 Eva Bru-Tari
Eva Bru-Tari Daniel Oropeza
Daniel Oropeza Pedro L. Herrera
Pedro L. Herrera- Department of Genetic Medicine and Development, Faculty of Medicine, University of Geneva, Geneva, Switzerland
The β-cell regeneration field has shown a strong knowledge boost in the last 10 years. Pluripotent stem cell differentiation and direct reprogramming from other adult cell types are becoming more tangible long-term diabetes therapies. Newly generated β-like-cells consistently show hallmarks of native β-cells and can restore normoglycemia in diabetic mice in virtually all recent studies. Nonetheless, these cells still show important compromises in insulin secretion, cell metabolism, electrical activity, and overall survival, perhaps due to a lack of signal integration from other islet cells. Mounting data suggest that diabetes is not only a β-cell disease, as the other islet cell types also contribute to its physiopathology. Here, we present an update on the most recent studies of islet cell heterogeneity and paracrine interactions in the context of restoring an integrated islet function to improve β-cell replacement therapies.
Introduction
The islets of Langerhans are complex micro-organs composed of different endocrine cell types whose principal function is the maintenance of glucose homeostasis and feeding behavior through coordinated hormone secretion and paracrine interactions. Different studies have estimated the human islet to be comprised mainly by insulin-secreting β-cells in the range of 52–75% (1–5). Following in number, glucagon-secreting α-cells and somatostatin-secreting δ-cells comprise some 40 and 10% of the islet. Pancreatic polypeptide (PP)-secreting γ-cells and ghrelin-secreting ϵ-cells are the minor cell types comprising about 5% and less than 1% of the islet, respectively. Islets cells are characterized by an exquisite secretory capacity and cell mass modulation that efficiently adapts to diverse metabolic stresses or pathologies like pregnancy and obesity. Defects in this adaptive capacity are at the core of certain impairments in nutrient metabolism and diabetes development (6–8).
The different islet cell types are arranged in an intricate network that facilitates cell proximity and direct contacts that fine-tune hormone secretion to robustly control glucose homeostasis. In human islets, there is a predominance of heterologous contacts between β- and α-cells, suggesting their direct interaction is crucial for glycaemia management (9). Indeed, paracrine α-cell signaling establishes a set point for insulin secretion and glycemia throughout different animal species (10). Dysregulation of islet paracrine interactions and non-β-cell function contribute substantially to diabetes symptomatology (11–17). This could be one of the reasons why conventional diabetes therapies relying solely on exogenous insulin do not maintain stable normoglycemia. It is becoming apparent that diabetes is a disease concerning the whole islet, yet regenerative approaches have for the most part focused mainly on restoring a functional β-cell mass and basal insulin secretion with little regard on achieving a balanced islet secretory output. Here, we present an update on recent studies highlighting the importance of islet paracrine interactions and cell heterogeneity for a highly malleable human islet function that achieves optimal glucose homeostasis and withstands the range of stresses present in the ever-changing physiologic and cellular environment.
Diabetes Affects All Islet Endocrine Cell Types
Mounting data shows that the complex islet cytoarchitecture, gene expression, and function of non-β-cells are also significantly compromised throughout diabetes progression. Both T1D and T2D patients present hyperglucagonemia in postprandial conditions or upon oral glucose challenge, that exacerbates hyperglycemia (11–15). The reasons are not fully understood, yet i) lack of intra-islet insulin, ii) dysfunctional α-cell glucose sensing, or iii) increases in the functional α-cell mass may be underlying mechanisms. A convoluted combination of these defects may also be possible, as T2D patients present hyperglucagonemia even in the fasting state (16, 17) while T1D patients present a defective α-cell response to hypoglycemia (18).
In T1D, α-cell mass is maintained in the early stages of the disease (19) while it clearly decreases in advanced stages (20). For T2D, most published studies do not specify the stage of the disease, and report conflicting results depending on the analytical method, with a higher α- to β-cell ratio in long-standing T2D pancreas (9, 21, 22) or a decrease of the total glucagon+ area in all regions of the pancreas (23), but no differences in α-cell mass (22). This suggests that α-cell defects may not be due to an increased cell mass. In concordance, recent studies with islets from T2D donors show no inhibition of glucagon secretion in vitro at high glucose concentrations (24). Moreover, transplantation of islets from T2D donors into a novel Glucagon knockout-NSG mouse model showed increased glucagon secretion during fasting and upon insulin-induced hypoglycemia, suggesting that hyperglucagonemia in T2D is caused by local islet defects that are not resolved when transplanted into a non-diabetic environment (25). Nonetheless, the effect of induced hyperglycemia was not tested in this study. In addition, single cell RNA sequencing (scRNA-seq) of α- and δ-cells from T2D donors showed a downregulation of energy metabolism and protein synthesis genes (26, 27). In contrast, islets from T1D donors show decreased glucagon secretion at low glucose concentrations in vitro (28), underlying the high risk of severe hypoglycemia after insulin administration in T1D patients (18, 28). α-cells from T1D donors also show differential gene expression, including in electrical activity and exocytosis, as well as master regulators of α-cell identity, ARX and MAFB (28).
Only few studies tested the effect of diabetes on the minor islet cell types, yet it is likely they are also significantly affected. ϵ-cells show a reduction in their number that could be linked to lower plasma ghrelin levels in T2D (29). Yet, this is unlikely since most ghrelin-secreting cells are extrapancreatic (gastric fundus). In δ-cells, islets from T2D donors showed blunted in vitro somatostatin secretion in response to glucose while some donors show hypersecretion at low glucose (30). While there are no reports on δ-cells from T1D donors, recent findings in diabetic mice indicate that increased somatostatin signaling may be reducing counter-regulatory glucagon secretion during insulin-induced hypoglycemia (31). Finally, T2D patients present high plasma levels of PP after an oral glucose challenge (32). As PP inhibits somatostatin secretion in human islets (33), is possible that increased levels of PP contribute to diabetic hyperglucagonemia by decreasing the somatostatin inhibitory effect on α-cells.
Collectively, these studies suggest that diabetes eventually becomes an islet disease affecting all islet cells or that the degree of initial non-β-cell dysfunction is a contributing factor accelerating the progression or the severity of diabetes. Whether non-β-cell defects are intrinsic or solely the result of the decrease in β-cells and local insulin, is still the focus of intense research. Little is known about the role of non-β-cell function in glucose intolerance, prediabetes or the initial stages of diabetes. δ-Cell electrical oscillatory activity in response to glucose stimulation is impaired in insulin resistant mice treated with high fat diet (34) and non-human primates show decreased proportion of δ-cells per islet that progresses with mounting hyperglycemia, possibly caused by δ-cell apoptosis (35). Alterations in δ-cell secretory function during the progression of type 2 diabetes may exacerbate β-cell exhaustion due to a lack of inhibitory signals exerted by somatostatin or could be an adaptation to the higher insulin demand during prediabetes. In the case of α-cells, insulin resistant and glucose intolerant mice under high fat diet present α-cell hypertrophy and lack of suppression of glucagon release upon intraperitoneal glucose injection (36). Studies with obese subjects also observed hyperglucagonemia upon postprandial conditions (37) and in non-human primates α-cell mass tends to increase with the duration and severity of obesity (38). It has been postulated that α-cell insulin resistance (39), intrinsic defects in α-cell glucose sensing or a reduced somatostatin signaling may lead to α-cell functional alterations at this stage (40). Nonetheless, these observations also highlight that defects in non-β-cells may appear in the eventual progression to T2D.
Analysis of non-β-cell numbers and circulating levels of their corresponding hormones in prediabetes or the initial stages of diabetes is needed to understand when these defects start and their contribution to diabetes before β-cell function is impaired. Likewise, whether defective α-cell function in diabetes is completely restored by the regeneration of a functional β-cell mass is still not clear. T1D recipients of islet transplants showed an absence or only partial restoration of glucagon secretion upon insulin-induced hypoglycemia (41–43). The combination of mouse diabetic models showing dysregulated glucagon secretion (44, 45) that allow the measurement of human plasmatic glucagon (25) and transplantation of purified human α-cells alone or in combination with other islets cells will be needed to address this issue.
Human Islet Architecture Favors Heterologous Contacts Between Endocrine Cells: Implications in The Counter-Regulatory Islet Response to Hypoglycemia
In 1982, pioneering studies described a poor responsiveness of isolated single rat β-cells to glucose, an effect that was linked to the lack of α-cell contacts and glucagon, revealing the crucial role of the islet architecture in the optimal functional cooperation between islet cells (46). Since then, most studies have focused on the core-mantle arrangement of rodent islets that clusters β-cells in the center surrounded by peripheral non-β-cells (4). This favors homologous β-to-β cell contacts shown to be critical in mice for regulating in vivo insulin secretory dynamics and glucose homeostasis through gap-junction coupling (47), which drives β-cell synchronization in terms of electrical activity and intracellular calcium concentration. However, it is broadly accepted that human islet cell types are distributed more randomly (3, 4). Recent studies show that the human islet involves a more intricate structure that depends on islet size. Small human islets (40-60 µm in diameter), which are more frequent during childhood, display the core-mantle structure of rodent islets, while large islets are formed by multiple subunits of β-cell clusters surrounded by non-β cells, containing a lower proportion of β-cells than the smaller ones (48). A similar trend is observed between juvenile and aged mice. This unique arrangement presents a higher rate of heterologous contacts, while maintaining homologous contacts between β-cells (9). In the case of humans, β-cells seem to be less synchronized than in mice in response to stimulatory glucose concentrations (4), possibly due to their organization within the islet, which can prime them to have a weaker β-cell electrical coupling. Indeed, synchronous intracellular calcium oscillations in response to stimulatory glucose concentrations have been recorded only in β-cells within the same islet region (49). The higher rate of heterologous contacts within the human islet suggests that counter-regulatory paracrine interactions might play a more important role in human than in mouse islets for the fine tuning of insulin secretion and glycemia maintenance. Figure 1 summarizes paracrine interactions between human islet cell types.
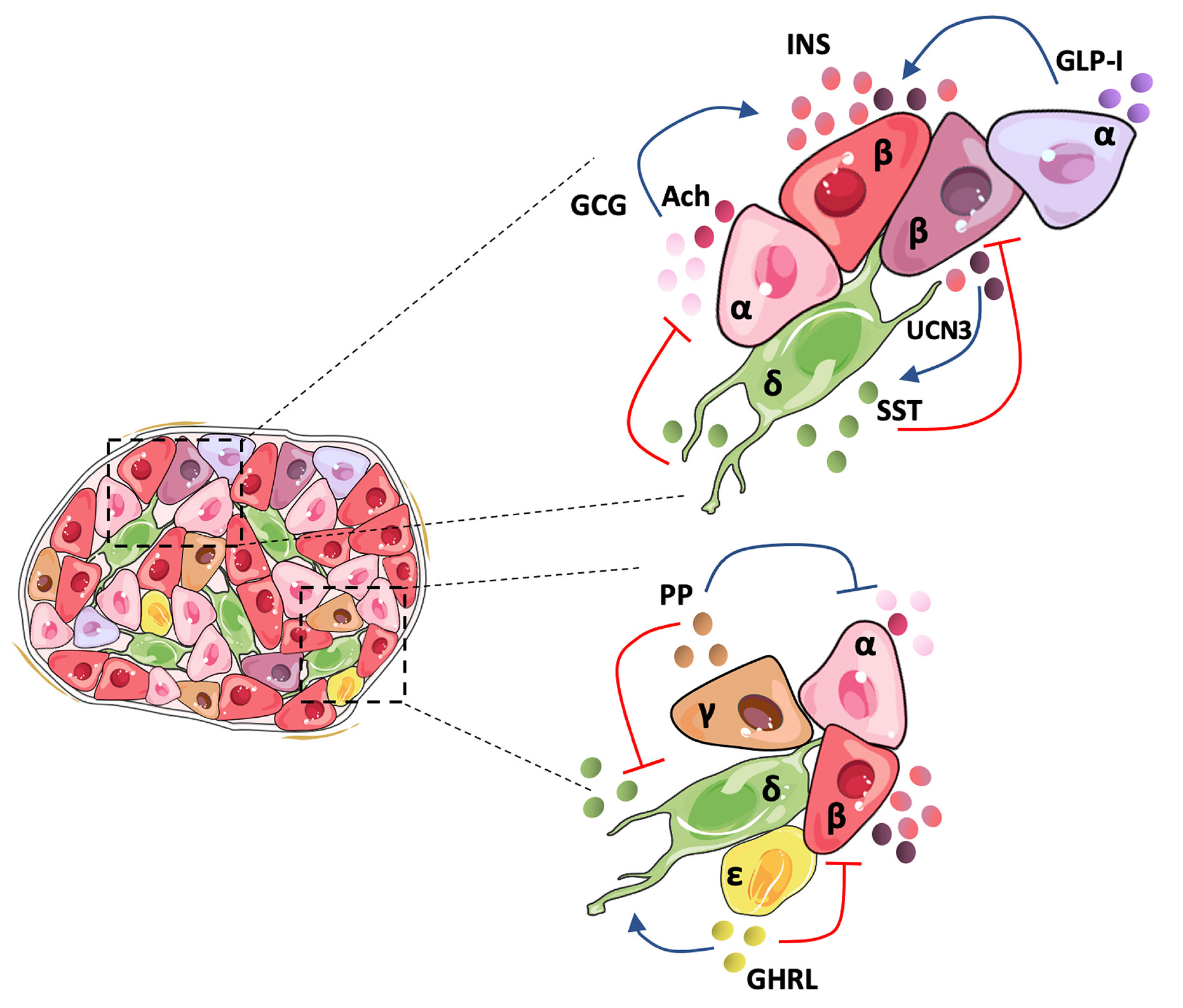
Figure 1 Human islet architecture favors heterologous contacts and holds a tightly regulated cellular inter-communication network. In the human islet, β-and non-β-cells present frequent contacts favoring paracrine signaling between the cells. Human α-cells secrete mainly glucagon (GCG), acetylcholine (Ach), and GLP-I, which activates insulin secretion by β-cells. It has been postulated that an α-cell subpopulation holds the intra-islet secretion of GLP-I. Human pancreatic β-cells comprise a heterogeneous population and may hold differences in the amount of insulin (INS) secreted depending on the subpopulation. UCN3 is secreted along with INS and activates somatostatin (SST) release by δ-cells. SST inactivates GLC and INS secretion, closing the loop of paracrine signaling between the main islet cell types. Although less studied, the minor islet cell types also contribute to the regulation of the islet hormone secretion. Pancreatic polypeptide (PP) secreted by γ-cells suppresses both GCG and SST release, and ghrelin (GHRL), produced by ϵ-cells, seems to activate SST and inhibit INS release.
In humans, α-cell signaling potentiates insulin secretion throughout a wide range of glucose concentrations (50), establishes the glycemic set point for insulin secretion (10) and enhances insulin secretion when β-cells are in contact with α-cells (51). This is classically known to be mediated by glucagon signaling (10), which activates human β-cell G protein-coupled receptors (GPCR) of class B, including glucagon receptor (GCGR), and glucagon-like peptide 1 receptor (GLP-1R), promoting insulin secretion by an increase in cyclic AMP and the recruitment of insulin granules (50). However, GLP-1 is also secreted by human α-cells (52–54) and necessary for insulin secretion in human islets, as GLP-1R antagonism blunted glucose stimulated insulin secretion (GSIS) in vitro (54). Moreover, GLP-1 can elicit synchronous intracellular calcium oscillations in human whole islets (55), suggesting an important role of this hormone and the location of α-cells within the islet to obtain pulsatile and synchronized insulin secretion. Human α-cells also amplify GSIS through the parasympathetic neurotransmitter acetylcholine, which is co-secreted with glucagon (56, 57) and acts through the activation of the muscarinic receptor M3 in β-cells (57). It has been postulated that this mechanism aids in maintaining β-cell responsiveness to the subsequent rise in glucose produced by glucagon action (56). Subsequently, excessive insulin release and hypoglycemia are avoided by a paracrine negative feedback loop between β- and δ-cells (58), mediated by urocortin-3 (UCN3), which is released by β-cells along with insulin. UCN3 activates type 2 corticotropin-releasing hormone receptors specifically borne by δ-cells within the human islet and stimulates somatostatin secretion (58), which directly inhibits insulin secretion through somatostatin receptor 2 (SSTR2) activation (59). Recent data shows that human δ-cells have long filopodia containing secretory granules that allow for direct contact with multiple β- and α-cells (34), and suggests that human β- and δ-cells are coupled by gap junctions (60), as somatostatin secretion follows the same pulsatile and coordinated response of insulin secretion in isolated islets (61).
Recent data is also starting to shed light on how paracrine interactions control α- and δ-cell secretory outputs. The activation of somatostatin secretion by β-cells, directly inhibits glucagon secretion through SSTR2 in α-cells (31) and in vitro chemical inhibition of insulin or somatostatin signaling in whole human islets induces glucagon secretion at non-stimulatory glucose concentrations (24). Isolated human α-cells also activate glucagon secretion at non-stimulatory glucose concentrations, which was corrected by reaggregation with purified β-cells but not by incubation with β-cell secreted factors (62). Lastly, ghrelin has been recently shown to suppress insulin secretion in human islets (29) and potentiate somatostatin release (63), thus suggesting a novel role for ϵ-cells in the control of hypoglycemia. Conversely, γ-cells seem to enhance human insulin secretion through an inhibition in somatostatin secretion caused by the PP activation of NPYR4 receptor in δ-cells (33). PP also activates the PPYR1 receptor (which is present in human α-cells) in mouse α-cells and inhibits glucagon secretion (64).
Knowledge about how human islet structure and the integrated input of paracrine signaling control synchronization of β-cells and islet hormone secretion is scarce and fragmented in comparison with the mouse islet. While mouse islets present tightly synchronized β-cell function and less heterologous cell contacts, non-β-cells might play a more important role in human islets. New techniques employing high yields of purified primary human islet cells will be necessary to study the contribution of each cell type to integrated islet function. Systematic and high-resolution secretory profiling under various metabolic stresses, using isolated as well as reaggregated human islet cells in different combinations, will help dissect the bewildering interplay of interactions that coordinate optimal islet secretion. A recent study employing reaggregated human islets and a microfluidic system that measures secretion concurrently with intracellular signaling dynamics highlights the importance of developing new tools to study human islet cells, as it revealed Gi GPCR signaling decreases insulin and glucagon secretion while Gq GPCRs stimulate glucagon secretion but have dual effects on insulin secretion (65).
Human Islets are Comprised of Heterogeneous Cell Populations: Relevance in β-Cell Function and Stress Adaptation
A higher level of complexity stems from recently identified β- and α-cell subpopulations based on physiological (66–68), transcriptomic (27, 67, 69–72), and proteomic differences (73–75). Upon stimulatory conditions, human β-cells located in discrete islet regions synchronize calcium flux and electrophysiology (4, 49, 76). Shedding light on whether this is due to their location in the islet, their specific β-β physical interactions or to intrinsic features, employing functional cell mapping with optogenetics, “hub” β-cells (10% of the human islet) were identified as first-responders that engage other β-cells into insulin secretion (66). These cells are considered immature based on low Pdx1 and Nkx6.1 expression and low insulin content (66). In mouse islets, “hub” β-cell function is not affected by the inhibition of glucagon signaling or by their location within the islet (66). However, these features have not been explored in human cells, neither whether “hub” cells have a higher number of contacts with α-cells or δ-cells, which could hint at which cell type has a bigger functional influence on this β-cell subpopulation.
Interestingly, “hub” β-cells are more susceptible to glucolipotoxicity, resulting in reduced numbers and high glucokinase protein levels (66). The genes involved in responses to different metabolic insults [including unfolded protein response (UPR), endoplasmic reticulum (ER) stress, and oxidative stress] efficiently cluster β-cells into subpopulations (69–71). This is one of the most relevant features across different scRNA-seq analyses of β-cell heterogeneity, although there is no consensus on the transcriptomic identity of β-cell subpopulations (27, 69–71). In one instance, three β-cell sub clusters where observed matching low UPR with low insulin expression, low UPR with high insulin, and high UPR with low insulin (69). These groups might be transiently moving between a state high insulin production and secretion to fulfill the requirements that maintain normoglycemia, and a state of UPR-mediated recovery from ER stress due to high insulin production, taking the role of hub cells that orchestrate secretion from neighboring cells (69). Importantly, the heterogeneity in UPR responses may have a significant impact on the survival of β-cells to metabolic insults as chronic UPR activation is present in islets from T2D donors and those at risk to suffer the disease (77). However, other studies have not observed differences in UPR-related genes or correlation of any of these β-cell subpopulations with obesity or T2D (27, 72).
In the case of human α-cells, several scRNA-seq analyses identified human α-cell subpopulations with a proliferative profile (27, 69, 72), which have also been reported in pancreatic sections of adolescents (78) and can be correlated with a lower expression of UPR genes (69). At the functional level, GLP-1 secretion has been linked to specific α-cell subgroups that are more prevalent in T2D, indicating a possible α-cell adaptation to higher insulin demand (68). At the structural level, α-cells can be divided into subsets containing different ranges of glucagon granules (75), which suggest distinct secretory properties, yet no heterogeneity in glucagon secretion has been reported (68). However, studies in mice have shown that α-cell subsets vary in calcium flux and membrane capacitance upon stimulatory conditions (45, 79), which may correlate with granule density.
Studies that connect transcriptionally distinct subpopulations with β- and α-cell function are scarce due to technical limitations. Initial reports linked the lack of cell surface markers CD9 and ST8SIA1 with a β-cell subpopulation showing decreased insulin secretion (73). More recently, a study combining scRNA-seq with patch-clamp electrophysiological measurements of vesicle exocytosis and ion-channel activity, found improved excitability properties in a subpopulation of low-expressing RBP4 β-cells in non-diabetic donors, as well as α-cell electrophysiological heterogeneity correlated with differential expression of ER stress markers in non-diabetic and T2D samples (67). Novel methods to inactivate specific subpopulations will unravel the role of heterogeneity in islet function.
Concluding Remarks: Targeting Islet Paracrine Interactions and Heterogeneity for Optimal and Robust Islet Function in β-Cell Regeneration Strategies
Available data suggests that diabetes affects all islet cell types. Islet paracrine interactions and heterogeneity are key features that allow adaptation to a wide spectrum of physiological challenges. Consequently, β-cell regeneration strategies must consider these factors to restore optimal islet secretory capacity. Indeed, islet transplantation, which would partially replenish a functional islet mass, restores circulating insulin to comparable levels of healthy individuals (80, 81), although efficient glucagon secretion upon hypoglycemia is only partially restored (41, 42). It is unclear if transplanting only β-cells to diabetic patients would give similar results, but analogous experiments could be performed in diabetic mice using different combinations of purified human islet populations. De novo generation of surrogate or replacement β-cells from stem cells (SC-β), or other cell sources, has focused in achieving insulin production and secretion comparable to native β-cells under stable conditions. Recent protocols yield SC-β cells that reverse hyperglycemia in mice for up to 45 days, with detectable human C-peptide within 3–14 days after transplantation (82–86). Despite this amazing progress, SC-β cells do not achieve the biphasic insulin release nor the magnitude of insulin secretion of cadaveric islets in vitro, possibly because of a disconnection in glucose sensing (87). Moreover, long-term analysis or the whole range of metabolic stresses including pregnancy, obesity, pathogenic infection, or extreme fasting [where β-cells undergo major modifications which must be quickly reversed after refeeding (88)] have not been explored. Besides, the impact of cell heterogeneity in SC-β strategies remains elusive, with only one report indicating that β-cell subpopulations were not detected after SC-β transplantation in mice (89). Although there are not such studies in human, heterogeneity is crucial in mice for β-cell adaptation to pathological stressors like obesogenic diets (90).
The signals coming from a diverse non-β-cell population might be pivotal to maintain robust insulin content and secretion throughout all these conditions. As described above, while mouse islets seem to rely on homologous β-cell contacts to achieve a synchronized function, in humans, non-β-cells may play a fundamental role as heterologous contacts are more prevalent and GLP-I signaling elicits a coordinated β-cell activation (55). Moreover, functional non-β-cells could also be required for the maturation, as glucagon receptor KO mice show lower expression of Pdx1, Glut2, and MafA in β-cells (91). Additionally, decreased insulin content occurs in glucagon-GFP knock-in mice that lack proglucagon derived peptides (92) and human insulin promoter activity is stimulated by GLP-1 (93). The implications of the cellular architecture and cell diversity in the generation of functional SC-β-cells has been studied recently by the generation of human islet-like organoids (94), containing some 60% of cells co-expressing insulin and other key β-cell markers, along with glucagon, somatostatin, and PP-positive cells (94). This improved functional maturation of SC-β-cells in terms of GSIS and, after the transplantation in mice, allowed for controlled insulin secretion upon a cycle of feeding, fasting, and refeeding (94). Although controls with only SC-β cells is needed to prove if this tuned insulin secretion is driven by adjacent non-β cells, the presence of UCN3 at protein level in β-cells, suggests that paracrine signaling may be restored (94).
Overall, non-β-cell paracrine signaling is key for optimal islet hormone secretion and any disruption to this balanced cell system may exacerbate diabetes symptomatology and compromise β-cell function. In diabetes, non-β-cells present defects that may be not rectified by the sole reestablishment of insulin signaling via β-cell regeneration approaches. This data supports the idea that non-β-cells should be included in the regenerative strategies to treat diabetes. Likewise, the capacity of adaptation of newly generated β-cells and the role cell heterogeneity may play in coping mechanisms that respond to different physiological and pathological metabolic challenges in vivo is also an open question. Experiments that resolve these matters would highlight pivotal pitfalls in β-cell regeneration approaches aimed at restoring integrated islet function.
Author Contributions
EB-T, DO, and PH wrote the mini-review. All authors contributed to the article and approved the submitted version.
Funding
DO was funded by the Hjelt Foundation and PH by the Swiss National Science Foundation (310030_L92496), the Fondation Aclon and the European Research Council (884449-Merlin).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab (2003) 88(5):2300–8. doi: 10.1210/jc.2002-020735
2. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes (2003) 52(1):102–10. doi: 10.2337/diabetes.52.1.102
3. Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem (2005) 53(9):1087–97. doi: 10.1369/jhc.5C6684.2005
4. Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A (2006) 103(7):2334–9. doi: 10.1073/pnas.0510790103
5. Pisania A, Weir GC, O’Neil JJ, Omer A, Tchipashvili V, Lei J, et al. Quantitative analysis of cell composition and purity of human pancreatic islet preparations. Lab Invest (2010) 90(11):1661–75. doi: 10.1038/labinvest.2010.124
6. Ellenbroek JH, Töns HAM, Hanegraaf MAJ, Rabelink TJ, Engelse MA, Carlotti F, et al. Pancreatic α-cell mass in obesity. Diabetes Obes Metab (2017) 19(12):1810–3. doi: 10.1111/dom.12997
7. Cohrs CM, Panzer JK, Drotar DM, Enos SJ, Kipke N, Chen C, et al. Dysfunction of Persisting β Cells Is a Key Feature of Early Type 2 Diabetes Pathogenesis. Cell Rep (2020) 31(1):107469. doi: 10.1016/j.celrep.2020.03.033
8. Baeyens L, Hindi S, Sorenson RL, German MS. β-Cell adaptation in pregnancy. Diabetes Obes Metab (2016) 18(Suppl 1):63–70. doi: 10.1111/dom.12716
9. Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, et al. Unique Arrangement of α- and β-Cells in Human Islets of Langerhans, in. Diabetes (2010) 1202–10. doi: 10.2337/db09-1177
10. Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, et al. Paracrine Interactions within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab (2018) 27(3):549–58.e4. doi: 10.1016/j.cmet.2018.01.015
11. Brown RJ, Sinaii N, Rother KI. Too much glucagon, too little insulin: time course of pancreatic islet dysfunction in new-onset type 1 diabetes. Diabetes Care (2008) 31(7):1403–4. doi: 10.2337/dc08-0575
12. Kramer CK, Borgoño CA, Van Nostrand P, Retnakaran R, Zinman B. Glucagon response to oral glucose challenge in type 1 diabetes: lack of impact of euglycemia. Diabetes Care (2014) 37(4):1076–82. doi: 10.2337/dc13-2339
13. Thivolet C, Marchand L, Chikh K. Inappropriate glucagon and GLP-1 secretion in individuals with long-standing type 1 diabetes: effects of residual C-peptide. Diabetologia (2019) 62(4):593–7. doi: 10.1007/s00125-018-4804-y
14. Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab (2000) 85(11):4053–9. doi: 10.1210/jc.85.11.4053
15. Bagger JI, Knop FK, Lund A, Holst JJ, Vilsbøll T. Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia (2014) 57(8):1720–5. doi: 10.1007/s00125-014-3264-2
16. Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, et al. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes (2000) 49(8):1367–73. doi: 10.2337/diabetes.49.8.1367
17. Demant M, Bagger JI, Suppli MP, Lund A, Gyldenløve M, Hansen KB, et al. Determinants of Fasting Hyperglucagonemia in Patients with Type 2 Diabetes and Nondiabetic Control Subjects. Metab Syndr Relat Disord (2018) 16(10):530–6. doi: 10.1089/met.2018.0066
18. Siafarikas A, Johnston RJ, Bulsara MK, O’Leary P, Jones TW, Davis EA. Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care (2012) 35(8):1757–62. doi: 10.2337/dc11-2010
19. Sayama K, Imagawa A, Okita K, Uno S, Moriwaki M, Kozawa J, et al. Pancreatic beta and alpha cells are both decreased in patients with fulminant type 1 diabetes: a morphometrical assessment. Diabetologia (2005) 48(8):1560–4. doi: 10.1007/s00125-005-1829-9
20. Bonnet-Serrano F, Diedisheim M, Mallone R, Larger E. Decreased α-cell mass and early structural alterations of the exocrine pancreas in patients with type 1 diabetes: An analysis based on the nPOD repository. PLoS One (2018) 13(1):e0191528. doi: 10.1371/journal.pone.0191528
21. Fujita Y, Kozawa J, Iwahashi H, Yoneda S, Uno S, Eguchi H, et al. Human pancreatic α- to β-cell area ratio increases after type 2 diabetes onset. J Diabetes Invest (2018) 9(6):1270–82. doi: 10.1111/jdi.12841
22. Henquin JC, Rahier J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia (2011) 54(7):1720–5. doi: 10.1007/s00125-011-2118-4
23. Kilimnik G, Zhao B, Jo J, Periwal V, Witkowski P, Misawa R, et al. Altered Islet Composition and Disproportionate Loss of Large Islets in Patients with Type 2 Diabetes. PLoS One (2011) 6(11):e27445. doi: 10.1371/journal.pone.0027445
24. Omar-Hmeadi M, Lund P-E, Gandasi NR, Tengholm A, Barg S. Paracrine control of α-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat Commun (2020) 11(1):1896. doi: 10.1038/s41467-020-15717-8
25. Tellez K, Hang Y, Gu X, Chang CA, Stein RW, Kim SK. In vivo studies of glucagon secretion by human islets transplanted in mice. Nat Metab (2020) 2(6):547–57. doi: 10.1038/s42255-020-0213-x
26. Lawlor N, George J, Bolisetty M, Kursawe R, Sun L, Sivakamasundari V, et al. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res (2017) 27(2):208–22. doi: 10.1101/gr.212720.116
27. Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab (2016) 24(4):593–607. doi: 10.1016/j.cmet.2016.08.020
28. Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep (2018) 22(10):2667–76. doi: 10.1016/j.celrep.2018.02.032
29. Lindqvist A, Shcherbina L, Prasad RB, Miskelly MG, Abels M, Martínez-Lopéz JA, et al. Ghrelin suppresses insulin secretion in human islets and type 2 diabetes patients have diminished islet ghrelin cell number and lower plasma ghrelin levels. Mol Cell Endocrinol (2020) 511:110835. doi: 10.1016/j.mce.2020.110835
30. Vergari E, Denwood G, Salehi A, Zhang Q, Adam J, Alrifaiy A, et al. Somatostatin secretion by Na(+)-dependent Ca(2+)-induced Ca(2+) release in pancreatic delta-cells. Nat Metab (2020) 2(1):32–40. doi: 10.1038/s42255-019-0158-0
31. Vergari E, Knudsen JG, Ramracheya R, Salehi A, Zhang Q, Adam J, et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat Commun (2019) 10(1):139. doi: 10.1038/s41467-018-08193-8
32. Chia CW, Odetunde JO, Kim W, Carlson OD, Ferrucci L, Egan JM. GIP contributes to islet trihormonal abnormalities in type 2 diabetes. J Clin Endocrinol Metab (2014) 99(7):2477–85. doi: 10.1210/jc.2013-3994
33. Kim W, Fiori JL, Shin YK, Okun E, Kim JS, Rapp PR, et al. Pancreatic polypeptide inhibits somatostatin secretion. FEBS Lett (2014) 588(17):3233–9. doi: 10.1016/j.febslet.2014.07.005
34. Arrojo e Drigo R, Jacob S, García-Prieto CF, Zheng X, Fukuda M, Nhu HTT, et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nat Commun (2019) 10(1):3700. doi: 10.1038/s41467-019-11517-x
35. Guardado Mendoza R, Perego C, Finzi G, La Rosa S, Capella C, Jimenez-Ceja LM, et al. Delta cell death in the islet of Langerhans and the progression from normal glucose tolerance to type 2 diabetes in non-human primates (baboon, Papio hamadryas). Diabetologia (2015) 58(8):1814–26. doi: 10.1007/s00125-015-3625-5
36. Merino B, Alonso-Magdalena P, Lluesma M, Ñeco P, Gonzalez A, Marroquí L, et al. Pancreatic alpha-cells from female mice undergo morphofunctional changes during compensatory adaptations of the endocrine pancreas to diet-induced obesity. Sci Rep (2015) 5(1):11622. doi: 10.1038/srep11622
37. Calanna S, Piro S, Di Pino A, Maria Zagami R, Urbano F, Purrello F, et al. Beta and alpha cell function in metabolically healthy but obese subjects: Relationship with entero-insular axis. Obesity (2013) 21(2):320–5. doi: 10.1002/oby.20017
38. Guardado-Mendoza R, Jimenez-Ceja L, Majluf-Cruz A, Kamath S, Fiorentino TV, Casiraghi F, et al. Impact of obesity severity and duration on pancreatic β- and α-cell dynamics in normoglycemic non-human primates. Int J Obes (2013) 37(8):1071–8. doi: 10.1038/ijo.2012.205
39. Tsuchiyama N, Takamura T, Ando H, Sakurai M, Shimizu A, Kato K, et al. Possible role of alpha-cell insulin resistance in exaggerated glucagon responses to arginine in type 2 diabetes. Diabetes Care (2007) 30(10):2583–7. doi: 10.2337/dc07-0066
40. Kellard JA, Rorsman NJG, Hill TG, Armour SL, van de Bunt M, Rorsman P, et al. Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol Metab (2020) 40:101021. doi: 10.1016/j.molmet.2020.101021
41. Rickels MR, Schutta MH, Mueller R, Markmann JF, Barker CF, Naji A, et al. Islet cell hormonal responses to hypoglycemia after human islet transplantation for type 1 diabetes. Diabetes (2005) 54(11):3205–11. doi: 10.2337/diabetes.54.11.3205
42. Rickels MR, Peleckis AJ, Markmann E, Dalton-Bakes C, Kong SM, Teff KL, et al. Long-Term Improvement in Glucose Control and Counterregulation by Islet Transplantation for Type 1 Diabetes. J Clin Endocrinol Metab (2016) 101(11):4421–30. doi: 10.1210/jc.2016-1649
43. Paty BW, Ryan EA, Shapiro AM, Lakey JR, Robertson RP. Intrahepatic islet transplantation in type 1 diabetic patients does not restore hypoglycemic hormonal counterregulation or symptom recognition after insulin independence. Diabetes (2002) 51(12):3428–34. doi: 10.2337/diabetes.51.12.3428
44. Bru-Tari E, Cobo-Vuilleumier N, Alonso-Magdalena P, Dos Santos RS, Marroqui L, Nadal A, et al. Pancreatic alpha-cell mass in the early-onset and advanced stage of a mouse model of experimental autoimmune diabetes. Sci Rep (2019) 9(1):9515. doi: 10.1038/s41598-019-45853-1
45. Huang YC, Rupnik MS, Karimian N, Herrera PL, Gilon P, Feng ZP, et al. In situ electrophysiological examination of pancreatic α cells in the streptozotocin-induced diabetes model, revealing the cellular basis of glucagon hypersecretion. Diabetes (2013) 62(2):519–30. doi: 10.2337/db11-0786
46. Pipeleers D, in’t Veld PI, Maes E, Van De Winkel M. Glucose-induced insulin release depends on functional cooperation between islet cells. Proc Natl Acad Sci U S A (1982) 79(23):7322–5. doi: 10.1073/pnas.79.23.7322
47. Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW, Benninger RKP. Connexin-36 Gap Junctions Regulate In Vivo First- and Second-Phase Insulin Secretion Dynamics and Glucose Tolerance in the Conscious Mouse. Diabetes (2012) 61(7):1700. doi: 10.2337/db11-1312
48. Bonner-Weir S, Sullivan BA, Weir CG. Human Islet Morphology Revisited: Human and Rodent Islets Are Not So Different After All. J Histochem Cytochem (2015) 63(8):604–12. doi: 10.1369/0022155415570969
49. Quesada I, Todorova MG, Alonso-Magdalena P, Beltrá M, Carneiro EM, Martin F, et al. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified alpha- and beta-cells within intact human islets of Langerhans. Diabetes (2006) 55(9):2463–9. doi: 10.2337/db06-0272
50. Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, et al. β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight (2019) 4(5):e126742. doi: 10.1172/jci.insight.126742
51. Wojtusciszyn A, Armanet M, Morel P, Berney T, Bosco D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia (2008) 51(10):1843. doi: 10.1007/s00125-008-1103-z
52. Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia (2012) 55(12):3262–72. doi: 10.1007/s00125-012-2716-9
53. Linnemann AK, Neuman JC, Battiola TJ, Wisinski JA, Kimple ME, Davis DB. Glucagon-Like Peptide-1 Regulates Cholecystokinin Production in β-Cells to Protect From Apoptosis. Mol Endocrinol (Baltimore Md) (2015) 29(7):978–87. doi: 10.1210/me.2015-1030
54. de Souza AH, Tang J, Yadev AK, Saghafi ST, Kibbe CR, Linnemann AK, et al. Intra-islet GLP-1, but not CCK, is necessary for β-cell function in mouse and human islets. Sci Rep (2020) 10(1):2823. doi: 10.1038/s41598-020-59799-2
55. Hodson DJ, Mitchell RK, Bellomo EA, Sun G, Vinet L, Meda P, et al. Lipotoxicity disrupts incretin-regulated human β cell connectivity. J Clin Invest (2013) 123(10):4182–94. doi: 10.1172/JCI68459
56. Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, et al. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med (2011) 17(7):888–92. doi: 10.1038/nm.2371
57. Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren PO, Caicedo A. Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes (2014) 63(8):2714–26. doi: 10.2337/db13-1371
58. van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing- Zitron C, Pound LD, et al. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat Med (2015) 21(7):769–76. doi: 10.1038/nm.3872
59. Kailey B, van de Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. Am J Physiol Endocrinol Metab (2012) 303(9):E1107–16. doi: 10.1152/ajpendo.00207.2012
60. Briant LJB, Reinbothe TM, Spiliotis I, Miranda C, Rodriguez B, Rorsman P. δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J Physiol (2018) 596(2):197–215. doi: 10.1113/JP274581
61. Hellman B, Salehi A, Gylfe E, Dansk H, Grapengiesser E. Glucose generates coincident insulin and somatostatin pulses and antisynchronous glucagon pulses from human pancreatic islets. Endocrinology (2009) 150(12):5334–40. doi: 10.1210/en.2009-0600
62. Liu W, Kin T, Ho S, Dorrell C, Campbell SR, Luo P, et al. Abnormal regulation of glucagon secretion by human islet alpha cells in the absence of beta cells. EBioMedicine (2019) 50:306–16. doi: 10.1016/j.ebiom.2019.11.018
63. DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab (2016) 5(7):449–58. doi: 10.1016/j.molmet.2016.04.007
64. Aragón F, Karaca M, Novials A, Maldonado R, Maechler P, Rubí B. Pancreatic polypeptide regulates glucagon release through PPYR1 receptors expressed in mouse and human alpha-cells. Biochim Biophys Acta (2015) 1850(2):343–51. doi: 10.1016/j.bbagen.2014.11.005
65. Walker JT, Haliyur R, Nelson HA, Ishahak M, Poffenberger G, Aramandla R, et al. Integrated human pseudoislet system and microfluidic platform demonstrate differences in GPCR signaling in islet cells. JCI Insight (2020) 5(10):e137017. doi: 10.1172/jci.insight.137017
66. Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab (2016) 24(3):389–401. doi: 10.1016/j.cmet.2016.06.020
67. Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab (2020) 31(5):1017–31.e4. doi: 10.1016/j.cmet.2020.04.005
68. Campbell SA, Golec DP, Hubert M, Johnson J, Salamon N, Barr A, et al. Human islets contain a subpopulation of glucagon-like peptide-1 secreting α cells that is increased in type 2 diabetes. Mol Metab (2020) 39:1010–4. doi: 10.1016/j.molmet.2020.101014
69. Xin Y, Dominguez Gutierrez G, Okamoto H, Kim J, Lee AH, Adler C, et al. Pseudotime Ordering of Single Human β-Cells Reveals States of Insulin Production and Unfolded Protein Response. Diabetes (2018) 67(9):1783–94. doi: 10.2337/db18-0365
70. Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst (2016) 3(4):346–60.e4. doi: 10.1016/j.cels.2016.08.011
71. Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst (2016) 3(4):385–94.e3. doi: 10.1016/j.cels.2016.09.002
72. Fang Z, Weng C, Li H, Tao R, Mai W, Liu X, et al. Single-Cell Heterogeneity Analysis and CRISPR Screen Identify Key β-Cell-Specific Disease Genes. Cell Rep (2019) 26(11):3132–44.e7. doi: 10.1016/j.celrep.2019.02.043
73. Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human islets contain four distinct subtypes of β cells. Nat Commun (2016) 7(1):11756. doi: 10.1038/ncomms11756
74. Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, et al. Single-Cell Mass Cytometry Analysis of the Human Endocrine Pancreas. Cell Metab (2016) 24(4):616–26. doi: 10.1016/j.cmet.2016.09.007
75. Ghazvini Zadeh EH, Huang Z, Xia J, Li D, Davidson HW. ZIGIR, a Granule-Specific Zn2+ Indicator, Reveals Human Islet α Cell Heterogeneity. Cell Rep (2020) 32(2):107904. doi: 10.1016/j.celrep.2020.107904
76. Almaça J, Liang T, Gaisano HY, Nam HG, Berggren PO, Caicedo A. Spatial and temporal coordination of insulin granule exocytosis in intact human pancreatic islets. Diabetologia (2015) 58(12):2810–8. doi: 10.1007/s00125-015-3747-9
77. Huang C-j, Lin C-y, Haataja L, Gurlo T, Butler AE, Rizza RA, et al. High Expression Rates of Human Islet Amyloid Polypeptide Induce Endoplasmic Reticulum Stress–Mediated β-Cell Apoptosis, a Characteristic of Humans With Type 2 but Not Type 1 Diabetes. Diabetes (2007) 56(8):2016. doi: 10.2337/db07-0197
78. Lam CJ, Cox AR, Jacobson DR, Rankin MM, Kushner JA. Highly Proliferative α-Cell-Related Islet Endocrine Cells in Human Pancreata. Diabetes (2018) 67(4):674–86. doi: 10.2337/db17-1114
79. Le Marchand SJ, Piston DW. Glucose suppression of glucagon secretion: metabolic and calcium responses from alpha-cells in intact mouse pancreatic islets. J Biol Chem (2010) 285(19):14389–98. doi: 10.1074/jbc.M109.069195
80. Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B, et al. Improvement in Outcomes of Clinical Islet Transplantation: 1999–2010. Diabetes Care (2012) 35(7):1436. doi: 10.2337/dc12-0063
81. Warnock GL, Thompson DM, Meloche RM, Shapiro RJ, Ao Z, Keown P, et al. A Multi-Year Analysis of Islet Transplantation Compared With Intensive Medical Therapy on Progression of Complications in Type 1 Diabetes. Transplantation (2008) 86(12):1762–66. doi: 10.1097/TP.0b013e318190b052
82. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol (2019) 21(2):263–74. doi: 10.1038/s41556-018-0271-4
83. Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, et al. Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Rep (2019) 12(2):351–65. doi: 10.1016/j.stemcr.2018.12.012
84. Ghazizadeh Z, Kao D-I, Amin S, Cook B, Rao S, Zhou T, et al. ROCKII inhibition promotes the maturation of human pancreatic beta-like cells. Nat Commun (2017) 8(1):298–8. doi: 10.1038/s41467-017-00129-y
85. Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic β cells in vitro. Cell (2014) 159(2):428–39. doi: 10.1016/j.cell.2014.09.040
86. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol (2014) 32(11):1121–33. doi: 10.1038/nbt.3033
87. Davis JC, Alves TC, Helman A, Chen JC, Kenty JH, Cardone RL, et al. Glucose Response by Stem Cell-Derived β Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. Cell Rep (2020) 31(6):107623–3. doi: 10.1016/j.celrep.2020.107623
88. Boland BB, Brown C Jr., Alarcon C, Demozay D, Grimsby JS, Rhodes CJ, et al. β-Cell Control of Insulin Production During Starvation-Refeeding in Male Rats. Endocrinology (2018) 159(2):895–906. doi: 10.1210/en.2017-03120
89. Redick SD, Leehy L, Rittenhouse AR, Blodgett DM, Derr AG, Kucukural A, et al. Recovery of viable endocrine-specific cells and transcriptomes from human pancreatic islet-engrafted mice. FASEB J (2020) 34(1):1901–11. doi: 10.1096/fj.201901022RR
90. Ellenbroek JH, Töns HA, de Graaf N, Loomans CJ, Engelse MA, Vrolijk H, et al. Topologically heterogeneous beta cell adaptation in response to high-fat diet in mice. PLoS One (2013) 8(2):e56922. doi: 10.1371/journal.pone.0056922
91. Vuguin PM, Kedees MH, Cui L, Guz Y, Gelling RW, Nejathaim M, et al. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology (2006) 147(9):3995–4006. doi: 10.1210/en.2005-1410
92. Takagi Y, Kinoshita K, Ozaki N, Seino Y, Murata Y, Oshida Y, et al. Mice Deficient in Proglucagon-Derived Peptides Exhibit Glucose Intolerance on a High-Fat Diet but Are Resistant to Obesity. PLoS One (2015) 10(9):e0138322–e0138322. doi: 10.1371/journal.pone.0138322
93. Hay CW, Sinclair EM, Bermano G, Durward E, Tadayyon M, Docherty K. Glucagon-like peptide-1 stimulates human insulin promoter activity in part through cAMP-responsive elements that lie upstream and downstream of the transcription start site. J Endocrinol (2005) 186(2):353–65. doi: 10.1677/joe.1.06205
Keywords: human islet, beta-cell regeneration, paracrine signaling, cell heterogeneity, glucose homeostasis
Citation: Bru-Tari E, Oropeza D and Herrera PL (2021) Cell Heterogeneity and Paracrine Interactions in Human Islet Function: A Perspective Focused in β-Cell Regeneration Strategies. Front. Endocrinol. 11:619150. doi: 10.3389/fendo.2020.619150
Received: 19 October 2020; Accepted: 14 December 2020;
Published: 03 February 2021.
Edited by:
Hanne Scholz, University of Oslo, NorwayReviewed by:
Weiping Han, Singapore Bioimaging Consortium (A*STAR), SingaporeHail Kim, Korea Advanced Institute of Science and Technology, South Korea
Copyright © 2021 Bru-Tari, Oropeza and Herrera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro L. Herrera, cGVkcm8uaGVycmVyYUB1bmlnZS5jaA==