 Esmael Besufikad Belachew
Esmael Besufikad Belachew Dareskedar Tsehay Sewasew
Dareskedar Tsehay Sewasew- 1Biology, Mizan Tepi University, Addis Ababa, Ethiopia
- 2Microbial, Cellular and Molecular Biology Department, Addis Ababa University, Addis Ababa, Ethiopia
- 3Immunology, Armauer Hansen Research Institute (AHRI), Addis Ababa, Ethiopia
The estrogen receptor is a vital receptor for therapeutic targets in estrogen receptor-positive breast cancer. The main strategy for the treatment of estrogen receptor-positive breast cancers is blocking the estrogen action on estrogen receptors by endocrine therapy but this can be restricted via endocrine resistance. Endocrine resistance occurs due to both de novo and acquired resistance. This review focuses on the mechanisms of the ligand-dependent and ligand-independent pathways and other coregulators, which are responsible for endocrine resistance. It concludes that combinatorial drugs that target different signaling pathways and coregulatory proteins together with endocrine therapy could be a novel therapeutic modality to stop endocrine resistance.
Introduction
The estrogen hormone is important in maintaining the function of the reproductive system, bone metabolism, cardiovascular maintenance, central nervous systems, and lubrication of the vaginal lining. Overexpression of the estrogen hormone is associated with an increased risk for breast cancer (1, 2).
The function of estrogen in estrogen receptor (ER) positive breast cancer is primarily mediated by ER. The estrogen receptor is a member of the nuclear receptor superfamily and is involved in various developmental and physiological processes (1). Two subdivisions of ER, estrogen receptor α (ERα) and estrogen receptor β (ERβ) are identified (3, 4). Estrogen receptor α (ERα) is predominantly expressed in the uterus and pituitary gland with highest levels in the liver, hypothalamus, bone, mammary gland, cervix, testis, kidney, heart, skeletal muscle, and vagina. Estrogen receptor β (Erβ) expression is high in the prostate and ovary and found exclusively in the granulosa cells (4, 5). Estrogen receptor α (ERα) activation promotes tumorigenesis in different types of cancer, including breast cancer and the role of ERβ is still unclear (6). Therefore, inhibition of the ERα has become one of the main strategies for the prevention and treatment of breast cancer (7). The main strategy for the treatment of ER-positive breast cancer is blocking the action of estrogen by endocrine therapy but limited by the development of resistance (2). Therefore, this review is mainly focused on the principal mechanisms involved in endocrine therapy resistance among ER-positive breast cancer.
Structure of the Estrogen Receptor
The structure of ERα and ERβ are similar to other nuclear receptor families, which have four structural and functional domains. These are amino-terminal (A/B domain), DNA binding domain (DBD; C-domain), hinge region (D-domain), and ligand-binding domain (LBD; E-domain) (Figure 1). The ERs have an additional fifth domain: the carboxyl-terminal domain (F-domain) whose function is still unclear. DNA binding domain (DBD) and LBD carry 96 and 60% of homology between ERα and ERβ, however, the amino-terminal, hinge region and carboxyl-terminal domains are divergent (4, 5, 8). The amino-terminal (A/B domain) encodes a hormone-independent transcriptional activation function 1 (AF1) (2), and acts synergistically with transcriptional activation function 2 (AF-2) to attain maximum transcriptional activity. The DBD is responsible for ER binding to estrogen response elements (EREs), and the D domain leads to nuclear transport. The LBD contains a dimerization surface and encodes ligand-dependent transcription activation through AF-2 (1, 2, 4, 7).
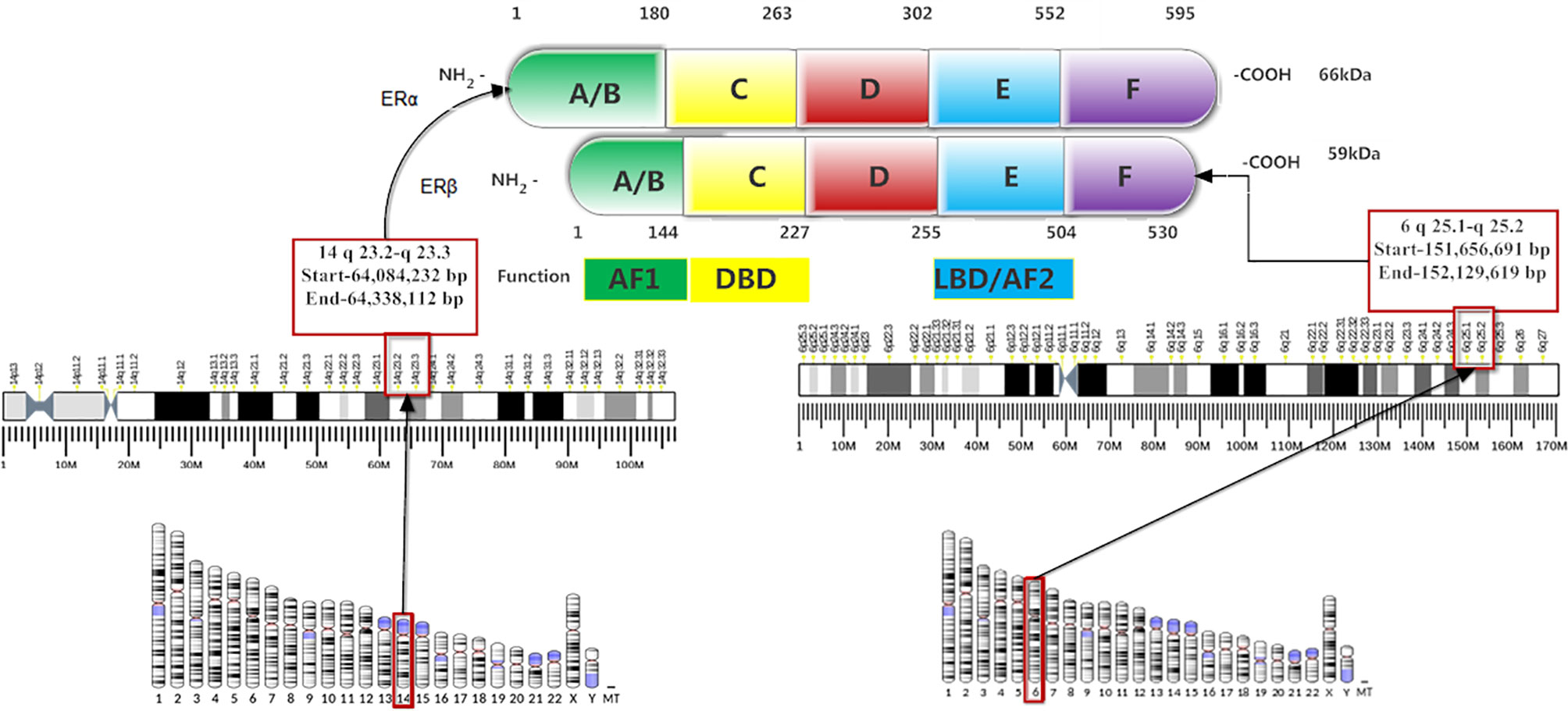
Figure 1 Structure and functional domains of the estrogen receptors.
The genes coding for ERα and ERβ are located on chromosome 14, locus 14q23.2, and chromosome 6, locus 6q25.1, respectively (Figure 1). The ERβ has 530 amino acids and 59 kDa molecular weight, while ERα has 595 amino acids and 66 kDa molecular weight (9). Five different isoforms of ERα, such as 62kDa, 53kDa, 46kDa, 45kDa, and 36kDa (10), and five ERβ variants (ERβ1-ERβ5) are detected in breast cancer (11). Both ERα and ERβ1 require ligand binding for ER target gene transcription (12). ERβ1 has gene transcriptional inhibition when signaling through the activator protein 1(AP-1) pathway and its binding with tamoxifen also promotes gene transcription (13). A low level of ERβ1 is an independent marker than ERα level to predict tamoxifen resistance (14).
Mechanism of Estrogen Receptor Action
The two main mechanisms of ER-dependent gene transcription are estrogen/ligand-dependent and estrogen/ligand-independent (2, 7, 15).
Ligand-Dependent
In ligand-dependent signaling mechanisms, the binding of estrogen with ER causes a conformational change, which allows various coregulators to stimulate transcription of ER-target genes. The ligand/estrogen-dependent mechanism is further classified into direct genomic or classical, indirect genomic or non-classical, and non-genomic mechanisms of activation (16–19).
Direct Genomic/Classical
The direct genomic or classical pathway regulates the expression of ER target genes by the direct binding of estrogen-activated ERs to DNA binding at EREs (Figure 2). During estrogen binding with ER, and the heat shock proteins (HSP70 and HSP90) dissociate ER from this binding in the cytosol, and change their conformation, then migrate as dimers into the nucleus to bind with EREs. This conformational change also allows helix 12 (H12) to accept coactivators and activate gene transcription (10, 20–24).
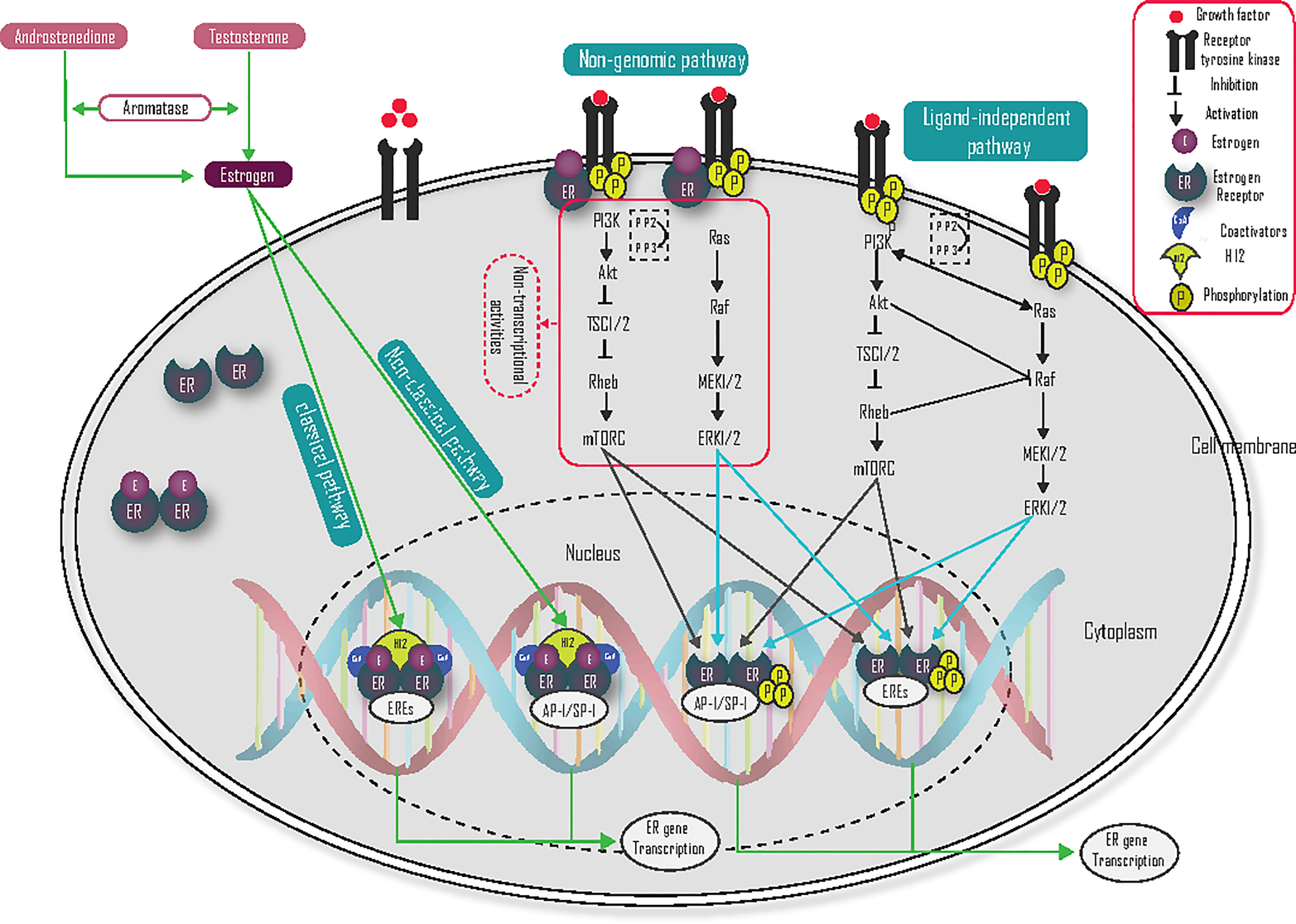
Figure 2 Ligand-dependent and ligand-independent mechanism of action of the estrogen receptor.
Indirect Genomic/Non-Classical
In indirect genomic/non-classical pathways, estrogen receptors regulate the transcription of genes that do not contain EREs through indirect binding to DNA. The indirect ER binding is mediated by different co-factors (like SP-1, AP-1, and NF-κB) that stimulate gene transcription through interaction with DNA (Figure 2) (20, 24). Specificity protein 1 (Sp-1) is the main transcriptional factor that binds with ER and contributes to coactivator recruitment (25). Several genes like low-density lipoprotein receptor, progesterone receptor B, endothelial nitric oxide synthase, GATA binding protein 1, signal transducer and activator of transcription 5, activating transcription factor-2, c-jun, c Fos, ATF-1/cAMP response element-binding protein, nuclear transcription factor-Y, cyclin D1 and the retinoic acid receptor-1α are induced by estrogen via the Sp-1 mechanism (10, 25). Activator protein 1 (AP-1) is also another main transcription co-factor that binds with ER and regulates target gene transcription. Genes like insulin-like growth factor-1 (IGF1), collagenase, IGF1-receptor, ovalbumin, and cyclin D1 are induced by the ER-AP-1 binding activation pathway (10), but ERβ inhibits the AP-1 dependent transcription of cyclin D1 (23).
Non-Genomic/Membrane-Initiated
The non-genomic ER pathway can occur very quickly and initially independent of genomic gene transcription. This rapid mechanism of action is mediated by the membrane-associated ER. Plasma membrane localization of ER is mediated by heat shock protein 27 (HSP27) (26, 27), and associates with the membrane at caveolae lipid rafts through interactions with caveolin-1, Src, and striatin. The binding of membrane-localized ER and estrogen interact directly with RTK, the p85 regulatory subunit of PI3K, Src, and Shc to activate RAS/RAF/MEK1/2 and ERK1/2, PI3K/Akt/mTOR) signaling pathway (Figure 2). These kinase pathways not only induce cell survival and cell proliferation but also phosphorylate ER and its coregulators, which result in the activation of nuclear genomic transcription. Estrogen activates growth factor signaling via non-genomic actions of ER and the growth factor signaling, in turn, activates ER, hence forming a vicious cycle (28). Coregulatory proteins such as proline-glutamic acid, leucine-rich protein 1, and metastasis-associated proteins are important to activate non-genomic activity (20, 26, 29, 30). G protein-coupled receptor 30 (GPR30) is also a membrane-localized receptor that has been observed to respond to estrogen to activate rapid signalings (27), such as PI3K and calcium signaling (23). ER-mediated, non-genomic signaling can also regulate nitric oxide, PKC, and calcium flux to promote autophagy, proliferation, apoptosis, survival, and differentiation. The calcium flux via membrane-localized ER leads to the activation of kinase pathways (27). As a result, targeting this pathway could be one of the possible treatment strategies to reduce endocrine resistance.
Ligand-Independent Activation of ER
Growth factors interact with activated receptor tyrosine kinases (RTK) like human epidermal growth factor receptors, insulin-like growth factor-1 receptor (IGF-1R), and the fibroblast growth factor receptor (FGFR), which leads to activation of the phosphatidylinositol 3 kinases (PI3K) signaling pathway (21, 25, 27). Phosphatidylinositol 3 kinase contains a catalytic domain (p110) and a regulatory domain (p85), and it phosphorylates phosphatidylinositol diphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3), which in turn facilitates the phosphorylation of the Akt. Then, Akt activates mTOR via the inhibition of tuberous sclerosis 1/2 (TSC1/2). Tuberous sclerosis 1/2 is a tumor suppressor and heterodimer of tuberin and hamartin, which acts as a guanosine triphosphatase activating protein and negatively regulates Rheb-GTP by converting it into its inactive guanosine diphosphate-bound state (31–33). The tumor suppressor gene phosphatase and tensin homolog deleted on chromosome ten (PTEN) have an inhibitory effect on PI3K by dephosphorylating PIP3 to PIP2, and inositol polyphosphate 4-phosphatase type II (INPP4B) is also dephosphorylated PIP3 to PIP26 (31, 32). Activation of the ER-target gene in the PI3K/Akt/mTOR pathway (Figure 2) is mediated by phosphorylation of ER on S167 (34). Taken together, activation of the PI3K/Akt/mTOR pathway plays a central role in breast cancer, and blocking of this pathway is an attractive treatment target, especially in endocrine-resistant ER-positive breast cancer.
Growth factors binding with the RTK receptors also lead to activation of the Ras/Raf/MEK/ERK signaling pathway (Figure 2) (21, 25, 27). The binding of growth factor with RTK activates RAS. Activated RAS can then bind with RAF and activate the downstream signaling pathway (35). When Raf is activated, its C-terminal catalytic domain can interact with MEK, and its catalytic VIII subregion is phosphorylated at the Ser218 and Ser222 activation loop, which activates MEK1/2. MEK1/2 is further activating ERK1/2 by phosphorylating the Tyr and Thr regulatory sites. Activated ERK1/2 are then translocated to the nucleus and promote phosphorylation of Ser 118 in the AF-1 domain of ER and activate its ER-target gene transcriptional activity (36). This pathway may be also a crucial target for the treatment of endocrine resistance ER-positive breast cancer.
Endocrine Therapy and Mechanism of Action
Endocrine therapy is the most efficacious drug for the treatment of ER-positive breast cancer patients (37). Based on their chemical structure, endocrine therapy (antiestrogen) is classified into steroidal and non-steroidal antiestrogen, which have no agonist and partial antagonist activity, respectively. The complete loss of estrogenic activity in all tissues by steroidal antiestrogens is not desirable, because estrogen is involved in many other physiological activities, therefore non-steroidal antiestrogen that has partial antagonist activity is preferred (7).
Estrogen receptor-targeted therapy for breast cancer was first used in 1896 by Beatson (3) and currently, at least six distinct therapeutic modalities are established, namely selective ER modulators (SERMs) (tamoxifen, raloxifene, and toremifene), selective ER down-regulators (SERDs), aromatase inhibitors (anastrozole, letrozole, and exemestane), mammalian target of rapamycin inhibitors in combination with aromatase inhibitors, and cyclin-dependent kinases 4 and 6 inhibitors in combination with aromatase inhibitors and cyclin-dependent kinases 4 and 6 inhibitors in combination with SERDs (4). Tamoxifen is the most commonly used SERM treatment and is mostly recommended for premenopausal women (38). It has shown a 31% reduction in the five-year mortality rate among hormone receptor-positive women (39) and around 500,000 women are alive today as a result of tamoxifen therapy alone (40). Aromatase inhibitors are competitive inhibitors of the aromatase enzyme and inhibit the synthesis of estrogen. It is mostly used among postmenopausal patients. Combination of everolimus (mTOR inhibitor) with endocrine therapy is also a breakthrough treatment strategy for previously aromatase inhibitor-treated advanced breast cancer (41–46).
Selective ER down-regulators like fulvestrant have antagonistic effects only and were approved by the FDA in 2007 for the treatment of ER-positive, metastatic breast cancer (46). Fulvestrant is shown to have a binding affinity, which is 100 times greater than tamoxifen (47).
There are two main strategies to block ER signaling in breast cancer. The first mechanism is through inhibition of estrogen action via ER antagonism. The second mechanism is through the reduction of estrogen levels. Selective ER modulators competitively inhibit estrogen action by binding and blocking ER (2, 37). This blocking prevents H12 from capping and causes H12 to occlude the coactivator recognition groove, which prevents coactivator proteins from binding and recruits corepressors that lead to termination of estrogen-activated gene transcription (38). Selective estrogen receptor down-regulators (SERDs) (fulvestrant) compete with estrogen for binding with ER, inhibit ER receptor dimerization, and induces proteasome-dependent degradation of the estrogen receptor (37, 44, 48) (Figure 3). However, a combination of endocrine therapy with other signaling pathways like PI3K/AKT/mTOR and Ras/Raf/MEK/ERK and coregulatory factors targeted agent should be the focus of future treatment, especially in endocrine-resistant breast cancer.
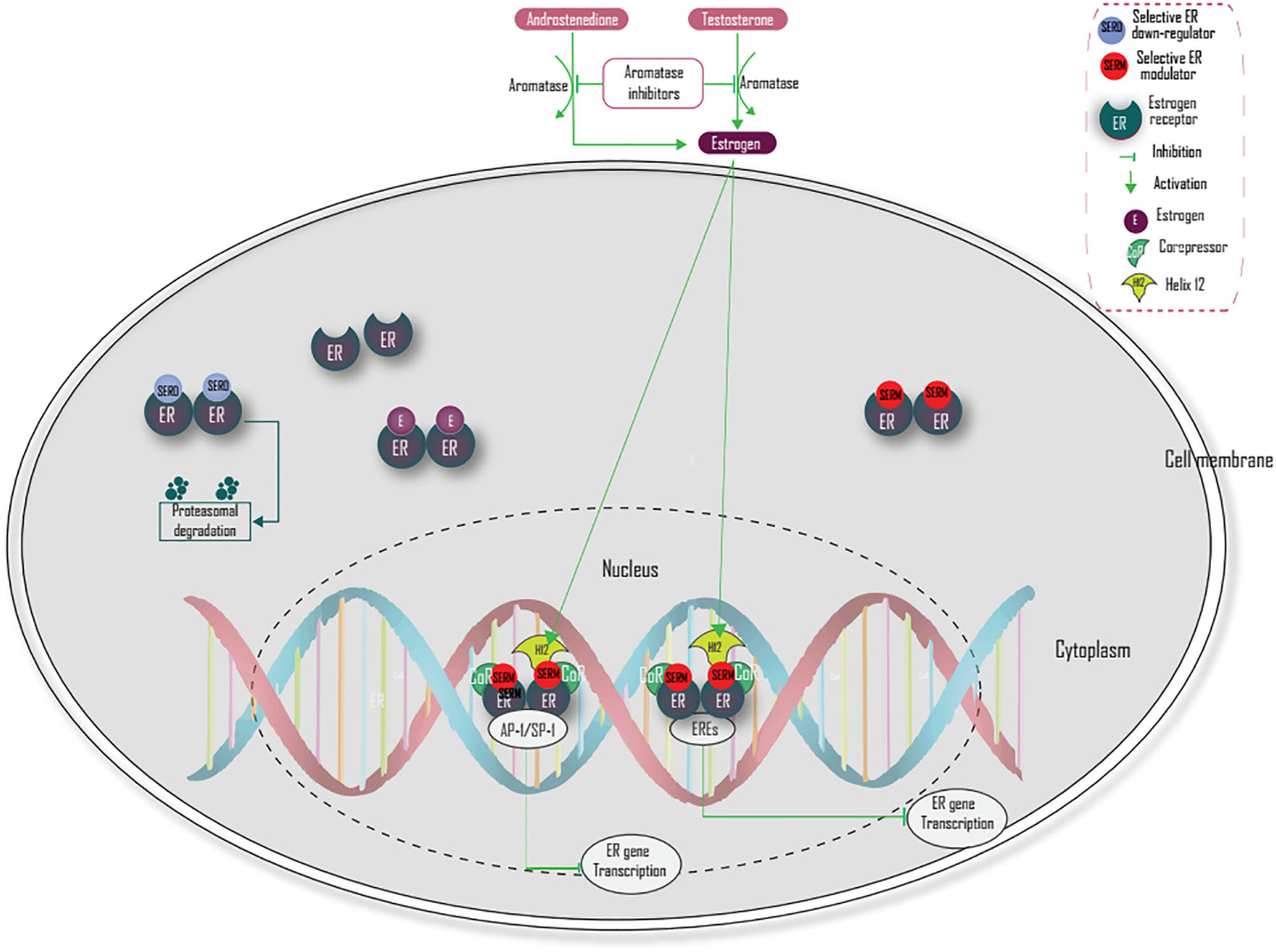
Figure 3 Mechanisms of action of main endocrine treatment.
Endocrine Resistance
Endocrine resistance occurs due to both de novo and acquired resistance. De novo resistance happens when endocrine resistance develops at the beginning of treatment, while acquired resistance occurs by non-responsive or stimulated growth after endocrine therapy (3).
De Novo or Intrinsic Resistance
Approximately, 30% of ER-positive tumors developed de novo resistance to tamoxifen therapy (49). The primary mechanism of de novo or intrinsic resistance to endocrine therapy particularly to tamoxifen, is due to lack of expression of ER (4). Recently, a second intrinsic mechanism has been documented in which patients carrying inactive alleles of cytochrome P450/2D6 (CYP2D6) fail to convert tamoxifen to its active metabolite, and are less responsive to tamoxifen (50).
Acquired Resistance
Acquired resistance occurs after endocrine treatment and the several factors listed below are responsible for this resistance (3).
Loss of ER Expression
Approximately 20% of breast cancer patients treated with endocrine therapy lose ER over time (37). Loss of ER expression involves a switch from an initially ER-positive to ER-negative phenotype, as a result, breast cancer is not suppressed by endocrine therapy that specifically targets ER. The transcriptional repression and population remodeling of the ER gene are responsible for the loss of ER expression (3). Transcriptional repression may be due to epigenetic changes, such as aberrant CpG island methylation of the ER promoter and histone deacetylation by histone deacetylase enzyme, resulting in a compact nucleosome structure that limits transcription (51). A small proportion of breast cancers presenting with nonexistent ER gene expression have an intrinsic gain in CpG site methylation (4).
Mutation of the ER Gene
Estrogen receptor gene mutations are seen in resistant breast cancer cells (52). The ER mutations are commonly found within the LBD, and the most common hot spot mutation is Tyr537Ser, Tyr537Asn, and Asp538Gly (Y537S, Y537N, D538G) (4, 53). These residues and their phosphorylation are essential in controlling the agonist state of the LBD domain of ER, conformational changes, and protein stability (54, 55), thus mutations at these residues lead to altering of the conformational dynamics of the loop connecting Helix 11 and Helix 12 in the LBD of ER, which leads to a stabilized agonist state, even though there is endocrine treatment (56). The Y537 and D538 ER mutants are also phosphorylated on S118 by the TFIIH kinase, cyclin-dependent kinase (CDK)7 in an estrogen-independent manner, which may be the possible reason for endocrine resistance by potentiating transcriptional activity of mutant ER-driven cancer (55). Mutations in the ER gene also confer loss of ER function, which is also associated with endocrine resistance (3). Targeting the transcriptional function of mutant ER proteins using BET inhibitor OTX015 is effective in reversing endocrine therapy resistance due to ER mutations (57). This indicates drugs that reverse ER mutation could be the possible treatment strategy in endocrine-resistant breast cancer resulting from ER mutation.
Altered Expression Patterns of Co-Regulatory Proteins
The transcriptional role of ER depends on co-regulatory proteins, which may activate (coactivators) or inhibit (corepressors) ER-driven transcription (3). The role of coactivators and corepressors in endocrine resistance is discussed below.
Coactivators
The SRC/p160 family of nuclear receptor coactivators are the best-characterized coactivators and consist of three members. These are SRC-1/NCoA-1, SRC-2/GRIP1/TIF2/NCoA-2, and SRC-3 (p/CIP, RAC3, ACTR, AIB1, and TRAM-1) (1, 58). These coactivators have several functional domains, such as NH2-terminal basic helix-loop-helix-Per/Ah receptor nuclear translocation/Sim domain, receptor-interacting domain (RID), and carboxyl-terminal activation domains 1 (AD1) and 2 (AD2). The RID is responsible for binding with ER and activates ER targeted gene transcription. The AD1 is capable of interacting with histone acetyltransferase (HAT) CBP and p300. The CBP and p300 are also involved in chromatin remodeling and able to activate ligand-induced ER function (59). The AD 2 can interact with protein arginine methyltransferases (PRMT), such as coactivator-associated arginine methyltransferase-1 (CARM-1) and PRMT-1 (59), which relax chromatin structure and increase the accessibility of basal components of the transcriptional machinery to ER target genes (60).
The SRC-1 initiates the transcription of endocrine-resistant genes independent of the ER (61). Coactivators like SRC-1 coordinate several signaling pathways, and that makes it an important player in tumor cells to escape endocrine therapy (62). SRC also promotes tamoxifen resistance by up-regulating SIRT1 (63).
The SRC-3 mRNA overexpression is also associated with tamoxifen resistance (37, 58, 64). Knockdown of SRC-3 can restore the antitumor effects of tamoxifen (65). The overexpression of AIB1-Δ3 isoform may contribute to antiestrogen resistance (66). The expression of SRC3 is also promoted by the general control non-derepressible 5 (GCN5), which leads to tamoxifen resistance by reducing p53 levels (67). Coactivators like SRC to play a significant role in the different signaling pathways, therefore using SRC inhibitors like small molecule inhibitors (SMIs) in combination with other treatments will be the best treatment strategy in endocrine-resistant ER-positive breast cancer.
Corepressors
Several transcriptional corepressors are involved in breast cancer, but nuclear corepressors (NCOR1), NCOR2, and the nuclear receptor subfamily 2, group F, member 2 (NR2F2) are the best-characterized corepressors (68).
Nuclear Receptor Corepressor 1
Nuclear receptor corepressor 1, also known as retinoid X receptor-interacting protein-13 (RIP-13) is a 270 kDa protein and has three RIDs found on the c-terminal region of NCOR1. The RIDs are responsible for the direct interactions between ERs and the repression domains (RI, RII, and RIII) found in the N-terminus region, which are responsible for the repressive functions. The binding of estrogen to ERs induces the movement of the H12 that stabilizes NCOR1 interactions (59, 69).
The downregulation of NCOR1 has been associated with tamoxifen resistance (6, 37, 70). Reduced levels of NCOR1 relieve the inhibition of MYC, CCND1, and SDF1 gene transcription and result in tamoxifen behaving as a partial agonist for cell cycle progression. It also regulates chromatin accessibility by recruiting histone deacetylase (HDAC)3, which leads to histone deacetylation, chromatin condensation, and loss of RNA polymerase II that induces repression of basal gene transcription (68, 71).
Nuclear Receptor Corepressor 2
Nuclear receptor corepressor 2 is also known as the silencing mediator for retinoid or thyroid hormone receptors (SMRTs) shows 41% amino acid sequence similarity with NCoR1 (68). It has four known repression domains within the N-terminal portion (RD1, RD2, RD3, and RD4) and two C-terminal nuclear receptor interaction domains (RID1 and RID2) (72). The RIDs are responsible for direct binding with ER and form a dimer on its 290-427 and 1788-1903 amino acids region and recruits other co-repressors such as GPS2, TBLR1, HDAC3, which suppress the pro-proliferative ER signaling pathway (72). Similar to NCOR1, the low-level expression of NCOR2 is related to tamoxifen resistance (68).
Nuclear Receptor Subfamily 2, Group F, Member 2
The NR2F2 gene that encodes chicken ovalbumin upstream promoter transcription factor 2 (COUP-TFII) displays copy number loss in 21% of ER-positive breast cancer. The direct interaction of COUP-TFII with dimerized ER prevents ER-dependent gene transcription and also allows it to recruit other corepressors, such as NCOR1 and NCOR2 that enhance repression. The antiproliferative effects of tamoxifen are enhanced by COUP-TFII overexpression (68). Similar to other corepressors, the expression of NR2F2 is also decreased in tamoxifen resistance (73).
Alteration of Transcriptional Factors
Transcriptional factors such as SP-1, AP-1, and NFκB are important for indirect/non-classical pathways to regulate the transcription of genes that do not contain EREs (20, 24). An increased expression of these transcriptional factors is associated with tamoxifen resistance in ER-positive breast cancer (74).
Role of MicroRNAs
MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate the expression of genes by degrading mRNA or suppressing translation (6). The oncogenic and tumor suppressor miRNAs are important for inducing and inhibiting cancer cell proliferation and invasion, respectively (75). Loss and modification of ER by miRNAs is the main mechanism of endocrine therapy resistance (75). They also inhibit the translation of the target mRNA (76). The miR-21 promotes tamoxifen resistance by targeting PTEN. Tamoxifen resistance due to miR-221/222 has also resulted via p27 and ER modulation, thereby enabling tumor growth in an ER-independent manner. The miR-155 also promotes tamoxifen resistance via suppression of cytokine signaling 6 (77). Other miRNAs that induce tamoxifen resistance include miR-181b, miR-101, miR-301and miR-519a, while miR-342, miR-116, miR-10a, miR-15a/16, miR-200b, miR-200c, miR-375, miR-261, miR-575, and miR-451 are also responsible for suppressing tamoxifen resistance (78). MiR-148a and miR-152 have also reduced tamoxifen resistance in ER breast cancer via downregulating ALCAM (79).
Role of Extracellular Vehicles
Extracellular vehicles are secreted particles that carry DNA, RNA, and protein, and are capable of transferring information and activities onto receptive cells. Extracellular vehicle treatment activates AP-1 and NF-κB transcription factors which were implicated in hormone therapy resistance in breast cancer. Exosome-mediated resistance is also achieved by the activation of the phosphoinositide 3-kinase (PI3K)/AKT pathway (6). Plasma circulating exosomes derived from obese women could also lead to tamoxifen resistance (80).
Extracellular vehicles also transport P-glycoprotein, which expels drugs located in the cytoplasm to the extracellular surface, and are the main mechanism of endocrine resistance. Transport of miR-221/222 by extracellular vehicles is also responsible for endocrine resistance (81, 82). Exporting larger quantities of drugs into the extracellular surface of the cell is seen in drug-resistant cells than drug-sensitive cells (83). The exosome mediated transfer of urothelial carcinoma-associated 1 (UCA1) also significantly increases tamoxifen resistance (84). Treatment of the parent MCF-7 cells with exosomes from the resistant cells also leads to the partial resistance of the MCF-7 cells to antiestrogen drugs (85).
Receptor Tyrosine Kinases
Human Epidermal Growth Factor 2 Signaling
Signaling through human epidermal growth factor 2 (HER2) influences the genomic actions mediated by ER. Increased crosstalk between ER and HER2 coupled with high expression of coactivator steroid receptor coactivator-3 (SRC3) is suggested as one of the endocrine drug resistance mechanisms (3). HER2, through its transcriptional regulator PEA3, contributes to endocrine resistance by potentiating steroid coactivator proteins (86).
Tamoxifen behaves as an estrogen agonist in breast cancer cells that express high levels of HER2 (87). Overexpression of HER2 and its downstream MAPK may contribute to the loss of ER, which is directly attributed to endocrine resistance (3). HER-2 overexpression also increases the anti-apoptotic Bcl-2 and BclxL proteins, which results in the reduction of tamoxifen-induced apoptosis and boosts tamoxifen resistance (44). The silencing of SRC3 or inhibiting the activity of HER2 can resensitize cells to tamoxifen treatment (3). Ubiquitin ligase c-Cbl can reverse tamoxifen resistance in HER2-overexpressing breast cancer cells by inhibiting the formation of the ER-SRC-HER2 complex (88).
Insulin-Like Growth Factor-1 Receptor
Insulin-like growth factor-1 receptor (IGF-1R) is indicated in breast cancer development, progression, and metastasis through its involvement in Ras/Raf/MEK1/2/ERK1/2 and PI3K/AKT/mTOR pathway (89). The interaction of this receptor with ER results in the redistribution of ER from the nucleus to extranuclear areas and increases ligand-independent activation of ER, which further activates Ras/Raf/MEK1/2/ERK1/2 and PI3K/AKT/mTOR pathway and results in acquired endocrine resistance (90).
Fibroblast Growth Factor Receptor
Similar to other RTK, fibroblast growth factor receptor (FGFR) families have also been implicated in breast cancer development and progression. High expression of fibroblast growth factor receptor 3 (FGFR3) is indicated in tamoxifen-resistant breast tumors by stimulating activation of the Ras/Raf/MEK1/2/ERK1/2 and PI3K/AKT/mTOR signaling pathways (91). Amplification of fibroblast growth factor receptor 1(FGFR1) also promotes cyclin D1 expression in ER-positive breast cancer, resulting in resistance to antiestrogen (92). Fibroblast growth factor receptor 2 (FGFR2) was also identified as a mediator of FGF7 action and associated with resistance to tamoxifen (93).
Cell Cycle Regulators
Over-Expression of Positive Regulators
The c-Myc is a well-known cell cycle regulator and oncogene frequently up-regulated in breast cancer (94). Over-expression of the positive regulators such as c-Myc and cyclins E1 and D1 are involved in endocrine resistance by activating cyclin-dependent kinases (37). The c-Myc expression is required for the estrogen-independent proliferation of breast cancer cells expressing ERαY537S and ERαD538G mutations and the c-Myc alone is sufficient to confer antiestrogen resistance in human breast cancer (95). The role of c-Myc in endocrine resistance may be linked with HSPC111 (HBV pre-S2 trans-regulated protein 3) (96, 97). The combinatorial use of cell cycle inhibitors along with hormonal therapy represents could be a novel therapeutic modality (94).
Reduced Expression of Negative Regulators
Reduced expression of a negative regulator such as p21 and p27 is associated with tamoxifen resistance (37). A recent study implicated loss of p21 function as one possible cause of tamoxifen-resistant (98). The loss of p21 was associated with a tamoxifen growth-inducing phenotype (99). Similar to p21, p27 inhibition has recently been associated with a tamoxifen-resistance (100). The absence of p21 enabled cyclin-CDK complexes to aberrantly phosphorylate ER when bound to tamoxifen, resulting in a growth-stimulatory phenotype (101).
Metabolic Resistance
The cytochrome P2D6 (CYP2D6) enzyme metabolizes a quarter of all prescribed drugs and is one of the main enzymes responsible for converting tamoxifen into its active metabolites (102). Endocrine therapy specifically tamoxifen is a prodrug that requires metabolism to form the pharmacologically active metabolites such as N-desmethyltamoxifen and 4-hydroxytamoxifen by cytochrome P450 (CYP)-mediated catalysis (3). Cytochrome P3A4/5 and 2D6 are major CYP isozymes involved in tamoxifen metabolism. Polymorphism of CYP2D6 also affects the metabolism of tamoxifen and leads to tamoxifen resistance (103).
Retrospective clinical data suggests that specific single nucleotide polymorphisms (SNPs) of CYP2D6 can lead to null or reduced enzyme activity resulting in worse outcomes for those individuals when treated with tamoxifen. Selective serotonin reuptake inhibitor antidepressant drugs such as paroxetine and fluoxetine have potently inhibited the metabolism of tamoxifen by CYP2D6 and thus potentially may lessen the efficacy of tamoxifen (104).
Increasing the efflux of the cell that leads to a decrease in intracellular concentrations of a drug is a general mechanism of drug resistance. Reduced uptake of tamoxifen from extracellular sources and the lower availability of intracellular tamoxifen could confer resistance (3). Extracts from resistant tumors had on average a 10-fold lower tamoxifen concentration than sensitive tumors (105). Although the precise mechanisms for lower intracellular tamoxifen levels remain unclear, potential mechanisms include the presence of microsomal antiestrogen binding sites (AEBSs) which bind tamoxifen with a similar high affinity as the ER and increase tamoxifen efflux via multi-drug resistance (MDR) P-glycoprotein drug pump (3, 106).
Immune System-Dependent Resistance
Increasing the level of immune cell-like tumor-associated macrophage and soluble mediators like interleukin-1B (IL-1B) and tumor necrosis factor α (TNFα) are important for predicting poor prognosis in breast cancer. The tumor necrosis factor α (TNFα) and Il-1B activates the nuclear factor kappa B (NF-κB) leads to endocrine resistance (4). The activity of NF‐κB is suppressed by COUP-TFII and decreased COUP-TFII expression results in an endocrine-resistant phenotype (107). The NF‐κB signaling pathway inhibitor (ACT001) in combination with tamoxifen hinders the proliferation of tamoxifen-resistant cells (108), hence NF-κB inhibition plays a promising approach to prevent tamoxifen resistance (109).
The IL-1b induces epithelial-mesenchymal transition by activation of the IL-1b/IL-1RI/b-catenin pathway, the up-regulation of TWIST1 leads to methylation of the estrogen receptor 1 gene promoter. This epigenetic modification produced a significant decrease of the ER receptor levels and increased resistance to tamoxifen (110). The expression of quiescent ALDH+ IL1R1+ cancer stem cell population that activates the IL1β signaling pathway is increasing following antiestrogen therapy and acts as an adaptive strategy that facilitates treatment resistance (111).
The chemokine (C-C motif) ligand 2 (CCL2) secreted by tumor-associated macrophages (TAM) activates PI3K/Akt/mTOR signaling and promotes an endocrine resistance feedback loop in the tumor microenvironment (TME), suggesting that CCL2 and TAM may be novel therapeutic targets for patients with endocrine-resistant breast cancer (112). The chemokine (C-C motif) ligand 2 (CCL2) secreted by tumor-associated macrophages activates PI3K/Akt/mTOR signaling and promotes an endocrine resistance feedback loop in the tumor microenvironment (TME), suggesting that CCL2 and tumor-associated macrophages may be novel therapeutic targets for patients with endocrine-resistant breast cancer (112). Transforming growth factor-beta (TGFβ) also contributes to the development of antiestrogen resistance (113).
Macrophage infiltration results in increased TNF and IL1B signaling, which stimulates aromatase expression, resulting in increased estrogen levels and increased ER signaling in ER-positive breast cancer and associated with tamoxifen resistance (114, 115).
Conclusions
The mechanisms of endocrine resistance to ER-positive breast cancer remain a mystery. Even though it is still at the early stage, several mechanisms of endocrine resistance are identified in the above review, which are basic and crucial insights for developing drugs that target interconnected pathways. Our review indicates that a combination of endocrine therapy with other drugs that target different molecular pathways and coregulators will be one of the most promising treatment approaches.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Mizan Tepi University, Addis Ababa University, and the Armauer Hansen Research Institute (AHRI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AD, Activation domains; AF1, Activation Function 1; AF-2, Activation Function 2; AIB1, Amplified in Breast Cancer 1; AP-1, Activator Protein; BLACAT1, Bladder Cancer-associated Transcript 1; CARM-1, Coactivator-associated arginine methyltransferase-1; CBP, CREB Binding Protein ; CCL2, Chemokine (C-C motif) Ligand 2; CDK, Cyclin-dependent kinase; CYP2D6, Cytochrome P450/2D6; DBD, DNA Binding Domain; ER, Estrogen receptor; EREs, Estrogen Response Elements; ERα, Estrogen Receptor α; ERβ, Estrogen Receptor β; FGFR, Fibroblast Growth Factor Receptor; GCN5, General control non-derepressible 5; GPR30, G protein-coupled receptor 30; H12, Helix 12; HAT, Histone acetyltransferase; HDAC, Histone deacetylase; HSP27, Heat shock protein 27; IGF, Insulin-like growth factor; IGF-1R, Insulin-like growth factor-1 receptor; IL-1B, Interleukin-1B; LBD, Ligand-binding Domain; MAPK, Mitogen-activated Protein Kinase; CoA, Coactivator; CoR, Corepressor; NCOR, Nuclear corepressors; NR2F2, Nuclear receptor subfamily 2, group F, member 2; PI3K, Phosphatidylinositol 3 kinases; PKA, Protein Kinase A; PRMT, Protein arginine methyltransferase; PTEN, Phosphatase and tensin homolog deleted on chromosome ten; RIDs, Receptor interaction domain; RTK, Receptor tyrosine kinase; SERDs, Selective ER Down-regulators; SERMs, Selective ER Modulators; SMRT, Silencing Mediator of Retinoic Acid and Thyroid Hormone Receptor; Sp1, Specificity Protein 1; TAM, tumor-associated macrophages; TGFβ, Transforming Growth Factor-beta; TME, Tumor microenvironment; TNFα, Tumor Necrosis Factor α; TSC, Tuberous sclerosis.
References
1. Shao W, Brown M. Advances in estrogen receptor biology: prospects for improvements in targeted breast cancer therapy. Breast Cancer Res (2003) 6(1):1–14. doi: 10.1186/bcr742
2. Lewis JS, Jordan VC. Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat Res/Fundamental Mol Mech Mutagenesis (2005) 591(1–2):247–63. doi: 10.1016/j.mrfmmm.2005.02.028
3. Chang M. Tamoxifen resistance in breast cancer. Biomolecules Ther (2012) 20(3):256. doi: 10.4062/biomolther.2012.20.3.256
4. Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer–from mechanism to therapy. Front Endocrinol (2019) 10:245. doi: 10.3389/fendo.2019.00245
5. Hamilton KJ, Hewitt SC, Arao Y, Korach KS. Chapter Four - Estrogen Hormone Biology. In: Current topics in developmental biology, vol. 125. Academic Press: Elsevier (2017). p. 109–46.
6. Haque M, Desai KV. Pathways to endocrine therapy resistance in breast cancer. Front Endocrinol (2019) 10:573. doi: 10.3389/fendo.2019.00573
7. Sommer S, Fuqua SA. Estrogen receptor and breast cancer. In: . Seminars in cancer biology, vol. 2001. Academic Press: Elsevier (2001). p. 339–52.
8. Haldosén L-A, Zhao C, Dahlman-Wright K. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol (2014) 382(1):665–72. doi: 10.1016/j.mce.2013.08.005
9. Pagano MT, Ortona E, Dupuis ML. A role for estrogen receptor alpha36 in cancer progression. Front Endocrinol (2020) 11. doi: 10.3389/fendo.2020.00506
10. Fuentes N, Silveyra P. Chapter Three - Estrogen receptor signaling mechanisms. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. vol. 116. Academic Press: Elsevier (2019). p. 135–70.
11. Zhou Y, Liu XJBR. The role of estrogen receptor beta in breast cancer. Curr Diabetes Rev (2020) 8(1):1–12. doi: 10.1186/s40364-020-00223-2
12. Omoto Y, Iwase H. Clinical significance of estrogen receptor β in breast and prostate cancer from biological aspects. Cancer Sci (2015) 106(4):337–43. doi: 10.1111/cas.12613
13. Balfe P, McCann A, McGoldrick A, McAllister K, Kennedy M, Dervan P, et al. Estrogen receptor α and β profiling in human breast cancer. Eur J Surg Oncol (2004) 30(5):469–74. doi: 10.1016/j.ejso.2004.02.010
14. Huang B, Omoto Y, Iwase H, Yamashita H, Toyama T, Coombes RC, et al. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc Natl Acad Sci U S A (2014) 111(5):1933–8. doi: 10.1073/pnas.1323719111
15. Szostakowska M, Trębińska-Stryjewska A, Grzybowska EA, Fabisiewicz A. Resistance to endocrine therapy in breast cancer: molecular mechanisms and future goals. Breast Cancer Res Treat (2019) 173(3):489–97. doi: 10.1007/s10549-018-5023-4
16. VanHook AM. Ligand-Independent ER Activation. Sci Signaling (2010) 3(117):ec112. doi: 10.1126/scisignal.3117ec112
17. Bai Z, Gust R. Breast cancer, estrogen receptor and ligands. Archiv der Pharmazie (2009) 342(3):133–49. doi: 10.1002/ardp.200800174
18. Castoria G, Migliaccio A, Giovannelli P, Auricchio F. Cell proliferation regulated by estradiol receptor: Therapeutic implications. Steroids (2010) 75(8-9):524–7. doi: 10.1016/j.steroids.2009.10.007
19. Giraldi T, Giovannelli P, Di Donato M, Castoria G, Migliaccio A, Auricchio F. Steroid signaling activation and intracellular localization of sex steroid receptors. J Cell Commun Signal (2010) 4(4):161–72. doi: 10.1007/s12079-010-0103-1
20. Puglisi R, Mattia G, Carè A, Marano G, Malorni W, Matarrese P. Non-genomic Effects of Estrogen on Cell Homeostasis and Remodeling With Special Focus on Cardiac Ischemia/Reperfusion Injury. Front Endocrinol (2019) 10:733. doi: 10.3389/fendo.2019.00733
21. Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res (2016) 118(6):994–1007. doi: 10.1161/CIRCRESAHA.115.305376
22. Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol (2005) 19(4):833–42. doi: 10.1210/me.2004-0486
23. Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med (2013) 19(3):197–209. doi: 10.1016/j.molmed.2012.12.007
24. Hayashi S-I, Yamaguchi Y. Estrogen signaling pathway and hormonal therapy. Breast Cancer (2008) 15(4):256–61. doi: 10.1007/s12282-008-0070-z
25. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics (2006) 7(8):497–508. doi: 10.2174/138920206779315737
26. Zilli M, Grassadonia A, Tinari N, Di Giacobbe A, Gildetti S, Giampietro J, et al. Molecular mechanisms of endocrine resistance and their implication in the therapy of breast cancer. Biochim Biophys Acta (2009) 1795(1):62–81. doi: 10.1016/j.bbcan.2008.08.003
27. Wilkenfeld SR, Lin C, Frigo DEJS. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids (2018) 133:2–7. doi: 10.1016/j.steroids.2017.11.005
28. Saxena NK, Sharma D. Epigenetic Reactivation of Estrogen Receptor: Promising Tools for Restoring Response to Endocrine Therapy. Mol Cell Pharmacol (2010) 2(5):191–202. doi: 10.4255/mcpharmacol.10.25
29. Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
30. Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol Rev (2017) 97(3):1045–87. doi: 10.1152/physrev.00024.2016
31. Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, et al. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev (2013) 39(8):935–46. doi: 10.1016/j.ctrv.2013.03.009
32. Liu J, Li H-Q, Zhou F-X, Yu J-W, Sun L, Han Z-H. Targeting the mTOR pathway in breast cancer. Tumour Biol (2017) 39(6):1010428317710825. doi: 10.1177/1010428317710825
33. Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol (2014) 6(4):154–66. doi: 10.1177/1758834014530023
34. Siersbæk R, Kumar S, Carroll JS. Signaling pathways and steroid receptors modulating estrogen receptor α function in breast cancer. Genes Dev (2018) 32(17-18):1141–54. doi: 10.1101/gad.316646.118
35. Degirmenci U, Wang M, Hu J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells (2020) 9(1):198. doi: 10.3390/cells9010198
36. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med (2020) 19(3):1997–2007. doi: 10.3892/etm.2020.8454
37. Giuliano M, Schiff R, Osborne CK, Trivedi MV. Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast (2011) 20:S42–9. doi: 10.1016/S0960-9776(11)70293-4
38. Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther (2018) 186:1–24. doi: 10.1016/j.pharmthera.2017.12.012
39. EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet (2005) 365(9472):1687–717. doi: 10.1016/S0140-6736(05)66544-0
40. Reinert T, de Paula B, Shafaee MN, Souza PH, Ellis MJ, Bines J. Endocrine therapy for ER-positive/HER2-negative metastatic breast cancer. Chin Clin Oncol (2018) 7(3):25–5. doi: 10.21037/cco.2018.06.06
41. Villarreal-Garza C, Cortes J, Andre F, Verma S. mTOR inhibitors in the management of hormone receptor-positive breast cancer: the latest evidence and future directions. Ann Oncol (2012) 23(10):2526–35. doi: 10.1093/annonc/mds075
42. Gnant M, Greil R, Hubalek M, Steger G. Everolimus in postmenopausal, hormone receptor-positive advanced breast cancer: summary and results of an austrian expert panel discussion. Breast Care (2013) 8(4):293–9. doi: 10.1159/000354121
43. Giuliano AE, Connolly JL, Edge SB, Mittendorf EA, Rugo HS, Solin LJ, et al. Breast Cancer-Major changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin (2017) 67(4):290–303. doi: 10.3322/caac.21393
44. Ali S, Rasool M, Chaoudhry H, Pushparaj PN, Jha P, Hafiz A, et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation (2016) 12(3):135. doi: 10.6026/97320630012135
45. Brufsky AM, Dickler MN. Estrogen receptor-positive breast cancer: exploiting signaling pathways implicated in endocrine resistance. Oncologist (2018) 23(5):528. doi: 10.1634/theoncologist.2017-0423
46. Ahmad I, Mathew S, Rahman S. Recent progress in selective estrogen receptor downregulators (SERDs) for the treatment of breast cancer. RSC Medicinal Chem (2020) 11(4):438–54. doi: 10.1039/C9MD00570F
47. Li J, Wang Z, Shao Z. : Fulvestrant in the treatment of hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: A review. Cancer Med (2019) 8(5):1943–57. doi: 10.1002/cam4.2095
48. McDonnell DP, Wardell SE, Norris JD. Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. In: . ACS Publications (2015).
49. Jordan VC, O’Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol (2007) 25(36):5815–24. doi: 10.1200/JCO.2007.11.3886
50. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer (2009) 9(9):631–43. doi: 10.1038/nrc2713
51. Viedma-Rodríguez R, Baiza-Gutman L, Salamanca-Gómez F, Diaz-Zaragoza M, Martínez-Hernández G, Ruiz Esparza-Garrido R, et al. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (Review). Oncol Rep (2014) 32(1):3–15. doi: 10.3892/or.2014.3190
52. Martin L-A, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun (2017) 8(1):1–15. doi: 10.1038/s41467-017-01864-y
53. Jeselsohn R. Are we ready to use ESR1 mutations in clinical practice. Breast Care (2017) 12(5):309–13. doi: 10.1159/000481428
54. Yudt MR, Vorojeikina D, Zhong L, Skafar DF, Sasson S, Gasiewicz TA, et al. Function of estrogen receptor tyrosine 537 in hormone binding, DNA binding, and transactivation. Biochemistry (1999) 38(43):14146–56. doi: 10.1021/bi9911132
55. Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, et al. Santos DB et al: Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene (2017) 36(16):2286–96. doi: 10.1038/onc.2016.382
56. Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. elife (2016) 5:e12792. doi: 10.7554/eLife.12792
57. Alluri P, Larios J, Malik R, Rae J, Chinnaiyan A. Targeting Estrogen Receptor (ER) Mutations for Treatment of Endocrine Therapy Resistance in Breast Cancer. Int J Radiat Oncology• Biology• Phys (2016) 96(2):S53. doi: 10.1016/j.ijrobp.2016.06.139
58. Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res (2003) 9(1 Pt 2):447s–54s.
59. Cottone E, Orso F, Biglia N, Sismondi P, De Bortoli M. Role of coactivators and corepressors in steroid and nuclear receptor signaling: potential markers of tumor growth and drug sensitivity. Int J Biol Markers (2001) 16(3):151–66. doi: 10.1177/172460080101600301
60. Karmakar S, Foster EA, Blackmore JK, Smith CL. Distinctive functions of p160 steroid receptor coactivators in proliferation of an estrogen-independent, tamoxifen-resistant breast cancer cell line. Endocr Rel Cancer (2011) 18(1):113–27. doi: 10.1677/ERC-09-0285
61. Browne AL, Charmsaz S, Varešlija D, Fagan A, Cosgrove N, Cocchiglia S, et al. Network analysis of SRC-1 reveals a novel transcription factor hub which regulates endocrine resistant breast cancer. Oncogene (2018) 37(15):2008–21. doi: 10.1038/s41388-017-0042-x
62. Walsh CA, Qin L, Tien JC-Y, Young LS, Xu J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int J Biol Sci (2012) 8(4):470. doi: 10.7150/ijbs.4125
63. Zhou J, Xu M, Le K, Ming J, Guo H, Ruan S, et al. SRC Promotes Tamoxifen Resistance in Breast Cancer via Up-Regulating SIRT1. OncoTargets Ther (2020) 13:4635–47. doi: 10.2147/OTT.S245749
64. Hou J, Liu J, Yuan M, Meng C, Liao J. The role of amplified in breast cancer 1 in breast cancer: A meta-analysis. Med (Baltimore) (2020) 99(46). doi: 10.1097/MD.0000000000023248
65. Su Q, Hu S, Gao H, Ma R, Yang Q, Pan Z, et al. Role of AIB1 for tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncology (2008) 75(3-4):159–68. doi: 10.1159/000159267
66. Reiter R, Oh AS, Wellstein A, Riegel AT. Impact of the nuclear receptor coactivator AIB1 isoform AIB1-Δ3 on estrogenic ligands with different intrinsic activity. Oncogene (2004) 23(2):403–9. doi: 10.1038/sj.onc.1207202
67. Oh JH, Lee J-Y, Kim KH, Kim CY, Cho Y, Nam KT, et al. Elevated GCN5 expression confers tamoxifen resistance by upregulating AIB1 expression in ER-positive breast cancer. Cancer Lett (2020) 495:145–55. doi: 10.1016/j.canlet.2020.09.017
68. Légaré S, Basik M. Minireview: the link between ERα corepressors and histone deacetylases in tamoxifen resistance in breast cancer. Mol Endocrinol (2016) 30(9):965–76. doi: 10.1210/me.2016-1072
69. Wong MM, Guo C, Zhang J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol (2014) 2(3):169.
70. Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen T-M, Schiff R, et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci (1998) 95(6):2920–5. doi: 10.1073/pnas.95.6.2920
71. Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer (2004) 11(4):643–58. doi: 10.1677/erc.1.00776
72. Gong C, Man EP, Tsoi H, Lee TK, Lee P, Ma S-T, et al. BQ323636. 1, a novel splice variant to NCOR2, as a predictor for tamoxifen-resistant breast cancer. Clin Cancer Res (2018) 24(15):3681–91. doi: 10.1158/1078-0432.CCR-17-2259
73. Erdős E, Bálint BL. NR2F2 Orphan Nuclear Receptor is Involved in Estrogen Receptor Alpha-Mediated Transcriptional Regulation in Luminal A Breast Cancer Cells. Int J Mol Sci (2020) 21(6):1910. doi: 10.3390/ijms21061910
74. Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NFκB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer (2007) 7(1):59. doi: 10.1186/1471-2407-7-59
75. Alamolhodaei NS, Behravan J, Mosaffa F, Karimi G. MiR 221/222 as New Players in Tamoxifen Resistance. Curr Pharm Des (2016) 22(46):6946–55. doi: 10.2174/1381612822666161102100211
76. Miller T, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro C, et al. MicroRNA-221/222 Confers Tamoxifen Resistance in Breast Cancer by Targeting p27Kip1. J Biol Chem (2008) 283:29897–903. doi: 10.1074/jbc.M804612200
77. Dobre E-G, Dinescu S, Costache M. Connecting the Missing Dots: ncRNAs as Critical Regulators of Therapeutic Susceptibility in Breast Cancer. Cancers (Basel) (2020) 12(9):2698. doi: 10.3390/cancers12092698
78. Zhang W, Xu J, Shi Y, Sun Q, Zhang Q, Guan X. The novel role of miRNAs for tamoxifen resistance in human breast cancer. Cell Mol Life Sci (2015) 72(13):2575–84. doi: 10.1007/s00018-015-1887-1
79. Chen M-J, Cheng Y-M, Chen C-C, Chen Y-C, Shen C-J. MiR-148a and miR-152 reduce tamoxifen resistance in ER+ breast cancer via downregulating ALCAM. Biochem Biophys Res Commun (2017) 483(2):840–6. doi: 10.1016/j.bbrc.2017.01.012
80. Sadegh-Nejadi S, Afrisham R, Emamgholipour S, Izadi P, Eivazi N, Tahbazlahafi B, et al. Influence of plasma circulating exosomes obtained from obese women on tumorigenesis and tamoxifen resistance in MCF-7 cells. IUBMB Life (2020) 72(9). doi: 10.1002/iub.2305
81. Cruz De los Santos M, Dragomir MP, Calin GA. The role of exosomal long non-coding RNAs in cancer drug resistance. Cancer Drug Resistance (2019) 2(4):1178–92. doi: 10.20517/cdr.2019.74
82. Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. Am J Pathol (2005) 167(4):913–20. doi: 10.1016/S0002-9440(10)61180-7
83. Xavier CPR, Caires HR, Barbosa MAG, Bergantim R, Guimarães JE, Vasconcelos MH. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells (2020) 9(5):1141. doi: 10.3390/cells9051141
84. Xu C, Yang M, Ren Y, Wu C, Wang L. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur Rev Med Pharmacol Sci (2016) 20(20):4362–8.
85. Semina SE, Scherbakov AM, Vnukova AA, Bagrov DV, Evtushenko EG, Safronova VM, et al. Exosome-mediated transfer of cancer cell resistance to antiestrogen drugs. Molecules (2018) 23(4):829. doi: 10.3390/molecules23040829
86. Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, et al. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol (2004) 57(10):1069–74. doi: 10.1136/jcp.2004.016733
87. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2–positive breast cancer. J Natl Cancer Institute (2004) 96(12):926–35. doi: 10.1093/jnci/djh166
88. Li W, Xu L, Che X, Li H, Zhang Y, Song N, et al. C-Cbl reverses HER2-mediated tamoxifen resistance in human breast cancer cells. BMC Cancer (2018) 18(1):1–13. doi: 10.1186/s12885-018-4387-5
89. Christopoulos PF, Msaouel P, Koutsilieris M. The role of the insulin-like growth factor-1 system in breast cancer. Mol Cancer (2015) 14:43–3. doi: 10.1186/s12943-015-0291-7
90. Fan P, Jordan VC. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resistance (Alhambra Calif) (2019) 2:198. doi: 10.20517/cdr.2019.13
91. Tomlinson D, Knowles M, Speirs V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer (2012) 130(12):2857–66. doi: 10.1002/ijc.26304
92. Zhou Y, Wu C, Lu G, Hu Z, Chen Q, Du X. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer (2020) 11(8):2000. doi: 10.7150/jca.40531
93. Turczyk L, Kitowska K, Mieszkowska M, Mieczkowski K, Czaplinska D, Piasecka D, et al. Romanska HM et al: FGFR2-Driven Signaling Counteracts Tamoxifen Effect on ERα-Positive Breast Cancer Cells. Neoplasia (2017) 19(10):791–804. doi: 10.1016/j.neo.2017.07.006
94. Nair BC, Vadlamudi RK. Regulation of hormonal therapy resistance by cell cycle machinery. Gene Ther Mol Biol (2008) 12:395.
95. Yu L, Wang L, Mao C, Duraki D, Kim JE, Huang R, et al. Estrogen-independent Myc over expression confers endocrine therapy resistance on breast cancer cells expressing ERαY537S and ERαD538G mutations. Cancer Lett (2019) 442:373–82. doi: 10.1016/j.canlet.2018.10.041
96. Venditti M, Iwasiow B, Orr FW, Shiu RP. C-myc gene expression alone is sufficient to confer resistance to antiestrogen in human breast cancer cells. Int J Cancer (2002) 99(1):35–42. doi: 10.1002/ijc.10269
97. Chen Z, Wang Y, Warden C, Chen S, Cross-talk between ER. and HER2 regulates c-MYC-mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. J Steroid Biochem Mol Biol (2015) 149:118–27. doi: 10.1016/j.jsbmb.2015.02.004
98. Jankevicius F, Goebell P, Kushima M, Schulz WA, Ackermann R, Schmitz-Dräger BJ. p21 and p53 immunostaining and survival following systemic chemotherapy for urothelial cancer. Urol Int (2002) 69(3):174–80. doi: 10.1159/000063949
99. Abukhdeir AM, Vitolo MI, Argani P, De Marzo AM, Karakas B, Konishi H, et al. Tamoxifen-stimulated growth of breast cancer due to p21 loss. Proc Natl Acad Sci U.S.A. (2008) 105(1):288–93. doi: 10.1073/pnas.0710887105
100. Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell (2007) 128(2):281–94. doi: 10.1016/j.cell.2006.11.049
101. Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med (2008) 10:e19. doi: 10.1017/S1462399408000744
102. Dean L. Tamoxifen therapy and CYP2D6 genotype. Bethesda (MD): National Center for Biotechnology Information (US). (2019).
103. Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anti Cancer Agents Medicinal Chem (Formerly Curr Medicinal Chemistry Anti Cancer Agents) (2009) 9(5):481–99. doi: 10.2174/187152009788451833
104. Singh MS, Francis PA, Michael M. Tamoxifen, cytochrome P450 genes and breast cancer clinical outcomes. Breast (2011) 20(2):111–8. doi: 10.1016/j.breast.2010.11.003
105. Osborne CK, Fuqua SA. Mechanisms of tamoxifen resistance. Breast Cancer Res Treat (1994) 32(1):49–55. doi: 10.1007/BF00666205
106. Lykkesfeldt AE. Mechanisms of tamoxifen resistance in the treatment of advanced breast cancer. Acta Oncol (1996) 35(sup5):9–14. doi: 10.3109/02841869609083961
107. Litchfield LM, Appana SN, Datta S, Klinge CM. COUP-TFII inhibits NFkappaB activation in endocrine-resistant breast cancer cells. Mol Cell Endocrinol (2014) 382(1):358–67. doi: 10.1016/j.mce.2013.10.010
108. Jin XH, Jia YS, Shi YH, Li QY, Bao SQ, Lu WP, et al. ACT001 can prevent and reverse tamoxifen resistance in human breast cancer cell lines by inhibiting NF-κB activation. J Cell Biochem (2019) 120(2):1386–97. doi: 10.1002/jcb.27146
109. Kastrati I, Joosten SE, Semina SE, Alejo LH, Brovkovych SD, Stender JD, et al. The NFκB pathway promotes tamoxifen tolerance and disease recurrence in estrogen receptor positive breast cancers. Mol Cancer Res (2020) 18(7). doi: 10.1158/1541-7786.MCR-19-1082
110. Jiménez-Garduño AM, Mendoza-Rodríguez MG, Urrutia-Cabrera D, Domínguez-Robles MC, Pérez-Yépez EA, Ayala-Sumuano JT, et al. IL-1β induced methylation of the estrogen receptor ERα gene correlates with EMT and chemoresistance in breast cancer cells. Biochem Biophys Res Commun (2017) 490(3):780–5. doi: 10.1016/j.bbrc.2017.06.117
111. Sarmiento-Castro A, Caamano-Gutierrez E, Sims A, James MI, Santiago-Gomez A, Eyre R, et al. A dormant sub-population expressing interleukin-1 receptor characterises anti-estrogen resistant ALDH+ breast cancer stem cells. Cancer Sci (2019) 111(1):821876. doi: 10.1101/821876
112. Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, et al. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci (2020) 111(1):47–58. doi: 10.1111/cas.14230
113. Joffroy CM, Buck MB, Stope MB, Popp SL, Pfizenmaier K, Knabbe C. Antiestrogens Induce Transforming Growth Factor β–Mediated Immunosuppression in Breast Cancer. Cancer Res (2010) 70(4):1314–22. doi: 10.1158/0008-5472.CAN-09-3292
114. Murray JI, West NR, Murphy LC, Watson PH. Intratumoural inflammation and endocrine resistance in breast cancer. Endocr Rel Cancer (2015) 22(1):R51–67. doi: 10.1530/ERC-14-0096
Keywords: acquired resistance, breast cancer, endocrine resistance, endocrine therapy, estrogen receptor, de novo resistance
Citation: Belachew EB and Sewasew DT (2021) Molecular Mechanisms of Endocrine Resistance in Estrogen-Receptor-Positive Breast Cancer. Front. Endocrinol. 12:599586. doi: 10.3389/fendo.2021.599586
Received: 28 September 2020; Accepted: 15 February 2021;
Published: 25 March 2021.
Edited by:
Antimo Migliaccio, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Zoran Culig, Innsbruck Medical University, AustriaPia Giovannelli, University of Campania Luigi Vanvitelli, Italy
Copyright © 2021 Belachew and Sewasew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esmael Besufikad Belachew, Z2V0YmIyMDA2QGdtYWlsLmNvbQ==