 Jenny E. Blau
Jenny E. Blau William F. Simonds
William F. Simonds- 1Early Clinical Development, Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gaithersburg, MD, United States
- 2Metabolic Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
Regulation of the serum calcium level in humans is achieved by the endocrine action of parathyroid glands working in concert with vitamin D and a set of critical target cells and tissues including osteoblasts, osteoclasts, the renal tubules, and the small intestine. The parathyroid glands, small highly vascularized endocrine organs located behind the thyroid gland, secrete parathyroid hormone (PTH) into the systemic circulation as is needed to keep the serum free calcium concentration within a tight physiologic range. Primary hyperparathyroidism (HPT), a disorder of mineral metabolism usually associated with abnormally elevated serum calcium, results from the uncontrolled release of PTH from one or several abnormal parathyroid glands. Although in the vast majority of cases HPT is a sporadic disease, it can also present as a manifestation of a familial syndrome. Many benign and malignant sporadic parathyroid neoplasms are caused by loss-of-function mutations in tumor suppressor genes that were initially identified by the study of genomic DNA from patients who developed HPT as a manifestation of an inherited syndrome. Somatic and inherited mutations in certain proto-oncogenes can also result in the development of parathyroid tumors. The clinical and genetic investigation of familial HPT in kindreds found to lack germline variants in the already known HPT-predisposition genes represents a promising future direction for the discovery of novel genes relevant to parathyroid tumor development.
Introduction
Typically evidenced by elevated serum calcium, primary hyperparathyroidism (HPT) is a disorder of mineral metabolism caused by the inappropriate or excessive secretion of parathyroid hormone (PTH) from one or several abnormal parathyroid glands (1). The majority of cases of HPT are sporadic and arise or occur randomly with no apparent predisposition (~95%). With respect to the remaining < 5% of patients with a familial predisposition to develop HPT, most carry germline mutation of a gene known to confer susceptibility to parathyroid tumor development (listed in Table 1). Even though these disorders are rare, study of the molecular genetics of these uncommon familial HPT syndromes has provided considerable insight into the molecular pathophysiology of both sporadic and familial parathyroid neoplasia. Since the release of parathyroid hormone (PTH) from parathyroid chief cells is tightly regulated by the calcium-sensing receptor (CASR), a cell surface-expressed G protein-coupled receptor (GPCR) belonging to GPCR family C (2), mutation in the germline of CASR or other genes transducing and propagating the CASR signal can also result in heritable syndromes associated with hypercalcemia and PTH levels that are high or inappropriately normal. This chapter will review and summarize current knowledge of the molecular pathophysiology and clinical genetics of familial syndromes that predispose to parathyroid gland neoplasia and HPT.
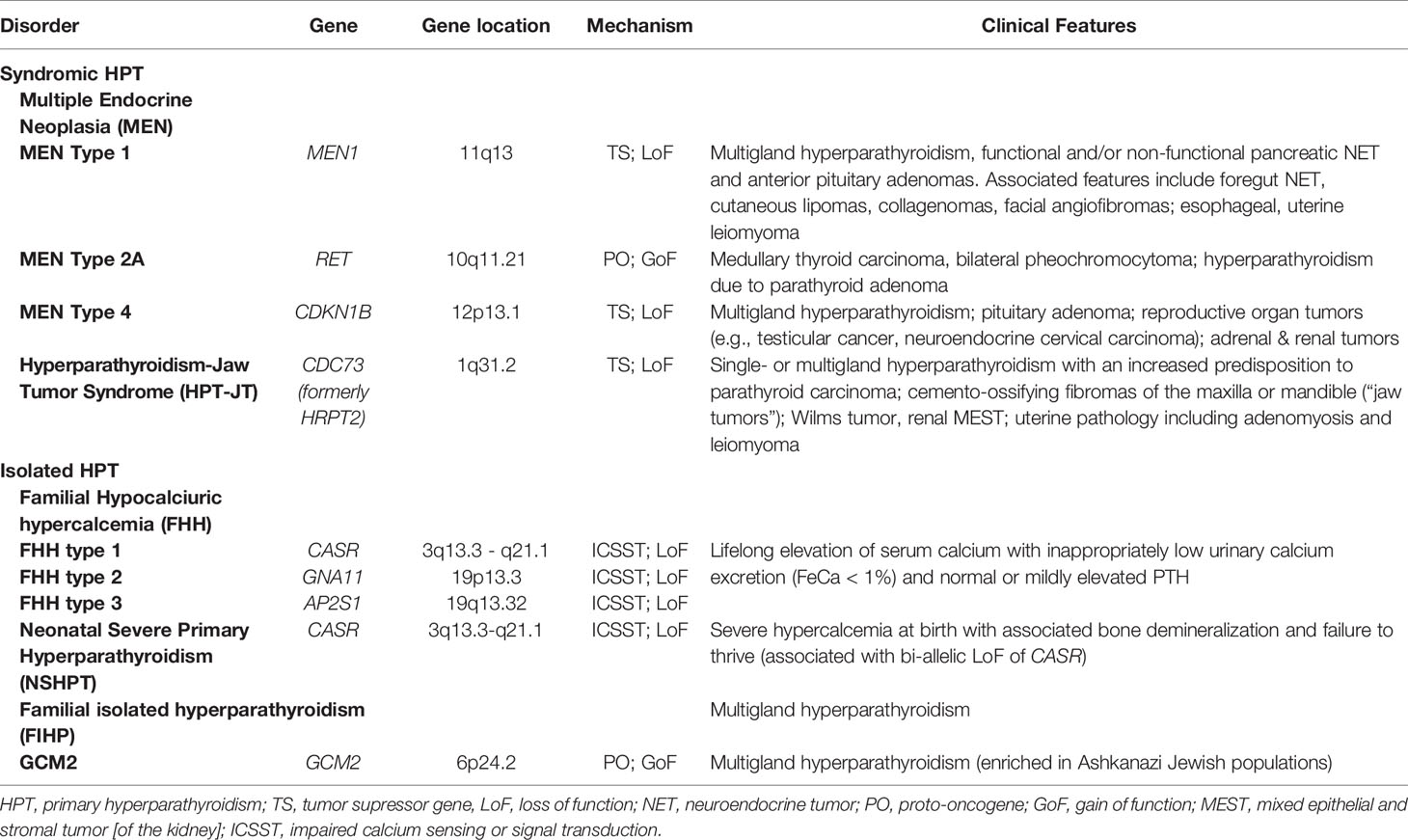
Table 1 Hereditary Hyperparathyroidism.
The Pathophysiology and Clinical Presentations of Primary Hyperparathyroidism
Mammals, like all vertebrates, depend on calcium for diverse cellular processes such as the actions of critical calcium-dependent enzymes, neuromuscular excitability, muscular contraction, intracellular second messenger signaling, neurotransmitter release, and membrane permeability. The ability of many proteins to reversibly bind calcium ions, which enables signaling events to be registered through the presence or absence of such binding (and associated protein conformational changes), is facilitated by calcium’s particular coordination chemistry (3). A system of physiologic calcium homeostasis, that depends on calcium reserves present in bone, ensures the continued normal functioning of the myriad intracellular processes that vitally depend on calcium.
In order to maintain the ambient calcium concentration within a defined physiologic range, parathyroid hormone (PTH) secretion from the parathyroid glands is tightly regulated in response to changes in the circulating ionized calcium level. Calcium sensing is mediated by the CASR, a GPCR situated on the plasma membrane of chief cells in the parathyroid glands and a critical regulator of PTH secretion (4, 5). In the bone, PTH binds to specific PTH1R receptors present on the surface of osteoblasts, stimulating the expression of receptor activator of nuclear factor kappa-B ligand (RANKL), which in turn recruits and activates osteoclasts whose bone resorptive activity mobilizes calcium from bone thus raising the ambient ionized calcium concentration (6). In another classic endocrine negative feedback loop, 1,25-dihydroxyvitamin D, the active form of cholecalciferol whose biosynthesis in proximal renal tubular cells is stimulated by PTH, inhibits the biosynthesis and release of PTH from parathyroid cells (7–10). A widely accepted clinical definition of HPT is the concurrent demonstration of hypercalcemia and an elevated or inappropriately normal PTH (1). The great majority of parathyroid tumors are adenomas (i.e. benign neoplasms), with parathyroid carcinoma making up less than 1% of cases of HPT in most clinical series.
Hypophosphatemia is a frequent concomitant of HPT. HPT figures prominently in the differential diagnosis of hypophosphatemia and the coincidence of hypercalcemia and hypophosphatemia is highly suggestive of HPT (11). The mechanism of relative or absolute hypophosphatemia in HPT involves the actions of PTH on PTH1R receptors present in the proximal renal tubule that promote the urinary excretion of phosphate. Activation of PTH1R by PTH downregulates the surface expression of type II sodium phosphate cotransporters (NPTIIa, SLC34A1; NPTIIc, SLC34A3) present on the apical membranes of proximal renal tubule cells resulting in PTH-stimulated phosphaturia and renal phosphate wasting (12, 13).
There is a wide spectrum of clinical presentations of HPT (1). This reflects the broad range of actions of PTH in target tissues as well as the myriad potential signs and symptoms of hypercalcemia per se. The metabolic abnormality is asymptomatic in as many as 70–80% of patients, often discovered incidentally on routine blood chemistry panels. Sporadic primary HPT occurs at all ages, but peaks in the sixth decade of life and is more common in women, with a female-to-male ratio between 2:1 and 3:1. Common symptomatic manifestations include fatigue, weakness, cognitive changes such as difficulty concentrating and impaired memory, gastrointestinal changes such as constipation and bloating, polyuria, nephrolithiasis, and osteoporosis. While the severity of symptoms may be proportional to the degree of hypercalcemia, for any degree of hypercalcemia, older patients tend to be more sensitive to the cognitive and neuromuscular manifestations. When severe, the renal phosphate wasting and subsequent hypophosphatemia associated with HPT may contribute to patients’ fatigue and weakness. Advanced HPT is classically characterized by osteitis fibrosa cystica, a syndrome of severe skeletal demineralization associated with cyst-like “brown tumors”, bone pain or tenderness, bone fractures, and skeletal deformities such as bowing of weight-bearing bones (14).
Parathyroid carcinoma is a rare cause of HPT, seen in fewer than 1% of sporadic cases (15, 16). Parathyroid carcinoma can be challenging to diagnose, as many of the histopathologic features are neither specific nor sensitive. Histopathologic findings include fibrous bands, elevated mitotic index, nuclear atypia, invasion of neighboring tissues, and perineural or angio-lymphatic invasion. Not infrequently the diagnosis is made only retrospectively, years after the initial parathyroid surgery, subsequent to the clinical manifestation of local or distant metastases. Clinical findings suggestive of carcinoma include a palpable neck mass, hoarseness (which may be indicative of vocal cord paresis and laryngeal nerve damage), serum calcium greater than 14 mg/dL, and overt bone and/or kidney disease at the time of presentation.
While most cases of HPT are sporadic, inherited forms of HPT represent approximately 2 to 5% of cases. As exemplified in Table 1, research into the genetics and molecular pathophysiology of the smaller subcategory of familial cases has nonetheless provided important insights regarding the genes and pathways that contribute to parathyroid tumor development. Multiple endocrine neoplasia type 1 (MEN1), the hyperparathyroidism-jaw tumor syndrome (HPT-JT), familial isolated hyperparathyroidism (FIHP), and multiple endocrine neoplasia type 2A (MEN2A), are the most frequently encountered inherited disorders that predispose to HPT (17–21). Familial hypocalciuric hypercalcemia (FHH), also known as familial benign hypercalcemia, is a related autosomal dominant condition often mis-diagnosed as HPT that is characterized by lifelong asymptomatic hypercalcemia that results from impaired calcium sensing or downstream signal transduction. In FHH, partial or even subtotal parathyroidectomy does not correct the hypercalcemia (22). The relevance of these rare familial disorders to the basic molecular pathogenesis of parathyroid neoplasia will be discussed in more detail in the sections to follow.
Tumor Suppressor Genes and the “Two-Hit” Hypothesis of Tumor Development
Cellular activation of tumorigenesis requires transformation of normal cell into a neoplastic derivative. This process is typically regulated by genes that encode proteins that help control cell growth and proliferation. Tumorigenesis initiates when the balance between cell growth and inhibition is lost, and genes that positively regulate growth (proto-oncogenes) or inhibit growth (tumor suppressor genes) are either constitutively activated or inactivated, respectively.
The most common mechanism for tumor growth in hereditary tumor syndromes, which account for ~5–10% of all cancer, is inactivation of tumor suppressor genes. Knudson’s “two‐hit hypothesis” has been fundamental for understanding tumor suppressor genes and familial tumor-predisposing syndromes. Before the advancement of molecular genetics, Knudson used mathematical modelling to compare the clinical presentation of sporadic and inherited cases of retinoblastoma (RB) in children to better understand the latency of the disease (23). By evaluating the time at which both eyes would be affected by retinoblastoma if one or two hits to DNA were required, Knudson accurately predicted the relative chance of this occurring. The conclusion that retinoblastoma required two-hits, one from the germline (inherited) and a subsequent second hit to inactivate the gene (termed “loss of heterozygosity” or LOH), was confirmed with the discovery of the RB gene in 1986 and identification of LOH in the tumor tissue (24). The most common tumor suppressor genes such as p53, RB, and PTEN are frequently identified in various tumor syndromes (25). A similar observation of the two-hit hypothesis holds true for inherited causes of hyperparathyroidism due to tumor suppressor genes such as MEN1 and CDKN1B (Table 1).
Proto-Oncogenes and Heritable Oncogenes
Another potential molecular mechanism for tumor development involves mutant genes called oncogenes that drive cell growth. Oncogenes derive from naturally occurring genes, called proto-oncogenes. Typically proto-oncogenes are genes which positively regulate cell division and/or cell growth under normal conditions (26). Examples of proto-oncogenes include SRC, HRAS, KRAS, NRAS, WNT1, and MYC. Oncogenes result from the mutational activation (e.g. via the acquisition of mutations that result in constitutive signaling activity) or overexpression of proto-oncogenes that can induce cell division and cell growth, causing tumor formation often in a tissue-specific fashion. The gene products encoded by proto-oncogenes often belong to mitogenic signaling pathways. With respect to the etiologies of currently understood inherited cancer syndromes, germline mutational gain-of-function of proto-oncogenes is rare compared to loss-of-function mutations in tumor suppressor genes (see below). Constitutive mitogenic signaling resulting from the germline gain-of-function of most proto-oncogenes is likely to be incompatible with normal embryonic and fetal development which requires extremely precise regulation of intercellular communication.
Multiple Endocrine Neoplasia Type 1 Syndrome (MEN1)
The initial identification of the co-occurrence of parathyroid, pancreas, and [anterior] pituitary tumors was published by Harvey Cushing in 1927 in a patient with acromegaly, hyperparathyroidism, and an islet cell tumor (27). Further case series emerged (28, 29) and recognition of the autosomal dominant inheritance of parathyroid hyperplasia, pancreatic islet cell tumors and anterior pituitary tumors was reported in 1954 by Paul Wermer (previously referred by the eponym Wermer’s syndrome) (30). Further understanding of the syndrome led to a hypothesis by Zollinger and Ellison who suspected an ulcerogenic hormone arising from a pancreatic islet cell tumor (31). Additional manifestations of the disease have emerged over time to create a complex picture of neoplasms affecting multiple organs (Table 1).
Linking the MEN1 trait to an unknown causative gene on chromosome 11q13 (32) and identification of loss of heterozygosity in the tumor tissue (33) ultimately paved the way for identification of the MEN1 gene in 1997 by positional cloning (34). Subsequent understanding of the protein product menin has evolved over the past 20 years since the discovery of the gene. However, many of its functions remain elusive. While menin is highly conserved in animal species, the amino acid sequence does not show homology with any known proteins. Menin is a nuclear protein with multiple functions, particularly transcription regulation and chromatin modification (35). In addition, menin functions as a DNA-repair protein in response to DNA damage, cell signaling, cytoskeletal structure, cell division, cell adhesion or cell motility (36).
MEN1 gene mutations have evolved from inherited tumor syndromes to recognizing the role of somatic MEN1 mutations in sporadic neuroendocrine tumors (37–39). Advances in tumor genetic profiling of pNETs have identified somatic MEN1 mutations in 30–44% of sporadic islet cell tumors (38, 40, 41). Key genetic regulators of the mTOR signaling pathway, histone modification, altered telomere length (ALT) and DNA damage repair pathways have also emerged as important pathogenic contributors to somatic pancreatic neuroendocrine tumor (pNET) development. In particular, up to 40% of somatic pNETs have mutations in either the apoptotic regulator DAXX (death-domain–associated protein) or the chromatin modifier ATRX (α thalassemia/intellectual disability syndrome X-linked) where they promote ALT and chromosomal instability (40, 42, 43). These alternations in MEN1, DAXX and ATRX have led to the hypothesis of molecular subtypes, which may confer both therapeutic interventions and possible survival benefit (40, 44).
MEN1 syndrome typically manifests with early onset of HPT. In known MEN1 kindreds, children and adolescents are recognized to have mild biochemical evidence of hyperparathyroidism with hypercalcemia prior to the onset of symptoms (45, 46). Penetrance of HPT is almost complete by age 50 (47). Nevertheless, some MEN1 patients may become symptomatic by the second decade with non-specific complaints (constipation, fatigue, abdominal pain, etc.) or present with end-organ effects such as nephrolithiasis or fracture (48). Similar to other familial HPT syndromes, parathyroid hyperplasia occurs in all four glands over time with asynchronous and often asymmetric growth (49). In contrast to patients with CDC73 mutations (as described below), parathyroid carcinoma is exceedingly rare in MEN1.
Epigenetic modulators also have been evaluated and linked to pathogenesis in HPT, although the data remains the strongest with links to parathyroid carcinoma. DNA methylation and chromatin modifications, for example, may contribute to the pathogenesis of HPT, although the MEN1 gene itself does not appear to be subject to methylation changes (50) and parathyroid adenomas do not appear to be affected by global methylation patterns. Rather, parathyroid tumors mainly demonstrate hypermethylation involving the CpG islands of the promoter region of specific genes in processes involved in cell cycle and transcription (e.g. CDKN2A/B, RB, WT1, etc.), Wnt/B-catenin (e.g. APC, SFRP1/2/4) and membrane transporters (e.g. MDR1). It may be that hypermethylation can link the frequency and prognosis (e.g. aggressive and/or malignant) of parathyroid tumors in the future. For a fuller review of this topic, please see the recent study by Corbetta and co-workers (51).
Other epigenetic regulators, including small non-coding microRNA, have also been postulated to contribute to the pathogenesis of tumor initiation among patients with MEN1. MicroRNA is known to contribute to multiple cellular processes, including cell proliferation, adhesion, cell death and differentiation (52) and negatively regulate post-transcriptional gene expression (53). One study evaluated parathyroid adenomas in MEN1 and suggests three main dysregulated microRNA: miR-1301, miR-4258 and miR-664. The latter two microRNAs were down-regulated in parathyroid cells that have lost both the wild type MEN1 alleles while miR-1301 was over-expressed after biallelic inactivation of wild type MEN1 (54). While further work needs to be done to explore the contribution of microRNA and other epigenetic regulators in tumorigenesis of MEN1, epigenetics may play an important role in the differentiation of phenotypic disease burden and may also help further inform diagnosis and prognosis in the future.
Anterior pituitary adenomas in MEN1 may be functional or non-functional. The genetic landscape of pituitary tumors has advanced over the last decade, with the identification of somatic (GNAS, USP8, GPR101) and germline (MEN1, cyclin dependent kinase inhibitor genes, AIP, DICER1, PRKAR1A, PRKACA, SDHx, and GPR101) drivers of pituitary tumorigenesis (55). Familial causes of pituitary adenomas account for ~5% of all cases, but should be considered especially in young patients, those with syndromic manifestations or a family history suggesting a germline mutation. Typically, MEN1 associated pituitary adenomas are benign, but may cause significant morbidity in the setting of hormonal hypersecretion, enlargement that impinges on the optic chiasm, pituitary apoplexy, or post-surgical hypopituitarism (56, 57). Prolactinomas represent the most common clinically presenting pituitary tumors, followed by non-functional, somatotroph pituitary tumors, and, rarely, corticotroph or thyrotroph adenomas (47) (Figure 1). Interestingly, while no reproducible variant has been identified in MEN1, two large kindreds followed over 30 years demonstrated a frequent prolactinoma and less frequent gastrinoma than typical MEN1 that was reproducible among kindreds and could not be explained by genetic mutation (58). Early and frequent (1–3 years) magnetic resonance imaging (MRI) screening identifies non-functional adenomas that may remain indolent (59).
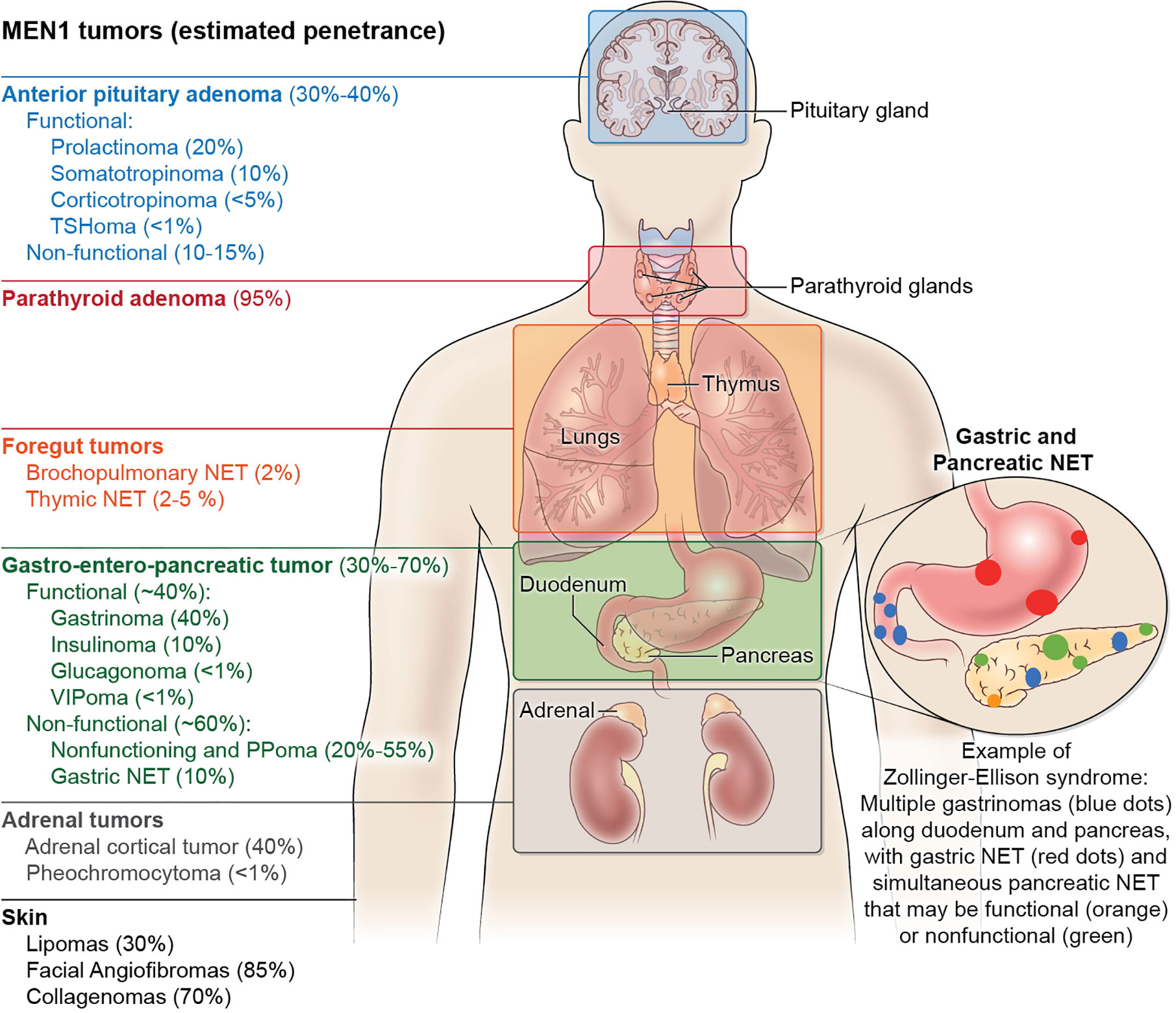
Figure 1 The clinical manifestations and estimated tumor penetrance of multiple endocrine neoplasia type 1 (MEN1) syndrome. The main manifestations of MEN1 include anterior pituitary adenomas, pancreatic neuroendocrine tumors, and primary hyperparathyroidism. Except for multi-gland hyperparathyroidism which approximates 95% penetrance by age 50, the manifestations of other tumors in MEN1 have no genotype-phenotype correlation and cannot be predicted, even within kindreds. Neuroendocrine tumor (NET) hormone secretion in the anterior pituitary and pancreas can be considered functional or non-functional, while duodenal NET and parathyroid adenomas are always functional with the secretion of gastrin and parathyroid hormone, respectively. As pictured, Zollinger-Ellison Syndrome can present with multiple submucosal duodenal gastrinomas with simultaneous pancreatic gastrinomas, non-functional pancreatic tumors, as well as the development of gastric NET typically within thickened gastric folds (not pictured). Less frequent manifestations of MEN1 include foregut bronchopulmonary and thymic neuroendocrine tumors, as well as adrenal cortical adenomas and pheochromocytomas. Skin manifestations of MEN1 include lipomas that can occur anywhere on the body, collagenomas mostly on the trunk, and facial angiofibromas on the nasal bridge and cheeks. Manifestations that have been described but are not pictured include meningiomas, leiomyoma (uterus, esophagus, ureter, bladder), café-au-lait spots, and malignant breast tumors.
The third hallmark feature of MEN1 includes gastroenteropancreatic neuroendocrine tumors (GEP-NET). Up to 80% of MEN1 patients have radiological evidence of GEP-NETs. Other complex hereditary syndromes also manifest particularly with pNETs, including >15% of patients von Hippel–Lindau disease (VHL), <10% of patients with neurofibromatosis 1 (NF1) and <1% of patients with tuberous sclerosis (60). The most common functional pancreatic NET in MEN1 is a gastrinoma, which can also occur in the duodenal mucosa as small multifocal tumors and leads to the Zollinger-Ellison Syndrome (ZES) (61) (Figure 1). About 40–60% of pNETs will secrete polypeptide hormones such as gastrin, insulin, glucagon or vasoactive intestinal peptide (VIP), while the others are non-functional. Hormonal excess induces clinical symptomatology that may occur simultaneously. The spectrum of symptoms includes multiple severe ulcers of the stomach and duodenum (ZES), hypoglycemia with seizures (insulinoma), glucose intolerance and watery diarrhea with hypokalemic alkalosis (WDHA) syndrome (VIPoma), or mass effect symptoms (62). The introduction of proton pump inhibitors in the late 1980s significantly shifted the mortality curve from gastrinoma-induced gastric ulcer perforation to non-functional pancreatic neuroendocrine tumors with unpredictable malignant potential. It is now recognized that thymic NETs, a rare but aggressive tumor, carry the highest odds ratio of death among MEN1 patients (63). Nevertheless, given the rarity of these thymic tumors, pancreatic NET are the main focus of therapeutic interventions to improve MEN1 mortality (64–66).
Several guidelines to assist the clinical practitioner in the management of patients with MEN1 have been published (47, 67). These aim to establish guiding principles for the evaluation, treatment, and genetic testing of patients with MEN1 and their family members.
Multiple Endocrine Neoplasia Type 2A (MEN2A)
MEN2A is an inherited endocrine cancer syndrome that includes HPT as one of its less penetrant and clinically less consequential features. Besides primary HPT, MEN2A is characterized by the predisposition to develop medullary thyroid cancer (MTC), a neoplasm of thyroid parafollicular C cells, and pheochromocytoma which, in the context of MEN2A, is typically benign and often bilateral (68). Occasionally patient with MEN2A develop cutaneous lichen amyloidosis. In the clinical setting of MEN2A, HPT resembles sporadic HPT and is usually mild and due to benign parathyroid disease. MEN2A is inherited in an autosomal dominant fashion and results from germline activating missense mutation in the RET proto-oncogene on the long arm of chromosome 10. The RET proto-oncogene encodes a receptor tyrosine kinase that binds, together with co-receptor glycosylphosphatidylinositol-anchored protein Gfra1 (69), members of the glial cell line-derived neurotrophic factor (GDNF) family of extracellular signaling molecules.
Germline gain-of-function mutations of the RET proto-oncogene are associated with three separate heritable endocrine neoplasia syndromes, all of which are associated with MTC: MEN2A also known as Sipple syndrome, multiple endocrine neoplasia type 2B (MEN2B) also known as the mucosal neuroma syndrome, and familial medullary thyroid cancer (FMTC). Usually parathyroid tumors and HPT are not part of the disease spectrum of MEN2B or FMTC. Distinct patterns of disease result from particular RET mutations giving rise to apparent genotype-phenotype correlations. Approximately 95% of MEN2A cases result from the presence in the germline of nonsynonymous codon substitutions affecting the cysteine-rich region within the RET receptor’s large extracellular domain, namely missense mutations of RET codons 609, 611, 618, 620, or 634 (70). Indeed, approximately 85% of cases of MEN2A are caused by germline missense substitution of RET residue cysteine-634 (68, 71). Somatic mutations in the RET proto-oncogene associated with MEN2 rarely cause sporadic parathyroid tumors (72, 73).
Based on the consensus view among an international group of endocrinologists and other expert practitioners, guidelines for the clinical management of patients with MEN2 have been published (67). The purpose of these guidelines is to establish principles for the evaluation, indications and timing of surgical intervention, and interpretation of the genotype-phenotype correlates of patients with MEN2 and others in their kindreds.
Multiple Endocrine Neoplasia Type 4 (MEN4)
Pellegata and coworkers first described MEN4 in a multi-generational kindred with manifestations overlapping those of MEN1 but whose affected members lacked germline MEN1 mutation (74, 75). A germline heterozygous stop-gain mutation in the cyclin dependent-kinase inhibitor p27(Kip1), encoded by CDKN1B, was identified in the proband with acromegaly and HPT and in several other members of this kindred (74). Genetic analysis of rats with a multi-tumor syndrome phenotype called multiple endocrine neoplasia X (MENX) had previously drawn attention to the Cdkn1b locus (74, 76). The rat MENX phenotype was discovered accidently when some rats in a Sprague–Dawley colony were observed to spontaneously develop multiple endocrine tumors (77). The MENX phenotype in rats was manifested by parathyroid hyperplasia, multifocal anterior pituitary adenoma, adrenal and extra-adrenal pheochromocytoma, thyroid C-cell hyperplasia, and pancreatic islet cells hyperplasia. MENX in rats was recessively inherited and caused by a frameshift mutation in Cdkn1b (74, 76). In the study by Pellegata et al., only the proband among members of the MEN4/MENX kindred described was reported to have HPT (74).
Prior to 2019, reports of kindreds demonstrating segregation of a mutation in CDKN1B with an MEN4 phenotype across multiple generations were quite limited. Several groups investigated for a possible role for CDKN1B mutation in parathyroid neoplasia following the original report by Pellegata et al. (74). Several studies examined MEN1 mutation-negative patients and kindreds expressing MEN1-like tumors and harboring germline mutation in CDKN1B and which thus could be considered as MEN4 (74, 78–85). Apart from the verification of HPT in association with CDKN1B mutation in a pair of monozygotic twins (79), none of these reports prior to 2019 had described kindreds with more than one individual with HPT proven to segregate with the CDKN1B mutation. In 2019 however, a report by Frederiksen et al. described a large multi-generational Danish MEN4 family in which HPT occurred in 13 members and segregated with a germline frameshift CDKN1B mutation (86). Molecular genetic analysis of sporadic parathyroid adenomas demonstrating that CDKN1B mutation can be both somatic and clonal further supports the characterization of CDKN1B as a gene predisposing to the development of primary parathyroid tumors (87, 88). A recent study suggests that germline CDKN1B mutation can also present as apparently sporadic, isolated pediatric Cushing’s disease (89).
The Hyperparathyroidism-Jaw Tumor Syndrome (HPT-JT)
HPT-JT was first recognized as a form of familial HPT that was genetically distinct from MEN1 and MEN2A in 1990 by Gene Jackson and co-workers (90). Central to this recognition was re-evaluation of a family, first described three decades earlier (91), in which occurrence of multiple ossifying fibromas of the maxilla and mandible in two affected members of the third generation was seen to be similar to the jaw tumors of four of five affected members of the first generation. As noted originally by Jackson and co-workers (90), the maxillary and mandibular tumors seen in HPT-JT are distinct from the “brown tumors” of HPT (often seen in the context of osteitis fibrosa cystica and metabolically severe HPT), because the former can appear and/or enlarge in affected members in the absence, or following surgical correction of, HPT. Furthermore, whereas the “brown tumors” of HPT can occur anywhere in the axial or appendicular skeleton, the jaw tumors found in HPT-JT kindreds are restricted to the maxilla and mandible. The jaw tumors in HPT-JT are histologically distinct fibro-osseous lesions without the abundant multinucleated giant cells seen in “brown tumors,” and have been formally classified as cemento-ossifying fibromas (92). The exact cell of origin from which cemento-ossifying fibromas derive in HPT-JT is not precisely known, but they are generally believed to be mesodermal odontogenic tumors derived from the mesenchymal blast cells of the periodontal ligament, with the potential to form fibrous tissue, cementum, and bone or a combination thereof (93, 94).
HPT-JT is an autosomal dominant inherited syndrome with variable penetrance and expressivity. HPT is the most penetrant feature of HPT-JT and is the manifestation that most often brings carriers to medical attention. Besides HPT, the key clinical features of HPT-JT include cemento-ossifying fibromas restricted to the maxilla and mandible (as described above), renal lesions, and uterine tumors in women (90, 95–97). In contrast to sporadic HPT and MEN1, parathyroid cancer is relatively frequent in the context of HPT-JT, and affects ~20% of those with HPT (90, 95, 96, 98, 99).
In the preponderance of HPT-JT kindreds, a germline inactivating mutation of the CDC73 gene (formerly called HRPT2) on the long arm of chromosome 1 can be identified (19, 100) The CDC73 gene encodes parafibromin, a protein of 531 residues, that is considered to be a tumor suppressor protein because germline mutation predicted to cause loss-of-function predisposes to the neoplastic expressions of HPT-JT (100). The majority of germline CDC73 mutations in kindreds with HPT-JT are predicted to inactivate gene function via frameshift or nonsense mutation, with only a minority of the variants encoding missense mutations (99, 101). Partial or complete deletion of the CDC73 gene has also been described in patients and kindreds with HPT-JT (102–105).
The high frequency of parathyroid cancer is a hallmark of HPT-JT. A genotype-phenotype correlation has been observed among CDC73-mutation carriers such that those with frameshift, nonsense or deletion mutations are nearly 7-fold more likely to develop parathyroid cancer than patients harboring missense CDC73 mutations (99). In apparently sporadic cases of parathyroid cancer, mutations of CDC73 are frequently identified (106–110). Interestingly, germline loss-of-function mutation in CDC73 may be found in some 25% of patients with seemingly sporadic parathyroid carcinoma, suggesting that such patients may have either de novo germline mutation in CDC73 or else a forme fruste of HPT-JT (19, 107, 108).
A potential genotype-phenotype correlation has also been observed with respect to certain renal manifestations of HPT-JT. Mixed epithelial and stromal tumor of the kidney (MEST), a rare type of renal tumor, has been associated with HPT-JT and/or CDC73 germline mutation in at least two kindreds (111, 112). MEST is a renal neoplasm, characterized by cystic structures lined by epithelium and admixed with ovarian-type stroma, that must be diagnosed differentially from cystic hamartoma of the renal pelvis, adult type mesoblastic nephroma, and leiomyomatous renal hamartoma (113, 114). MEST in the context of HPT-JT and/or CDC73 germline mutation appears to correlate with a specific CDC73 missense mutation in which the initiator methionine of parafibromin is replaced with isoleucine, i.e. the Met1Ile genotype (100, 111, 112). Wilms tumor has been identified in members of several families with HPT-JT (115, 116). Renal cysts are also reported to be part of the clinical spectrum of HPT-JT and CDC73 germline mutation (19). No genotype-phenotype correlation has been reported for Wilms tumor and renal cysts in the context of CDC73 germline mutation.
The presence of uterine manifestations of HPT-JT was first elucidated by Bradley et al. who recognized a high frequency of menorrhagia often leading to early hysterectomy among affected women and adult female carriers (95). There was a range of uterine pathology among women with HPT-JT who underwent hysterectomy that included adenosarcomas, adenofibromas, leiomyomas, adenomyosis, and endometrial hyperplasia (95). The uterine manifestations of HPT-JT significantly reduced the reproductive fitness of affected women. Lifelong monitoring for uterine tumors with routine gynecologic care and pelvic ultrasound examination as clinically indicated has been recommended for women with HPT-JT, starting at reproductive age (19).
A subset of kindreds classified as FIHP have been shown to harbor germline CDC73 mutation, suggesting that incompletely penetrant HPT-JT can phenocopy FIHP (see below and Figure 2). Approximately 20% of obligate or genetically confirmed CDC73 mutation-positive subjects lack any clinical manifestations of HPT-JT at the time of kindred ascertainment (99), in line with the variable penetrance and expressivity of CDC73 mutation. Lifelong surveillance of initially asymptomatic CDC73 mutation carriers is recommended since the penetrance of the manifestations of HPT-JT increases with age (117).
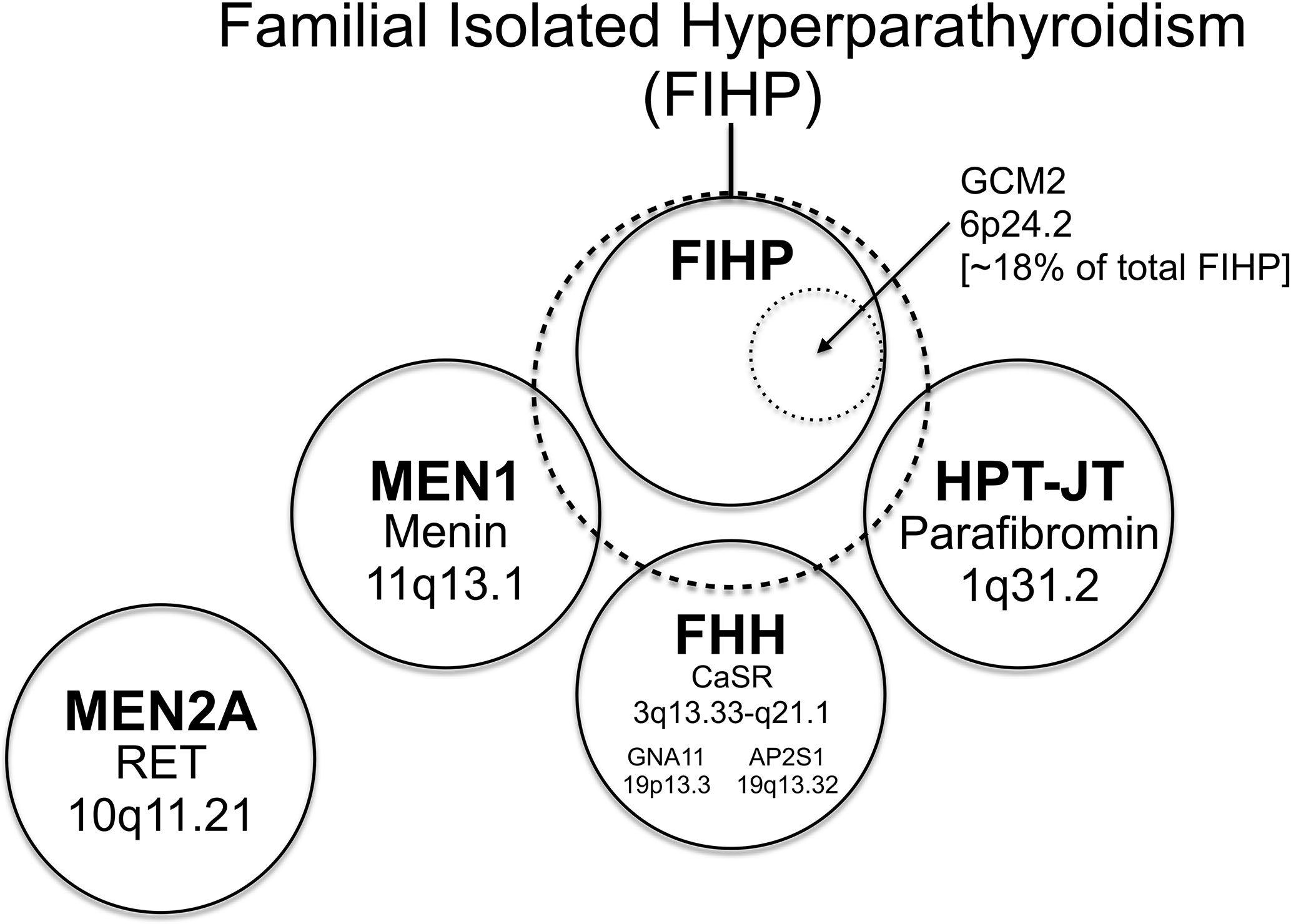
Figure 2 The relationship among familial forms of hyperparathyroidism that may present as familial isolated hyperparathyroidism (FIHP) as a Venn diagram. The large dashed circle represents the set of patients that can present with a provisional diagnosis of FIHP at the time of initial ascertainment. This includes patients with FIHP who have been evaluated for, but lack findings diagnostic of, MEN1, FHH, and HPT-JT (nonsyndromic FIHP; in a solid circle). Approximately 18% of nonsyndromic FIHP kindreds harbor germline gain-of-function mutations in GCM2 (inner dotted circle) (see text), whereas the remainder have currently unknown genetic etiologies. Subsets of patients with incomplete expression of MEN1, FHH and HPT-JT (the total set of patients in each syndrome represented by a solid circle) can also present with the FIHP phenotype (and thus overlap with the large dashed circle). The distinction between the nonsyndromic FIHP category and the syndromic categories arbitrarily depends on the depth and rigor of evaluation and the sensitivity of diagnostic tests used to detect the syndrome, testing that can include germline gene mutational analysis. MEN2A is a familial form of hyperparathyroidism that seldom if ever presents as FIHP, with patients usually coming to medical attention for signs and symptoms of medullary thyroid cancer and/or pheochromocytoma. Within each circle representing a defined syndrome are included the genetic locus (or loci in the case of FHH; see text) of the syndromic trait and the associated gene product. The causative gene for HPT-JT that encodes parafibromin is CDC73, formerly called HRPT2. The relationship among the patient sets illustrated as circles in this diagram is intended to be qualitative and neither the area of each circle nor the area of overlap between circles has any quantitative significance.
Familial Isolated Hyperparathyroidism (FIHP)
As is evident from its appellation, FIHP defines a category of familial HPT that encompasses kindreds containing two or more members with HPT but that lack other, specific features of a familial HPT syndrome: MEN1, MEN2A, HPT-JT or FHH (Figure 2) (118). FIHP is a diagnosis of exclusion with the strength of the diagnosis proportional to the depth and rigor of clinical and genetic evaluation. Following the initial ascertainment and clinical evaluation of possible FIHP kindreds, genetic testing may reveal that germline mutation of MEN1, CDC73, or CASR accounts for a fraction of the FIHP kindreds under investigation (20, 119–122). Nevertheless, following careful clinical and genetic analysis, the majority of FIHP kindreds have been shown to lack germline mutation in the established HPT-susceptibility genes (Figure 2) (20, 119, 123). Because sporadic HPT is not uncommon (1), some small kindreds with a tentative diagnosis of FIHP may in fact reflect the coincidental clustering of sporadic cases among close relatives.
Genetic variants that result in missense mutations in GCM2, which is a nuclear transcription factor required for parathyroid gland organogenesis named for its homology to the “glial cells missing” (gcm) gene in Drosophila, were recently discovered in the germline DNA of eight different families with FIHP (21). This gene had already been implicated in parathyroid gland dysfunction, since it had been previously shown that germline loss-of-function and dominant-negative mutations of GCM2 were associated, respectively, with autosomal recessive and autosomal dominant familial isolated hypoparathyroidism (124, 125). Testing in vitro has shown that the two germline variants in GCM2 associated with FIHP act as gain-of-function mutations (21). The FIHP-associated missense mutations in GCM2 map to its C-terminal conserved inhibitory domain (CCID) and increase its transcriptional activity, presumably through a mechanism of dis-inhibition, suggesting that in the context of FIHP GCM2 is a parathyroid proto-oncogene. Specific variants affecting the CCID of GCM2 are enriched among FIHP kindreds and sporadic HPT patients with an Ashkenazi Jewish background (126). Genetic screening of multiple FIHP kindreds suggests that approximately 18% harbor germline activating GCM2 mutations (21), leaving ~80% of carefully curated FIHP families without a currently-defined genetic basis (118). Clinical investigators in other centers have also documented rare germline GCM2 variants that map to the CCID domain and segregate with the disease in a subcategory of FIHP kindreds (127). Activating germline variants in GCM2 that map to the CCID domain have also been found in low frequency among patients with typically-presenting sporadic HPT who underwent parathyroid tumor excision and appear to be of low penetrance (128).
Familial Hypocalciuric Hypercalcemia (FHH)
FHH describes a condition of PTH-dependent hypercalcemia, resembling and in the differential diagnosis of HPT, that is typically benign (Table 1) (22, 129). The condition, also known as “familial benign hypercalcemia”, is genetically heterogeneous and results from mutations that cause parathyroid gland insensitivity to extracellular calcium with a resulting rightward shift of the set point for suppression by calcium of PTH secretion. As a result of this intrinsic insensitivity of the parathyroid cells to suppression by circulating calcium, affected patients from FHH kindreds almost always remain hypercalcemic following partial or subtotal parathyroidectomy. FHH is inherited in an autosomal dominant manner and usually results in mild hypercalcemia, non-suppressed or mildly elevated PTH, and relative hypocalciuria. The hypercalcemia observed in FHH is highly penetrant across the age spectrum, including in neonates and infants (22, 130). The majority of cases of FHH results from heterozygous germline loss-of-function mutation of the calcium-sensing receptor, encoded by the CASR gene on the long arm of chromosome 3 (4, 131). FHH due to inactivating mutation in the CASR gene is classified as type 1 FHH (FHH1) and is the most common type (132). Neonatal severe primary hyperparathyroidism (NSHPT) that presents as severe hypercalcemia, typically occurring in the first 6 months of life, is a rare autosomal recessive disorder that most often is a consequence of the compound heterozygous or homozygous inheritance of two mutationally-inactivated CASR alleles (133). While true parathyroid tumors demonstrate cellular monoclonality, the hyperfunctioning parathyroid tissue removed from a patient with NSHPT demonstrated generalized polyclonal hyperplasia by molecular genetic analysis, illustrating the non-neoplastic quality of the abnormal parathyroids that result from CASR loss-of-function mutation (134).
Reduced cell surface expression of the CASR protein has been demonstrated in parathyroid adenomas and may explain the rightward shift of the calcium set point and the impaired calcium-mediated suppression of parathyroid hormone release typical of such adenomas. Decreased CASR transcript expression, but not loss-of-heterozygosity at the CASR chromosomal locus, has been demonstrated in benign parathyroid tumors (135). In molecular genetic studies of sporadic parathyroid tumors reported to date, somatic inactivation of the CASR gene has not been described (136, 137).
A minority of families with FHH result from germline mutations in genes other than CASR on chromosome 3q. Type 2 FHH (FHH2) results from inactivating mutation in the germline of the G protein α11 subunit encoded by GNA11 (138, 139). GNA11 encodes the α-subunit of the signal-transducing G protein downstream of the CASR (140). The CASR allosteric activator cinacalcet has been reported to correct the hypercalcemia of patients with FHH2 due to GNA11 mutation (141). Type 3 FHH (FHH3) results from germline loss-of-function mutation in adaptor related protein complex 2 subunit sigma 1 (AP2S1), an adaptor protein involved in clathrin-mediated endocytosis and encoded by AP2S1 on chromosome 19q (142–145). In FHH3 the vast majority of inactivating mutations in AP2S1 involve missense substitutions of codon Arg-15, but analysis of large scale exome datasets suggests that other residues may be more rarely involved (146). Mutant forms of AP2S1 involving codon Arg-15 that result in FHH3 may exert dominant-negative effects on CASR signaling (144). In molecular genetic analyses of sporadic parathyroid tumors, somatic loss-of-function mutation in the genes encoding GNA11 and AP2S1 have thus far not been described.
Differential Diagnosis of Familial Forms of Hyperparathyroidism
When a familial form HPT is suspected based on patient or family medical history, and/or one or more suspicious non-parathyroid features, certain key clinical or laboratory findings can help in the differential diagnosis. A history of pituitary adenomas, pNETs, bronchial carcinoid, or duodenal endocrine tumors in the patient or first-degree relatives favor MEN1. Suspicion of MEN2A is heightened if a history of MTC or pheochromocytoma is documented in the patient or a family member. History of parathyroid cancer, maxillary or mandibular tumors, Wilms tumor, or uterine abnormalities requiring early hysterectomy in the patient or another member of the kindred, should elevate the possibility of HPT-JT. Persistent hypercalcemia following parathyroidectomy, hypocalciuria, hypermagnesemia, and hypercalcemia in family members younger than 10 years of age could be clues to the diagnosis of FHH. Genetic testing, conveniently performed with a gene panel that includes MEN1, CDC73, CASR, GNA11, AP2S1, CDKN1B, and GCM2, can help clarify the diagnosis and establish the genetic etiology. Recommendations for the testing of younger patients with seemingly sporadic HPT are described below.
Discussion
Even though inherited forms of HPT represent only a small proportion of total cases (< 5%), investigation into the molecular basis of these rare familial syndromes has resulted in considerable insight into the genetics and pathophysiology of both sporadic and familial HPT and spotlighted the importance of genes such as MEN1, CDC73, CASR, GNA11, AP2S1, CDKN1B, and GCM2. It seems very likely that gain- or loss-of-function mutation of other genes, currently unrecognized, can also promote parathyroid tumor formation. As an illustration of this, the risk predisposing to the development of parathyroid tumors in most FIHP kindreds appears to result from the germline mutation of genes currently not recognized to play a role in parathyroid neoplasia. This inference follows from the fact that nearly 70% of families initially characterized with an FIHP phenotype in several clinical studies that screened for germline MEN1, CASR, and CDC73/HRPT2 gene mutation, were found to have no currently recognized syndromic or genetic basis (Figure 2) (20, 119–121). From the FIHP families which are MEN1, CASR, and CDC73/HRPT2 mutation-negative, estimates are that approximately 20% carry germline gain-of-function mutations in the GCM2 proto-oncogene (21). The clear implication is that some 80% of FIHP families have no currently defined genetic etiology for their predisposition to HPT.
The analysis of DNA extracted from parathyroid tumors using methods such as comparative genomic hybridization (CGH) to highlight particular chromosomal regions that have lost or gained segments of DNA also implies the presence of currently unidentified parathyroid tumor suppressors and oncogenes. Several research groups have demonstrated recurrent DNA loss at chromosomal locations 1p, 6q, 9p, and 13q in parathyroid tumors, indicating the possible presence there of currently unrecognized parathyroid tumor suppressor genes (147–150). On the other hand, the demonstration of specific chromosomal gain at chromosomal loci 9q, 16p, 19p, and Xq in benign or malignant parathyroid tumors suggests the possible presence at these loci of novel parathyroid oncogenes (147, 149–151).
What clinical, demographic, or historical considerations should persuade the practitioner to screen individual patients for a genetic basis for their HPT, even if they lack obvious features of a syndrome? As was discussed above in the case of tumor suppressor genes, germline mutation predisposes to earlier onset of disease so very often it is the younger cohort of seemingly sporadic patients with HPT who are considered for such genetic screening. Skandarajah and co-workers performed a retrospective genetic analysis of 21 patients who had undergone surgery for HPT before the age of 40. Although none had suspicious personal or family histories suggestive of MEN1, one patient, who had a double parathyroid adenoma at surgery, was found to have a germline MEN1 frameshift mutation (152). In another study from Carling and colleagues, 86 patients ≤ 45 years of age with seemingly sporadic HPT underwent genetic analysis for genes predisposing to HPT. Eight of the 86 patients (~ 9%) were found to harbor germline loss-of-function mutations in known HPT susceptibility genes: four MEN1, three CASR, and one CDC73 (153). An expert panel of physicians, surgeons, and geneticists assembled to provide clinical practice guidelines for MEN1 suggested that gene mutational testing be performed on patients presenting with seemingly sporadic HPT younger than 40 years of age due to multi-gland parathyroid disease (47). A recent retrospective study of 121 patients screened for familial forms of HPT found that with respect to sole risk factors, a positive family history, but not age at diagnosis or the finding of multiglandular parathyroid disease, was strongly predictive of a pathogenic germline variant in a HPT susceptibility gene (132).
Author Contributions
JB and WS conceived, planned, and executed the writing of this chapter. All authors contributed to the article and approved the submitted version.
Funding
The Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (ZIA DK043012-18) supported this research.
Conflict of Interest
JB is employed by the company AstraZeneca.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the authors JB and WS.
Acknowledgments
The authors wish to thank past and present members of the Metabolic Diseases Branch, NIDDK for many informative and inspirational discussions and suggestions.
References
1. Bilezikian JP. Primary Hyperparathyroidism. J Clin Endocrinol Metab (2018) 103(11):3993–4004. doi: 10.1210/jc.2018-01225
2. Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets (2007) 8(1):169–84. doi: 10.2174/138945007779315614
3. Carafoli E, Krebs J. Why Calcium? How Calcium Became the Best Communicator. J Biol Chem (2016) 291(40):20849–57. doi: 10.1074/jbc.R116.735894
4. Brown EM. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract Res Clin Endocrinol Metab (2013) 27(3):333–43. doi: 10.1016/j.beem.2013.02.006
5. Zhang C, Miller CL, Gorkhali R, Zou J, Huang K, Brown EM, et al. Molecular Basis of the Extracellular Ligands Mediated Signaling by the Calcium Sensing Receptor. Front Physiol (2016) 7:441:441. doi: 10.3389/fphys.2016.00441
6. Silva BC, Bilezikian JP. Parathyroid hormone: anabolic and catabolic actions on the skeleton. Curr Opin Pharmacol (2015) 22:41–50. doi: 10.1016/j.coph.2015.03.005
7. Cantley LK, Russell J, Lettieri D, Sherwood LM. 1,25-Dihydroxyvitamin D3 suppresses parathyroid hormone secretion from bovine parathyroid cells in tissue culture. Endocrinology (1985) 117(5):2114–9. doi: 10.1210/endo-117-5-2114
8. Russell J, Lettieri D, Sherwood LM. Suppression by 1,25(OH)2D3 of transcription of the pre-proparathyroid hormone gene. Endocrinology (1986) 119(6):2864–6. doi: 10.1210/endo-119-6-2864
9. Silver J, Naveh-Many T, Mayer H, Schmelzer HJ, Popovtzer MM. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J Clin Invest (1986) 78(5):1296–301. doi: 10.1172/JCI112714
10. Silver J, Russell J, Sherwood LM. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci USA (1985) 82(12):4270–3. doi: 10.1073/pnas.82.12.4270
11. Weber TJ, Quarles LD. Molecular Control of Phosphorus Homeostasis and Precision Treatment of Hypophosphatemic Disorders. Curr Mol Biol Rep (2019) 5(2):75–85. doi: 10.1007/s40610-019-0118-1
12. Jacquillet G, Unwin RJ. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflugers Arch (2019) 471(1):83–98. doi: 10.1007/s00424-018-2231-z
13. Farrow EG, White KE. Recent advances in renal phosphate handling. Nat Rev Nephrol (2010) 6(4):207–17. doi: 10.1038/nrneph.2010.17
14. Silva BC, Bilezikian JP. Skeletal abnormalities in Hypoparathyroidism and in Primary Hyperparathyroidism. Rev Endocr Metab Disord (2020). doi: 10.1007/s11154-020-09614-0
15. Sharretts JM, Kebebew E, Simonds WF. Parathyroid cancer. Semin Oncol (2010) 37(6):580–90. doi: 10.1053/j.seminoncol.2010.10.013
16. Byrd C, Kashyap S, Kwartowitz G. Parathyroid Cancer. In: StatPearls. Treasure Island, FL (2020).
17. Insogna KL. Primary Hyperparathyroidism. N Engl J Med (2018) 379(11):1050–9. doi: 10.1056/NEJMcp1714213
18. Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat Rev Cancer (2005) 5(5):367–75. doi: 10.1038/nrc1610
19. Hyde SM, Rich TA, Waguespack SG, Perrier ND, Hu MI. CDC73-Related Disorders. University of Washington, Seattle: Seattle (WA (1993-2019).
20. Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, et al. Familial isolated hyperparathyroidism: Clinical and genetic characteristics of thirty-six kindreds. Med (Baltimore) (2002) 81:1–26. doi: 10.1097/00005792-200201000-00001
21. Guan B, Welch JM, Sapp JC, Ling H, Li Y, Johnston JJ, et al. GCM2-Activating Mutations in Familial Isolated Hyperparathyroidism. Am J Hum Genet (2016) 99(5):1034–44. doi: 10.1016/j.ajhg.2016.08.018
22. Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs RW Jr., Lasker RD. The hypocalciuric or benign variant of familial hypercalcemia: clinical and biochemical features in fifteen kindreds. Med (Baltimore) (1981) 60(6):397–412. doi: 10.1097/00005792-198111000-00002
23. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U.S.A. (1971) 68(4):820–3. doi: 10.1073/pnas.68.4.820
24. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature (1986) 323(6089):643–6. doi: 10.1038/323643a0
25. Lu KH, Wood ME, Daniels M, Burke C, Ford J, Kauff ND, et al. American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers. J Clin Oncol (2014) 32(8):833–40. doi: 10.1200/JCO.2013.50.9257
26. Harris TJ, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol (2010) 7(5):251–65. doi: 10.1038/nrclinonc.2010.41
27. Cushing H, Davidoff LM. Studies in acromegaly V. The pathological finding in four autopsied cases of acromegaly with a discussion of their significance (Monographs of the Rockefeller Institute for Medical Research). New York, NY: The Rockefeller UniversityPress (1927), p. 131.
28. Underdahl LO, Woolner LB, Black BM. Multiple endocrine adenomas; report of 8 cases in which the parathyroids, pituitary and pancreatic islets were involved. J Clin Endocrinol Metab (1953) 13(1):20–47. doi: 10.1210/jcem-13-1-20
29. Moldawer MP, Nardi GL, Raker JW. Concomitance of multiple adenomas of the parathyroids and pancreatic islets with tumor of the pituitary: a syndrome with a familial incidence. Am J Med Sci (1954) 228(2):190–206. doi: 10.1097/00000441-195408000-00008
30. Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med (1954) 16(3):363–71. doi: 10.1016/0002-9343(54)90353-8
31. Zollinger RM, Ellison EH. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann Surg (1955) 142(4):709–23. discussion, 24-8.
32. Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature (1988) 332(6159):85–7. doi: 10.1038/332085a0
33. Bale AE, Norton JA, Wong EL, Fryburg JS, Maton PN, Oldfield EH, et al. Allelic loss on chromosome 11 in hereditary and sporadic tumors related to familial multiple endocrine neoplasia type 1. Cancer Res (1991) 51(4):1154–7.
34. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science (1997) 276(5311):404–7. doi: 10.1126/science.276.5311.404
35. Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA (1998) 95(4):1630–4. doi: 10.1073/pnas.95.4.1630
36. Agarwal SK. The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer (2017) 24(10):T119–T34. doi: 10.1530/ERC-17-0199
37. Bergman L, Boothroyd C, Palmer J, Grimmond S, Walters M, Teh B, et al. Identification of somatic mutations of the MEN1 gene in sporadic endocrine tumours. Br J Cancer (2000) 83(8):1003–8. doi: 10.1054/bjoc.2000.1385
38. Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature (2017) 543(7643):65–71. doi: 10.1038/nature21063
39. Yang MA, Lee WK, Shin HS, Park SH, Kim BS, Kim JW, et al. Neuroendocrine Tumors in the Stomach, Duodenum, and Pancreas Accompanied by Novel MEN1 Gene Mutation. Korean J Gastroenterol (2017) 69(3):181–6. doi: 10.4166/kjg.2017.69.3.181
40. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science (2011) 331(6021):1199–203. doi: 10.1126/science.1200609
41. Raj N, Shah R, Stadler Z, Mukherjee S, Chou J, Untch B, et al. Real-Time Genomic Characterization of Metastatic Pancreatic Neuroendocrine Tumors Has Prognostic Implications and Identifies Potential Germline Actionability. JCO Precis Oncol (2018) 2018:1–18. doi: 10.1200/po.17.00267
42. Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol (2010) 28(2):245–55. doi: 10.1200/jco.2008.21.5988
43. Chan CS, Laddha SV, Lewis PW, Koletsky MS, Robzyk K, Da Silva E, et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun (2018) 9(1):4158. doi: 10.1038/s41467-018-06498-2
44. Lee ME, Tepede AA, Mandl A, Weinstein LS, Del Rivero J, Agarwal SK, et al. c-MET inhibition: novel treatment for sporadic and MEN1-associated GEP NETs. J Mol Endocrinol (2020) 65(2):R1–r17. doi: 10.1530/jme-20-0020
45. Newey PJ, Jeyabalan J, Walls GV, Christie PT, Gleeson FV, Gould S, et al. Asymptomatic children with multiple endocrine neoplasia type 1 mutations may harbor nonfunctioning pancreatic neuroendocrine tumors. J Clin Endocrinol Metab (2009) 94(10):3640–6. doi: 10.1210/jc.2009-0564
46. Vannucci L, Marini F, Giusti F, Ciuffi S, Tonelli F, Brandi ML. MEN1 in children and adolescents: Data from patients of a regional referral center for hereditary endocrine tumors. Endocrine (2018) 59(2):438–48. doi: 10.1007/s12020-017-1322-5
47. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97(9):2990–3011. doi: 10.1210/jc.2012-1230
48. Lourenço DM Jr., Coutinho FL, Toledo RA, Montenegro FL, Correia-Deur JE, Toledo SP. Early-onset, progressive, frequent, extensive, and severe bone mineral and renal complications in multiple endocrine neoplasia type 1-associated primary hyperparathyroidism. J Bone Miner Res (2010) 25(11):2382–91. doi: 10.1002/jbmr.125
49. Marx SJ, Menczel J, Campbell G, Aurbach GD, Spiegel AM, Norton JA. Heterogeneous size of the parathyroid glands in familial multiple endocrine neoplasia type 1. Clin Endocrinol (1991) 35(6):521–6. doi: 10.1111/j.1365-2265.1991.tb00938.x
50. Sulaiman L, Juhlin CC, Nilsson IL, Fotouhi O, Larsson C, Hashemi J. Global and gene-specific promoter methylation analysis in primary hyperparathyroidism. Epigenetics (2013) 8(6):646–55. doi: 10.4161/epi.24823
51. Guarnieri V, Muscarella LA, Verdelli C, Corbetta S. Alterations of DNA methylation in parathyroid tumors. Mol Cell Endocrinol (2018) 469:60–9. doi: 10.1016/j.mce.2017.05.010
52. Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer (2006) 94(6):776–80. doi: 10.1038/sj.bjc.6603023
53. Ghafouri-Fard S, Shoorei H, Anamag FT, Taheri M. The Role of Non-Coding RNAs in Controlling Cell Cycle Related Proteins in Cancer Cells. Front Oncol (2020) 10:608975:608975. doi: 10.3389/fonc.2020.608975
54. Luzi E, Ciuffi S, Marini F, Mavilia C, Galli G, Brandi ML. Analysis of differentially expressed microRNAs in MEN1 parathyroid adenomas. Am J Transl Res (2017) 9(4):1743–53.
55. Barry S, Korbonits M. Update on the Genetics of Pituitary Tumors. Endocrinol Metab Clin North Am (2020) 49(3):433–52. doi: 10.1016/j.ecl.2020.05.005
56. O’Brien T, O’Riordan DS, Gharib H, Scheithauer BW, Ebersold MJ, van Heerden JA. Results of treatment of pituitary disease in multiple endocrine neoplasia, type I. Neurosurgery (1996) 39(2):273–8. doi: 10.1097/00006123-199608000-00008 discussion 8-9.
57. Syro LV, Scheithauer BW, Kovacs K, Toledo RA, Londoño FJ, Ortiz LD, et al. Pituitary tumors in patients with MEN1 syndrome. Clinics (Sao Paulo Brazil) (2012) 67 Suppl 1(Suppl 1):43–8. doi: 10.6061/clinics/2012(sup01)09
58. Hao W, Skarulis MC, Simonds WF, Weinstein LS, Agarwal SK, Mateo C, et al. Multiple endocrine neoplasia type 1 variant with frequent prolactinoma and rare gastrinoma. J Clin Endocrinol Metab (2004) 89(8):3776–84. doi: 10.1210/jc.2003-031511
59. de Laat JM, Dekkers OM, Pieterman CR, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab (2015) 100(9):3288–96. doi: 10.1210/jc.2015-2015
60. Anlauf M, Garbrecht N, Bauersfeld J, Schmitt A, Henopp T, Komminoth P, et al. Hereditary neuroendocrine tumors of the gastroenteropancreatic system. Virchows Arch (2007) 451(Suppl 1):S29–38. doi: 10.1007/s00428-007-0450-3
61. Pipeleers-Marichal M, Somers G, Willems G, Foulis A, Imrie C, Bishop AE, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med (1990) 322(11):723–7. doi: 10.1056/nejm199003153221103
62. Yates CJ, Newey PJ, Thakker RV. Challenges and controversies in management of pancreatic neuroendocrine tumours in patients with MEN1. Lancet Diabetes Endocrinol (2015) 3(11):895–905. doi: 10.1016/s2213-8587(15)00043-1
63. Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg (2010) 34(2):249–55. doi: 10.1007/s00268-009-0290-1
64. Frost M, Lines KE, Thakker RV. Current and emerging therapies for PNETs in patients with or without MEN1. Nat Rev Endocrinol (2018) 14(4):216–27. doi: 10.1038/nrendo.2018.3
65. Iyer S, Agarwal SK. Epigenetic regulation in the tumorigenesis of MEN1-associated endocrine cell types. J Mol Endocrinol (2018) 61(1):R13–r24. doi: 10.1530/jme-18-0050
66. Niederle B, Selberherr A, Bartsch D, Brandi ML, Doherty GM, Falconi M, et al. Multiple Endocrine Neoplasia Type 1 (MEN1) and the Pancreas - Diagnosis and Treatment of Functioning and Non-Functioning Pancreatic and Duodenal Neuroendocrine Neoplasia within the MEN1 Syndrome - An International Consensus Statement. Neuroendocrinology (2020). doi: 10.1159/000511791
67. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab (2001) 86(12):5658–71. doi: 10.1210/jcem.86.12.8070
68. Eng C. Multiple Endocrine Neoplasia Type 2. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. Multiple Endocrine Neoplasia Type 2. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle (1999). (1993-2020).
69. Wells SA Jr., Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res (2009) 15(23):7119–23. doi: 10.1158/1078-0432.CCR-08-2742
70. Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res (2015) 204:139–56. doi: 10.1007/978-3-319-22542-5_6
71. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA (1996) 276(19):1575–9.
72. Willeke F, Hauer MP, Buchcik R, Gebert JF, Hahn M, Fitze G, et al. Multiple endocrine neoplasia type 2-associated RET proto-oncogene mutations do not contribute to the pathogenesis of sporadic parathyroid tumors. Surgery (1998) 124(3):484–90.
73. Pausova Z, Soliman E, Amizuka N, Janicic N, Konrad EM, Arnold A, et al. Role of the RET proto-oncogene in sporadic hyperparathyroidism and in hyperparathyroidism of multiple endocrine neoplasia type 2. J Clin Endocrinol Metab (1996) 81(7):2711–8. doi: 10.1210/jcem.81.7.8675600
74. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA (2006) 103(42):15558–63. doi: 10.1073/pnas.0603877103
75. Alrezk R, Hannah-Shmouni F, Stratakis CA. MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer (2017) 24(10):T195–208. doi: 10.1530/ERC-17-0243
76. Fritz A, Walch A, Piotrowska K, Rosemann M, Schaffer E, Weber K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res (2002) 62(11):3048–51.
77. Pellegata NS. MENX and MEN4. Clinics (Sao Paulo Brazil) (2012) 67(Suppl 1):13–8. doi: 10.6061/clinics/2012(sup01)04
78. Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab (2007) 92(8):3321–5. doi: 10.1210/jc.2006-2843
79. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab (2009) 94(5):1826–34. doi: 10.1210/jc.2008-2083
80. Molatore S, Marinoni I, Lee M, Pulz E, Ambrosio MR, degli Uberti EC, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat (2010) 31(11):E1825–35. doi: 10.1002/humu.21354
81. Belar O, De La Hoz C, Perez-Nanclares G, Castano L, Gaztambide S, Spanish MENG. Novel mutations in MEN1, CDKN1B and AIP genes in patients with multiple endocrine neoplasia type 1 syndrome in Spain. Clin Endocrinol (2012) 76(5):719–24. doi: 10.1111/j.1365-2265.2011.04269.x
82. Malanga D, De Gisi S, Riccardi M, Scrima M, De Marco C, Robledo M, et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol (2012) 166(3):551–60. doi: 10.1530/EJE-11-0929
83. Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PloS Genet (2013) 9(3):e1003350. doi: 10.1371/journal.pgen.1003350
84. Tonelli F, Giudici F, Giusti F, Marini F, Cianferotti L, Nesi G, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol (2014) 171(2):K7–K17. doi: 10.1530/EJE-14-0080
85. Elston MS, Meyer-Rochow GY, Dray M, Swarbrick M, Conaglen JV. Early Onset Primary Hyperparathyroidism Associated with a Novel Germline Mutation in CDKN1B. Case Rep Endocrinol (2015) 2015:510985. doi: 10.1155/2015/510985
86. Frederiksen A, Rossing M, Hermann P, Ejersted C, Thakker RV, Frost M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J Clin Endocrinol Metab (2019) 104(9):3637–46. doi: 10.1210/jc.2019-00082
87. Costa-Guda J, Marinoni I, Molatore S, Pellegata NS, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab (2011) 96(4):E701–6. doi: 10.1210/jc.2010-1338
88. Costa-Guda J, Arnold A. Genetic and epigenetic changes in sporadic endocrine tumors: parathyroid tumors. Mol Cell Endocrinol (2014) 386(1-2):46–54. doi: 10.1016/j.mce.2013.09.005
89. Chasseloup F, Pankratz N, Lane J, Faucz FR, Keil MF, Chittiboina P, et al. Germline CDKN1B Loss-of-Function Variants Cause Pediatric Cushing’s Disease With or Without an MEN4 Phenotype. J Clin Endocrinol Metab (2020) 105(6):1983–2005. doi: 10.1210/clinem/dgaa160
90. Jackson CE, Norum RA, Boyd SB, Talpos GB, Wilson SD, Taggart RT, et al. Hereditary hyperparathyroidism and multiple ossifying jaw fibromas: a clinically and genetically distinct syndrome. Surgery (1990) 108(6):1006–12.
91. Jackson CE. Hereditary hyperparathyroidism associated with recurrent pancreatitis. Ann Intern Med (1958) 49(4):829–36. doi: 10.7326/0003-4819-49-4-829
92. Kramer IR, Pindborg JJ, Shear M. The WHO Histological Typing of Odontogenic Tumours. A commentary on the Second Edition. Cancer (1992) 70(12):2988–94. doi: 10.1002/1097-0142(19921215)70:12<2988::aid-cncr2820701242>3.0.co;2-v
93. Ram R, Singhal A, Singhal P. Cemento-ossifying fibroma. Contemp Clin Dent (2012) 3(1):83–5. doi: 10.4103/0976-237X.94553
94. Liu Y, Wang H, You M, Yang Z, Miao J, Shimizutani K, et al. Ossifying fibromas of the jaw bone: 20 cases. Dentomaxillofac Radiol (2010) 39(1):57–63. doi: 10.1259/dmfr/96330046
95. Bradley KJ, Hobbs MR, Buley ID, Carpten JD, Cavaco BM, Fares JE, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med (2005) 257(1):18–26. doi: 10.1111/j.1365-2796.2004.01421.x
96. Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT. Hyperparathyroidism-jaw tumour syndrome. J Intern Med (2003) 253(6):634–42. doi: 10.1046/j.1365-2796.2003.01168.x
97. Torresan F, Iacobone M. Clinical Features, Treatment, and Surveillance of Hyperparathyroidism-Jaw Tumor Syndrome: An Up-to-Date and Review of the Literature. Int J Endocrinol (2019) 2019:1761030. doi: 10.1155/2019/1761030
98. Mehta A, Patel D, Rosenberg A, Boufraqech M, Ellis RJ, Nilubol N, et al. Hyperparathyroidism-jaw tumor syndrome: Results of operative management. Surgery (2014) 156(6):1315–24. doi: 10.1016/j.surg.2014.08.004
99. Li Y, Zhang J, Adikaram PR, Welch J, Guan B, Weinstein LS, et al. Genotype of CDC73 germline mutation determines risk of parathyroid cancer. Endocr Relat Cancer (2020) 27(9):483–94. doi: 10.1530/ERC-20-0149
100. Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet (2002) 32(4):676–80. doi: 10.1038/ng1048
101. Newey PJ, Bowl MR, Thakker RV. Parafibromin–functional insights. J Intern Med (2009) 266(1):84–98. doi: 10.1111/j.1365-2796.2009.02107.x
102. Domingues R, Tomaz RA, Martins C, Nunes C, Bugalho MJ, Cavaco BM. Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clin Endocrinol (2012) 76(1):33–8. doi: 10.1111/j.1365-2265.2011.04184.x
103. Cascon A, Huarte-Mendicoa CV, Javier Leandro-Garcia L, Leton R, Suela J, Santana A, et al. Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer (2011) 50(11):922–9. doi: 10.1002/gcc.20911
104. Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, Salenave S, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab (2013) 98(2):E403–8. doi: 10.1210/jc.2012-2789
105. Guarnieri V, Seaberg RM, Kelly C, Jean Davidson M, Raphael S, Shuen AY, et al. Large intragenic deletion of CDC73 (exons 4-10) in a three-generation hyperparathyroidism-jaw tumor (HPT-JT) syndrome family. BMC Med Genet (2017) 18(1):83. doi: 10.1186/s12881-017-0445-0
106. Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet (2003) 40(9):657–63. doi: 10.1136/jmg.40.9.657
107. Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, Cianferotti L, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab (2004) 89(11):5583–91. doi: 10.1210/jc.2004-0294
108. Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med (2003) 349(18):1722–9. doi: 10.1056/NEJMoa031237
109. Yu W, McPherson JR, Stevenson M, van Eijk R, Heng HL, Newey P, et al. Whole-exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC-catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. J Clin Endocrinol Metab (2015) 100(2):E360–4. doi: 10.1210/jc.2014-3238
110. Pandya C, Uzilov AV, Bellizzi J, Lau CY, Moe AS, Strahl M, et al. Genomic profiling reveals mutational landscape in parathyroid carcinomas. JCI Insight (2017) 2(6):e92061. doi: 10.1172/jci.insight.92061
111. Teh BT, Farnebo F, Kristoffersson U, Sundelin B, Cardinal J, Axelson R, et al. Autosomal dominant primary hyperparathyroidism and jaw tumor syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21-q32 and loss of the wild type allele in renal hamartomas. J Clin Endocrinol Metab (1996) 81(12):4204–11. doi: 10.1210/jcem.81.12.8954016
112. Vocke CD, Ricketts CJ, Ball MW, Schmidt LS, Metwalli AR, Middelton LA, et al. CDC73 Germline Mutation in a Family With Mixed Epithelial and Stromal Tumors. Urology (2019) 124:91–7. doi: 10.1016/j.urology.2018.11.013
113. Michal M, Syrucek M. Benign mixed epithelial and stromal tumor of the kidney. Pathol Res Pract (1998) 194(6):445–8. doi: 10.1016/S0344-0338(98)80038-1
114. Michal M, Hes O, Bisceglia M, Simpson RH, Spagnolo DV, Parma A, et al. Mixed epithelial and stromal tumors of the kidney. A report of 22 cases. Virchows Arch (2004) 445(4):359–67. doi: 10.1007/s00428-004-1060-y
115. Kakinuma A, Morimoto I, Nakano Y, Fujimoto R, Ishida O, Okada Y, et al. Familial primary hyperparathyroidism complicated with Wilms’ tumor. Intern Med (1994) 33(2):123–6. doi: 10.2169/internalmedicine.33.123
116. Szabo J, Heath B, Hill VM, Jackson CE, Zarbo RJ, Mallette LE, et al. Hereditary hyperparathyroidism-jaw tumor syndrome: the endocrine tumor gene HRPT2 maps to chromosome 1q21-q31. Am J Hum Genet (1995) 56(4):944–50.
117. van der Tuin K, Tops CMJ, Adank MA, Cobben JM, Hamdy NAT, Jongmans MC, et al. CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J Clin Endocrinol Metab (2017) 102(12):4534–40. doi: 10.1210/jc.2017-01249
118. Marx SJ. New Concepts About Familial Isolated Hyperparathyroidism. J Clin Endocrinol Metab (2019) 104:4058–66. doi: 10.1210/jc.2018-02789
119. Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab (2004) 89(1):96–102. doi: 10.1210/jc.2003-030675
120. Warner J, Epstein M, Sweet A, Singh D, Burgess J, Stranks S, et al. Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet (2004) 41(3):155–60. doi: 10.1136/jmg.2003.016725
121. Cetani F, Pardi E, Ambrogini E, Lemmi M, Borsari S, Cianferotti L, et al. Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clin Endocrinol (2006) 64(2):146–52. doi: 10.1111/j.1365-2265.2006.02438.x
122. Vierimaa O, Villablanca A, Alimov A, Georgitsi M, Raitila A, Vahteristo P, et al. Mutation analysis of MEN1, HRPT2, CASR, CDKN1B, and AIP genes in primary hyperparathyroidism patients with features of genetic predisposition. J Endocrinol Invest (2009) 32(6):512–8. doi: 10.1007/BF03346498
123. Pontikides N, Karras S, Kaprara A, Anagnostis P, Mintziori G, Goulis DG, et al. Genetic basis of familial isolated hyperparathyroidism: a case series and a narrative review of the literature. J Bone Miner Metab (2014) 32(4):351–66. doi: 10.1007/s00774-013-0551-9
124. Baumber L, Tufarelli C, Patel S, King P, Johnson CA, Maher ER, et al. Identification of a novel mutation disrupting the DNA binding activity of GCM2 in autosomal recessive familial isolated hypoparathyroidism. J Med Genet (2005) 42(5):443–8. doi: 10.1136/jmg.2004.026898
125. Canaff L, Zhou X, Mosesova I, Cole DE, Hendy GN. Glial cells missing-2 (GCM2) transactivates the calcium-sensing receptor gene: effect of a dominant-negative GCM2 mutant associated with autosomal dominant hypoparathyroidism. Hum Mutat (2009) 30(1):85–92. doi: 10.1002/humu.20827
126. Guan B, Welch JM, Vemulapalli M, Li Y, Ling H, Kebebew E, et al. Ethnicity of Patients With Germline GCM2-Activating Variants and Primary Hyperparathyroidism. J Endocr Soc (2017) 1(5):488–99. doi: 10.1210/js.2017-00043
127. Cetani F, Pardi E, Aretini P, Saponaro F, Borsari S, Mazoni L, et al. Whole exome sequencing in familial isolated primary hyperparathyroidism. J Endocrinol Invest (2020) 43(2):231–45. doi: 10.1007/s40618-019-01107-5
128. Riccardi A, Aspir T, Shen L, Kuo CL, Brown TC, Korah R, et al. Analysis of Activating GCM2 Sequence Variants in Sporadic Parathyroid Adenomas. J Clin Endocrinol Metab (2019) 104(6):1948–52. doi: 10.1210/jc.2018-02517
129. Lee JY, Shoback DM. Familial hypocalciuric hypercalcemia and related disorders. Best Pract Res Clin Endocrinol Metab (2018) 32(5):609–19. doi: 10.1016/j.beem.2018.05.004
130. Papadopoulou A, Gole E, Melachroinou K, Meristoudis C, Siahanidou T, Papadimitriou A. Identification and Functional Characterization of a Calcium-Sensing Receptor Mutation in an Infant with Familial Hypocalciuric Hypercalcemia. J Clin Res Pediatr Endocrinol (2016) 8(3):341–6. doi: 10.4274/jcrpe.2800
131. Brown EM. Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metab Clin North Am (2000) 29(3):503–22. doi: 10.1016/s0889-8529(05)70148-1
132. Mariathasan S, Andrews KA, Thompson E, Challis BG, Wilcox S, Pierce H, et al. Genetic testing for hereditary hyperparathyroidism and familial hypocalciuric hypercalcaemia in a large UK cohort. Clin Endocrinol (2020) 93: 409–18. doi: 10.1111/cen.14254
133. Brown EM. Mutations in the calcium-sensing receptor and their clinical implications. Horm Res (1997) 48(5):199–208. doi: 10.1159/000185516
134. Corrado KR, Andrade SC, Bellizzi J, D’Souza-Li L, Arnold A. Polyclonality of Parathyroid Tumors in Neonatal Severe Hyperparathyroidism. J Bone Miner Res (2015) 30(10):1797–802. doi: 10.1002/jbmr.2516
135. Farnebo F, Enberg U, Grimelius L, Backdahl M, Schalling M, Larsson C, et al. Tumor-specific decreased expression of calcium sensing receptor messenger ribonucleic acid in sporadic primary hyperparathyroidism. J Clin Endocrinol Metab (1997) 82(10):3481–6. doi: 10.1210/jcem.82.10.4300
136. Hosokawa Y, Pollak MR, Brown EM, Arnold A. Mutational analysis of the extracellular Ca(2+)-sensing receptor gene in human parathyroid tumors. J Clin Endocrinol Metab (1995) 80(11):3107–10. doi: 10.1210/jcem.80.11.7593409
137. Cetani F, Pinchera A, Pardi E, Cianferotti L, Vignali E, Picone A, et al. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J Bone Miner Res (1999) 14(6):878–82. doi: 10.1359/jbmr.1999.14.6.878
138. Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T, et al. Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med (2013) 368(26):2476–86. doi: 10.1056/NEJMoa1300253
139. Gorvin CM, Cranston T, Hannan FM, Rust N, Qureshi A, Nesbit MA, et al. A G-protein Subunit-alpha11 Loss-of-Function Mutation, Thr54Met, Causes Familial Hypocalciuric Hypercalcemia Type 2 (FHH2). J Bone Miner Res (2016) 31(6):1200–6. doi: 10.1002/jbmr.2778
140. Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium-sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol (2016) 57(3):R127–42. doi: 10.1530/JME-16-0124
141. Gorvin CM, Hannan FM, Cranston T, Valta H, Makitie O, Schalin-Jantti C, et al. Cinacalcet Rectifies Hypercalcemia in a Patient With Familial Hypocalciuric Hypercalcemia Type 2 (FHH2) Caused by a Germline Loss-of-Function Galpha11 Mutation. J Bone Miner Res (2018) 33(1):32–41. doi: 10.1002/jbmr.3241
142. Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet (2013) 45(1):93–7. doi: 10.1038/ng.2492
143. Hendy GN, Canaff L, Newfield RS, Tripto-Shkolnik L, Wong BY, Lee BS, et al. Codon Arg15 mutations of the AP2S1 gene: common occurrence in familial hypocalciuric hypercalcemia cases negative for calcium-sensing receptor (CASR) mutations. J Clin Endocrinol Metab (2014) 99(7):E1311–5. doi: 10.1210/jc.2014-1120
144. Hannan FM, Howles SA, Rogers A, Cranston T, Gorvin CM, Babinsky VN, et al. Adaptor protein-2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype-phenotype correlations, codon bias and dominant-negative effects. Hum Mol Genet (2015) 24(18):5079–92. doi: 10.1093/hmg/ddv226
145. Vargas-Poussou R, Mansour-Hendili L, Baron S, Bertocchio JP, Travers C, Simian C, et al. Familial Hypocalciuric Hypercalcemia Types 1 and 3 and Primary Hyperparathyroidism: Similarities and Differences. J Clin Endocrinol Metab (2016) 101(5):2185–95. doi: 10.1210/jc.2015-3442
146. Gorvin CM, Metpally R, Stokes VJ, Hannan FM, Krishnamurthy SB, Overton JD, et al. Large-scale exome datasets reveal a new class of adaptor-related protein complex 2 sigma subunit (AP2sigma) mutations, located at the interface with the AP2 alpha subunit, that impair calcium-sensing receptor signalling. Hum Mol Genet (2018) 27(5):901–11. doi: 10.1093/hmg/ddy010
147. Palanisamy N, Imanishi Y, Rao PH, Tahara H, Chaganti RS, Arnold A. Novel chromosomal abnormalities identified by comparative genomic hybridization in parathyroid adenomas. J Clin Endocrinol Metab (1998) 83(5):1766–70. doi: 10.1210/jcem.83.5.4806
148. Agarwal SK, Schrock E, Kester MB, Burns AL, Heffess CS, Ried T, et al. Comparative genomic hybridization analysis of human parathyroid tumors. Cancer Genet Cytogenet (1998) 106(1):30–6. doi: 10.1016/s0165-4608(98)00049-1
149. Farnebo F, Kytola S, Teh BT, Dwight T, Wong FK, Hoog A, et al. Alternative genetic pathways in parathyroid tumorigenesis. J Clin Endocrinol Metab (1999) 84(10):3775–80. doi: 10.1210/jcem.84.10.6057
150. Kytola S, Farnebo F, Obara T, Isola J, Grimelius L, Farnebo LO, et al. Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol (2000) 157(2):579–86. doi: 10.1016/S0002-9440(10)64568-3
151. Garcia JL, Tardio JC, Gutierrez NC, Gonzalez MB, Polo JR, Hernandez JM, et al. Chromosomal imbalances identified by comparative genomic hybridization in sporadic parathyroid adenomas. Eur J Endocrinol (2002) 146(2):209–13. doi: 10.1530/eje.0.1460209
152. Skandarajah A, Barlier A, Morlet-Barlat N, Sebag F, Enjalbert A, Conte-Devolx B, et al. Should routine analysis of the MEN1 gene be performed in all patients with primary hyperparathyroidism under 40 years of age? World J Surg (2010) 34(6):1294–8. doi: 10.1007/s00268-009-0388-5
Keywords: tumor suppressor, oncogene, multiple endocrine neoplasia, MEN1, jaw tumor syndrome, CASR, CDC73, GCM2
Citation: Blau JE and Simonds WF (2021) Familial Hyperparathyroidism. Front. Endocrinol. 12:623667. doi: 10.3389/fendo.2021.623667
Received: 30 October 2020; Accepted: 13 January 2021;
Published: 25 February 2021.
Edited by:
Naris Nilubol, National Institutes of Health (NIH), United StatesReviewed by:
Daniela Pasquali, University of Campania Luigi Vanvitelli, ItalySabrina Corbetta, University of Milan, Italy
Copyright © 2021 Blau and Simonds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William F. Simonds, YmlsbHNAbmlkZGsubmloLmdvdg==