 Lucas Massier
Lucas Massier Matthias Blüher1,3
Matthias Blüher1,3 Peter Kovacs
Peter Kovacs Rima M. Chakaroun
Rima M. Chakaroun- 1Medical Department III – Endocrinology, Nephrology, Rheumatology, University of Leipzig Medical Center, Leipzig, Germany
- 2Department of Medicine (H7), Karolinska Institutet, Karolinska University Hospital, Huddinge, Stockholm, Sweden
- 3Helmholtz Institute for Metabolic, Obesity and Vascular Research (HI-MAG) of the Helmholtz Zentrum München, University Hospital Leipzig, University of Leipzig, Leipzig, Germany
An intact intestinal barrier, representing the interface between inner and outer environments, is an integral regulator of health. Among several factors, bacteria and their products have been evidenced to contribute to gut barrier impairment and its increased permeability. Alterations of tight junction integrity - caused by both external factors and host metabolic state - are important for gut barrier, since they can lead to increased influx of bacteria or bacterial components (endotoxin, bacterial DNA, metabolites) into the host circulation. Increased systemic levels of bacterial endotoxins and DNA have been associated with an impaired metabolic host status, manifested in obesity, insulin resistance, and associated cardiovascular complications. Bacterial components and cells are distributed to peripheral tissues via the blood stream, possibly contributing to metabolic diseases by increasing chronic pro-inflammatory signals at both tissue and systemic levels. This response is, along with other yet unknown mechanisms, mediated by toll like receptor (TLR) transduction and increased expression of pro-inflammatory cytokines, which in turn can further increase intestinal permeability leading to a detrimental positive feedback loop. The modulation of gut barrier function through nutritional and other interventions, including manipulation of gut microbiota, may represent a potential prevention and treatment target for metabolic diseases.
Introduction
While the obesity and type 2 diabetes (T2D) pandemics are increasing at a fast pace (1), new factors relevant to the lack of adaptation toward the increasingly rapid changes in our environment have been proposed as possible perpetrators. Among those, the intestinal microbiota and its interactions with host metabolism and the immune system have been acknowledged to influence and contribute to several diseases including gastrointestinal disorders such as inflammatory bowel diseases (2) and more recently the plethora of metabolic and cardiovascular diseases (3, 4). However, the underlying mechanisms are still unknown (5–7). One poorly understood feature of metabolic disease is the alteration and dysfunction of the intestinal barrier, accompanied with an increase in intestinal permeability. The healthy gut barrier constitutes a crucial boundary protecting the host from external stimuli and pathogens by providing spatial compartmentalization and various defense mechanisms, while concurrently allowing the uptake of necessary nutrients (8). While the influx of microbial products has been suggested to underlie chronic inflammation observed in metabolic disease and more specifically T2D (9), clinical features of metabolic diseases such as hyperglycemia in T2D have been associated with increased influx of microbial products in humans reflective of glucotoxicity (10). First studies in the 1950s suggested that increased endotoxins, which are lipopolysaccharides of gram-negative bacteria in the circulation result from an increased intestinal permeability (11). Although still under debate, independent studies in mice and humans could confirm these results (12–16). In 1984, the term “leaky gut” was first introduced when Bjarnason et al. reported that patients with excessive alcohol consumption had increased intestinal permeability (17), which is also referred to as hyperpermeability (18). In the following years research on intestinal permeability focused on autoimmune and inflammatory diseases such as celiac disease (19, 20) or Crohn’s disease (21). Only recently, possible associations between intestinal permeability and characteristics of a metabolic disease like obesity or T2D have emerged as plausible and attractive targets in the field of obesity research.
In this review, we aim to highlight underlying mechanisms of an impaired intestinal barrier and its possible impact on metabolic health. We specifically discuss recent findings on how endotoxemia and translocation of bacteria, bacterial genetic material and products may cause tissue and organ dysfunction subsequently contributing to metabolic diseases.
The Intestinal Barrier
Components of Intestinal Barrier
The intestine represents an active interface, where external environmental factors, such as diet, medication and the billions of symbionts inhabiting our gut co-exist and interact – usually peacefully – with host factors including the immune system (Figure 1A). The intestinal barrier allows and facilitates the uptake of nutrients and water from the intestinal lumen, whilst at the same time providing effective mechanisms to combat harmful substances and pathogens and prevent their translocation into the host circulation and peripheral tissues (22) (Figure 1B). These checkpoints comprise an intricate network of mechanical and immunological factors including the mucus, the epithelium and the underlying lamina propria (22) as well as humoral and immunological factors. The goblet cells produced mucus layer constitutes of mucin and forms a protective barrier, limiting the amount of bacteria reaching the epithelial cells (23). Epithelial cells form microvilli, protrusions in the range of 100 nm width, that help to increase the area for absorptions of ions and nutrients using specified receptors (24). This line of densely arranged microvilli is termed brush border, which, on the one hand, physically prevents bacteria coming in contact with the cell soma, but otherwise help to actively combat pathogenic bacteria (25). For example, native luminal vesicles containing intestinal alkaline phosphatase are released from the tips of the microvilli, helping to dephosphorylate lipopolysacchradies (LPS), hinder bacterial growth and prevent adherence of enteropathogenic E. coli to the epithelial cell layer (26). Epithelial cells are connected by tight junctions (TJ), which hinder the entry of pathogens while regulating the paracellular flow of water, small ions and nutrients (27). Tight junction complexes mainly consist of occludin and members of the claudin family. Expression of the latter is tissue-specific and the claudin composition defines the kind and function of a specific tight junction, e.g., claudin-7 reduces permeability to anions and claudin-1 reduces permeability to cations (28, 29). Claudins and occludin are connected to the cytoskeleton by scaffolding proteins including zonula occludens (ZO) proteins (30) (Figure 1C). Additionally, cells are connected by adherence junctions (for cell-cell signaling) (31) and desmosomes (for cell stability) (32). The mechanical gut barrier interacts constantly with and is highly impacted by local immune components consisting of soluble Immunoglobulin A (IgA), epithelial and Paneth cell produced, and secreted antimicrobial peptides such as alpha-defensins, lysozyme, and C-type lectin (33) providing additional protection to the epithelial layer and the crypts of Lieberkühn (34). Not less important are cells belonging to the innate and adaptive immunity system such as macrophages, dendritic cells (35), T-regulatory cells (36) and the highly dense lymphoid tissues inhabiting the lining of the gut but also highly active in adjunct lymph nodes (37). Disruptions in this intricately balanced system through toxins, microorganisms, nutrients, or food contaminants can lead to an alteration of the intestinal permeability, by an increased and inadequately controlled influx via the paracellular and transcellular pathway.
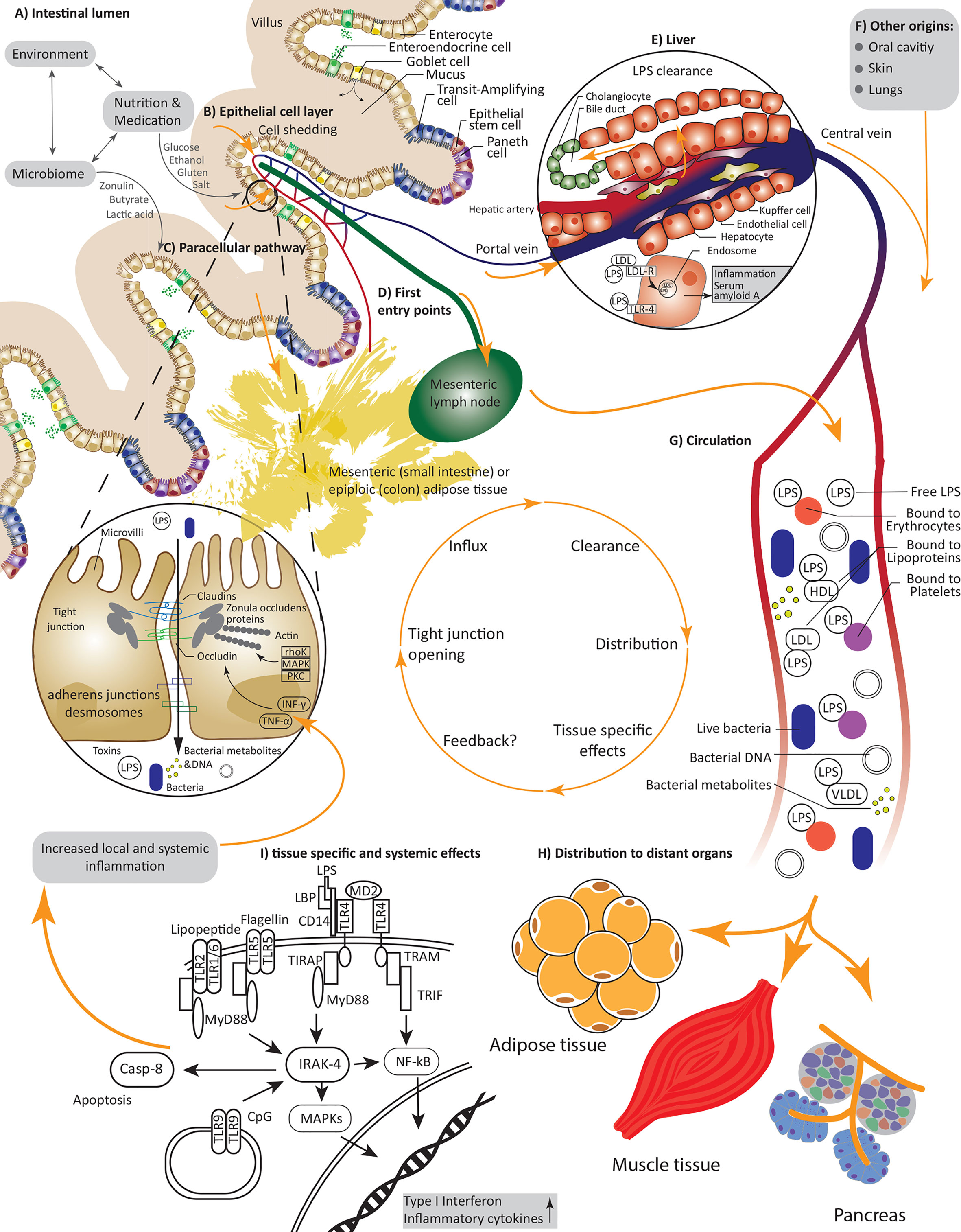
Figure 1 Causes and consequences of increased intestinal permeability; (A) An interplay between environmental factors, nutrients, drugs and the microbiome defines our intestinal lumen and can directly or indirectly alter permeability of the epithelial cell layer; (B) Increased cell shedding under pathophysiological conditions or the reversible interaction with and opening of tight junctions under allow an influx of pathogens including bacteria or their nucleic acids, metabolites, and lipopolysaccharides as well as further substances and toxins; (C) Paracellular transport via tight junctions is regulated by various kinases and inflammatory cytokines, nutrients and bacteria can interfere with claudin and occludin expression; (D) First entry points of invading substances are capillaries and lymph vessels of the villi and tissues in close proximity such as mesenteric adipose tissue; (E) Endotoxin is rapidly and constantly cleared in the liver; (F) Next to the gut lumen, pathogens could also invade through the oral cavity, the skin and the lung; (G) Under constant interactions and clearance with the immune system the LPS and bacteria are transported in the circulation, thereby LPS can be bound to various proteins and cells including erythrocytes and lipoproteins; (H) Effected distant organs include adipose tissue, pancreas tissue and possibly muscle tissue; (I) Pathogens interact with many receptors, i.e., LPS is recruited to TLR-4 leading to increased expression and secretion of inflammatory cytokines.
The regulation of this system can, under both physiological and pathophysiological conditions, be altered by various endogenous and exogenous factors as discussed in the following paragraph.
Endogenous Regulation of the Intestinal Permeability
A constant challenge for upholding the integrity of the intestinal barrier is the continuous cell shedding on the tip of the villi, with a turnaround time of about five days (38). Tight junctions are redistributed to block the gap under involvement of rho kinase (39). Other signal cascades involved in tight junctions regulation under physiological conditions include protein kinase C’s (PKC) and mitogen-activated protein kinase (MAPK) pathways, which regulate protein phosphorylation and expression allowing a selective transport of molecules (40). In disease, increased proinflammatory cytokines such as interferon-γ (INF γ) and tumor necrosis factor α (TNF-α) shift the homeostatic balance (27) by downregulating the expression of claudin-1 (27) and increasing occludin and claudins-1 and -4 internalization, leading to an increasingly compromised gut permeability, as shown in T84 cells via immunofluorescence microscopy (41). Further mechanisms of INF-γ induced permeability in T84 cells involve phosphatiodylinositol-3-kinase/protein kinase B (PI3-K/Akt) activation and a delayed but prolonged NF-kB response (42) (Figure 1C). This is of particular interest, as metabolic diseases are usually attended by a chronic low grade inflammation (43), possibly initiating a detrimental positive feedback loop.
Mechanisms leading to whole bacterial translocation through the mucosal barrier remain unclear. Several studies suggest transcytosis via M-cells (44), which uptake large antigens and particulate matter such as viruses, bacteria, and protozoa through receptor mediated endocytosis (45). The transcellular pathway can be triggered and upheld by low concentrations of INF-γ (that are insufficient to alter the paracellular pathway), as shown by Clark et al. in Caco-2 cells (46). This uptake is also dependent on extracellular signal-regulated kinase (ERK) 1/2 and ADP-ribosylation factor (ARF)-6 signaling (25, 47).
Exogenous Perturbation
Several external factors, including bacteria, alcohol consumption, nutrition and medication have an impact on the intestinal permeability (34). A high caloric “western” diet, in particular due to high-fat/low-fiber content (48) and food additives (49), is associated with adverse changes in the microbiome (50) and both are independently important factors in the development of metabolic diseases. Whereas pathogenic bacterial species such as enteropathogenic E. coli (51–53), H. pylori (54), S. Typhimurium (55), and V. cholera (56) increase intestinal permeability, the totality of the gut microbiota contributes equally to upholding the gut barrier health by shaping tolerogenic mechanisms controlling the responses of resident dendritic cells and macrophages (57). Pathogenic bacteria can use their type III secretion system to alter the host cytoskeleton in their favor to facilitate entry to epithelial cells (58), but also non-invasive parasites like G. lamblia can introduce adverse changes, in this case by causing microvilli shortening and TJ disruption (25, 59). Moreover, beneficial effects were reported for gut bacteria produced short chain fatty acids (60, 61), in particular butyrate (61, 62), which has been shown to act as an energy source for colonocytes (63), regulate hypoxia induced factor 1α (HIF-1α) dependent expression of many pro-barrier genes in intestinal epithelial cells (64), facilitate tight junctions assembly (61) and induce colonic regulatory T-cells, leading to the suppression of inflammatory and allergic responses (62). Taxonomic groups-wise, A. muciniphila (65), Bifidobacterium (66), and lactic acid producing bacteria like L. plantarum (67) and L. reuteri (68) were, among others, also reported to improve intestinal permeability (Figure 1A).
Experimental evidence for the effect of food components mostly stems from rodent and cell culture experiments. Glucose, well known for its many adverse effects in metabolic disease (69), is partially transported paracellularly and enhances small intestinal permeability (70), increases Caco-2 cell permeability and leads to altered TJ arrangements (71). Recently, Thaiss et al. demonstrated a key role for hyperglycemia in impairing the barrier function by increasing retrograde uptake of glucose into epithelial cells via the glucose transporter (GLUT)-2 transporter, leading to changes in intracellular glucose metabolism and to transcriptional reprogramming. The latter includes, among others, alterations in the expression of genes involved in N-glycan biosynthesis and pentose-glucuronate interconversion, two pathways linked to the maintenance of epithelial barrier function (10). Further food components increasing TJ permeability include salt (72) and various fatty acids (73), all of which are known to contribute to obesity development (74, 75). Another important nutritional factor affecting the intestinal permeability is gliadin, a component of gluten found in wheat. In mouse experiments, Lammers et al. found that gliadin increased permeability by binding to the chemokine receptor 3 (Cxcr3) (76) (Figure 1A). Beneficial effects on intestinal permeability were reported for casein (77) and other cheese peptides (78), vitamin D (79) as well as polyphenols (80, 81), which is in line with findings that polyphenol rich diets ameliorate metabolic disease (82). Furthermore, addition of apple-derived pectin to a high fat diet (HFD) counteracted some negative effects of HFD by preventing microbial shifts and improving barrier function due to increased expression of claudin-1 and abating metabolic endotoxemia (83).
Moreover, occludin and claudin expression are subject to circadian control, associated with regulation of intestinal permeability and susceptibility to colitis (84). This is further substantiated by the increase in intestinal permeability after circadian disruption in mice leading to promotion of alcohol induced liver disease and inflammation (85). Similarly, gut bacterial signatures related to diurnal oscillation also enable the risk stratification and prediction of T2D within 5 years of sampling (86) linking circadian control with gut microbiota profiles and intestinal permeability regulation and downstream metabolic sequalae.
Intestinal Permeability in Metabolic Disease
Animal Studies
An increased intestinal permeability is the key factor for the migration of toxins and bacterial components to the circulation. First evidence for a link between obesity and increased permeability was found in mouse studies. Brun et al. used Ussing chambers – a physiological system to measure the transport of ions, nutrients, and drugs across epithelial tissues including the gut (87, 88) to analyze the barrier integrity in leptin-deficient (ob/ob) and hyperleptinemic, but leptin-receptor mutant (db/db) mice, a widely used animal models in obesity-induced T2D (89). They observed a significantly increased permeability, which was more prominent in db/db mice. This was accompanied by an increase in portal endotoxemia and systemic inflammation parameters (90). Later, changes in the microbiome were found to be an important factor linking intestinal permeability to inflammation (91, 92). In a key study, Cani et al. could further show that a HFD resulted in increased levels of plasma endotoxin as well as a reduced expression of TJ genes ZO-1 and occludin. This effect was reduced when mice on HFD were treated with antibiotics and plasma endotoxin levels were positively correlated with markers of inflammation (Plasminogen-Aktivator-Inhibitor 1, PAI-1) and oxidative stress (six transmembrane protein of prostate 2, STAMP2) (9). It was further shown that the shift in the intestinal permeability of mice on HFD could be attributed to claudin switching, which leads to tight junction restructuring. In this case, the expression of claudin-1,-3,-4,-7, and -15 decreased, whereas expression of claudin-2 increased (93). However, as shown in db/db diabetic mice with induced C. rodentium infection, physiological countermeasures are in place to overcome initial intestinal barrier impairment. In particular, exogenous IL-22 restored mucosal host defense as indicated by histological evaluation (94).
Human Studies
Although some studies have linked intestinal permeability with metabolic disease in humans, they should be seen with caution, as they did not employ reproducible and robust methods to test for gut permeability such as lactulose/mannitol (La/Ma) or similar tests and Ussing chambers. In 2011 Gummesson et al. reported an association between intestinal permeability and increased visceral obesity in 67 otherwise healthy women (95) and in 2012, Teixeira et al. corroborated these finding by showing that increased permeability is associated with increased BMI and insulin resistance in 40 patients with obesity and T2D (96). Damms-Machado et al. similarly reported a correlation between intestinal permeability and Homeostasis Model Assessment for insulin resistance (HOMA-IR) in 27 individuals with obesity of which 18 suffered from the metabolic syndrome (97). Furthermore, Luther et al. reported in a meta-analysis showing that nonalcoholic fatty liver disease (NAFLD) was associated with increased intestinal permeability (odds ratio increased intestinal permeability=5.1), which was even more prominent in nonalcoholic steatohepatitis (NASH) (odds ratio=7.2) (98). Challenging this hypothesis with interventions has seldom been done: A low-caloric diet of 800 kcal/day was able to significantly reduce the intestinal permeability, indicating the relevance of nutrition as a fast acting player influencing intestinal permeability (99). Further interventions leading to mechanistic insights in the link between metabolism and intestinal permeability are highly warranted.
Metabolic Endotoxemia and the Translocation of Bacterial Genetic Material and Live Bacteria in Human Tissues: A Compartment Centered Approach
The evidence related to translocation of bacterial components, live bacteria, and bacterial metabolites into the circulation and beyond is highly dependent on the tissue studied leading to diverging degrees of confidence in the relationship between gut permeability, organ dysfunction and overall modulation of host metabolism. On their way from the gut, bacteria are enriched in mesenteric lymph nodes (MLN) (Figure 1D) both in health and in disease as evidenced in translocation of enteric microorganisms to MLN in patients with advanced cirrhosis (prevalence 30.8%) and comparable prevalence of bacterial translocation (~8%) in patients and healthy controls after selective intestinal decontamination (100). Consequently, it is postulated that immune cells interacting with bacteria are activated in the MLN after which they are redistributed in the blood (Figure 1H) (101). Similarly, bacterial products have also been implicated as mediators between the gut microbiota and peripheral organs: A major part of consumed dietary fibers are fermented by bacteria in the colon, which results in various metabolites, including short chain fatty acids (SCFA) (102). SCFA are readily absorbed by the colonocytes and only a small part is secreted (103). Production of SCFA increases with a fiber-rich diet (102), and increased circulating levels of SCFA are associated with improvements in insulin sensitivity and decreased levels of lipolysis, triacylglycerols, and free fatty acids in humans (104).
Beyond the blood, there seem to be unexpected and promising niches of bacterial translocation and bacterial targeted action are currently drawing increasing multidisciplinary attention leading to a paradigm shift in the study and definition of metabolic diseases as “non-communicable” diseases.
Impact of Blood Borne Bacteria and Bacterial Components on Metabolic Disease
Lipopolysaccharides in Health and Disease
The measurement of bacterial components in the circulation allows an indirect verification of increased intestinal permeability. This link has emanated from the observation that sepsis is associated with an acute but reversible state of insulin resistance (105). To this end, most studies have focused on the measurement of LPS, i.e., endotoxins, which are molecules on the outer membrane of gram-negative bacteria. The condition of increased exposure to bacterial LPS in the blood in obesity or metabolic disease is referred to as “metabolic endotoxemia”. Fat intake is associated with an increase in postprandial LPS levels (106), which have been shown to be distributed from the gut into the circulation via the mesenteric lymph nodes using freshly formed chylomicrons (Figure 1D) (107). LPS is recognized and bound by both LPS-binding protein (LBP) and soluble cluster of differentiation (sCD14), which can also be used as marker proteins, and recruited to the TLR 4 (108). The highest proportion of LPS in the circulation is bound to lipoproteins, whereby most of LPS is bound to high density lipoprotein (HDL) (109). Smaller fractions of LPS are also bound to platelets (110, 111), monocytes (112) and erythrocytes (113, 114) or are present as free LPS (115) (Figure 1G). However, these observations mostly stem from models of sepsis. Endotoxin and other bacterial components taken up from the gut due to increased intestinal permeability will presumably enter the portal vein and, as a first step be transported to the liver, where they are rapidly cleared. Clearance mostly takes place in the liver after uptake via – among other routes – various lipoprotein receptors, e.g., LDL-R (115) and is mainly accomplished by hepatocytes and Kupffer cells to be then excreted to the bile (Figure 1E). Moreover, since binding of LPS with lipoproteins and chylomicrons has been shown to prevent endotoxin induced monocytes activation and secretion of proinflammatory cytokines (116, 117), the redistribution and increased content of phospholipids among the different lipoproteins has been suggested as a potential mechanism for the attenuation of the immunostimulatory effects of LPS.
LPS is detectable in low concentrations even in healthy subjects, and a single meal with high fat content already increases LPS levels (118, 119). In the last years, many studies in large cohorts reported increased levels of LPS and LBP in subjects with metabolic syndrome or T2D (120–123). Pussinen et al. analyzed LPS levels of patients with prevalent (n=537) and incident T2D (n=462) and compared them to a control group (n=6,170), of which about 20% had the metabolic syndrome. Endotoxin was significantly increased in individuals with T2D and endotoxin activity was associated with an increased risk for incident T2D, independently of other metabolic risk factors (121). Furthermore, serum endotoxin was linearly associated with the number of metabolic syndrome traits (121). Direct administration of LPS induced insulin resistance and systemic inflammation (124) and an 8-weeks long overfeeding intervention was associated with increased endotoxemia, linking overnutrition with endotoxemia and insulin resistance (125). A more recent study by Cox et al. used LPS, LBP as well as intestinal fatty acid binding protein (iFABP) to calculate a permeability risk score, which was increased in individuals with type 2 diabetes (123). In secondary complications, such as NAFLD, even higher endotoxin levels are reported (126). Mechanistic insights in the cascades underlying the effects of LPS on gut permeability and subsequent metabolic impact stem from mice studies, where subcutaneous LPS infusion has been shown to lead to an obese phenotype, comparable to that of mice on high-fat diet, including increased glucose and insulin levels as well as whole body and adipose tissue weight gain (106). Moreover, LPS application in vitro has been shown to result in an increase in gut permeability through tight junction dysfunction via a TLR4-dependent process (127), which is the most specific LPS receptor (128, 129) (Figure 1I).
As for the impacts of metabolic intervention on LPS: Roux-en-Y gastric bypass (RYGB) surgery has been shown to reduce LPS levels by 20 ± 5% after a 180 days follow-up period in patients with obesity and T2D (122), which could also be confirmed for sleeve gastrectomy and duodenal switch procedures (130, 131).
Most of the studies looking at LPS however neglect the fact, that LPS from different bacteria has a high heterogeneity in its chemical structure, mostly due to variation in the O-antigen polysaccharide (132, 133), and consequently, biological function (134). The latter was nicely demonstrated by Vatanen et al., showing that LPS derived from B. dorei counteracts the immunostimulatory activity of E. coli LPS (134). To our knowledge, there are no studies looking at LPS variation in metabolic disease published so far.
That being said, a reduction of intestinal permeability has been evidenced both in rat and human models of bariatric surgery (135). In rats undergoing duodenojejunal bypass surgery (DJB), intestinal permeability as assessed by Ussing chamber studies and dextran-FITC through mucosa-to-serosa flux was reduced in the alimentary and common limb as well as the colon, consistently supported by increased mucosal expression of occludin in both the alimentary and common limbs after DJB. LPS and LBP levels were not different between DJB and sham operated rats, possibly due to increased bacterial counts and compensatory increased intestinal surface in the remaining intestine. This is in contrast to data from humans, where mucosal surface was significantly reduced after surgery and occludin as well as zonula occludens expression was reduced. Congruently though, intestinal permeability as assessed by Ussing chambers was decreased owing to an increased expression of claudin-3-expression (136), further tying up reduction of microbiota-derived inflammatory mediators and rearrangement of gut barrier regulators after bariatric surgery (135).
Bacteria and Bacterial Fragments
In addition to LPS measurement, other bacterial components such as bacteria itself or bacterial DNA have been suggested as possible contributors to metabolic disease. The amplification of bacterial DNA allows quantification of bacterial load while subsequent sequencing allows the assessment of microbial composition and identification of specific bacterial perpetrators. There is increasing evidence for the presence of bacteria in blood, even in healthy subjects, as extensively discussed by Castillo et al. (100). Briefly, there are multiple studies reporting concurrent bacterial phyla in blood with Proteobacteria and Firmicutes being the most dominant phyla (100, 137). The first study to relate bacterial presence in the blood with metabolic disease was published by Amar et al. in 2011 and included subjects of the D.E.S.I.R. cohort within a longitudinal study aimed at understanding the complex pathophysiology of the metabolic syndrome (138, 139). 16S rRNA gene quantities were measured in 3,280 subjects at baseline and after nine years. Subjects who developed T2D during the duration of the study had significantly higher amounts of bacterial DNA at baseline (odds ratio=1.35, p=0.002). Moreover, bacterial DNA was significantly elevated in subjects with abdominal obesity (odds ratio=1.18, p=0.01). Additionally, pyrosequencing showed the dominant phylum to be Proteobacteria (139), whose amount was found to be an independent risk factor of cardiovascular disease in a subsequent analysis in 3,936 participants of the same cohort (140).
Similarly, blood bacterial DNA composition reminiscent of that observed for the gut microbiome was found in a Japanese cohort of 100 subjects including 50 with T2D. In addition, the detection rate for bacterial DNA was significantly higher in subjects with T2D compared with healthy subjects (28% vs. 4%, p<0.01) (141). In line with above mentioned studies, patients with obesity and liver fibrosis seem to have elevated concentrations of bacterial DNA compared to patients with obesity alone (142). Most recently, Qiu et al. showed that subjects carrying Bacteroides had a reduced risk while patients carrying Sediminibacterium an increased risk to develop T2D (143). Lammers et al. analyzed the effect of bacterial DNA of eight probiotic strains including Bifidobacterium and Lactobacillus on peripheral blood mononuclear cells (PBMCs) of healthy donors. All strains increased expression of IL-1, 6, and 10 at high concentrations of 70 µg/ml with largest changes observed for B. infantis showing that bacterial DNA can indeed induce strain specific immune alterations in the host (144).
In a small cohort of 58 subjects with obesity, who had undergone bariatric surgery, Ortiz et al. analyzed the translocation of bacterial DNA before and up to 12 months post-surgery. Bacterial DNA was detected in 32.8% of the patients prior to surgery, but only in 13.8% and 5.2% of patients after 3 and 12 months, respectively (145). More importantly, inflammation, LPS levels and insulin resistance persisted in subjects with a reduced clearance of bacterial DNA in the blood even after significant weight loss following bariatric surgery with multivariate analyses revealing bacterial DNA presence as an independent predictor (145). In summary, there is increasing evidence not only for the presence of bacterial DNA and potentially intact bacteria even in healthy subjects but also for the association between the amount of bacterial DNA and bacterial composition with obesity and related metabolic traits. However, current methods have considerable limitations (see Limitations of the review) and further, more elaborative studies are warranted to substantiate initial results.
Bacteria and Bacterial Products Regulate Adipose Tissue Inflammation Locally
The human adipose tissue is a metabolically active organ with large heterogeneity in cellular composition, function, and expression signatures depending on its anatomic location (146).
LPS, Adipocytes, and Adipose Tissue Macrophages
Although there are only a few studies providing evidence for the existence of bacterial components in adipose tissue, their potential impact has been repeatedly demonstrated in cell culture experiments. Toll-like receptors, especially TLR4, which are needed for the recognition of LPS are expressed on adipose tissue macrophages as well as adipocytes (147). Stimulation of adipose tissue macrophages with LPS induces fibrosis via TLR4 and the induction of the profibrotic factor Transforming growth factor beta 1 (TGFß1) (148). LPS can also act directly on adipocytes, as demonstrated in numerous studies (149–153). Creely et al. could show that IL-6 and TNF-α are secreted in response to LPS in mature subcutaneous adipocytes from lean individuals and subjects with obesity, whereas inhibition of NF-kB reduced the levels of secreted IL-6 after LPS stimulation (149). These results could be confirmed by Vitseva et al. using subcutaneous abdominal fat from subjects with obesity (150). Further works suggest a role for LPS on modifying glycerol permeability and metabolism of murine 3T3-L1 adipocytes (153) and hint to a role of adiponectin in regulating local inflammation by inhibiting LPS-induced NF-kB activation using primary pig adipocytes (152).
Bacterial DNA and Live Bacteria
First evidence that bacteria are indeed able to translocate from the gut to adipose tissue stems from mouse experiments. In a pioneering work, Amar et al. used, among other methods, green fluorescent protein (GFP)-labeled E. coli to demonstrate the transmucosal passage in mice on high-fat diet (154). Although some initial studies suggested the presence of bacterial DNA in human adipose tissue as well (155), others could not confirm these results (156). In their work, Zulian et al. isolated DNA from whole adipose tissue and mature adipocytes of 14 subjects with obesity. Bacterial PCR products were only observed in the enzymatically isolated adipocytes, but sequencing revealed that 90% of this bacterial DNA belonged to C. histolyticum, from which the collagenase enzyme used for adipocytes isolation is derived. All subsequent experiments including culturing experiments were negative (156). In 2017, Udayappan et al. reported the presence of bacterial DNA, mainly stemming from Actinobacter and Ralstonia, in mesenteric adipose tissue of twelve patients with obesity. However, as they did not include negative controls, contamination as a source of these observed genera could not be excluded (157). Another noteworthy study published by Nakatsuji et al., investigated the subepidermal adipose tissue of six healthy subjects and suggested that the skin microbiome extends to this specific skin-adjacent adipose tissue depot (158). Furthermore, bacterial DNA belonging to several bacteria was observed in the epicardial adipose tissue in six patients with acute coronary syndrome (Streptophyta and Rickettsiale) and stable angina (Pseudomonas and Moracellaceae) as compared to subjects with isolated mitral insufficiency. The authors related coronary heart disease with increased bacterial colonization of epicardial adipose tissue and ensuing inflammasone activation (159). More recently, Anhê et al. reported data on contaminant-controlled presence of bacterial DNA in mesenteric, subcutaneous and visceral adipose as well as liver tissue in 40 patients with obesity. Hereby, highest abundance was observed in visceral adipose tissue and liver samples, and patients with T2D had a decreased bacterial diversity. Diversity was especially high in patients with obesity but without T2D in mesenteric adipose tissue (160). Data recently presented by our group could extend and go beyond the proof of bacterial DNA by showing the presence of living bacteria in adipose tissue using catalyzed reporter deposition (CARD) - fluorescence in situ hybridization (FISH) methods. Furthermore 16S rRNA gene content of 75 patients with obesity was quantified and sequenced, showing that both bacterial quantity and taxonomy were associated with markers of inflammation and insulin resistance (137). Moreover, miniscule amounts of bacterial DNA (E. coli) were sufficient to induce a proinflammatory response in human subcutaneous adipocytes, as observed by a dose-dependent increase of IL6 and TNF expression. Unexpectedly, expression of neither TLR4 and TLR9, nor the NLR family pyrin domain containing 3 (NLRP3) inflammasome increased at any concentration (137). Similar direct evidence for the translocation of viable gut bacteria to adipose tissue, in this case mesenteric adipose tissue, was presented by Ha et al. using human biopsies, gnobiotic mice as well as primary cells (161). Bacterial translocation occurred in both healthy subjects and patients with Crohn`s disease (CD), but taxonomic profiles differed. Using gnobiotic mice treated with C. innocuum, the bacterium most frequently isolated in CD, Ha et al. evidenced translocation of these bacteria into adipose tissue. This was accompanied by increases in adiponectin and Peroxisome proliferator-activated receptor Gamma (PPARG) expression and adipose tissue expansion. These results suggest that, at least in the case of CD, specific bacteria including C. innocuum can restructure mesenteric adipose tissue leading to the expansion and fibrosis of creeping fat. This might be a defense mechanism to prevent systemic translocation of bacteria beyond this compartment (161).
Impact of Bacterial Metabolites on Adipose Tissue Function
SCFA are recognized by multiple G-protein coupled receptors (GPR), and receptors GPR41, 43 and 81 are known to be expressed in adipose tissue (162). First evidence for an adipose tissue specific effect of SCFA was observed by Kimura et al., who showed that mice with a global GPR43 knockout were obese compared to wild type controls whereas mice with a selective overexpression of GPR43 in adipose tissue were leaner (163). The authors further demonstrated that GPR43 stimulation with acetate improved glucose and lipid metabolism in adipose tissue, but not in muscle and liver (163), thereby confirming prior results by Robertson et al., that showed reduced lipolysis in adipose tissue after stimulation of GPR43 with acetate (164). These results were recently strengthened by experimental treatment of multipotent human adipose tissue-derived stem cells with either mixtures of butyrate, propionate and acetate or the individual SCFA in varying concentration between 1 µmol/l and 1 mmol/l. Whereas mixtures high in butyrate had no effect on glycerol release, mixture with high concentration of acetate and propionate decreased basal glycerol release (165). Treatment with only butyrate even increased glycerol release slightly but significantly, whereas isolated treatment with acetate reduced glycerol release (165). Next to their effects on lipolysis, acetate and propionate also seem to influence adipogenesis via GPR43, as shown by Hong et al. in 3T3-L1 cells (166). The authors found no GPR43 expression in the stromal vascular fraction (SVF) or preadipocytes, but expression levels increased gradually with adipogenesis. Furthermore, siRNA mediated silencing of GPR43 blocked adipocyte differentiation (166). A further study analyzed the effect of SCFA-rich HFD compared to normal HFD diet on adipose tissue biology: Supplementation of SCFA increased expression of GPR43 in adipose tissue and reduced its expression in the colon. Next to decreased leptin and increased adiponectin expression in adipose tissue, the authors showed an increase in adipose tissue beigeing under SCFA treatment (167).
Pancreas and Liver: Bacterial Actions on Central Metabolic Organs
LPS Effects on Liver and Pancreas
The liver has been shown to participate in active LPS clearance. Increased amounts of endotoxin are still not without consequence for the liver: As a response to LPS uptake and stimulation, hepatocytes express acute phase protein serum amyloid A, which further promotes LPS clearance (168). However, hepatocytes can also utilize the TLR4 cascade to take up LPS (169, 170). That being said, it is assumed that altered TLR4 signaling is a key factor in metabolic liver diseases including NAFLD (171). TLR4 is expressed on various liver cell types including hepatocytes, monocytes, Kupffer cells and stellate cells (172). Using primary human liver biopsies and human hepatocytes (HepaRG), it was shown that LPS increases NF-kB translocation and activity by ~2.5 fold, although the observed effect was only partially mediated by TLR4 as demonstrated by siRNA knockdown and chemical blocking of TLR4 (173). Kheder et al. stimulated macrophages (J774) and HepG2 hepatocytes with LPS, which led to increased TNF-α expression (Figure 1E). Co-treatment with increasing doses of vitamin D3 and docosahexaenoic acid (DHA) reduced these effects in a dose dependent manner (174). Lee et al. used mouse hepatocytes deficient for TLR4, Myeloid differentiation primary response 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) to show that LPS induces hepcidin expression, an antimicrobial and iron regulating protein, via TLR4 in a MyD88 dependent manner (175).
On the other hand, endotoxins have been shown to exert varying effects on various pancreatic cells: Endocrine cells of the human pancreatic islets express CD14 and TLR4. CD14 was synthesized and secreted by SV40-transformed islet cells (HP62) after treatment with LPS. LPS was shown to regulate glucose-dependent insulin secretion and induced an inflammatory response (176). By analyzing isolated human and rodent islets, Amyot et al. showed that LPS impairs insulin expression, whereby human islets are more sensitive to this effect. LPS further decreases the expression of pancreatic and duodenal homeobox 1 (PDX-1) and Transcription factor Maf (MafA) and this inhibition is prevented by blocking NF-kB but not p38 MAPK signaling, linking bacterial induced inflammation with pancreatic dysfunction (177).
Bacterial Translocation to the Liver
Bacterial DNA has also been evidenced in liver tissue under contamination control (160). More recently, Sookoian et al. were able to localize LPS derived from Gram-negative bacteria in the portal tract and evidenced a diverse repertoire of bacterial DNA in the liver, the composition of which was driven in part by obesity. This metataxonomic signature was furthermore related to histological findings in NAFLD, with expansion of proteobacterial DNA being associated with lobular and portal inflammation as well as liver cirrhosis in non-morbidly obese subjects. Morbid obesity on the other hand displayed wider associations to several taxa thought to contribute to more detrimental findings (178).
SCFA Impact on Liver Function and Pancreas
Most of the SCFA taken up from the colon are metabolized by the liver and only small proportions enter the peripheral circulation (about 20% for butyrate and propionate) (179–181). Treatment of HepG2 cells with SCFA ranging from C3 to C6 increased Apolipoprotein A (ApoA-I) expression in a dose-dependent matter whereas secreted ApoA-I to medium was reduced. Additionally, butyric acid increased carnitine palmitoyltransferase I (CPT1) expression, a key protein of beta-oxidation, and increased activity of PPARA (180). Butyrate was also shown to improve insulin sensitivity and increase energy expenditure in mice, and mice on HFD treated with butyrate exhibited higher protein levels of cyclic 5’ AMP-activated protein kinase (cAMPK), P38 and PGC-1a in the liver compared to HFD mice without treatment (182). Sahuri-Arisoylu et al. analyzed the effect of acetate, showing that acetate decreased lipid accumulation and improved hepatic function. The authors observed decreased levels of circulating aspartate transaminase and alkaline phosphatase, as well as reduced expression of genes involved in lipogenesis, such as fatty acid synthetase (183). Acetate also protects rodent models against diet-induced weight increase by altering liver metabolism, as Kondo et al. showed by increased expression levels of fatty acid oxidation enzymes as well as PPARA in the liver (184).
As in adipose tissue, GPR43 is also expressed on pancreatic β-cells, and expression is increased in mice fed with HFD. GPR43 knockout in mice leads to impaired insulin secretion and treatment of murine and human islet cells with a GPR43 agonist leads to increased intracellular Ca2+ and inositol triphosphate levels as well as increased insulin secretion (185). In contrast, a study concurrently did not evidence any effects on glucose homeostasis in GPR43-/- mice on normal chow or HFD compared to wild type mice. In ex vivo studies, presence of acetate potentiated insulin secretion (186). Similarly, it was shown that propionate potentiated dynamic glucose-stimulated insulin secretion in vitro and further protected human islets from inflammatory cytokine and sodium palmitate induced apoptosis (179).
Impact of Bacteria and Bacterial Products on Skeletal Muscle Metabolism
Evidence on the role of endotoxin on muscle tissue insulin sensitivity mainly stems from septic models. During hyperinsulinemic euglycemic clamps, 4 ng LPS per kg body weight were administered to healthy donors, resulting in an increased peripheral glucose uptake as well as increased circulating concentrations of norepinephrine and cytokines (180). Concurrently Reyna et al. evidenced an elevated TLR4 expression in muscle tissue of subjects with obesity and T2D and concentrations of TLR4 protein isolated from muscle tissue correlated positively with insulin resistance (181). Furthermore, patients exhibited lower IκBα content and increased expression of IL6 and SOD2 and these results could be replicated in primary human myotubes by treatment with palmitate (181). More functional approaches were, among other performed by Liang et al., who similarly differentiated primary human muscle cells into myotubes and treated them with LPS. Endotoxin induced inflammation and reduced insulin signaling; effects which were counteracted by a treatment with the TLR4-Inhibitor (182). These results are supported by data published by Frisard et al. on treatment of mice and human myotubes with high and low concentrations of LPS, thereby reflecting septic or metabolic endotoxemia conditions and showing an increased glucose utilization and reduced fatty acid oxidation in skeletal muscle (183). Low levels of endotoxin are sufficient to modulate mitochondrial consumption and substrate oxidation preferences, an effect which was absent in the presence of specific antioxidants, thereby suggesting a role of reactive oxygen species in mediating observed effects (184).
Houghton et al. tested the effect of various microbiome catabolites, including phenolic compounds, bacterial metabolites, and their phenolic conjugates. Several of these compounds increased glucose uptake to differentiated human skeletal myoblasts (LHCN-M2) as well as overall metabolism rates (187). Most prominent effects were observed for isovanillic acid 3-O-sulfate (IVAS) and dihydroferulic acid 4-O-sulfate. IVAS furthermore promoted upregulation of GLUT1, GLUT4, and phosphoinositid-3-kinase (Pi3K) p85α proteins (187).
The effect of SCFA on skeletal muscle metabolism and function was recently reviewed in an excellent publication by Frampton et al. (188). Studies examining direct effect on skeletal muscle cells are mainly using rat-derived L6 myotubes or mouse-derived C2C12 myotubes and no human studies were found. Briefly, treatment with SCFA increased fatty acid oxidation (about 30% for 0.5 mM butyrate (189)) and fatty acid uptake (i.e., using 0.5 mM acetate) in L6 myotubes (190). Furthermore, stimulation with acetate and propionate promotes enhanced insulin-independent uptake of glucose to both mouse and rat myotubes (190, 191). Maruta et al. also report an upregulation of GLUT4 gene expression as well as protein levels upon treatment with 0.5 mM acetate (190).
Limitations
It can be assumed that subjects not suffering from sepsis have lower bacterial load and display lower endotoxemia making careful handling of samples to avoid contamination, careful data interpretation to avoid over-reporting of false positive results as well as method standardization allowing comparability and reproducibility, paramount aspects for planning studies contributing to the subject as well as interpretation of the available literature. Here, we highlight some concerns, which should be considered in interpretation of relevant literature.
Measurement of Intestinal Permeability
There are various methods to measure the integrity of the intestinal barrier with differing degrees of sensitivity and specificity: Methods include Ussing chambers (87), histology and electron microscopy for biopsies and transepithelial/transendothelial electrical resistance (TEER) for cell culture experiments (192). The gold standard for in vivo studies is the lactulose/mannitol (La/Ma) or similar dual sugar tests, which assess the flow of indigestible sugars from gastrointestinal tract to the circulation and the combination of different sugars allows a location specific measurement (34, 193). In addition, biomarkers such as calprotectin, alpha-1-trypsin, fatty acid binding protein, and zonulin are used. There is extensive literature on benefits and disadvantages of each method (34, 193–196). However, in our opinion, studies employing biomarkers are generally lacking in strength due to various reasons: a) correlations with dynamic permeability tests such as dual sugar tests or Ussing chambers are not present or very weak; b) most of these markers are also acute phase proteins and are therefore increased in inflammation; c) their actions are not limited to the gut paracellular pathway; d) commercially available ELISAs are often not validated, as we and others recently demonstrated that the preferred ELISA for the most commonly used biomarker zonulin measures different products (197, 198). Consequently, relevant literature should be interpreted carefully in this regard (199).
Endotoxin
Measurement of endotoxin, in particular at low concentrations, is very challenging and prone to errors, leading to considerable discussion pertaining to the significance of the test and its interpretation. One major aspect to be considered is that LPS are not actively excreted by bacteria but that release of LPS only takes place after gram negative bacterial cell death and lysis, which in turn does not justify the wide use of LPS as a surrogate marker for “live” bacterial translocation. However, there could still be an intrinsic value for the increased exposition of the host to bacteria in general. Measurement of LPS has routinely been done using the Limulus Amoebocyte Lysate (LAL) assay. Although the FDA has recognized the validity of approaches using the LAL method, LAL reagents are not required to be obtained from an FDA-licensed manufacturer when used in the context of research, making standardization between methods reported in the literature almost impossible. Beyond this, conducting the LAL assay is highly challenging. This is related to the magnitude of chemical and physical products and factors, which are known to interfere with the test’s ability to detect LPS. These factors include pre-analytical conditions such as sampling, where plastic or siliconized ware can lead to endotoxin absorption (200) and render LAL assay ineffective. Moreover LPS quantification presumes sampling in pyrogen/LPS free ware, which can be done via dry heat sterilization at high temperature, a widely ignored factor possibly contributing to the immense range of LPS from 0.01 to 60 EU/ml reported in healthy subjects in the literature (120, 201). Furthermore, various factors within the blood can interfere with LPS testing, including bile salts, lipoproteins, EDTA and heparin (202), which makes pretreatments of samples to measure LPS unavoidable. Moreover, using different units impedes the interpretation of findings from different studies (203): whereas some studies report LPS levels in weight per water volume (pg/mL), others report endotoxin units per unit volume (EU/mL), which reflects LPS activity. Additionally, when considering that host-derived proteins binding of LPS largely modulates its activity and clearance from the circulation (204–206), it becomes evident that whatever is measured after pretreatment of the samples to overcome “low LPS recovery” might not reflect in vivo conditions at all. This is best exemplified by the fact that there is poor concordance between endotoxemia and gram-negative bacteremia and that endotoxemia is detected in less than 50% of subjects with gram-negative sepsis (207). Independent from units, most studies reported a 0.5 to 2-fold increase of LPS in subjects with obesity compared to subjects with normal weight, as clarified by Boutagy et al. (203). Consequently, they propose using fold changes (in disease vs. controls) instead of absolute values to describe the contribution of endotoxemia to the study setting until unified and comparable methods are established.
In addition, LPS derived from different bacteria can show large heterogeneity in the O-antigen polysaccharide with consequence on the inflammatory potential and biological function of endotoxin (133, 134). This point is normally neglected in studies on endotoxin and should therefore be considered more often in new studies.
Bacterial DNA
In studies analyzing bacterial DNA in samples with low bacterial biomass, be it quantification or sequencing, contamination is a major problem, which is well-highlighted by the recent debate about a possible placenta microbiome (208–211). Contamination can arise from multiple sources (air, skin, reagents) and during all experimental steps. Furthermore, even sterile lab ware can contain traces of bacterial DNA, as per definition a sterile environment is only defined by the absence of living microorganisms (212). The use of adequate negative controls and the deployment of subsequent bioinformatic tools to address them become eminent to avoid reporting false positive results (213). Although these problems are increasingly addressed by the research community, a set of consistent and widely accepted approaches would help for the general comparability and reproducibility of data.
Data Interpretation
In most works on metabolic endotoxemia it is assumed that LPS is the consequence of impaired intestinal barrier. However, other locations can contribute to a metabolic endotoxemia as well. For instance, it was shown that oral interventions like extraction of teeth or periodontal probing but also everyday actions such as chewing and oral hygiene can lead to an influx of endotoxin and bacteria into the circulation (214, 215). It was also observed that subjects with obesity are more prone to suffer from gingivitis, which is most likely due to increased insulin resistance (216, 217). It is therefore likely that LPS and bacteria from the oral cavity contribute more to systemic ‘bacterial burden’ and possibly inflammation than has been previously assumed. Additionally, the skin microbiome extends to various compartments, thus implicating a potential impact for this compartment in translocation to subsequent tissues (158). However further studies are desirable to support these findings. Furthermore, since LPS is rapidly cleared from the circulation in the liver, which is also the first organ reached by LPS taken up from the gastrointestinal tract, whatever measureable endotoxin under various conditions points toward a chronic influx and the additional contribution of other origins like oral cavity, lungs, or the skin (Figure 1F).
Summary/Conclusion
In this review, we focused on causes and possible consequences of impaired intestinal permeability, thereby focusing on obesity and components of metabolic diseases. Consequences include translocation of endotoxin, bacterial DNA, or live bacteria as well as bacterial metabolites to the circulation, a process often associated with the term metabolic endotoxemia or bacteremia. Starting with animal studies, there now is also compelling evidence for an impairment of the human intestinal barrier in diseases such as T2D and obesity. Consequently, increased circulating bacterial load is now a well-established hallmark of metabolic diseases and published data suggest that impairment of the gut barrier can trigger and further aggravate metabolic impairment. A less studied subject is the presence and effect of bacteria in other host tissues, including liver, muscle, pancreas, and adipose tissue.
However, as shown in this review, several studies suggest that tissue bacteria or components and metabolites thereof either reflect or directly contribute to the development and progression of metabolic diseases. Underlying mechanisms such as TLR4 dependent activation of NF-kB have been introduced but a holistic approach encompassing the complexity of host immune factors is widely lacking. There has also been compelling underreporting of shortcomings in the methods used such as LPS measurement, making claims toward causality rather elusive. Furthermore, only a few studies were published on quantification and sequencing of bacterial DNA in host tissues, which have been similarly plagued with false positive results partly due to contamination.
In conclusion, it seems unavoidable that our bacterial inhabitants also contribute to the modulation of their metabolic environment shaping the body’s responses to nutrients and contributing ultimately to disease as has been shown for the gut microbiota in recent years (218). Considering the current relevance of microbiome research in understanding health and nutrition and the promising avenues for therapy and prevention, it will be inevitable to revisit many of the notions introduced here to allow for robust and reliable mechanistic approaches.
Author Contributions
LM, PK, and RC designed the review. LM and RC interpreted literature and drafted the manuscript. LM designed the figure. MB and PK critically reviewed and edited the manuscript. All the authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
LM was funded, and this work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation—Projektnummer 209933838—SFB 1052; B03). The authors acknowledge support from the German Research Foundation (DFG) and Universität Leipzig within the program of Open Access Publishing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary

References
1. Bhupathiraju SN, Hu FB. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ Res (2016) 118:1723–35. doi: 10.1161/CIRCRESAHA.115.306825
2. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
3. Arora T, Backhed F. The gut microbiota and metabolic disease: current understanding and future perspectives. J Internal Med (2016) 280:339–49. doi: 10.1111/joim.12508
4. Howitt MR, Garrett WS. A complex microworld in the gut: gut microbiota and cardiovascular disease connectivity. Nat Med (2012) 18:1188–9. doi: 10.1038/nm.2895
5. Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut (2014) 63:1513–21. doi: 10.1136/gutjnl-2014-306928
6. Parekh PJ, Balart LA, Johnson DA. The Influence of the Gut Microbiome on Obesity, Metabolic Syndrome and Gastrointestinal Disease. Clin Trans Gastroenterol (2015) 6:e91. doi: 10.1038/ctg.2015.16
7. Mazidi M, Rezaie P, Kengne AP, Mobarhan MG, Ferns GA. Gut microbiome and metabolic syndrome. Diabetes Metab Syndrome (2016) 10:S150–7. doi: 10.1016/j.dsx.2016.01.024
8. Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, et al. Homeostasis of the gut barrier and potential biomarkers. Am J Physiol Gastrointestinal Liver Physiol (2017) 312:G171–93. doi: 10.1152/ajpgi.00048.2015
9. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes (2008) 57:1470–81. doi: 10.2337/db07-1403
10. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Sci (N Y NY) (2018) 359:1376–83. doi: 10.1126/science.aar3318
11. Ravin HA, Rowley D, Jenkins C, Fine J. On the absorption of bacterial endotoxin from the gastro-intestinal tract of the normal and shocked animal. J Exp Med (1960) 112:783–92. doi: 10.1084/jem.112.5.783
12. Menzies I. Absorption of Intact Oligosaccharide in Health and Disease. Biochem Soc Trans (1974) 2:1042–7. doi: 10.1042/bst0021042
13. Berry LJ, Smythe DS, Young LG. Effects of bacterial endotoxin on metabolism. I. Carbohydrate depletion and the protective role of cortisone. J Exp Med (1959) 110:389–405. doi: 10.1084/jem.110.3.389
14. Jackson PG, Baker RWR, Lessof MH, Ferrett J, Macdonald DM. Intestinal Permeability In Patients With Eczema And Food Allergy. Lancet (1981) 317:1285–6. doi: 10.1016/S0140-6736(81)92459-4
15. Nolan JP, Hare DK, McDevitt JJ, Vilayat Ali M. In Vitro Studies of Intestinal Endotoxin Absorption. Gastroenterology (1977) 72:434–9. doi: 10.1016/S0016-5085(77)80253-9
16. Gans H, Matsumoto K. Are enteric endotoxins able to escape from the intestine? Proc Soc Exp Biol Med Soc Exp Biol Med (N Y NY) (1974) 147:736–9. doi: 10.3181/00379727-147-38428
17. Bjarnason I, Ward K, Peters T. The Leaky Gut Of Alcoholism: Possible Route Of Entry For Toxic Compounds. Lancet (London England) (1984) 323:179–82. doi: 10.1016/S0140-6736(84)92109-3
18. Zhou Q, Verne GN. Intestinal hyperpermeability: a gateway to multi-organ failure? J Clin Invest (2018) 128:4764–6. doi: 10.1172/JCI124366
19. Ukabam SO, Cooper BT. Small intestinal permeability as an indicator of jejunal mucosal recovery in patients with celiac sprue on a gluten-free diet. J Clin Gastroenterol (1985) 7:232–6. doi: 10.1097/00004836-198506000-00009
20. Heyman M, Abed J, Lebreton C, Cerf-Bensussan N. Intestinal permeability in coeliac disease: insight into mechanisms and relevance to pathogenesis. Gut (2012) 61:1355–64. doi: 10.1136/gutjnl-2011-300327
21. Michielan A, D’Incà R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflammation (2015) 2015:628157. doi: 10.1155/2015/628157
22. Groschwitz KR, Hogan SP. Intestinal Barrier Function: Molecular Regulation and Disease Pathogenesis. J Allergy Clin Immunol (2009) 124:3–22. doi: 10.1016/j.jaci.2009.05.038
23. Pelaseyed T, Bergström JH, Gustafsson JK, Ermund A, Birchenough GM, Schütte A, et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev (2014) 260:8–20. doi: 10.1111/imr.12182
24. Walton KD, Freddo AM, Wang S, Gumucio DL. Generation of intestinal surface: an absorbing tale. Dev (Cambridge England) (2016) 143:2261–72. doi: 10.1242/dev.135400
25. Yu LC-H. Commensal bacterial internalization by epithelial cells: An alternative portal for gut leakiness. Tissue Barriers (2015) 3:e1008895. doi: 10.1080/21688370.2015.1008895
26. Shifrin DA, McConnell RE, Nambiar R, Higginbotham JN, Coffey RJ, Tyska MJ. Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr Biol CB (2012) 22:627–31. doi: 10.1016/j.cub.2012.02.022
27. Hu Y-J, Wang Y-D, Tan F-Q, Yang W-X. Regulation of paracellular permeability: factors and mechanisms. Mol Biol Rep (2013) 40:6123–42. doi: 10.1007/s11033-013-2724-y
28. Günzel D, Yu AS. Claudins and the Modulation of Tight Junction Permeability. Physiol Rev (2013) 93:525–69. doi: 10.1152/physrev.00019.2012
29. Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta (2008) 1778:631–45. doi: 10.1016/j.bbamem.2007.10.018
30. Günzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol (2012) 2:1819–52. doi: 10.1002/cphy.c110045
31. Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol (2010) 11:502–14. doi: 10.1038/nrm2927
32. Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta (2008) 1778:572–87. doi: 10.1016/j.bbamem.2007.07.014
33. Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep (2012) 13:684–98. doi: 10.1038/embor.2012.96
34. Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke J-D, Serino M, et al. Intestinal permeability – a new target for disease prevention and therapy. BMC Gastroenterol (2014) 14:189. doi: 10.1186/s12876-014-0189-7
35. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature (2012) 483:345–9. doi: 10.1038/nature10863
36. Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, Huehn J. Microbiome Dependent Regulation of Tregs and Th17 Cells in Mucosa. Front Immunol (2019) 10:426. doi: 10.3389/fimmu.2019.00426
37. Butler JE, Sinkora M. The enigma of the lower gut-associated lymphoid tissue (GALT). J Leukocyte Biol (2013) 94:259–70. doi: 10.1189/jlb.0313120
38. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature (2009) 459:262–5. doi: 10.1038/nature07935
39. Williams JM, Duckworth CA, Burkitt MD, Watson AJ, Campbell BJ, Pritchard DM. Epithelial cell shedding and barrier function: a matter of life and death at the small intestinal villus tip. Veterinary Pathol (2015) 52:445–55. doi: 10.1177/0300985814559404
40. Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol (2010) 20:142–9. doi: 10.1016/j.tcb.2009.12.002
41. Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol (Baltimore Md 1950) (2003) 171:6164–72. doi: 10.4049/jimmunol.171.11.6164
42. Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY. Mechanism of Interferon-γ–Induced Increase in T84 Intestinal Epithelial Tight Junction. J Interferon Cytokine Res (2009) 29:45–54. doi: 10.1089/jir.2008.0128
43. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest (2017) 127:1–4. doi: 10.1172/JCI92035
44. Regoli M, Borghesi C, Bertelli E, Nicoletti C. Uptake of a gram-positive bacterium (Streptococcus pneumoniae R36a) by the M cells of rabbit Peyer’s patches. Ann Anat Anatomischer Anzeiger (1995) 177:119–24. doi: 10.1016/S0940-9602(11)80057-9
45. Guerville M, Boudry G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am J Physiol Gastrointestinal Liver Physiol (2016) 311:G1–15. doi: 10.1152/ajpgi.00098.2016
46. Clark E, Hoare C, Tanianis-Hughes J, Carlson GL, Warhurst G. Interferon gamma induces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterology (2005) 128:1258–67. doi: 10.1053/j.gastro.2005.01.046
47. Smyth D, McKay CM, Gulbransen BD, van Phan C, Wang A, McKay DM. Interferon-gamma signals via an ERK1/2-ARF6 pathway to promote bacterial internalization by gut epithelia. Cell Microbiol (2012) 14:1257–70. doi: 10.1111/j.1462-5822.2012.01796.x
48. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (New York NY) (2011) 334:105–8. doi: 10.1126/science.1208344
49. Zinöcker MK, Lindseth IA. The Western Diet–Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients (2018) 10:365. doi: 10.3390/nu10030365
50. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Trans Med (2009) 1:6ra14. doi: 10.1126/scitranslmed.3000322
51. Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol Microbiol (2004) 54:665–75. doi: 10.1111/j.1365-2958.2004.04308.x
52. Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol (2004) 2:123–40. doi: 10.1038/nrmicro818
53. Viswanathan VK, Koutsouris A, Lukic S, Pilkinton M, Simonovic I, Simonovic M, et al. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect Immun (2004) 72:3218–27. doi: 10.1128/IAI.72.6.3218-3227.2004
54. Caron TJ, Scott KE, Fox JG, Hagen SJ. Tight junction disruption: Helicobacter pylori and dysregulation of the gastric mucosal barrier. World J Gastroenterol (2015) 21:11411–27. doi: 10.3748/wjg.v21.i40.11411
55. Tafazoli F, Magnusson K-E, Zheng L. Disruption of epithelial barrier integrity by Salmonella enterica serovar typhimurium requires geranylgeranylated proteins. Infect Immun (2003) 71:872–81. doi: 10.1128/IAI.71.2.872-881.2003
56. Fasano A, Baudry B, Pumplin DW, Wasserman SS, Tall BD, Ketley JM, et al. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci USA (1991) 88:5242–6. doi: 10.1073/pnas.88.12.5242
57. Rossi O, van Baarlen P, Wells JM, Dobrindt U, Hacker JH, Svanborg C. Between Pathogenicity and Commensalism. Berlin, Heidelberg: Springer (2013). p. 291–321.
58. Tosi T, Pflug A, Discola KF, Neves D, Dessen A. Structural basis of eukaryotic cell targeting by type III secretion system (T3SS) effectors. Res Microbiol (2013) 164:605–19. doi: 10.1016/j.resmic.2013.03.019
59. Scott KG-E, Yu LC, Buret AG. Role of CD8+ and CD4+ T lymphocytes in jejunal mucosal injury during murine giardiasis. Infect Immun (2004) 72:3536–42. doi: 10.1128/IAI.72.6.3536-3542.2004
60. Ohata A, Usami M, Miyoshi M. Short-chain fatty acids alter tight junction permeability in intestinal monolayer cells via lipoxygenase activation. Nutr (Burbank Los Angeles County Calif) (2005) 21:838–47. doi: 10.1016/j.nut.2004.12.004
61. Peng L, Li Z-R, Green RS, Holzman IR, Lin J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J Nutr (2009) 139:1619–25. doi: 10.3945/jn.109.104638
62. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
63. Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, et al. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metab (2011) 13:517–26. doi: 10.1016/j.cmet.2011.02.018
64. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
65. Naito Y, Uchiyama K. Takagi T. A next-generation beneficial microbe: Akkermansia muciniphila. J Clin Biochem Nutr (2018) 63:33–5. doi: 10.3164/jcbn.18-57
66. Ling X, Linglong P, Weixia Du, Hong W. Protective Effects of Bifidobacterium on Intestinal Barrier Function in LPS-Induced Enterocyte Barrier Injury of Caco-2 Monolayers and in a Rat NEC Model. PLoS One (2016) 11:e0161635. doi: 10.1371/journal.pone.0161635
67. Wang L, Li L, Lv Y, Chen Q, Feng J, Zhao X. Lactobacillus plantarum Restores Intestinal Permeability Disrupted by Salmonella Infection in Newly-hatched Chicks. Sci Rep (2018) 8:2229. doi: 10.1038/s41598-018-20752-z
68. Ahrne S, Hagslatt M-LJ. Effect of lactobacilli on paracellular permeability in the gut. Nutrients (2011) 3:104–17. doi: 10.3390/nu3010104
69. Stanhope KL. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit Rev Clin Lab Sci (2016) 53:52–67. doi: 10.3109/10408363.2015.1084990
70. Pappenheimer JR, Reiss KZ. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J Membrane Biol (1987) 100:123–36. doi: 10.1007/bf02209145
71. Yu Q, Wang Z, Li P, Yang Q. The effect of various absorption enhancers on tight junction in the human intestinal Caco-2 cell line. Drug Dev Ind Pharm (2013) 39:587–92. doi: 10.3109/03639045.2012.692376
72. Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol (1997) 273:C1378–85. doi: 10.1152/ajpcell.1997.273.4.C1378
73. Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci CMLS (2013) 70:631–59. doi: 10.1007/s00018-012-1070-x
74. Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, et al. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci USA (2018) 115:3138–43. doi: 10.1073/pnas.1713837115
75. Feng R, Luo C, Li C, Du S, Okekunle AP, Li Y, et al. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: a case - control study. Lipids Health Dis (2017) 16:165. doi: 10.1186/s12944-017-0551-1
76. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology (2008) 135:194–204. doi: 10.1053/j.gastro.2008.03.023
77. Yasumatsu H, Tanabe S. The casein peptide Asn-Pro-Trp-Asp-Gln enforces the intestinal tight junction partly by increasing occludin expression in Caco-2 cells. Br J Nutr (2010) 104:951–6. doi: 10.1017/S0007114510001698
78. Isobe N, Suzuki M, Oda M, Tanabe S. Enzyme-modified cheese exerts inhibitory effects on allergen permeation in rats suffering from indomethacin-induced intestinal inflammation. Biosci Biotechnol Biochem (2008) 72:1740–5. doi: 10.1271/bbb.80042
79. Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointestinal Liver Physiol (2008) 294:G208–16. doi: 10.1152/ajpgi.00398.2007
80. Suzuki T, Hara H. Quercetin enhances intestinal barrier function through the assembly of zonula corrected occludens-2, occludin, and claudin-1 and the expression of claudin-4 in Caco-2 cells. J Nutr (2009) 139:965–74. doi: 10.3945/jn.108.100867
81. Suzuki T, Tanabe S, Hara H. Kaempferol enhances intestinal barrier function through the cytoskeletal association and expression of tight junction proteins in Caco-2 cells. J Nutr (2011) 141:87–94. doi: 10.3945/jn.110.125633
82. Amiot MJ, Riva C, Vinet A. Effects of dietary polyphenols on metabolic syndrome features in humans: a systematic review. Obes Rev An (2016) 17:573–86. doi: 10.1111/obr.12409
83. Jiang T, Gao X, Wu C, Tian F, Lei Q, Bi J, et al. Apple-Derived Pectin Modulates Gut Microbiota, Improves Gut Barrier Function, and Attenuates Metabolic Endotoxemia in Rats with Diet-Induced Obesity. Nutrients (2016) 8:126. doi: 10.3390/nu8030126
84. Kyoko O-o, Kono H, Ishimaru K, Miyake K, Kubota T, Ogawa H, et al. Expressions of tight junction proteins Occludin and Claudin-1 are under the circadian control in the mouse large intestine: implications in intestinal permeability and susceptibility to colitis. PLoS One (2014) 9:e98016. doi: 10.1371/journal.pone.0098016
85. Summa KC, Voigt RM, Forsyth CB, Shaikh M, Cavanaugh K, Tang Y, et al. Disruption of the Circadian Clock in Mice Increases Intestinal Permeability and Promotes Alcohol-Induced Hepatic Pathology and Inflammation. PLoS One (2013) 8:e67102. doi: 10.1371/journal.pone.0067102
86. Reitmeier S, Kiessling S, Clavel T, List M, Almeida EL, Ghosh TS, et al. Arrhythmic Gut Microbiome Signatures Predict Risk of Type 2 Diabetes. Cell Host Microbe (2020) 28:258–72. doi: 10.1016/j.chom.2020.06.004
87. HH U. ZERAHN K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol Scandinavica (1951) 23:110–27. doi: 10.1111/j.1748-1716.1951.tb00800.x
88. Clarke LL. A guide to Ussing chamber studies of mouse intestine. Am J Physiol Gastrointestinal Liver Physiol (2009) 296:G1151–66. doi: 10.1152/ajpgi.90649.2008
89. Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev (2014) 10:131–45. doi: 10.2174/1573399810666140508121012
90. Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palu G, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointestinal Liver Physiol (2007) 292:G518–25. doi: 10.1152/ajpgi.00024.2006
91. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut (2009) 58:1091–103. doi: 10.1136/gut.2008.165886
92. Lam YY, Ha CW, Campbell CR, Mitchell AJ, Dinudom A, Oscarsson J, et al. Increased Gut Permeability and Microbiota Change Associate with Mesenteric Fat Inflammation and Metabolic Dysfunction in Diet-Induced Obese Mice. PLoS One (2012) 7:e34233. doi: 10.1371/journal.pone.0034233
93. Ahmad R, Rah B, Bastola D, Dhawan P, Singh AB. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci Rep (2017) 7:5125. doi: 10.1038/s41598-017-04989-8
94. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature (2014) 514:237–41. doi: 10.1038/nature13564
95. Gummesson A, Carlsson LM, Storlien LH, Backhed F, Lundin P, Lofgren L, et al. Intestinal permeability is associated with visceral adiposity in healthy women. Obes (Silver Spring Md) (2011) 19:2280–2. doi: 10.1038/oby.2011.251
96. Teixeira TF, Souza NC, Chiarello PG, Franceschini SC, Bressan J, Celia LLF, et al. Intestinal permeability parameters in obese patients are correlated with metabolic syndrome risk factors. Clin Nutr (Edinburgh Scotland) (2012) 31:735–40. doi: 10.1016/j.clnu.2012.02.009
97. Damms-Machado A, Louis S, Schnitzer A, Volynets V, Rings A, Basrai M, et al. Gut permeability is related to body weight, fatty liver disease, and insulin resistance in obese individuals undergoing weight reduction. Am J Clin Nutr (2017) 105:127–35. doi: 10.3945/ajcn.116.131110
98. Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol (2015) 1:222–32. doi: 10.1016/j.jcmgh.2015.01.001
99. Ott B, Skurk T, Hastreiter L, Lagkouvardos I, Fischer S, Büttner J, et al. Effect of caloric restriction on gut permeability, inflammation markers, and fecal microbiota in obese women. Sci Rep (2017) 7:11955. doi: 10.1038/s41598-017-12109-9
100. Castillo DJ, Rifkin RF, Cowan DA, Potgieter M. The Healthy Human Blood Microbiome: Fact or Fiction? Front Cell Infect Microbiol (2019) 9:148. doi: 10.3389/fcimb.2019.00148
101. Úbeda M, Muñoz L, Borrero M-J, Díaz D, Francés R, Monserrat J, et al. Critical role of the liver in the induction of systemic inflammation in rats with preascitic cirrhosis. Hepatol (Baltimore Md) (2010) 52:2086–95. doi: 10.1002/hep.23961
102. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54:2325–40. doi: 10.1194/jlr.R036012
103. Ruppin H, Bar-Meir S, Soergel KH, Wood CM, Schmitt MG. Absorption of Short-Chain Fatty Acids by the Colon. Gastroenterology, 78(6), 1500-1507. Gastroenterology (1980) 78:1500–7. doi: 10.1016/S0016-5085(19)30508-6
104. Müller M, Hernández MA, Goossens GH, Reijnders D, Holst JJ, Jocken JW, et al. Circulating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and GLP-1 concentrations in humans. Sci Rep (2019) 9:12515. doi: 10.1038/s41598-019-48775-0
105. White RH, Frayn KN, Little RA, Threlfall CJ, Stoner HB, Irving MH. Hormonal and metabolic responses to glucose infusion in sepsis studied by the hyperglycemic glucose clamp technique. JPEN J Parenteral Enteral Nutr (1987) 11:345–53. doi: 10.1177/0148607187011004345
106. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56:1761–72. doi: 10.2337/db06-1491
107. Ghoshal S, Witta J, Zhong J, de VW, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res (2009) 50:90–7. doi: 10.1194/jlr.M800156-JLR200
108. Park BS, Lee J-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med (2013) 45:e66. doi: 10.1038/emm.2013.97
109. Levels JH, Abraham PR, van den Ende A, van Deventer SJ. Distribution and kinetics of lipoprotein-bound endotoxin. Infect Immun (2001) 69:2821–8. doi: 10.1128/IAI.69.5.2821-2828.2001
110. Salden HJ, Bas BM. Endotoxin binding to platelets in blood from patients with a sepsis syndrome. Clin Chem (1994) 40:1575–9. doi: 10.1093/clinchem/40.8.1575
111. Stahl A-l, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood (2006) 108:167–76. doi: 10.1182/blood-2005-08-3219
112. Takeshita S, Nakatani K, Tsujimoto H, Kawamura Y, Sekine I. Detection of circulating lipopolysaccharide-bound monocytes in children with gram-negative sepsis. J Infect Dis (2000) 182:1549–52. doi: 10.1086/315884
113. Roth RI, Levin FC, Levin J. Distribution of bacterial endotoxin in human and rabbit blood and effects of stroma-free hemoglobin. Infect Immun (1993) 61:3209–15. doi: 10.1128/IAI.61.8.3209-3215.1993
114. Poschl JM, Leray C, Ruef P, Cazenave JP, Linderkamp O. Endotoxin binding to erythrocyte membrane and erythrocyte deformability in human sepsis and in vitro. Crit Care Med (2003) 31:924–8. doi: 10.1097/01.CCM.0000055366.24147.80
115. Topchiy E, Cirstea M, Kong HJ, Boyd JH, Wang Y, Russell JA, et al. Lipopolysaccharide Is Cleared from the Circulation by Hepatocytes via the Low Density Lipoprotein Receptor. PLoS One (2016) 11:e0155030. doi: 10.1371/journal.pone.0155030
116. Cavaillon JM, Fitting C, Haeffner-Cavaillon N, Kirsch SJ, Warren HS. Cytokine response by monocytes and macrophages to free and lipoprotein-bound lipopolysaccharide. Infect Immun (1990) 58:2375–82. doi: 10.1128/IAI.58.7.2375-2382.1990
117. Harris HW, Grunfeld C, Feingold KR, Read TE, Kane JP, Jones AL, et al. Chylomicrons alter the fate of endotoxin, decreasing tumor necrosis factor release and preventing death. J Clin Invest (1993) 91:1028–34. doi: 10.1172/JCI116259
118. Erridge C, Attina T, Spickett CM. Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr (2007) 86:1286–92. doi: 10.1093/ajcn/86.5.1286
119. Harte AL, Varma MC, Tripathi G, McGee KC, Al-Daghri NM, Al-Attas OS, et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care (2012) 35:375–82. doi: 10.2337/dc11-1593
120. Lassenius MI, Pietilainen KH, Kaartinen K, Pussinen PJ, Syrjanen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care (2011) 34:1809–15. doi: 10.2337/dc10-2197
121. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care (2011) 34:392–7. doi: 10.2337/dc10-1676
122. Monte SV, Caruana JA, Ghanim H, Sia CL, Korzeniewski K, Schentag JJ, et al. Reduction in endotoxemia, oxidative and inflammatory stress, and insulin resistance after Roux-en-Y gastric bypass surgery in patients with morbid obesity and type 2 diabetes mellitus. Surgery (2012) 151:587–93. doi: 10.1016/j.surg.2011.09.038
123. Cox AJ, Zhang P, Bowden DW, Devereaux B, Davoren PM, Cripps AW, et al. Increased intestinal permeability as a risk factor for type 2 diabetes. Diabetes Metab (2017) 43:163–6. doi: 10.1016/j.diabet.2016.09.004
124. Agwunobi AO, Reid C, Maycock P, Little RA, Carlson GL. Insulin resistance and substrate utilization in human endotoxemia. J Clin Endocrinol Metab (2000) 85:3770–8. doi: 10.1210/jcem.85.10.6914
125. Krogh-Madsen R, Plomgaard P, Akerstrom T, Møller K, Schmitz O, Pedersen BK. Effect of short-term intralipid infusion on the immune response during low-dose endotoxemia in humans. Am J Physiol Endocrinol Metab (2008) 294:E371–9. doi: 10.1152/ajpendo.00507.2007
126. Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (London England) (2010) 7:15. doi: 10.1186/1476-9255-7-15
127. Guo S, Nighot M, Al-Sadi R, Alhmoud T, Nighot P, Ma TY. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J Immunol (Baltimore Md 1950) (2015) 195:4999–5010. doi: 10.4049/jimmunol.1402598
128. Janssens S, Beyaert R. Role of Toll-like receptors in pathogen recognition. Clin Microbiol Rev (2003) 16:637–46. doi: 10.1128/cmr.16.4.637-646.2003
129. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol (2005) 17:1–14. doi: 10.1093/intimm/dxh186
130. Trøseid M, Nestvold TK, Rudi K, Thoresen H, Nielsen EW, Lappegård KT. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: evidence from bariatric surgery. Diabetes Care (2013) 36:3627–32. doi: 10.2337/dc13-0451
131. Clemente-Postigo M, Roca-Rodriguez Mdel M, Camargo A, Ocaña-Wilhelmi L, Cardona F, Tinahones FJ. Lipopolysaccharide and lipopolysaccharide-binding protein levels and their relationship to early metabolic improvement after bariatric surgery. Surg Obes Related Dis (2015) 11:933–9. doi: 10.1016/j.soard.2014.11.030
132. Maldonado RF, Sá-Correia I, Valvano MA. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol Rev (2016) 40:480–93. doi: 10.1093/femsre/fuw007
133. Lerouge I. Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol Rev (2002) 26:17–47. doi: 10.1111/j.1574-6976.2002.tb00597.x
134. Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
135. Yang P-J, Yang W-S, Nien H-C, Chen C-N, Lee P-H, Yu LC-H, et al. Duodenojejunal Bypass Leads to Altered Gut Microbiota and Strengthened Epithelial Barriers in Rats. Obes Surg (2016) 26:1576–83. doi: 10.1007/s11695-015-1968-0
136. Casselbrant A, Elias E, Fändriks L, Wallenius V. Expression of tight-junction proteins in human proximal small intestinal mucosa before and after Roux-en-Y gastric bypass surgery. Surg Obes Related Dis (2015) 11:45–53. doi: 10.1016/j.soard.2014.05.009
137. Massier L, Chakaroun R, Tabei S, Crane A, Didt KD, Fallmann J, et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut (2020) 69:1796–806. doi: 10.1136/gutjnl-2019-320118
138. Balkau B, Lange C, Fezeu L, Tichet J, Lauzon-Guillain B, de Czernichow S, et al. Predicting diabetes: clinical, biological, and genetic approaches: data from the Epidemiological Study on the Insulin Resistance Syndrome (DESIR). Diabetes Care (2008) 31:2056–61. doi: 10.2337/dc08-0368
139. Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, et al. Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia (2011) 54:3055–61. doi: 10.1007/s00125-011-2329-8
140. Amar J, Lange C, Payros G, Garret C, Chabo C, Lantieri O, et al. Blood Microbiota Dysbiosis Is Associated with the Onset of Cardiovascular Events in a Large General Population: The D.E.S.I.R. Study. PLoS One (2012) 8:e54461. doi: 10.1371/journal.pone.0054461
141. Sato J, Kanazawa A, Ikeda F, Yoshihara T, Goto H, Abe H, et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care (2014) 37:2343–50. doi: 10.2337/dc13-2817
142. Lelouvier B, Servant F, Paisse S, Brunet A-C, Benyahya S, Serino M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatol (Baltimore Md) (2016) 64:2015–27. doi: 10.1002/hep.28829
143. Qiu J, Zhou H, Jing Y, Dong C. Association between blood microbiome and type 2 diabetes mellitus: A nested case-control study. J Clin Lab Anal (2019) 33:e22842. doi: 10.1002/jcla.22842
144. Lammers KM, Brigidi P, Vitali B, Gionchetti P, Rizzello F, Caramelli E, et al. Immunomodulatory effects of probiotic bacteria DNA: IL-1 and IL-10 response in human peripheral blood mononuclear cells. FEMS Immunol Med Microbiol (2003) 38:165–72. doi: 10.1016/S0928-8244(03)00144-5
145. Ortiz S, Zapater P, Estrada JL, Enriquez P, Rey M, Abad A, et al. Bacterial DNA translocation holds increased insulin resistance and systemic inflammatory levels in morbid obese patients. J Clin Endocrinol Metab (2014) 99:2575–83. doi: 10.1210/jc.2013-4483
146. Zwick RK, Guerrero-Juarez CF, Horsley V, Plikus MV. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab (2018) 27:68–83. doi: 10.1016/j.cmet.2017.12.002
147. Poulain-Godefroy O, Le Bacquer O, Plancq P, Lecoeur C, Pattou F, Frühbeck G, et al. Inflammatory role of Toll-like receptors in human and murine adipose tissue. Mediators Inflamm (2010) 2010:823486. doi: 10.1155/2010/823486
148. Vila IK, Badin P-M, Marques M-A, Monbrun L, Lefort C, Mir L, et al. Immune Cell Toll-like Receptor 4 Mediates the Development of Obesity- and Endotoxemia-Associated Adipose Tissue Fibrosis. Cell Rep (2014) 7:1116–29. doi: 10.1016/j.celrep.2014.03.062
149. Creely SJ, McTernan PG, Kusminski CM, Fisher FM, Da Silva NF, Khanolkar M, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab (2007) 292:E740–7. doi: 10.1152/ajpendo.00302.2006
150. Vitseva OI, Tanriverdi K, Tchkonia TT, Kirkland JL, McDonnell ME, Apovian CM, et al. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obes (Silver Spring Md) (2008) 16:932–7. doi: 10.1038/oby.2008.25
151. Hoareau L, Bencharif K, Rondeau P, Murumalla R, Ravanan P, Tallet F, et al. Signaling pathways involved in LPS induced TNFalpha production in human adipocytes. J Inflamm (London England) (2010) 7:1. doi: 10.1186/1476-9255-7-1
152. Ajuwon KM, Spurlock ME. Adiponectin inhibits LPS-induced NF-kappaB activation and IL-6 production and increases PPARgamma2 expression in adipocytes. Am J Physiol Regulatory Integr Comp Physiol (2005) 288:R1220–5. doi: 10.1152/ajpregu.00397.2004
153. Chiadak JD, Gena P, Gregoire F, Bolaky N, Delforge V, Perret J, et al. Lipopolysaccharide Modifies Glycerol Permeability and Metabolism in 3T3-L1 Adipocytes. Int J Mol Sci (2017) 18:2566. doi: 10.3390/ijms18122566
154. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez-Humaran LG, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol Med (2011) 3:559–72. doi: 10.1002/emmm.201100159
155. Burcelin R, Serino M, Chabo C, Garidou L, Pomie C, Courtney M, et al. Metagenome and metabolism: the tissue microbiota hypothesis. Diabetes Obes Metab (2013) 15 Suppl 3:61–70. doi: 10.1111/dom.12157
156. Zulian A, Cancello R, Cesana E, Rizzi E, Consolandi C, Severgnini M, et al. Adipose tissue microbiota in humans: An open issue. Int J Obes (2005) (2016) 40:1643–8. doi: 10.1038/ijo.2016.111
157. Udayappan SD, Kovatcheva-Datchary P, Bakker GJ, Havik SR, Herrema H, Cani PD, et al. Intestinal Ralstonia pickettii augments glucose intolerance in obesity. PLoS One (2017) 12:e0181693. doi: 10.1371/journal.pone.0181693
158. Nakatsuji T, Chiang H-I, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun (2013) 4:1431. doi: 10.1038/ncomms2441
159. Pedicino D, Severino A, Ucci S, Bugli F, Flego D, Giglio AF, et al. Epicardial adipose tissue microbial colonization and inflammasome activation in acute coronary syndrome. Int J Cardiol (2017) 236:95–9. doi: 10.1016/j.ijcard.2017.02.040
160. Anhê FF, Jensen BA, Varin TV, Servant F, van Blerk S, Richard D, et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat Metab (2020) 374:788. doi: 10.1038/s42255-020-0178-9
161. Ha CW, Martin A, Sepich-Poore GD, Shi B, Wang Y, Gouin K, et al. Translocation of Viable Gut Microbiota to Mesenteric Adipose Drives Formation of Creeping Fat in Humans. Cell (2020) 183:666–83.e17. doi: 10.1016/j.cell.2020.09.009
162. Koh A, de VF, Kovatcheva-Datchary P, Bäckhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
163. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun (2013) 4:1829. doi: 10.1038/ncomms2852
164. Robertson MD, Bickerton AS, Dennis AL, Vidal H, Frayn KN. Insulin-sensitizing effects of dietary resistant starch and effects on skeletal muscle and adipose tissue metabolism. Am J Clin Nutr (2005) 82:559–67. doi: 10.1093/ajcn.82.3.559
165. Jocken JW, González Hernández MA, Hoebers NT, van der Beek CM, Essers YP, Blaak EE, et al. Short-Chain Fatty Acids Differentially Affect Intracellular Lipolysis in a Human White Adipocyte Model. Front Endocrinol (2017) 8:372. doi: 10.3389/fendo.2017.00372
166. Hong Y-H, Nishimura Y, Hishikawa D, Tsuzuki H, Miyahara H, Gotoh C, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology (2005) 146:5092–9. doi: 10.1210/en.2005-0545
167. Lu Y, Fan C, Li P, Lu Y, Chang X, Qi K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci Rep (2016) 6:37589. doi: 10.1038/srep37589
168. Cheng N, Liang Y, Du X, Ye RD. Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep (2018) 19(10):e45517. doi: 10.15252/embr.201745517
169. Scott MJ, Billiar TR. Beta2-integrin-induced p38 MAPK activation is a key mediator in the CD14/TLR4/MD2-dependent uptake of lipopolysaccharide by hepatocytes. J Biol Chem (2008) 283:29433–46. doi: 10.1074/jbc.M803905200
170. Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, et al. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol (Baltimore Md 1950) (2013) 190:5152–60. doi: 10.4049/jimmunol.1300496
171. Soares J-B, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int (2010) 4:659–72. doi: 10.1007/s12072-010-9219-x
172. Kiziltas S. Toll-like receptors in pathophysiology of liver diseases. World J Hepatol (2016) 8:1354–69. doi: 10.4254/wjh.v8.i32.1354
173. Sharifnia T, Antoun J, Verriere TG, Suarez G, Wattacheril J, Wilson KT, et al. Hepatic TLR4 signaling in obese NAFLD. American journal of physiology. Gastrointestinal Liver Physiol (2015) 309:G270–8. doi: 10.1152/ajpgi.00304.2014
174. Kheder RK, Hobkirk J, Stover CM. In vitro Modulation of the LPS-Induced Proinflammatory Profile of Hepatocytes and Macrophages- Approaches for Intervention in Obesity? Front Cell Dev Biol (2016) 4:61. doi: 10.3389/fcell.2016.00061
175. Lee Y-S, Kim Y-H, Jung YS, Kim K-S, Kim D-K, Na S-Y, et al. Hepatocyte toll-like receptor 4 mediates lipopolysaccharide-induced hepcidin expression. Exp Mol Med (2017) 49:e408. doi: 10.1038/emm.2017.207
176. Vives-Pi M, Somoza N, Fernández-Alvarez J, Vargas F, Caro P, Alba A, et al. Evidence of expression of endotoxin receptors CD14, toll-like receptors TLR4 and TLR2 and associated molecule MD-2 and of sensitivity to endotoxin (LPS) in islet beta cells. Clin Exp Immunol (2003) 133:208–18. doi: 10.1046/j.1365-2249.2003.02211.x
177. Amyot J, Semache M, Ferdaoussi M, Fontes G, Poitout V. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via Toll-Like Receptor-4 and NF-kappaB signalling. PLoS One (2012) 7:e36200. doi: 10.1371/journal.pone.0036200
178. Sookoian S, Salatino A, Castaño GO, Landa MS, Fijalkowky C, Garaycoechea M, et al. Intrahepatic bacterial metataxonomic signature in non-alcoholic fatty liver disease. Gut (2020) 69:1483–91. doi: 10.1136/gutjnl-2019-318811
179. Pingitore A, Chambers ES, Hill T, Maldonado IR, Liu B, Bewick G, et al. The diet-derived short chain fatty acid propionate improves beta-cell function in humans and stimulates insulin secretion from human islets in vitro. Diabetes Obes Metab (2017) 19:257–65. doi: 10.1111/dom.12811
180. van der Crabben SN, Blümer RM, Stegenga ME, Ackermans MT, Endert E, Tanck MW, et al. Early endotoxemia increases peripheral and hepatic insulin sensitivity in healthy humans. J Clin Endocrinol Metab (2009) 94:463–8. doi: 10.1210/jc.2008-0761
181. Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes (2008) 57:2595–602. doi: 10.2337/db08-0038
182. Liang H, Hussey SE, Sanchez-Avila A, Tantiwong P, Musi N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS One (2013) 8:e63983. doi: 10.1371/journal.pone.0063983
183. Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, et al. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab (2010) 298:E988–98. doi: 10.1152/ajpendo.00307.2009
184. Frisard MI, Wu Y, McMillan RP, Voelker KA, Wahlberg KA, Anderson AS, et al. Low levels of lipopolysaccharide modulate mitochondrial oxygen consumption in skeletal muscle. Metabol: Clin Exp (2015) 64:416–27. doi: 10.1016/j.metabol.2014.11.007
185. McNelis JC, Lee YS, Mayoral R, van der Kant R, Johnson AM, Wollam J, et al. GPR43 Potentiates β-Cell Function in Obesity. Diabetes (2015) 64:3203–17. doi: 10.2337/db14-1938
186. Priyadarshini M, Villa SR, Fuller M, Wicksteed B, Mackay CR, Alquier T, et al. FFAR2, Regulates Insulin Secretion. Mol Endocrinol (Baltimore Md) (2015) 29:1055–66. doi: 10.1210/me.2015-1007
187. Houghton MJ, Kerimi A, Mouly V, Tumova S, Williamson G. Gut microbiome catabolites as novel modulators of muscle cell glucose metabolism. FASEB J (2019) 33:1887–98. doi: 10.1096/fj.201801209R
188. Frampton J, Murphy KG, Frost G, Chambers ES. Short-chain fatty acids as potential regulators of skeletal muscle metabolism and function. Nat Metab (2020) 2:840–8. doi: 10.1038/s42255-020-0188-7
189. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes (2009) 58:1509–17. doi: 10.2337/db08-1637
190. Maruta H, Yoshimura Y, Araki A, Kimoto M, Takahashi Y, Yamashita H. Activation of AMP-Activated Protein Kinase and Stimulation of Energy Metabolism by Acetic Acid in L6 Myotube Cells. PLoS One (2016) 11:e0158055. doi: 10.1371/journal.pone.0158055
191. Han J-H, Kim I-S, Jung S-H, Lee S-G, Son H-Y, Myung C-S. The effects of propionate and valerate on insulin responsiveness for glucose uptake in 3T3-L1 adipocytes and C2C12 myotubes via G protein-coupled receptor 41. PLoS One (2014) 9:e95268. doi: 10.1371/journal.pone.0095268
192. Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. J Lab Automation (2015) 20:107–26. doi: 10.1177/2211068214561025
193. Wang L, Llorente C, Hartmann P, Yang A-M, Chen P, Schnabl B. Methods to determine intestinal permeability and bacterial translocation during liver disease. J Immunol Methods (2015) 421:44–53. doi: 10.1016/j.jim.2014.12.015
194. Vancamelbeke M, Vermeire S. The intestinal barrier: a fundamental role in health and disease. Expert Rev Gastroenterol Hepatol (2017) 11:821–34. doi: 10.1080/17474124.2017.1343143
195. González-González M, Díaz-Zepeda C, Eyzaguirre-Velásquez J, González-Arancibia C, Bravo JA, Julio-Pieper M. Investigating Gut Permeability in Animal Models of Disease. Front Physiol (2019) 9:1962. doi: 10.3389/fphys.2018.01962
196. Galipeau HJ, Verdu EF. The complex task of measuring intestinal permeability in basic and clinical science. Neurogastroenterol Motil (2016) 28:957–65. doi: 10.1111/nmo.12871
197. Scheffler L, Crane A, Heyne H, Tonjes A, Schleinitz D, Ihling CH, et al. Widely Used Commercial ELISA Does Not Detect Precursor of Haptoglobin2, but Recognizes Properdin as a Potential Second Member of the Zonulin Family. Front Endocrinol (2018) 9:22. doi: 10.3389/fendo.2018.00022
198. Ajamian M, Steer D, Rosella G, Gibson PR. Serum zonulin as a marker of intestinal mucosal barrier function: May not be what it seems. PLoS One (2019) 14:e0210728. doi: 10.1371/journal.pone.0210728
199. Massier L, Chakaroun R, Kovacs P, Heiker JT. Blurring the picture in leaky gut research: how shortcomings of zonulin as a biomarker mislead the field of intestinal permeability. Gut (2020) gutjnl-2020-323026. doi: 10.1136/gutjnl-2020-323026
200. Novitsky TJ, Schmidt-Gengenbach J, Remillard JF. Factors affecting recovery of endotoxin adsorbed to container surfaces. J Parenteral Sci Technol Publ Parenteral Drug Assoc (1986) 40:284–6.
201. Nádházi Z, Takáts A, Offenmüller K, Bertók L. Plasma endotoxin level of healthy donors. Acta Microbiol Immunol Hungarica (2002) 49:151–7. doi: 10.1556/AMicr.49.2002.1.15
202. Hurley JC. Endotoxemia: methods of detection and clinical correlates. Clin Microbiol Rev (1995) 8:268–92. doi: 10.1128/CMR.8.2.268
203. Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie (2016) 124:11–20. doi: 10.1016/j.biochi.2015.06.020
204. Elsbach P, Weiss J. The bactericidal/permeability-increasing protein (BPI), a potent element in host-defense Against gram-negative bacteria and lipopolysaccharide. Immunobiology (1993) 187:417–29. doi: 10.1016/S0171-2985(11)80354-2
205. Mathison J, Tobias P, Wolfson E, Ulevitch R. Regulatory mechanisms of host responsiveness to endotoxin (lipopolysaccharide). Pathobiol J Immunopathol Mol Cell Biol (1991) 59:185–8. doi: 10.1159/000163641
206. Wright SD. Multiple receptors for endotoxin. Curr Opin Immunol (1991) 3:83–90. doi: 10.1016/0952-7915(91)90082-C
207. Hurley JC. Concordance of endotoxemia with gram-negative bacteremia in patients with gram-negative sepsis: a meta-analysis. J Clin Microbiol (1994) 32:2120–7. doi: 10.1128/JCM.32.9.2120-2127.1994
208. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The Placenta Harbors a Unique Microbiome. Sci Trans Med (2014) 6:237ra65. doi: 10.1126/scitranslmed.3008599
209. de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, SJ P, et al. Human placenta has no microbiome but can contain potential pathogens. Nature (2019) 572:329–34. doi: 10.1038/s41586-019-1451-5
210. Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome (2016) 4:1–11. doi: 10.1186/s40168-016-0172-3
211. Leiby JS, McCormick K, Sherrill-Mix S, Clarke EL, Kessler LR, Taylor LJ, et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome (2018) 6:1–11. doi: 10.1186/s40168-018-0575-4
212. Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol (2014) 12:87. doi: 10.1186/s12915-014-0087-z
213. Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome (2018) 6:226. doi: 10.1186/s40168-018-0605-2
214. Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol (2006) 33:401–7. doi: 10.1111/j.1600-051X.2006.00924.x
215. Parahitiyawa NB, Jin LJ, Leung WK, Yam WC, Samaranayake LP. Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev (2009) 22:46–64. doi: 10.1128/CMR.00028-08
216. Chaffee BW, Weston SJ. Association between chronic periodontal disease and obesity: a systematic review and meta-analysis. J Periodontol (2010) 81:1708–24. doi: 10.1902/jop.2010.100321
217. Franchini R, Petri A, Migliario M, Rimondini L. Poor oral hygiene and gingivitis are associated with obesity and overweight status in paediatric subjects. J Clin Periodontol (2011) 38:1021–8. doi: 10.1111/j.1600-051X.2011.01770.x
Keywords: obesity, metabolic disease, intestinal permeability, microbiome, zonulin, adipose tissue microbiota, type 2 diabetes, endotoxemia
Citation: Massier L, Blüher M, Kovacs P and Chakaroun RM (2021) Impaired Intestinal Barrier and Tissue Bacteria: Pathomechanisms for Metabolic Diseases. Front. Endocrinol. 12:616506. doi: 10.3389/fendo.2021.616506
Received: 12 October 2020; Accepted: 02 February 2021;
Published: 09 March 2021.
Edited by:
Mora Murri, University of Málaga, SpainReviewed by:
Linda Chia-Hui Yu, National Taiwan University, TaiwanHemraj B. Dodiya, University of Chicago, United States
Copyright © 2021 Massier, Blüher, Kovacs and Chakaroun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rima M. Chakaroun, cmltYS5jaGFrYXJvdW5AbWVkaXppbi51bmktbGVpcHppZy5kZQ==