 Neha Shrestha
Neha Shrestha Elisa De Franco
Elisa De Franco Peter Arvan
Peter Arvan Miriam Cnop
Miriam Cnop- 1Department of Molecular and Integrative Physiology, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Institute of Biomedical and Clinical Science, University of Exeter College of Medicine and Health, Exeter, United Kingdom
- 3Division of Metabolism, Endocrinology & Diabetes, Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI, United States
- 4ULB Center for Diabetes Research, Medical Faculty, Université Libre de Bruxelles, Brussels, Belgium
- 5Division of Endocrinology, Erasmus Hospital, Université Libre de Bruxelles, Brussels, Belgium
The notion that in diabetes pancreatic β-cells express endoplasmic reticulum (ER) stress markers indicative of increased unfolded protein response (UPR) signaling is no longer in doubt. However, what remains controversial is whether this increase in ER stress response actually contributes importantly to the β-cell failure of type 2 diabetes (akin to ‘terminal UPR’), or whether it represents a coping mechanism that represents the best attempt of β-cells to adapt to changes in metabolic demands as presented by disease progression. Here an intercontinental group of experts review evidence for the role of ER stress in monogenic and type 2 diabetes in an attempt to reconcile these disparate views. Current evidence implies that pancreatic β-cells require a regulated UPR for their development, function and survival, as well as to maintain cellular homeostasis in response to protein misfolding stress. Prolonged ER stress signaling, however, can be detrimental to β-cells, highlighting the importance of “optimal” UPR for ER homeostasis, β-cell function and survival.
Overview
Pancreatic islet β-cells are specialized secretory cells designed for massive insulin storage. β-cells have a huge biosynthetic capability, which is further upregulated in insulin resistant states. The endoplasmic reticulum (ER) is the major organelle responsible for secretory protein synthesis, folding and quality control. To maintain ER homeostasis during the stresses associated with secretory protein synthesis and folding, cells activate the intracellular transduction pathway termed the unfolded protein response (UPR) (1).
The UPR signals through three branches involving PERK, ATF6 and IRE1 (Figure 1) (2). PERK phosphorylates eIF2α, thereby attenuating protein translation and activating transcriptional responses mediated by transcription factors ATF4, CHOP and ATF3, with downstream induction of PPP1R15A and -B to dephosphorylate phosho-eIF2α, thereby limiting overshoot. ATF6 upregulates chaperones (including BiP) and foldases. IRE1 splices XBP1 mRNA to synthesize an active transcription factor that induces ER folding enzymes and chaperones, ER size expansion, and ER-associated degradation (ERAD). IRE1’s endoribonuclease activity also degrades ER-localized mRNAs in a process called regulated IRE1-dependent decay (RIDD), thereby indirectly decreasing translational demand placed upon the organelle. Besides their role in ER stress response, the tripartite limbs of the UPR play important roles in proinsulin production, folding and trafficking as well as β-cell expansion (3, 4).
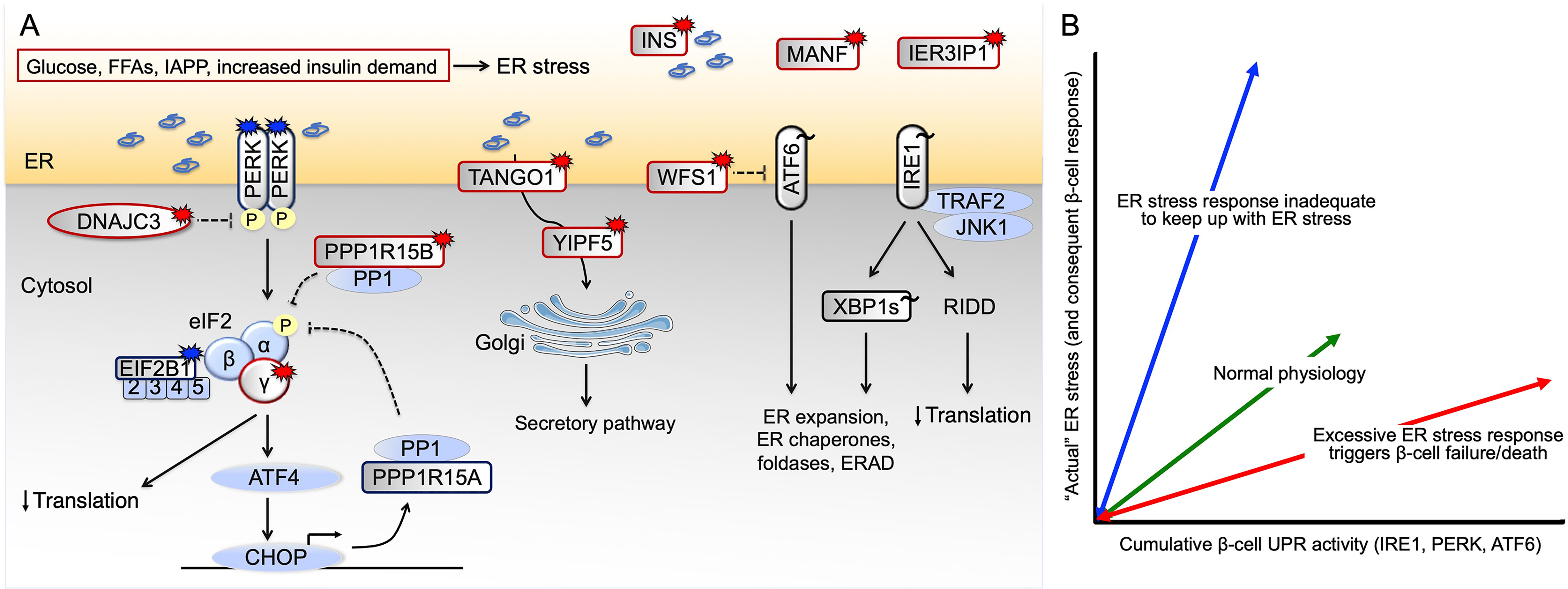
Figure 1 Pathways leading to β-cell ER stress and failure. (A) Genetic and non-genetic causes of human β-cell ER stress in diabetes. Patients with EIF2AK3 and EIF2B1 mutations that result in impaired signaling downstream of PERK develop young-onset diabetes, as do patients with EIF2S3, DNAJC3 and PPP1R15B mutations that result in excessive PERK signaling. Homozygous WFS1, YIPF5, TANGO1, MANF and IER3IP1 mutations also cause excessive UPR. Burst symbol indicates loss-of-function mutation causing monogenic diabetes, with red color indicating excessive and blue insufficient UPR signaling. For GWAS variants associated with T2D risk (indicated by tilde), the direction of effect is most often not known (black color). (B) Potential dysregulated β-cell responses to nutritional/metabolic challenge. UPR function is required for normal β-cell physiology, sustaining β-cell health and survival (green arrow). In conditions of insufficient UPR signaling relative to the prevailing ER stress (blue arrow), β-cells will fail. Conversely, excessive UPR signaling will cause β-cell dysfunction and death (red arrow). Currently available data provide evidence for both red and blue scenarios taking place in β-cells in T2D. ERAD ER-associated degradation, FFAs free fatty acids, IAPP islet amyloid polypeptide, RIDD regulated IRE1-dependent decay.
Despite the critical roles of UPR in promoting β-cell function, sustained and unresolved UPR signaling (i.e., exuberant ER stress response) can be detrimental to β-cells. Chronic PERK activation increases expression of proapoptotic C/EBP homologous protein (CHOP, also known as growth arrest and DNA-damage inducible-153, GADD153) (5). Chronic IRE1 activation can lead to the recruitment of TRAF2 with subsequent activation of proapoptotic JNK and MAPK pathways. Prolonged IRE1 activation may also promote apoptosis through RIDD (6).
Perspectives of the UPR are thus divided between whether UPR signaling is actually beneficial or harmful for β-cell function. Accumulating studies suggest that both too much and too little UPR is detrimental to β-cells, highlighting the importance of UPR-mediated ER homeostasis. Here, we cite examples of monogenic diabetes, type 2 diabetes (T2D) genome-wide association studies (GWAS) and non-genetic triggers of diabetes to provide evidence both for and against UPR influences in β-cell physiology and T2D pathogenesis. We also discuss some caveats of ER stress hypotheses in establishing cause-and-effect on β-cell dysfunction.
Monogenic Forms of Diabetes Highlight β-Cell Sensitivity to UPR Dysregulation, Suggesting That a Fine-Tuned ER Stress Response Is Needed for Maintenance of Functional β-Cell Mass
Monogenic diabetes is caused by highly penetrant single gene mutations, with the unique potential to reveal genes necessary for human β-cell development, function and survival. Of >40 genes with mutations reported to cause diabetes, 11 directly or indirectly result in β-cell ER stress. Discussing all of them would be beyond the scope of this review; here we consider several that reveal specific mechanisms by which dysregulation of ER stress affects human β-cells (Figure 1).
Mutations in the WFS1 gene, encoding Wolframin (7), are known to result in a spectrum of phenotypes including β-cell dysfunction and diabetes. Wolframin has multiple proposed roles in β-cells, including in insulin production, processing and secretion, regulation of ER calcium levels, and suppression of ER stress-mediated cell death. Biallelic WFS1 loss-of-function mutations result in childhood-onset diabetes [median age of onset 6 years, range 1-32 (8)], with dysregulated β-cell UPR, plus other phenotypes including optic atrophy, deafness, and diabetes insipidus. Heterozygous WFS1 mutations may also be linked to isolated presentation of each of these phenotypes as well as isolated nuclear cataracts, with distinct genotype-phenotype relationships (9, 10). Recently, de novo heterozygous WFS1 mutations were identified as causing a neonatal syndrome characterized by diabetes with onset in the first months of life and other congenital features (9). Functional in vitro studies suggest that these de novo WFS1 mutations cause protein aggregation accompanied by a strong, dysregulated ER stress response and likely cell death (9), highlighting a separate disease mechanism from WFS1 loss-of-function.
Other ER-resident proteins implicated in monogenic diabetes include mesencephalic astrocyte-derived neurotrophic factor (MANF) (11) and immediate early response 3 interacting protein 1 (IER3IP1) (12); they may directly or indirectly modulate the UPR.
Genetic defects in at least 5 genes regulating the UPR through the PERK branch cause early onset β-cell failure and diabetes. Biallelic mutations in the EIF2AK3 gene (encoding PERK) and dominant mutations in EIF2B1 (resulting in loss of sensitivity of the EIF2B complex to eIF2α phosphorylation) result in syndromic forms of neonatal/early onset diabetes and likely β-cell death (13, 14). In these two diseases, signaling downstream of PERK is impaired. Recessive loss-of-function mutations in EIF2S3 (15), DNAJC3 [encoding a BiP co-chaperone (16)] and PPP1R15B [encoding a constitutive repressor of eIF2α phosphorylation (17)] all result in syndromic early-onset monogenic diabetes subtypes with excessive signaling downstream of PERK. These findings highlight β-cell sensitivity to perturbations of eIF2α phosphorylation and regulated mRNA translation in response to ER stress (18).
Recent studies suggest that ER stress caused by impaired ER-to-Golgi trafficking is another pathway resulting in monogenic diabetes. Recessive YIPF5 missense mutations have been reported in patients with neonatal diabetes, microcephaly and epilepsy (19). Functional studies showed that β-cells are specifically sensitive to YIPF5 loss-of-function, increasing ER stress and apoptosis. Complete YIPF5 loss-of-function (by CRISPR/Cas9 knockout) resulted in proinsulin retention and β-cell ER stress, but did not affect α-cells (19). In keeping with this, a homozygous loss-of-function mutation in TANGO1, encoding an ER transmembrane protein that plays an essential role in ER-to-Golgi transport of bulky cargo, was reported in one family with syndromic diabetes (20).
Collectively, these monogenic forms of diabetes highlight the sensitivity of human β-cells to dysregulation of ER stress response or ER-to-Golgi trafficking, and are consistent with the notion that β-cell dysfunction may result from either an inadequate or an excessive ER stress response (21).
GWAS of T2D Identify Variants in Genes Essential for ER Stress Regulation
In more complex and common diseases such as T2D, clues to the genetic and physiological mechanisms may be inferred from large-scale GWAS. To date, >400 genetic loci have been associated with T2D risk (22). Some of these are in proximity of UPR genes such as the well-established WFS1 risk variants (23) that are associated with reduced insulin secretion (24). Of these, the WFS1 rs10010131 intronic variant is traditionally recognized as the variant characterizing the linkage block in this region, which includes both coding and non-coding variants (23). However, over 10 years after their initial genetic discovery, it is still unclear which variant primarily causes the T2D risk, highlighting the challenges that remain to define the biological mechanisms underlying genetic predisposition.
Variants in ERN1 (encoding IRE1α) exhibit a strong genetic association with T2D, possibly caused by dysfunctional alleles or diminished IRE1 protein expression. XBP1 and ATF6 exhibit a weaker association with T2D (25). ATF6 variants have been suggested to be associated with increased T2D risk in East-Asians (26, 27). An upstream variant in XBP1 has been suggested to be associated with increased T2D risk, increased fasting plasma glucose levels and fasting insulin levels in Han Chinese (28). Two common variants in the CREBRF gene, encoding a transcriptional repressor of the UPR, have been recently found to be associated with obesity and T2D risk in Pacific Highlanders (29). Variants in PDX1 have a strong genetic association with random and fasting glucose (25). Interestingly, besides its role in β-cell development (30), PDX1 also maintains ER health through regulation of the ER calcium pump SERCA (31, 32).
These data highlight defects in ER stress regulation as a potential biological mechanism contributing to T2D predisposition, but additional studies are needed to confirm this hypothesis. Of course, T2D is a complex disease that develops as a result of the interplay of “lifestyle” (e.g., diet, exercise) with a myriad of genetic variants — thus it is unrealistic to expect that a single β-cell stress response can explain the spectrum of human T2D.
Non-Genetic Triggers of β-Cell ER Stress in T2D Provide Evidence for the Importance of Subthreshold ER Stress in β-Cell Function
In vitro and in vivo studies have shown that β-cells activate the UPR following exposure to high glucose (33, 34); fatty acids - especially when saturated (35, 36); increased functional demand (as in the context of insulin resistance) (37, 38); or insulin and islet amyloid polypeptide (IAPP) misfolding/aggregation (39–41) (Figure 1).
With regards to nutrient stimulation, high glucose induces a moderate ER load, thereby activating subthreshold ER stress that drives insulin biosynthesis through IRE1 (42) as well as β-cell proliferation through ATF6 (34). Saturated free fatty acids, on the other hand, induce sustained activation of IRE1, PERK and ATF6 pathways (35, 43), resulting in β-cell apoptosis. Misfolded proinsulin in the ER lumen triggers the UPR to enhance protein folding and degradation but in case of INS mutations (causing mutant INS-gene-induced diabetes of youth, MIDY), the misfolding continues, resulting in persistent activation of the UPR (44). In such cases, β-cell apoptosis may be seen, most likely triggered by the transcriptional activation of CHOP (45). Finally, increased insulin demand also increases expression of UPR and chaperone proteins as exemplified by animal models of obesity (ob/ob mice) and T2D (db/db) (38). The increase in UPR in these models may ultimately be both beneficial and maladaptive. The UPR in these models of increased insulin demand is frequently upregulated during β-cell expansion/adaptation phases (34, 38, 46–48) to promote β-cell proliferation, yet the further benefit to β-cell mass and insulin secretion seen upon CHOP deletion in db/db models seems to diminish what would otherwise be a detrimental UPR contribution (49).
Crucially, ER stress may be triggered in β-cells during the development and/or after the onset of T2D in a vicious circle in which ER stress will impair β-cell function while the metabolic consequences may amplify the stress. The relative importance of ER stress in the chicken or egg dilemma may vary between different T2D patients considering the heterogeneity of disease pathogenesis. It is conceivable that in some patients, ER stress caused by genetic or environmental risk begets β-cell failure, while in others it is the hyperglycemia, dyslipidemia and increased insulin demand that begets β-cell ER stress.
Studies of human T2D include ultrastructural evidence of ER volume expansion in β-cells compared to non-diabetic control β-cells, suggesting response to ER stress (50). Although BiP and sXBP1 mRNAs were found to be decreased in islets of T2D individuals, they were induced in T2D islets upon high glucose exposure (50). However, in the largest RNA-sequencing study of human islets from T2D vs non-diabetic organ donors published to date (n=28 vs 58), no significant differences in expression of these genes have been detected (36). Engin et al. reported decreased sXBP1, ATF6, and P-eIF2α immunostaining in T2D islets compared with non-diabetic controls (46). Increased immunostaining for BiP, DNAJC3, ATF3 and CHOP was observed in human islets from T2D patients compared with islets from non-diabetic individuals; some of the staining was β-cell-specific (39, 51, 52). Clearly, more studies are needed on human islets from T2D patients, such as the transplantation of human islets from non-diabetic and T2D organ donors in immunocompromised mice subsequently exposed to high fat diet (53, 54), in order to longitudinally follow ER stress, function and survival of human β-cells in real time. Importantly, while these studies show correlative evidence to suggest changes in the total levels of β-cell UPR-related proteins, no studies have been able to report the bioactivity of these proteins (and thus the biological significance of these changes). Thus, much work is needed to determine whether, in vivo, exuberant ER stress response contributes to β-cell death in T2D.
Caveats of the “ER Stress Hypothesis”
ER stress response is often cited as a double-edged sword which, upon chronic or excessive signaling, triggers apoptosis (55, 56). While in vitro studies clearly support these ideas, there is generally a lack of direct evidence to support this hypothesis in vivo. It may be that in vivo, signaling crosstalk within and among different organelles, and within and among different cells, allows cells and tissues to cope with and survive chronic ER stress in ways that are not easily clarified from in vitro studies. Indeed, we note that a sustained high level of UPR signaling is often not observed in vivo, unlike most in vitro studies that tend to use large acute doses of non-physiological ER stressors. Importantly, UPR pathways upregulate expression of ERAD and autophagy genes for degradation of misfolded proteins — in turn, ERAD (in particular the Sel1L-Hrd1 complex) can downregulate protein expression of IRE1α via its degradation, thereby limiting excessive UPR signaling (57). Another study showed that β-cells have endogenous mechanisms involving N-myc interactor and UbiquitinD to provide negative feedback signal on IRE-induced JNK activation in response to cytokines (58, 59). WFS1 is yet another stress-induced gene with a potential to inhibit ATF6 and tone down the UPR (60). These studies point to the very real possibility that excessive UPR signaling may be more restricted in vivo. Indeed, recent single cell analysis showed that healthy β-cells constantly undergo cycles of high and low UPR signaling, highlighting the importance of UPR dynamics in normal β-cell physiology (61), which is certainly not contrary to the notion that increased UPR activity during diabetes development represents a physiological response that may provide overall benefit to β-cells.
The human β-cell capital is probably fixed by young adulthood and human β-cells age with the body (62, 63). Unlike in type 1 diabetes, β-cell death is generally not considered a major player in the initial onset of T2D, which is primarily characterized by β-cell dysfunction (64) and even dedifferentiation (65). In fact, many studies have now demonstrated that ER dysfunction in β-cells may lead to dedifferentiation irrespective of ER stress response. For instance, dysfunction of ERAD induced by β-cell-specific genetic deletion of Sel1L, results in rapid and severe β-cell dedifferentiation. Importantly, these effects appeared independent of UPR, and instead altered the expression of TGFβ receptors — the activity of which promotes β-cell de-differentiation (66). Indeed, several studies have now shown that loss of insulin expression followed by dedifferentiation in cultured human β-cells is associated with induction of epithelial-mesenchymal transition-related pathways (67, 68). These studies point to the idea that signals emanating from the ER may directly contribute to β-cell failure by alternative mechanisms to that of apoptosis. In fact, IRE1 (which, as noted above, is an essential component of the UPR) also appears to play an important role in β-cell differentiation, as shown in a recent study (69) in which deletion of Ire1 in NOD mice prior to insulitis resulted in β-cell dedifferentiation that could potentially help to limit antigen presentation to the immune system.
Conclusions
The levels of UPR signaling in β-cells normally shift back and forth, which is likely to be important for proinsulin biosynthesis and folding during the metabolic/nutritional fluctuations that represent normal physiology. However, many studies show correlation of increased UPR and ER stress-associated genes with β-cell dysfunction and apoptosis. The data are particularly convincing from in vitro studies with pharmacologic ER stressors, as well as with high glucose and/or fatty acid exposure. However, animal models and a large number of human monogenic forms of diabetes (and to some extent, human GWAS) support that it is a dysregulated ER stress response that is more likely to predispose to diabetes onset, suggesting that a functional, homeostatic ER stress response is generally β-cell protective in vivo. Thus our consensus view is that UPR function is required for normal β-cell health and survival, although it may go awry in conditions of excessive or unresolvable ER stress or dysregulated signaling (Figure 1). Other aspects of ER homeostasis, beyond the UPR, may be equally (or even more) important in determining β-cell phenotype(s) in T2D. Ultimately, a systematic and detailed study of the time-course of UPR signaling during different stages of β-cell dysfunction in T2D will be needed in order to provide greater clarity about the role(s) of ER stress response/UPR in the β-cell failure of “garden-variety” T2D, and the subclassifications that will continue to emerge from a growing understanding of personalized medicine.
Author Contributions
All authors: conceptualization. MC: investigation, PA, NS, EF, MC: writing—original draft, and PA, NS, MC: writing—review and editing. PA, NS, EF, MC: resources and funding acquisition. PA and MC: supervision. All authors contributed to the article and approved the submitted version.
Funding
PA is supported by NIH R01 DK48280, and R01 DK111174. EF is a Diabetes UK RD Lawrence Fellow (19/0005971). The MC lab is funded by the Fonds National de la Recherche Scientifique (FNRS), the Fonds Erasme for Medical Research, the Brussels Region Innoviris project DiaType, the Walloon Region SPW-EER Win2Wal project BetaSource, the Francophone Foundation for Diabetes Research (FFRD, that is sponsored by the French Diabetes Federation, Abbott, Eli Lilly, Merck Sharp & Dohme and Novo Nordisk) and the Innovative Medicines Initiative 2 Joint Undertaking Rhapsody, under grant agreement No 115881, supported by the European Union’s Horizon 2020 research and innovation programme, EFPIA and the Swiss State Secretariat for Education, Research and Innovation (SERI) under contract number 16.0097.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Bill and Dee Brehm for their support of the Brehm Center for Diabetes Research at the University of Michigan, and for the University of Michigan Protein Folding Diseases Program.
Abbreviations
ER, endoplasmic reticulum; ERAD, ER-associated degradation; GWAS, genome-wide association studies; MIDY, mutant INS-gene-induced diabetes of youth; RIDD, regulated IRE1-dependent decay; T2D, type 2 diabetes; UPR, unfolded protein response.
References
1. Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science (2011) 334(6059):1081–6. doi: 10.1126/science.1209038
2. Ron D, Walter P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat Rev Mol Cell Biol (2007) 8(7):519–29. doi: 10.1038/nrm2199
3. Eizirik DL, Cardozo AK, Cnop M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr Rev (2008) 29(1):42–61. doi: 10.1210/er.2007-0015
4. Shrestha N, Reinert RB, Qi L. Endoplasmic Reticulum Protein Quality Control in β Cells. Semin Cell Dev Biol (2020) 103:59–67. doi: 10.1016/j.semcdb.2020.04.006
5. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat Cell Biol (2013) 15(5):481–90. doi: 10.1038/ncb2738
6. Ghosh R, Colon-Negron K, Papa FR. Endoplasmic Reticulum Stress, Degeneration of Pancreatic Islet Beta-Cells, and Therapeutic Modulation of the Unfolded Protein Response in Diabetes. Mol Metab (2019) 27S:S60–S8. doi: 10.1016/j.molmet.2019.06.012
7. Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, et al. A Gene Encoding a Transmembrane Protein is Mutated in Patients With Diabetes Mellitus and Optic Atrophy (Wolfram Syndrome). Nat Genet (1998) 20(2):143–8. doi: 10.1038/2441
8. de Heredia ML, Cleries R, Nunes V. Genotypic Classification of Patients With Wolfram Syndrome: Insights Into the Natural History of the Disease and Correlation With Phenotype. Genet Med (2013) 15(7):497–506. doi: 10.1038/gim.2012.180
9. De Franco E, Flanagan SE, Yagi T, Abreu D, Mahadevan J, Johnson MB, et al. Dominant ER Stress-Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes (2017) 66(7):2044–53. doi: 10.2337/db16-1296
10. Bonnycastle LL, Chines PS, Hara T, Huyghe JR, Swift AJ, Heikinheimo P, et al. Autosomal Dominant Diabetes Arising From a Wolfram Syndrome 1 Mutation. Diabetes (2013) 62(11):3943–50. doi: 10.2337/db13-0571
11. Montaser H, Patel KA, Balboa D, Ibrahim H, Lithovius V, Naatanen A, et al. Loss of MANF Causes Childhood-Onset Syndromic Diabetes Due to Increased Endoplasmic Reticulum Stress. Diabetes (2021) 70(4):1006–18. doi: 10.2337/db20-1174
12. Poulton CJ, Schot R, Kia SK, Jones M, Verheijen FW, Venselaar H, et al. Microcephaly With Simplified Gyration, Epilepsy, and Infantile Diabetes Linked to Inappropriate Apoptosis of Neural Progenitors. Am J Hum Genet (2011) 89(2):265–76. doi: 10.1016/j.ajhg.2011.07.006
13. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, Encoding Translation Initiation Factor 2-α Kinase 3, is Mutated in Patients With Wolcott-Rallison Syndrome. Nat Genet (2000) 25(4):406–9. doi: 10.1038/78085
14. De Franco E, Caswell R, Johnson MB, Wakeling MN, Zung A, Dung VC, et al. De Novo Mutations in EIF2B1 Affecting EIF2 Signaling Cause Neonatal/Early-Onset Diabetes and Transient Hepatic Dysfunction. Diabetes (2020) 69(3):477–83. doi: 10.2337/db19-1029
15. Skopkova M, Hennig F, Shin BS, Turner CE, Stanikova D, Brennerova K, et al. EIF2S3 Mutations Associated With Severe X-Linked Intellectual Disability Syndrome MEHMO. Hum Mutat (2017) 38(4):409–25. doi: 10.1002/humu.23170
16. Synofzik M, Haack TB, Kopajtich R, Gorza M, Rapaport D, Greiner M, et al. Absence of Bip Co-Chaperone DNAJC3 Causes Diabetes Mellitus and Multisystemic Neurodegeneration. Am J Hum Genet (2014) 95(6):689–97. doi: 10.1016/j.ajhg.2014.10.013
17. Abdulkarim B, Nicolino M, Igoillo-Esteve M, Daures M, Romero S, Philippi A, et al. A Missense Mutation in PPP1R15B Causes a Syndrome Including Diabetes, Short Stature, and Microcephaly. Diabetes (2015) 64(11):3951–62. doi: 10.2337/db15-0477
18. Cnop M, Toivonen S, Igoillo Esteve M, Salpea P. Endoplasmic Reticulum Stress and Eif2α Phosphorylation: The Achilles Heel of Pancreatic β Cells. Mol Metab (2017) 6:1024–39. doi: 10.1016/j.molmet.2017.06.001
19. De Franco E, Lytrivi M, Ibrahim H, Montaser H, Wakeling MN, Fantuzzi F, et al. YIPF5 Mutations Cause Neonatal Diabetes and Microcephaly Through Endoplasmic Reticulum Stress. J Clin Invest (2020) 130(12):6338–53. doi: 10.1172/JCI141455
20. Lekszas C, Foresti O, Raote I, Liedtke D, Konig EM, Nanda I, et al. Biallelic TANGO1 Mutations Cause a Novel Syndromal Disease Due to Hampered Cellular Collagen Secretion. Elife (2020) 9:e51319. doi: 10.7554/eLife.51319
21. Herbert TP, Laybutt DR. A Reevaluation of the Role of the Unfolded Protein Response in Islet Dysfunction: Maladaptation or a Failure to Adapt? Diabetes (2016) 65(6):1472–80. doi: 10.2337/db15-1633
22. Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, et al. Discovery of 318 New Risk Loci for Type 2 Diabetes and Related Vascular Outcomes Among 1.4 Million Participants in a Multi-Ancestry Meta-Analysis. Nat Genet (2020) 52(7):680–91. doi: 10.1101/19012690
23. Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, et al. Common Variants in WFS1 Confer Risk of Type 2 Diabetes. Nat Genet (2007) 39(8):951–3. doi: 10.1038/ng2067
24. Wood AR, Jonsson A, Jackson AU, Wang N, van Leewen N, Palmer ND, et al. A Genome-Wide Association Study of IVGTT-Based Measures of First-Phase Insulin Secretion Refines the Underlying Physiology of Type 2 Diabetes Variants. Diabetes (2017) 66(8):2296–309. doi: 10.2337/db16-1452
25. Accelerating Medicines Partnership (AMP). Type 2 Diabetes Knowledge Portal. Available at: http://www.type2diabetesgenetics.org2020.
26. Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, et al. Meta-Analysis of Genome-Wide Association Studies Identifies Eight New Loci for Type 2 Diabetes in East Asians. Nat Genet (2012) 44(1):67–72. doi: 10.1038/ng.1019
27. Chu WS, Das SK, Wang H, Chan JC, Deloukas P, Froguel P, et al. Activating Transcription Factor 6 (ATF6) Sequence Polymorphisms in Type 2 Diabetes and Pre-Diabetic Traits. Diabetes (2007) 56(3):856–62. doi: 10.2337/db06-1305
28. Liu S, Ma G, Yao S, Chen Z, Wang C, Zhao B, et al. Polymorphism - 116C/G of the Human X Box Binding Protein 1 Gene is Associated With Risk of Type 2 Diabetes in a Chinese Han Population. Gene (2016) 575(1):71–4. doi: 10.1016/j.gene.2015.08.037
29. Hanson RL, Safabakhsh S, Curtis JM, Hsueh WC, Jones LI, Aflague TF, et al. Association of CREBRF Variants With Obesity and Diabetes in Pacific Islanders From Guam and Saipan. Diabetologia (2019) 62(9):1647–52. doi: 10.1007/s00125-019-4932-z
30. Spaeth JM, Gupte M, Perelis M, Yang YP, Cyphert H, Guo S, et al. Defining a Novel Role for the Pdx1 Transcription Factor in Islet β-Cell Maturation and Proliferation During Weaning. Diabetes (2017) 66(11):2830–9. doi: 10.2337/db16-1516
31. Johnson JS, Kono T, Tong X, Yamamoto WR, Zarain-Herzberg A, Merrins MJ, et al. Pancreatic and Duodenal Homeobox Protein 1 (Pdx-1) Maintains Endoplasmic Reticulum Calcium Levels Through Transcriptional Regulation of Sarco-Endoplasmic Reticulum Calcium ATPase 2b (SERCA2b) in the Islet Beta Cell. J Biol Chem (2014) 289(47):32798–810. doi: 10.1074/jbc.M114.575191
32. Sachdeva MM, Claiborn KC, Khoo C, Yang J, Groff DN, Mirmira RG, et al. Pdx1 (MODY4) Regulates Pancreatic β Cell Susceptibility to ER Stress. Proc Natl Acad Sci U S A (2009) 106(45):19090–5. doi: 10.1073/pnas.0904849106
33. Elouil H, Bensellam M, Guiot Y, Vander Mierde D, Pascal SM, Schuit FC, et al. Acute Nutrient Regulation of the Unfolded Protein Response and Integrated Stress Response in Cultured Rat Pancreatic Islets. Diabetologia (2007) 50(7):1442–52. doi: 10.1007/s00125-007-0674-4
34. Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, et al. Insulin Demand Regulates β Cell Number Via the Unfolded Protein Response. J Clin Invest (2015) 125(10):3831–46. doi: 10.1172/JCI79264
35. Cunha DA, Hekerman P, Ladrière L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and Execution of Lipotoxic ER Stress in Pancreatic β-Cells. J Cell Sci (2008) 121(Pt 14):2308–18. doi: 10.1242/jcs.026062
36. Marselli L, Piron A, Suleiman M, Colli ML, Yi X, Khamis A, et al. Persistent or Transient Human β Cell Dysfunction Induced by Metabolic Stress: Specific Signatures and Shared Gene Expression With Type 2 Diabetes. Cell Rep (2020) 33(9):108466. doi: 10.1016/j.celrep.2020.108466
37. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science (2004) 306(5695):457–61. doi: 10.1126/science.1103160
38. Chan JY, Luzuriaga J, Bensellam M, Biden TJ, Laybutt DR. Failure of the Adaptive Unfolded Protein Response in Islets of Obese Mice is Linked With Abnormalities in β-Cell Gene Expression and Progression to Diabetes. Diabetes (2013) 62(5):1557–68. doi: 10.2337/db12-0701
39. Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, et al. High Expression Rates of Human Islet Amyloid Polypeptide Induce Endoplasmic Reticulum Stress Mediated β-Cell Apoptosis, a Characteristic of Humans With Type 2 But Not Type 1 Diabetes. Diabetes (2007) 56(8):2016–27. doi: 10.2337/db07-0197
40. Casas S, Gomis R, Gribble FM, Altirriba J, Knuutila S, Novials A. Impairment of the Ubiquitin-Proteasome Pathway is a Downstream Endoplasmic Reticulum Stress Response Induced by Extracellular Human Islet Amyloid Polypeptide and Contributes to Pancreatic B-Cell Apoptosis. Diabetes (2007) 56(9):2284–94. doi: 10.2337/db07-0178
41. Arunagiri A, Haataja L, Cunningham CN, Shrestha N, Tsai B, Qi L, et al. Misfolded Proinsulin in the Endoplasmic Reticulum During Development of Beta Cell Failure in Diabetes. Ann N Y Acad Sci (2018) 1418(1):5–19. doi: 10.1111/nyas.13531
42. Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, et al. Regulation of Insulin Biosynthesis in Pancreatic β Cells by an Endoplasmic Reticulum-Resident Protein Kinase IRE1. Cell Metab (2006) 4(3):245–54. doi: 10.1016/j.cmet.2006.07.007
43. Cnop M, Ladrière L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, et al. Selective Inhibition of Eukaryotic Translation Initiation Factor 2α Dephosphorylation Potentiates Fatty Acid-Induced Endoplasmic Reticulum Stress and Causes Pancreatic β-Cell Dysfunction and Apoptosis. J Biol Chem (2007) 282(6):3989–97. doi: 10.1074/jbc.M607627200
44. Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F, et al. INS-gene Mutations: From Genetics and Beta Cell Biology to Clinical Disease. Mol Aspects Med (2015) 42:3–18. doi: 10.1016/j.mam.2014.12.001
45. Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted Disruption of the Chop Gene Delays Endoplasmic Reticulum Stress-Mediated Diabetes. J Clin Invest (2002) 109(4):525–32. doi: 10.1172/JCI0214550
46. Engin F, Nguyen T, Yermalovich A, Hotamisligil GS. Aberrant Islet Unfolded Protein Response in Type 2 Diabetes. Sci Rep (2014) 4:4054. doi: 10.1038/srep04054
47. Omikorede O, Qi C, Gorman T, Chapman P, Yu A, Smith DM, et al. ER Stress in Rodent Islets of Langerhans is Concomitant With Obesity and β-Cell Compensation But Not With β-Cell Dysfunction and Diabetes. Nutr Diabetes (2013) 3:e93. doi: 10.1038/nutd.2013.35
48. Chan JY, Luzuriaga J, Maxwell EL, West PK, Bensellam M, Laybutt DR. The Balance Between Adaptive and Apoptotic Unfolded Protein Responses Regulates Beta-Cell Death Under ER Stress Conditions Through XBP1, CHOP and JNK. Mol Cell Endocrinol (2015) 413:189–201. doi: 10.1016/j.mce.2015.06.025
49. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop Deletion Reduces Oxidative Stress, Improves β Cell Function, and Promotes Cell Survival in Multiple Mouse Models of Diabetes. J Clin Invest (2008) 118(10):3378–89. doi: 10.1172/JCI34587
50. Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, et al. The Endoplasmic Reticulum in Pancreatic Beta Cells of Type 2 Diabetes Patients. Diabetologia (2007) 50(12):2486–94. doi: 10.1007/s00125-007-0816-8
51. Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, et al. Role for Activating Transcription Factor 3 in Stress-Induced β-Cell Apoptosis. Mol Cell Biol (2004) 24(13):5721–32. doi: 10.1128/MCB.24.13.5721-5732.2004
52. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic Reticulum Stress Contributes to Beta Cell Apoptosis in Type 2 Diabetes. Diabetologia (2007) 50(4):752–63. doi: 10.1007/s00125-006-0590-z
53. Gargani S, Thevenet J, Yuan JE, Lefebvre B, Delalleau N, Gmyr V, et al. Adaptive Changes of Human Islets to an Obesogenic Environment in the Mouse. Diabetologia (2013) 56(2):350–8. doi: 10.1007/s00125-012-2775-y
54. Dai C, Kayton NS, Shostak A, Poffenberger G, Cyphert HA, Aramandla R, et al. Stress-Impaired Transcription Factor Expression and Insulin Secretion in Transplanted Human Islets. J Clin Invest (2016) 126(5):1857–70. doi: 10.1172/JCI83657
55. Papa FR. Endoplasmic Reticulum Stress, Pancreatic β-Cell Degeneration, and Diabetes. Cold Spring Harb Perspect Med (2012) 2(9):a007666. doi: 10.1101/cshperspect.a007666
56. Ghosh R, Colon-Negron K, Papa FR. Endoplasmic Reticulum Stress, Degeneration of Pancreatic Islet β-Cells, and Therapeutic Modulation of the Unfolded Protein Response in Diabetes. Mol Metab (2019) 27S:S60–S8. doi: 10.1016/j.molmet.2019.06.012
57. Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, et al. Ire1α is an Endogenous Substrate of Endoplasmic-Reticulum-Associated Degradation. Nat Cell Biol (2015) 17(12):1546–55. doi: 10.1038/ncb3266
58. Brozzi F, Gerlo S, Grieco FA, Juusola M, Balhuizen A, Lievens S, et al. Ubiquitin D Regulates IRE1α/c-Jun N-Terminal Kinase (JNK) Protein-Dependent Apoptosis in Pancreatic Beta Cells. J Biol Chem (2016) 291(23):12040–56. doi: 10.1074/jbc.M115.704619
59. Brozzi F, Gerlo S, Grieco FA, Nardelli TR, Lievens S, Gysemans C, et al. A Combined “Omics” Approach Identifies N-Myc Interactor as a Novel Cytokine-Induced Regulator of IRE1 Protein and c-Jun N-Terminal Kinase in Pancreatic Beta Cells. J Biol Chem (2014) 289(30):20677–93. doi: 10.1074/jbc.M114.568808
60. Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. WFS1-deficiency Increases Endoplasmic Reticulum Stress, Impairs Cell Cycle Progression and Triggers the Apoptotic Pathway Specifically in Pancreatic B-Cells. Hum Mol Genet (2006) 15(10):1600–9. doi: 10.1093/hmg/ddl081
61. Xin Y, Dominguez Gutierrez G, Okamoto H, Kim J, Lee AH, Adler C, et al. Pseudotime Ordering of Single Human β-Cells Reveals States of Insulin Production and Unfolded Protein Response. Diabetes (2018) 67(9):1783–94. doi: 10.2337/db18-0365
62. Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, et al. The Long Lifespan and Low Turnover of Human Islet Beta Cells Estimated by Mathematical Modelling of Lipofuscin Accumulation. Diabetologia (2010) 53(2):321–30. doi: 10.1007/s00125-009-1562-x
63. Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, et al. Significant Human β-Cell Turnover is Limited to the First Three Decades of Life as Determined by in Vivo Thymidine Analog Incorporation and Radiocarbon Dating. J Clin Endocrinol Metab (2010) 95(10):E234–9. doi: 10.1210/jc.2010-0932
64. Eizirik DL, Pasquali L, Cnop M. Pancreatic β-Cells in Type 1 and Type 2 Diabetes Mellitus: Different Pathways to Failure. Nat Rev Endocrinol (2020) 16(7):349–62. doi: 10.1038/s41574-020-0355-7
65. Bensellam M, Jonas JC, Laybutt DR. Mechanisms of Beta-Cell Dedifferentiation in Diabetes: Recent Findings and Future Research Directions. J Endocrinol (2018) 236(2):R109–R43. doi: 10.1530/JOE-17-0516
66. Shrestha N, Liu T, Ji Y, Reinert RB, Torres M, Li X, et al. Sel1L-Hrd1 ER-associated Degradation Maintains β Cell Identity Via TGF-β Signaling. J Clin Invest (2020) 130(7):3499–510. doi: 10.1172/JCI134874
67. Russ HA, Ravassard P, Kerr-Conte J, Pattou F, Efrat S. Epithelial-Mesenchymal Transition in Cells Expanded in Vitro From Lineage-Traced Adult Human Pancreatic Beta Cells. PloS One (2009) 4(7):e6417. doi: 10.1371/journal.pone.0006417
68. Efrat S. Beta-Cell Dedifferentiation in Type 2 Diabetes: Concise Review. Stem Cells (2019) 37(10):1267–72. doi: 10.1002/stem.3059
Keywords: endoplasmic reticulum, stress, unfolded protein response, insulin, PERK (PKR-like endoplasmic reticulum kinase), ATF6 (activating transcription factor 6), IRE1 (inositol-requiring enzyme 1)
Citation: Shrestha N, De Franco E, Arvan P and Cnop M (2021) Pathological β-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front. Endocrinol. 12:650158. doi: 10.3389/fendo.2021.650158
Received: 06 January 2021; Accepted: 06 April 2021;
Published: 22 April 2021.
Edited by:
Michele Solimena, Technische Universität Dresden, GermanyReviewed by:
Fumihiko Urano, Washington University School of Medicine, United StatesAmna Khamis, UMR8199 Génomique Intégrative et Modélisation des Maladies Métaboliques, France
Martine Vaxillaire, Institut Pasteur de Lille, France
Copyright © 2021 Shrestha, De Franco, Arvan and Cnop. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Arvan, cGFydmFuQHVtaWNoLmVkdQ==; Miriam Cnop, bWNub3BAdWxiLmFjLmJl
†These authors have contributed equally to this work
‡These authors share senior authorship