 Seyed M. Ghiasi
Seyed M. Ghiasi Guy A. Rutter
Guy A. Rutter- Section of Cell Biology and Functional Genomics, Division of Diabetes, Endocrinology and Metabolism, Department of Metabolism, Digestion and Reproduction, Faculty of Medicine, Imperial College London, London, United Kingdom
Mounting evidence suggests a role for alternative splicing (AS) of transcripts in the normal physiology and pathophysiology of the pancreatic β-cell. In the apparent absence of RNA repair systems, RNA decay pathways are likely to play an important role in controlling the stability, distribution and diversity of transcript isoforms in these cells. Around 35% of alternatively spliced transcripts in human cells contain premature termination codons (PTCs) and are targeted for degradation via nonsense-mediated decay (NMD), a vital quality control process. Inflammatory cytokines, whose levels are increased in both type 1 (T1D) and type 2 (T2D) diabetes, stimulate alternative splicing events and the expression of NMD components, and may or may not be associated with the activation of the NMD pathway. It is, however, now possible to infer that NMD plays a crucial role in regulating transcript processing in normal and stress conditions in pancreatic β-cells. In this review, we describe the possible role of Regulated Unproductive Splicing and Translation (RUST), a molecular mechanism embracing NMD activity in relationship to AS and translation of damaged transcript isoforms in these cells. This process substantially reduces the abundance of non-functional transcript isoforms, and its dysregulation may be involved in pancreatic β-cell failure in diabetes.
Introduction
Diabetes mellitus currently affects ~460 m adults worldwide and its incidence is expected to exceed 700 m by 2045 (1). Both T1D and T2D – the latter being the predominant form – involve defective pancreatic β-cell function, and a contribution of inflammatory processes which is most acute in the former. β-Cell death is marked in T1D: typically >80% of the cell mass is lost, though this figure is lower in some patients, particularly those with later disease onset (2), but more limited in T2D (3) where dysfunction predominates (4). The molecular mechanisms involved in β-cell dysfunction and loss in both settings are only partly understood.
Insults associated with diabetes including challenge with inflammatory cytokines, hyperglycaemia and gluco(lipo)toxicity downregulate selected β-cell specific transcripts and up-regulate others (4–6). This is followed by a profound up-regulation of mRNA surveillance systems (7). The up-regulation of splicing factors, and of proteins involved in pre-mRNA processing, gives rise to alternative splicing (AS) events, which in turn deregulate the balance and turnover of transcript isoforms. Interestingly, most human genes exhibit alternative splicing, but not all alternatively spliced transcripts are translated into functional proteins and so targeted for degradation. This varies in a cell-specific manner and depends on the capacity of cells to cope with damaged transcripts (7–9).
At least four types of mRNA decay pathways have been studied in mammalian cells and which scrutinize transcript quality: nonsense-mediated decay (NMD), Staufen1 (STAU1)-mediated mRNA decay (SMD), no-go decay (NGD), and nonstop-decay (NSD) (10–12). Here, we focus on how the NMD pathway interacts with alternative splicing to regulate transcript isoform expression. We also consider how as its deregulation may contribute to β-cell dysfunction, vulnerability, and destruction in diabetes.
Alternative Splicing (AS); Implications In β-Cells
Non-coding interspaced sequences, namely introns, are removed by the splicesome, a dynamic RNA-protein complex, during transcription from precursor-mRNAs (13, 14). Around 90%–95% of human transcripts are thought to leave the nucleoplasm as pre-mRNAs that need further splicing and processing to become a mature mRNA (15). Alternate (or alternative) isoforms created either by switching the usual promoter to alternative promoter of a gene and/or pre-mRNA alternative splicing (Figure 1A). Alternative splicing, an evolutionarily post-transcriptional pre-mRNA processing process, produces multiple distinct transcript variants of most human genes (16, 17). The mechanisms and biology of splicing and alternative splicing have been extensively reviewed (18–20). As described previously (15) and in Figure 1B, AS can involve inclusion or skipping (exclusion) of an exon, mutually exclusive exons, alternative 5′ donor splice sites, and alternative 3′ acceptor splice sites and intron retention in mRNAs (15). AS is a consequence of interactions between RNA binding proteins (RBPs) and splicing regulatory elements (SREs) in pre-mRNAs. The Serine Arginine rich proteins (SRs) are a family of constitutive or regulatory RNA binding proteins recognize pre-mRNA SREs through interaction with the N-terminal and C-terminal domains enriched with arginine (R) and serine (S) sequences. These then interact with other proteins and/or SREs to enhance splicing by recruiting the spliceosome (21). On the other hand, the second class of RBPs, members of the heterogeneous nuclear ribonucleoprotein (hnRNP) protein family, have been shown to antagonize SR functions by competing for binding to exonic splicing enhancers (ESE) or intronic splicing suppressors (ISS) (21, 22).
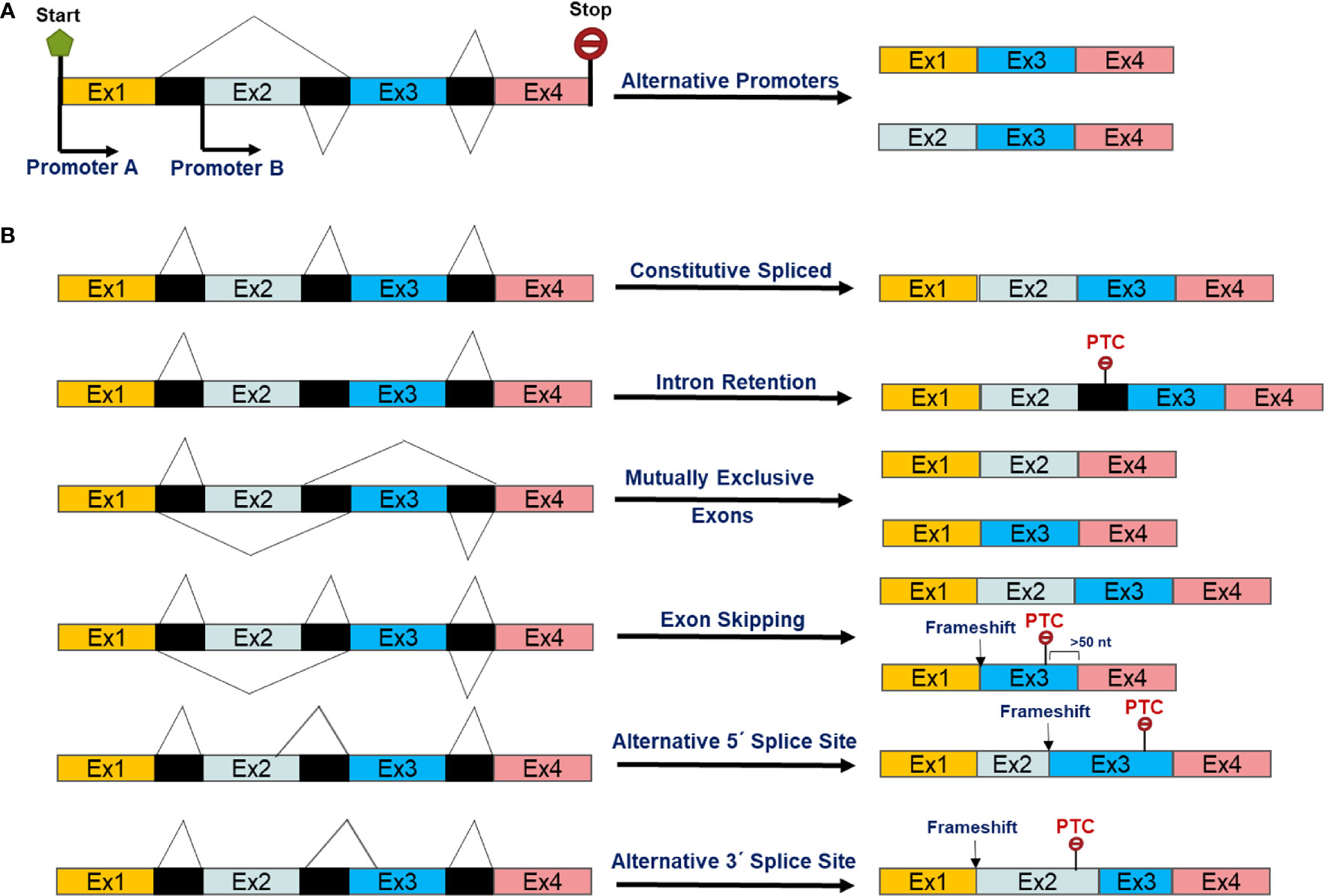
Figure 1 Scheme of alternative transcript isoforms as a result of (A) alternative promoters and post-transcriptional alternative splicing events of pre-mRNAs (B).
Alternative splicing is observed in all human tissues, but has been most extensively studied in neurons (23, 24). Thus, aberrant alternative splicing of the pre-mRNAs encoding calcium signalling transducers affects neuronal function and causes neurodegenerative diseases (25–27). In the past decade, pancreatic β-cell transcriptomic analyses have revealed differential expression profiles of the RBPs and splicing factors which are abundantly expressed in neurons, and whose genetic ablation can lead to impairments in insulin secretion and reduced β-cell viability (28–30).
It is now established that alternative splicing plays an important role in β-cell function and viability. Glucose, a major regulator of pancreatic β-cell function (31), strongly affects insulin gene expression, biosynthesis, and secretion, through multiple mechanisms including changes in transcription, pre-mRNA alternative splicing, translation and mRNA stability (32–34). Insulin intron-2-containing pre-mRNA levels increased six-fold within an hour of a human islet exposure to high glucose, whereas increases in mature mRNA did not occur before 48 h of exposure (35), suggesting that substantial of insulin production is exerted at the level of pre-mRNA alternative splicing. Another study (36) showed that alternative splicing of the insulin receptor is regulated by insulin signalling and modulates β-cell survival in an autocrine pathway involving insulin secretion, binding to and activation of insulin receptors in human and mouse islets and in clonal MIN6 cells.
Several lines of evidence support the importance of regulated AS in inflammatory stresses in pancreatic β-cells, as reviewed previously (28, 37). The pro-inflammatory cytokines interleukin-1β (IL-1β) and interferon-ɣ (IFNɣ) upregulate >30 splicing factors, affecting alternative splicing of 35% of genes in the human islet transcriptome (9, 37). Genetic manipulation of several RBP candidates involved in alternative splicing, as listed in Table 1, impaired insulin secretion and sensitised β-cells to basal and/or cytokine-induced toxicity. These changes may reflect deregulation of transcript isoforms encoding anti/pro-apoptotic proteins as well as those annotated in exocytosis and secretory pathways in pancreatic β-cells (28, 40, 41). Finally, genome-wide RNA sequencing revealed that three transcript isoform variants of CD137 were associated with T1D development in the NOD mice (42).
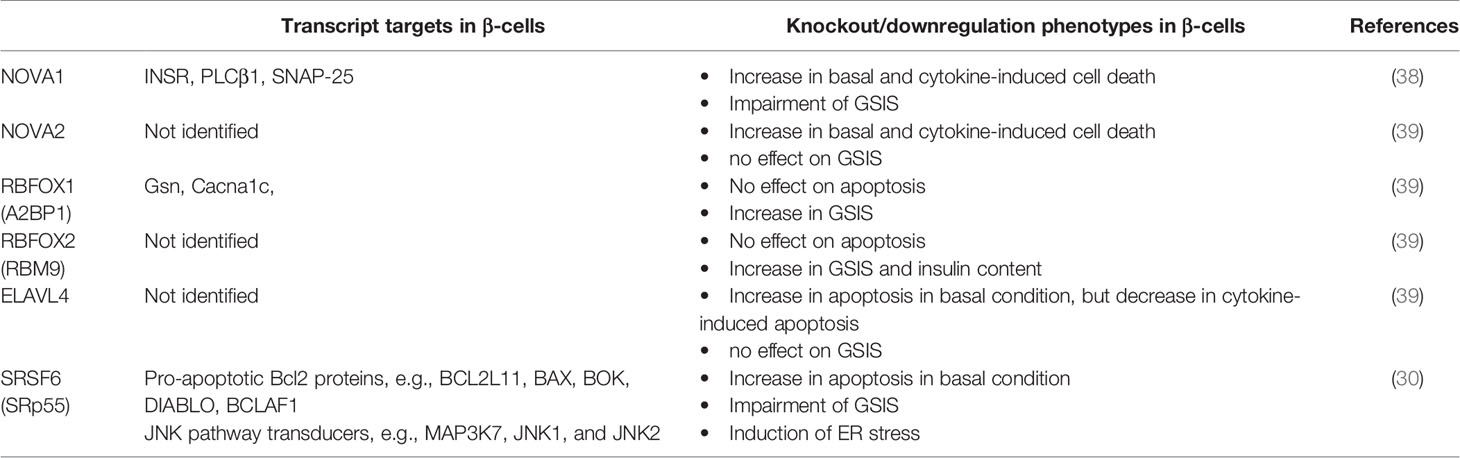
Table 1 Examples of the function of alternative splicing regulators in β-cells.
At present, there is little direct evidence that genetic variants in the genes involved in components of the NMD pathway are involved in altered T2D risk (43). Nevertheless, alternative splicing of genes which are implicated may be relevant. For example, variants in the Transcription factor 7–like 2 (TCF7L2) gene are strongly associated with T2D risk in man (44–47). Mapping of TCF7L2 splice variants revealed a specific pattern in pancreatic islets, with variants carrying exons 4 and 15 correlated with glycated haemoglobin A1c (HbA1c) (48). The presence of deleterious TCF7L2 splice variants (i.e., exons 13-16) was also suggested to be a mechanism of β-cell failure in T2D mouse models (49). Although deletion of TCF7L2 selectively in the pancreas (50) or β-cell (51) in mice lowers β-cell function, increased levels of TCF7L2 mRNA are associated with elevated diabetes risk in man (52, 53), with the latter study reporting increased levels of the 3’ exon (and 15 o 18 exons overall) in islets of carriers of the risk rs7903146 allele. However, and as previously discussed (51), risk allele-dependent alternative splicing of the TCF7L2 gene in β-cells may affect the inclusion of a “CRARF” motif in the expressed protein and, as such, may impact the transcriptional activity of this factor (i.e., lowered transcriptional activity despite an increased overall transcript load).
The circadian clock has recently been shown to modulate synchronicity of insulin secretion in dark-light phases by regulating the alternative splicing of pre-mRNAs coding for proteins involved in insulin biosynthesis and exocytosis in primary mouse β-cells. The circadian clock core transcription factors CLOCK and BMAL1 autonomously determine oscillatory regulation of ~27% of the β-cell transcript isoforms corresponding to genes coding for proteins that are involved in the assembly, trafficking, and fusion of secretory vesicles at the plasma membrane (54). Disruption of the CLOCK and BMAL1 genes perturbs rhythmic genome-wide alternative splicing of pre-mRNAs encoding regulators of insulin biosynthesis and secretion in murine insulin-producing cell lines and primary β-cells (41, 54). A later exploration of the underlying mechanisms revealed that thyroid hormone receptor-associated protein 3 (THRAP3), an RNA-binding protein, modulates circadian clock-dependent alternative splicing of calcium/calmodulin-dependent serine protein kinase (Cask) and MAP kinase-activating death domain (Madd). Consistent with findings of exon skipping due to circadian clock perturbation, CRISPR-Cas9-mediated deletion of exons-11 and -26 of Cask and Madd pre-mRNAs, respectively, impairs insulin secretion in murine insulin-producing β-cells (41). Madd knockout mice developed hyperglycaemia associated with impaired insulin secretion in mice (55).
Alternatively spliced transcript variants can produce functionally different protein isoforms with altered amino acid sequences and protein domains, resulting in modification of activity. This, in turn, may drive alterations in protein localization, interaction with binding partners or post-translational polypeptide processing (56, 57). A substantial number of alternatively spliced variants contain a premature termination codon (PTC) or other mRNA “discrediting” features such as an upstream open reading frame (uORF), long 3’ untranslated region (UTR) or the retention of introns after stop codons (58, 59). Any of these could potentially render the mRNA a target forf nonsense-mediated decay (60–62). Whether these isoforms are (mis-)expressed in pancreatic β-cells in diabetes – for example as a result of inflammatory or metabolic stresses – remains to be explored.
The NMD Pathway: Biology and Emerging Role in β-Cells
The nonsense-mediated decay pathway, originally identified as an RNA surveillance mechanism, eliminates aberrant RNAs harbouring PTCs (63). Computational and experimental results indicate that roughly a third of reliably inferred alternative splicing events in humans result in mRNA isoforms that harbour a PTC (64, 65). PTCs can arise in cells through various mechanisms: germline or somatic mutations in DNA; errors in transcription; or post-transcriptional mRNA damage or errors in processing, notably including alternative splicing (66). PTCs have been implicated in approximately 30% of all inherited diseases, indicating that the NMD pathway plays a vital role in survival and health (11, 67). Failure to recognize and eliminate these unproductive transcripts seems likely to result in the production of truncated dysfunctional proteins that directly perturb cell function or lead to an accumulation of misfolded proteins that accumulate in the ER to cause ER stress.
The human NMD machinery is complex and involves multiple proteins including Upf1, Upf2, Upf3a, Upf3b, Smg1, Smg5, Smg6, And Smg7 (See Table 2). Together, these are responsible for the detection and decay of PTC-containing transcripts (Figure 2A). ATPase-dependent RNA helicases play a central role in NMD activity. Thus, the ability of UPF1 to selectively target PTC-containing mRNAs depends on its ATPase and helicase activities (59, 72). Additionally, activation of NMD requires an interaction between Upf1 and protein partners on the targeted mRNA. These partners consist of UPF2 that forms a bridge between Upf1 and Upf3, forming the Upf1–Upf2–Upf3 complex (66, 73). However, in addition to canonical NMD pathway in which all key NMD components function on target transcripts, NMD is also activated (in)dependent of some of its key factors including Upf2 and Upf3 with a cell-type specific manner. Thus, NMD should be seen as a “branched pathway”, with the different branches defined by autoregulatory feedback loops (Figure 2B) (58, 66, 74).
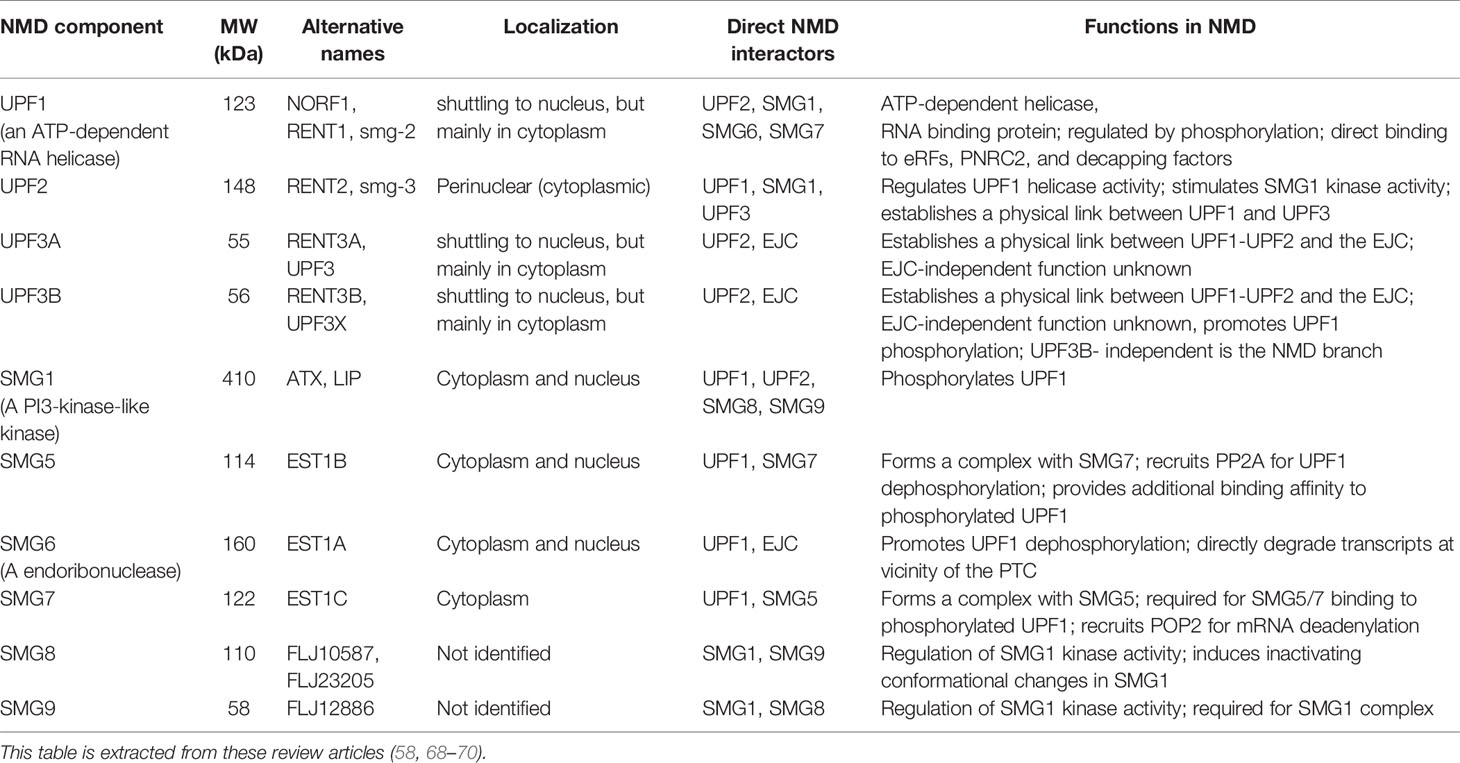
Table 2 Characteristics of core machinery and effective NMD components.

Figure 2 Simplified canonical model of NMD pathway activation (A) in human cells which is controlled by three different ways so far and yet to be identified (B). Inter-individual variation of NMD efficiency due to transcript variants modifies the presentation of clinical phenotypes and response to the PTC read-through drug PTC124 treatment (B). At early ER stress, UPR suppresses NMD to provide maximum capacity of UPR proteins replenishment. Once ER stress is being resolved, the UPR is downregulated, while NMD is supposed to return to its normal activity to eliminate unproductive transcript isoforms, thereby leading to further downregulation of the UPR as UPR transcripts isoforms are NMD specific targets. Unresolved ER stress induces apoptosis which in turn suppresses NMD pathway to execute the cell to avoid deleterious outcomes (C) (59, 68, 69, 71).
Upf1 promiscuously binds to both NMD-targeted and non-targeted mRNAs undergoing translation (75). On PTC-containing mRNAs, Upf1 and its associated phosphoinositide 3-kinase (PI3K)-like kinase, Smg1 act as a clamp to bind to eukaryotic releasing factors 2 and 3 (eRF2 and eRF3) to form a surveillance complex. The exon-exon junctions at least 50 nucleotides downstream of stop codons possesses a nucleation point where the EJC, Upf2, and Upf3b bind as a foundation of a decay-inducing complex whose Interaction with the surveillance complex triggers Upf1 phosphorylation, dissociation of eRF1 and eRF3, and conformational remodelling of NMD. This adopts Upf1 activity to resolve mRNA secondary structure by its helicase activity, allowing access to the mRNA of the NMD effector proteins Smg5-7 (59, 72–77). The decay of targeted transcripts then takes place through the following steps: recruitment of the endoribonuclease Smg6, which catalyses PTC-proximal mRNA cleavage, producing 5ʹ and 3ʹ cleavage fragments that are degraded by exoribonucleases (73, 78, 79); recruitment of the Smg5–Smg7 heterodimer, which bridges an interaction with the carbon catabolite repressor protein 4 (CCR4)-NOT deadenylase complex, thereby shortening the poly(A) tail to stimulate mRNA decapping by the general decapping complex (80–82); recruitment of the decapping enhancer Proline-rich nuclear receptor co-activator 2 (PNRC2), possibly in a complex with SMG5, which recruits the general decapping complex (83); and/or direct recruitment of the general decapping complex (73, 79, 84). Decapped mRNA is in turn degraded from the 5΄-end by the Xrn1 exoribonuclease and from the 3’ terminus by the Dis3L1 and/or Dis3L2 exosome complex (85, 86). Xrn1 has also been shown to promote general 5΄-3΄ co-translational mRNA decay following the last translating ribosome (85). These degradation pathways are not mutually exclusive, and their balance varies depending on the particular mRNA or organism (73, 86).
NMD pathways have been studied in different human and mouse tissues, and most recently by one of us (SMG) in human and rodent islets (7). These studies revealed that NMD components are differentially up/down-regulated by inflammatory cytokines and glucolipotoxicity. Genetic suppression of the key NMD component Smg6, an endoribonuclease which cleaves NMD targeted transcripts at the proximate location of the PTC (Table 2), alleviated cytokine-mediated toxicity associated with increased insulin biosynthesis and glucose–induced insulin secretion in INS-1 cells, a rat insulinoma-derived insulin-producing cell line. This study also revealed that nitroxidative stress is mechanistically involved in cytokine-mediated up-regulation of NMD components, since chemical inhibition of inducible nitric oxide (iNOS) by N-methyl-L-argenine (NMA), normalized cytokine upregulation of the NMD components in INS-1 cells (7). Whether this up-regulation of NMD components culminates in activation of the pathway remains to be elucidated.
Buffering mechanisms appear to have evolved in a cell-type specific manner to control NMD activity, reflecting its important role in regulating normal transcripts as well as eliminating unproductive transcript isoforms. Three different mechanisms include, firstly, microRNAs such as mir-128 targeting NMD component transcripts; another RNA decay pathway called STAU1-mediated mRNA decay (SMD) whose the RNA-binding protein Staufen 1 competes with Upf1 of the NMD, and auto-regulatory feedback (Figure 2B) (87–90). The feedback loop has been reported to be exerted at both the mRNA and protein levels of the NMD factors. At the mRNA level, NMD controls the rate-limiting mRNA levels of its components. At the protein level, control is also exerted by protein stabilization of proteins involved in regulation such as Upf3a, an RNA-binding protein suppressing NMD activity (91, 92). It is therefore is not surprising that different branches of the NMD pathway have different efficiency profiles in different tissues (58, 93). Nevertheless, it will be important to determine what branches of NMD control pathway activity in stress conditions in primary pancreatic β-cells.
Could NMD transcript variations such as genetic mutations, SNPs and environmental factors affecting NMD activity be associated with cause and development of diabetes? Considering the emerging role of NMD in regulating transcript processing in pancreatic β-cells, we sought to understand whether transcript variants of NMD components are associated with T2D. Our interrogation of publicly-available GWAS data (type2diabetesgenes.org) reveals that an accumulated list of the NMD transcript variants with surprisingly high burden of natural loss-of-function variation including stop-gained, essential splice, and frameshift variants are significantly associated with T2D (p<0.05) (Figure 3A). In addition, chromatin analysis using ATAC-sequencing, a popular method for determining chromatin accessibility across the genome, indicates high chromatin accessibility to enhancers and subsequent strong transcription upon the key NMD components, e.g., Upf1 and Upf2 and Upf3a (Figure 3B).
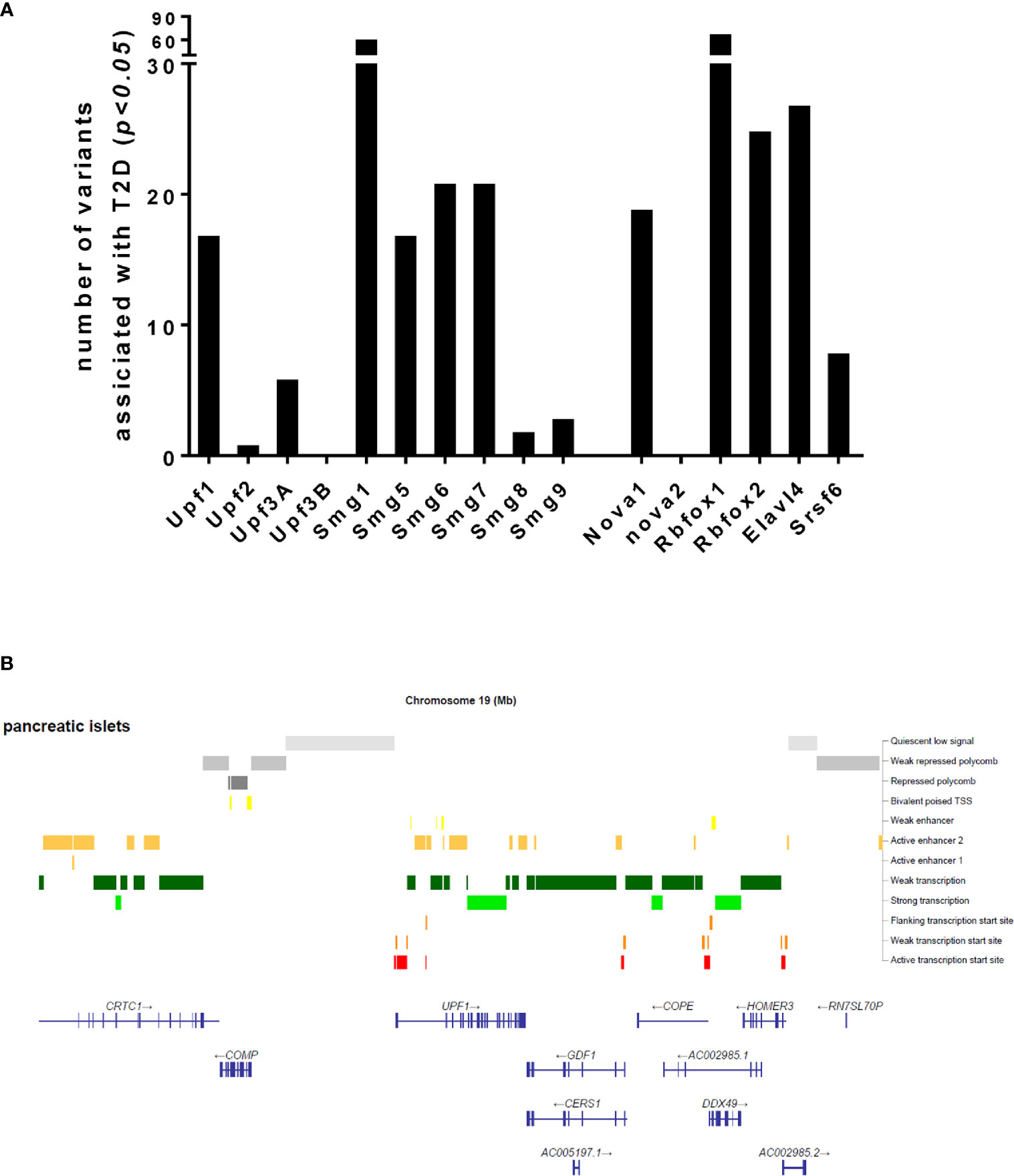
Figure 3 Number of transcript variants of the NMD pathway components and alternative splicing regulators associated with type 2 diabetes (A) and an ATAC-sequencing analysis for chromatin accessibility of the Upf1 gene (B), extracted from publicly available type 2 diabetes datasets.
Transcript Processing in the β-Cell Transcriptome
The integrity and accuracy of transcript processing is likely to be crucially important to shape the transcriptome of the β-cell and, in turn, to meet the physiological demands and pathophysiological challenges it faces.
In addition to degrading PTC-containing transcripts (i.e., “unproductive” transcripts) (63), NMD is also involved in normal physiology and in the transcriptional regulation of normal transcripts (i.e., “productive” transcripts), functioning as a fine-tuning mechanism of gene expression (59, 71). In fact, early embryonic lethality of mice depleted of the NMD factors Upf1, Upf2, Upf3a, Smg1, and Smg6 suggests that NMD is important for normal development and growth of the cell (60, 94–97). Whether impaired transcript processing due to deregulated NMD pathway may induce the dedifferentiation of β-cells, as may occur in T2D (98) and possibly T1D (99), or hinders interactions between β- with other cell types in the islet (100, 101) are important questions.
In T2D, a compensatory increase in insulin secretion in response to insulin resistance can stimulate a sequence of “stressful” events in the β-cell (the most important being; ER stress, inflammasome activation with subsequent β-cell-driven cytokine (e.g., IL-1β) production and NF-κB activation and nitroxidative/oxidative stress). Together, these may then initiate low-grade inflammation in the islet microenvironment (102, 103). In T1D, a cascade of inflammatory cytokines secreted from the immune cells leads to autoimmune destruction of β-cells (104). Cellular senescence may also be involved (105, 106). The overt loss of functional β-cell mass in T1D is thought, ultimately, to result from accelerated apoptosis (3). ER stress is a common upstream culprit in both T1D and T2D (102, 104). The unfolded protein response, which may lead to ER stress, on the one hand, and NMD on the other, mutually regulate each other in mouse and human tissues and cell lines (71). Thus, UPR transcripts may be NMD-specific targets (58).
The NMD plays a central role in the RUST mechanism to eliminate the PTC-containing transcript isoforms generated due to perturbed AS (107). The degree and magnitude of NMD activity differs among studied mice and human cells and tissues, as reviewed previously (89). In transgenic mice ubiquitously expressing the Men1 gene, the ratio of PTC-containing versus wild-type transcripts was significantly different between adult mouse tissues. Among the tested tissues, testis, ovary, brain, and heart exhibited high NMD activity, measured by strong downregulation, and lung, intestine, and thymus exhibited weak downregulation of the mutant Men1 transcripts compared to wild-type transcripts (108). Unfortunately, neither this study nor others reported NMD efficacy in the pancreas.
Other studies have suggested that NMD efficiency varies among individuals with nonsense mutations e.g., in the cystic fibrosis gene CFTR in response to the drug PTC124, forcing read-through of mutated mRNAs (Figure 2B) (109). The RUST mechanism was first proposed by Lewis and colleagues in 2003 (64). These authors found that 35% (i.e., 1,989 out of 5,693) of alternatively spliced transcript isoforms in the human cell transcriptome were NMD targets since they contained PTCs (64). Several subsequent studies identified a role for incomplete RUST in regulating transcript processing in breast and myelodysplastic syndrome (MDS) cancers, and in neurological disorders such as Alzheimer’s disease and multiple sclerosis (16). However, the role of RUST is completely unknown in transcript processing in pancreatic β-cells. Knowing that pro-inflammatory cytokines regulate alternative splicing events and the NMD pathway in human and rodent primary β-cells (9, 28, 37, 40, 41), we propose a model (Figure 4) in which incomplete RUST leads to accumulation of unproductive transcripts whose translation into unfolded, truncated polypeptides overwhelms ER capacity and consequently drives unresolved ER stress. Consistent with this view, enforcing ribosomal read-through of such PTC-containing mRNAs with the drug PTC124 aggravates cytokine-induced apoptosis and is associated with an increase in ER stress in human islets and INS-1 cells (7).
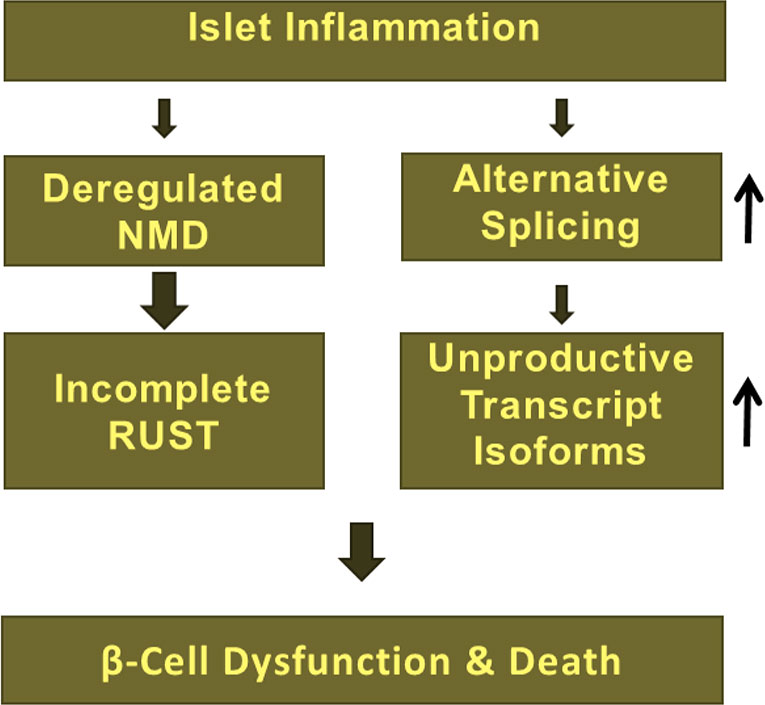
Figure 4 Deregulation of NMD due to islet inflammation largely influence RUST mechanism of transcript processing and subsequent accumulation of unproductive transcript isoforms, which implicates in β-cell dysfunction, vulnerability, and death.
Perspective: A New Type of Unproductive Transcript?
The recent discovery of a special class of bifunctional RNAs, namely coding-noncoding RNAs (cncRNA), implicates another culprit in the pathogenesis of diseases such as Alzheimer’s disease. Amongst the cncRNAs, certain noncoding mRNA isoforms (ncimRNA) of (usually) protein-coding genes quantitatively predominate (110, 111). The precise function and underlying molecular mechanism(s) of action of cncRNAs has been investigated in a few cases (110). Insulin receptor substrate 1 (IRS1), a major substrate and cytoplasmic docking protein for the insulin receptor and insulin-like growth factor receptor, is involved in insulin signalling. The level of IRS1 is highly increased in proliferative cells such as human and mouse cancer cells (110), whereas profoundly decreased in differentiated cells. In addition, whole body Irs1 knockout mice exhibited severe insulin resistance in skeletal muscle and liver, with compensatory β-cell hyperplasia (112). Surprisingly, a further study (113) found that the 5′UTR of Irs1 mRNA acts as an antisense mRNA to the cell cycle regulator retinoblastoma (Rb).
Intronic or exonic Circular RNAs (circRNAs) are a type of single-stranded noncoding RNAs whose 5’ and 3’ termini are covalently linked by back-splicing of exons from a single pre-mRNA and they are, therefore, stable and resistant to exonuclease degradation (114). With a feature of cell-specificity and being conserved between species, circRNAs play important roles in the development of diseases by modulating post-transcriptional regulation of gene expression (114, 115). Recently, two intronic circRNA borne from murine insulin genes, ci-Ins2 and ci-INS have been reported to control insulin secretion. Thus, silencing of ci-Ins2 in pancreatic islets deceases in the expression of key components of the secretory machinery of β-cells, resulting in impaired pulsatile insulin secretion and calcium signalling (116). Interestingly, these circRNAs were shown to interact with the RNA-binding protein TAR DNA-binding protein 43 KDa (TDP43) (116), indicating a possible correlation with alternative splicing and pre-mRNA turnover, and eventually NMD activity.
Discussion
Pancreatic β-cells must fine tune protein synthesis given large fluctuations between low and very high demands for insulin secretion. Post-transcriptional regulation plays an important role (33, 117). Transcript processing is, however, not limited only to alternative splicing events. Other homeostatic pathways including NMD directly, and UPR indirectly, are involved in this fine-tuning. Inflammatory cytokines and glucolipotoxicity are major drivers of ER stress in pancreatic β-cells leading to UPR and NMD activation and are likely needed to efficiently and accurately eliminate unproductive transcript isoforms (Figure 2C). If these remained intact, the production of truncated proteins may exert deleterious effects on β-cell function and viability.
With respect to the identified role of RBPs and alternative splicing factors in pancreatic β-cell function and viability, and given that over 90% of human genes transcribed into at least four transcript isoforms (17, 118–120), we suggest that transcript processing by the RUST mechanism may be mandatory to guarantee functional accuracy and integrity of pancreatic β-cells. In regard to the development, differentiation, function and resilience of pancreatic β-cells in health and in diabetes, we suggest that the following questions represent important areas for future research:
1. What is the role of the NMD in regulating normal transcripts?
2. Can we identify NMD-specific targets?
3. What is the contribution of the NMD to the RUST mechanism of eliminating unproductive transcript isoforms?
4. Do inflammatory and glucolipotoxic stresses exerts adverse effects on insulin biosynthesis and secretion, as well as cell viability, through changes in NMD activity?
5. If so, which branches of NMD are involved and are the key components indispensable in normal, stress and disease conditions?
Author Contributions
The first draft was written by SG and edited by GR and SG. All authors contributed to the article and approved the submitted version.
Funding
This study was supported within the independent postdoctoral grant (international mobility, grant number: 9034-00001B) for SMG by the Independent Research Fund Denmark (DFF-Medical Council). GR was supported by a Wellcome Trust Senior Investigator (WT098424AIA) and Investigator (WT212625/Z/18/Z) Awards, MRC Programme grants (MR/R022259/1, MR/J0003042/1, MR/L020149/1, MR/R022259/1) and Experimental Challenge Grant (DIVA, MR/L02036X/1), MRC (MR/N00275X/1), Diabetes UK (BDA/11/0004210, BDA/15/0005275, BDA 16/0005485), and Imperial Confidence in Concept (ICiC) grants. This project has received funding from the European Union’s Horizon 2020 research and innovation programme via the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115881 (RHAPSODY) to GR.
Conflict of Interest
GR has received grant funding and consultancy fees from Sun Pharmaceuticals and Les Laboratories Servier for unrelated studies.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
NMD, nonsense-mediated decay; AS, alternative splicing; PTCs, premature termination codons; T1D, type 1 diabetes; T2D, type 2 diabetes; RUST, Regulated Unproductive Splicing and Translation; SMD, Staufen1 (STAU1)-mediated mRNA decay; NGD, no-go decay; NSD, nonstop-decay; RBPs, RNA binding proteins; SREs, splicing regulatory elements; SRs, Serine Arginine rich proteins; hnRNP, heterogeneous nuclear ribonucleoprotein; ESE, exonic splicing enhancers; ISS, intronic splicing suppressors; IL-1β, interleukin-1β; IFNɣ, interferon-ɣ; NOD mice, Non-obese diabetic mice; HbA1c, haemoglobin A1c; TCF7L2, Transcription factor 7–like 2; Cask, calcium/calmodulin-dependent serine protein kinase; Madd, MAP kinase-activating death domain; uORF, upstream open reading frame; 3’-UTR, long 3’ untranslated region; eRF2, eukaryotic releasing factors 2; eRF3, eukaryotic releasing factors 3; EJC, exon-junction complex; PI3K, phosphoinositide 3-kinase; PNRC2, Proline-rich nuclear receptor co-activator 2; iNOS, inducible nitric oxide; NMA, N-methyl-L-arginine; MDS, myelodysplastic syndrome; ATAC-sequencing, assay for transposase-accessible chromatin with sequencing; cncRNA, coding-noncoding RNA; ncimRNA, noncoding mRNA isoforms; IRS1, Insulin receptor substrate 1; circRNA, circular RNA; TDP43, TAR DNA-binding protein 43 KDa; RB, retinoblastoma; Gsn, gelsolin; Cacna1c, calcium channel 1C; INSR, insulin receptor; PLC β1, Phospholipase Cβ1; SNAP-25, synaptosomal-associated protein 25; SNPs, Single nucleotide polymorphisms; UPF1-3, Up-Frameshift Suppressor 1 Homolog.
References
1. The International Diabetes Federation (IDF). Available at: https://idf.org/ (Accessed October 26, 2020).
2. Morgan NG, Richardson SJ. Fifty years of pancreatic islet pathology in human type 1 diabetes: insights gained and progress made. Diabetologia (2018) 61:2499–506. doi: 10.1007/s00125-018-4731-y
3. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab (2008) 10 Suppl 4:32–42. doi: 10.1111/j.1463-1326.2008.00969.x
4. Rutter GA, Georgiadou E, Martinez-Sanchez A, Pullen TJ. Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia (2020) 63:1990–8. doi: 10.1007/s00125-020-05205-5
5. Lytrivi M, Castell AL, Poitout V, Cnop M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J Mol Biol (2020) 432:1514–34. doi: 10.1016/j.jmb.2019.09.016
6. Eizirik DL, Pasquali L, Cnop M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol (2020) 16:349–62. doi: 10.1038/s41574-020-0355-7
7. Ghiasi SM, Krogh N, Tyrberg B, Mandrup-Poulsen T. The No-Go and Nonsense-Mediated RNA Decay Pathways Are Regulated by Inflammatory Cytokines in Insulin-Producing Cells and Human Islets and Determine beta-Cell Insulin Biosynthesis and Survival. Diabetes (2018) 67:2019–37. doi: 10.2337/db18-0073
8. Le K, Mitsouras K, Roy M, Wang Q, Xu Q, Nelson SF, et al. Detecting tissue-specific regulation of alternative splicing as a qualitative change in microarray data. Nucleic Acids Res (2004) 32:e180. doi: 10.1093/nar/gnh173
9. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PloS Genet (2012) 8:e1002552. doi: 10.1371/journal.pgen.1002552
10. Jamar NH, Kritsiligkou P, Grant CM. The non-stop decay mRNA surveillance pathway is required for oxidative stress tolerance. Nucleic Acids Res (2017) 45:6881–93. doi: 10.1093/nar/gkx306
11. Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F. and Dietz HC Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet (2004) 36:1073–8. doi: 10.1038/ng1429
12. Park E, Maquat LE. Staufen-mediated mRNA decay. Wiley Interdiscip Rev RNA (2013) 4:423–35. doi: 10.1002/wrna.1168
13. Chen W, Moore MJ. The spliceosome: disorder and dynamics defined. Curr Opin Struct Biol (2014) 24:141–9. doi: 10.1016/j.sbi.2014.01.009
14. Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol (2011) 3(7):a003707. doi: 10.1101/cshperspect.a003707
15. Ramanouskaya TV, Grinev VV. The determinants of alternative RNA splicing in human cells. Mol Genet Genomics (2017) 292:1175–95. doi: 10.1007/s00438-017-1350-0
16. da Costa PJ, Menezes J, Romao L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int J Biochem Cell Biol (2017) 91:168–75. doi: 10.1016/j.biocel.2017.07.013
17. Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science (2003) 302:2141–4. doi: 10.1126/science.1090100
18. Briese M, Haberman N, Sibley CR, Faraway R, Elser AS, Chakrabarti AM, et al. A systems view of spliceosomal assembly and branchpoints with iCLIP. Nat Struct Mol Biol (2019) 26:930–40. doi: 10.1038/s41594-019-0300-4
19. Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev (2004) 18:210–22. doi: 10.1101/gad.1163204
20. Jamison SF, Crow A, Garcia-Blanco MA. The spliceosome assembly pathway in mammalian extracts. Mol Cell Biol (1992) 12:4279–87. doi: 10.1128/mcb.12.10.4279
21. Jeong S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol Cells (2017) 40:1–9. doi: 10.14348/molcells.2017.2319
22. Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet (2016) 135:851–67. doi: 10.1007/s00439-016-1683-5
23. Stamm S, Zhu J, Nakai K, Stoilov P, Stoss O, Zhang MQ. An alternative-exon database and its statistical analysis. DNA Cell Biol (2000) 19:739–56. doi: 10.1089/104454900750058107
24. Xu Q, Modrek B, Lee C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res (2002) 30:3754–66. doi: 10.1093/nar/gkf492
25. Lipscombe D. Neuronal proteins custom designed by alternative splicing. Curr Opin Neurobiol (2005) 15:358–63. doi: 10.1016/j.conb.2005.04.002
26. Ule J, Darnell RB. RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr Opin Neurobiol (2006) 16:102–10. doi: 10.1016/j.conb.2006.01.003
27. Xie J. Control of alternative pre-mRNA splicing by Ca(++) signals. Biochim Biophys Acta (2008) 1779:438–52. doi: 10.1016/j.bbagrm.2008.01.003
28. Alvelos MI, Juan-Mateu J, Colli ML, Turatsinze JV, Eizirik DL. When one becomes many-Alternative splicing in beta-cell function and failure. Diabetes Obes Metab (2018) 20 Suppl 2:77–87. doi: 10.1111/dom.13388
29. Dlamini Z, Mokoena F, Hull R. Abnormalities in alternative splicing in diabetes: therapeutic targets. J Mol Endocrinol (2017) 59:R93–R107. doi: 10.1530/JME-17-0049
30. Juan-Mateu J, Alvelos MI, Turatsinze JV, Villate O, Lizarraga-Mollinedo E, Grieco FA, et al. SRp55 Regulates a Splicing Network That Controls Human Pancreatic beta-Cell Function and Survival. Diabetes (2018) 67:423–36. doi: 10.2337/db17-0736
31. Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic beta-cell identity, glucose sensing and the control of insulin secretion. Biochem J (2015) 466:203–18. doi: 10.1042/BJ20141384
32. Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS. Regulation of the insulin gene by glucose and fatty acids. J Nutr (2006) 136:873–6. doi: 10.1093/jn/136.4.873
33. Magro MG, Solimena M. Regulation of beta-cell function by RNA-binding proteins. Mol Metab (2013) 2:348–55. doi: 10.1016/j.molmet.2013.09.003
34. Knoch KP, Nath-Sain S, Petzold A, Schneider H, Beck M, Wegbrod C, et al. PTBP1 is required for glucose-stimulated cap-independent translation of insulin granule proteins and Coxsackieviruses in beta cells. Mol Metab (2014) 3:518–30. doi: 10.1016/j.molmet.2014.05.002
35. Evans-Molina C, Garmey JC, Ketchum R, Brayman KL, Deng S, Mirmira RG. Glucose regulation of insulin gene transcription and pre-mRNA processing in human islets. Diabetes (2007) 56:827–35. doi: 10.2337/db06-1440
36. Malakar P, Chartarifsky L, Hija A, Leibowitz G, Glaser B, Dor Y, et al. Insulin receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci Rep (2016) 6:31222. doi: 10.1038/srep31222
37. Juan-Mateu J, Villate O, Eizirik DL. MECHANISMS IN ENDOCRINOLOGY: Alternative splicing: the new frontier in diabetes research. Eur J Endocrinol (2016) 174:R225–38. doi: 10.1530/EJE-15-0916
38. Villate O, Turatsinze JV, Mascali LG, Grieco FA, Nogueira TC, Cunha DA, et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res (2014) 42:11818–30. doi: 10.1093/nar/gku861
39. Juan-Mateu J, Rech TH, Villate O, Lizarraga-Mollinedo E, Wendt A, Turatsinze JV, et al. Neuron-enriched RNA-binding Proteins Regulate Pancreatic Beta Cell Function and Survival. J Biol Chem (2017) 292:3466–80. doi: 10.1074/jbc.M116.748335
40. Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PloS Genet (2013) 9:e1003532. doi: 10.1371/journal.pgen.1003532
41. Marcheva B, Perelis M, Weidemann BJ, Taguchi A, Lin H, Omura C, et al. A role for alternative splicing in circadian control of exocytosis and glucose homeostasis. Genes Dev (2020) 34:1089–105. doi: 10.1101/gad.338178.120
42. Kachapati K, Bednar KJ, Adams DE, Wu Y, Mittler RS, Jordan MB, et al. Recombinant soluble CD137 prevents type one diabetes in nonobese diabetic mice. J Autoimmun (2013) 47:94–103. doi: 10.1016/j.jaut.2013.09.002
43. Type 2 diabetes knowledge portal. Available at: http://t2d.hugeamp.org/ (Accessed October 26, 2020).
44. Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet (2006) 38:320–3. doi: 10.1038/ng1732
45. Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature (2007) 445:881–5. doi: 10.1038/nature05616
46. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science (2007) 316:889–94. doi: 10.1126/science.1141634
47. Groves CJ, Zeggini E, Minton J, Frayling TM, Weedon MN, Rayner NW, et al. Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes (2006) 55:2640–4. doi: 10.2337/db06-0355
48. Osmark P, Hansson O, Jonsson A, Ronn T, Groop L, Renstrom E. Unique splicing pattern of the TCF7L2 gene in human pancreatic islets. Diabetologia (2009) 52:850–4. doi: 10.1007/s00125-009-1293-z
49. Le Bacquer O, Shu L, Marchand M, Neve B, Paroni F, Kerr Conte J, et al. TCF7L2 splice variants have distinct effects on beta-cell turnover and function. Hum Mol Genet (2011) 20:1906–15. doi: 10.1093/hmg/ddr072
50. da Silva Xavier G, Mondragon A, Sun G, Chen L, McGinty JA, French PM, et al. Abnormal glucose tolerance and insulin secretion in pancreas-specific Tcf7l2-null mice. Diabetologia (2012) 55:2667–76. doi: 10.1007/s00125-012-2600-7
51. Mitchell RK, Mondragon A, Chen L, McGinty JA, French PM, Ferrer J, et al. Selective disruption of Tcf7l2 in the pancreatic beta cell impairs secretory function and lowers beta cell mass. Hum Mol Genet (2015) 24:1390–9. doi: 10.1093/hmg/ddu553
52. Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med (2008) 359:2220–32. doi: 10.1056/NEJMoa0801869
53. Vinuela A, Varshney A, van de Bunt M, Prasad RB, Asplund O, Bennett A, et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat Commun (2020) 11:4912. doi: 10.1038/s41467-020-18581-8
54. Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, et al. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science (2015) 350:aac4250. doi: 10.1126/science.aac4250
55. Li LC, Wang Y, Carr R, Haddad CS, Li Z, Qian L, et al. IG20/MADD plays a critical role in glucose-induced insulin secretion. Diabetes (2014) 63:1612–23. doi: 10.2337/db13-0707
56. Kalyna M, Simpson CG, Syed NH, Lewandowska D, Marquez Y, Kusenda B, et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res (2012) 40:2454–69. doi: 10.1093/nar/gkr932
57. Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. Function of alternative splicing. Gene (2013) 514:1–30. doi: 10.1016/j.gene.2012.07.083
58. Huang L, Wilkinson MF. Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip Rev RNA (2012) 3:807–28. doi: 10.1002/wrna.1137
59. Hug N, Longman D, Caceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res (2016) 44:1483–95. doi: 10.1093/nar/gkw010
60. McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, et al. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA (2010) 107:12186–91. doi: 10.1073/pnas.1007336107
61. Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res (2010) 20:45–58. doi: 10.1101/gr.093302.109
62. Muhlemann O, Eberle AB, Stalder L, Zamudio Orozco R. Recognition and elimination of nonsense mRNA. Biochim Biophys Acta (2008) 1779:538–49. doi: 10.1016/j.bbagrm.2008.06.012
63. Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc Natl Acad Sci USA (1979) 76:5134–7. doi: 10.1073/pnas.76.10.5134
64. Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A (2003) 100:189–92. doi: 10.1073/pnas.0136770100
65. Green RE, Lewis BP, Hillman RT, Blanchette M, Lareau LF, Garnett AT, et al. Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics (2003) 19 Suppl 1:i118–21. doi: 10.1093/bioinformatics/btg1015
66. Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem (2007) 76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909
67. Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet (2004) 36:801–8. doi: 10.1038/ng1403
68. Fatscher T, Boehm V, Gehring NH. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell Mol Life Sci (2015) 72:4523–44. doi: 10.1007/s00018-015-2017-9
69. Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci Biobehav Rev (2014) 46 Pt 2:175–86. doi: 10.1016/j.neubiorev.2013.10.016
70. Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science (2007) 318:798–801. doi: 10.1126/science.1147182
71. Goetz AE, Wilkinson M. Stress and the nonsense-mediated RNA decay pathway. Cell Mol Life Sci (2017) 74:3509–31. doi: 10.1007/s00018-017-2537-6
72. Franks TM, Singh G, Lykke-Andersen J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell (2010) 143:938–50. doi: 10.1016/j.cell.2010.11.043
73. Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol (2015) 16:665–77. doi: 10.1038/nrm4063
74. Jaffrey SR, Wilkinson MF. Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat Rev Neurosci (2018) 19:715–28. doi: 10.1038/s41583-018-0079-z
75. Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol (2019) 20:406–20. doi: 10.1038/s41580-019-0126-2
76. Melero R, Uchiyama A, Castano R, Kataoka N, Kurosawa H, Ohno S, et al. Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure (2014) 22:1105–19. doi: 10.1016/j.str.2014.05.015
77. Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, et al. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev (2006) 20:355–67. doi: 10.1101/gad.1389006
78. Boehm V, Haberman N, Ottens F, Ule J, Gehring NH. 3’ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep (2014) 9:555–68. doi: 10.1016/j.celrep.2014.09.012
79. Schmidt SA, Foley PL, Jeong DH, Rymarquis LA, Doyle F, Tenenbaum SA, et al. Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res (2015) 43:309–23. doi: 10.1093/nar/gku1258
80. Karam R, Wengrod J, Gardner LB, Wilkinson MF. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta (2013) 1829:624–33. doi: 10.1016/j.bbagrm.2013.03.002
81. Unterholzner L, Izaurralde E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell (2004) 16:587–96. doi: 10.1016/j.molcel.2004.10.013
82. Yamashita A, Kashima I, Ohno S. The role of SMG-1 in nonsense-mediated mRNA decay. Biochim Biophys Acta (2005) 1754:305–15. doi: 10.1016/j.bbapap.2005.10.002
83. Cho H, Han S, Choe J, Park SG, Choi SS, Kim YK. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res (2013) 41:1319–28. doi: 10.1093/nar/gks1222
84. Fiorini F, Bagchi D, Le Hir H, Croquette V. Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nat Commun (2015) 6:7581. doi: 10.1038/ncomms8581
85. Pelechano V, Wei W, Steinmetz LM. Widespread Co-translational RNA Decay Reveals Ribosome Dynamics. Cell (2015) 161:1400–12. doi: 10.1016/j.cell.2015.05.008
86. Siwaszek A, Ukleja M, Dziembowski A. Proteins involved in the degradation of cytoplasmic mRNA in the major eukaryotic model systems. RNA Biol (2014) 11:1122–36. doi: 10.4161/rna.34406
87. Gong C, Kim YK, Woeller CF, Tang Y, Maquat LE. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev (2009) 23:54–66. doi: 10.1101/gad.1717309
88. Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell (2011) 42:500–10. doi: 10.1016/j.molcel.2011.04.018
89. Huang L, Lou CH, Chan W, Shum EY, Shao A, Stone E, et al. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell (2011) 43:950–61. doi: 10.1016/j.molcel.2011.06.031
90. Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Muhlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci (2010) 67:677–700. doi: 10.1007/s00018-009-0177-1
91. Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, et al. The Antagonistic Gene Paralogs Upf3a and Upf3b Govern Nonsense-Mediated RNA Decay. Cell (2016) 165:382–95. doi: 10.1016/j.cell.2016.02.046
92. Neu-Yilik G, Raimondeau E, Eliseev B, Yeramala L, Amthor B, Deniaud A, et al. Dual function of UPF3B in early and late translation termination. EMBO J (2017) 36:2968–86. doi: 10.15252/embj.201797079
93. Chan WK, Bhalla AD, Le Hir H, Nguyen LS, Huang L, Gecz J, et al. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat Struct Mol Biol (2009) 16:747–53. doi: 10.1038/nsmb.1612
94. Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, et al. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J (2015) 34:1630–47. doi: 10.15252/embj.201489947
95. Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet (2001) 10:99–105. doi: 10.1093/hmg/10.2.99
96. Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, et al. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev (2008) 22:1381–96. doi: 10.1101/gad.468808
97. Lou CH, Dumdie J, Goetz A, Shum EY, Brafman D, Liao X, et al. Nonsense-Mediated RNA Decay Influences Human Embryonic Stem Cell Fate. Stem Cell Rep (2016) 6:844–57. doi: 10.1016/j.stemcr.2016.05.008
98. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell (2012) 150:1223–34. doi: 10.1016/j.cell.2012.07.029
99. Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. beta Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab (2017) 25:727–38. doi: 10.1016/j.cmet.2017.01.005
100. Rutter GA, Hodson DJ. Author Correction: Beta cell connectivity in pancreatic islets: a type 2 diabetes target? Cell Mol Life Sci (2018) 75:1303–5. doi: 10.1007/s00018-018-2767-2
101. Cui K, Han Z. Association between disaster experience and quality of life: the mediating role of disaster risk perception. Qual Life Res (2019) 28:509–13. doi: 10.1007/s11136-018-2011-4
102. Ghiasi SM, Hansen JB, Christensen DP, Tyrberg B, Mandrup-Poulsen T. The Connexin 43 Regulator Rotigaptide Reduces Cytokine-Induced Cell Death in Human Islets. Int J Mol Sci (2020) 21(12):4311. doi: 10.3390/ijms21124311
103. Ghiasi SM, Dahllof MS, Osmai Y, Osmai M, Jakobsen KK, Aivazidis A, et al. Regulation of the beta-cell inflammasome and contribution to stress-induced cellular dysfunction and apoptosis. Mol Cell Endocrinol (2018) 478:106–14. doi: 10.1016/j.mce.2018.08.001
104. Eizirik DL, Mandrup-Poulsen T. A choice of death–the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia (2001) 44:2115–33. doi: 10.1007/s001250100021
105. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab (2019) 29:1045–1060 e10. doi: 10.1016/j.cmet.2019.01.021
106. Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, et al. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab (2019) 30:129–142 e4. doi: 10.1016/j.cmet.2019.05.006
107. McGlincy NJ, Smith CW. Alternative splicing resulting in nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends Biochem Sci (2008) 33:385–93. doi: 10.1016/j.tibs.2008.06.001
108. Zetoune AB, Fontaniere S, Magnin D, Anczukow O, Buisson M, Zhang CX, et al. Comparison of nonsense-mediated mRNA decay efficiency in various murine tissues. BMC Genet (2008) 9:83. doi: 10.1186/1471-2156-9-83
109. Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet (2008) 372:719–27. doi: 10.1016/S0140-6736(08)61168-X
110. Sampath K, Ephrussi A. CncRNAs: RNAs with both coding and non-coding roles in development. Development (2016) 143:1234–41. doi: 10.1242/dev.133298
111. Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, Tiao G, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature (2020) 578:102–11. doi: 10.1038/s41586-020-1965-x
112. Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, et al. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest (2000) 105:199–205. doi: 10.1172/JCI7917
113. Nagano H, Yamagishi N, Tomida C, Yano C, Aibara K, Kohno S, et al. A novel myogenic function residing in the 5’ non-coding region of Insulin receptor substrate-1 (Irs-1) transcript. BMC Cell Biol (2015) 16:8. doi: 10.1186/s12860-015-0054-8
114. Santer L, Bar C, Thum T. Circular RNAs: A Novel Class of Functional RNA Molecules with a Therapeutic Perspective. Mol Ther (2019) 27:1350–63. doi: 10.1016/j.ymthe.2019.07.001
115. Yu CY, Kuo HC. The emerging roles and functions of circular RNAs and their generation. J BioMed Sci (2019) 26:29. doi: 10.1186/s12929-019-0523-z
116. Stoll L, Rodriguez-Trejo A, Guay C, Brozzi F, Bayazit MB, Gattesco S, et al. A circular RNA generated from an intron of the insulin gene controls insulin secretion. Nat Commun (2020) 11:5611. doi: 10.1038/s41467-020-19381-w
117. Rhodes CJ. and White MF Molecular insights into insulin action and secretion. Eur J Clin Invest (2002) 32 Suppl 3:3–13. doi: 10.1046/j.1365-2362.32.s3.2.x
118. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature (2001) 409:860–921. doi: 10.1038/35057062
119. Davuluri RV, Suzuki Y, Sugano S, Plass C, Huang TH. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet (2008) 24:167–77. doi: 10.1016/j.tig.2008.01.008
Keywords: β-cell, insulin secretion, transcript, nonsense-mediated decay, RNA decay, RNA processing
Citation: Ghiasi SM and Rutter GA (2021) Consequences for Pancreatic β-Cell Identity and Function of Unregulated Transcript Processing. Front. Endocrinol. 12:625235. doi: 10.3389/fendo.2021.625235
Received: 02 November 2020; Accepted: 26 January 2021;
Published: 08 March 2021.
Edited by:
Anca Dana Dobrian, Eastern Virginia Medical School, United StatesReviewed by:
Scott Soleimanpour, University of Michigan, United StatesHelena Cristina Barbosa, State University of Campinas, Brazil
Copyright © 2021 Ghiasi and Rutter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed M. Ghiasi, cy5naGlhc2lAaW1wZXJpYWwuYWMudWs=; Guy A. Rutter, Zy5ydXR0ZXJAaW1wZXJpYWwuYWMudWs=