 Taylor Landry
Taylor Landry Daniel Shookster
Daniel Shookster Hu Huang
Hu Huang- 1East Carolina Diabetes and Obesity Institute, East Carolina University, Greenville, NC, United States
- 2Department of Kinesiology, East Carolina University, Greenville, NC, United States
- 3Human Performance Laboratory, College of Human Performance and Health, East Carolina University, Greenville, NC, United States
- 4Department of Physiology, East Carolina University, Greenville, NC, United States
Rho-kinase 1 (ROCK1) has been implicated in diverse metabolic functions throughout the body, with promising evidence identifying ROCK1 as a therapeutic target in diabetes and obesity. Considering these metabolic roles, several pharmacological inhibitors have been developed to elucidate the mechanisms underlying ROCK1 function. Y27632 and fasudil are two common ROCK1 inhibitors; however, they have varying non-specific selectivity to inhibit other AGC kinase subfamily members and whole-body pharmacological approaches lack tissue-specific insight. As a result, interpretation of studies with these inhibitors is difficult, and alternative approaches are needed to elucidate ROCK1’s tissue specific metabolic functions. Fortunately, recent technological advances utilizing molecular carriers or genetic manipulation have facilitated discovery of ROCK1’s tissue-specific mechanisms of action. In this article, we review the tissue-specific roles of ROCK1 in the regulation of energy balance and substrate utilization. We highlight prominent metabolic roles in liver, adipose, and skeletal muscle, in which ROCK1 regulates energy expenditure, glucose uptake, and lipid metabolism via inhibition of AMPK2α and paradoxical modulation of insulin signaling. Compared to ROCK1’s roles in peripheral tissues, we also describe contradictory functions of ROCK1 in the hypothalamus to increase energy expenditure and decrease food intake via leptin signaling. Furthermore, dysregulated ROCK1 activity in either of these tissues results in metabolic disease phenotypes. Overall, tissue-specific approaches have made great strides in deciphering the many critical metabolic functions of ROCK1 and, ultimately, may facilitate the development of novel treatments for metabolic disorders.
Introduction
Rho-kinase (ROCK) belongs to the protein kinase A/G/C (AGC) subfamily of serine/threonine protein kinases and is a major downstream effector of small GTPase RhoA (1). The two ROCK isoforms, ROCK1 and ROCK2, each contain a kinase domain at its N-terminus, a central coiled-coil domain, and a pleckstrin-homology domain split by a cysteine-rich region at its C-terminus (2–4). ROCK1 and ROCK2 share 65% amino acid homology and have been implicated in a variety of cellular functions, including smooth-muscle contraction and actin cytoskeleton arrangement; however, these isoforms also perform independent functions due to differences in their structure, subcellular localization, and gene distribution (5, 6). For example, ROCK1 has uniquely been identified as an important regulator of energy balance and substrate metabolism. Pharmacological ROCK1 inhibitors, like Y-27632 and fasudil (HA1077), have been valuable to elucidating ROCK1’s diverse metabolic roles (7–12); however, both these compounds have varying selectivity to also inhibit ROCK2 and other AGC kinase subfamily members. The nonspecific and systemic effects of these inhibitors make interpretation of studies in which they are used challenging (13); therefore, alternative approaches to investigating ROCK1 function are required to understand its tissue-specific metabolic functions.
In this article, we review the physiological roles of ROCK1 in the regulation of energy balance and substrate utilization. We describe novel insights into ROCK1’s tissue-specific functions facilitated by recent technological advances and highlight prominent roles in liver, adipose, skeletal muscle, and hypothalamus. Furthermore, emerging evidence suggests ROCK1 is a molecular mediator underlying the pathogenesis of diabetes and obesity.
ROCK1 in Liver
Hepatic ROCK1 Overactivity Is Associated With Metabolic Disease States-
To date, the most well-documented metabolic roles of ROCK1 are observed in liver, with clear connections demonstrated between hepatic ROCK1 overactivity and humans or rodents with metabolic disorders (14–21). For example, in humans, liver ROCK1 protein content positively correlates with BMI, liver triglycerides (TG’s), and markers of liver damage including alanine transaminase and aspartate transaminase (14). Moreover, elevated hepatic ROCK1 activity has consistently been observed in a plethora of models of disordered metabolism including: humans with fatty liver disease (14), DIO mice (14), db/db mice (14), ob/ob mice (14), TNFα-treated hepatocytes (15), endothelial nitric oxide synthase (eNOS) deficient mice (16), palmitate-treated hepG2 cells (21), palmitate metabolite lysophosphatidylcholine (LPS) -treated Huh7 cells (20), LPS-treated mice (15), and DIO streptozotocin-treated rats (18). Consequently, ROCK1’s role in homeostatic and disordered liver metabolism has been an important subject of investigation.
The causal relationship between hepatic ROCK1 and metabolic disease has been investigated using a constitutively active ROCK1-specific mutant in the liver (L-CA-ROCK1), which increases ROCK1 activity 2-fold (14). In chow-fed L-CA-ROCK1 mice, body weight is normal, however fasting glucose levels and lipogenic gene expression including fatty acid synthase (FAS) and stearoyl-CoA desaturase (SCD1) is elevated. DIO L-CA-ROCK1 mice experience a more striking phenotype, characterized by accelerated obesity, insulin resistance, hepatic steatosis, hyperglycemia, and dyslipidemia. These mice also have decreased thermogenic gene expression indicated by decreased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and uncoupling protein 1 (UCP1) mRNA in brown adipose tissue (BAT) and/or white adipose tissue (WAT). Overall, the L-CA-ROCK1 phenotype demonstrates a possible causal role of hepatic ROCK1 overactivity in metabolic disease pathologies, identifying a potential therapeutic target in liver ROCK1 to treat obesity and diabetes.
Inhibition of Liver ROCK1 Protects Against Metabolic Disease Pathologies-
Several studies have investigated the therapeutic potential of genetically or chemically inhibiting ROCK1 in various models of disordered liver metabolism (14–17, 20). One mouse model of liver ROCK1-deficiency (L-ROCK1-/-), in which hepatic ROCK1 activity was specifically knocked down 80%, resulted in significant protection from DIO and related comorbidities (14). In chow-fed L-ROCK1-/- mice, there are no differences in body weight, body composition, or food intake; however, in DIO L-ROCK1-/- mice, body weight and adiposity are reduced, at least in part, due to elevated energy expenditure and locomotor activity. Increases in energy expenditure may be due to augmented thermogenic gene expression in BAT (PGC1α, UCP1, COX7a1, COX8b, and ELOVL3) and WAT (COX8b) (14).
ROCK1 inhibition also improves insulin sensitivity, glucose clearance, fatty liver, and circulating lipid levels (14–17, 22). Chow-fed, DIO, and db/db L-ROCK1-/- mice experience improved glucose clearance and insulin sensitivity, as well as decreased liver weight, TG’s, and cholesterol content (14). Supporting these findings, Y-27632 treatment in primary mouse hepatocytes abolishes TNFα-induced insulin resistance (15). L-ROCK1-/- mice also have decreased lipogenic gene expression (FAS, SCD1, SREBP1c, and ELOVL2), despite no observed differences in gene expression involved glucose metabolism (14). Overall, studies have observed encouraging therapeutic potential of liver-specific ROCK1 inhibition in metabolic disease models, demonstrated by increased energy expenditure, improved insulin sensitivity, and attenuated lipid accumulation. These results underscore the value of determining the molecular mechanisms underlying ROCK1 function to further understand the pathology of diabetes and obesity.
Hepatic ROCK1 Negatively Regulates AMPK Activity-
The effects of ROCK1 inhibition on energy balance and lipid metabolism are abolished in AMPKα2-/- mice, suggesting a mechanistic relationship between liver ROCK1 and AMPK in metabolic regulation (16, 17). Hepatic ROCK1 decreases phosphorylation (thr172) and activity of AMPK, which is the proposed mechanism through which ROCK1 increases gene expression and decreases phosphorylation of ACCser79 and SREBP1ser372 to increase lipogenesis (14, 16, 17). Interestingly, therapeutic agents metformin and paeoniflorin target hepatic ROCK1/AMPK signaling to improve steatosis and dyslipidemia in DIO mice (14) and palmitate treated HepG2 cells, respectively (21). In summary, hepatic ROCK1 appears to have a prominent role in promoting lipogenesis via suppression of AMPK activity and subsequent elevations in AMPK’s downstream targets SREBP1c and ACC. At this time, the upstream molecular mediators of ROCK1 pathologies are less clear, but a recent study determining TNFα stimulates NF-κB to activate hepatic ROCK1 in primary hepatocytes and LPS-treated mice may suggest the involvement of inflammatory pathways.
Interestingly, the ability of hepatic ROCK1 inhibition to improve glucose metabolism was found to be AMPKα2 independent, suggesting an alternative, not yet discovered mechanism (14, 16, 17). The role of ROCK1 in the regulation of insulin signaling is complex and tissue specific, with some studies reporting ROCK1 directly reduces phosphorylation of the tyrosine612 residue (23, 24) and induces phosphorylation of the serine632/635 residues (25, 26) on insulin receptor substrate 1 (IRS1). Consequently, ROCK1 activity has been associated with impaired insulin signaling in smooth muscle (23, 27), fibroblasts (28), adipose tissue (24), heart (29, 30), and leukocytes (31). Conversely, some studies observe ROCK1 to facilitate glucose uptake in adipocytes (25, 32, 33) and skeletal muscle (25, 33–35). These convoluted results are most likely due to differences in experimental models and the tissue-specific differences in ROCK1-mediated regulation of insulin signaling should be considered when developing therapeutic agents.
ROCK1 and Adipose Tissue
Adipocyte-Specific ROCK1 Inhibition Is Therapeutic in Models of Metabolic Disease-
ROCK1 activity is elevated in the adipose tissue of DIO and db/db mice, and adipocyte-specific inhibition of ROCK1 rescues many metabolic disease pathologies (24, 36). While adipose-specific ROCK1 disruption by 50% has no obvious phenotype in healthy mice (24), DIO mice experience improved insulin sensitivity and glucose clearance, despite no changes in adipogenesis, energy balance, or inflammation (24). The benefits are even greater when adipocyte-specific ROCK1 activity is reduced by ~83% (36), resulting in attenuated HFD-induced weight gain and improved insulin sensitivity independent of body weight changes. Furthermore, fasting insulin, fasting glucose, FFA’s, adipocyte growth, and macrophage infiltration are all reduced. Despite this encouraging therapeutic potential, the mechanisms underlying ROCK1-mediated adipose pathologies remain relatively unexplored.
ROCK1 Is Critical to Adipose Insulin Signaling-
Despite the glucose-lowering effects of ROCK1 inhibition in mouse models of metabolic disease (24, 36), ROCK1 inhibition in cultured adipocytes impairs insulin-stimulated glucose uptake (25, 32, 33). ROCK1 activity is critical to insulin-stimulated phosphorylation of IRS1ser632/635 and PI3kinase activity (25, 32). Interestingly, insulin directly stimulates rho membrane translocation via PI3kinase in adipocytes (37), and PI3kinase inhibition abolishes ROCK1-mediated glucose transport (33). This suggests a circuitous, poorly understood, regulatory mechanism of insulin signaling, in which ROCK1 is both downstream and upstream of PI3kinase. Overall, ROCK1’s role in insulin signaling is complex, and the opposing effects of adipocyte-specific ROCK1 inhibition in healthy vs. pathological models, indicates an importance of basal ROCK1 activity to glucose homeostasis but also implicates its overactivity in metabolic disease pathologies.
Adipose ROCK1 Is Involved in Adipocyte Differentiation and Lipid Metabolism-
Studies utilizing primary human and rodent adipocytes have revealed ROCK1 also regulates adipocyte differentiation and storage. For example, silencing of the ROCK1 antagonist “deleted in liver cancer 1” in both white and brown cultured adipocytes, and subsequent overactivation of ROCK1, results in decreased adipocyte differentiation, lipid accumulation, and adipogenic gene expression (fatty acid binding protein 4;FABP4 and adiponectin) (38, 39). Additional impairments in thermogenic (UCP1 and ELOVL3) and mitochondrial (cox7a1 and cox5b) gene expression are observed in BAT, as well as reduced mitochondrial respiration (38). Increased ROCK1 activity in cultured human adipocytes has also been shown to impair lipolysis and reduce protein levels of phosphorylated hormone sensitive lipaseser660 and adipose TG lipase (40). Overall, while these studies lack in-depth mechanistic insight, their findings suggest adipose ROCK1 is a physiological negative regulator of adipogenesis, lipolysis, and thermogenesis.
ROCK1 and Skeletal Muscle Metabolism
Skeletal Muscle ROCK1 Overactivity Is Associated With Metabolic Disease States-
Similar to adipose and liver, skeletal muscle ROCK1 expression and activity are elevated in rodent models of metabolic disease (26, 41–43). Conversely, one study observed no differences in basal vastus lateralis (VL) ROCK1 protein expression or activity between obese and lean humans (35), highlighting the importance of considering potential differences between rodent and human ROCK1 function. Further supporting the hypothesis that overactive ROCK1 is involved in metabolic disease pathologies, at least in mice, constitutively active skeletal muscle-specific ROCK1 (SM-CA-ROCK1) results in early-onset obesity, even when eating a normal diet (44). These mice exhibit reduced physical activity, decreased energy expenditure, impaired glucose clearance and insulin sensitivity, elevated fasting TG’s and cholesterol, and increased respiratory exchange ratio suggesting decreased fat utilization. They also experience decreased thermogenic gene expression in BAT and WAT, as well as reduced mitochondrial size and content specifically in Type I muscle fibers. Lastly, myogenic gene expression is altered in these mice including reduced irisin and IL13 mRNA by 60% and 25%, respectively.
Skeletal Muscle ROCK1 Paradoxically Regulates Insulin Signaling-
The metabolic dysfunctions of SM-CA-ROCK1 mice may be due to impaired insulin signaling (41, 42). Lipid-induced geranylgeranyl diphosphate synthase 1 (GGPPS), a branchpoint enzyme in the mevalonate pathway involved in cholesterol synthesis, activates RhoA/ROCK1 signaling in muscle, which then increases inhibitory phosphorylation of IRS1ser307 to inhibit downstream signaling (41). This phenomenon is rescued in muscle-specific GGPPS knockout mice, as is insulin sensitivity and glucose homeostasis (41). ROCK1 also activates phosphatase and tensin homolog to inhibit phosphorylation of AKT in cultured myotubes, providing another mechanism for ROCK1-mediated negative regulation of insulin signaling (43). Interestingly, in L6 myotubes, insulin inhibits ROCK1 to promote AMPK2α activity and subsequently inhibit the lipogenic transcription factor SREBP-1c (42). This suggests a mechanism in which insulin may inhibit ROCK1 activity to prevent inhibitory IRS1 phosphorylation and ultimately facilitate downstream insulin signaling.
The association between skeletal muscle ROCK1 and metabolic dysfunction has been well-documented in animal models; however, much like ROCK1 in adipose tissue, basal ROCK1 activity may be essential to skeletal muscle glucose uptake. In humans, VL ROCK1 activity positively correlates with glucose disposal in lean subjects, while insulin-stimulated ROCK1 activity is impaired in those with diabetes or obesity, possibly due to elevated levels of the ROCK1 antagonist RhoE (35). Furthermore, systemic ROCK1 knockout impairs skeletal muscle insulin signaling, and ROCK1 suppression in myoblasts blunts glucose uptake in a PI3kinase-dependent manner (25, 33, 34). Overall, ROCK1-modulated glucose uptake in skeletal muscle is similarly paradoxical to its role in adipose tissue, both regarding mechanisms and complexity (Section 3.2) (25, 33).
Summary of ROCK1 in Peripheral Tissues
To date, similar metabolic roles of ROCK1 have been identified in liver, skeletal muscle, and adipose tissue. ROCK1 in peripheral tissues inhibits AMPK2α, which results in changes in gene expression and downstream phosphorylation events to ultimately decrease energy expenditure and increase lipogenesis. ROCK1 also interferes with insulin signaling to increase blood glucose levels and ROCK1 overactivity is associated with metabolic disease states and related comorbidities including obesity, insulin resistance, and dyslipidemia. Despite this, increasing evidence suggests basal ROCK1 activity is also paradoxically essential to glucose disposal and insulin/PI3kinase signaling (Figure 1). Overall homeostatic ROCK1 function in peripheral tissues appears to be critical to metabolic health and future studies should focus on the differences between healthy and pathological ROCK1 activity.
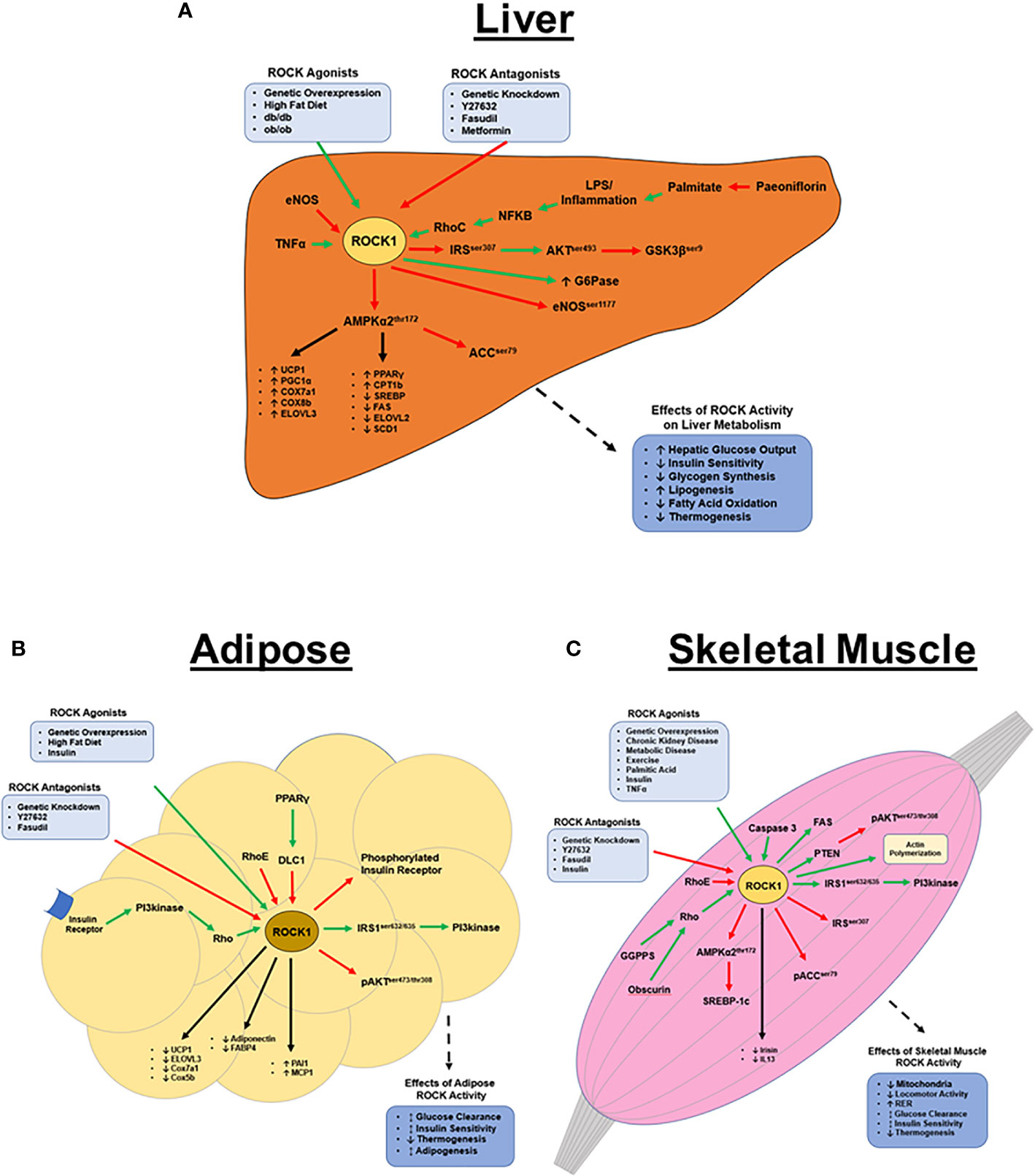
Figure 1 ROCK1’s metabolic functions in peripheral tissues. (A) Hepatic ROCK1 overactivity is associated with disordered metabolic regulation; conversely, downregulation of ROCK1 is therapeutic in metabolic disease models. ROCK1 primarily regulates lipid metabolism and thermogenesis via AMPK signaling, however the mechanisms underlying ROCK1’s role in insulin signaling remain unclear. The pathophysiology of ROCK1 overactivity in metabolic disease is poorly understood, however sustained inflammation and subsequent NF-κB signaling may be an upstream ROCK1 agonist. (B) Basal adipose ROCK1 activity is critical to homeostatic glucose metabolism; however, overactivity of ROCK1 is associated with metabolic disease phenotypes. ROCK1 is a negative regulator of thermogenic, adipogenic, and mitochondrial gene expression. Mechanistically, insulin and PI3kinase signaling are upstream activators of ROCK, while RhoE and DLC1 are antagonists. Downstream of ROCK1 includes a paradoxical insulin signaling mechanism, where ROCK1 activates IRS1 and PI3kinase, but also attenuates activation of insulin receptor and AKT. (C) Similar to in adipose, ROCK1 paradoxically regulates glucose metabolism in skeletal muscle. Basal ROCK1 function is critical to glucose regulation, but overactivity is associated with metabolic disease. ROCK1 regulates metabolism in skeletal muscle by downregulating AMPK, ACC, and AKT signaling, but also activates PI3kinase, FAS, and SREBP-1c. (Green arrows indicate activation; red arrows indicate inhibition; black arrows indicate regulation of gene expression).
ROCK1 in the Central Nervous System (CNS)
Hypothalamic ROCK1 Regulates Metabolism-
Unlike liver, adipose, and skeletal muscle, ROCK1 activity in the hypothalamus is reduced in db/db and DIO mice (45). Furthermore, hypothalamic ROCK1 knockout in healthy mice results in excessive food intake, dyslipidemia, and obesity, while ROCK1 overexpression has opposite effects (45, 46). One study observed fasudil treatment to increase food intake and gene expression of the orexigenic neuropeptide, neuropeptide Y, which is predominantly expressed in the arcuate nucleus of the hypothalamus (ARC) (47). Considering this, studies have identified novel roles for hypothalamic ROCK1 to regulate energy balance via ARC neuron populations.
ROCK1 Regulates ARC Neurons-
The ARC, located in the medio-basal hypothalamus, contains both the orexigenic neuropeptide Y/agouti-related peptide (NPY/AgRP) -expressing and the anorexic proopiomelanocortin-expressing neuron populations (48–50). Disruption of ROCK1 in either of these neuron populations results in disordered neuronal activity and metabolism (45, 46). For example, deletion of ROCK1 in NPY/AgRP neurons results in increased NPY/AgRP activity and accelerated weight gain in chow-fed and DIO mice. These mice exhibit decreased resting energy expenditure and locomotor activity with increased serum TG’s (45, 46). Similarly, ROCK1 deletion in POMC neurons leads to POMC hypoactivity and obesity due to reduced locomotor activity, while whole-ARC ROCK1 deletion has even greater effects (46).
Tyrosine hydroxylase is the rate-limiting enzyme in dopamine synthesis (51), and activation of tyrosine hydroxylase-expressing (TH) neurons in the ARC has recently been shown to increase food intake and body weight (52). While RhoA deletion in TH neurons (RhoA-TH-/-) has no effects on energy balance in chow-fed mice, DIO RhoA-TH-/- mice experience accelerated weight gain and adiposity due to increased food intake, despite no differences in energy expenditure or glucose regulation (53). Additionally, hypothalamic NPY and AgRP mRNA is elevated in RhoA-TH-/- mice, suggesting RhoA/ROCK1 in TH neurons likely regulates other post-synaptic neuron populations as well (53). Overall, studies in the hypothalamus highlight a prominent role for ROCK1 to regulate ARC neurons involved in metabolic regulation; however, nonspecific ROCK1 knockdown in the hypothalamus results in a much more robust metabolic phenotype (45, 46, 53). Thus, ROCK1 likely regulates other, currently unidentified, neuron populations, in addition to NPY/AgRP, POMC, and TH neurons.
ROCK1 Facilitates Hypothalamic Leptin Signaling-
Leptin is a potent adipokine that regulates ARC neurons to increase energy expenditure and suppress food intake (48, 53, 54). Deficiency in leptin, its receptor (LepR), or its downstream signaling results in hyperphagia, hyperglycemia, and obesity (48, 55). Interestingly, RhoA or ROCK1 deletion in NPY/AgRP, POMC, or TH neurons impairs leptin-mediated signaling and regulation of these respective neurons (45, 46, 53). Furthermore, hyper-leptinemia is observed in hypothalamic ROCK1 knockout mice, suggesting the involvement of ROCK1 in the development of leptin resistance seen in metabolic disease states (45, 46, 56).
Considering the strong association between ROCK1 and leptin activity, ROCK1 has been identified as a cell signaling molecule directly involved in LepR action. Following leptin binding to LepR, ROCK1 phosphorylates JAK2, which stimulates dimerization and phosphorylation of STAT3 (46). Phosphorylated STAT3 stimulates nuclear translocation and transcription of target genes, including POMC and signal of cytokine signaling 3 (SOCS3), each of which act to maintain energy homeostasis (57, 58). In addition to this leptin➔ROCK1➔JAK2➔STAT3 signaling mechanism, ROCK1 likely functions via other signaling pathways as well. For example, RhoA deletion in TH neurons also increases sensitivity to the hunger-inducing hormone ghrelin through unknown mechanisms (53). Insulin also modulates NPY/AgRP and POMC neurons and, like in peripheral tissues, may also facilitate hypothalamic ROCK1 function (54, 59–61).
Summary of ROCK1 in the CNS-
In summary, hypothalamic ROCK1 regulates various neuron populations, including NPY/AgRP, POMC, and TH neurons, to decrease food intake and increase energy expenditure, with no obvious effects on glucose metabolism. Mechanistically, ROCK1 directly mediates leptin signaling and impairs ghrelin signaling through unknown mechanisms and the importance of these functions is underscored by obesity manifesting when hypothalamic ROCK1 function is impaired (Figure 2). The seemingly conflicting functions of central and peripheral ROCK1 are teleologically perplexing, and the reasons for these differences are unclear. Despite this. the various tissue-specific models described in this review cumulatively indicate both hypo- and hyper-ROCK1 activity have drastic metabolic effects, clearly demonstrating the critical nature of maintaining homeostatic ROCK1 function.
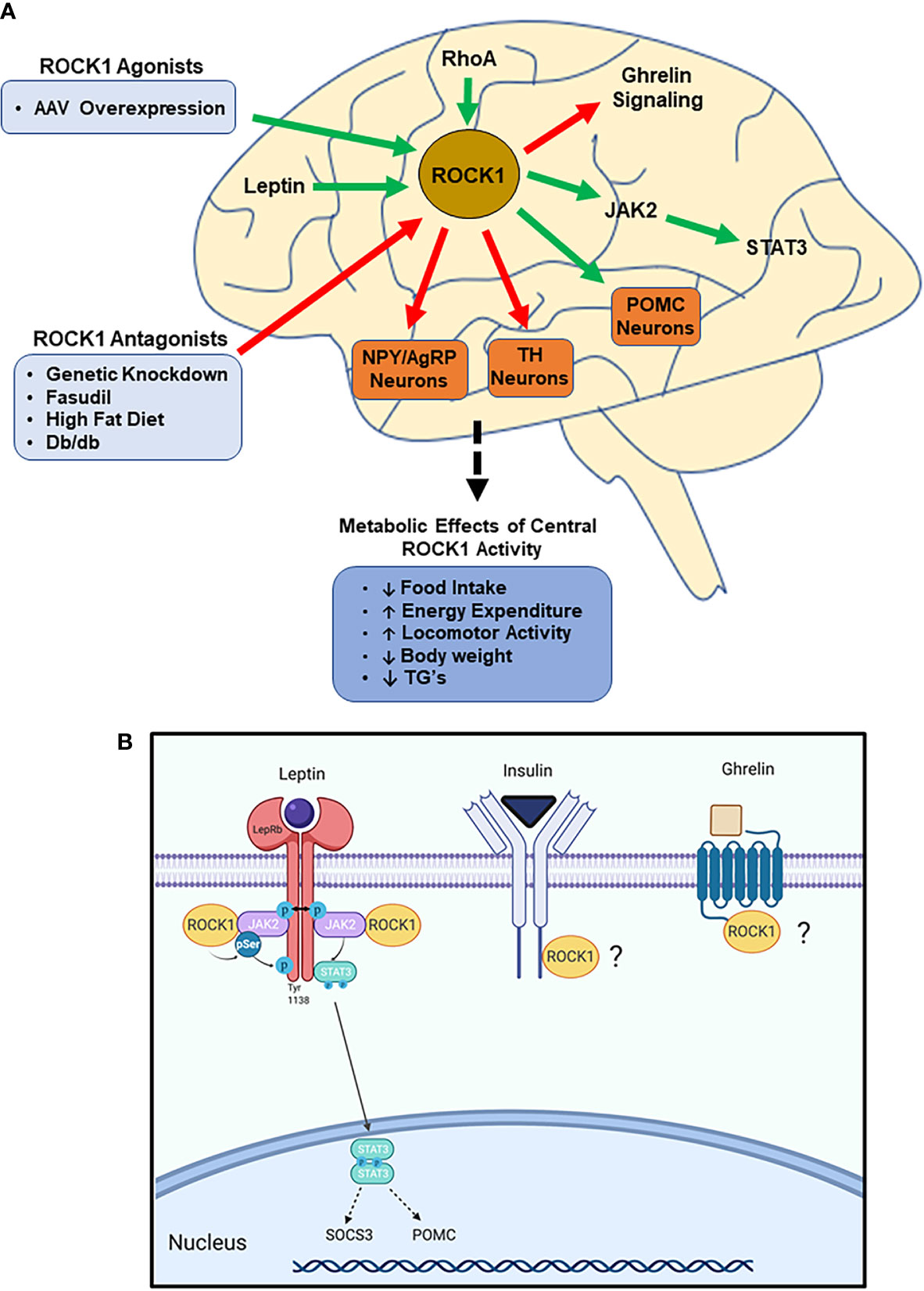
Figure 2 ROCK1 is critical to central nervous system (CNS)-mediated regulation of energy homeostasis. (A) Impaired ROCK1 activity in the hypothalamus is associated with disordered energy homeostasis. ROCK1 inhibits NPY/AgRP and TH neurons, while stimulating POMC neurons. Mechanistically, ROCK1 facilitates leptin signaling and attenuates ghrelin signaling. (Green arrows indicate activation; red arrows indicate inhibition). (B) Following leptin binding to LepR, ROCK1 phosphorylates JAK2, which stimulates dimerization and phosphorylation of STAT3. Subsequent STAT3 nuclear translocation elicits transcriptional changes including increased POMC and SOCS3 mRNA. Other potential mediators of hypothalamic ROCK1 action may be ghrelin and insulin receptor signaling.
Concluding Remarks
Many studies have used chemical inhibitors and whole-body genetic manipulation to identify ROCK1 as a prominent homeostatic regulator of diverse metabolic functions; however, these studies are limited in their isoform and tissue-specific insight. Recently, technological advances have facilitated development of novel models utilizing tissue-specific approaches, which have greatly enhanced our understanding of ROCK1’s functions. These studies have observed critical functions for ROCK1 in various metabolic tissues, including tissue-specific action in liver, adipose tissue, skeletal muscle, and hypothalamus to regulate food intake, thermogenesis, locomotor activity, glucose metabolism, and/or lipid metabolism. The molecular mechanisms underlying these functions are complex, underscored by disease states manifesting in response to ROCK1 overactivity, despite basal ROCK1 activity being critical to homeostatic maintenance of many physiological functions. Additionally, elevated ROCK1 activity consistently is associated with various metabolic disease states, suggesting ROCK1 may be useful as a preclinical marker of diabetes and obesity. Nonspecific ROCK1 inhibitors fasudil and Y-27632 demonstrate inhibitor pharmacotherapy is beneficial for these diseases; however, adverse effects such as hypotension, insulin resistance, and obesity are observed when ROCK expression/activity is non-specifically altered or systemically downregulated (25, 32–35, 62, 63). This once again highlights the importance of tissue-specific targeting of ROCK1, for example, via mannose-6-phosphate carriers (62, 64, 65), vitamin-A-coupled lysosomes (66), or genetic engineering. Overall, these tissue-specific approaches will greatly facilitate deciphering the many critical metabolic functions of ROCK1 and, ultimately, may result in the development of novel treatments for metabolic 2974disorders.
Author Contributions
TL and DS wrote this manuscript. HH supervised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J (1996) 15:2208–16. doi: 10.1002/j.1460-2075.1996.tb00574.x
2. Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett (1996) 392:189–93. doi: 10.1016/0014-5793(96)00811-3
3. Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J (1996) 15:1885–93. doi: 10.1002/j.1460-2075.1996.tb00539.x
4. Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem (1995) 270:29051–4. doi: 10.1074/JBC.270.49.29051
5. Amano M, Fukata Y, Kaibuchi K. Regulation and Functions of Rho-Associated Kinase. Exp Cell Res (2000) 261:44–51. doi: 10.1006/excr.2000.5046
6. Riento K, Ridley AJ. ROCKs: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol (2003) 4:446–56. doi: 10.1038/nrm1128
7. Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and Safety of Fasudil in Patients With Stable Angina. J Am Coll Cardiol (2005) 46:1803–11. doi: 10.1016/j.jacc.2005.07.047
8. Liao JK, Seto M, Noma K. Rho Kinase (ROCK) Inhibitors. J Cardiovasc Pharmacol (2007) 50:17–24. doi: 10.1097/FJC.0b013e318070d1bd
9. Narumiya S, Ishizaki T, Uehata M. Use and properties of ROCK-specific inhibitor Y-27632. Methods Enzymol (2000) 325:273–84. doi: 10.1016/s0076-6879(00)25449-9
10. Ono-Saito N, Niki I, Hidaka H. H-Series Protein Kinase Inhibitors and Potential Clinical Applications. Pharmacol Ther (1999) 82:123–31. doi: 10.1016/S0163-7258(98)00070-9
11. Hirooka Y, Shimokawa H. Therapeutic Potential of Rho-Kinase Inhibitors in Cardiovascular Diseases. Am J Cardiovasc Drugs (2005) 5:31–9. doi: 10.2165/00129784-200505010-00005
12. Hu E, Lee D. Rho kinase as potential therapeutic target for cardiovascular diseases: opportunities and challenges. Expert Opin Ther Targ (2005) 9:715–36. doi: 10.1517/14728222.9.4.715
13. Tamura M, Nakao H, Yoshizaki H, Shiratsuchi M, Shigyo H, Yamada H, et al. Development of specific Rho-kinase inhibitors and their clinical application. Biochim Biophys Acta Proteins Proteom (2005) 1754:245–52. doi: 10.1016/J.BBAPAP.2005.06.015
14. Huang H, Lee S-H, Sousa-Lima I, Kim SS, Hwang WM, Dagon Y, et al. Rho-kinase/AMPK axis regulates hepatic lipogenesis during overnutrition. J Clin Invest (2018) 128:5335–50. doi: 10.1172/JCI63562
15. Okin D, Medzhitov R. The Effect of Sustained Inflammation on Hepatic Mevalonate Pathway Results in Hyperglycemia. Cell (2016) 165:343–56. doi: 10.1016/J.CELL.2016.02.023
16. Noda K, Godo S, Saito H, Tsutsui M, Shimokawa H. Opposing Roles of Nitric Oxide and Rho-Kinase in Lipid Metabolism in Mice. Tohoku J Exp Med (2015) 235:171–83. doi: 10.1620/tjem.235.171
17. Noda K, Nakajima S, Godo S, Saito H, Ikeda S, Shimizu T, et al. Rho-Kinase Inhibition Ameliorates Metabolic Disorders through Activation of AMPK Pathway in Mice. PloS One (2014) 9:e110446. doi: 10.1371/journal.pone.0110446
18. Gupta J, Gaikwad AB, Tikoo K. Hepatic expression profiling shows involvement of PKC epsilon, DGK eta, Tnfaip, and Rho kinase in type 2 diabetic nephropathy rats. J Cell Biochem (2010) 111:944–54. doi: 10.1002/jcb.22783
19. Zhou H, Fang C, Zhang L, Deng Y, Wang M, Meng F. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, ameliorates hepatic fibrosis in rats with type 2 diabetes. Chin Med J (Engl) (2014) 127:225–31. doi: 10.3760/cma.j.issn.0366-6999.20131917
20. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology (2016) 150:956–67. doi: 10.1053/j.gastro.2015.12.037
21. Ma Z, Liu H, Wang W, Guan S, Yi J, Chu L. Paeoniflorin suppresses lipid accumulation and alleviates insulin resistance by regulating the Rho kinase/IRS-1 pathway in palmitate-induced HepG2Cells. Biomed Pharmacother (2017) 90:361–7. doi: 10.1016/J.BIOPHA.2017.03.087
22. Ma Z, Zhang J, Ji E, Cao G, Li G, Chu L. Rho kinase inhibition by fasudil exerts antioxidant effects in hypercholesterolemic rats. Clin Exp Pharmacol Physiol (2011) 38:688–94. doi: 10.1111/j.1440-1681.2011.05561.x
23. Begum N, Sandu OA, Ito M, Lohmann SM, Smolenski A. Active Rho kinase (ROK-alpha ) associates with insulin receptor substrate-1 and inhibits insulin signaling in vascular smooth muscle cells. J Biol Chem (2002) 277:6214–22. doi: 10.1074/jbc.M110508200
24. Lee S-H, Huang H, Choi K, Lee DH, Shi J, Liu T, et al. ROCK1 isoform-specific deletion reveals a role for diet-induced insulin resistance. Am J Physiol Metab (2014) 306:E332–43. doi: 10.1152/ajpendo.00619.2013
25. Furukawa N, Ongusaha P, Jahng WJ, Araki K, Choi CS, Kim H-J, et al. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab (2005) 2:119–29. doi: 10.1016/J.CMET.2005.06.011
26. Kanda T, Wakino S, Homma K, Yoshioka K, Tatematsu S, Hasegawa K, et al. Rho-kinase as a molecular target for insulin resistance and hypertension. FASEB J (2006) 20:169–71. doi: 10.1096/fj.05-4197fje
27. Song P, Zhang M, Wang S, Xu J, Choi HC, Zou M-H. Thromboxane A2 Receptor Activates a Rho-associated Kinase/LKB1/PTEN Pathway to Attenuate Endothelium Insulin Signaling. J Biol Chem (2009) 284:17120–8. doi: 10.1074/jbc.M109.012583
28. Sordella R, Classon M, Hu K-Q, Matheson SF, Brouns MR, Fine B, et al. Modulation of CREB Activity by the Rho GTPase Regulates Cell and Organism Size during Mouse Embryonic Development. Dev Cell (2002) 2:553–65. doi: 10.1016/S1534-5807(02)00162-4
29. Lin G, Craig G, Zhang L, Yuen V, Allard M, Mcneill J, et al. Acute inhibition of Rho-kinase improves cardiac contractile function in streptozotocin-diabetic rats. Cardiovasc Res (2007) 75:51–8. doi: 10.1016/j.cardiores.2007.03.009
30. Wingard C, Fulton D, Husain S. Altered Penile Vascular Reactivity and Erection in the Zucker Obese-Diabetic Rat. J Sex Med (2007) 4:348–63. doi: 10.1111/j.1743-6109.2007.00439.x
31. Liu P-Y, Chen J-H, Lin L-J, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol (2007) 49:1619–24. doi: 10.1016/j.jacc.2006.12.043
32. Standaert M, Bandyopadhyay G, Galloway L, Ono Y, Mukai H, Farese R. Comparative effects of GTPgammaS and insulin on the activation of Rho, phosphatidylinositol 3-kinase, and protein kinase N in rat adipocytes. Relationship to glucose transport. J Biol Chem (1998) 273:7470–7. doi: 10.1074/jbc.273.13.7470
33. Chun K-H, Araki K, Jee Y, Lee D-H, Oh B-C, Huang H, et al. Regulation of Glucose Transport by ROCK1 Differs from That of ROCK2 and Is Controlled by Actin Polymerization. Endocrinology (2012) 153:1649–62. doi: 10.1210/en.2011-1036
34. Lee DH, Shi J, Jeoung NH, Kim MS, Zabolotny JM, Lee SW, et al. Targeted disruption of ROCK1 causes insulin resistance in vivo. J Biol Chem (2009) 284:11776–80. doi: 10.1074/jbc.C900014200
35. Chun K-H, Choi K-D, Lee D-H, Jung Y, Henry RR, Ciaraldi TP, et al. In vivo activation of ROCK1 by insulin is impaired in skeletal muscle of humans with type 2 diabetes. Am J Physiol Metab (2011) 300:E536–42. doi: 10.1152/ajpendo.00538.2010
36. Hara Y, Wakino S, Tanabe Y, Saito M, Tokuyama H, Washida N, et al. Rho and Rho-kinase activity in adipocytes contributes to a vicious cycle in obesity that may involve mechanical stretch. Sci Signal (2011) 4:ra3–3. doi: 10.1126/scisignal.2001227
37. Karnam P, Standaert ML, Galloway L, Farese RV. Activation and translocation of Rho (and ADP ribosylation factor) by insulin in rat adipocytes. Apparent involvement of phosphatidylinositol 3-kinase. J Biol Chem (1997) 272:6136–40. doi: 10.1074/JBC.272.10.6136
38. Sim CK, Kim S-Y, Brunmeir R, Zhang Q, Li H, Dharmasegaran D, et al. Regulation of white and brown adipocyte differentiation by RhoGAP DLC1. PloS One (2017) 12:e0174761. doi: 10.1371/journal.pone.0174761
39. Takahashi N, Nobusue H, Shimizu T, Sugihara E, Yamaguchi-Iwai S, Onishi N, et al. ROCK inhibition induces terminal adipocyte differentiation and suppresses tumorigenesis in chemoresistant osteosarcoma cells. Cancer Res (2019) 79:3088–99. doi: 10.1158/0008-5472.CAN-18-2693
40. Dankel SN, Røst TH, Kulyté A, Fandalyuk Z, Skurk T, Hauner H, et al. The Rho GTPase RND3 regulates adipocyte lipolysis. Metabolism (2019) 101:153999. doi: 10.1016/j.metabol.2019.153999
41. Tao W, Wu J, Xie BX, Zhao YY, Shen N, Jiang S, et al. Lipid-induced muscle insulin resistance is mediated by GGPPS via modulation of the RhoA/Rho kinase signaling pathway. J Biol Chem (2015) 290:20086–97. doi: 10.1074/jbc.M115.657742
42. Tang S, Wu W, Tang W, Ge Z, Wang H, Hong T, et al. Suppression of Rho-kinase 1 is responsible for insulin regulation of the AMPK/SREBP-1c pathway in skeletal muscle cells exposed to palmitate. Acta Diabetol (2017) 54:635–44. doi: 10.1007/s00592-017-0976-z
43. Peng H, Cao J, Yu R, Danesh F, Wang Y, Mitch WE, et al. CKD stimulates muscle protein loss via rho-associated protein kinase 1 activation. J Am Soc Nephrol (2016) 27:509–19. doi: 10.1681/ASN.2014121208
44. Zhou X, Li R, Liu X, Wang L, Hui P, Chan L, et al. ROCK1 reduces mitochondrial content and irisin production in muscle suppressing adipocyte browning and impairing insulin sensitivity. Sci Rep (2016) 6:1–14. doi: 10.1038/srep29669
45. Huang H, Lee SH, Ye C, Lima IS, Oh B-C, Lowell BB, et al. ROCK1 in AgRP Neurons Regulates Energy Expenditure and Locomotor Activity in Male Mice. Endocrinology (2013) 154:3660–70. doi: 10.1210/en.2013-1343
46. Huang H, Kong D, Byun KH, Ye C, Koda S, Lee DH, et al. Rho-kinase regulates energy balance by targeting hypothalamic leptin receptor signaling. Nat Neurosci (2012) 15:1391–8. doi: 10.1038/nn.3207
47. Butruille L, Mayeur S, Duparc T, Knauf C, Moitrot E, Fajardy I, et al. Prenatal fasudil exposure alleviates fetal growth but programs hyperphagia and overweight in the adult male rat. Eur J Pharmacol (2012) 689:278–84. doi: 10.1016/j.ejphar.2012.05.040
48. Huo L, Gamber K, Greeley S, Silva J, Huntoon N, Leng XH, et al. Leptin-Dependent Control of Glucose Balance and Locomotor Activity by POMC Neurons. Cell Metab (2009) 9:537–47. doi: 10.1016/j.cmet.2009.05.003
49. Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest (2011) 121:1424–8. doi: 10.1172/JCI46229
50. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature (2006) 443:289–95. doi: 10.1038/nature05026
51. Daubner SC, Le T, Wang S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem Biophys (2011) 508:1–12. doi: 10.1016/j.abb.2010.12.017
52. Zhang X, Van Den Pol AN. Hypothalamic arcuate nucleus tyrosine hydroxylase neurons play orexigenic role in energy homeostasis. Nat Neurosci (2016) 19:1341–7. doi: 10.1038/nn.4372
53. Skov LJ, Ratner C, Hansen NW, Thompson JJ, Egerod KL, Burm H, et al. RhoA in tyrosine hydroxylase neurones regulates food intake and body weight via altered sensitivity to peripheral hormones. J Neuroendocrinol (2019) 31:e12761. doi: 10.1111/jne.12761
54. Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep (2012) 13:1079–86. doi: 10.1038/embor.2012.174
55. Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, et al. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest (2001) 108:1113–21. doi: 10.1172/JCI13914
56. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat Med (1995) 1:1311–4. doi: 10.1038/nm1295-1311
57. Gong L, Yao F, Hockman K, Heng HH, Morton GJ, Takeda K, et al. Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology (2008) 149:3346–54. doi: 10.1210/en.2007-0945
58. Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased Pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology (2007) 148:72–80. doi: 10.1210/en.2006-1119
59. Al-Qassab H, Smith MA, Irvine EE, Guillermet-Guibert J, Claret M, Choudhury AI, et al. Dominant Role of the p110β Isoform of PI3K over p110α in Energy Homeostasis Regulation by POMC and AgRP Neurons. Cell Metab (2009) 10:343–54. doi: 10.1016/J.CMET.2009.09.008
60. Könner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab (2007) 5:438–49. doi: 10.1016/j.cmet.2007.05.004
61. Mayer CM, Belsham DD. Insulin directly regulates NPY and AgRP gene expression via the MAPK MEK/ERK signal transduction pathway in mHypoE-46 hypothalamic neurons. Mol Cell Endocrinol (2009) 307:99–108. doi: 10.1016/j.mce.2009.02.031
62. Klein S, Van Beuge MM, Granzow M, Beljaars L, Schierwagen R, Kilic S, et al. HSC-specific inhibition of Rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J Hepatol (2012) 57:1220–7. doi: 10.1016/J.JHEP.2012.07.033
63. Anegawa G, Kawanaka H, Yoshida D, Konishi K, Yamaguchi S, Kinjo N, et al. Defective endothelial nitric oxide synthase signaling is mediated by rho-kinase activation in rats with secondary biliary cirrhosis. Hepatology (2008) 47:966–77. doi: 10.1002/hep.22089
64. Klein S, Frohn F, Magdaleno F, Reker-Smit C, Schierwagen R, Schierwagen I, et al. Rho-kinase inhibitor coupled to peptide-modified albumin carrier reduces portal pressure and increases renal perfusion in cirrhotic rats. Sci Rep (2019) 9:2256. doi: 10.1038/s41598-019-38678-5
65. van Beuge MM, Prakash J, Lacombe M, Gosens R, Post E, Reker-Smit C, et al. Reduction of fibrogenesis by selective delivery of a Rho kinase inhibitor to hepatic stellate cells in mice. J Pharmacol Exp Ther (2011) 337:628–35. doi: 10.1124/jpet.111.179143
Keywords: Rho-kinase, metabolism, energy balance, glucose metabolism, lipid metabolism
Citation: Landry T, Shookster D and Huang H (2021) Tissue-Specific Approaches Reveal Diverse Metabolic Functions of Rho-Kinase 1. Front. Endocrinol. 11:622581. doi: 10.3389/fendo.2020.622581
Received: 28 October 2020; Accepted: 22 December 2020;
Published: 09 February 2021.
Edited by:
Lee E. Eiden, National Institutes of Health (NIH), United StatesReviewed by:
Klaas Poelstra, University of Groningen, NetherlandsJin Kwon Jeong, George Washington University, United States
Copyright © 2021 Landry, Shookster and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hu Huang, aHVhbmdoQGVjdS5lZHU=