 Fatin Athirah Pauzi
Fatin Athirah Pauzi Elena Aisha Azizan
Elena Aisha Azizan- Department of Medicine, Faculty of Medicine, Universiti Kebangsaan Malaysia Medical Centre, Cheras, Malaysia
Primary aldosteronism (PA) is one of the most frequent curable forms of secondary hypertension. It can be caused by the overproduction of aldosterone in one or both adrenal glands. The most common subtypes of PA are unilateral aldosterone over-production due to aldosterone-producing adenomas (APA) or bilateral aldosterone over-production due to bilateral hyperaldosteronism (BHA). Utilizing the immunohistochemical (IHC) detection of aldosterone synthase (CYP11B2) has allowed the identification of aldosterone-producing cell clusters (APCCs) with unique focal localization positive for CYP11B2 expression in the subcapsular portion of the human adult adrenal cortex. The presence of CYP11B2 supports that synthesis of aldosterone can occur in these cell clusters and therefore might contribute to hyperaldosteronism. However, the significance of the steroidogenic properties of APCCs especially in regards to PA remains unclear. Herein, we review the available evidence on the presence of APCCs in normal adrenals and adrenal tissues adjacent to APAs, their aldosterone-stimulating somatic gene mutations, and their accumulation during the ageing process; raising the possibility that APCCs may play a role in the development of PA and age-related hypertension.
Introduction
Arterial hypertension is a worldwide health problem that affects approximately 1.13 billion of the global population (1) and is estimated to cause 7.5 million deaths or approximately 12.8% of the total all deaths (2). In more than 90% of patients, there is not a single cause for arterial hypertension, known as essential or primary hypertension (3). However, in certain cases, hypertension may emerge from a specific disease and therefore known as secondary hypertension, which includes endocrine hypertension that develops following a dysregulation of one or more hormones that are implicated in blood pressure regulation (4). Primary aldosteronism (PA), also known as Conn’s syndrome, is one of the most frequent curable forms of secondary/endocrine hypertension (5). This syndrome accounts for 7–10% of all referred hypertensive patients (6–8), 4% in primary care (9), and 15–20% of patients with resistant hypertension (10, 11). PA is caused by the autonomous aldosterone production in one or both adrenal glands, in which patients with PA are clinically associated with high levels of aldosterone despite being under suppressed renin conditions (12).
Under physiological conditions, the two primary systems that regulate aldosterone production are the renin-angiotensin-aldosterone system (RAAS) and the hypothalamic-pituitary-adrenal axis, in addition to blood potassium levels (13, 14). However, in PA, excess aldosterone independent of the RAAS acts on the distal tubule and medullary collecting duct to increase sodium reabsorption as well as potassium excretion (13). Water reabsorption follows salt, causing volume expansion that develops into hypertension and in some cases hypokalemic metabolic alkalosis (14). Hence, clinical presentation for hypokalemia, though not specific for PA, may also present as cramping and muscle weakness, palpitations, polydipsia, and polyuria among others (13). Furthermore, chronic inappropriate elevations of aldosterone levels in PA patients can increase pro-inflammatory cytokines (15), causing oxidative stress (16), which in the long-term leads to tissue damage and fibrosis (17). Compared to the age-, sex-, and blood pressure-matched essential hypertension patients, the risk for cardiovascular complications is significantly higher in PA patients (18, 19). This suggests the impact of autonomous aldosterone production on cardiovascular complications is beyond the increase in blood pressure (20–22).
Due to the high prevalence of PA among hypertensive patients and the increased risk for cardiovascular complications, early diagnosis as well as targeted treatment is of importance (12). The biochemical picture of patients with PA comprises of high aldosterone to renin ratio which has become one of the primary PA’s screening tools alongside different confirmatory tests, that includes fludrocortisone suppression test, intravenous saline infusion test, oral salt-loading test, or captopril test, which are all to be performed after a positive screening test (i.e. high aldosterone to renin ratio) (13, 18). The current guidelines recommend that when aldosterone is overproduced unilaterally, as detected by adrenal vein sampling (AVS), or in some exceptions when CT-scan detects a unilateral adenoma in a young hypokalemic PA patient (<35 years old), unilateral adrenalectomy is the treatment of choice. Conversely, mineralocorticoid receptor antagonists are utilized when AVS implies bilateral hyperaldosteronism or when the patient is not suitable for surgery (18). As identifying the most suitable therapy for PA is heavily influenced by the laterality of aldosterone overproduction, AVS, the gold standard to determine unilateralization (13), should be used where possible.
The majority of unilateral PA is due to an aldosterone-producing adenoma (APA), whereas bilateral PA, also known as bilateral hyperaldosteronism (BHA), are mostly due to adrenal zona glomerulosa hyperplasia or bilateral micronodular hyperplasia (23, 24). Bilateral PA can be further categorized into CT-negative idiopathic hyperaldosteronism (IHA) and rarely CT-positive bilateral APA (25–27). Approximately, APA and BHA account for 35%–40% and 60% of PA cases, respectively (28). In addition, there are less common subtypes of PA that include adrenal carcinoma, rare familial subtypes, and unilateral adrenal hyperplasia (29). It is postulated that bilateral PA could be caused by the accumulation of aldosterone-producing cell clusters (APCCs) (23), i.e. clusters of cells autonomously expressing aldosterone synthase (CYP11B2). Interestingly, these APCCs are not only found in the cells adjacent to an APA of which excised tissue is available (24, 30–33) but also in normal adrenal glands (30, 34, 35), with the prevalence seeming to increase with age (34–36). Hence, it is of importance to understand whether these clusters could contribute to the state of hyperaldosteronism or affect the increased prevalence of hypertension occurring with age.
Characteristics of Aldosterone-Producing Cell Clusters (APCCs)
Physiologically, aldosterone synthesis occurs in the most outer zone of the adrenal, the zona glomerulosa (ZG) (23). This is because the cells in this zone can express CYP11B2, the enzyme aldosterone synthase, which is essential for the final steps of aldosterone synthesis. The synthesis of aldosterone is primarily stimulated by the activation of intracellular calcium signaling in the ZG that can be mediated either by angiotensin II from the renin-angiotensin system or by extracellular potassium levels. Similar to aldosterone, adrenal cortisol steroid production physiologically occurs in distinct zones of the adrenal cortex, the zona fasciculata (ZF), due to the expression of 11β-hydroxylase (CYP11B1), the enzyme responsible for the final reaction in cortisol production. The synthesis of cortisol is primarily regulated by the hypothalamus-pituitary-adrenal axis mainly through the adrenocorticotropic hormone (ACTH) (4). Hence, in conventional adrenal zonation, CYP11B2 is only expressed in the ZG but not in ZF, whereas CYP11B1 is expressed in the ZF but not in the ZG (30, 37–40). However, immunohistochemical characterization of the adrenal gland by Nishimoto et al. (30) has led to the novel discovery of microscopic clusters of subcapsular adrenal cells that express CYP11B2 that extends from the ZG into the ZF which have been termed as APCCs. However, as the term “cell cluster” in APCCs does not describe the histology and pathology of the feature a recent consensus on histopathology of PA adrenals recommends the usage of the term “aldosterone-producing micronodule” instead of APCCs (41).
Nevertheless, whether termed APCCs or aldosterone-producing micronodule, the definition of this feature varies between research groups and a clear description that can distinguish this unique structure from APA and adjacent normal ZG cells has yet to be established. The size of APCCs has been reported to be smaller, approximately 0.2–1.5 mm in length, than APAs that are typically more than 3 mm in length (30, 36, 42). Further, APCCs have been characterized as having non-adenomatous features unlike that seen frequently with APA, e.g. not having a fibrous capsule or having cellular/tissue atypia (12). APCCs have also been described to be composed of morphological ZG cells in contact with the capsule and inner columnar ZF like cells forming cords along sinusoids (30). The current common definition for APCCs is to have positive CYP11B2 expression in a unique focal localization (30). As the morphology of APCC cells appears to be remarkably close to those of non-APCCs (33, 35), they cannot be easily distinguished from adjacent normal cortical cells by routine hematoxylin and eosin staining (30, 36, 42). Instead, immunohistochemistry (IHC) of CYP11B2 has been the method of choice to identify the presence of APCCs (30, 43).
IHC analysis has shown that compared to ZF cells that are generally negative for CYP11B2 and unrelated to aldosterone production, ZF-like looking cells of APCCs in the ZF has high levels of CYP11B2 and low levels of CYP11B1 or CYP17A1, both enzymes being involved in cortisol production (33, 35, 37, 38). However, according to Lim & Rainey (44), APCCs may manifest various patterns of enzyme expression with some exhibiting homogeneous expression of CYP11B2 while others exhibit polarity with a declining expression of CYP11B2 aligned with the development of CYP17A1 expression. This pattern of expression indicates that some APCCs cells may go through a transition to a more ZF-like phenotype, at least partially (44), thus making CYP11B1 and CYP17A1 IHC useful to characterize APCCs (33, 35). Another published method to detect APCCs is in situ hybridization of CYP11B2. Utilizing this method, Boulkroun et al. (45) distinguished three CYP11B2-expressing structures beneath the adrenal capsule that were termed as foci, megafoci and APCCs based on the size of cell clusters and their relative expression of a glomerulosa marker, Disable-2 (DAB2). In this study, APCCs were described as large clusters of CYP11B2 cells that did not express DAB2 indicating that APCCs contain cells presenting with an intermediate phenotype between ZG and ZF cells (45). However, on the transcriptomic level, Omata et al. (12) reported APCCs to be more similar to the adrenal ZG transcriptome than ZF or zona reticularis transcriptomes.
Physiological and Pathological Aspects of APCCs
APCCs are now accepted as a common feature in adrenal tissues adjacent to an APA (24, 30–33). Analysis of the adrenal glands of PA patients demonstrated that 40% to 50% of adrenals with APAs contained APCCs in the non-tumor portions of APA (30, 31). Interestingly, in Kometani et al. (32) study that found a 94% prevalence of APCCs in 16 adrenal tissue adjacent to the APA; the number and summed-area of APCCs in APAs were significantly higher in patients with discordant AVS results whose diagnosis changed to bilateral PA post-adrenocorticotropic hormone (ACTH) stimulation. The authors therefore proposed that APA patients with multiple APCCs might have similar adrenocortical pathological conditions in the contralateral adrenal gland. This assumption would explain their finding that in the pre-ACTH group having multiple APCCs, the post- to pre-ACTH plasma aldosterone concentrations (PACs) ratio was markedly higher on the non-dominant than the dominant side. Although the aldosterone responsiveness of APCCs towards ACTH has not been thoroughly described, it is conceivable that ACTH stimulation triggers the synthesis of aldosterone by APCCs in the contralateral adrenal gland, resulting in a decreased lateralization index (32). As RAAS is systemically suppressed in PA patients, these findings indicate that APCCs might play a role in autonomous aldosterone production (12). Concurringly, APCCs were also reported to be increased in adrenals of unilateral CT-negative PA compared to normotensive adrenals, implying that the elevated number of adrenal CYP11B2-expressing nodules might be the cause of hyperaldosteronism in these patients (33). Moreover, in one study that investigated the presence of APCCs in adrenals from IHA PA patients, all IHA adrenals were found to have at least one APCC or a microAPA (24). The number of APCCs in IHA adrenals were markedly larger compared to the cohorts of age-matched normotensive adrenals (24). Thus, these cell clusters could be the cause of hyperaldosteronism in these patients. This was proved to be the case in four patients with unilateral APCCs (aldosterone-producing micronodules not more than 3 mm in greatest dimension) whereby unilateral adrenalectomy resulted in a complete amelioration of blood pressure and hormonal abnormalities (46). Due to the small sizes of APCCs, patients with this feature may often be misdiagnosed as IHA as standard imaging test may not identify tiny anomalies of adrenals. Thus AVS, or if possible segmental selective AVS, should be used as part of the diagnostic procedure (47).
On the other hand, APCCs have not only been found in pathological adrenal tissues but also in normal adrenals where no tumors were present (30, 34, 35). In situ hybridization studies have found that APCCs in normal adrenals have a very similar profile to APCCs found in adrenal tissue adjacent to an APA (48, 49). In a study of nine Japanese patients with renal cell carcinoma (RCC) or upper urinary tract urothelial carcinoma (n=1), the presence of APCCs was found in eight of nine normal adrenals (30). Concurringly, another study from Japan on normotensive adrenal glands procured from an autopsy cohort found that out of 107 adrenals, 61 APCCs were detected in 31 autopsy cases (35). Similarly, a larger study by Nanba et al. (34) that analyzed the relationship between age and adrenal CYP11B2 expression, reported 69% of normal adrenal glands (88/127) had at least one APCC. Interestingly, this study also reported that although the total CYP11B2-expressing area negatively correlated with age (r=-0.431, P<0.0001), the total APCCs area positively correlated with age (r=0.390, P<0.0001) (34).
Significance of APCCs to Age-Related Hypertension
Owing to the fact that APCCs are frequently seen in non-hypertensive as well as pathological human adrenal glands expressing CYP11B2, several studies have investigated if these so-called APCCs detected through CYP11B2 expression could produce aldosterone. By utilizing in situ hybridization method, Shigematsu et al. (49) reported the presence of APCC-like subcapsular micronodules, showing intense transcript expression for HSD3B2, CYP11B1, and CYP11B2, but not CYP17A1, implying that the nodules have the steroidogenic enzymatic property needed to synthesize aldosterone and thus might be responsible for hyperaldosteronism. This was supported by Sugiura et al. (50) study of PA adrenal sections using Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) and tandem mass spectrometry imaging that demonstrated high levels of aldosterone and 18-oxocortisol, a potential serum marker of APA, in APCCs. Similarly, a recent study using tandem mass spectrometry imaging verified Suguira et al’s findings in normal adrenals as elevated levels of aldosterone and 18-oxocortisol were found in APCC’s relative to the adjacent adrenal tissue in an adrenal obtained from a patient that had undergone radical nephrectomy for renal cell carcinoma (51). These studies support the steroidogenic activity of APCCs in producing aldosterone in normal adrenals as well as under suppressed renin conditions, as shown in PA patients’ adrenals (44). Accordingly, APCCs has been proposed to be the transitioning step to APAs (50).
Based on aldosterone studies from their groups and others, Sugiura et al. (50) suggested that with ageing, APCCs accumulate aldosterone-driver mutations causing autonomous aldosterone production that can lead to APCC-to-APA translational lesions (pAATL). This is as with age, there is a loss of the classical subcapsular CYP11B2-positive ZG due to the effects of trophic hormones that lead to the decline in total aldosterone production. Thus within this background of decreased ZG CYP11B2 expression, the number of CYP11B2-positive APCCs grows (34, 44). Finally, the expansion of the mutation bearing APCCs leads to the generation of an aldosterone-producing adrenal lesion. The age-related progressive pattern of APCCs has been validated by several studies that showed the number of adrenal APCCs increased with ageing in normal adrenals from kidney donors (34) and non-hypertensive Japanese cohorts (33), signifying the contribution of APCCs in the early stages of PA in ageing adults. Altogether, the findings of APCCs in normal adrenals and the adrenal glands of PA patients with APAs, as well as their accumulation during the ageing process, has brought up the possibility that APCCs might play a role in the development of APAs and therefore is a continuum of PA.
Somatic Gene Mutations of APCCs
Whole exome sequencing studies on APAs have defined recurrent somatic mutations in genes coding for ion channels (KCNJ5, CACNA1H, and CACNA1D) and ATPases (ATP1A1 and ATP2B3) that either caused cytosolic acidification (for ATP1A1) or activated intracellular calcium signaling by depolarization of ZG cell membrane that opens the voltage-gated calcium channels or by affecting the intracellular calcium recycling (4, 52–56). This ultimately results in an increase of CYP11B2 expression that leads to aldosterone overproduction. The promising discovery of recurrent somatic mutations in APAs through the application of next-generation sequencing (52–55) has driven several studies to utilize this platform to determine the prevalence of aldosterone-driving somatic mutations in APCCs. From a cohort of 42 normal adrenals among kidney donors, 8 of 23 APCCs (35%) were found to harbor known APA-associated driver mutations, predominantly in CACNA1D (26%) followed by ATP1A1 (9%) (43). However, this study did not find any APCCs to harbor the KCNJ5 mutations, the most common mutations found in APAs (43). Similarly, in another study that performed next-generation sequencing analysis on CYP11B2-expressing nodules from the adrenals of PA patients that were CT-negative for adenomas, APCC-like micronodules were found to pre-dominantly harbor CACNAID somatic mutations (33).
Compared to KCNJ5 mutant APAs that have been described as more prevalent in young women with higher levels of plasma aldosterone and larger ZF-like APAs (12, 57–60), CACNA1D mutations are associated with smaller ZG-like APAs in older men that are more difficult to be diagnosis (43, 53, 60). CACNA1D encodes for the voltage-dependent L-type (long-lasting) calcium channel subunit alpha-1D (the Cav1.3 calcium channel) (52, 53). This calcium channel is composed of 4 homologous repeated domains with 6 transmembrane segments (S1-S6) each and a membrane-associated loop between S5 and S6 (52, 53, 61). Mutations occurring in CACNA1D are gain of function mutations that lead to a shift of the voltage-dependent channel activation to more negative voltages or delay the inactivation of the channel. The resulting net effect is increased intracellular calcium concentrations and thereby induction of excessive aldosterone synthesis (52, 53). Hence, the high prevalence of APCCs with CACNA1D mutations suggests that an intracellular increase of calcium is causal for the high CYP11B2 expression in APCCs (62).
A study on 107 adrenal glands of normotensive Japanese patients from an 837 consecutive autopsy cohort was conducted to test the hypothesis that hyperaldosteronism in the group of CT-negative PA patients was induced by the elevated number of adrenal CYP11B2-expressing nodules (33, 35). The study in adrenals from non-hypertensive Japanese patients demonstrated that APCCs were common with 34% harboring known aldosterone-driver somatic mutations, CACNA1D being the most frequent mutations while there being no detections of KCNJ5 mutant APCCs (35). The mutations were found to be present in both the ZG and ZF-like components of APCCs and absent in neighboring cells negative for CYP11B2 (35). The similarity of APCCs somatic mutation spectrum observed in this normotensive cohort to that found in PA patients that had adrenals that were CT-negative for adenomas implied that normal adrenal glands may progress to PA through an increase in the APCCs frequency (12). Concurringly, a study on the IHA subtype of PA found that in 15 IHA adrenals, 99 APCCs were detected of which 58% had a CACNA1D mutation thus supporting the notion that PA in IHA may result from the accumulation or enlargement of CT-undetectable APCCs harboring APA-associated somatic mutations that increases aldosterone production (24).
The high presence of aldosterone-driver mutations in APCCs, especially in CACNA1D and ATP1A1 (as shown in Table 1), supports the suggestion that APCCs are capable of autonomous aldosterone production and therefore could be the origin of APAs. Nishimoto et al. (42, 43) proposed that the series of events contributing to emergence of APAs from APCCs arises through the formation of possible APCC-to-APA transitional lesions (pAATLs) that has been characterized to consist of an outer APCC-like portion and an inner micro-APA (mAPA)-like portion (42). Interestingly, in one pAATL, the same ATP1A1 mutation has been found in the APCC-like portion and the mAPA-like portion of the pAATL, indicating that these portions have a clonal origin (42). However, the genetic characteristic of pAATL is heterogeneous. In one adrenal gland, Nishimoto et al. (42) found the mAPA-like portions of pAATLs examined harbored KCNJ5 mutations whereas their corresponding APCC-like portions did not, with the mutation being different from that identified in the APA within the same adrenal. This indicates that the APA and the pAATL had different origins and that the mAPA-like portion arises from existing APCCs due to the introduction of a KCNJ5 mutation that differentiated the mAPA portion from the APCC. The lack of detection of KCNJ5 mutation in the APCC-like portion could be due to a rapid progression to APA, therefore hard to be witnessed prior to its development (43, 63). However, some authors postulate that KCNJ5 mutation could rather be a second hit mutation (than a causal mutation of pAATL), as only pAATL nodules larger than 3 mm have been reported to harbor KCNJ5 mutations (63). Thus, these observational findings require further functional analysis to better elucidate the genomic events involved in the transition of APCCs to APA.
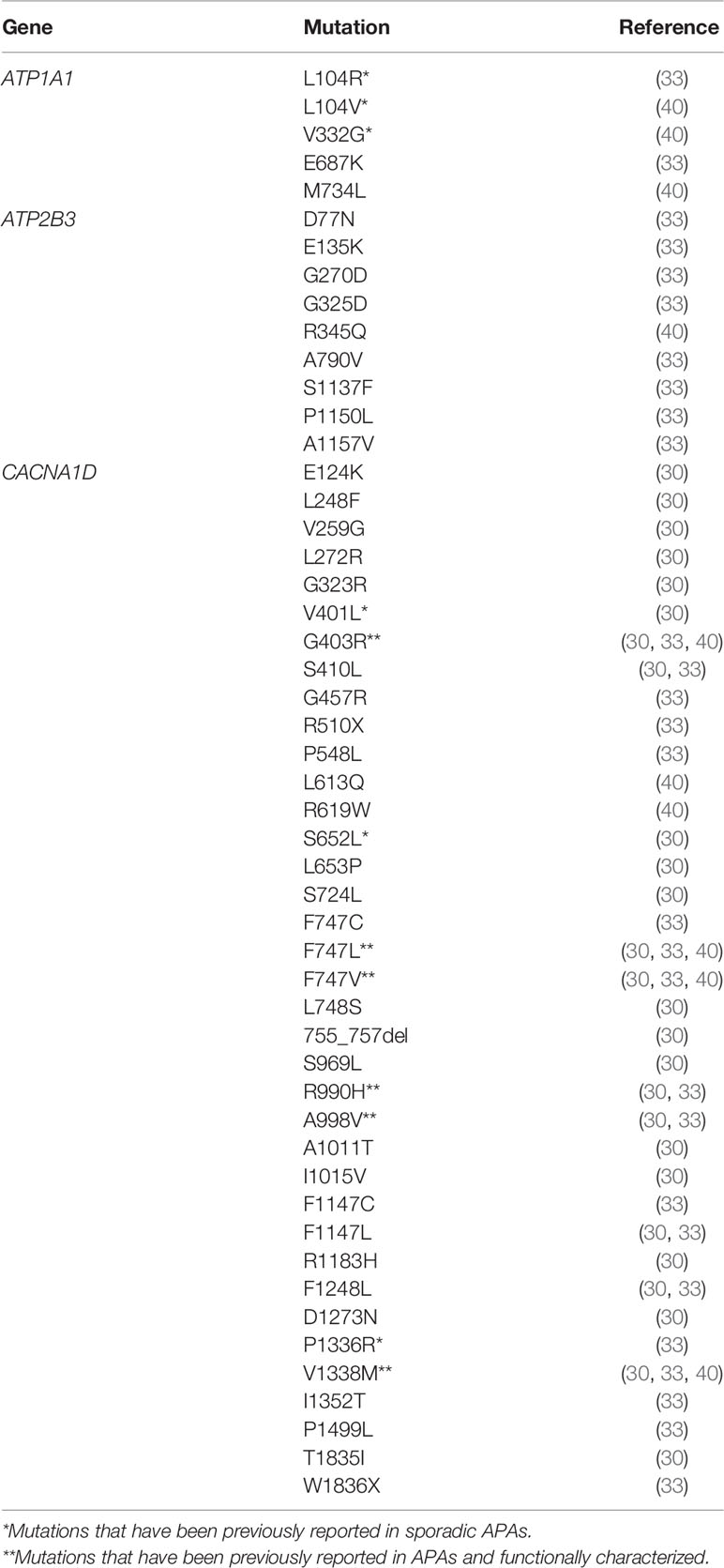
Table 1 Somatic gene mutations detected in APCCs.
Conclusions
In conclusion, several studies have found the presence of aldosterone-driving somatic gene mutations, similar to those found in APAs, among APCCs which supports the autonomous secretion of aldosterone by these clusters of cells and its role in the pathology of PA. The high frequency of CACNA1D and ATP1A1 mutations, but not KCNJ5 mutations in APCCs, suggest that these cells could be a potential precursor for ZG-like APAs rather than ZF-like APAs. Further, there is supporting evidence to suggest that accumulation of APCCs with age might contribute to the increased prevalence of hypertension occurring with age. More studies on APCCs are necessary to clarify the pathological mechanisms (and perhaps physiological as also found in “normal” adrenals) in regulating aldosterone production in these clusters of cells. As technology develops, whole-genome sequencing may be the important step forward to reveal novel gene mutations that lead to the development of aldosterone-driving somatic mutation. Identifications of germline variants may help to elucidate the potential mechanisms and pathways that lead to accumulation of aldosterone-driving somatic mutations and thus may consequently provide novel personalized treatment for age-related hypertension.
Author Contributions
EA and FP conceived the presented idea. EA supervised and verified the concept. FP prepared the original draft. EA and FP reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
FP is supported by the program Malaysia Partnership and Alliances in Research (MyPAiR) Newton-MRC UK-MY (NEWTON-MRC/2020/002). EA is supported by the Royal Society-Newton Advanced Fellowship (FF-2018-033/NA170257).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Suerialoasan Navanesan for proofreading this article.
References
1. Zhou B, Bentham J, Di Cesare M, Bixby H, Danaei G, Cowan MJ, et al. Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19·1 million participants. Lancet (2017) 389(10064):37–55. doi: 10.1016/S0140-6736(16)31919-5
2. Mendis S. World Health Organization. Global status report on non communicable diseases 2010. (2010). http://www.who.int/nmh/publications/ncd_report2010/en.
3. Zahedmehr A. “Chapter 17 - Hypertension”. In: Maleki M, Alizadehasl A, Haghjoo M.B.T.-P.C., editors. Practical Cardiology. St. Louis, Missouri: Eds.; Elsevier (2018). p. 291–302.
4. El Zein RM, Boulkroun S, Fernandes-Rosa FL, Zennaro MC. Molecular genetics of Conn adenomas in the era of exome analysis. Presse Med (2018) 47(7-8P2):e151–8. doi: 10.1016/j.lpm.2018.07.006
5. Mohler ER, Townsend RR. “Elevated Aldosterone Renin Ratio”. In: Mohler ER, Townsend RR, editors. Advanced Therapy in Hypertension and Vascular Disease. Hamilton, Ontario: Eds.; B.C. Decker (2006). p. 321–8.
6. Käyser SC, Dekkers T, Groenewoud HJ, van der Wilt GJ, Carel Bakx J, van der Wel MC, et al. Study Heterogeneity and Estimation of Prevalence of Primary Aldosteronism: A Systematic Review and Meta-Regression Analysis. J Clin Endocrinol Metab (2016) 101(7):2826–35. doi: 10.1210/jc.2016-1472
7. Monticone S, D’Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol (2018) 6(1):41–50. doi: 10.1016/S2213-8587(17)30319-4
8. Williams B, MacDonald TM, Morant SV, Webb DJ, Sever P, McInnes GT, et al. Endocrine and haemodynamic changes in resistant hypertension, and blood pressure responses to spironolactone or amiloride: the PATHWAY-2 mechanisms substudies. Lancet Diabetes Endocrinol (2018) 6(6):464–75. doi: 10.1016/S2213-8587(18)30071-8
9. Hannemann A, Wallaschofski H. Prevalence of primary aldosteronism in patient’s cohorts and in population-based studies–a review of the current literature. Hormone Metab Res (2012) 44(3):157–62. doi: 10.1055/s-0031-1295438
10. Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism Among Black and White Subjects With Resistant Hypertension. Hypertension (2002) 40(6):892–6. doi: 10.1161/01.HYP.0000040261.30455.B6
11. Douma S, Petidis K, Doumas M, Papaefthimiou P, Triantafyllou A, Kartali N, et al. Prevalence of primary hyperaldosteronism in resistant hypertension: a retrospective observational study. Lancet (2008) 371(9628):1921–6. doi: 10.1016/S0140-6736(08)60834-X
12. Omata K, Tomlins SA, Rainey WE. Aldosterone-producing cell clusters in normal and pathological states. Hormone Metab Res (2017) 49(12):951–6. doi: 10.1055/s-0043-122394
13. Deipolyi AR, Oklu R. Adrenal vein sampling in the diagnosis of aldosteronism. J Vasc Diagn Interventions (2015) 3:17–23. doi: 10.2147/JVD.S79302
14. Bassett MH, White PC, Rainey WE. The regulation of aldosterone synthase expression. Mol Cell Endocrinol (2004) 217(1–2):67–74. doi: 10.1016/j.mce.2003.10.011
15. Lim JS, Park S, Park S II, Oh YT, Choi E, Kim JY, et al. Cardiac Dysfunction in Association with Increased Inflammatory Markers in Primary Aldosteronism. Endocrinol Metab (Seoul Korea) (2016) 31(4):567–76. doi: 10.3803/EnM.2016.31.4.567
16. Zia AA, Kamalov G, Newman KP, McGee JE, Bhattacharya SK, Ahokas RA, et al. From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation. Hypertens Res (2010) 33(11):1091–101. doi: 10.1038/hr.2010.159
17. Herrada AA, Campino C, Amador CA, Michea LF, Fardella CE, Kalergis AM. Aldosterone as a modulator of immunity: implications in the organ damage. J Hypertens (2011) 29(9):1684–92. doi: 10.1097/HJH.0b013e32834a4c75
18. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2016) 101(5):1889–916. doi: 10.1210/jc.2015-4061
19. Milliez P, Girerd X, Plouin P-F, Blacher J, Safar M, Mourad J-J. Evidence for an Increased Rate of Cardiovascular Events in Patients With Primary Aldosteronism. J Am Coll Cardiol (2005) 45(8):1243–8. doi: 10.1016/j.jacc.2005.01.015
20. Yang Y, Reincke M, Williams TA. Prevalence, diagnosis and outcomes of treatment for primary aldosteronism. Best Pract Res Clin Endocrinol Metab (2019) 34(2):101365. doi: 10.1016/j.beem.2019.101365
21. Rossi GP. Primary Aldosteronism. J Am Coll Cardiol (2019) 74(22):2799–811. doi: 10.1016/j.jacc.2019.09.057
22. Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol (2018) 6(1):51–9. doi: 10.1016/S2213-8587(17)30367-4
23. Gomez-Sanchez C, Kuppusamy M, Reincke M, Williams T. Disordered CYP11B2 Expression in Primary Aldosteronism. Hormone Metab Res (2017) 49:957–62. doi: 10.1055/s-0043-122238
24. Omata K, Satoh F, Morimoto R, Ito S, Yamazaki Y, Nakamura Y, et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension (2018) 72(4):874–80. doi: 10.1161/HYPERTENSIONAHA.118.11086
25. Satoh F, Morimoto R, Seiji K, Satani N, Ota H, Iwakura Y, et al. Is there a role for segmental adrenal venous sampling and adrenal sparing surgery in patients with primary aldosteronism? Eur J Endocrinol (2015) 173(4):465–77. doi: 10.1530/EJE-14-1161
26. Wu VC, Chueh SC, Chang HW, Lin WC, Liu KL, Li HY, et al. Bilateral aldosterone-producing adenomas: differentiation from bilateral adrenal hyperplasia. QJM: Int J Med (2008) 101(1):13–22. doi: 10.1093/qjmed/hcm101
27. Sonoyama T, Sone M. Regulatory Role of ACTH on Aldosterone in Aldosterone-Producing Adenoma. J Rare Dis Res Treat (2017) 2(3):30–3. doi: 10.29245/2572-9411/2017/3.1101
28. Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (2007) 66(5):607–18. doi: 10.1111/j.1365-2265.2007.02775.x
29. Vonend O, Ockenfels N, Gao X, Allolio B, Lang K, Mai K, et al. Adrenal Venous Sampling Evaluation of the German Conn’s Registry. Hypertension (2011) 57(5):990–5. doi: 10.1161/HYPERTENSIONAHA.110.168484
30. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical Zonation in Humans under Normal and Pathological Conditions. J Clin Endocrinol Metab (2010) 95(5):2296–305. doi: 10.1210/jc.2009-2010
31. Nanba K, Tsuiki M, Sawai K, Mukai K, Nishimoto K, Usui T, et al. Histopathological diagnosis of primary aldosteronism using CYP11B2 immunohistochemistry. J Clin Endocrinol Metab (2013) 98(4):1567–74. doi: 10.1210/jc.2012-3726
32. Kometani M, Yoneda T, Aono D, Karashima S, Demura M, Nishimoto K, et al. Impact of aldosterone-producing cell clusters on diagnostic discrepancies in primary aldosteronism. Oncotarget (2018) 9(40):26007–18. doi: 10.18632/oncotarget.25418
33. Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, et al. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J Clin Endocrinol Metab (2017) 102(4):1182–92. doi: 10.1210/jc.2016-2986
34. Nanba K, Vaidya A, Williams GH, Zheng I, Else T, Rainey WE. Age-Related Autonomous Aldosteronism. Circulation (2017) 136(34):347–55. doi: 10.1161/CIRCULATIONAHA.117.028201
35. Omata K, Anand SK, Hovelson DH, Liu C-J, Yamazaki Y, Nakamura Y, et al. Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate With Age in Normal Adrenals. J Endocrine Soc (2017) 1(7):787–99. doi: 10.1210/js.2017-00134
36. Nishimoto K, Seki T, Hayashi Y, Mikami S, Al-eyd G, Nakagawa K, et al. Human Adrenocortical Remodeling Leading to Aldosterone-Producing Cell Cluster Generation. Int J Endocrinol (2016) 2016(7834356):6. doi: 10.1155/2016/7834356
37. Nakamura Y, Maekawa T, Felizola SJA, Satoh F, Qi X, Velarde-Miranda C, et al. Adrenal CYP11B1/2 expression in primary aldosteronism: Immunohistochemical analysis using novel monoclonal antibodies. Mol Cell Endocrinol (2014) 392(1):73–9. doi: 10.1016/j.mce.2014.05.002
38. Gomez-Sanchez C, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol (2014) 383(1–2):111–7. doi: 10.1016/j.mce.2013.11.022
39. Miller WL, Auchus RJ. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocrine Rev (2011) 32(1):81–151. doi: 10.1210/er.2010-0013
40. Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol (2012) 350(2):151–62. doi: 10.1016/j.mce.2011.07.034
41. Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, et al. International histopathology consensus for unilateral primary aldosteronism. J Gerontol Ser A: Biol Sci Med Sci (2021) 106(1):42–54. doi: 10.1210/clinem/dgaa484
42. Nishimoto K, Seki T, Kurihara I, Yokota K, Omura M, Nishikawa T, et al. Case Report: Nodule Development From Subcapsular Aldosterone-Producing Cell Clusters Causes Hyperaldosteronism. J Clin Endocrinol Metab (2016) 101(1):6–9. doi: 10.1210/jc.2015-3285
43. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A (2015) 112(33):E4591–9. doi: 10.1073/pnas.1505529112
44. Lim JS, Rainey WE. The Potential Role of Aldosterone-Producing Cell Clusters in Adrenal Disease. Hormone Metab Res (2020) 52(6):427–34. doi: 10.1055/a-1128-0421
45. Boulkroun S, Samson-Couterie B, Golib Dzib JF, Lefebvre H, Louiset E, Amar L, et al. Adrenal cortex remodeling and functional zona glomerulosa hyperplasia in primary aldosteronism. Hypertension (2010) 56(5):885–92. doi: 10.1161/HYPERTENSIONAHA.110.158543
46. Omura M, Sasano H, Fujiwara T, Yamaguchi K, Nishikawa T. Unique cases of unilateral hyperaldosteronemia due to multiple adrenocortical micronodules, which can only be detected by selective adrenal venous sampling. Metabol: Clin Exp (2002) 51(3):350–5. doi: 10.1053/meta.2002.30498
47. Kitamoto T, Kitamoto KK, Omura M, Takiguchi T, Tsurutani Y, Kubo H, et al. Precise Mapping of Intra-Adrenal Aldosterone Activities Provides a Novel Surgical Strategy for Primary Aldosteronism. Hypertension (2020) 76(3):976–84. doi: 10.1161/HYPERTENSIONAHA.119.14341
48. Enberg U, Volpe C, Hoog A, Wedell A, Farnebo LO, Thoren M, et al. Postoperative differentiation between unilateral adrenal adenoma and bilateral adrenal hyperplasia in primary aldosteronism by mRNA expression of the gene CYP11B2. Eur J Endocrinol (2004) 151(1):73–85. doi: 10.1530/eje.0.1510073
49. Shigematsu K, Kawai K, Irie J, Sakai H, Nakashima O, Iguchi A, et al. Analysis of Unilateral Adrenal Hyperplasia with Primary Aldosteronism from the Aspect of Messenger Ribonucleic Acid Expression for Steroidogenic Enzymes: A Comparative Study with Adrenal Cortices Adhering to Aldosterone-Producing Adenoma. Endocrinology (2006) 147(2):999–1006. doi: 10.1210/en.2005-0765
50. Sugiura Y, Takeo E, Shimma S, Yokota M, Higashi T, Seki T, et al. Aldosterone and 18-oxocortisol coaccumulation in aldosterone-producing lesions. Hypertension (2018) 72(6):1345–54. doi: 10.1161/HYPERTENSIONAHA.118.11243
51. Takeo E, Sugiura Y, Uemura T, Nishimoto K, Yasuda M, Sugiyama E, et al. Tandem Mass Spectrometry Imaging Reveals Distinct Accumulation Patterns of Steroid Structural Isomers in Human Adrenal Glands. Analyt Chem (2019) 91(14):8918–25. doi: 10.1021/acs.analchem.9b00619
52. Scholl UI, Goh G, Oliveira RC, De Choi M, Overton JD, Fonseca AL, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45: (9):1050–4. doi: 10.1038/ng.2695
53. Azizan EAB, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45(9):1055–60. doi: 10.1038/ng.2716
54. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (New York NY) (2011) 331(6018):768–72. doi: 10.1126/science.1198785
55. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45(4):440–4. doi: 10.1038/ng.2550
56. Stindl J, Tauber P, Sterner C, Tegtmeier I, Warth R, Bandulik S. Pathogenesis of Adrenal Aldosterone-Producing Adenomas Carrying Mutations of the Na+/K+-ATPase. Endocrinology (2015) 156(12):4582–91. doi: 10.1210/en.2015-1466
57. Boulkroun S, Beuschlein F, Rossi G-P, Golib-Dzib J-F, Fischer E, Amar L, et al. Prevalence, Clinical, and Molecular Correlates of KCNJ5 Mutations in Primary Aldosteronism. Hypertension (2012) 59(3):592–8. doi: 10.1161/HYPERTENSIONAHA.111.186478
58. Rossi GP, Cesari M, Letizia C, Seccia TM, Cicala MV, Zinnamosca L, et al. KCNJ5 gene somatic mutations affect cardiac remodelling but do not preclude cure of high blood pressure and regression of left ventricular hypertrophy in primary aldosteronism. J Hypertens (2014) 32(7):1514–21. doi: 10.1097/HJH.0000000000000186
59. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GPA. Meta-Analysis of Somatic KCNJ5 K+ Channel Mutations In 1636 Patients With an Aldosterone-Producing Adenoma. J Clin Endocrinol Metab (2015) 100(8):E1089–95. doi: 10.1210/jc.2015-2149
60. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic Spectrum and Clinical Correlates of Somatic Mutations in Aldosterone-Producing Adenoma. Hypertension (2014) 64(2):354–61. doi: 10.1161/HYPERTENSIONAHA.114.03419
61. Catterall WA. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett (2010) 486(2):107–16. doi: 10.1016/j.neulet.2010.08.085
62. Zennaro M-C, Boulkroun S, Fernandes-Rosa F. Genetic Causes of Functional Adrenocortical Adenomas. Endocrine Rev (2017) 38(6):516–37. doi: 10.1210/er.2017-00189
Keywords: primary aldosteronism, aldosterone, adrenal, aldosterone-producing cell clusters, hypertension
Citation: Pauzi FA and Azizan EA (2021) Functional Characteristic and Significance of Aldosterone-Producing Cell Clusters in Primary Aldosteronism and Age-Related Hypertension. Front. Endocrinol. 12:631848. doi: 10.3389/fendo.2021.631848
Received: 23 November 2020; Accepted: 01 February 2021;
Published: 03 March 2021.
Edited by:
Vin-Cent Wu, National Taiwan University, TaiwanReviewed by:
Silvia Monticone, University of Turin, ItalyTetsuo Nishikawa, Yokohama Rosai Hospital, Japan
Celso E. Gomez-Sanchez, University of Mississippi Medical Center School of Dentistry, United States
Copyright © 2021 Pauzi and Azizan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Aisha Azizan, ZWxlbmEuYXppemFuQHVrbS5lZHUubXk=