MJ, Mitchell M, Corbett M, et al. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J (2013) 27:4226–43. doi: 10.1096/fj.12-224048
59. Morita S, Horii T, Kimura M, Arai Y, Kamei Y, Ogawa Y, et al. Paternal allele influences high fat diet-induced obesitye85477. PLoS One (2014) 9(1):e85477. doi: 10.1371/journal.pone.0085477
60. Desai M, Jellyman JK, Han G, Beall M, Lane RH, Ross MG. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am J Obstet Gynecol (2014) 211:237 e1–237 e13. doi: 10.1016/j.ajog.2014.03.025
61. Poon K, Barson JR, Fagan SE, Leibowitz SF. Developmental changes in embryonic hypothalamic neurons during prenatal fat exposure. Am J Physiol Endocrinol Metab (2012) 303:E432–41. doi: 10.1152/ajpendo.00238.2012
62. Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology (2012) 153:2823–30. doi: 10.1210/en.2011-2161
63. Rouschop SH, Karl T, Risch A, van Ewijk PA, Schrauwen-Hinderling VB, Opperhuizen A, et al. Gene expression and DNA methylation as mechanisms of disturbed metabolism in offspring after exposure to a prenatal HF diet. J Lipid Res (2019) 60:1250–9. doi: 10.1194/jlr.M092593
64. Stump M, Guo DF, Lu KT, Mukohda M, Cassell MD, Norris AW, et al. Nervous System Expression of PPARgamma and Mutant PPARgamma Has Profound Effects on Metabolic Regulation and Brain Development. Endocrinology (2016) 157:4266–75. doi: 10.1210/en.2016-1524
65. Poon K, Mandava S, Chen K, Barson JR, Buschlen S, Leibowitz SF. Prenatal exposure to dietary fat induces changes in the transcriptional factors, TEF and YAP, which may stimulate differentiation of peptide neurons in rat hypothalamus. PLoS One (2013) 8:e77668. doi: 10.1371/journal.pone.0077668
66. Poon K, Alam M, Karatayev O, Barson JR, Leibowitz SF. Regulation of the orexigenic neuropeptide, enkephalin, by PPARdelta and fatty acids in neurons of the hypothalamus and forebrain. J Neurochem (2015) 135:918–31. doi: 10.1111/jnc.13298
67. Kao YC, Wei WY, Tsai KJ, Wang LC. High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra. Int J Mol Sci (2019) 21(1):207. doi: 10.3390/ijms21010207
68. Chang GQ, Karatayev O, Lukatskaya O, Leibowitz SF. Prenatal fat exposure and hypothalamic PPAR beta/delta: Possible relationship to increased neurogenesis of orexigenic peptide neurons. Peptides (2016) 79:16–26. doi: 10.1016/j.peptides.2016.03.007
69. Liu R, Jagannathan R, Li F, Lee J, Balasubramanyam N, Kim BS, et al. Tead1 is required for perinatal cardiomyocyte proliferation. PLoS One (2019) 14:e0212017. doi: 10.1371/journal.pone.0212017
70. Mukhtar T, Breda J, Grison A, Karimaddini Z, Grobecker P, Iber D, et al. Tead transcription factors differentially regulate cortical development. Sci Rep (2020) 10:4625. doi: 10.1038/s41598-020-61490-5
71. Chen X, Li Y, Luo J, Hou N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front Physiol (2020) 11:389. doi: 10.3389/fphys.2020.00389
72. Rojek KO, Krzemien J, Dolezyczek H, Boguszewski PM, Kaczmarek L, Konopka W, et al. Amot and Yap1 regulate neuronal dendritic tree complexity and locomotor coordination in mice. PLoS Biol (2019) 17:e3000253. doi: 10.1371/journal.pbio.3000253
73. Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev (2008) 22:3320–34. doi: 10.1101/gad.1726608
74. Zhang H, Deo M, Thompson RC, Uhler MD, Turner DL. Negative regulation of Yap during neuronal differentiation. Dev Biol (2012) 361:103–15. doi: 10.1016/j.ydbio.2011.10.017
75. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev (2010) 24:1106–18. doi: 10.1101/gad.1903310
76. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta (2011) 1812:1007–22. doi: 10.1016/j.bbadis.2011.02.014
77. Waise TMZ, Toshinai K, Naznin F, NamKoong C, Md Moin AS, Sakoda H, et al. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem Biophys Res Commun (2015) 464:1157–62. doi: 10.1016/j.bbrc.2015.07.097
78. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol (2008) 8:923–34. doi: 10.1038/nri2449
79. de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol (2010) 299:G440–8. doi: 10.1152/ajpgi.00098.2010
80. Lionetti L, Mollica MP, Sica R, Donizzetti I, Gifuni G, Pignalosa A, et al. Differential effects of high-fish oil and high-lard diets on cells and cytokines involved in the inflammatory process in rat insulin-sensitive tissues. Int J Mol Sci (2014) 15:3040–63. doi: 10.3390/ijms15023040
81. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi: 10.1172/JCI200319246
82. Takanabe-Mori R, Ono K, Sowa N, Wada H, Takaya T, Horie T, et al. Lectin-like oxidized low-density lipoprotein receptor-1 is required for the adipose tissue expression of proinflammatory cytokines in high-fat diet-induced obese mice. Biochem Biophys Res Commun (2010) 398:576–80. doi: 10.1016/j.bbrc.2010.06.123
83. Barbarroja N, Lopez-Pedrera R, Mayas MD, Garcia-Fuentes E, Garrido-Sanchez L, Macias-Gonzalez M, et al. The obese healthy paradox: is inflammation the answer? Biochem J (2010) 430:141–9. doi: 10.1042/BJ20100285
84. Galland L. Diet and inflammation. Nutr Clin Pract (2010) 25:634–40. doi: 10.1177/0884533610385703
85. Fernandez-Real JM, Broch M, Vendrell J, Ricart W. Insulin resistance, inflammation, and serum fatty acid composition. Diabetes Care (2003) 26:1362–8. doi: 10.2337/diacare.26.5.1362
86. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids (2018) 132:41–8. doi: 10.1016/j.plefa.2018.03.004
87. Poon K, Barson JR, Ho HT, Leibowitz SF. Relationship of the Chemokine, CXCL12, to Effects of Dietary Fat on Feeding-Related Behaviors and Hypothalamic Neuropeptide Systems. Front Behav Neurosci (2016) 10:51. doi: 10.3389/fnbeh.2016.00051
88. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci (2009) 29:359–70. doi: 10.1523/JNEUROSCI.2760-08.2009
89. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146:4192–9. doi: 10.1210/en.2004-1520
90. Deng SL, Chen JG, Wang F. Microglia: A Central Player in Depression. Curr Med Sci (2020) 40:391–400. doi: 10.1007/s11596-020-2193-1
91. Wittenberg GM, Greene J, Vertes PE, Drevets WC, Bullmore ET. Major Depressive Disorder Is Associated With Differential Expression of Innate Immune and Neutrophil-Related Gene Networks in Peripheral Blood: A Quantitative Review of Whole-Genome Transcriptional Data From Case-Control Studies. Biol Psychiatry (2020) 88(8):625–37. doi: 10.1016/j.biopsych.2020.05.006
92. Almeida-Suhett CP, Graham A, Chen Y, Deuster P. Behavioral changes in male mice fed a high-fat diet are associated with IL-1beta expression in specific brain regions. Physiol Behav (2017) 169:130–40. doi: 10.1016/j.physbeh.2016.11.016
93. Wu H, Liu Q, Kalavagunta PK, Huang Q, Lv W, An X, et al. Normal diet Vs High fat diet - A comparative study: Behavioral and neuroimmunological changes in adolescent male mice. Metab Brain Dis (2018) 33:177–90. doi: 10.1007/s11011-017-0140-z
94. Yi CX, Tschop MH, Woods SC, Hofmann SM. High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis Model Mech (2012) 5:686–90. doi: 10.1242/dmm.009464
95. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest (2012) 122:153–62. doi: 10.1172/JCI59660
96. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
97. Engstrom L, Engblom D, Blomqvist A. Systemic immune challenge induces preproenkephalin gene transcription in distinct autonomic structures of the rat brain. J Comp Neurol (2003) 462:450–61. doi: 10.1002/cne.10770
98. Van Koughnet K, Smirnova O, Hyman SE, Borsook D. Proenkephalin transgene regulation in the paraventricular nucleus of the hypothalamus by lipopolysaccharide and interleukin-1beta. J Comp Neurol (1999) 405:199–215. doi: 10.1002/(SICI)1096-9861(19990308)405:2<199::AID-CNE5>3.0.CO;2-W
99. Senaris RM, Trujillo ML, Navia B, Comes G, Ferrer B, Giralt M, et al. Interleukin-6 regulates the expression of hypothalamic neuropeptides involved in body weight in a gender-dependent way. J Neuroendocrinol (2011) 23:675–86. doi: 10.1111/j.1365-2826.2011.02158.x
100. Schele E, Fekete C, Egri P, Fuzesi T, Palkovits M, Keller E, et al. Interleukin-6 receptor alpha is co-localised with melanin-concentrating hormone in human and mouse hypothalamus. J Neuroendocrinol (2012) 24:930–43. doi: 10.1111/j.1365-2826.2012.02286.x
101. Oh IS, Thaler JP, Ogimoto K, Wisse BE, Morton GJ, Schwartz MW. Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. Am J Physiol Endocrinol Metab (2010) 299:E47–53. doi: 10.1152/ajpendo.00026.2010
102. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab (2009) 296:E1003–12. doi: 10.1152/ajpendo.90377.2008
103. Buckman LB, Hasty AH, Flaherty DK, Buckman CT, Thompson MM, Matlock BK, et al. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun (2014) 35:33–42. doi: 10.1016/j.bbi.2013.06.007
104. Baufeld C, Osterloh A, Prokop S, Miller KR, Heppner FL. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol (2016) 132:361–75. doi: 10.1007/s00401-016-1595-4
105. Douglass JD, Dorfman MD, Thaler JP. Glia: silent partners in energy homeostasis and obesity pathogenesis. Diabetologia (2017) 60:226–36. doi: 10.1007/s00125-016-4181-3
106. Lee CH, Suk K, Yu R, Kim MS. Cellular Contributors to Hypothalamic Inflammation in Obesity. Mol Cells (2020) 43:431–7. doi: 10.1438/molcells.2020.0055
107. Chen J, He X, Huang J. Diet effects in gut microbiome and obesity. J Food Sci (2014) 79:R442–51. doi: 10.1111/1750-3841.12397
108. Shen J, Obin MS, Zhao L. The gut microbiota, obesity and insulin resistance. Mol Aspects Med (2013) 34:39–58. doi: 10.1016/j.mam.2012.11.001
109. Hassan AM, Mancano G, Kashofer K, Liebisch G, Farzi A, Zenz G, et al. Anhedonia induced by high-fat diet in mice depends on gut microbiota and leptin. Nutr Neurosci (2020), 1–14. doi: 10.1080/1028415X.2020.1751508
110. Agusti A, Moya-Perez A, Campillo I, Montserrat-de la Paz S, Cerrudo V, Perez-Villalba A, et al. Bifidobacterium pseudocatenulatum CECT 7765 Ameliorates Neuroendocrine Alterations Associated with an Exaggerated Stress Response and Anhedonia in Obese Mice. Mol Neurobiol (2018) 55:5337–52. doi: 10.1007/s12035-017-0768-z
111. Bleau C, Karelis AD, St-Pierre DH, Lamontagne L. Crosstalk between intestinal microbiota, adipose tissue and skeletal muscle as an early event in systemic low-grade inflammation and the development of obesity and diabetes. Diabetes Metab Res Rev (2015) 31:545–61. doi: 10.1002/dmrr.2617
112. Gaillard R, Rifas-Shiman SL, Perng W, Oken E, Gillman MW. Maternal inflammation during pregnancy and childhood adiposity. Obes (Silver Spring) (2016) 24:1320–7. doi: 10.1002/oby.21484
113. Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab (2007) 92:969–75. doi: 10.1210/jc.2006-2083
114. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature (2018) 563:347–53. doi: 10.1038/s41586-018-0698-6
115. Racicot K, Kwon JY, Aldo P, Silasi M, Mor G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol (2014) 72:107–16. doi: 10.1111/aji.12289
116. Aghaeepour N, Ganio EA, McIlwain D, Tsai AS, Tingle M, Van Gassen S, et al. An immune clock of human pregnancy. Sci Immunol (2017) 2(15):eaan2946. doi: 10.1126/sciimmunol.aan2946
117. Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta (2008) 29:274–81. doi: 10.1016/j.placenta.2007.12.010
118. Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, et al. Placental structure and inflammation in pregnancies associated with obesity. Placenta (2011) 32:247–54. doi: 10.1016/j.placenta.2010.12.023
119. Gohir W, Kennedy KM, Wallace JG, Saoi M, Bellissimo CJ, Britz-McKibbin P, et al. High-fat diet intake modulates maternal intestinal adaptations to pregnancy and results in placental hypoxia, as well as altered fetal gut barrier proteins and immune markers. J Physiol (2019) 597:3029–51. doi: 10.1113/JP277353
120. Zheng J, Zhang Q, Mul JD, Yu M, Xu J, Qi C, et al. Maternal high-calorie diet is associated with altered hepatic microRNA expression and impaired metabolic health in offspring at weaning age. Endocrine (2016) 54:70–80. doi: 10.1007/s12020-016-0959-9
121. Griffiths PS, Walton C, Samsell L, Perez MK, Piedimonte G. Maternal high-fat hypercaloric diet during pregnancy results in persistent metabolic and respiratory abnormalities in offspring. Pediatr Res (2016) 79:278–86. doi: 10.1038/pr.2015.226
122. Poret JM, Souza-Smith F, Marcell SJ, Gaudet DA, Tzeng TH, Braymer HD, et al. High fat diet consumption differentially affects adipose tissue inflammation and adipocyte size in obesity-prone and obesity-resistant rats. Int J Obes (Lond) (2018) 42:535–41. doi: 10.1038/ijo.2017.280
123. Yang XF, Qiu YQ, Wang L, Gao KG, Jiang ZY. A high-fat diet increases body fat mass and up-regulates expression of genes related to adipogenesis and inflammation in a genetically lean pig. J Zhejiang Univ Sci B (2018) 19:884–94. doi: 10.1631/jzus.B1700507
124. Zacarias MF, Collado MC, Gomez-Gallego C, Flinck H, Aittoniemi J, Isolauri E, et al. Pregestational overweight and obesity are associated with differences in gut microbiota composition and systemic inflammation in the third trimester. PLoS One (2018) 13:e0200305. doi: 10.1371/journal.pone.0200305
125. Rohr MW, Narasimhulu CA, Rudeski-Rohr TA, Parthasarathy S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv Nutr (2020) 11:77–91. doi: 10.1093/advances/nmz061
126. Guo Y, Wang Z, Chen L, Tang L, Wen S, Liu Y, et al. Diet induced maternal obesity affects offspring gut microbiota and persists into young adulthood. Food Funct (2018) 9:4317–27. doi: 10.1039/C8FO00444G
127. Zhou L, Xiao X. The role of gut microbiota in the effects of maternal obesity during pregnancy on offspring metabolism. Biosci Rep (2018) 38(2):BSR20171234. doi: 10.1042/BSR20171234
128. Soderborg TK, Clark SE, Mulligan CE, Janssen RC, Babcock L, Ir D, et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat Commun (2018) 9:4462. doi: 10.1038/s41467-018-06929-0
129. Pimentel GD, Lira FS, Rosa JC, Oliveira JL, Losinskas-Hachul AC, Souza GI, et al. Intake of trans fatty acids during gestation and lactation leads to hypothalamic inflammation via TLR4/NFkappaBp65 signaling in adult offspring. J Nutr Biochem (2012) 23:265–71. doi: 10.1016/j.jnutbio.2010.12.003
130. Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology (2010) 151:1622–32. doi: 10.1210/en.2009-1019
131. Kim DW, Glendining KA, Grattan DR, Jasoni CL. Maternal Obesity in the Mouse Compromises the Blood-Brain Barrier in the Arcuate Nucleus of Offspring. Endocrinology (2016) 157:2229–42. doi: 10.1210/en.2016-1014
132. Kim DW, Glendining KA, Grattan DR, Jasoni CL. Maternal obesity leads to increased proliferation and numbers of astrocytes in the developing fetal and neonatal mouse hypothalamus. Int J Dev Neurosci (2016) 53:18–25. doi: 10.1016/j.ijdevneu.2016.06.005
133. Teo JD, Morris MJ, Jones NM. Maternal obesity increases inflammation and exacerbates damage following neonatal hypoxic-ischaemic brain injury in rats. Brain Behav Immun (2017) 63:186–96. doi: 10.1016/j.bbi.2016.10.010
134. Stofkova A, Skurlova M, Kiss A, Zelezna B, Zorad S, Jurcovicova J. Activation of hypothalamic NPY, AgRP, MC4R, AND IL-6 mRNA levels in young Lewis rats with early-life diet-induced obesity. Endocr Regul (2009) 43:99–106.
135. White CL, Purpera MN, Morrison CD. Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am J Physiol Regul Integr Comp Physiol (2009) 296:R1464–72. doi: 10.1152/ajpregu.91015.2008
136. Bilbo SD, Tsang V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J (2010) 24:2104–15. doi: 10.1096/fj.09-144014
137. Duan Y, Zeng L, Zheng C, Song B, Li F, Kong X, et al. Inflammatory Links Between High Fat Diets and Diseases. Front Immunol (2018) 9:2649. doi: 10.3389/fimmu.2018.02649
138. Bolton JL, Bilbo SD. Developmental programming of brain and behavior by perinatal diet: focus on inflammatory mechanisms. Dialog Clin Neurosci (2014) 16:307–20. doi: 10.31887/DCNS.2014.16.3/jbolton
139. Dudele A, Hougaard KS, Kjolby M, Hokland M, Winther G, Elfving B, et al. Chronic maternal inflammation or high-fat-feeding programs offspring obesity in a sex-dependent manner. Int J Obes (Lond) (2017) 41:1420–6. doi: 10.1038/ijo.2017.136
140. Hsu MH, Chen YC, Sheen JM, Huang LT. Maternal Obesity Programs Offspring Development and Resveratrol Potentially Reprograms the Effects of Maternal Obesity. Int J Environ Res Public Health (2020) 17(5). doi: 10.3390/ijerph17051610
141. Tsai TA, Tsai CK, Huang LT, Sheen JM, Tiao MM, Tain YL, et al. Maternal Resveratrol Treatment Re-Programs and Maternal High-Fat Diet-Induced Retroperitoneal Adiposity in Male Offspring. Int J Environ Res Public Health (2020) 19(1):174. doi: 10.3390/ijerph17082780
142. Melik-Parsadaniantz S, Rostene W. Chemokines and neuromodulation. J Neuroimmunol (2008) 198:62–8. doi: 10.1016/j.jneuroim.2008.04.022
143. Guyon A, Massa F, Rovere C, Nahon JL. How cytokines can influence the brain: a role for chemokines? J Neuroimmunol (2008) 198:46–55. doi: 10.1016/j.jneuroim.2008.04.009
144. Dawson J, Miltz W, Mir AK, Wiessner C. Targeting monocyte chemoattractant protein-1 signalling in disease. Expert Opin Ther Targets (2003) 7:35–48. doi: 10.1517/14728222.7.1.35
145. Trousse F, Jemli A, Silhol M, Garrido E, Crouzier L, Naert G, et al. Knockdown of the CXCL12/CXCR7 chemokine pathway results in learning deficits and neural progenitor maturation impairment in mice. Brain Behav Immun (2019) 80:697–710. doi: 10.1016/j.bbi.2019.05.019
146. Hillmer RE, Boisvert JP, Cucciare MJ, Dwinell MB, Joksimovic M. Generation and characterization of mice harboring a conditional CXCL12 allele. Int J Dev Biol (2015) 59:205–9. doi: 10.1387/ijdb.140348mj
147. Takabatake Y, Sugiyama T, Kohara H, Matsusaka T, Kurihara H, Koni PA, et al. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol (2009) 20:1714–23. doi: 10.1681/ASN.2008060640
148. Chen WC, Tzeng YS, Li H, Tien WS, Tsai YC. Lung defects in neonatal and adult stromal-derived factor-1 conditional knockout mice. Cell Tissue Res (2010) 342:75–85. doi: 10.1007/s00441-010-1035-z
149. Plata-Salaman CR, Borkoski JP. Chemokines/intercrines and central regulation of feeding. Am J Physiol (1994) 266:R1711–5. doi: 10.1152/ajpregu.1994.266.5.R1711
150. Sanchez-Alcaniz JA, Haege S, Mueller W, Pla R, Mackay F, Schulz S, et al. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron (2011) 69:77–90. doi: 10.1016/j.neuron.2010.12.006
151. Wang Y, Li G, Stanco A, Long JE, Crawford D, Potter GB, et al. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron (2011) 69:61–76. doi: 10.1016/j.neuron.2010.12.005
152. Bodea GO, Spille JH, Abe P, Andersson AS, Acker-Palmer A, Stumm R, et al. Reelin and CXCL12 regulate distinct migratory behaviors during the development of the dopaminergic system. Development (2014) 141:661–73. doi: 10.1242/dev.099937
153. Chen Q, Zhang M, Li Y, Xu D, Wang Y, Song A, et al. CXCR7 Mediates Neural Progenitor Cells Migration to CXCL12 Independent of CXCR4. Stem Cells (2015) 33:2574–85. doi: 10.1002/stem.2022
154. Abe P, Wust HM, Arnold SJ, van de Pavert SA, Stumm R. CXCL12-mediated feedback from granule neurons regulates generation and positioning of new neurons in the dentate gyrus. Glia (2018) 66:1566–76. doi: 10.1002/glia.23324
155. Wang Y, Xu P, Qiu L, Zhang M, Huang Y, Zheng JC. CXCR7 Participates in CXCL12-mediated Cell Cycle and Proliferation Regulation in Mouse Neural Progenitor Cells. Curr Mol Med (2016) 16:738–46. doi: 10.2174/1566524016666160829153453
156. Poon K, Barson JR, Shi H, Chang GQ, Leibowitz SF. Involvement of the CXCL12 System in the Stimulatory Effects of Prenatal Exposure to High-Fat Diet on Hypothalamic Orexigenic Peptides and Behavior in Offspring. Front Behav Neurosci (2017) 11:91. doi: 10.3389/fnbeh.2017.00091
157. Chen A, Mumick S, Zhang C, Lamb J, Dai H, Weingarth D, et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes Res (2005) 13:1311–20. doi: 10.1038/oby.2005.159
158. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116:115–24. doi: 10.1172/JCI24335
159. Poon K, Ho HT, Barson JR, Leibowitz SF. Stimulatory role of the chemokine CCL2 in the migration and peptide expression of embryonic hypothalamic neurons. J Neurochem (2014) 131:509–20. doi: 10.1111/jnc.12827
160. Banisadr G, Gosselin RD, Mechighel P, Kitabgi P, Rostene W, Parsadaniantz SM. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol (2005) 489:275–92. doi: 10.1002/cne.20598
161. Chang GQ, Karatayev O, Boorgu D, Leibowitz SF. CCL2/CCR2 system in neuroepithelial radial glia progenitor cells: involvement in stimulatory, sexually dimorphic effects of maternal ethanol on embryonic development of hypothalamic peptide neurons. J Neuroinflam (2020) 17:207. doi: 10.1186/s12974-020-01875-5
162. Le Thuc O, Cansell C, Bourourou M, Denis RG, Stobbe K, Devaux N, et al. Central CCL2 signaling onto MCH neurons mediates metabolic and behavioral adaptation to inflammation. EMBO Rep (2016) 17:1738–52. doi: 10.15252/embr.201541499
163. Poon K, Abramova D, Ho HT, Leibowitz S. Prenatal fat-rich diet exposure alters responses of embryonic neurons to the chemokine, CCL2, in the hypothalamus. Neuroscience (2016) 324:407–19. doi: 10.1016/j.neuroscience.2016.03.017
164. Morari J, Anhe GF, Nascimento LF, de Moura RF, Razolli D, Solon C, et al. Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes (2014) 63:3770–84. doi: 10.2337/db13-1495
165. Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab (2017) 26:185–197 e3. doi: 10.1016/j.cmet.2017.05.015
166. Marques P, Collado A, Martinez-Hervas S, Domingo E, Benito E, Piqueras L, et al. Systemic Inflammation in Metabolic Syndrome: Increased Platelet and Leukocyte Activation, and Key Role of CX3CL1/CX3CR1 and CCL2/CCR2 Axes in Arterial Platelet-Proinflammatory Monocyte Adhesion. J Clin Med (2019) 8:708. doi: 10.3390/jcm8050708
167. Polyak A, Ferenczi S, Denes A, Winkler Z, Kriszt R, Pinter-Kubler B, et al. The fractalkine/Cx3CR1 system is implicated in the development of metabolic visceral adipose tissue inflammation in obesity. Brain Behav Immun (2014) 38:25–35. doi: 10.1016/j.bbi.2014.01.010
168. Chou SY, Ajoy R, Changou CA, Hsieh YT, Wang YK, Hoffer B. CCL5/RANTES contributes to hypothalamic insulin signaling for systemic insulin responsiveness through CCR5. Sci Rep (2016) 6:37659. doi: 10.1038/srep37659
169. Le Thuc O, Stobbe K, Cansell C, Nahon JL, Blondeau N, Rovere C. Hypothalamic Inflammation and Energy Balance Disruptions: Spotlight on Chemokines. Front Endocrinol (Lausanne) (2017) 8:197. doi: 10.3389/fendo.2017.00197
170. Fioravante M, Bombassaro B, Ramalho AF, de Moura RF, Haddad-Tovolli R, Solon C, et al. Hypothalamic expression of the atypical chemokine receptor ACKR2 is involved in the systemic regulation of glucose tolerance. Biochim Biophys Acta Mol Basis Dis (2019) 1865:1126–37. doi: 10.1016/j.bbadis.2019.01.001
Keywords: high-fat diet, hypothalamus, maternal high-fat diet, chemokines, orexigenic neuropeptides, inflammation
Citation: Poon K (2020) Behavioral Feeding Circuit: Dietary Fat-Induced Effects of Inflammatory Mediators in the Hypothalamus. Front. Endocrinol. 11:591559. doi: 10.3389/fendo.2020.591559
Received: 04 August 2020; Accepted: 23 October 2020;
Published: 26 November 2020.
Edited by:
Claudia Torres-Farfan, Austral University of Chile, ChileReviewed by:
Jessica R. Barson, Drexel University, United StatesDiego Halabi, Austral University of Chile, Chile
Copyright © 2020 Poon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kinning Poon, cG9vbmtAb2xkd2VzdGJ1cnkuZWR1