 Milan Obradovic1
Milan Obradovic1 Emina Sudar-Milovanovic1
Emina Sudar-Milovanovic1 Sanja Soskic1
Sanja Soskic1 Magbubah Essack2Swati Arya3Alan J. Stewart3
Magbubah Essack2Swati Arya3Alan J. Stewart3 Takashi Gojobori2,4
Takashi Gojobori2,4 Esma R. Isenovic1*
Esma R. Isenovic1*- 1Department of Radiobiology and Molecular Genetics, “VINČA” Institute of Nuclear Sciences - National Institute of the Republic of Serbia, University of Belgrade, Belgrade, Serbia
- 2Computer, Electrical and Mathematical Sciences and Engineering Division (CEMSE), Computational Bioscience Research Center, Computer (CBRC), King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 3School of Medicine, University of St Andrews, St Andrews, United Kingdom
- 4Biological and Environmental Sciences and Engineering Division (BESE), King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
The peptide hormone leptin regulates food intake, body mass, and reproductive function and plays a role in fetal growth, proinflammatory immune responses, angiogenesis and lipolysis. Leptin is a product of the obese (ob) gene and, following synthesis and secretion from fat cells in white adipose tissue, binds to and activates its cognate receptor, the leptin receptor (LEP-R). LEP-R distribution facilitates leptin’s pleiotropic effects, playing a crucial role in regulating body mass via a negative feedback mechanism between adipose tissue and the hypothalamus. Leptin resistance is characterized by reduced satiety, over-consumption of nutrients, and increased total body mass. Often this leads to obesity, which reduces the effectiveness of using exogenous leptin as a therapeutic agent. Thus, combining leptin therapies with leptin sensitizers may help overcome such resistance and, consequently, obesity. This review examines recent data obtained from human and animal studies related to leptin, its role in obesity, and its usefulness in obesity treatment.
Introduction
Obesity-associated co-morbidities such as hypertension, dyslipidemia, type 2 diabetes mellitus, fatty liver disease, heart disease, and some types of cancer cause about 3.4 million adults (over age 18) deaths in 2016, according to the World Health Organization (1). They further reported that an alarming 1.9 billion adults are overweight, and over 650 million overweight adults are obese. Hyperleptinemia and resistance to a reduction of body mass are two common characteristics of obesity (2). In this regard, studies report a strong positive association between serum leptin levels and the percentage of body fat (3, 4). Thus, pharmaceutical companies are pursuing the idea of using leptin-based drugs as a therapeutic strategy for weight loss (5, 6).
In 1994 Zhang et al. identified leptin as the product of the obese (ob) gene after characterizing genetically obese (ob/ob) mice (7). This factor was coined leptin the following year, derived from the Greek word leptos, meaning thin (8). Leptin regulates food intake, body mass, reproductive functioning and plays a vital role in fetal growth, proinflammatory immune responses, angiogenesis, and lipolysis (2, 9, 10). Studies demonstrated that the concentration of circulating leptin decreases during fasting (11) or energy restriction (12) but increases during refeeding (13), overfeeding (14), as well as during surgical stress (15). These effects provide an overview of how various pathways regulate the leptin signaling system to maintain body mass. For example, when the fat cells increase, leptin levels increase proportionally, then bind to leptin receptors (LEP-R) in the brain that send signals to inhibit food intake and increase energy expenditure (16, 17). However, when a positive energy balance (i.e., caloric intake exceeds energy expenditure) is sustained for critical periods, weight is gained (3, 16, 17). Here we review the literature to collate and provide a comprehensive summary of the relationship between leptin signaling and obesity.
Leptin and Its Cognate Receptor
The leptin molecule is 16 kDa in size and comprises 167 amino acids (including a 21 amino acid secretory signal sequence), and it exhibits the tertiary structure of a globular protein (18, 19). Leptin acts via its transmembrane receptors, the LEP-R, that exhibit structural similarity to the class I family of cytokine receptors, which include receptors for interleukins (IL), leukemia inhibitory factor (LIF), colony-stimulating factor 3 (CSF-3), growth hormone (GH), prolactin and erythropoietin (20–23). These family members have characteristic extracellular motifs, including four cysteine residues, a Trp-Ser-Xaa-Trp-Ser motif, and fibronectin type III (FN III) domains (24). LEP-R exists in several alternatively spliced variants labeled as LEP-Ra, LEP-Rb, LEP-Rc, LEP-Rd, LEP-Re, and LEP-Rf and the extracellular and transmembrane domains comprise over 800-amino acids and 34-amino acid, respectively, while a variable intracellular domain characteristic for each of the LEP-R isoforms (21–23, 25). The isoforms are classified into three classes: short, long, and secretive (23).
The Role of Leptin in the Regulation of Energy Balance
Brain lesion and stimulation research led to the discovery of the “satiety center” in the ventromedial hypothalamic nucleus (VMH) and the “hunger center” in the lateral hypothalamic nuclei (LH). This defines the dual-center model for feeding, proposing that energy input is provided through eating (26). Thus, energy balance is maintained when energy from food intake is equal to energy expenditure. About one year after the discovery of the leptin gene, it is shown that leptin regulates appetite and metabolism by inhibiting the synthesis and release of neuropeptide Y (NPY) in the arcuate nucleus (ARC) (27). Subsequently, it was discovered the LEP-R isoform b (LEP-Rb) in the VMH, ARC, LH, and the dorsomedial hypothalamic nucleus (DMH), which plays a crucial role in the regulation of energy balance and body mass (28). Earlier studies revealed that lesions of the ARC, VMH, or DMH could lead to hyperphagia and obesity in rats (29, 30), and lesions of the LH can lead to anaphylaxis (31). Later studies have demonstrated that leptin can inhibit neural pathways activated by appetite stimulants (orexigenic) to reduce energy intake and activate pathways targeted by anorexigenic to suppress appetite (32, 33). Examples of orexigenic neuropeptides include NPY and the agouti-related protein (AgRP). The product of proopiomelanocortin (POMC), alpha-melanocyte-stimulating hormone (α-MSH), is an anorexigenic (34). Neurons that express AgRP, POMC, and melanocortin include those in the central melanocortin system involved in energy balance regulation (34, 35).
The interaction between the signaling of leptin and the dominant feeding regulation constitutes a simple model: leptin affects the transcription of POMC, whose α-MSH product is released into the synapse to activate neurons via binding to the melanocortin receptor (MCR) and leads to appetite-suppression (36, 37). Also, leptin inhibits NPY/AgRP synthesis in neurons, which, in turn, reduces the agonistic effect of AgRP on MCR (Figure 1) (36, 37).
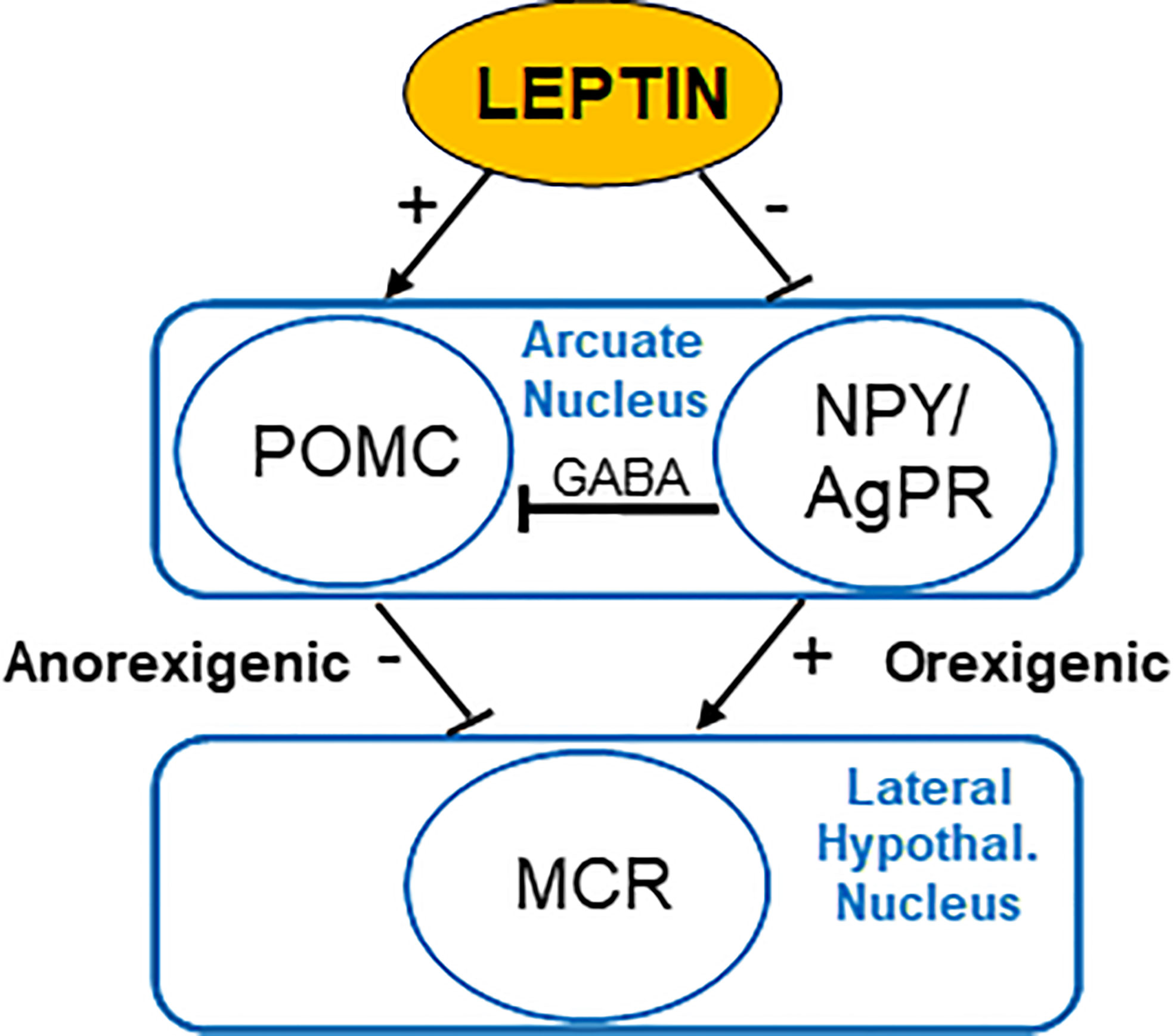
Figure 1 Regulation of appetite by leptin acting on the nucleus arcuatus of the hypothalamus. POMC, proopiomelanocortin; NPY, neuropeptide Y; AgRP, agouti-related protein; MCR, melanocortin receptors; GABA, γ-aminobutyric acid.
The significance of the melanocortin system is not only due to the direct action of leptin in the hypothalamus but also the fact that the loss of melanocortin 4 receptor (MC4R) function, a key MCR expressed in the hypothalamus, is the most common genetic cause of obesity in humans and occurs in 3-5% people with extreme obesity (38, 39). In brief, leptin regulates energy balance by modulating the activity of NPY/AgRP and POMC neurons in the ARC nucleus (34). Another mechanism of energy balance regulation was discovered by identifying rapid regeneration of the ARC nucleus’ neural circuits using leptin (40). Among ob/ob mice and wild-type mice are different synapses extended on NPY/AgRP and POMC neurons (40). Furthermore, leptin treatment normalized the synaptic density on NPY/AgRP and POMC neurons 6 hours after treatment, a few hours before it affected food intake (40). These findings indicate that leptin acts on the hypothalamus by regulating neuronal plasticity (34, 41).
Regulation of Leptin Secretion
Leptin is primarily produced in white adipose tissue. Still, smaller quantities have been detected in other body tissues, including the brown adipose tissue (BAT), placenta, fetal tissue, stomach, muscles, bone marrow, teeth, and brain (42, 43). Leptin circulates in the blood in both free and protein-bound forms, where the free form of leptin is the biologically active form (43). The equilibrium between free and bound leptin regulates leptin bioavailability (39). Leptin can enter the central nervous system (CNS) (in the area of the choroid plexus) by receptor-mediated transport (44). The LEP-R isoform plays a particularly significant role in transporting leptin through the blood-brain barrier (BBB) (44). A complex array of endocrine, neuroendocrine, and paracrine signals governs leptin synthesis and secretion (45). The secretion of leptin is proportional to body mass and nutritional status. The serum leptin levels decrease during starvation, associated with an adaptive physiological response to the state of starvation (45). Furthermore, leptin secretion is higher in subcutaneous than in visceral adipose tissue (46, 47).
Food intake, total body fat, as well as several hormones regulate leptin secretion (45). Insulin and, to a lesser extent, other pancreatic peptide hormones, including amylin, glucagon, and pancreatic polypeptides, reduce food intake and affect leptin secretion (48). Insulin is the primary regulator of leptin production (49). Prolonged hyperinsulinemia leads to an increase in leptin’s plasma concentration, while short-term hyperinsulinemia does not cause such a change (49). Moreover, insulin infusion increases plasma leptin concentration in humans (50), and rodents with type 1 diabetes exhibit significantly reduced leptin levels (51). Based on such in vitro studies, it is assumed that insulin stimulates leptin production via glucose metabolism (51–53). The blockade of glucose transports or glycolysis in the presence of high insulin levels inhibits the expression and secretion of leptin in adipocytes (51, 53). Changes in glucose metabolism due to the application of a high-fat diet for 24 hours explain the reduced level of leptin in human circulation and thus contribute to a high-fat diet in promoting weight gain and obesity (54). The reduced level of leptin in the circulation observed during high energy consumption is associated with humans’ hunger (45). Therefore, leptin flows from the adipocyte into the bloodstream, passes through the BBB, and arrives in areas of the brain involved in regulating the hypothalamus’s energy balance (55). Unlike insulin, catecholamines bind to β2- and β3-adrenergic receptors to inhibit leptin synthesis (52), indicating a link between neuroendocrine and sympathetic control of adipose tissue endocrine function, i.e., the existence of negative feedback between the brain and adipose tissue (56). Corticosteroids and tumor necrosis factor α (TNF-α) stimulate leptin synthesis, while thyroid hormones are likely to decrease it (49).
Molecular Mechanisms of Leptin Action
The distribution of the LEP-R facilitates the pleiotropic effects of leptin (23). The binding of leptin to its receptor initiates numerous signal transduction pathways and, as a result, regulates a range of cellular functions in the body (19, 23). LEP-R, as a member of the type I cytokine receptor family, signals via the Janus kinase family (Figure 2) of tyrosine kinases (57). The intracellular domain of all LEP-R isoforms contains in the juxtamembrane region a “box” 1 -JAK-binding domain, while LEP-Rb also includes a “box” 2 motif and a signal transducer and activator of transcription (STAT)-binding sites (23, 58–60). Usually, functional receptors for cytokines contain the box 1 motif required for the interaction and activation of JAK (61). Box 2 also plays a role in the interactions and selectivity of JAK isoforms. However, for leptin signaling, only box 1 and an Ala-Ala motif in the immediate environment are essential for JAK activation (62, 63). Although initially only LEP-Rb was observed as an isoform involved in signaling, it has also been demonstrated for the short isoforms (64–66). Mainly JAK2 members of the JAK family proteins are associated with membrane-proximal sequences of the intracellular receptor domain, which is phosphorylated after binding the ligand.

Figure 2 Leptin signaling. L- leptin; LEP-R- leptin receptor; IRS 1/2, insulin receptor substrate 1/2; JAK 2, Janus kinase 2; PI3K, phosphoinositide 3-kinase; SH2, Src-like homology 2; SHP-2, SH2 domain-containing protein tyrosine phosphatase; SOCS3, suppressor of cytokine signaling 3; STAT, signal transducer and activator of transcription.
LEP-R and other cytokine receptors do not have kinase activity but couples with tyrosine kinases. After LEP-R binds leptin, LEP-R undergoes a conformational change, critical for leptin signaling and activation of the associated JAK2. JAK2 autophosphorylates and simultaneously phosphorylates tyrosine residues on the functional LEP-R’s intracellular domain, allowing binding of STAT proteins and their subsequent translocation to the nucleus where they act as transcription factors (23). Also, cytokine signaling 3 (SOCS3) and protein tyrosine phosphatase 1B (PTP1B) can act as suppressors of the JAK-STAT pathway (23, 67, 68). PTP1B is a known negative modulator of leptin signal transduction via the de-phosphorylation of JAK2. Excessive expression of PTPB1 reduces phosphorylation of JAK2 and inhibits the transcription of SOCS3 and c-fos, which are induced by leptin (23). Furthermore, isoforms of LEP-R with a long intracellular domain may also activate other signaling pathways. The binding of leptin to LEP-R also activates phosphoinositol-3 kinase (PI3K) (69) and mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK) (70) signaling cascades. The activation of each of these pathways contributes to leptin’s anorexigenic effects (suppressing appetite, stimulating weight loss, and increasing thermogenesis) (69–71).
It is important to note that distinct signal transduction pathways are responsible for mediating the leptin’s metabolic effects compared with its cardiovascular effects. For example, the JAK2/STAT3 pathway is primarily responsible for regulating gene expression changes, while the PI3K pathway often signals more rapidly through phosphorylation of cytoplasmic proteins. The PI3K pathway plays an important role in leptin’s acute effects, such as regulating food intake and arterial hypertension (72). However, the Jak/STAT3, MAPK, and PI3K pathways appear to collectively regulate energy balance (72).
The effects of leptin are similar to other acute phase reactants; it increases the secretion of multiple inflammatory cytokines such as IL-6, IL-12, and TNF-α (73). In turn, exposure to inflammatory stimuli such as TNF-α and IL-1 increases leptin expression in the adipose tissue and circulating leptin, which creates a feedback loop that promotes inflammation (74, 75). This feedback loop emphasizes how leptin promotes low-grade inflammation since the proinflammatory mediators increase leptin expression and other acute phase reactants that promote chronic inflammation.
The effects of leptin are manifold; it stimulates the expression of IL-1Rα, a cluster of differentiation (CD) 25, CD39, CD69, and CD71 (76), and the production of proinflammatory cytokines TNF-α and IL-6 (77) in macrophages. The number of macrophages present in white adipose tissue correlates directly with obesity, i.e., obese individuals have more macrophages in adipose tissue (78, 79). The adipocyte-produced cytokines, CC-chemokine ligand 2 (CCL2), contribute to this macrophage infiltration process. The macrophages and adipocytes in adipose tissue are major TNFα and IL-6 sources in obese individuals, respectively. Thus, together these adipose tissue cells are also involved in a feedback loop that perpetuates macrophage recruitment and production of proinflammatory cytokines. These feedback loops explain why obesity is associated with chronic pro-inflammatory signaling pathways, abnormal cytokine production, and increased acute-phase reactants (80) and why obesity increases an individual’s risk of developing inflammatory-based diseases and immune-mediated disorders (80–82).
Leptin and Obesity
Leptin Expression in Obesity
Severe early obesity develops from rare genetic mutations that affect leptin signaling (2, 83). Such mutations often lead to congenital leptin deficiency or high but ineffective leptin and leptin resistance (84). Hyperleptinemia and resistance to reducing body mass are two characteristics of typical obesity (2, 3, 85). Leptin is overexpressed at the gene level in the adipose tissue of individuals with obesity (86). Furthermore, strong positive associations exist between plasma leptin levels and body fat percentage (87, 88). Other studies point towards leptin resistance. For example, plasma leptin levels and ob mRNA content decrease in individuals with obesity at the initial time of weight loss but increases as they continue to lose weight (88). Also, despite the expectation, leptin therapy’s termination does not result in weight gain and hyperleptinemia (89). There is also evidence that hyperleptinemia does not mimic the CNS consequences of chronic weight gain in diet-induced obese (DIO) mice (2, 89).
Different areas within the brain may be involved in the temporal and spatial dysregulation of neurological functioning associated with leptin under nutrient excess conditions (90). In this regard, Matheny et al. demonstrated that consuming a diet rich in high-fat induced leptin resistance in the ARC and ventral tegmental area (VTA), while medial basal hypothalamic regions stayed sensitive to leptin (91). Subsequently, the selective downregulation of Ob-Rb using lentivirus in ARC promoted diet-induced obesity in rats (92), demonstrating the ARC region has a role when leptin resistance develops in obesity. Interestingly, DIO is induced by the differential expression of leptin in brain regions, which may result from the various experimental methods used to regulate leptin expression. Moreover, these studies show the anorectic effects of leptin are not brain-specific. The ARC and VTA appear to be the main areas for the responsiveness of leptin. When the response to leptin is decreased in one region of the brain, it may be overcompensated by another, which suggests coordinated functioning. A high-fat diet may cause SOCS3 expression and activation of STAT3 resistance by leptin in POMC (93), ARC (94, 95), and AgRP neurons in rodents. Also, in AgRP neurons, the expression of SOCS3 decreases after shifting from high-fat to low-fat diets, indicating that those neurons may be more sensitive to leptin than POMC neurons (90, 96).
Experiments on obese mice confirmed a polymorphism in the ob gene (97, 98). This polymorphism alters the leptin protein function such that mice become morbidly obese (97, 98). Similarly, mice with a polymorphism in the gene encoding LEP-R, display altered leptin signaling that leads to obesity (99, 100). A single-nucleotide polymorphism identified in the 5’-untranslated region of the leptin gene (LEP -2548 G/A polymorphism) and its association with obesity is the most studied in humans. Still, the literature data are inconsistent (101–105). Carayol et al. designed and performed the first protein quantitative trait locus (pQTL) analysis in obesity and examined the role of genetic variations in determining protein level variation (106). They identified cis-pQTL and trans-pQTL signals associated with BMI at baseline and after the intervention and concluded that in human adipose tissue, human NTases belonging to the FAM46A family (family-with-sequence-similarity-46) was a negative regulator of leptin signaling (106).
A range of studies has investigated genetic and epigenetic factors that control leptin expression. For example, a distant leptin enhancer 1 (LE1) sequence has been identified 16 kb upstream from the transcription start site (TSS) of the ob gene. The LE1 contains a 17-bp non-canonical peroxisome proliferator-activated receptor gamma (PPARγ)/retinoid X receptor alpha (RXRα)-binding site, named leptin regulatory element 1 (LepRE1) that is essential for fat-regulated expression (107). In the same study, a functionally analogous LepRE1 site was also found in a second DNA regulatory element 13 kb downstream from the TSS of the ob gene. Non-coding RNAs have been implicated in the regulation of leptin gene expression, with its dysregulation linked to obesity (108) and in the development of hypothalamic leptin insensitivity (109). In addition, leptin has been shown to modulate the expression of miRNAs that target POMC mRNA (110). Epigenetic mechanisms linked to obesity that impact leptin and LEP-R expression are also at play. A study investigating DNA methylation in promoter sequences in bariatric surgery patients found higher Ob gene promoter methylation patterns in pre bariatric surgery patients compared to postoperative patients. Whilst DNA methylation of the LEP-R gene promoter was significantly higher in the postoperative group (111).
Leptin Resistance in Obesity
The term “leptin resistance” was coined shortly after discovering leptin in 1994 (7, 112, 113). The concept of leptin resistance implies the processes that result from a state of obesity impair the effects of leptin, thereby contributing to the formation of obesity and obstructing the potential efficacy of therapy with the use of exogenous leptin (3, 113). Leptin resistance occurs due to the leptin’s inability to reach the target cells, reduced LEP-R expression, or disturbed LEP-R signaling (3, 113). There are likely a number of molecular and genetic mechanisms that can lead to leptin resistance. Although relatively rare, loss of function mutations has been identified in genes encoding leptin and its receptors (28, 114, 115). More common mechanisms likely include defects in the pathways that regulate leptin synthesis. Leptin concentrations are directly dependent upon Ob gene transcription, which correlates with adipocyte size and lipid content. A complete understanding of how these factors are mechanistically linked or how such pathways are altered to trigger leptin resistance remains unclear. However, additional external stimuli, including eating behaviors and the circadian rhythm, modulate leptin expression and may play a role (116). It has been demonstrated that the decreased transport of leptin across BBB can lead to leptin resistance. Microcapillary vessels at the BBB express short truncated LEP-R forms that bind leptin and transport it to the nervous system (21, 117). It has been shown that even if plasma leptin levels rise above the range of 25–30 ng/mL, the concentration of leptin in cerebrospinal fluid does not increase further (118). Furthermore, it appears that excessive plasma leptin levels can result in decreased BBB permeability (119, 120).
A more nuanced or selective form of leptin resistance (SLR) has also been described, where the effects of leptin on appetite (and body mass) are absent. Still, the results of leptin on the sympathetic nervous system are preserved (3, 113). Interestingly, SLR characterizes preservation of sympathetic nerve activity (SNA) in the kidney and normal blood pressure (BP) responses to leptin action in obesity, despite alterations in responses to leptin in appetite, thermogenesis, and body mass (121). Two potential overlapping pathogenic mechanisms for SLR development have been proposed. Firstly, defects in differential leptin molecular signaling pathways that mediate selective as opposed to universal leptin action and secondly, defects in processes that regulate brain site-specific leptin actions (121).
Moreover, the latest studies unexpectedly propose that the brain renin-angiotensin system (RAS) mediates the leptin effects on renal and BAT thermogenic SNA with the absence of the effects of leptin on food intake (121). These findings imply that elevation or reduction of brain RAS activity may regulate leptin actions on BP and energy expenditure with no impact on the leptin-induced reduction in food intake (121). BAT thermogenesis is stimulated by leptin via central LEP-R, acting primarily through the sympathetic nervous system (122–124). A few hypothalamic areas (DMH, preoptic area (POA), paraventricular nucleus (PVN), VMH, ARC), but also some extra-hypothalamic regions as the nucleus tractus solitarius (NTS) participate in leptin-induced thermogenesis (125). The sympathetic regulation of BAT implicates neurons of the NTS that obtain vagal information and project nearby in the hypothalamic areas and the brainstem (126). Since NTS neurons have LEP-R, a specific administration of leptin to NTS leads to a decline of body mass accompanied with a decrease in food intake (124, 127).
Besides leptin actions/resistance on neurons in the hypothalamus, an SLR that extends to some extra-hypothalamic brain areas has been described. SLR in ARC of DIO mice has been shown, whereas other hypothalamic and extrahypothalamic nuclei remain leptin responsive (33, 95). Although DIO induced site-specific leptin resistance, constant overexpression of leptin in CNS induced leptin resistance in every brain region investigated. This suggests that SLR is distinctive to DIO and is not a nonspecific central neural response induced by high leptin exposure (91, 121).
Furthermore, in contrast to insulin, which induces improvement in SNA by acting in ARC as the only specific site, leptin takes action in few hypothalamic sites, all of which seem to interact in PVN (128). The main effects of insulin and leptin in states of obesity include sexually dimorphic alterations. The latest observations regarding the link between sexual dimorphism and sympathetic in the obese human population reveal that several variations exist in lean females that restrict the effects of leptin and insulin to increase SNA and/or BP (128). The first is that only during proestrus leptin increases SNA by the synergistic effects of raised concentrations of estrogen. The second one is that leptin and insulin do not induce the rise in SNA, leading to vasoconstriction and BP elevation in females while induced in males (128).
Furthermore, in obese males, sympathoexcitatory response to insulin is increased, unlike in obese females, where it is eliminated. Regarding leptin and its sympathoexcitatory response, it is also preserved or increased in obese males. In contrast, in obese females, the reproductive cycle is disturbed, and the sympathoexcitatory response to leptin is limited. This is probably due to the sexually dimorphic changes in NPY and POMC entrants to PVN. In obese males, stimulant PVN and NPY sympathoinhibitory response is abolished, and POMC entrant to PVN is elevated, probably due to increased cellular signaling of ARC and POMC induced by insulin.
Conversely, in obese females, stimulant NPY sympathoinhibitory response is preserved and not inhibited by insulin, and POMC insulin sensitivity may also be reduced. Until now, the mechanisms for obesity-induced sexually dimorphic alterations are not fully elucidated. There is a hypothesis that a considerable suppression of NPY via hypertensive, as opposed to a non-hypertensive branch of RAS, and a considerable POMC excitation, in obese males concerning obese females might be important. Nonetheless, the precise mechanisms in the base of insulin and leptin actions on ARC, NPY, or POMC and silenced in obese females have not yet been fully discovered (128).
Clinical Trials Examining the Effectiveness of Leptin-Based Interventions in Obesity
Combining therapies of leptin and leptin sensitizers can overcome leptin resistance (16, 129). Table 1 summarizes some essential clinical trials investigating the use of such agents. The first clinical study observing common polygenic or simple obesity with recombinant methionyl human leptin (r-metHuLeptin), also known as metreleptin, was carried out by Heymsfield and colleagues in 1999 (130). As the leptin dose increases, the group with obesity exhibits mean weight changes ranging from 0.7 kg to 7.1 kg over 24 weeks (130). The administration of pegylated human recombinant leptin (PEG-OB) was studied in men with obesity (131, 132) using weekly doses combined with a moderate diet. These pilot 12-week clinical studies demonstrated no difference in weight between the PEG-OB group and a placebo group (131, 132).
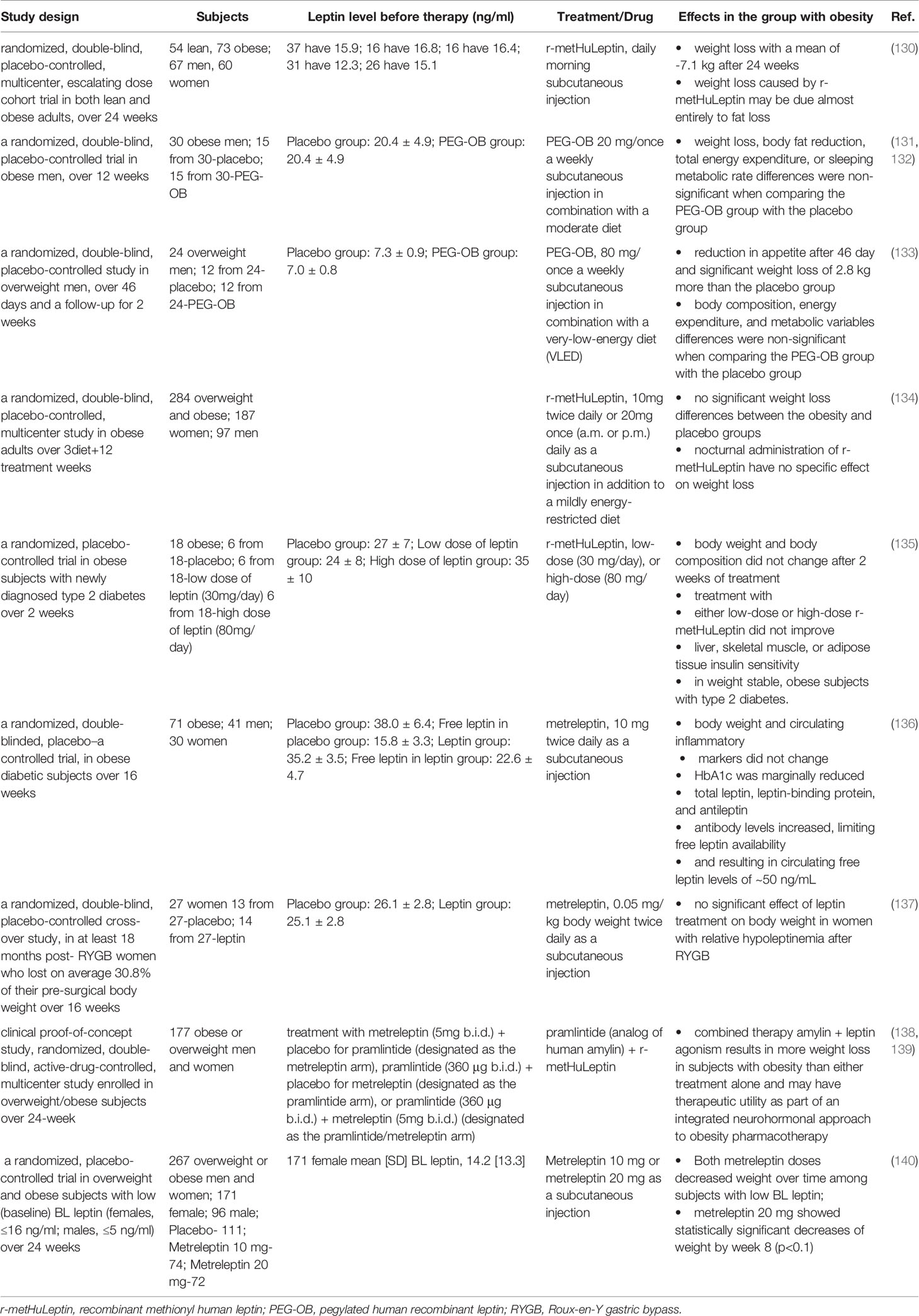
Table 1 Summary of some clinical trials involving the use of leptin-based therapies to treat obesity.
Similarly, Bartness et al. found that Fc-leptin’s weekly administration (engineered leptin) did not lead to weight loss than a placebo group (141). Hukshorn et al. investigated leptin’s influence in combination with a very low-calorie diet using PEG-OB treatment (80 mg administered weekly). They found that PEG-OB treatment resulted in significant additional weight loss in severely energy-restricted, overweight men. It suggests that a decrease in leptin concentrations during starvation increases appetite in humans (133). Also, Zelissen et al. carried out a study with calorific intake restricted to 500 kcal/day coupled with 10 mg of recombinant leptin administered daily (once or twice) for 12 weeks (134). This trial did not show significant weight loss differences between groups with obesity and placebo groups (134). Mittendorfer et al. conducted a clinical study to determine whether leptin treatment has weight loss–independent effects on insulin action in obese subjects with type 2 diabetes. They evaluated the impact of a low and high dose of r-metHuLeptin treatment on insulin action, glucose uptake, and lipolysis (135). The study results showed that r-metHuLeptin does not have weight-loss–independent, clinically important effects on insulin sensitivity in obese subjects with newly diagnosed type 2 diabetes (135).
Furthermore, r-metHuLeptin/metreleptin treatment did not alter body weight or circulating inflammatory markers but marginally reduced HbA1c in obese hyperleptinemic patients with type 2 diabetes (136). Also, total leptin, leptin-binding protein, and antileptin antibody levels increased, limiting free leptin availability (136). Korner et al. investigated whether leptin treatment to post-Roux-en-Y gastric bypass (RYGB) patients promotes further weight loss and shows no significant effect of leptin treatment on women’s body weight with relative hypoleptinemia after RYGB (137). Also, no changes were shown in percent fat mass, resting energy expenditure, thyroid hormones, or cortisol levels (137). A few clinical trials have reported a reduced tendency to regain weight after caloric restriction or weight loss coupled with recombinant leptin’s daily administration. Those studies examined effects on skeletal muscle and autonomic and neuroendocrine adaptation to mass body maintenance (142) and reproductive hormonal regulation (143). Potential mediators of weight regain, including the cortisol, growth hormone, and thyroid axes were not systematically affected (144–147).
Synergistic effects of leptin and amylin promote weight loss while preventing the compensatory reduction in energy expenditure associated with weight loss (138, 148). The combined therapy of leptin and pramlintide (an amylin analog) results in more weight loss in subjects with obesity than either treatment alone. This effect seems to be additive rather than synergistic, suggesting that amylin and its analog cannot increase leptin sensitivity (138, 139). The signaling pathways induced by leptin and amylin overlap and exert an additive effect in humans’ peripheral tissues (149).
Ravussin et al. administered metreleptin and pramlintide to 177 subjects with obesity, which resulted in a mean weight loss of 12.7% after 20 weeks (139). Unfortunately, some subjects developing anti-metreleptin antibodies that led to suspending the study. Later Chan et al. carried out a larger clinical trial with metreleptin and pramlintide on 579 patients with obesity and 134 patients with lipodystrophy for 20-52 weeks (150). Antibody development in patients with obesity or lipodystrophy was associated with higher leptin concentration, and higher antibody titers were associated with higher leptin concentration (150). Other studies have shown that exercise increases leptin sensitivity in human skeletal muscle (151), which may provide an alternative to pharmacological sensitizers.
Despite the remarkable results of leptin-based therapy on weight loss in genetically predisposed obese subjects (mutations in the leptin gene), this approach has a limited or completely absent effect on weight loss in subjects with common obesity, especially in hyperleptinemic patients (3, 152). Different responses to leptin-based therapy on weight loss in obese subjects in clinical studies may be explained by differences in treated population, study design, and administered therapy (leptin type, dosage, etc.). Also, leptin resistance and increased blood leptin level are significant factors that influence leptin-based therapy’s success (3, 152). Indeed, further clinical trials are needed to assess the selectivity and effectiveness of leptin-based therapy on weight loss regarding obesity, particularly defined the threshold of endogenous leptin level as a predictive factor for therapy response to determining the dose-response ratio of leptin-based therapy.
Development of New Leptin-Based Therapies for Obesity
As previously mentioned, leptin administration combined with a leptin sensitizer is a potential pharmacological strategy for weight loss (5, 6). To avoid difficulties associated with leptin’s short half-life and low stability, leptin analogs capable of binding and activating LEP-R are often used as another approach (6). A few studies have examined blocking negative regulators of the leptin signaling pathway, including SOCS3 and PTP1B, to enhance leptin administration effects in individuals with obesity (5, 153, 154). Inhibitors of PTP1B, such as thiazolidinedione and trodusquemine, suppress weight gain and decrease food intake and body weight in DIO mice (155).
Thus, modulation of endocytosis and the intracellular trafficking of LEP-R (6) may be ways to treat obesity. Leptin must cross the BBB through a specific and saturable transporter (156) to bind LEP-R in the hypothalamus. In obesity, high leptin levels lead to leptin resistance, which the transporter’s hyperactivation may cause by the high levels of leptin (6). Thus, another possible way to improve leptin therapy is to enhance its ability to cross the BBB, potentially fusing it with another molecule to improve uptake by vesicular endocytosis (6).
Although leptin reduces food intake and body mass and stimulates energy expenditure, obese subjects that develop leptin resistance did not respond to leptin-based clinical therapies (157, 158). However, several leptin-sensitizing compounds have been described to influence leptin action and promote beneficial effects in DIO hyperleptinemic mice (159–163). Leptin-sensitizing compounds may be divided into two groups (160). Compounds that enhance the anorectic effect of exogenous leptin but minimally affect weight loss, including meta-chlorophenylpiperazine (164), metformin (165), and betulinic acid (166). The other group comprises compounds that induce weight loss in obese animals with hyperleptinaemia and restore endogenous leptin signaling, such as glucagon-like peptide-1 (167) and heat shock protein 90 inhibitors (168, 169). Some of these leptin sensitizers are in clinical use for diabetes therapy, such as amylin and pramlintide, that enhance leptin action, probably increasing IL-6 production in microglia ventromedial hypothalamic nucleus that in turn activates pSTAT3 signaling in LepR neurons (170, 171). It was found that resveratrol attenuates the expression of leptin in adipocytes, elevates phosphorylation of STAT3 in the hypothalamus, and restores leptin resistance in adult offspring from HF rat mothers attenuating obesity (172). Ozcan and colleagues identified the natural compound celastrol as a potential leptin sensitizer and anti-obesity agent (161). They found that celastrol suppresses food intake, increases energy expenditure, and reduces body weight up to 45% in hyperleptinemic DIO mice (161). Although celastrol’s molecular mechanism regulates leptin sensitivity remains obscure, it was found that celastrol mediates leptin sensitization and exerts anti-obesity effects through increasing interleukin-1 receptor 1 (IL1R1) expression in the hypothalamus (173). Furthermore, celastrol promotes leptin sensitivity through inhibition of 6-phosphofructokinase (PFK) in skeletal muscle and activation of adenosine 5’monophosphate-activated protein kinase (AMPK), which leads to alterations in energy demand from glycolysis to the free fatty acid oxidation in skeletal muscle and increases energy expenditure (174). Another natural compound that acts as a potential leptin sensitizer with additional anti-diabetic actions is withaferin A (162). Treatment of DIO mice with withaferin A reduces body mass by 23%, fat mass by 35%, endoplasmic reticulum stress, hepatic steatosis, leptin level in the blood, and increases the potency of leptin and energy expenditure (162). These effects of withaferin A are exerted at least partly by sensitizing LEP-R signaling and increasing STAT3 phosphorylation in the hypothalamus of DIO mice (162). A partial reduction of plasma leptin level by leptin neutralizing antibody in obesity state improved leptin sensitivity and effectively led to weight loss and enhanced insulin sensitivity (159). Despite the impressive leptin sensitizing effects, using celastrol or withaferin A as an anti-obesity drug has some adverse effects (175–177). Also, these compounds minimally affect the body weight and metabolic disorders in genetically predisposed obesity, such as ob/ob and db/db mice, which lack leptin or the LEP-R (162).
Zhao et al., using leptin neutralizing antibodies in diverse mouse models, reported that hyperleptinemia triggers developing metabolic diseases (178). Partial leptin reduction has been characterized by returning leptin sensitivity in the hypothalamus, improving insulin sensitivity, and successfully diminishing weight gain (178). The same author suggested that increased leptin sensitivity resulting from partial leptin reduction is a new promising therapeutic tool for treating obesity (178). Another study by Ottaway et al. treated lean and obese mice with an antagonist of the leptin receptor. Regarding (diet-induced obese) DIO mice, antagonist improved body weight (BW) and feeding in lean mice (179). This improvement is related to the decline of expression of Socs3 in the hypothalamus (179). There is an estimation that DIO mice that have hyperleptinemia maintain leptin-feeding inhibition similar to lean mice and oppose an attitude that the stability of DIO in mice is based on resistance to endogenous leptin action (179).
Conclusions
The discovery of leptin has provided new insight into how to control obesity. The altered expression of leptin and its receptor leads to leptin resistance, which plays a critical role in obesity-related complications (3, 4). Despite knowing that leptin is one of the principal suppressors of appetite and leptin’s link with obesity, the treatment of obesity using leptin-based therapeutics remains to be fully explored (3, 4). The focus of further studies should be identifying new mechanisms of leptin regulation at the whole-body level to design new drugs that reverse leptin resistance. In this regard, understanding the pathogenesis of obesity-related disorders and the regulation of energy homeostasis by leptin should provide new alternatives in obesity treatment.
Author Contributions
MO, ES-M, and ERI designed, wrote and supervised the manuscript. SS, ME, and SA wrote the manuscript. AJS and TG critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is part of the collaboration between the Department of Radiobiology and Molecular Genetics, “VINČA” Institute of Nuclear Sciences - National Institute of the Republic of Serbia, University of Belgrade, Belgrade, Serbia, and Computational Bioscience Research Center (CBRC) at King Abdullah University of Science and Technology (KAUST). This work was funded by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Contract No#451-03-9/2021-14/200017) and KAUST grant OSR#4129 (awarded to EI and V.B.B.), which also supported MO and ES-M. ME has been supported by the KAUST Office of Sponsored Research (OSR) Award no. FCC/1/1976-17-01, and TG by the King Abdullah University of Science and Technology (KAUST) Base Research Fund (BAS/1/1059-01-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AgRP, agouti-related protein; AMPK, adenosine 5’monophosphate-activated protein kinase; ARC, arcuate nucleus; BAT, brown adipose tissue; BBB, blood-brain barrier; CCL2, CC-chemokine ligand 2; CD, cluster of differentiation; CNS, central nervous system; CSF-3, colony-stimulating factor 3; DIO, diet-induced obese; DMH, dorsomedial hypothalamic nucleus; FN III, fibronectin type III; GH, growth hormone; IL, interleukin; IL1R1, interleukin 1 receptor 1; JAK, Janus-associated kinase; LEP-R, leptin receptor; LEP-Rb, LEP-R isoform b; LH, lateral hypothalamic nuclei; LIF, leukemia inhibitory factor; MAPK/ERK - mitogen-activated protein kinases/extracellular signal-regulated kinase; MCR, melanocortin receptor; NPY, neuropeptide Y; NTS, nucleus tractus solitarius; ob, obese gene; PFK, 6-phosphofructokinase; PI3K, phosphoinositol-3 kinase; POMC, proopiomelanocortin; POA, preoptic area; pQTL, protein quantitative trait locus; PTP1B, protein tyrosine phosphatase 1B; PVNparaventricular nucleus; RYGB, Roux-en-Y gastric bypass; SLR, selective leptin resistance; SOCS3, cytokine signaling 3; STAT, signal transducer and activator of transcription; TNFα, tumor necrosis factor α; VMH, ventromedial hypothalamic nucleus; VTA, ventral tegmental area; WHO, World Health Organization; α-MSH, alpha-melanocyte-stimulating hormone.
References
1. WHO. World Health Statistics Overview 2019: Monitoring Health for the SDGs, Sustainable Development Goals. Geneva: World Health Organization (2019) (WHO/DAD/2019.1). Licence: CC BY-NC-SA 3.0 IGO.
2. Farr OM, Gavrieli A, Mantzoros CS. Leptin Applications in 2015: What Have We Learned About Leptin and Obesity? Curr Opin Endocrinol Diabetes Obes (2015) 22:353–9. doi: 10.1097/MED.0000000000000184
3. Izquierdo AG, Crujeiras AB. Leptin, Obesity, and Leptin Resistance: Where are We 25 Years Later? Nutrients (2019) 11:2704. doi: 10.3390/nu11112704
4. Landecho MF, Tuero C, Valenti V, Bilbao I. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients (2019) 11:2664. doi: 10.3390/nu11112664
5. Roujeau C, Jockers R, Dam J. New Pharmacological Perspectives for the Leptin Receptor in the Treatment of Obesity. Front Endocrinol (Lausanne) (2014) 5:167. doi: 10.3389/fendo.2014.00167
6. Crujeiras AB, Carreira MC, Cabia B, Andrade S, Amil M, Casanueva FF. Leptin Resistance in Obesity: An Epigenetic Landscape. Life Sci (2015) 140:57–63. doi: 10.1016/j.lfs.2015.05.003
7. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional Cloning of the Mouse Obese Gene and its Human Homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
8. Abate N, Garg A, Peshock RM, Stray-Gundersen J, Grundy SM. Relationships of Generalized and Regional Adiposity to Insulin Sensitivity in Men. J Clin Invest (1995) 96:88–98. doi: 10.1172/JCI118083
9. De Vos P, Saladin R, Auwerx J, Staels B. Induction of Ob Gene Expression by Corticosteroids is Accompanied by Body Weight Loss and Reduced Food Intake. J Biol Chem (1995) 270:15958–61. doi: 10.1074/jbc.270.27.15958
10. Saladin R, De Vos P, Guerre-Millo M, Leturque A, Girard J, Staels B, et al. Transient Increase in Obese Gene Expression After Food Intake or Insulin Administration. Nature (1995) 377:527–9. doi: 10.1038/377527a0
11. Boden G, Chen X, Mozzoli M, Ryan I. Effect of Fasting on Serum Leptin in Normal Human Subjects. J Clin Endocrinol Metab (1996) 81:3419–23. doi: 10.1210/jcem.81.9.8784108
12. Dubuc GR, Phinney SD, Stern JS, Havel PJ. Changes of Serum Leptin and Endocrine and Metabolic Parameters After 7 Days of Energy Restriction in Men and Women. Metabolism (1998) 47:429–34. doi: 10.1016/S0026-0495(98)90055-5
13. Kolaczynski JW, Considine RV, Ohannesian J, Marco C, Opentanova I, Nyce MR, et al. Responses of Leptin to Short-Term Fasting and Refeeding in Humans: A Link With Ketogenesis But Not Ketones Themselves. Diabetes (1996) 45:1511–5. doi: 10.2337/diabetes.45.11.1511
14. Kolaczynski JW, Ohannesian JP, Considine RV, Marco CC, Caro JF. Response of Leptin to Short-Term and Prolonged Overfeeding in Humans. J Clin Endocrinol Metab (1996) 81:4162–5. doi: 10.1210/jcem.81.11.8923877
15. Hernandez C, Simo R, Chacon P, Sabin P, Baena JA, Castellanos JM, et al. Influence of Surgical Stress and Parenteral Nutrition on Serum Leptin Concentration. Clin Nutr (2000) 19:61–4. doi: 10.1054/clnu.1999.0075
16. Park HK, Ahima RS. Physiology of Leptin: Energy Homeostasis, Neuroendocrine Function and Metabolism. Metabolism (2015) 64:24–34. doi: 10.1016/j.metabol.2014.08.004
17. Papathanasiou AE, Nolen-Doerr E, Farr OM, Mantzoros CS. Geoffrey Harris Prize Lecture 2018: Novel Pathways Regulating Neuroendocrine Function, Energy Homeostasis and Metabolism in Humans. Eur J Endocrinol (2019) 180:R59–r71. doi: 10.1530/EJE-18-0847
18. Glaum SR, Hara M, Bindokas VP, Lee CC, Polonsky KS, Bell GI, et al. Leptin, the Obese Gene Product, Rapidly Modulates Synaptic Transmission in the Hypothalamus. Mol Pharmacol (1996) 50:230–5.
19. Munzberg H, Morrison CD. Structure, Production and Signaling of Leptin. Metabolism (2015) 64:13–23. doi: 10.1016/j.metabol.2014.09.010
20. Bazan JF. A Novel Family of Growth Factor Receptors: A Common Binding Domain in the Growth Hormone, Prolactin, Erythropoietin and IL-6 Receptors, and the P75 IL-2 Receptor Beta-Chain. Biochem Biophys Res Commun (1989) 164:788–95. doi: 10.1016/0006-291X(89)91528-3
21. Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, et al. Identification and Expression Cloning of a Leptin Receptor, OB-R. Cell (1995) 83:1263–71. doi: 10.1016/0092-8674(95)90151-5
22. Lollmann B, Gruninger S, Stricker-Krongrad A, Chiesi M. Detection and Quantification of the Leptin Receptor Splice Variants Ob-Ra, B, and, E in Different Mouse Tissues. Biochem Biophys Res Commun (1997) 238:648–52. doi: 10.1006/bbrc.1997.7205
23. Fruhbeck G. Intracellular Signalling Pathways Activated by Leptin. Biochem J (2006) 393:7–20. doi: 10.1042/BJ20051578
24. Bazan JF. Structural Design and Molecular Evolution of a Cytokine Receptor Superfamily. Proc Natl Acad Sci U S A (1990) 87:6934–8. doi: 10.1073/pnas.87.18.6934
25. Myers MG Jr. Leptin Receptor Signaling and the Regulation of Mammalian Physiology. Recent Prog Horm Res (2004) 59:287–304. doi: 10.1210/rp.59.1.287
27. Stephens TW, Basinski M, Bristow PK, Bue-Valleskey JM, Burgett SG, Craft L, et al. The Role of Neuropeptide Y in the Antiobesity Action of the Obese Gene Product. Nature (1995) 377:530–2. doi: 10.1038/377530a0
28. Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, et al. Anatomic Localization of Alternatively Spliced Leptin Receptors (Ob-R) in Mouse Brain and Other Tissues. Proc Natl Acad Sci U S A (1997) 94:7001–5. doi: 10.1073/pnas.94.13.7001
29. Hetherington AW RS. The Relation of Various Hypothalamic Lesions to Adiposity in the Rat. J Comp Neurol (1942) 76:475. doi: 10.1002/cne.900760308
30. Brobeck JR. Mechanism of the Development of Obesity in Animals With Hypothalamic Lesions. Physiol Rev (1946) 26:541–59. doi: 10.1152/physrev.1946.26.4.541
31. Anand BK, Brobeck JR. Localization of a “Feeding Center” in the Hypothalamus of the Rat. Proc Soc Exp Biol Med (1951) 77:323–4. doi: 10.3181/00379727-77-18766
32. Ahima RS. Revisiting Leptin’s Role in Obesity and Weight Loss. J Clin Invest (2008) 118:2380–3. doi: 10.1172/JCI36284
33. Fruhwürth S, Vogel H, Schürmann A, Williams KJ. Novel Insights Into How Overnutrition Disrupts the Hypothalamic Actions of Leptin. Front Endocrinol (2018) 9:89. doi: 10.3389/fendo.2018.00089
34. Li MD. Leptin and Beyond: An Odyssey to the Central Control of Body Weight. Yale J Biol Med (2011) 84:1–7.
35. Diano S. New Aspects of Melanocortin Signaling: A Role for PRCP in Alpha-MSH Degradation. Front Neuroendocrinol (2011) 32:70–83. doi: 10.1016/j.yfrne.2010.09.001
36. Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, et al. Leptin Differentially Regulates NPY and POMC Neurons Projecting to the Lateral Hypothalamic Area. Neuron (1999) 23:775–86. doi: 10.1016/s0896-6273(01)80035-0
37. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, et al. Leptin Activates Anorexigenic POMC Neurons Through a Neural Network in the Arcuate Nucleus. Nature (2001) 411:480–4. doi: 10.1038/35078085
38. Hinney A, Schmidt A, Nottebom K, Heibult O, Becker I, Ziegler A, et al. Several Mutations in the Melanocortin-4 Receptor Gene Including a Nonsense and a Frameshift Mutation Associated With Dominantly Inherited Obesity in Humans. J Clin Endocrinol Metab (1999) 84:1483–6. doi: 10.1210/jcem.84.4.5728
39. Lahlou N, Clement K, Carel JC, Vaisse C, Lotton C, Le Bihan Y, et al. Soluble Leptin Receptor in Serum of Subjects With Complete Resistance to Leptin: Relation to Fat Mass. Diabetes (2000) 49:1347–52. doi: 10.2337/diabetes.49.8.1347
40. Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid Rewiring of Arcuate Nucleus Feeding Circuits by Leptin. Science (2004) 304:110–5. doi: 10.1126/science.1089459
41. Toda C, Santoro A, Kim JD, Diano S. Pomc Neurons: From Birth to Death. Annu Rev Physiol (2017) 79:209–36. doi: 10.1146/annurev-physiol-022516-034110
42. MacDougald OA, Hwang CS, Fan H, Lane MD. Regulated Expression of the Obese Gene Product (Leptin) in White Adipose Tissue and 3T3-L1 Adipocytes. Proc Natl Acad Sci U S A (1995) 92:9034–7. doi: 10.1073/pnas.92.20.9034
43. Chan JL, Bluher S, Yiannakouris N, Suchard MA, Kratzsch J, Mantzoros CS. Regulation of Circulating Soluble Leptin Receptor Levels by Gender, Adiposity, Sex Steroids, and Leptin: Observational and Interventional Studies in Humans. Diabetes (2002) 51:2105–12. doi: 10.2337/diabetes.51.7.2105
44. Di Spiezio A, Sandin ES, Dore R, Müller-Fielitz H, Storck SE, Bernau M, et al. The Lepr-Mediated Leptin Transport Across Brain Barriers Controls Food Reward. Mol Metab (2018) 8:13–22. doi: 10.1016/j.molmet.2017.12.001
45. Fried SK, Ricci MR, Russell CD, Laferrère B. Regulation of Leptin Production in Humans. J Nutr (2000) 130:3127S–31S. doi: 10.1093/jn/130.12.3127S
46. Wajchenberg BL. Subcutaneous and Visceral Adipose Tissue: Their Relation to the Metabolic Syndrome. Endocr Rev (2000) 21:697–738. doi: 10.1210/edrv.21.6.0415
47. Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the Release of Adipokines by Adipose Tissue, Adipose Tissue Matrix, and Adipocytes From Visceral and Subcutaneous Abdominal Adipose Tissues of Obese Humans. Endocrinology (2004) 145:2273–82. doi: 10.1210/en.2003-1336
48. Tsai M, Asakawa A, Amitani H, Inui A. Stimulation of Leptin Secretion by Insulin. Indian J Endocrinol Metab (2012) 16:S543–8. doi: 10.4103/2230-8210.105570
49. Nogueiras R, Wilson H, Rohner-Jeanrenaud F, Tschop MH. Central Nervous System Regulation of Adipocyte Metabolism. Regul Pept (2008) 149:26–31. doi: 10.1016/j.regpep.2007.09.034
50. Saad MF, Khan A, Sharma A, Michael R, Riad-Gabriel MG, Boyadjian R, et al. Physiological Insulinemia Acutely Modulates Plasma Leptin. Diabetes (1998) 47:544–9. doi: 10.2337/diabetes.47.4.544
51. Mueller WM, Gregoire FM, Stanhope KL, Mobbs CV, Mizuno TM, Warden CH, et al. Evidence That Glucose Metabolism Regulates Leptin Secretion From Cultured Rat Adipocytes. Endocrinology (1998) 139:551–8. doi: 10.1210/endo.139.2.5716
52. Wabitsch M, Jensen PB, Blum WF, Christoffersen CT, Englaro P, Heinze E, et al. Insulin and Cortisol Promote Leptin Production in Cultured Human Fat Cells. Diabetes (1996) 45:1435–8. doi: 10.2337/diabetes.45.10.1435
53. Casabiell X, Piñeiro V, De la Cruz LF, Gualillo O, Folgar L, Diéguez C, et al. Dual Effect of Insulin on In Vitro Leptin Secretion by Adipose Tissue. Biochem Biophys Res Commun (2000) 276:477–82. doi: 10.1006/bbrc.2000.3506
54. Havel PJ. Role of Adipose Tissue in Body-Weight Regulation: Mechanisms Regulating Leptin Production and Energy Balance. Proc Nutr Soc (2000) 59:359–71. doi: 10.1017/S0029665100000410
55. Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, et al. Obesity is Associated With a Decreased Leptin Transport Across the Blood-Brain Barrier in Rats. Diabetes (2000) 49:1219–23. doi: 10.2337/diabetes.49.7.1219
56. Trayhurn P, Bing C. Appetite and Energy Balance Signals From Adipocytes. Philos Trans R Soc Lond B Biol Sci (2006) 361:1237–49. doi: 10.1098/rstb.2006.1859
57. Ghilardi N, Skoda RC. The Leptin Receptor Activates Janus Kinase 2 and Signals for Proliferation in a Factor-Dependent Cell Line. Mol Endocrinol (1997) 11:393–9. doi: 10.1210/mend.11.4.9907
58. Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, et al. The Full-Length Leptin Receptor has Signaling Capabilities of Interleukin 6-Type Cytokine Receptors. Proc Natl Acad Sci U S A (1996) 93:8374–8. doi: 10.1073/pnas.93.16.8374
59. Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT Signaling by the Leptin Receptor in Diabetic Mice. Proc Natl Acad Sci U S A (1996) 93:6231–5. doi: 10.1073/pnas.93.13.6231
60. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of Interleukin (IL)-6-Type Cytokine Signalling and its Regulation. Biochem J (2003) 374:1–20. doi: 10.1042/bj20030407
61. Ihle JN, Kerr IM. Jaks and Stats in Signaling by the Cytokine Receptor Superfamily. Trends Genet (1995) 11:69–74. doi: 10.1016/S0168-9525(00)89000-9
62. Bahrenberg G, Behrmann I, Barthel A, Hekerman P, Heinrich PC, Joost HG, et al. Identification of the Critical Sequence Elements in the Cytoplasmic Domain of Leptin Receptor Isoforms Required for Janus Kinase/Signal Transducer and Activator of Transcription Activation by Receptor Heterodimers. Mol Endocrinol (2002) 16:859–72. doi: 10.1210/mend.16.4.0800
63. Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG Jr. Regulation of Jak Kinases by Intracellular Leptin Receptor Sequences. J Biol Chem (2002) 277:41547–55. doi: 10.1074/jbc.M205148200
64. Murakami T, Yamashita T, Iida M, Kuwajima M, Shima K. A Short Form of Leptin Receptor Performs Signal Transduction. Biochem Biophys Res Commun (1997) 231:26–9. doi: 10.1006/bbrc.1996.6030
65. Bjorbaek C, Elmquist JK, Michl P, Ahima RS, van Bueren A, McCall AL, et al. Expression of Leptin Receptor Isoforms in Rat Brain Microvessels. Endocrinology (1998) 139:3485–91. doi: 10.1210/endo.139.8.6154
66. Hileman SM, Tornoe J, Flier JS, Bjorbaek C. Transcellular Transport of Leptin by the Short Leptin Receptor Isoform Obra in Madin-Darby Canine Kidney Cells. Endocrinology (2000) 141:1955–61. doi: 10.1210/endo.141.6.7450
67. Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced Leptin Sensitivity and Attenuation of Diet-Induced Obesity in Mice With Haploinsufficiency of Socs3. Nat Med (2004) 10:734–8. doi: 10.1038/nm1072
68. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 Deficiency in the Brain Elevates Leptin Sensitivity and Confers Resistance to Diet-Induced Obesity. Nat Med (2004) 10:739–43. doi: 10.1038/nm1071
69. Rahmouni K, Haynes WG, Morgan DA, Mark AL. Intracellular Mechanisms Involved in Leptin Regulation of Sympathetic Outflow. Hypertension (2003) 41:763–7. doi: 10.1161/01.HYP.0000048342.54392.40
70. Rahmouni K, Sigmund CD, Haynes WG, Mark AL. Mitogen Activated Protein Kinase: A Newly Discovered Mediator of Selective Leptin Actions. Hypertension (2005) 46:867. doi: 10.1161/hyp.46.5.814
71. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, et al. STAT3 Signalling is Required for Leptin Regulation of Energy Balance But Not Reproduction. Nature (2003) 421:856–9. doi: 10.1038/nature01388
72. Harlan SM, Rahmouni K. PI3K Signaling: A Key Pathway in the Control of Sympathetic Traffic and Arterial Pressure by Leptin. Mol Metab (2013) 2:69–73. doi: 10.1016/j.molmet.2013.03.004
73. La Cava A, Matarese G. The Weight of Leptin in Immunity. Nat Rev Immunol (2004) 4:371–9. doi: 10.1038/nri1350
74. Faggioni R, Fantuzzi G, Fuller J, Dinarello CA, Feingold KR, Grunfeld C. Il-1 Beta Mediates Leptin Induction During Inflammation. R204-208: Am J Physiol. 274 (1998).
75. Landman RE, Puder JJ, Xiao E, Freda PU, Ferin M, Wardlaw SL. Endotoxin Stimulates Leptin in the Human and Nonhuman Primate. J Clin Endocrinol Metab (2003) 88:1285–91. doi: 10.1210/jc.2002-021393
76. Gabay C, Dreyer M, Pellegrinelli N, Chicheportiche R, Meier CA. Leptin Directly Induces the Secretion of Interleukin 1 Receptor Antagonist in Human Monocytes. J Clin Endocrinol Metab (2001) 86:783–91. doi: 10.1210/jc.86.2.783
77. Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cell Immunol (1999) 194:6–11. doi: 10.1006/cimm.1999.1490
78. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is Associated With Macrophage Accumulation in Adipose Tissue. J Clin Invest (2003) 112:1796–808. doi: 10.1172/JCI200319246
79. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J Clin Invest (2003) 112:1821–30. doi: 10.1172/JCI200319451
80. Wellen KE, Hotamisligil GS. Inflammation, Stress, and Diabetes. J Clin Invest (2005) 115:1111–9. doi: 10.1172/JCI25102
81. Calle EE, Kaaks R. Overweight, Obesity and Cancer: Epidemiological Evidence and Proposed Mechanisms. Nat Rev Cancer (2004) 4:579–91. doi: 10.1038/nrc1408
82. Mannino DM, Mott J, Ferdinands JM, Camargo CA, Friedman M, Greves HM, et al. Boys With High Body Masses Have an Increased Risk of Developing Asthma: Findings From the National Longitudinal Survey of Youth (Nlsy). Int J Obes (Lond) (2006) 30:6–13. doi: 10.1038/sj.ijo.0803145
83. Yazdi FT, Clee SM, Meyre D. Obesity Genetics in Mouse and Human: Back and Forth, and Back Again. PeerJ (2015) 3:e856. doi: 10.7717/peerj.856
84. Dubern B, Clement K. Leptin and Leptin Receptor-Related Monogenic Obesity. Biochimie (2012) 94:2111–5. doi: 10.1016/j.biochi.2012.05.010
85. Liu W, Zhou X, Li Y, Zhang S, Cai X, Zhang R, et al. Serum Leptin, Resistin, and Adiponectin Levels in Obese and non-Obese Patients With Newly Diagnosed Type 2 Diabetes Mellitus: A Population-Based Study. Med (Baltimore) (2020) 99:e19052. doi: 10.1097/MD.0000000000019052
86. Lonnqvist F, Arner P, Nordfors L, Schalling M. Overexpression of the Obese (Ob) Gene in Adipose Tissue of Human Obese Subjects. Nat Med (1995) 1:950–3. doi: 10.1038/nm0995-950
87. Hamilton BS, Paglia D, Kwan AY, Deitel M. Increased Obese Mrna Expression in Omental Fat Cells From Massively Obese Humans. Nat Med (1995) 1:953–6. doi: 10.1038/nm0995-953
88. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N Engl J Med (1996) 334:292–5. doi: 10.1056/NEJM199602013340503
89. Ravussin Y, LeDuc CA, Watanabe K, Mueller BR, Skowronski A, Rosenbaum M, et al. Effects of Chronic Leptin Infusion on Subsequent Body Weight and Composition in Mice: can Body Weight Set Point be Reset? Mol Metab (2014) 3:432–40. doi: 10.1016/j.molmet.2014.02.003
90. Sainz N, Barrenetxe J, Moreno-Aliaga MJ, Martinez JA. Leptin Resistance and Diet-Induced Obesity: Central and Peripheral Actions of Leptin. Metabolism (2015) 64:35–46. doi: 10.1016/j.metabol.2014.10.015
91. Matheny M, Shapiro A, Tumer N, Scarpace PJ. Region-Specific Diet-Induced and Leptin-Induced Cellular Leptin Resistance Includes the Ventral Tegmental Area in Rats. Neuropharmacology (2011) 60:480–7. doi: 10.1016/j.neuropharm.2010.11.002
92. Bian J, Bai XM, Zhao YL, Zhang L, Liu ZJ. Lentiviral Vector-Mediated Knockdown of Lrb in the Arcuate Nucleus Promotes Diet-Induced Obesity in Rats. J Mol Endocrinol (2013) 51:27–35. doi: 10.1530/JME-12-0212
93. Gamber KM, Huo L, Ha S, Hairston JE, Greeley S, Bjorbaek C. Over-Expression of Leptin Receptors in Hypothalamic POMC Neurons Increases Susceptibility to Diet-Induced Obesity. PloS One (2012) 7:e30485. doi: 10.1371/journal.pone.0030485
94. El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two Defects Contribute to Hypothalamic Leptin Resistance in Mice With Diet-Induced Obesity. J Clin Invest (2000) 105:1827–32. doi: 10.1172/JCI9842
95. Munzberg H, Flier JS, Bjorbaek C. Region-Specific Leptin Resistance Within the Hypothalamus of Diet-Induced Obese Mice. Endocrinology (2004) 145:4880–9. doi: 10.1210/en.2004-0726
96. Olofsson LE, Unger EK, Cheung CC, Xu AW. Modulation of Agrp-Neuronal Function by SOCS3 as an Initiating Event in Diet-Induced Hypothalamic Leptin Resistance. Proc Natl Acad Sci U S A (2013) 110:E697–706. doi: 10.1073/pnas.1218284110
97. Hirasawa T, Ohara T, Makino S. Genetic Typing of the Mouse Ob Mutation by PCR and Restriction Enzyme Analysis. Exp Anim (1997) 46:75–8. doi: 10.1538/expanim.46.75
98. Igel M, Becker W, Herberg L, Joost H-G. Hyperleptinemia, Leptin Resistance, and Polymorphic Leptin Receptor in the New Zealand Obese Mouse*. Endocrinology (1997) 138:4234–9. doi: 10.1210/endo.138.10.5428
99. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal Splicing of the Leptin Receptor in Diabetic Mice. Nature (1996) 379:632–5. doi: 10.1038/379632a0
100. Vaisse C, Halaas JL, Horvath CM, Darnell JE Jr, Stoffel M, Friedman JM. Leptin Activation of Stat3 in the Hypothalamus of Wild-Type and Ob/Ob Mice But Not Db/Db Mice. Nat Genet (1996) 14:95–7. doi: 10.1038/ng0996-95
101. Riestra P, Garcia-Anguita A, Viturro E, Schoppen S, de Oya M, Garces C. Influence of the Leptin G-2548A Polymorphism on Leptin Levels and Anthropometric Measurements in Healthy Spanish Adolescents. Ann Hum Genet (2010) 74:335–9. doi: 10.1111/j.1469-1809.2010.00586.x
102. Soskic S, Stokic E, Obradovic M, Sudar E, Tanic N, Kupusinac A, et al. Association of Leptin Gene Polymorphism G-2548A With Metabolic and Anthropometric Parameters in Obese Patients in a Serbian Population: Pilot Study. Clin Lipidol (2014) 9:505–13. doi: 10.2217/clp.14.42
103. Zhang L, Yuan LH, Xiao Y, Lu MY, Zhang LJ, Wang Y. Association of Leptin Gene -2548 G/a Polymorphism With Obesity: A Meta-Analysis. Ann Nutr Metab (2014) 64:127–36. doi: 10.1159/000363392
104. Paolini B, Maltese PE, Del Ciondolo I, Tavian D, Missaglia S, Ciuoli C, et al. Prevalence of Mutations in LEP, LEPR, and MC4R Genes in Individuals With Severe Obesity. Genet Mol Res (2016) 15. doi: 10.4238/gmr.15038718
105. Nesrine Z, Haithem H, Imen B, Fadoua N, Asma O, Fadhel NM, et al. Leptin and Leptin Receptor Polymorphisms, Plasma Leptin Levels and Obesity in Tunisian Volunteers. Int J Exp Pathol (2018) 99:121–30. doi: 10.1111/iep.12271
106. Carayol J, Chabert C, Di Cara A, Armenise C, Lefebvre G, Langin D. Protein Quantitative Trait Locus Study in Obesity During Weight-Loss Identifies a Leptin Regulator. Nat Commun (2017) 8:2084. doi: 10.1038/s41467-017-02182-z
107. Zhang Y, Dallner OS, Nakadai T, Fayzikhodjaeva G, Lu YH, Lazar MA, et al. A Noncanonical Pparγ/Rxrα-Binding Sequence Regulates Leptin Expression in Response to Changes in Adipose Tissue Mass. Proc Natl Acad Sci U S A (2018) 115:E6039–e6047. doi: 10.1073/pnas.1806366115
108. Dallner OS, Marinis JM, Lu YH, Birsoy K. Dysregulation of a Long Noncoding RNA Reduces Leptin Leading to a Leptin-Responsive Form of Obesity. Nat Med (2019) 25:507–16. doi: 10.1038/s41591-019-0370-1
109. Derghal A, Djelloul M, Azzarelli M, Degonon S, Tourniaire F, Landrier J-F, et al. Micrornas are Involved in the Hypothalamic Leptin Sensitivity. Epigenetics (2018) 13:1127–40. doi: 10.1080/15592294.2018.1543507
110. Derghal A, Astier J, Sicard F, Couturier C, Landrier J-F, Mounien L. Leptin Modulates the Expression of Mirnas-Targeting POMC Mrna by the JAK2-STAT3 and PI3K-Akt Pathways. J Clin Med (2019) 8:2213. doi: 10.3390/jcm8122213
111. Wilhelm J, Birkenstock A, Buchholz V, Müller A, Aly SA, Gruner-Labitzke K, et al. Promoter Methylation of LEP and LEPR Before and After Bariatric Surgery: A Cross-Sectional Study. Obes Facts (2021) 14:1–7. doi: 10.1159/000511918
112. Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin Levels Reflect Body Lipid Content in Mice: Evidence for Diet-Induced Resistance to Leptin Action. Nat Med (1995) 1:1311–4. doi: 10.1038/nm1295-1311
113. Myers MG Jr, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, et al. Challenges and Opportunities of Defining Clinical Leptin Resistance. Cell Metab (2012) 15:150–6. doi: 10.1016/j.cmet.2012.01.002
114. Friedman J. 20 Years of Leptin: Leptin at 20: An Overview. J Endocrinol (2014) 223:T1–8. doi: 10.1530/JOE-14-0405
115. Wabitsch M, Funcke JB, Lennerz B, Kuhnle-Krahl U, Lahr G, Debatin KM, et al. Biologically Inactive Leptin and Early-Onset Extreme Obesity. N Engl J Med (2015) 372:48–54. doi: 10.1056/NEJMoa1406653
116. Mainardi M, Pizzorusso T, Maffei M. Environment, Leptin Sensitivity, and Hypothalamic Plasticity. Neural Plast (2013) 2013:438072. doi: 10.1155/2013/438072
117. Chumakova G, Ott A, Veselovskaya N, Gritsenko O, Shenkova N. Pathogenetic Mechanisms of Leptin Resistance. Russ J Cardiol (2015) 4:107–10. doi: 10.15829/1560-4071-2015-4-107-110
118. Holtkamp K, Hebebrand J, Mika C, Heer M, Heussen N, Herpertz-Dahlmann B. High Serum Leptin Levels Subsequent to Weight Gain Predict Renewed Weight Loss in Patients With Anorexia Nervosa. Psychoneuroendocrinology (2004) 29:791–7. doi: 10.1016/S0306-4530(03)00143-4
119. Mantzoros CS. The Role of Leptin in Human Obesity and Disease: A Review of Current Evidence. Ann Intern Med (1999) 130:671–80. doi: 10.7326/0003-4819-130-8-199904200-00014
120. Philbrick KA, Wong CP, Branscum AJ, Turner RT, Iwaniec UT. Leptin Stimulates Bone Formation in Ob/Ob Mice at Doses Having Minimal Impact on Energy Metabolism. J Endocrinol (2017) 232:461–74. doi: 10.1530/JOE-16-0484
121. Mark AL. Selective Leptin Resistance Revisited. J Physiol Regul Integr Comp Physiol (2013) 305:R566–81. doi: 10.1152/ajpregu.00180.2013
122. Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-Mediated Regional Sympathetic Nerve Activation by Leptin. J Clin Invest (1997) 100:270–8. doi: 10.1172/JCI119532
123. Richards RJ, Blalock A, Liao J, Reisin E. Leptin: Sympathetic and Cardiovascular Effects. Curr Cardiol Rep (2003) 5:453–8. doi: 10.1007/s11886-003-0106-0
124. Seoane-Collazo P, Martinez-Sanchez N, Milbank E, Contreras C. Incendiary Leptin. Nutrients (2020) 12:472. doi: 10.3390/nu12020472
125. Kwon O, Kim KW, Kim MS. Leptin Signalling Pathways in Hypothalamic Neurons. Cell Mol Life Sci (2016) 73:1457–77. doi: 10.1007/s00018-016-2133-1
126. Geerling JC, Loewy AD. Aldosterone-Sensitive Neurons in the Nucleus of the Solitary Tract: Bidirectional Connections With the Central Nucleus of the Amygdala. J Comp Neurol (2006) 497:646–57. doi: 10.1002/cne.21019
127. Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG. Evidence That the Caudal Brainstem is a Target for the Inhibitory Effect of Leptin on Food Intake. Endocrinology (2002) 143:239–46. doi: 10.1210/endo.143.1.8589
128. Shi Z, Wong J, Brooks VL. Obesity: Sex and Sympathetics. Biol Sex Differ (2020) 11:10. doi: 10.1186/s13293-020-00286-8
129. DePaoli AM. 20 Years of Leptin: Leptin in Common Obesity and Associated Disorders of Metabolism. J Endocrinol (2014) 223:T71–81. doi: 10.1530/JOE-14-0258
130. Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant Leptin for Weight Loss in Obese and Lean Adults: A Randomized, Controlled, Dose-Escalation Trial. JAMA (1999) 282:1568–75. doi: 10.1001/jama.282.16.1568
131. Hukshorn CJ, Saris WH, Westerterp-Plantenga MS, Farid AR, Smith FJ, Campfield LA. Weekly Subcutaneous Pegylated Recombinant Native Human Leptin (PEG-OB) Administration in Obese Men. J Clin Endocrinol Metab (2000) 85:4003–9. doi: 10.1210/jcem.85.11.6955
132. Westerterp-Plantenga MS, Saris WH, Hukshorn CJ, Campfield LA. Effects of Weekly Administration of Pegylated Recombinant Human OB Protein on Appetite Profile and Energy Metabolism in Obese Men. Am J Clin Nutr (2001) 74:426–34. doi: 10.1093/ajcn/74.4.426
133. Hukshorn CJ, Westerterp-Plantenga MS, Saris WH. Pegylated Human Recombinant Leptin (PEG-OB) Causes Additional Weight Loss in Severely Energy-Restricted, Overweight Men. Am J Clin Nutr (2003) 77:771–6. doi: 10.1093/ajcn/77.4.771
134. Zelissen PM, Stenlof K, Lean ME, Fogteloo J, Keulen ET, Wilding J, et al. Effect of Three Treatment Schedules of Recombinant Methionyl Human Leptin on Body Weight in Obese Adults: A Randomized, Placebo-Controlled Trial. Diabetes Obes Metab (2005) 7:755–61. doi: 10.1111/j.1463-1326.2005.00468.x
135. Mittendorfer B, Horowitz JF, DePaoli AM, McCamish MA, Patterson BW, Klein S. Recombinant Human Leptin Treatment Does Not Improve Insulin Action in Obese Subjects With Type 2 Diabetes. Diabetes (2011) 60:1474–7. doi: 10.2337/db10-1302
136. Moon HS, Matarese G, Brennan AM, Chamberland JP, Liu X, Fiorenza CG, et al. Efficacy of Metreleptin in Obese Patients With Type 2 Diabetes: Cellular and Molecular Pathways Underlying Leptin Tolerance. Diabetes (2011) 60:1647–56. doi: 10.2337/db10-1791
137. Korner J, Conroy R, Febres G, McMahon DJ, Conwell I, Karmally W, et al. Randomized Double-Blind Placebo-Controlled Study of Leptin Administration After Gastric Bypass. Obes (Silver Spring) (2013) 21:951–6. doi: 10.1002/oby.20433
138. Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin Responsiveness Restored by Amylin Agonism in Diet-Induced Obesity: Evidence From Nonclinical and Clinical Studies. Proc Natl Acad Sci U S A (2008) 105:7257–62. doi: 10.1073/pnas.0706473105
139. Ravussin E, Smith SR, Mitchell JA, Shringarpure R, Shan K, Maier H, et al. Enhanced Weight Loss With Pramlintide/Metreleptin: An Integrated Neurohormonal Approach to Obesity Pharmacotherapy. Obes (Silver Spring) (2009) 17:1736–43. doi: 10.1038/oby.2009.184
140. Depaoli A, Long A, Fine GM, Stewart M, Rahilly S. Efficacy of Metreleptin for Weight Loss in Overweight and Obese Adults With Low Leptin Levels. Diabetes (2018) 67:296–LB. doi: 10.2337/db18-296-LB
141. Bartness TJ. North American Association for the Study of Obesity Annual Meeting. IDrugs (2001) 4:41–3.
142. Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, et al. Low-Dose Leptin Reverses Skeletal Muscle, Autonomic, and Neuroendocrine Adaptations to Maintenance of Reduced Weight. J Clin Invest (2005) 115:3579–86. doi: 10.1172/JCI25977
143. Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, et al. Recombinant Human Leptin in Women With Hypothalamic Amenorrhea. N Engl J Med (2004) 351:987–97. doi: 10.1056/NEJMoa040388
144. Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low Dose Leptin Administration Reverses Effects of Sustained Weight-Reduction on Energy Expenditure and Circulating Concentrations of Thyroid Hormones. J Clin Endocrinol Metab (2002) 87:2391–4. doi: 10.1210/jcem.87.5.8628
145. Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The Role of Falling Leptin Levels in the Neuroendocrine and Metabolic Adaptation to Short-Term Starvation in Healthy Men. J Clin Invest (2003) 111:1409–21. doi: 10.1172/JCI200317490
146. Schurgin S, Canavan B, Koutkia P, Depaoli AM, Grinspoon S. Endocrine and Metabolic Effects of Physiologic R-Methuleptin Administration During Acute Caloric Deprivation in Normal-Weight Women. J Clin Endocrinol Metab (2004) 89:5402–9. doi: 10.1210/jc.2004-1102
147. Gavrila A, Chan JL, Miller LC, Heist K, Yiannakouris N, Mantzoros CS. Circulating Melanin-Concentrating Hormone, Agouti-Related Protein, and Alpha-Melanocyte-Stimulating Hormone Levels in Relation to Body Composition: Alterations in Response to Food Deprivation and Recombinant Human Leptin Administration. J Clin Endocrinol Metab (2005) 90:1047–54. doi: 10.1210/jc.2004-1124
148. Trevaskis JL, Coffey T, Cole R, Lei C, Wittmer C, Walsh B, et al. Amylin-Mediated Restoration of Leptin Responsiveness in Diet-Induced Obesity: Magnitude and Mechanisms. Endocrinology (2008) 149:5679–87. doi: 10.1210/en.2008-0770
149. Moon HS, Chamberland JP, Diakopoulos KN, Fiorenza CG, Ziemke F, Schneider B, et al. Leptin and Amylin Act in an Additive Manner to Activate Overlapping Signaling Pathways in Peripheral Tissues: In Vitro and Ex Vivo Studies in Humans. Diabetes Care (2011) 34:132–8. doi: 10.2337/dc10-0518
150. Chan JL, Koda J, Heilig JS, Cochran EK, Gorden P, Oral EA, et al. Immunogenicity Associated With Metreleptin Treatment in Patients With Obesity or Lipodystrophy. Clin Endocrinol (Oxf) (2016) 85:137–49. doi: 10.1111/cen.12980
151. Guerra B, Olmedillas H, Guadalupe-Grau A, Ponce-Gonzalez JG, Morales-Alamo D, Fuentes T, et al. is Sprint Exercise a Leptin Signaling Mimetic in Human Skeletal Muscle? J Appl Physiol (1985) (2011) 111:715–25. doi: 10.1152/japplphysiol.00805.2010
152. Friedman JM. Leptin and the Endocrine Control of Energy Balance. Nat Metab (2019) 1:754–64. doi: 10.1038/s42255-019-0095-y
153. Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, et al. Protein Tyrosine Phosphatase 1B Negatively Regulates Leptin Signaling in a Hypothalamic Cell Line. Mol Cell Endocrinol (2002) 195:109–18. doi: 10.1016/S0303-7207(02)00178-8
154. Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG Jr, Xu AW. Functional Role of Suppressor of Cytokine Signaling 3 Upregulation in Hypothalamic Leptin Resistance and Long-Term Energy Homeostasis. Diabetes (2010) 59:894–906. doi: 10.2337/db09-1024
155. Lantz KA, Hart SG, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, et al. Inhibition of PTP1B by Trodusquemine (MSI-1436) Causes Fat-Specific Weight Loss in Diet-Induced Obese Mice. Obes (Silver Spring) (2010) 18:1516–23. doi: 10.1038/oby.2009.444
156. Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin Enters the Brain by a Saturable System Independent of Insulin. Peptides (1996) 17:305–11. doi: 10.1016/0196-9781(96)00025-3
157. Zhu A, Kaneshiro M, Kaunitz JD. Evaluation and Treatment of Iron Deficiency Anemia: A Gastroenterological Perspective. Dig Dis Sci (2010) 55:548–59. doi: 10.1007/s10620-009-1108-6
158. Domenica Cappellini M, Motta I. Anemia in Clinical Practice—Definition and Classification: Does Hemoglobin Change With Aging? Semin Hematol (2015) 52:261–9. doi: 10.1053/j.seminhematol.2015.07.006
159. Ahlquist DA, McGill DB, Schwartz S, Taylor WF, Owen RA. Fecal Blood Levels in Health and Disease. A Study Using Hemoquant. N Engl J Med (1985) 312:1422–8. doi: 10.1056/NEJM198505303122204
160. McIntyre AS, Long RG. Prospective Survey of Investigations in Outpatients Referred With Iron Deficiency Anaemia. Gut (1993) 34:1102–7. doi: 10.1136/gut.34.8.1102
161. Raju GS, Gerson L, Das A, Lewis B. American Gastroenterological Association (Aga) Institute Medical Position Statement on Obscure Gastrointestinal Bleeding. Gastroenterology (2007) 133:1694–6. doi: 10.1053/j.gastro.2007.06.008
162. Kim BS, Li BT, Engel A, Samra JS, Clarke S, Norton ID, et al. Diagnosis of Gastrointestinal Bleeding: A Practical Guide for Clinicians. World J Gastrointest Pathophysiol (2014) 5:467–78. doi: 10.4291/wjgp.v5.i4.467
163. Stein J, Connor S, Virgin G, Ong DEH, Pereyra L. Anemia and Iron Deficiency in Gastrointestinal and Liver Conditions. World J Gastroenterol (2016) 22:7908–25. doi: 10.3748/wjg.v22.i35.7908
164. Niv E, Elis A, Zissin R, Naftali T, Novis B, Lishner M. Iron Deficiency Anemia in Patients Without Gastrointestinal Symptoms–a Prospective Study. Fam Pract (2005) 22:58–61. doi: 10.1093/fampra/cmh705
165. Freeman HJ. Iron Deficiency Anemia in Celiac Disease. World J Gastroenterol (2015) 21:9233–8. doi: 10.3748/wjg.v21.i31.9233
166. Jackson CS, Strong R. Gastrointestinal Angiodysplasia: Diagnosis and Management. Gastrointest Endosc Clin N Am (2017) 27:51–62. doi: 10.1016/j.giec.2016.08.012
167. Peyrin-Biroulet L, Lopez A. Review Article: Treating-to-Target for Inflammatory Bowel Disease-Associated Anaemia. Aliment Pharmacol Ther (2018) 48:610–7. doi: 10.1111/apt.14922
168. Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory Diagnosis of Iron-Deficiency Anemia: An Overview. J Gen Intern Med (1992) 7:145–53. doi: 10.1007/BF02598003
169. Jolobe OM. Prevalence of Hypochromia (Without Microcytosis) vs Microcytosis (Without Hypochromia) in Iron Deficiency. Clin Lab Haematol (2000) 22:79–80. doi: 10.1046/j.1365-2257.2000.00293.x
170. Gomollón F, Gisbert JP. Anemia and Inflammatory Bowel Diseases. World J Gastroenterol (2009) 15:4659–65. doi: 10.3748/wjg.15.4659
171. Bager P, Dahlerup JF. Randomised Clinical Trial: Oral vs. Intravenous Iron After Upper Gastrointestinal Haemorrhage–a Placebo-Controlled Study. Aliment Pharmacol Ther (2014) 39:176–87. doi: 10.1111/apt.12556
172. Bizzaro N, Antico A. Diagnosis and Classification of Pernicious Anemia. Autoimmun Rev (2014) 13:565–8. doi: 10.1016/j.autrev.2014.01.042
173. Rockey DC, Altayar O, Falck-Ytter Y, Kalmaz D. Aga Technical Review on Gastrointestinal Evaluation of Iron Deficiency Anemia. Gastroenterology (2020) 159:1097–119. doi: 10.1053/j.gastro.2020.06.045
174. Rockey DC. Occult Gastrointestinal Bleeding. N Engl J Med (1999) 341:38–46. doi: 10.1056/NEJM199907013410107
175. Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of Anemia in Persons 65 Years and Older in the United States: Evidence for a High Rate of Unexplained Anemia. Blood (2004) 104:2263–8. doi: 10.1182/blood-2004-05-1812
176. Rockey DC. Occult Gastrointestinal Bleeding. Gastroenterol Clin North Am (2005) 34:699–718. doi: 10.1016/j.gtc.2005.08.010
178. Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab (2019) 30:706–19.e706. doi: 10.1016/j.cmet.2019.08.005
Keywords: leptin-based therapies, obesity, leptin resistance, leptin receptor, leptin
Citation: Obradovic M, Sudar-Milovanovic E, Soskic S, Essack M, Arya S, Stewart AJ, Gojobori T and Isenovic ER (2021) Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 12:585887. doi: 10.3389/fendo.2021.585887
Received: 21 July 2020; Accepted: 30 April 2021;
Published: 18 May 2021.
Edited by:
Hendrik Lehnert, University of Salzburg, AustriaReviewed by:
Valeria Guglielmi, University of Rome Tor Vergata, ItalyRiccardo Dore, University of Lübeck, Germany
Copyright © 2021 Obradovic, Sudar-Milovanovic, Soskic, Essack, Arya, Stewart, Gojobori and Isenovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esma R. Isenovic, aXNlbm92aWNAeWFob28uY29t