 Inbal Halabi1,2
Inbal Halabi1,2 Marie Noufi Barohom1,2,3Sarit Peleg4Phillippe Trougouboff5
Marie Noufi Barohom1,2,3Sarit Peleg4Phillippe Trougouboff5 Ghadir Elias-Assad1,6Rhania Agbaria7
Ghadir Elias-Assad1,6Rhania Agbaria7 Yardena Tenenbaum-Rakover1,6*
Yardena Tenenbaum-Rakover1,6*- 1Pediatric Endocrine Institute, Ha’Emek Medical Center, Afula, Israel
- 2Pediatric Health Center, Clalit Health Services, Naharia, Israel
- 3Faculty of Medicine, Bar Ilan University, Safed, Israel
- 4Pediatric Health Center, Clalit Health Services, Hadera, Israel
- 5Tissue Diagnosis and Cancer Research Department, Ha’Emek Medical Center, Afula, Israel
- 6The Ruth & Bruce Rappaport Faculty of Medicine, Technion, Haifa, Israel
- 7Pediatric Gastroenterology Unit, Ha’Emek Medical Center, Afula, Israel
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare monogenic disorder, associated with endocrine deficiencies and non-endocrine involvement. Gastrointestinal (GI) manifestations appear in approximately 25% of patients and are the presenting symptom in about 10% of them. Limited awareness among pediatricians of autoimmune enteropathy (AIE) caused by destruction of the gut endocrine cells in APECED patients delays diagnosis and appropriate therapy. We describe an 18-year-old female presenting at the age of 6.10 years with hypoparathyroidism, oral candidiasis and vitiligo. The clinical diagnosis of APECED was confirmed by sequencing the autoimmune regulator-encoding (AIRE) gene. Several characteristics of the disease—Hashimoto’s thyroiditis, Addison’s disease, diabetes mellitus type 1 and primary ovarian insufficiency—developed over the years. She had recurrent episodes of severe intractable hypocalcemia. Extensive GI investigations for possible malabsorption, including laboratory analyses, imaging and endoscopy with biopsies were unremarkable. Revision of the biopsies and chromogranin A (CgA) immunostaining demonstrated complete loss of enteroendocrine cells in the duodenum and small intestine, confirming the diagnosis of AIE. Management of hypocalcemia was challenging. Only intravenous calcitriol maintained calcium in the normal range. Between hypocalcemic episodes, the proband maintained normal calcium levels, suggesting a fluctuating disease course. Repeated intestinal biopsy revealed positive intestinal CgA immunostaining. The attribution of severe hypocalcemic episodes to AIE emphasizes the need for increased awareness of this unique presentation of APECED. The fluctuating disease course and repeated intestinal biopsy showing positive CgA immunostaining support a reversible effect of GI involvement. CgA immunostaining is indicated in patients with APECED for whom all other investigations have failed to reveal an explanation for the malabsorption.
Introduction
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), also known as autoimmune polyendocrine syndrome type 1, is a rare monogenic disorder caused by mutations in the autoimmune regulator gene (AIRE; OMIM 240300). AIRE plays a critical role in central immune tolerance (1, 2). The classic triad manifestations of APECED are chronic mucocutaneous candidiasis, hypoparathyroidism and adrenal insufficiency. Developing any two of these three major components of the disease establishes the diagnosis clinically. Multiple organs with autoimmune involvement have been reported in patients with APECED (3–8).
Gastrointestinal (GI) manifestations appear in approximately 25% of APECED patients and in about 10%, they are the presenting symptom. GI symptoms include malabsorption, constipation, watery diarrhea or steatorrhea (9). Autoimmune enteropathy (AIE) as part of APECED was first described in 1982 (9) but since then, only small series and case reports have been published (10–12). The mechanism leading to GI involvement in AIE is still not completely understood. Reduction or absence of enteroendocrine (EE) cells has been shown to be the main specific contributor to intestinal dysfunction (10–12).
The EE cells are scattered throughout the GI tract and constitute the gut endocrine system. Different EE cell types produce different gut hormones. These cells have important roles in gut growth, blood flow, motility, and secretion of pancreatic enzymes, bile and bicarbonate-rich fluid (13–15). The most common type of EE cell is the enterochromaffin cell, which secretes mainly serotonin and is dispersed throughout the GI system. Serotonin is synthesized by the hydroxylation and decarboxylation of tryptophan (14), and tryptophan hydroxylase has been identified as the intestinal autoantigen in APECED (16); moreover, anti-tryptophan hydroxylase antibodies are a highly specific marker for autoimmune GI involvement (16, 17).
Chromogranin A (CgA) colocalizes with almost all EE cell types in the different parts of the human GI tract, and it is therefore regarded as a surrogate marker for these cells (18). EE cells, which are lacking in AIE as part of APECED syndrome, are not affected in other intestinal diseases (10). Moreover, except for the absence of EE cells, intestinal histology is normal in APECED (9).
Herein, we describe an 18-year-old female diagnosed with APECED who first presented with hypoparathyroidism, vitiligo and chronic mucocutaneous candidiasis; over the years, she developed Hashimoto’s thyroiditis, diabetes mellitus type 1, Addison’s disease and primary ovarian insufficiency. The patient’s disease course, which involved recurrent admissions to the hospital with GI symptoms and severe persistent hypocalcemia that required prolonged hospitalizations, led to the diagnosis of AIE confirmed by intestinal CgA immunostaining. This report highlights AIE as one of the unrecognized factors of GI involvement in APECED and discusses the challenges of managing hypocalcemia associated with AIE.
Case Description
The proband was an 18-year-old female of Christian–Arab descent, whose parents were first-degree cousins. At the age of 6.10 years, she presented to the rheumatological clinic with episodes of muscle spasm, leg pain, recurrent fever, intermittent abdominal pain and diarrhea. Laboratory evaluation revealed no remarkable findings. Ten months later, she was admitted to our medical center due to severe hypocalcemia (4.5 mg/dL; normal range, 8.1–10.6), elevated phosphorus (9.11 mg/dL; normal range, 2.5–4.5) and undetectable serum parathyroid hormone levels (less than 1 pg/mL; normal range, 12–65). These findings were consistent with the diagnosis of acquired hypoparathyroidism. On examination, she had oral candidiasis and vitiligo on the knees, left elbow, and around the mouth and eyes. Presence of the two major components of the disease (hypoparathyroidism and chronic mucocutaneous candidiasis) and one minor component (vitiligo) resulted in a clinical diagnosis of APECED. Administration of calcium supplement (40 mg/kg elemental calcium daily) and α-hydroxycholecalciferol (alpha D3; 0.25 µg daily) was initiated and at these doses, calcium was maintained at the desired level (above 7.5 mg/dL). At the age of 9 years, positive thyroid antibodies were detected with normal thyroid function and the diagnosis of euthyroid Hashimoto’s thyroiditis was made (Tables 1 and 2). On follow up, the proband underwent recurrent hospitalizations in the intensive care unit due to severe hypocalcemia which was difficult to normalize. At the age of 9.6 years, she was admitted to our hospital due to fatty acholic diarrhea, abdominal pain and a fever lasting 10 days. She had not gained weight for 12 months prior to her admission (Figure 1). Laboratory evaluation revealed severe hypocalcemia. Despite high doses of intravenously (IV) administered calcium (180 mg/kg elemental calcium daily) along with oral calcium supplements of 56 mg/kg elemental calcium per day, alpha D3 (5 µg daily; 0.17 µg/kg daily) and a magnesium supplement, calcium levels remained low. It was only after initiation of calcitriol, administered IV at a dose of 0.5 µg three times a week that calcium levels were maintained. Calcitriol (1 microgram/ml) Kern Pharma, S.L.Spain. N° Reg.: 68.934. This hospitalization lasted 1 month. The episodes of hypocalcemia that required hospitalization were preceded by GI manifestations, including abdominal pain, bloating, diarrhea, constipation and fatty stool, and therefore an extensive GI investigation was carried out with unremarkable findings. Imaging of the upper GI tract, including a radiological GI series, demonstrated normal gastric emptying and normal bowel motility without any pathological findings for inflammatory bowel disease. Ultrasonographic scans of the abdomen were without liver or pancreatic anomalies. Upper GI endoscopic biopsies showed mild lymphocytic esophagitis, and the gastric biopsy demonstrated a picture of autoimmune atrophic gastritis with reduction in acid-secreting parietal cells, without intestinal metaplasia. There was no evidence of Helicobacter pylori and no villous atrophy of the duodenal or terminal ileal mucosa, and the number of intraepithelial lymphocytes was within the normal range. No active inflammation or granulomas were seen in the biopsies of the terminal ileum or colonic mucosa. Colonoscopy with biopsies and magnetic resonance elastography of the bowel excluded inflammatory bowel disease. Liver needle biopsy revealed no significant histopathological changes. Following these extensive GI investigations, which excluded exocrine pancreas insufficiency, inflammatory bowel disease, celiac disease, GI infections and autoimmune hepatitis, the diagnosis of AIE was raised, and specific CgA immunostaining of the stomach, duodenal and ileal biopsies was performed. This revealed a complete absence of EE cells in the duodenum and the small intestine with almost normal staining of the stomach, confirming the diagnosis of AIE (Figure 2). After this long hospitalization, during the follow-up period, she maintained calcium serum levels within 7.5 to 10.0 mg/dL with some fluctuations, with calcium supplement of around 20 mg/kg/d and α-D3 of 2 µg daily. At the age of 13.8 years, following 4 weeks of polyuria and polydipsia, blood glucose was 450 mg/dL and HbA1c was 9.1% (normal range <5.6%). The diagnosis of diabetes mellitus type 1 was made (Table 2), and insulin injections were initiated. Two months later, adrenocorticotropic hormone stimulation test revealed peak cortisol of 10.53 µg/dL—consistent with partial adrenal insufficiency—and oral glucocorticoid administration (10 mg/m2 per day, hydrocortisone) was initiated (Table 3). At the same age, the proband had no signs of pubertal development and growth deceleration was seen, with a decrease from the 60th to 10th centile for height (Figure 1). Elevated gonadotropins confirmed a diagnosis of primary ovarian insufficiency (Table 3), and supplemental estradiol treatment was initiated. At the age of 18 years, she was admitted to our Medical Center due to coffee ground vomiting, loss of appetite and weight loss. Laboratory evaluation revealed hypercalcemia of 13.11 mg/dL that was normalized by IV 0.9% NaCl solution. Repeated intestinal biopsy revealed normal numbers of positive CgA-immunostained cells in the duodenum. Stomach biopsies demonstrated a Helicobacter pylori-positive active gastritis and an associated lymphocytic gastritis, possibly related to H. pylori as well. In a biopsy from the stomach body, no cells were present with CgA immunostaining. Sequencing of AIRE identified a previously described homozygous missense mutation (c.47C>T, p.Thr16Met), confirming the diagnosis of APECED. She is currently 20 years old and she graduated high school with distinguish.
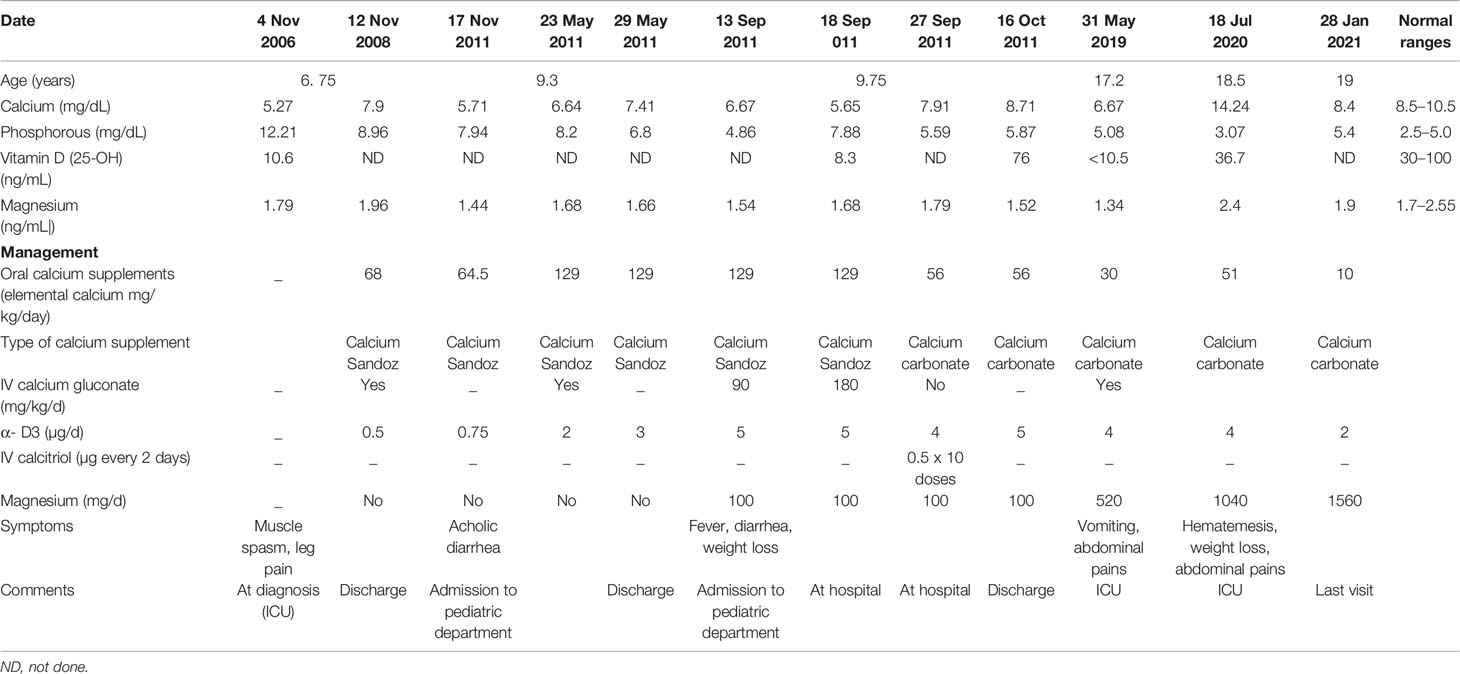
Table 1 Summary of the biochemical results and the treatment modalities over the years in the proband.
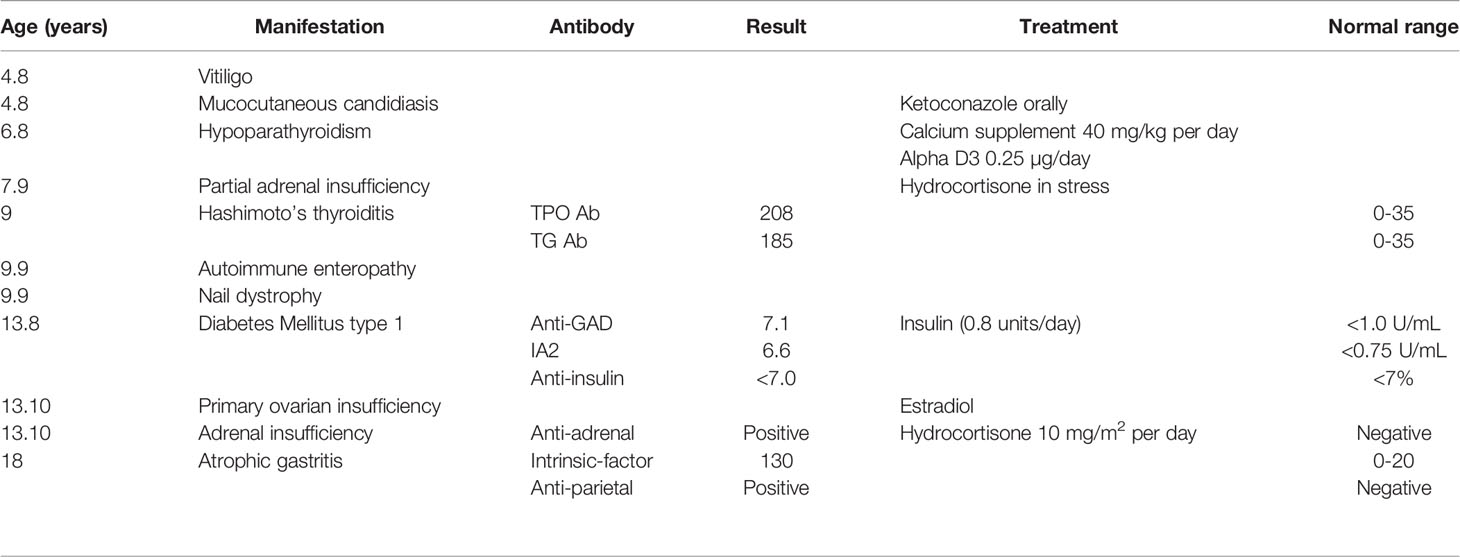
Table 2 Disease manifestation, age at onset and treatment.
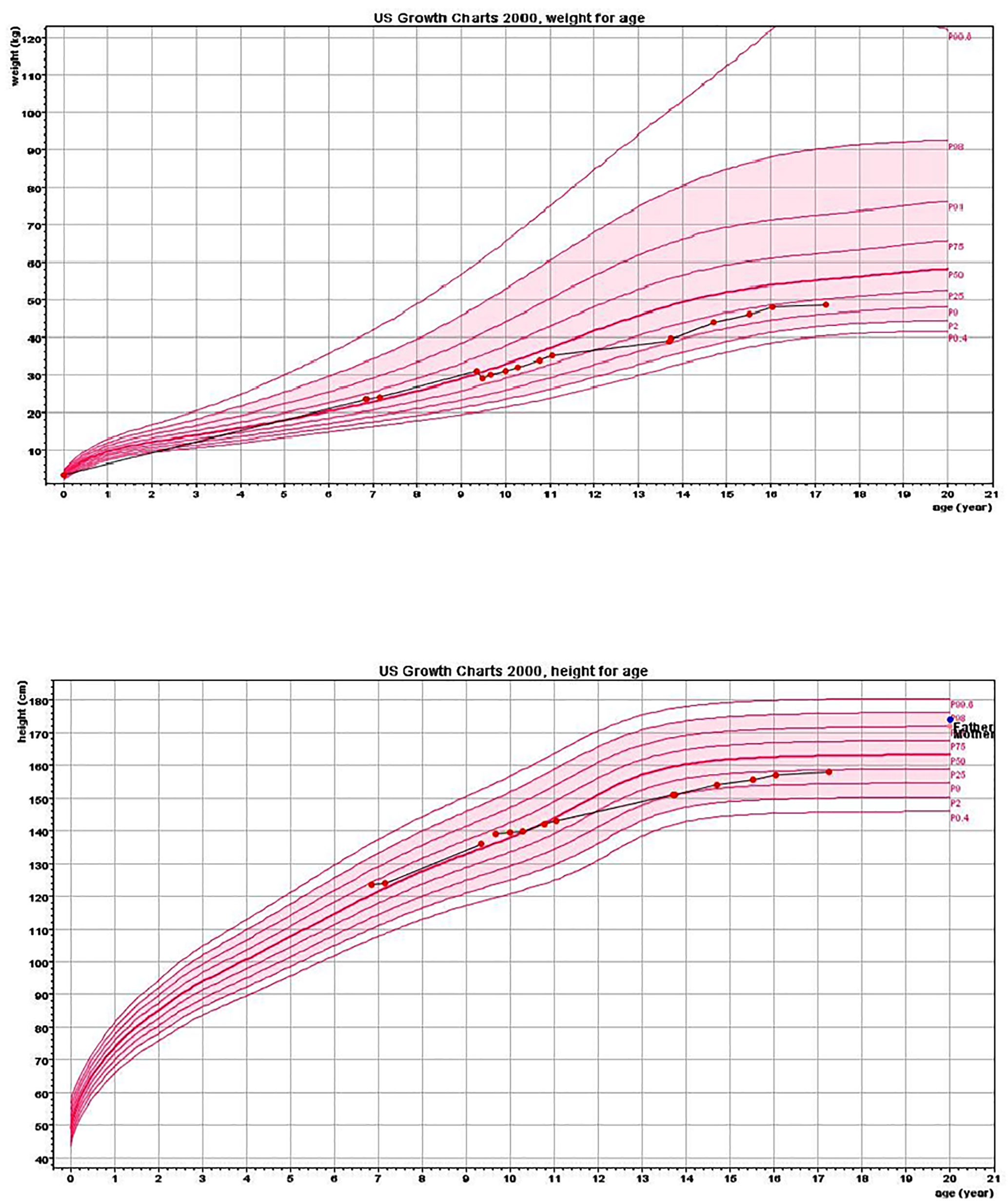
Figure 1 Height and weight charts. Charts show growth deceleration from the 60th to 10th centile and a lack of weight gain from the age of 9.5 years.
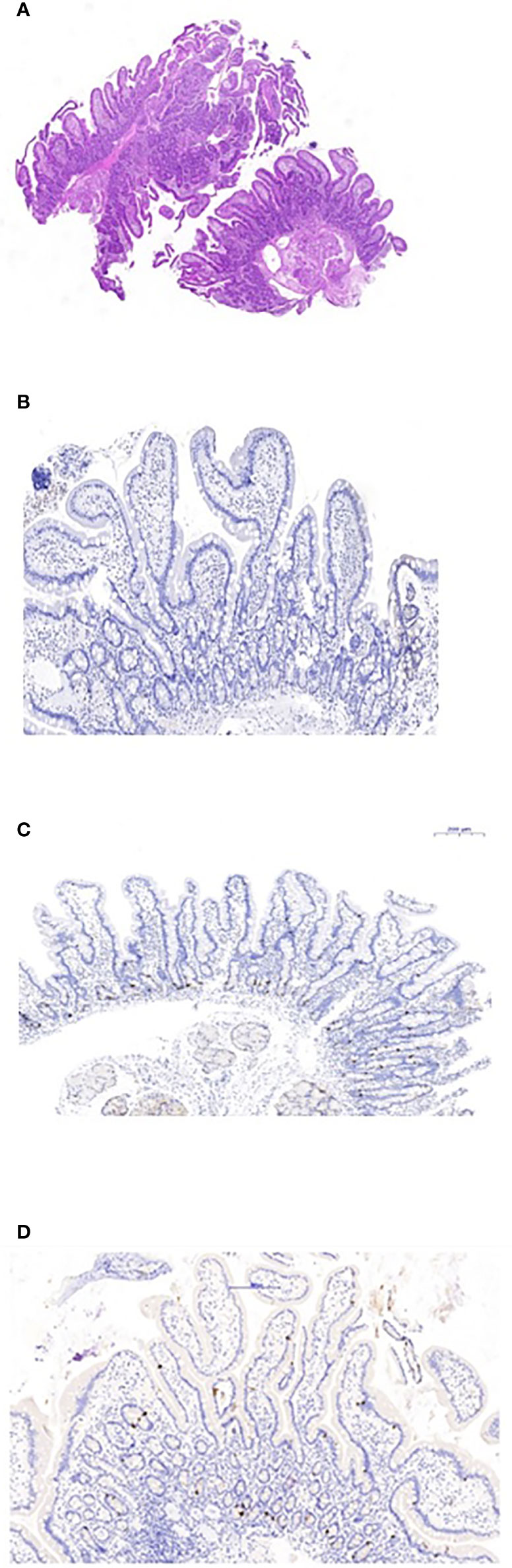
Figure 2 Duodenal biopsies. (A) Hematoxylin and eosin staining shows normal duodenal mucosa without villous atrophy. (B) CgA immunostaining of the duodenal biopsy shows complete loss of EE cells. (C) Normal control duodenal mucosa immunostained with CgA, showing EE cell distribution in the crypt epithelium. (D) Repeated biopsy shows normal immunostained CgA duodenal mucosa.
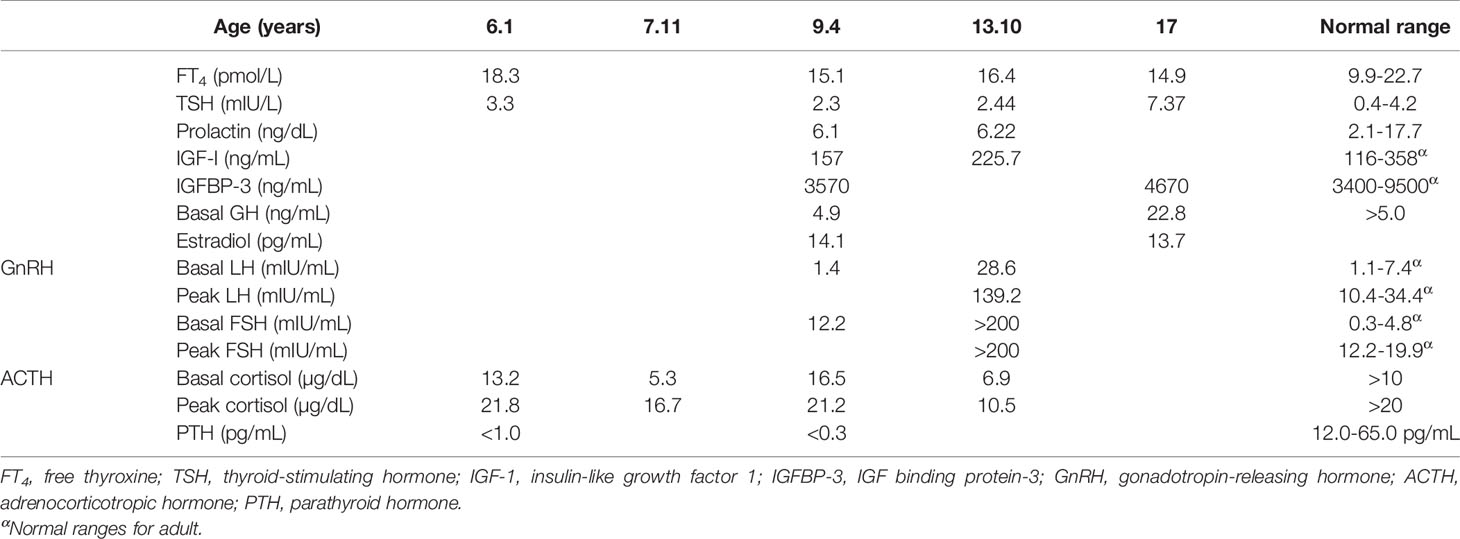
Table 3 Hormonal results.
Discussion
We describe a girl with APECED who first presented with hypoparathyroidism, and then developed several characteristics of the disease over the years. Intestinal involvement due to AIE caused recurrent episodes of severe, difficult to manage hypocalcemia. There is limited awareness of GI involvement in APECED (3, 9). Our knowledge of AIE as part of APECED was gathered from small series and case reports (9–12, 19). The precise mechanism of autoimmune intestinal involvement is not well understood. More than 90% of patients with intestinal dysfunction have been found positive for anti-tryptophan hydroxylase antibodies (20).
We demonstrated a total loss of EE cells in the duodenal and small bowel mucosa by lack of CgA staining in intestinal biopsies. The recurrent episodes of persistent, severe hypocalcemia following the GI manifestations suggested that the hypocalcemic episodes are a consequence of intestinal malabsorption. However, extensive malabsorption workup excluded exocrine pancreatic insufficiency as a cause. The pathophysiology of the EE cell loss’s interference with calcium and/or vitamin D absorption is unclear. Infants with NEUROG3 mutations, which result in congenital dysgenesis of EE cells, present with severe, life-threatening watery diarrhea (21). The absence of EE cells in this congenital disease emphasizes their important role in intestinal absorption (10). An animal model suggests that Neurog3 is an important transcription factor in the development and differentiation of EE cells (15). The small intestine, and mainly the duodenum, are primarily responsible for calcium absorption, through active transport stimulated by 1,25-dihydroxyvitamin D (22). EE cells have been recently shown to play an important role in the digestion of macronutrients into their component parts: glucose, amino acids, and fatty acids. These small molecules are then detected by specific transporters and receptors located on the EE cells, and stimulate hormone secretion predominantly at the sites where nutrient absorption is maximal. It seems that malabsorptive diarrhea results from the loss of EE cells (15) and hypocalcemic episodes resulted from malabsorption of oral calcium and vitamin D. Moreover, in each such hypocalcemic episode, a stress dose of hydrocortisone was given due to adrenal insufficiency, which might reduce intestinal calcium absorption, renal calcium resorption, and bone remodeling, further decreasing the proband’s serum calcium levels (23). Furthermore, hypocalcemia itself can cause malabsorption by reducing cholecystokinin secretion, decreasing gall bladder emptying, and causing pancreatic insufficiency (7, 23). The association between loss of EE cells and diarrhea in patients with APECED and severe hypocalcemia that is unresponsive to conventional therapy has been reported by others as well (19, 23–25).
Management of hypocalcemia in such cases is extremely challenging. Despite a high dose of calcium supplements and alpha D3, calcium levels remained very low in the proband. Only IV calcitriol maintained calcium in the normal range. It has been shown that parenteral vitamin D analogs are superior to oral calcitriol for the long-term control of hyperparathyroidism in dialysis patients (26). However, no data has been reported on the efficacy of IV calcitriol in patients with hypocalcemia related to other conditions. Our experience indicates that IV calcitriol should be consider in unresponsive hypocalcemia related to AIE.
At present, there are no clear guidelines for the treatment of hypoparathyroidism in children and adolescents in general, and even less so in hypoparathyroidism associated with AIE (27).
Immunosuppressive therapy in a patient with APECED presenting with unresponsive severe hypocalcemia led to improved calcium absorption and maintenance of normal calcium levels (25). However, this improvement could also be attributed to the fluctuating course of AIE, and be independent of the effect of the immunosuppressive therapy; further studies are therefore needed to justify the benefit of such aggressive therapy (10). Interestingly, our patient demonstrated a fluctuating disease course and fluctuating serum calcium levels, suggesting reversible involvement of EE cells. Indeed, in contrast to the first biopsy, repeated intestinal biopsy revealed normal CgA immunostaining in the duodenum, indicating a regenerative process; however, no CgA staining was demonstrated in the stomach. This discrepancy may attributed to different regeneration time between the stomach and the duodenum as was demonstrated in mice model (28). Contrary to this concept, Barrett et al., have shown that endocrine cells of the gut in human have no proliferative capacity (29). This seems to contrast with the classic endocrine manifestations of APECED, such as hypoparathyroidism, Addison’s disease and primary ovarian insufficiency, which are permanent (3, 19).
Recently, treatment involving injections of recombinant parathyroid hormone has been recommended in hypoparathyroidism that does not respond to conventional therapy. This treatment is not yet approved in children, and the possible risk of osteosarcoma in childhood has to be taken into consideration (30).
Finally, sequencing of AIRE revealed a homozygous missense mutation (c.47C>T, p.Thr16Met) that has been previously described in patients of Russian (31) and Greek (32) origin, and is predicted to be pathogenic according to bioinformatics tools. This is a common mutation in the Israeli Christian–Arab population, known as a founder mutation.
Conclusions
We report on a patient with APECED in which severe episodes of hypocalcemia were related to calcium and vitamin D malabsorption due to AIE, and discuss the challenging management of this presentation. CgA staining is indicated in patients with APECED and GI involvement when all other investigations fail to reveal an explanation for the malabsorption. This report highlights the need for more awareness among pediatricians of the entity of AIE as an important clinical presentation in APECED.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the proband’s family for their cooperation, and Camille Vainstein for professional English editing.
Abbreviations
AIE, autoimmune enteropathy; AIRE, autoimmune regulator; APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; CgA, chromogranin A; EE, enteroendocrine; GI, gastrointestinal.
References
1. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional Cloning of the APECED Gene. Nat Genet (1997) 17:393–8. doi: 10.1038/ng1297-393
2. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an Immunological Self-Shadow Within the Thymus by the Aire Protein. Science (2002) 298:1395–401. doi: 10.1126/science.1075958
3. Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical Variation of Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) in a Series of 68 Patients. N Engl J Med (1990) 322:1829–36. doi: 10.1056/NEJM199006283222601
4. Perheentupa J. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. J Clin Endocrinol Metab (2006) 91:2843–50. doi: 10.1210/jc.2005-2611
5. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined Clinical Features and Diagnostic Criteria in Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. JCI Insight (2016) 1:e88782. doi: 10.1172/jci.insight.88782
6. Capalbo D, De Martino L, Giardino G, Di Mase R, Di Donato I, Parenti G, et al. Autoimmune Polyendocrinopathy Candidiasis Ectodermal Dystrophy: Insights Into Genotype-Phenotype Correlation. Int J Endocrinol (2012) 2012:353250. doi: 10.1155/2012/353250
7. Husebye ES, Perheentupa J, Rautemaa R, Kämpe O. Clinical Manifestations and Management of Patients With Autoimmune Polyendocrine Syndrome Type I. J Intern Med (2009) 265:514–29. doi: 10.1111/j.1365-2796.2009.02090.x
8. Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N Engl J Med (2018) 378:1132–41. doi: 10.1056/NEJMra1713301
9. Kluger N, Jokinen M, Krohn K, Ranki A. Gastrointestinal Manifestations in APECED Syndrome. J Clin Gastroenterol (2013) 47:112–20. doi: 10.1097/MCG.0b013e31827356e1
10. Posovszky C, Lahr G, von Schnurbein J, Buderus S, Findeisen A, Schröder C, et al. Loss of Enteroendocrine Cells in Autoimmune-Polyendocrine-Candidiasis-Ectodermal-Dystrophy (APECED) Syndrome With Gastrointestinal Dysfunction. J Clin Endocrinol Metab (2012) 97:E292–300. doi: 10.1210/jc.2011-2044
11. Oliva-Hemker M, Berkenblit GV, Anhalt GJ, Yardley JH. Pernicious Anemia and Widespread Absence of Gastrointestinal Endocrine Cells in a Patient With Autoimmune Polyglandular Syndrome Type I and Malabsorption. J Clin Endocrinol Metab (2006) 91:2833–8. doi: 10.1210/jc.2005-2506
12. Sköldberg F, Portela-Gomes GM, Grimelius L, Nilsson G, Perheentupa J, Betterle C, et al. Histidine Decarboxylase, a Pyridoxal Phosphate-Dependent Enzyme, is an Autoantigen of Gastric Enterochromaffin-Like Cells. J Clin Endocrinol Metab (2003) 88:1445–52. doi: 10.1210/jc.2002-021761
13. Rindi G, Leiter AB, Kopin AS, Bordi C, Solcia E. The “Normal” Endocrine Cell of the Gut: Changing Concepts and New Evidences. Ann NY Acad Sci (2004) 1014:1–12. doi: 10.1196/annals.1294.001
14. Gunawardene AR, Corfe BM, Staton CA. Classification and Functions of Enteroendocrine Cells of the Lower Gastrointestinal Tract. Int J Exp Pathol (2011) 92:219–31. doi: 10.1111/j.1365-2613.2011.00767.x
15. Worthington JJ, Reimann F, Gribble FM. Enteroendocrine Cells – Sensory Sentinels of the Intestinal Environment and Orchestrators of Mucosal Immunity. Mucosal Immunol (2018) 11:3–20. doi: 10.1038/mi.2017.73
16. Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J, et al. Identification of Tryptophan Hydroxylase as an Intestinal Autoantigen. Lancet (1998) 352:279–83. doi: 10.1016/S0140-6736(97)11050-9
17. Ekwall O, Sjöberg K, Mirakian R, Rorsman F, Kämpe O. Tryptophan Hydroxylase Autoantibodies and Intestinal Disease in Autoimmune Polyendocrine Syndrome Type 1. Lancet (1999) 354:568. doi: 10.1016/S0140-6736(99)01989-3
18. Portela-Gomes GM, Stridsberg M, Johansson H, Grimelius L. Complex Co-Localization of Chromogranins and Neurohormones in the Human Gastrointestinal Tract. J Histochem Cytochem (1997) 45:815–22. doi: 10.1177/002215549704500606
19. Högenauer C, Meyer RL, Netto GJ, Bell D, Little KH, Ferries L, et al. Malabsorption Due to Cholecystokinin Deficiency in a Patient With Autoimmune Polyglandular Syndrome Type I. N Engl J Med (2001) 344:270–4. doi: 10.1056/NEJM200101253440405
20. Kashem SW, Binstadt BA. Pathogenic and Protective Autoantibodies in Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED). Antibodies (Basel) (2017) 6:1. doi: 10.3390/antib6010001
21. Wang J, Cortina G, Wu SV, Tran R, Cho JH, Tsai MJ, et al. Mutant Neurogenin-3 in Congenital Malabsorptive Diarrhea. N Engl J Med (2006) 355:270–80. doi: 10.1056/NEJMoa054288
22. Fleet JC, Schoch RD. Molecular Mechanisms for Regulation of Intestinal Calcium Absorption by Vitamin D and Other Factors. Crit Rev Clin Lab Sci (2010) 47:181–95. doi: 10.3109/10408363.2010.536429
23. Geyer M, Fairchild J, Moore D, Moore L, Henning P, Tham E. Recalcitrant Hypocalcaemia in Autoimmune Enteropathy. Pediatrics (2014) 134:e1720–6. doi: 10.1542/peds.2013-3308
24. Padeh S, Theodor R, Jonas A, Passwell JH. Severe Malabsorption in Autoimmune Polyendocrinopathy-Candidosis-Ectodermal Dystrophy Syndrome Successfully Treated With Immunosuppression. Arch Dis Child (1997) 76:532–4. doi: 10.1136/adc.76.6.532
25. Ward L, Paquette J, Seidman E, Huot C, Alvarez F, Crock P, et al. Severe Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy in an Adolescent Girl With a Novel AIRE Mutation: Response to Immunosuppressive Therapy. J Clin Endocrinol Metab (1999) 84:844–52. doi: 10.1210/jc.84.3.844
26. Andress LA. Intravenous Versus Oral Vitamin D Therapy in Dialysis Patients: What is the Question? Am J Kidnet Dis (2001) 38(Suppl 5):S41–S4. doi: 10.1053/ajkd.2001.28108
27. Di Maio S, Soliman AT, De Sanctis V, Kattamis CC. Current Treatment of Hypoparathyroidism: Theory Versus Reality Waiting Guidelines for Children and Adolescents. Acta Biomed (2018) 89:122–31. doi: 10.23750/abm.v89i1.7118
28. Thompson EM, Price YE, Wright NA. Kinetics of Enteroendocrine Cells With Implications for Their Origin: A Study of the Cholecystokinin and Gastrin Subpopulations Combining Tritiated Thymidine Labelling With Immunocytochemistry in the Mouse. Gut (1990) 31:406–11. doi: 10.1136/gut.31.4.406
29. Barrett P, Hobbs RC, Coates PJ, Risdon RA, Wright NA, Hall PA. Endocrine Cells of the Human Gastrointestinal Tract Have No Proliferative Capacity. Histochem J (1995) 27:482–6. doi: 10.1007/BF02388805
30. Cihakova D, Trebusak K, Heino M, Fadeyev V, Tiulpakov A, Battelino T, et al. Novel AIRE Mutations and P450 Cytochrome Autoantibodies in Central and Eastern European Patients With APECED. Hum Mutat (2001) 18:225–32. doi: 10.1002/humu.1178
31. Kollios K, Tsolaki A, Antachopoulos C, Moix I, Morris MA, Papadopoulou M, et al. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Syndrome (APECED) Due to AIRET16M Mutation in a Consanguineous Greek Girl. J Pediatr Endocrinol Metab (2011) 24:599–601. doi: 10.1515/jpem.2011.012
Keywords: autoimmune polyglandular type 1 syndrome, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, autoimmune regulator gene, autoimmune enteropathy, hypocalcemia, enteroendocrine cell, chromogranin A
Citation: Halabi I, Barohom MN, Peleg S, Trougouboff P, Elias-Assad G, Agbaria R and Tenenbaum-Rakover Y (2021) Case Report: Severe Hypocalcemic Episodes Due to Autoimmune Enteropathy. Front. Endocrinol. 12:645279. doi: 10.3389/fendo.2021.645279
Received: 22 December 2020; Accepted: 19 May 2021;
Published: 14 June 2021.
Edited by:
Aaron Hanukoglu, Tel Aviv University, IsraelReviewed by:
David Gillis, Hebrew University Hadassah Medical School, IsraelZvi Zadik, Kaplan Pediatric Endocrinology, Israel
Copyright © 2021 Halabi, Barohom, Peleg, Trougouboff, Elias-Assad, Agbaria and Tenenbaum-Rakover. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yardena Tenenbaum-Rakover, cmFrb3Zlcl95QGNsYWxpdC5vcmcuaWw=