 Terry J. Smith
Terry J. Smith- Department of Ophthalmology and Visual Sciences, Kellogg Eye Center, Division of Metabolism, Endocrinology, and Diabetes, Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI, United States
The insulin-like growth factor (IGF) pathway comprises two activating ligands (IGF-I and IGF-II), two cell-surface receptors (IGF-IR and IGF-IIR), six IGF binding proteins (IGFBP) and nine IGFBP related proteins. IGF-I and the IGF-IR share substantial structural and functional similarities to those of insulin and its receptor. IGF-I plays important regulatory roles in the development, growth, and function of many human tissues. Its pathway intersects with those mediating the actions of many cytokines, growth factors and hormones. Among these, IGFs impact the thyroid and the hormones that it generates. Further, thyroid hormones and thyrotropin (TSH) can influence the biological effects of growth hormone and IGF-I on target tissues. The consequences of this two-way interplay can be far-reaching on many metabolic and immunologic processes. Specifically, IGF-I supports normal function, volume and hormone synthesis of the thyroid gland. Some of these effects are mediated through enhancement of sensitivity to the actions of TSH while others may be independent of pituitary function. IGF-I also participates in pathological conditions of the thyroid, including benign enlargement and tumorigenesis, such as those occurring in acromegaly. With regard to Graves’ disease (GD) and the periocular process frequently associated with it, namely thyroid-associated ophthalmopathy (TAO), IGF-IR has been found overexpressed in orbital connective tissues, T and B cells in GD and TAO. Autoantibodies of the IgG class are generated in patients with GD that bind to IGF-IR and initiate the signaling from the TSHR/IGF-IR physical and functional protein complex. Further, inhibition of IGF-IR with monoclonal antibody inhibitors can attenuate signaling from either TSHR or IGF-IR. Based on those findings, the development of teprotumumab, a β-arrestin biased agonist as a therapeutic has resulted in the first medication approved by the US FDA for the treatment of TAO. Teprotumumab is now in wide clinical use in North America.
Introduction
Among the most ubiquitous regulatory factors governing functions in the mammalian body are those belonging to the insulin and insulin-like growth factor-I (IGF-I) family, including their respective receptors, binding and related proteins, and extensive signaling pathways that mediate/modulate their actions. Discovery of insulin has been attributed to several individuals, but most prominent among them are Paulescu [cited in reference (1)] in France and Banting, Best and colleagues (2) in Toronto, Canada, working independently. Therapeutic insulin was first administered to patients with diabetes mellitus in 1922, representing a seismic event ranking among the most impactful in modern medicine. In aggregate, this body of work concerning the discovery and characterization of insulin opened the door to our current understanding of normal and pathological carbohydrate metabolism with implications extending far beyond. Its “first cousin”, sulfation factor, was first described nearly four decades later by Daughaday and colleagues (3). Sulfation factor underwent a name change to somatomedin C in 1972 (4) and finally to its current designation, IGF-I, in the early 1980s (5). Subsequent to their discovery, the actions of both insulin and IGF-I have been characterized extensively and found to be overlapping in many regards. Their physiological roles and impact on target tissues and cells have been slowly disentangled but their functional promiscuity and that of their respective receptors continue to intrigue students of the field.
In this article, I focused on the growth hormone (GH) and IGF-I pathways and have attempted to briefly review their numerous intersections in the hypothalamic-pituitary-thyroid axis. The relevance of IGF-IR in the pathogenesis of thyroid-associated ophthalmopathy (TAO), the most serious extra-thyroidal autoimmune manifestation of Graves’ disease (GD), has only come into prominence over the past 2 decades (6–8). As I will describe, IGF-IR plays not only a critical role in the pathogenesis of TAO, but can be effectively targeted as a therapeutic strategy for managing the disease. The concept of IGF-IR playing an important role in the development of TAO or targeting the protein as a strategy for treating the disease had been met with overwhelming skepticism when it was first proposed (9, 10). Despite these dismissive views, teprotumumab, an inhibitory monoclonal antibody directed at IGF-IR, was recently approved specifically for the treatment of TAO. It is now in clinical use in North America; however, considerably more information will be required if we are to fully understand its mechanism of action in ameliorating the signs and symptoms of TAO and the entirety of its off-target consequences on the human body.
IGF-I and Its Associated Pathway
The molecular structure of IGF-I is closely related to insulin (11) (Figure 1). IGF-I comprises 70 amino acids and mediates growth during childhood and adolescence (12). IGF-II contains 67 amino acids. Both IGF-I and IGF-II have three intramolecular disulfide bridges. While IGF-I synthesis was initially considered to be driven by GH as a systemically active mediator, recent decades have witnessed increasing insight into its roles as a paracrine and autocrine factor. Mediating the actions of insulin and IGF-I are their respective, closely related receptors (IGF-IR and IR, respectively), both belonging to the tyrosine kinase family (Figure 2). The cell surface receptors of both ligands also exhibit substantial structural and functional relatedness and may have evolved from gene duplication of a common precursor (13). They form IR/IR and IGF-IR/IGF-IR as well as IR/IGF-IR hybrids, the particular protein pairings of which might be determined stochastically. Both possess heterotetrameric protein structures comprising extracellular domains containing ligand binding sites located in two α subunits. Two β subunits consist of extracellular, transmembrane and intracellular domains containing the kinase domains, the ATP binding site, and multiple potential tyrosine phosphorylation sites (14). The α and β subunits are linked by two disulfide bonds. Both insulin and IGF-I engage in promiscuous interactions with each other’s receptor, causing substantial overlap in the physiological and pathological consequences of activating the IGF-IR and IR pathways. While IGF-IR primarily functions as an integral membrane receptor the activation of which provokes tyrosine kinase autophosphorylation, recent studies have revealed the nuclear translocation of IGF-IR as well as other components of the IGF-I pathway (15). Similar to many other cell-surface receptors, the density of IGF-IR determines in part the pattern of signaling downstream from that receptor (16). It has been proposed that unligated IGF-IR can initiate signaling mediated through microRNAs and imprinted genes (17). Several factors can upregulate IGF-IR gene expression while others reduce target gene transcriptional activity (8). In addition to IGF-IR, a second receptor, IGF-IIR, aka the mannose-6-phosphate receptor, can bind IGF-II (18) and plays roles in trafficking molecules to the Golgi and the endosomal-lysosomal system (19). IGF-IIR does not mediate tyrosine phosphorylation-dependent signaling analogous to that attributed to IGF-IR and IR. Besides the two receptors, the IGF-I pathway includes six IGF-I binding proteins and nine IGFBP related proteins (20, 21). Their potential relevance to the thyroid has been reviewed recently in detail (8).
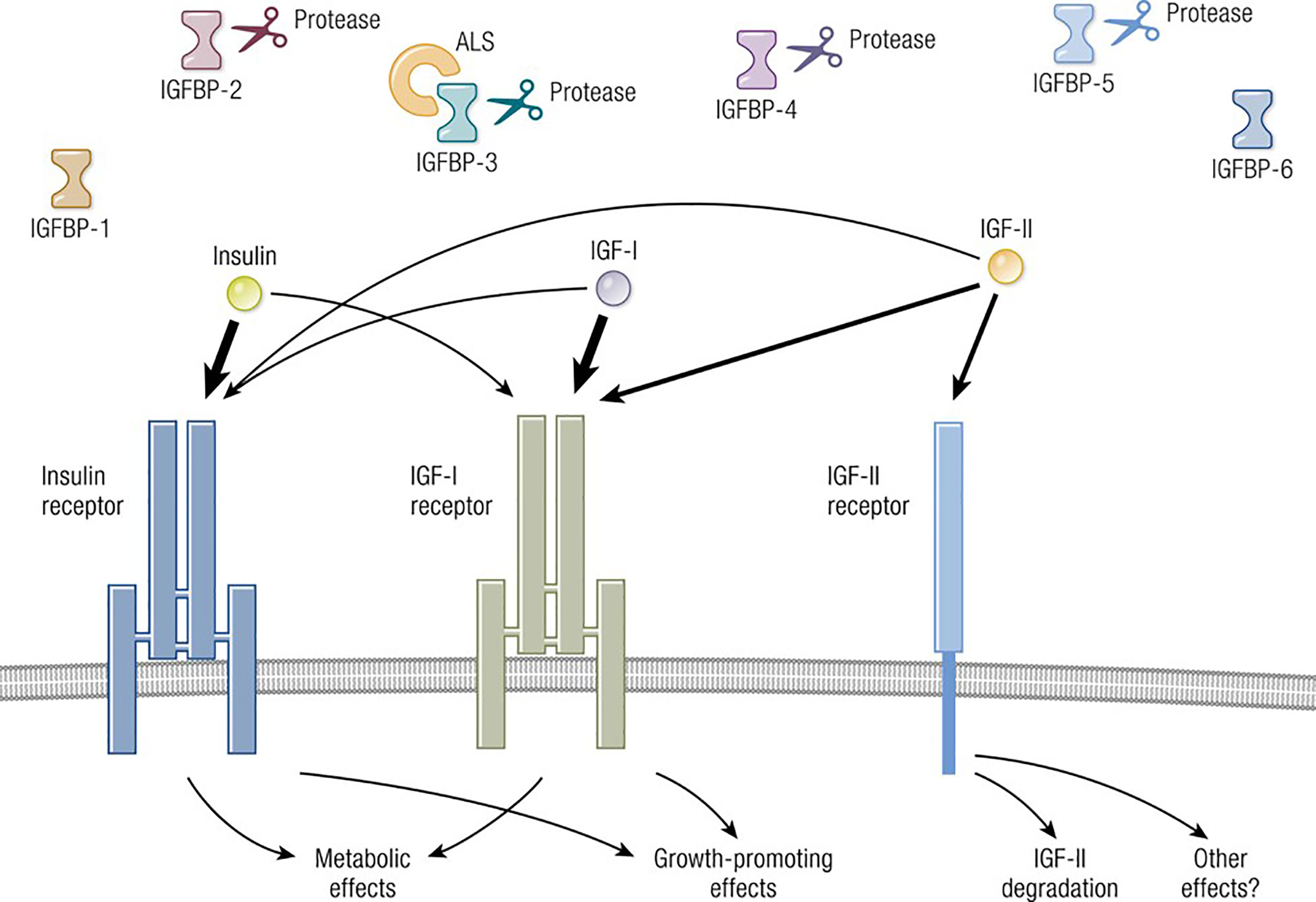
Figure 1 Overview of the IGF-I pathway, including its multiple ligands, modulatory binding proteins and receptors. Ligands including IGF-I, IGF-II, and insulin share important structural and functional relationships including the promiscuous utilization of multiple receptors, such as IGF-IR, IGF-IIR, and IR. The pathway also contains IGF-binding proteins (IGFBPs) which can function either independently or may require ligation. IGF-IR plays a central role in mediating growth regulation. In contrast, the insulin receptor is the primary regulator of metabolism and carbohydrate handling. IGF-IIR (mannose-6-phosphate receptor) influences IGF-II degradation and participates in the biology of the Golgi apparatus. Biological impact of both IGF-I and IGF-II action is modulated by six thus-far identified IGFBPs. Adapted from Lowe WL. Insulin-like growth factors. Science & Medicine 1996; 3; 65.
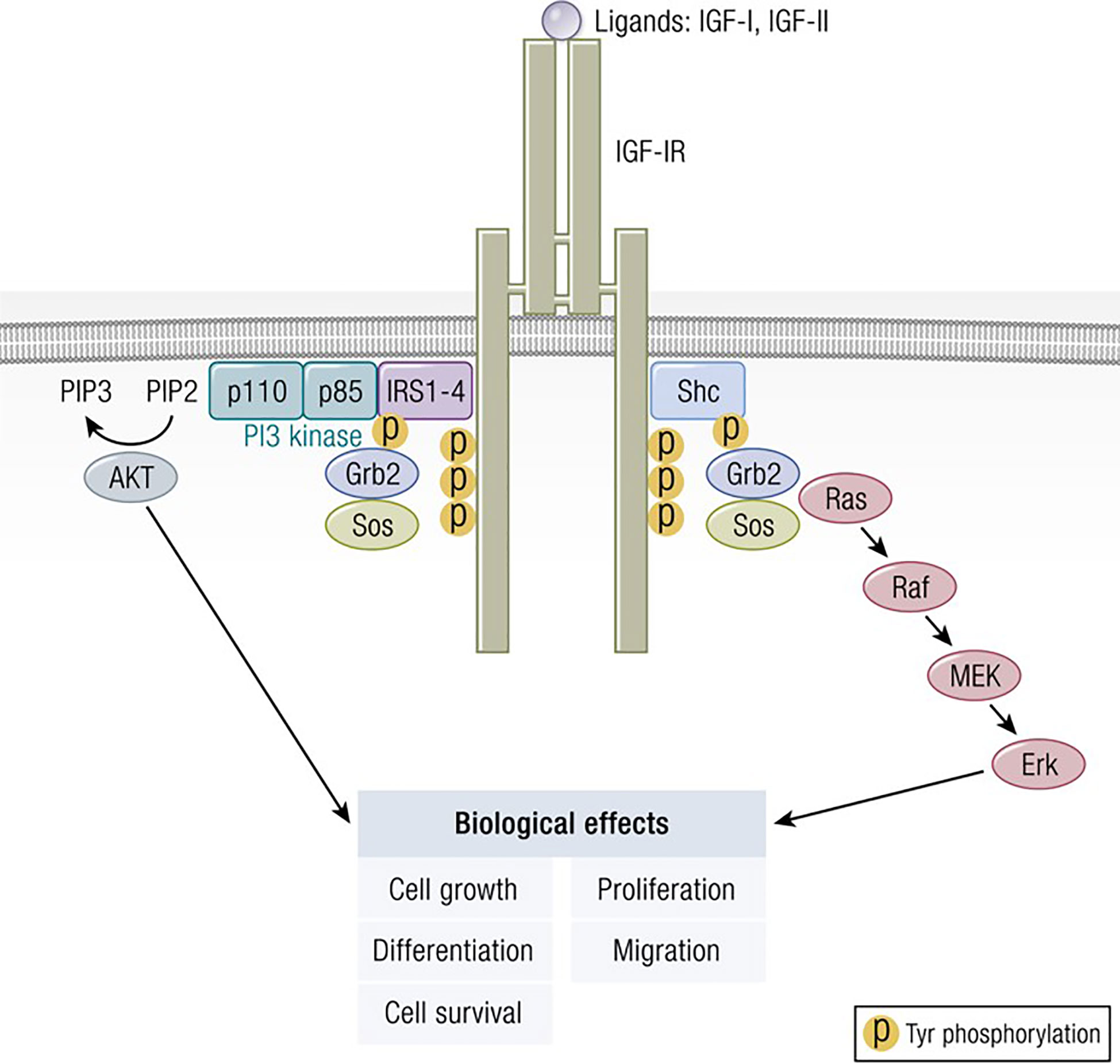
Figure 2 Classical model of signaling through the IGF-IR. IGF-I ligation of IGF-IR most commonly results in canonical mitogen activated protein kinase (MAPK)/Ras-Raf-Erk and phosphatidylinositol-3-kinase/AKT/mTOR (PI3K/AKT) phosphorylation, critical to downstream signaling and the activation of targeted genes associated with IGF-I responses. Adapted from Worrall et al. Novel Mechanisms of Regulation of IGF-1R Action: Functional and Therapeutic Implications. Pediatr Endocrinol Rev. 2013 10:473-484.
Impact of GH and its Primary Effector, IGF-I, on the Thyroid Gland
The relationship between GH, IGF-I and the thyroid gland is complex, frequently reciprocal and temporally spans the interval during fetal development throughout adult life (22, 23). Much of what we understand about the impact of the IGF-I pathway on the thyroid derives from observations made in states of hormone excess such as acromegaly (22, 24). Porcine thyroid cells express both IGF-IR and IR (25) as do human thyroid cancer cells and normal thyroid (26). Thyrospheres derived from healthy tissue express both isoforms of IR, namely IR-A and IR-B, as well as IGF-IR, IGF-I and IGF-II, levels which decline following cell differentiation. Interestingly, between the two insulin receptor isoforms, IR-A expression dominates thyrospheres while IR-B is more abundant in differentiated thyrocytes. IGF-I binding is saturable and Scatchard analysis discloses a single binding class with a Ka 4.3 X 10-10 and 49 X 103 sites per cell. IGF-I stimulates DNA synthesis and thyroid cell proliferation, actions synergistic with those of epidermal growth factor. IGF-I and insulin have important roles in regulating not only proliferation but also thyroid cell differentiation. In studies of FRTL-5 rat thyroid cells, IGF-II but not IGF-I was found to be synthesized and released (27). IGF-II promotes DNA synthesis in FRTL-5. On the other hand, medullary thyroid carcinoma cells were found to express both IGF-I and IGF-II (28, 29). A p21 Ras mutation can induce IGF-I expression and release in immortalized human thyroid epithelial cells (30). In porcine thyroid follicles, TSH and iodine were found to regulate IGF-I mRNA levels (31). In those studies, IGF-I transcript was undetectable in the absence of TSH, effects that were mimicked by forskolin. In contrast, Iodine downregulated the levels of this mRNA. Similar TSH-dependent effects on IGF-I were demonstrated in another report (32). Interestingly, differentiation of mouse embryonic stem cells to thyroid epithelial cells requires insulin/IGF-I, suggesting the complexities of these factors in thyroid development (33). The incidence of thyroid nodules was found to be increased substantially in children with extreme insulin resistance (34) bringing clinical relevance to the basic observations made in the laboratory.
Signaling through both IGF-IR and IR is considered essentially indispensable in the normal function of the thyroid gland, both through their independent support of thyroid epithelial survival and vitality and also by virtue of the interactions of their downstream pathways with those of the thyrotropin receptor (TSHR) (35). The patterns of effects in FRTL-5 cells suggest that IGF-I and insulin have distinct actions on specific gene expression, both independent of and in concert with TSH (36). Among the IGF-IR/IR-activated pathways in thyroid cells are Erk 1/2, and Akt, while TSHR is G protein coupled and results in the generation of cAMP. Examination of IGF-IR and insulin receptor docking proteins has revealed a role for insulin receptor substrate 2 (IRS-2) in mediating the proliferative actions of IGF-I, both in vitro and in vivo (37). Iodide uptake and organification are among the specific aspects of thyroid metabolism under dual TSH and IGF-I/insulin control. These processes in turn are mediated through the activities of the sodium iodide symporter and thyroperoxidase, respectively. In a study of mice with conditional double-thyroid knockout of IGF-IR and IR, neonatal thyroid glands were smaller, exhibited repressed FOXE1 expression, and manifested defective folliculogenesis (38). At postnatal day 14, mTOR-dependent epithelial cell proliferation and serum TSH were detectable. By week 50, lesions resembling papillary carcinoma had developed, coincident with ErbB activation. The authors concluded that both IGF-IR and IR are critical to follicle formation in the developing thyroid. IGF-IR engagement in cultured thyroid epithelial cells has been shown to activate the expression of chemokines and cytokines such as IL-16 and RANTES (39). Both insulin and IGF-I can regulate the rate of major histocompatibility complex class 1 (MHC class 1) gene transcription (40). These effects are similar to those of TSH in FRTL-5 cells. IGF-I and insulin in combination with TSH can suppress the transcription of the Mac-2BP gene in these cells by decreasing the binding of an upstream specific factor to a gene promoter site (41). IGF-I and cAMP appear to differentially activate the PI3 kinase pathway in FRTL-5 cells, leading to G1 cyclin-cyclin dependent kinase activation and DNA synthesis (42). IGF-I induces several anti-apoptotic proteins in thyroid cells, including Fas-associated death domain-like interleukin-1-converting enzyme-inhibitory protein, actions mediated through activation of the NF-κB pathway (43). Quercetin inhibition of FRTL-5 proliferation is mediated by alterations in insulin and IGF-I signaling involving the Akt pathway (41). It has been suggested that lithium might influence IGF-IR coupled Gi-proteins during the G1 phase of FRTL-5 cell cycling (44). With regard to thyroid cancer, the IGF-I pathway has been directly implicated in the transformation of thyroid epithelial cells (45). It remains uncertain whether the relationship between IGF-I action, IGF-IR signaling, or any of the other components of the IGF-I pathway differ in their relationship to thyroid cancers when compared to other forms of neoplastic disease. Elevated levels of the IGF-IEc isoform have been reported in differentiated papillary thyroid carcinoma and are associated with advanced disease (46).
The Thyroid in Acromegaly
The hypothalamic-pituitary-thyroid axis can be affected at several levels in acromegaly, including those imposed directly and indirectly on the gland itself. For instance, interpretation of laboratory thyroid testing in this condition can be complicated. Both basal and pulsatile TSH secretion are often reduced while thyroxine levels can remain unaffected (47). This phenomenon may result from enhanced thyroid sensitivity to the actions of TSH caused by either growth hormone, IGF-I, or both. In their very recent report, Natchev and colleagues studied a cross-section of 146 acromegalic patients and found that secondary hypothyroidism and hyperthyroidism are commonly encountered (48). Thyroid gland volume is frequently impacted (49) and frank thyroid enlargement in acromegaly is not unusual. Thyromegaly was first recognized in acromegaly by Rolleston in 1936 (24). Gland volume in acromegaly correlates with serum GH and IGF-I levels. Distinguishing which of these hormones might be impacting thyroid enlargement in a particular case is frequently not possible (50). The incidence of both euthyroid and hyperthyroid goiters is increased in acromegaly, a consequence potentially independent of TSH (51). In the absence of TSH, it appears that GH has little or no effect on thyroid size (52). Surgical cure of acromegaly can result in reduced thyroid volume (50). Sera from patients manifesting untreated acromegaly can induce [3H] thymidine incorporation into the DNA of FRTL-5 cells more than sera from healthy control donors (53). The mitogenic activities of both normal and acromegalic sera can be attenuated by pretreating them with neutralizing monoclonal anti-IGF-I antibodies. Thyroid enlargement in acromegaly can be nodular, multinodular or diffuse (48, 54). The nodules in this condition are described as “stiff”, a quality thought to result from localized fibrosis (55). Besides benign goiter, the incidence of thyroid carcinoma is said to be increased in acromegaly (56, 57). In fact, both benign and malignant thyroid nodules are commonplace in these patients (54). Patients with acromegaly are at increased risk for a variety of cancers besides those of the thyroid. These include those of the colon, prostate, and breast, but thyroid cancers may be the most common malignancy associated with the disease (58). IGF-I can promote tumor progression and perhaps facilitate neoplastic initiation (59). McCune-Albright syndrome occurring in adults can be associated with toxic multinodular goiter and an increased risk of thyroid cancer (60). Of the thyroid cancers associated with acromegaly, papillary tumors can occur, especially in those patients manifesting other endocrine and non-endocrine neoplasms. These include pheochromocytoma, growth hormone-producing pituitary adenoma, and duodenal adenocarcinoma (61, 62).
Interplay Between IGF-I and Thyroid Hormone Metabolism and Pathways
As suggested above, entanglement between the actions of GH, IGF-I and thyroid hormones is complex and can become unmasked with excesses or deficiencies in one or both pathways. Childhood deficiency of GH can occur as an isolated defect, whereas those occurring in adults are typically associated with a constellation of pituitary-hypothalamic insufficiencies and are mostly associated with anatomic lesions. Exogenous GH administration in GH-deficient children has been shown to reduce circulating levels of T4, although the mechanism(s) involved has yet to be established. Some studies have revealed a GH-dependent increase in T4 to T3 conversion (63) and that GH may support physiological T4 to T3 conversion. Further, GH deficiency may impair T3 generation (64). GH may also work centrally, either through direct actions or those mediated through IGF-I to reduce the synthesis and release of TSH from the pituitary (65). T3 and IGF-I influence divergently GH synthesis and release from cultured pituitary cells in monolayer (66). The enhancement of GH synthesis by T3 is mediated through enhanced gene transcription while the inhibition by IGF-I of GH induction appears to reflect changes in posttranscriptional events. Insulin and thyroid hormones can act synergistically by mutual enhancement. For instance, T3 can upregulate the expression of glutamic acid decarboxylase in pure cortical neuronal cultures, actions requiring the presence of insulin (67). T3 (and thyroxine to a lesser extent) enhances the increased sulfation of embryonic cartilage by sera from either intact or hypophysectomized rats, effects mediated at least in part by IGF-I (68). IGF-I and thyroid hormones interact on growth plate chondrocytes where IGF-I enhances Wnt-4 expression and β-catenin activation (69). In that study, T3 was shown to enhance IGF-1R signaling and its upregulation of PI3K/Akt/GSK-3β signaling. IGF-I increases levels of thyroid transcription factor-2, actions similar to those of insulin (70). T3 and IGF-I have synergistic, enhancing effects on the expression of fast-type sarcoplasmic-reticulum Ca++-ATPase in L6 myocytes (71). T3 acts on the rate of gene transcription while IGF-I enhances mRNA and protein stabilities. These interactions can translate into clinically relevant events. For instance, exogenous GH attenuates thyroid hormone action in patients with Turner syndrome (72).
Impact of Thyroidal Status on the GH/IGF-I Axis
Normal growth requires intactness of both GH/IGF-I and thyroid hormone pathways. Thyroid hormones influence IGF-I levels in the pituitary and in peripheral tissue compartments (73), including expression and release of GH from the pituitary (74, 75) and the circulating levels of both GH and IGF-I (76). Animals harboring the mutant transcription factor, Pit-1, (Snell dwarf mice), exhibit multiple deficiencies in anterior pituitary hormones resulting in defective B cell development (77). This defect can be partially ameliorated with exogenous GH or IGF-I but a more complete remediation can be accomplished with administration of exogenous thyroxine. Thus, the regulatory actions of GH/IGF-I and TSH/thyroid hormones appear to overlap in bone marrow cells. Evidence suggests that not all thyroid hormone effects on the IGF-I pathway are mediated through GH expression and release but that other, GH-independent mechanisms may also be involved (78). Some contradictory findings have been reported in human dysthyroidemia. Plasma levels of IGF-I were found to be reduced in hypothyroid individuals; these were increased substantially with adequate thyroid hormone replacement. Basal IGF-I levels are lower in hypothyroid patients compared to those who are euthyroid (79). In another report, serum IGF-I levels were lower in hyperthyroid patients while levels trended toward elevation in hypothyroid individuals (80). Other components of the IGF-I pathway are influenced by thyroid hormone. Thyroxine replacement therapy appears to increase serum levels of IGFBP1 (81). In 18 day old rats, thyroid hormones can regulate the expression of hepatic IGFBP2 mRNA and serum protein levels through a mechanism independent of GH (78). Thus thyroid hormone effects on IGFBP2 in these animals diverge from those of IGF-I where its effects are mediated through GH. Serum IGFBP3 and IGFBP4 levels are reduced in hypothyroid animals (82). Thyroidectomized patients in whom thyroid hormone replacement has been withdrawn exhibit a reduction in circulating IGFBP1 levels (83) while thyroxine treatment increases them (81). It appears that the impact of thyroid hormone status on the GH/IGF pathway may change with maturation; further some of these developmental-stage sensitive effects are mediated through GH (78). Propylthiouracil treatment in rats results in substantially increased IGF-II binding site density on thyroid epithelial cells (84). In contrast, thyroid hormones induce IGF-IR mRNA expression in rat epiphyseal chondrocytes (85) and enhance IGF-I binding in rat pituitary cells (86). They regulate IGF-IR expression in rat heart and lung, both during animal development and in adults (87). In aggregate, these findings suggest the complex interactions shared by the two pathways. They support the important roles played by each in the maintenance of both endocrine functions at multiple levels.
IGF-I and Insulin Enhance the Actions of TSH and Thyroid-Stimulating Immunoglobulins: Evidence for Interplaying Pathways
Several factors are necessary for thyroid epithelial cells to function normally. Among these are IGF-I and TSH, both of which are critical to thyroid hormone synthesis (88). Of these two molecules, TSH and its cognate receptor, TSHR, represent the primary regulatory pathway for thyroid development, growth and function. Several ‘well-travelled” IGF-IR signaling pathways cross-talk with those downstream from TSHR in thyroid epithelial cells. These interactions can either enhance or modulate TSHR-mediated biosynthetic events, depending on their context. Among the intersecting signaling cascades, the p42/44 mitogen-activated protein kinase (MAPK) pathway is pivotal (89). TSHR signaling through p42/44 MAPK is independent of cAMP but dependent on the receptor’s coupling to G13 protein (89). So too, is this pathway of central importance to the signaling initiated through IGF-I. The interplay between the actions of IGF-I, insulin and TSH in the thyroid involves interactions between tyrosine kinase receptors (RTKs) and G protein coupled receptors (GPCRs) (Figure 3). These receptor classes share scaffolding proteins, namely β-arrestin 1 and 2. The apparently critical role played by β-arrestin 1 in downregulating IGF-IR expression through ubiquination is mediated by an MDM2 E3 Ligase dependent mechanism (90). β-arrestin also mediates IGF-IR signaling through the Erk pathway (91). Studies in Ewing’s sarcoma cells treated with figitumumab, an IGF-IR antagonist, revealed that this antibody functioned as an IGF-1R-biased agonist through β-arrestin1 recruitment to the receptor, enhancing IGF-IR ubiquination and provoking Erk phosphorylation in the absence of Akt activation (92). The scope of potential RTK/GPCR hybrid formation is not limited to that involving the IGF-I/insulin family. These hybrids have been observed with other RTKs, including the epidermal growth factor receptor (93). Initial observations that IGF-I and insulin can amplify effects of TSH on thyroid epithelial cells were conducted in FRTL-5 cells (94). Ingbar and colleagues found that both IGF-IR- and IR- activating ligands could provoke proliferation and DNA synthesis in these cultured cells. Their initial report was soon followed by more complete characterizations from that group as well as studies emanating from other laboratories (95). Those studies disclosed that IGF-I could consistently enhance TSH actions in thyroid epithelial cells and demonstrated synergism with regard to cell proliferation and tyrosine kinase activation. As an example, TSH and IGF-I synergistically enhance levels of 1,2-diacylglycerol in FRTL-5 cells, resulting in increased DNA synthesis (96). On the other hand, TSH and IGF-I have distinctly different effects on immediate early gene expression in rat thyroid epithelial cells (97). Thus, despite similarities, the two pathways should be viewed as non-equivalent but frequently intersecting. Tsui et al. subsequently reported that signaling initiated by both rhTSH and GD-IgG could be attenuated in vitro by 1H7, a monoclonal anti-IGF-IR-inhibitory antibody (98). Those investigators also reported that TSHR and IGF-IR co-localize in orbital fibroblasts, in situ in orbital fat and in primary human thyroid epithelial cells. Further, the two receptor proteins co-precipitate in monoclonal antibody-utilizing pull-down studies (98). Those studies were followed by others demonstrating that the conditional knock-out of IGF-IR in thyroid diminishes its responsiveness to TSH (99). Conversely, selective, combined over-expression of IGF-I and IGF-IR in thyroid of transgenic mice exhibits enhanced sensitivity to endogenous TSH (100). Extensive crosstalk of the multiple downstream signal transduction pathways utilized by the two receptors has been identified (101–104). Those pathways include Erk 1/2 (98). Further, TSH can enhance IGF-I signaling in thyroid, actions mediated through the generation of cAMP (105). Thus, there exists substantial molecular rationale for considering the importance of a functional interplay between IGF-IR and TSHR and for targeting this protein complex therapeutically. Congruent with that possibility, Chen et al. (106) found that the fully human IGF-IR inhibiting antibody, teprotumumab, examined earlier in clinical trials for multiple cancers (107–111), could also attenuate the actions of both IGF-I and TSH in cultured CD34+ fibrocytes. IGF-I was recently found to enhance the expression of TSHR in orbital fibroblasts (112).
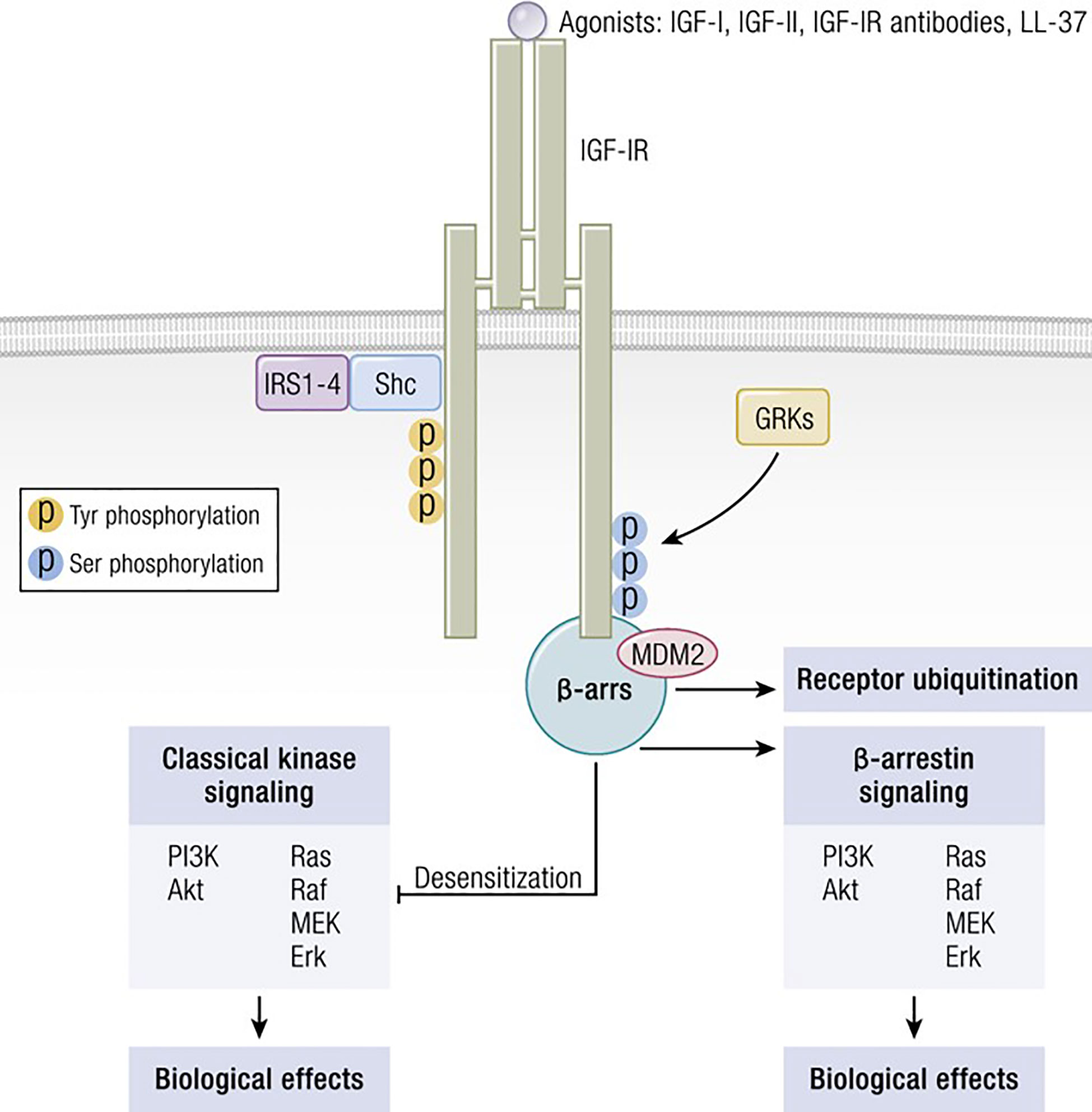
Figure 3 IGF-IR can form functional RTK/GPCR hybrids, thus aggregating tyrosine kinase and GPCR signaling. Ligand-dependent activation of classical kinase-dependent signaling with β-arrestin recruitment provokes GRK-dependent serine phosphorylation located in the IGF-IR C-domain. β-arrestin activates kinase desensitization and ubiquitination and initiates kinase-independent signaling through MAPK. Adapted from Worrall et al. Novel Mechanisms of Regulation of IGF-1R Action: Functional and Therapeutic Implications. Pediatr Endocrinol Rev. 2013 10:473-484.
Emerging Relevance of IGF-IR in the Pathogenesis of GD and TAO
The development of GD remains an only partially understood process. These deficits have resulted historically in suboptimal medical management of both the thyroid dysfunction and TAO components of this vexing condition (113) (Figure 4). Initial insights that the IGF-I pathway might be involved in the pathogenesis of TAO emerged from the study of Weightman et al. (114). These investigators reported detecting IgGs collected from patients with GD (GD-IgG) that were capable of displacing radiolabeled IGF-I from binding sites on the surface of orbital fibroblasts coming from these individuals (GD-OF). Pritchard et al. demonstrated subsequently that these bindings sites were IGF-IR rather than one or more of the IGFBPs (9). Further, Pritchard et al. reported that GD-IgGs can initiate signaling in GD-OF, resulting in the activation of the PI3 kinase/FRAP/mTOR/p70s6 kinase pathway and the induction of target genes encoding IL-16 and “Regulated Upon Activation, Normal T Cell Expressed and Presumably Secreted” (RANTES), two T cell chemoattractants (115). A major issue remaining to be clarified concerns whether the agonistic autoantibodies generated in TAO that induce responses in GD-OF and fibrocytes act directly through TSHR, IGF-IR or both. Whether the anti-IGF-IR antibodies represent those that stimulate or block the receptor’s activation or do neither (neutral) remains another open question.
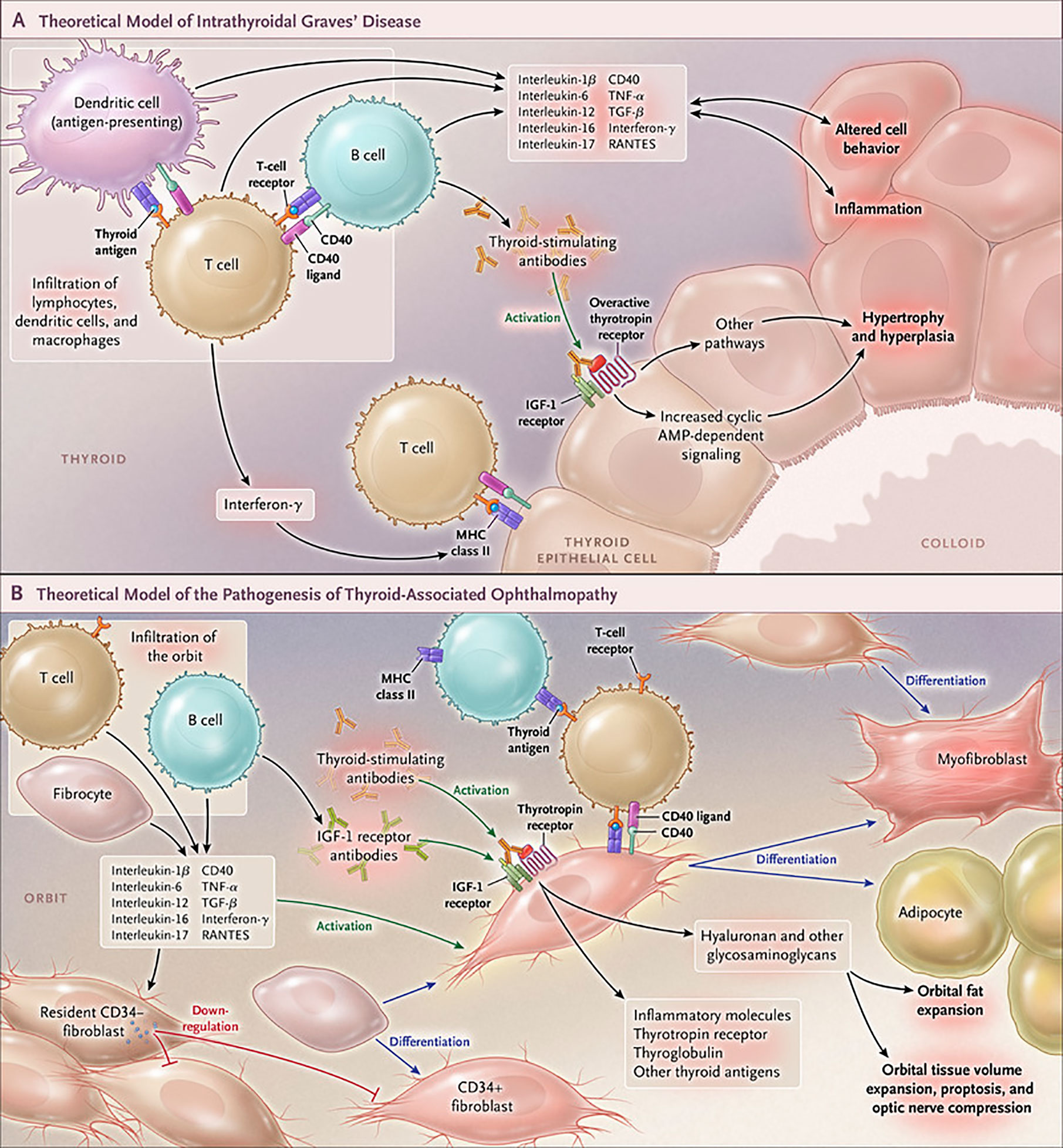
Figure 4 Cartoon proposed model of Graves’ disease and thyroid-associated ophthalmopathy (TAO) pathogenesis. (A) Thyroid-stimulating immunoglobulins (TSI) drive the over-production of thyroid hormones by activating the thyrotropin receptor (TSHR), thus overriding the normal regulatory role of thyrotropin on the process. B and T cells and antigen-presenting cells which infiltrate the gland produce interleukins 1β, 6, 12, 13, interferon γ, tumor necrosis factor α, and CD40 ligand. These cytokines then activate thyroid epithelial cells, promote inflammation, and induce genes ordinarily unexpressed by these cells such as major histocompatibility complex II (MHC II). Anti-thyroid drugs are useful therapeutically by reducing excess thyroid hormone production as well as modulating the expression of pathogenic intrathyroidal cytokines. (B) The pathogenesis of TAO also involves the infiltration of professional immune cells. Orbital fibroblasts exhibit particularly robust responses to inflammatory mediators. Among these cells are CD34+ fibroblasts which we propose derive from fibrocytes, monocyte-derived progenitor cells that traffic from bone marrow. Fibrocytes circulate in Graves’ disease at higher frequency than that found in healthy individuals. When cultivated from the peripheral circulation, fibrocytes express several thyroid-specific proteins, including thyrotropin receptor (TSHR), thyroglobulin, thyroperoxidase and sodium-iodide symporter. They also express MHC constitutively and can present antigens. When exposed to the appropriate culture conditions, they undergo differentiation into myofibroblasts (through Smad pathway activation by TGF-β) and adipocytes (through the activation of PPAR-γ). Many of the genes expressed by fibrocytes are detected at considerably lower levels in CD34+ orbital fibroblasts. We have found recently that these lower levels of expression result from the actions of Slit2 which acts through its cognate receptor, Roundabout 1 (ROBO1). When activated, CD34+ fibrocytes and CD34+ fibroblasts generate several pro-inflammatory or anti-inflammatory cytokines, including interleukins 1β, 6, 8, 10, 12, 16, tumor necrosis factor α, and regulated on activation, normal T expressed and secreted (RANTES), CXCL-12 and CD40-CD154. Both CD34+ and CD34- orbital fibroblasts cell-surface display insulin-like growth factor-I receptor (IGF-IR). Orbital fibroblasts express three mammalian hyaluronan synthase (HAS) isoenzymes and UDP glucose dehydrogenase and synthesize hyaluronan, the glycosaminoglycan associated with expanding orbital tissue in TAO. The vast majority of hyaluronan synthesis occurs in CD34- orbital fibroblasts. From N. Engl. J. Med, Smith T.J. and Hegedus L., Graves’ Disease, 375; 1552-1565. Copyright © (2016) Massachusetts Medical Society. Reprinted with permission.
IGF-IR is over-expressed by GD-OF (9) as well as T cells (116) and B cells (117). This increased IGF-IR expression in patients with GD is undetectable in the unaffected monozygotic twin of a sibling with GD, strongly suggesting that non-genetic factors are responsible, at least in part, for the increased receptor levels associated with the disease (118). IGF-I and GD-IgG purified from patients with GD were found to enhance hyaluronan accumulation in cultured GD-OF but had no effect in orbital fibroblasts from healthy donors (119). In contrast, rhTSH failed to influence glycosaminoglycan synthesis. Further, IGF-I appears to skew the accumulating HA molecules toward higher molecular weight species and to promote the proliferation of perimysial GD-OF (120). The effects of hyaluronan on proliferation of these fibroblasts was found to involve differential effects on membrane polarization, where high molecular weight hyaluronan results in depolarization and low molecular weight hyaluronan causes membrane hyperpolarization and inhibits proliferation.
As mentioned above, subsequent studies have disclosed the physical and functional interactions between IGF-IR and TSHR occurring in thyroid epithelial cells, GD-OF and in situ in TAO orbital fat (98). Those studies of Tsui et al. also demonstrated that inhibiting IGF-IR could attenuate Erk 1/2 p42/44 activation, regardless of whether the signaling was initiated by either TSHR or IGF-IR. They revealed that the actions of rhTSH, rhIGF-I and GD-IgG could be inhibited, strongly suggesting that the two receptors are functionally linked. Based on those findings, we proffered that IGF-IR might be targeted therapeutically with either monoclonal antibody or small molecule inhibitors of IGF-IR for TAO (10). The capacity for IGF-IR to crosstalk with additional proteins is becoming increasingly recognized (121). Several years after the initial observation of Tsui et al., β-arrestin was found to function as a scaffold for both TSHR and IGF-IR (122), This association potentially accounts for protein:protein crosstalk as underlying the apparent importance of IGF-IR activity in TSHR signaling. Similar associations have been identified in other receptor complexes.
Culmination of Evidence That IGF-IR Represents A Clinically Important Therapeutic Target in TAO
To test the central hypothesis that IGF-IR represents not only a critical component in the pathogenesis of TAO, but can also be therapeutically targeted, two placebo-controlled, multicenter clinical trials of teprotumumab have been conducted (123, 124). The two studies were designed similarly. The drug was developed as an antineoplastic agent and had already been administered to hundreds of patients with a broad range of cancers (107–111). In general, teprotumumumab (AKA R1507) was well-tolerated in those earlier studies, frequently involving fragile patients; however, the drug was not devoid of adverse events which were more severe in younger patients (125). Like other biologicals targeting IGF-IR simultaneously under development at several other pharmaceutical companies, the efficacy of teprotumumab was found inadequate for sustaining its development program by Roche (107, 109–111, 126). The failure of teprotumumab in the cancer space had made it available for potential repurposing in TAO.
The initial trial of teprotumumab in TAO was organized by River Vision Development starting in 2010. This study, a multicenter phase 2 trial, involved the recruitment of 88 patients within 9 months of developing ocular manifestations of GD (123). The trial enrolled patients with moderate to severe, active TAO between 18 and 75 years of age between July 2, 2013 and September 23, 2015. Patients were clinically euthyroid (within 50% above or below the normative range for serum thyroxine and triiodothyronine levels) at baseline. None had undergone orbital radiotherapy or remedial surgery for TAO. Further, none had received > 1 gram of prednisolone or equivalent for the treatment of TAO. For those with systemic exposure, a uniform steroid washout period of 6 weeks was required prior to study enrolment. Each patient was randomized to receive either placebo or teprotumumab in a 1:1 ratio. Doses were administered as 8 infusions, each at 3 week intervals over a 24-week treatment phase. The initial (partial) dose (10 mg/Kg B.W.) was followed by doses of 20 mg/Kg B.W. The primary response endpoint was the aggregate of 1) ≥ 2-point improvement in clinical activity score (CAS) using a 7-point scale AND 2) ≥ 2 mm proptosis reduction. Both responses must have occurred in the more severely affected (study) eye assessed at 24 weeks. This response must have occurred in the absence of a similar worsening in the contralateral (fellow) eye. Secondary responses included reduction from baseline in proptosis ≥ 2 mm, improvement from baseline in CAS ≥ 2 points, (both measured as continuous independent variables), improved subjective diplopia, and improved quality of life using a validated instrument (GO-QOL) (127). The results of the study were as follows: Twenty-nine of 42 patients in the intention to treat cohort receiving teprotumumab achieved the primary response at 24 weeks compared to 9/45 individuals in the placebo group (p<0.001). The differences between the two treatment groups emerged at week 6 of treatment (p<0.001). These highly significant differences continued over the duration of the treatment phase (p<0.001 at all clinical assessments). The time to first response was significantly shorter in those patients receiving teprotumumab. Further, more subjects receiving the active drug achieved a “high” primary response (≥3 mm proptosis reduction AND CAS reduction ≥ 3 points, p<0.001). With regard to the secondary endpoints, reduction in CAS and proptosis from baseline was significantly different in the two treatment groups as was improvement in the visual function scale of GO-QOL. Subjective diplopia response rates were significantly higher in those receiving teprotumumab compared with the placebo group. The drug safety profile from that trial was encouraging (123). The most common adverse events were muscle cramping and hyperglycemia, most commonly seen in patients with baseline abnormalities in glycemic control or frank diabetes mellitus. The worsening of glycemic control was easily managed with adjustment of diabetes medication. Further, these changes reverted to baseline following completion of the treatment phase of the trial. At analysis it was discovered that an imbalance of smokers occurred with more representation of tobacco users in the placebo group.
A second, phase 3 trial, was initiated by Horizon Pharmaceuticals (now Horizon Therapeutics) after a licensing agreement was established with River Vision in 2017. Eighty-three patients with moderate to severe TAO who had disease and demographic characteristics very similar to those included in the phase 2 trial were enrolled at performance sites in North America and Europe. This occurred from October 24, 2017 until August 31, 2018 (124) (Figure 5). The participating investigators represented a subset of those enrolling patients in the phase 2 trial. Subjects aged 18-80 years, were randomized to receive either teprotumumab (n=41) or placebo (n=42). Like the initial study, this trial was also placebo-controlled, double-masked, and all patients were clinically euthyroid, and were judged to manifest active, moderate to severe TAO. All had eye disease ≤ 9 months in duration. Individuals who had previously undergone orbital surgery, had received tocilizumab or rituximab or who had been treated with high-dose glucocorticoids for TAO (excepting those receiving < 1 gm prednisone equivalent following a 6-week washout period) were excluded. The primary outcome was changed from that in the initial study to a reduction in proptosis ≥ 2 mm in the study eye at week 24. The aggregate of ≥ 2 mm reduction in proptosis and improved CAS ≥ 2 points (the overall response and the primary response in the phase 2 trial) was among the secondary end points. Others included improved CAS > 2 points, reduction in proptosis, both measured as independent variables from baseline, reduced diplopia ≥ 1 Gorman scale grade and mean change in the GO-QOL questionnaire score. Trial results of the phase 3 study were congruent with those observed in phase 2. In addition, the skewed distribution of smokers in the two treatment groups in the earlier trial was successfully corrected. More patients receiving active drug experienced a ≥ 2mm proptosis reduction at week 24 when compared to those receiving placebo (teprotumumab 83% versus placebo controls 10%, p<0.001) (Figure 6). The necessary number to treat was 1.36. Further, all secondary endpoints were more frequently achieved in patients receiving teprotumumab than those in the placebo group. A few patients underwent orbital imaging at a single performance site at baseline and again at week 24. Those studies were conducted off protocol and revealed that both orbital fat and extraocular muscle volumes were reduced in 6 patients undergoing imaging (Figure 7). The phase 3 trial included an extension where all non-responders were offered teprotumumab as an open label, regardless of whether or not they had received the active drug or placebo during the 24-week treatment phase. A similar fraction of patients responded to the drug as did those in the initial intervention phase. Follow-up data, including those from the extension study of this phase 3 trial, reveal that a majority of both proptosis and diplopia responders at Week 24 maintained their responses (56% and 58%, respectively). The aggregate results from the two trials indicate that clinical improvement of moderate to severe, active TAO is very similar to the best outcomes of the ophthalmic remedial surgeries thus far reported in the literature.
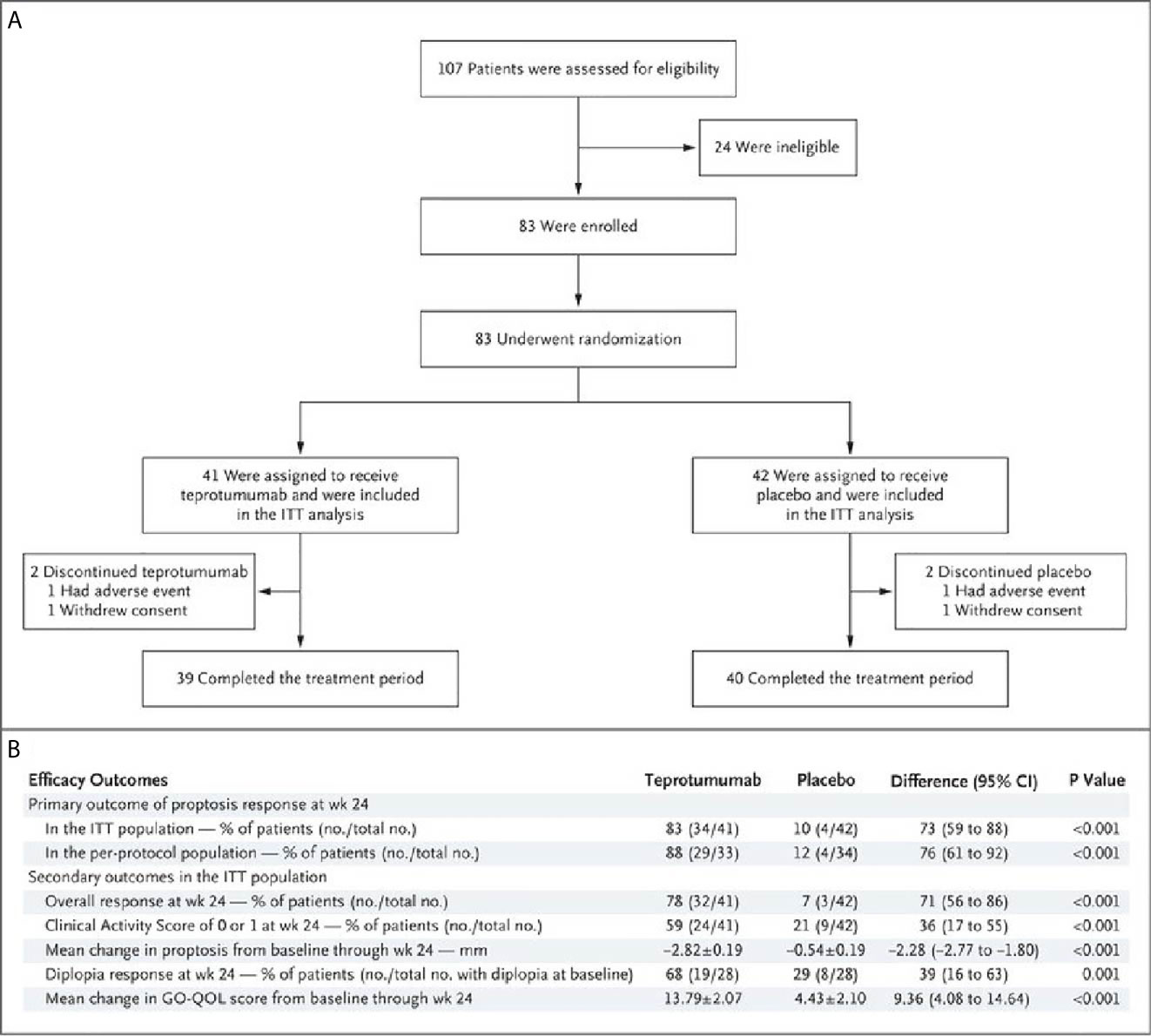
Figure 5 (A) Trial profile describing enrollment, randomization, and follow-up. Random assignment to subgroups receiving intravenous infusions of either teprotumumab (10 mg/Kg B.W. for test infusion and 20 mg/Kg for subsequent infusions) or placebo. Infusions administered every 3 weeks for 21 weeks eight infusions in total. (B) Efficacy Endpoints describing the phase 3 trial of teprotumumab in patients with moderate to severe active TAO. CMH weighting was used to estimate the common risk difference and the 95% Confidence Interval (95% CI) of the common risk difference for the primary and secondary endpoints of overall responder, percent with CAS 0 or 1, and diplopia responder; least squares mean difference was calculated for secondary endpoints of change in proptosis from baseline and change in Graves’ Orbitopathy quality of life (GO-QOL) questionnaire from baseline using the Mixed-Model Repeated-Measures (MMRM) analysis of covariance (ANCOVA). From N. Engl. J. Med, Douglas R.S, Kahaly G.J., Patel A., Sile E.H.Z., Thompson R. et al. Teprotumumab for the treatment of active thyroid eye disease. 382; 341-352. Copyright © (2020) Massachusetts Medical Society.
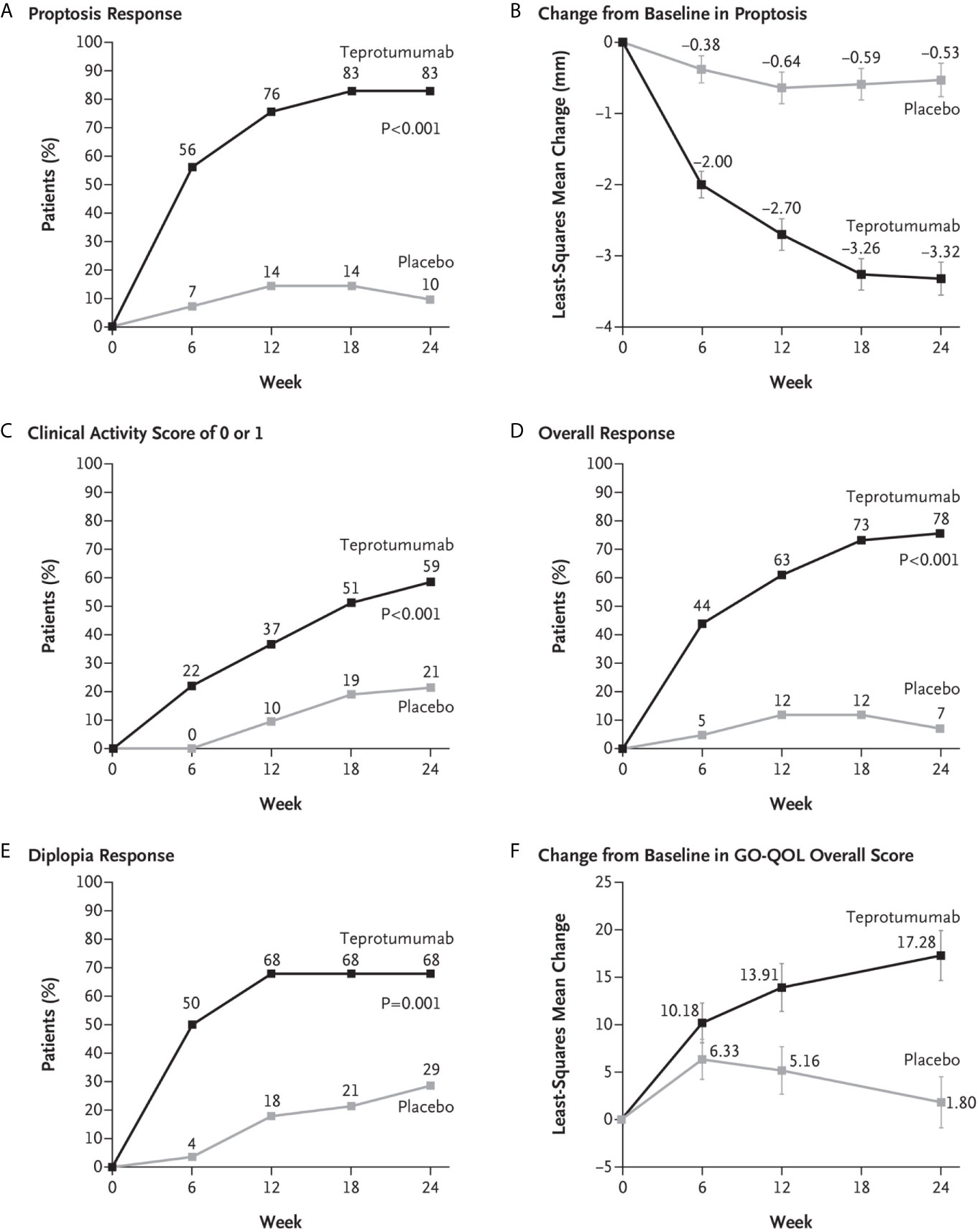
Figure 6 (A) Proptosis responder analysis (percent of patients with ≥2 mm reduction in proptosis from baseline in study eye). (B) Change from baseline in proptosis (least squares mean ± standard error). (C) Percent of patients with clinical activity score (CAS) of 0 or 1 in study eye. (D) Overall responder rate (percent of patients with ≥2-point reduction in CAS and ≥2 mm reduction in proptosis from baseline in study eye). (E) Diplopia response (percent of patients with improvement of at least 1 grade from baseline). (F) Change from baseline in transformed GO-QOL score (least squares mean ± standard error). From N. Engl. J. Med, Douglas R.S, Kahaly G.J., Patel A., Sile E.H.Z., Thompson R. et al. Teprotumumab for the treatment of active thyroid eye disease. 382; 341-352. Copyright © (2020) Massachusetts Medical Society. Reprinted with permission.
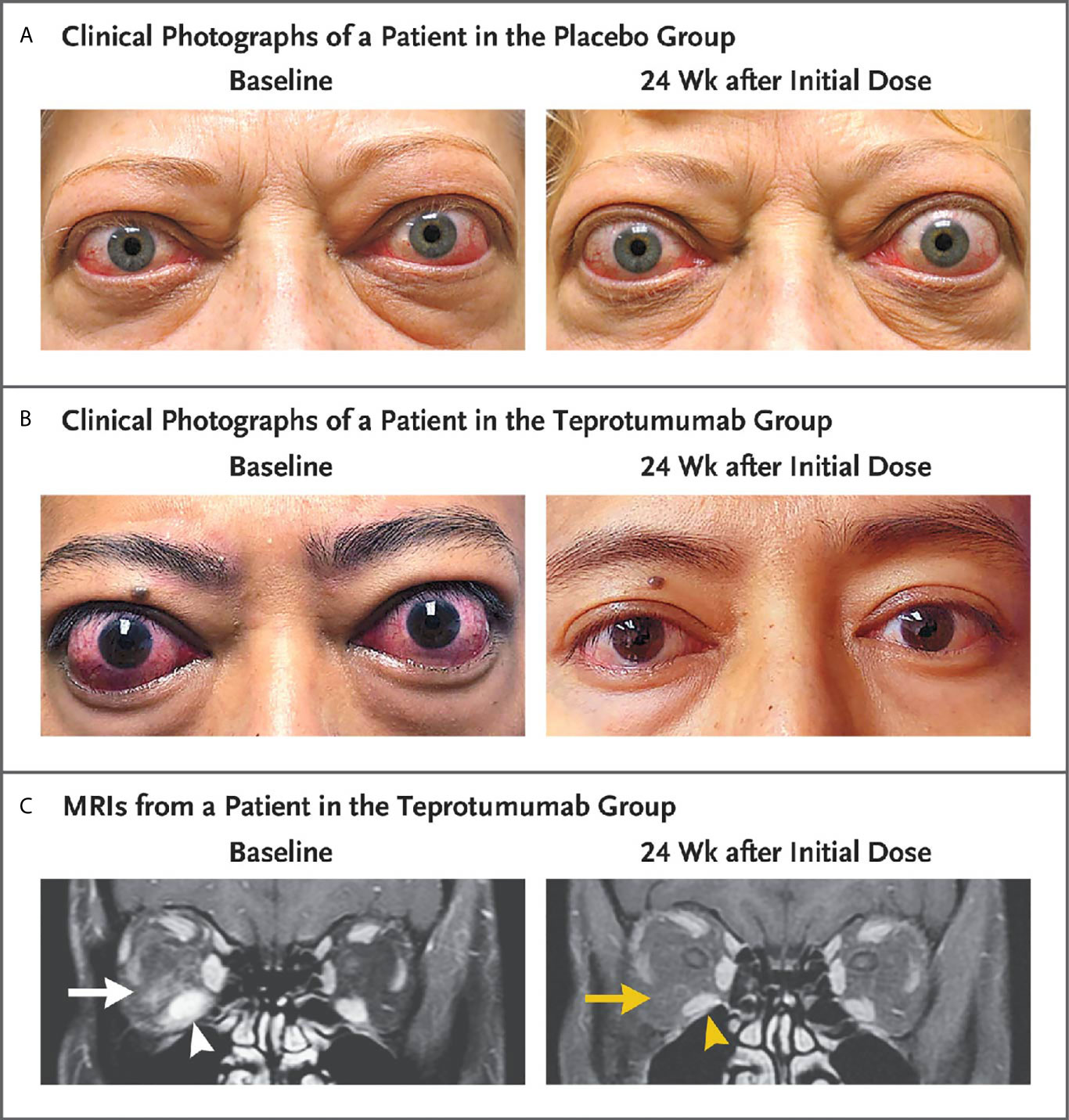
Figure 7 Facial photographic images and MRIs at Baseline and 24 Weeks following treatment with either placebo or teprotumumab in patients enrolled in the Phase 3 trial. Panel (A) Clinical photographs of a patient receiving placebo. At baseline, the patient exhibits substantial proptosis (left eye, 29 mm and right eye, 27 mm) as well as multiple inflammatory signs (left eye Clinical Activity Score of 7 and right eye 5). At week 24, considerable proptosis and inflammatory signs remain. (B) Images of a teprotumumab-treated patient. Baseline proptosis (both eyes 24 mm), edema, upper and lower eyelid retraction, and multiple inflammatory signs (CAS 5 bilaterally). At week 24, considerable bilateral reductions in proptosis (−5 mm) and CAS (−4 points). (C) Coronal, contrast-enhanced, fat-saturated, T1-weighted MRIs in a single patient receiving teprotumumab at baseline and at week 24. Note marked enhancement of the inferior rectus muscle (white arrowhead) and orbital fat (white arrow) as well as inferior rectus muscle enlargement. At week 24, resolved inferior rectus muscle (yellow arrowhead) enhancement and orbital fat (yellow arrow). The muscle volume was reduced by 49% (yellow arrowhead). Proptosis reduction decreased from 23 mm at baseline to 18 mm at week 24. From N. Engl. J. Med, Douglas R.S, Kahaly G.J., Patel A., Sile E.H.Z., Thompson R. et al. Teprotumumab for the treatment of active thyroid eye disease. 382; 341-352. Copyright © (2020) Massachusetts Medical Society. Reprinted with permission.
Aggregate safety data from the two trials suggest that teprotumumab was well-tolerated. Several adverse events, most mild to moderate in severity, were identified. Among the most common was hyperglycemia, especially in individuals who were diabetic or glucose intolerant at baseline. Grade 2-3 hyperglycemia developed in some patients with pre-study diabetes mellitus who were receiving teprotumumab. These were managed by increasing diabetes medications. No ketoacidosis occurred in this group of patients. Baseline diabetes medication requirements returned to pre-study levels following the completion of the 24-week treatment phase. A few patients in both treatment arms not having histories of carbohydrate intolerance developed grade 1 hyperglycemia. Other adverse events include hearing abnormalities, muscle cramps, hair loss, dysgeusia, and diarrhea. These uniformly resolved or improved substantially after the treatment phase of the trials had been completed.
Teprotumumab Becomes First Ever FDA-Approved Medical Therapy for TAO
Based on the results of the two clinical trials conducted for teprotumumab in TAO, the FDA approved its use in that disease in January, 2020 (128). Several important exclusions in the profiles of eligible patients who were enrolled in those studies have resulted in additional questions needing answers during the post-approval period. For instance, all trial subjects must have manifested TAO for ≤ 9 months prior to their study enrolment, leaving uncertain whether more chronic, less active disease might respond to teprotumumab. Enrollment criteria were stringently skewed toward early disease since a priori reasoning suggested that the most active patients were more likely to respond. The relatively short term follow-up of the studies has left the question of long-term therapeutic durability of the drug. The efficacy of the drug in stable disease is being addressed, not only in the formal study currently under development but also by monitoring real-world experience. Potential effectiveness of teprotumumab in vision threatening TAO resulting from compressive optic neuropathy is unknown since patients with signs of impending vision loss were excluded from both trials. Those open questions are also under study. Single case reports are now appearing suggesting that the drug may be beneficial in apparently stable, longer term disease (129) and in optic neuropathy (130, 131). Clearly more extensive clinical experience with the drug will be necessary before teprotumumab can be considered a reliable treatment option in long-standing or vision-threatening TAO. An important goal of this medical therapy is to lessen reliance on either routine surgical rehabilitation for chronic disease or urgent surgical intervention in sight-threatening TAO.
Are Disease Indications Beyond TAO in Store for Teprotumumab?
The IGF-I pathway regulates a vast array of physiological and pathological processes in most mammalian tissues. The effective and well-tolerated treatment with teprotumumab of TAO suggests that this pathway might be beneficially targeted in many other diseases. With regard to GD, pretibial myxedema, a potentially debilitating and disfiguring condition seen in a subset of those with TAO, might also improve with the drug. In fact, a recent case report from Varma et al. suggests that long-standing pretibial myxedema might also respond to teprotumumab (132). Shortly after the original observations concerning anti-IGF-IR antibody involvement in TAO (9, 115), similar findings were reported in rheumatoid arthritis (RA) (133). That study demonstrated that synovial fibroblasts from patients with RA responded to their own IgGs as well as to GD-IgG in inducing IL-16 and RANTES expression. Thus, it remains possible that teprotumumab and other IGF-IR inhibitors might prove effective in in the treatment of RA and allied autoimmune diseases. Investigators have speculated that teprotumumab might prove effective in arresting the deleterious lung tissue remodeling associated with coronavirus-19 (134). It remains possible that additional indications for the therapeutic targeting of IGF-I and its pathway will become identified as the experience with teprotumumab broadens.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares US Patents covering the use of IGF-1 receptor inhibitors in autoimmune disease and consultancy for Horizon Therapeutics Consultant Immunovant.
Acknowledgments
The author is grateful to Ms. Dana Barnhart and Ms. Linda Polonsky for their outstanding help in the preparation of this manuscript. This work was supported in part by the National Institutes of Health grants EY008976, EY11708, DK063121, 5UMIA110557, a Core Center for Research grant EY007003 from the NEI, an unrestricted grant from Research to Prevent Blindness and by the Bell Charitable Family Foundation.
References
1. Pavel I, Sdrobici D. N. Paulescu, Discoverer of Insulin. 50th Anniversary of the Discovery of That Hormone. Med Int (1972) 24:1285–94.
2. Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J (1922) 12:141–6.
3. Daughaday WH, Salmon WD Jr, Alexander F. Sulfation Factor Activity of Sera From Patients With Pituitary Disorders. J Clin Endocrinol Metab (1959) 19:743–58. doi: 10.1210/jcem-19-7-743
4. Daughaday WH, Hall K, Raben MS, Salmon WD Jr., van den Brande JL, van Wyk JJ. Somatomedin: Proposed Designation for Sulphation Factor. Nature (1972) 235:107. doi: 10.1038/235107a0
5. Blethen SL, White NH, Santiago JV, Daughaday WH. Plasma Somatomedins, Endogenous Insulin Secretion, and Growth in Transient Neonatal Diabetes Mellitus. J Clin Endocrinol Metab (1981) 52:144–7. doi: 10.1210/jcem-52-1-144
6. Smith TJ. Pathogenesis of Graves’ Orbitopathy: A 2010 Update. J Endocrinol Invest (2010) 33:414–21. doi: 10.1007/BF03346614
7. Smith TJ. Challenges in Orphan Drug Development: Identification of Effective Therapy for Thyroid-Associated Ophthalmopathy. Annu Rev Pharmacol Toxicol (2019) 59:129–48. doi: 10.1146/annurev-pharmtox-010617-052509
8. Smith TJ, Janssen J. Insulin-Like Growth Factor-I Receptor and Thyroid-Associated Ophthalmopathy. Endocr Rev (2019) 40:236–67. doi: 10.1210/er.2018-00066
9. Pritchard J, Han R, Horst N, Cruikshank WW, Smith TJ. Immunoglobulin Activation of T Cell Chemoattractant Expression in Fibroblasts From Patients With Graves’ Disease is Mediated Through the Insulin-Like Growth Factor I Receptor Pathway. J Immunol (Baltimore Md: 1950) (2003) 170:6348–54. doi: 10.4049/jimmunol.170.12.6348
10. Smith TJ. Insulin-Like Growth Factor-I Regulation of Immune Function: A Potential Therapeutic Target in Autoimmune Diseases? Pharmacol Rev (2010) 62:199–236. doi: 10.1124/pr.109.002469
11. De Meyts P, Palsgaard J, Sajid W, Theede AM, Aladdin H. Structural Biology of Insulin and IGF-1 Receptors. Novartis Foundation Symposium (2004) 262:160–71. doi: 10.1002/0470869976.ch10
12. Furlanetto RW, Cara JF. Somatomedin-C/insulin-like Growth Factor-I as a Modulator of Growth During Childhood and Adolescence. Horm Res (1986) 24:177–84. doi: 10.1159/000180557
13. De Meyts P, Sajid W, Palsgaard J, Theede A, Gaugain L, Aladdin H, et al. Insulin and IGF-I Receptor Structure and Binding Mechanism. In: Saltiel AR, Pessin JE, editors. Mechanisms of Insulin Action. Austin, TX: Landes Bioscience (2007). p. 1–32. doi: 10.1007/978-0-387-72204-7_1
14. LeRoith D, Werner H, Beitner-Johnson D, Roberts CT Jr. Molecular and Cellular Aspects of the Insulin-Like Growth Factor I Receptor. Endocr Rev (1995) 16:143–63. doi: 10.1210/edrv-16-2-143
15. Poreba E, Durzynska J. Nuclear Localization and Actions of the Insulin-Like Growth Factor 1 (IGF-1) System Components: Transcriptional Regulation and DNA Damage Response. Mutat Res (2020) 784:108307. doi: 10.1016/j.mrrev.2020.108307
16. Roberts CT Jr. Control of Insulin-Like Growth Factor (IGF) Action by Regulation of IGF-I Receptor Expression. Endocr J (1996) 43 Suppl:S49–55. doi: 10.1507/endocrj.43.Suppl_S49
17. Boucher J, Charalambous M, Zarse K, Mori MA, Kleinridders A, Ristow M, et al. Insulin and Insulin-Like Growth Factor 1 Receptors are Required for Normal Expression of Imprinted Genes. Proc Natl Acad Sci USA (2014) 111:14512–7. doi: 10.1073/pnas.1415475111
18. Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth RA, et al. Insulin-Like Growth Factor II Receptor as a Multifunctional Binding Protein. Nature (1987) 329:301–7. doi: 10.1038/329301a0
19. Ghosh P, Dahms NM, Kornfeld S. Mannose 6-Phosphate Receptors: New Twists in the Tale. Nat Rev Mol Cell Biol (2003) 4:202–12. doi: 10.1038/nrm1050
20. Jones JI, Clemmons DR. Insulin-Like Growth Factors and Their Binding Proteins: Biological Actions. Endocr Rev (1995) 16:3–34. doi: 10.1210/edrv-16-1-3
21. Hwa V, Oh Y, Rosenfeld RG. The Insulin-Like Growth Factor-Binding Protein (IGFBP) Superfamily. Endocr Rev (1999) 20:761–87. doi: 10.1210/edrv.20.6.0382
22. Grosvenor CE, Turner CW. Effect of Growth Hormone Upon Thyroid Secretion Rate in the Rat. Proceedings of the Society for Experimental Biology and Medicine. Soc Exp Biol Med (N Y NY) (1959) 100:70–2. doi: 10.3181/00379727-100-24527
23. Solomon J, Greep RO. The Effect of Alterations in Thyroid Function on the Pituitary Growth Hormone Content and Acidophil Cytology. Endocrinology (1959) 65:158–64. doi: 10.1210/endo-65-2-158
24. Rolleston HD. The Endocrine Organs in Health and Disease: With an Historical Review. Milford H, editor. Oxford, England: Oxford University Press (1936).
25. Saji M, Tsushima T, Isozaki O, Murakami H, Ohba Y, Sato K, et al. Interaction of Insulin-Like Growth Factor I With Porcine Thyroid Cells Cultured in Monolayer. Endocrinology (1987) 121:749–56. doi: 10.1210/endo-121-2-749
26. Malaguarnera R, Frasca F, Garozzo A, Gianì F, Pandini G, Vella V, et al. Insulin Receptor Isoforms and Insulin-Like Growth Factor Receptor in Human Follicular Cell Precursors From Papillary Thyroid Cancer and Normal Thyroid. J Clin Endocrinol Metab (2011) 96:766–74. doi: 10.1210/jc.2010-1255
27. Maciel RM, Moses AC, Villone G, Tramontano D, Ingbar SH. Demonstration of the Production and Physiological Role of Insulin-Like Growth Factor II in Rat Thyroid Follicular Cells in Culture. J Clin Invest (1988) 82:1546–53. doi: 10.1172/JCI113764
28. Höppener JW, Steenbergh PH, Slebos RJ, de Pagter-Holthuizen P, Roos BA, Jansen M, et al. Expression of Insulin-Like Growth Factor-I and -II Genes in Rat Medullary Thyroid Carcinoma. FEBS Lett (1987) 215:122–6. doi: 10.1016/0014-5793(87)80125-4
29. Bethel C, Saadi H, Hout W, Vitullo J, Aron D. Secretion and Binding of Insulin-Like Growth Factor-I by a Human Medullary-Thyroid Carcinoma Cell-Line. Int J Oncol (1993) 2:961–7. doi: 10.3892/ijo.2.6.961
30. Dawson TP, Radulescu A, Wynford-Thomas D. Expression of Mutant p21ras Induces Insulin-Like Growth Factor 1 Secretion in Thyroid Epithelial Cells. Cancer Res (1995) 55:915–20.
31. Hofbauer LC, Rafferzeder M, Janssen OE, Gärtner R. Insulin-Like Growth Factor I Messenger Ribonucleic Acid Expression in Porcine Thyroid Follicles Is Regulated by Thyrotropin and Iodine. Eur J Endocrinol (1995) 132:605–10. doi: 10.1530/eje.0.1320605
32. Roger PP, Dumont JE. Thyrotropin-Dependent Insulin-Like Growth Factor I mRNA Expression in Thyroid Cells. Eur J Endocrinol (1995) 132:601–2. doi: 10.1530/eje.0.1320601
33. Arufe MC, Lu M, Lin RY. Differentiation of Murine Embryonic Stem Cells to Thyrocytes Requires Insulin and Insulin-Like Growth Factor-1. Biochem Biophys Res Commun (2009) 381:264–70. doi: 10.1016/j.bbrc.2009.02.035
34. Kushchayeva YS, Kushchayev SV, Startzell M, Cochran E, Auh S, Dai Y, et al. Thyroid Abnormalities in Patients With Extreme Insulin Resistance Syndromes. J Clin Endocrinol Metab (2019) 104:2216–28. doi: 10.1210/jc.2018-02289
35. Eggo MC, Bachrach LK, Burrow GN. Interaction of TSH, Insulin and Insulin-Like Growth Factors in Regulating Thyroid Growth and Function. Growth Factors (1990) 2:99–109. doi: 10.3109/08977199009071497
36. Lee YJ, Park DJ, Shin CS, Park KS, Kim SY, Lee HK, et al. Microarray Analysis of Thyroid Stimulating Hormone, Insulin-Like Growth factor-1, and Insulin-Induced Gene Expression in FRTL-5 Thyroid Cells. J Korean Med Sci (2007) 22:883–90. doi: 10.3346/jkms.2007.22.5.883
37. Iglesias-Osma MC, Blanco EJ, Carretero-Hernandez M, Catalano-Iniesta L, Sanchez-Robledo V, Garcia-Barrado MJ, et al. The Influence of the Lack of Insulin Receptor Substrate 2 (IRS2) on the Thyroid Gland. Sci Rep (2019) 9:5673. doi: 10.1038/s41598-019-42198-7
38. Ock S, Ahn J, Lee SH, Kim HM, Kang H, Kim YK, et al. Thyrocyte-Specific Deletion of Insulin and IGF-1 Receptors Induces Papillary Thyroid Carcinoma-Like Lesions Through EGFR Pathway Activation. Int J Cancer (2018) 143:2458–69. doi: 10.1002/ijc.31779
39. Gianoukakis AG, Douglas RS, King CS, Cruikshank WW, Smith TJ. Immunoglobulin G From Patients With Graves’ Disease Induces interleukin-16 and RANTES Expression in Cultured Human Thyrocytes: A Putative Mechanism for T-Cell Infiltration of the Thyroid in Autoimmune Disease. Endocrinology (2006) 147:1941–9. doi: 10.1210/en.2005-1375
40. Giuliani C, Saji M, Bucci I, Fiore G, Liberatore M, Singer DS, et al. Transcriptional Regulation of Major Histocompatibility Complex Class I Gene by Insulin and IGF-I in FRTL-5 Thyroid Cells. J Endocrinol (2006) 189:605–15. doi: 10.1677/joe.1.06486
41. Grassadonia A, Tinari N, Fiorentino B, Nakazato M, Chung HK, Giuliani C, et al. Upstream Stimulatory Factor Regulates Constitutive Expression and Hormonal Suppression of the 90K (Mac-2BP) Protein. Endocrinology (2007) 148:3507–17. doi: 10.1210/en.2007-0024
42. Fukushima T, Nedachi T, Akizawa H, Akahori M, Hakuno F, Takahashi S. Distinct Modes of Activation of Phosphatidylinositol 3-Kinase in Response to Cyclic Adenosine 3’, 5’-Monophosphate or Insulin-Like Growth Factor I Play Different Roles in Regulation of Cyclin D1 and p27Kip1 in FRTL-5 Cells. Endocrinology (2008) 149:3729–42. doi: 10.1210/en.2007-1443
43. Ren M, Guan Q, Zhong X, Gong B, Sun Y, Xin W, et al. Phosphatidylinositol 3-Kinase/Nuclear Factor-Kappa B Signaling Pathway is Involved in the Regulation of IGF-I on Fas-Associated Death Domain-Like interleukin-1-Converting Enzyme-Inhibitory Protein Expression in Cultured FRTL Thyroid Cells. J Mol Endocrinol (2007) 38:619–25. doi: 10.1677/JME-07-0020
44. Takada K, Tada H, Takano T, Nishiyama S, Amino N. Functional Regulation of GTP-binding Protein Coupled to Insulin-Like Growth Factor-I Receptor by Lithium During G1 Phase of the Rat Thyroid Cell Cycle. FEBS Lett (1993) 318:245–8. doi: 10.1016/0014-5793(93)80521-U
45. Manzella L, Massimino M, Stella S, Tirrò E, Pennisi MS, Martorana F, et al. Activation of the IGF Axis in Thyroid Cancer: Implications for Tumorigenesis and Treatment. Int J Mol Sci (2019) 20:3258. doi: 10.3390/ijms20133258
46. Karagiannis AK, Philippou A, Tseleni-Balafouta S, Zevolis E, Nakouti T, Tsopanomichalou-Gklotsou M, et al. IGF-Iec Expression Is Associated With Advanced Differentiated Thyroid Cancer. Anticancer Res (2019) 39:2811–9. doi: 10.21873/anticanres.13409
47. Roelfsema F, Biermasz NR, Frolich M, Keenan DM, Veldhuis JD, Romijn JA. Diminished and Irregular Thyrotropin Secretion With Preserved Diurnal Rhythm in Patients With Active Acromegaly. J Clin Endocrinol Metab (2009) 94:1945–50. doi: 10.1210/jc.2009-0174
48. Natchev E, Vandeva S, Kovatcheva R, Kirilov G, Kalinov K, Zacharieva S. Thyroid Gland Changes in Patients With Acromegaly. Arch Endocrinol Metab (2020) 64:269–75. doi: 10.20945/2359-3997000000247
49. Dogan S, Atmaca A, Dagdelen S, Erbas B, Erbas T. Evaluation of Thyroid Diseases and Differentiated Thyroid Cancer in Acromegalic Patients. Endocrine (2014) 45:114–21. doi: 10.1007/s12020-013-9981-3
50. Chen Z, Jiang X, Feng Y, Li X, Chen D, Mao Z, et al. Decrease In Acromegaly-Associated Thyroid Enlargement After Normalization Of Igf-1 Levels: A Prospective Observation and In Vitro Study. Endocrine Practice: Off J Am Coll Endocrinol Am Assoc Clin Endocrinol (2020) 26:369–77. doi: 10.4158/EP-2019-0353
51. Wüster C, Steger G, Schmelzle A, Gottswinter J, Minne HW, Ziegler R. Increased Incidence of Euthyroid and Hyperthyroid Goiters Independently of Thyrotropin in Patients With Acromegaly. Hormone Metab Res Hormon Und Stoffwechselforschung Hormones Metabol (1991) 23:131–4. doi: 10.1055/s-2007-1003632
52. Cheung NW, Lou JC, Boyages SC. Growth Hormone Does Not Increase Thyroid Size in the Absence of Thyrotropin: A Study in Adults With Hypopituitarism. J Clin Endocrinol Metab (1996) 81:1179–83. doi: 10.1210/jc.81.3.1179
53. Misaki T, Maciel RM, Tramontano D, Moses AC, Lombardi A, Ingbar SH. Supranormal Stimulation of Deoxyribonucleic Acid Synthesis in FRTL5 Cells by Serum From Patients With Untreated Acromegaly. J Clin Endocrinol Metab (1988) 66:1227–32. doi: 10.1210/jcem-66-6-1227
54. Rogozinski A, Furioso A, Glikman P, Junco M, Laudi R, Reyes A, et al. Thyroid Nodules in Acromegaly. Arquivos Brasileiros Endocrinol E Metabol (2012) 56:300–4. doi: 10.1590/S0004-27302012000500004
55. Andrioli M, Scacchi M, Carzaniga C, Vitale G, Moro M, Poggi L, et al. Thyroid Nodules in Acromegaly: The Role of Elastography. J Ultrasound (2010) 13:90–7. doi: 10.1016/j.jus.2010.09.008
56. Ruchala M, Skiba A, Gurgul E, Uruski P, Wasko R, Sowinski J. The Occurrence of Thyroid Focal Lesions and a Need for Fine Needle Aspiration Biopsy in Patients With Acromegaly Due to an Increased Risk of Thyroid Cancer. Neuro Endocrinol Lett (2009) 30:382–6.
57. dos Santos MC, Nascimento GC, Nascimento AG, Carvalho VC, Lopes MH, Montenegro R, et al. Thyroid Cancer in Patients With Acromegaly: A Case-Control Study. Pituitary (2013) 16:109–14. doi: 10.1007/s11102-012-0383-y
58. Gullu BE, Celik O, Gazioglu N, Kadioglu P. Thyroid Cancer is the Most Common Cancer Associated With Acromegaly. Pituitary (2010) 13:242–8. doi: 10.1007/s11102-010-0224-9
59. Loeper S, Ezzat S. Acromegaly: Re-Thinking the Cancer Risk. Rev Endocrine Metab Disord (2008) 9:41–58. doi: 10.1007/s11154-007-9063-z
60. Chanson P, Salenave S, Orcel P. McCune-Albright Syndrome in Adulthood. Pediatr Endocrinol Rev (2007) 4 Suppl 4:453–62.
61. Sisson JC, Giordano TJ, Avram AM. Three Endocrine Neoplasms: An Unusual Combination of Pheochromocytoma, Pituitary Adenoma, and Papillary Thyroid Carcinoma. Thyroid (2012) 22:430–6. doi: 10.1089/thy.2011.0345
62. Kato K, Takeshita Y, Misu H, Ishikura K, Kakinoki K, Sawada-Kitamura S, et al. Duodenal Adenocarcinoma With Neuroendocrine Features in a Patient With Acromegaly and Thyroid Papillary Adenocarcinoma: A Unique Combination of Endocrine Neoplasia. Endocr J (2012) 59:791–6. doi: 10.1507/endocrj.EJ11-0324
63. Sato T, Suzukui Y, Taketani T, Ishiguro K, Masuyama T. Enhanced Peripheral Conversion of Thyroxine to Triiodothyronine During hGH Therapy in GH Deficient Children. J Clin Endocrinol Metab (1977) 45:324–9. doi: 10.1210/jcem-45-2-324
64. Portes ES, Oliveira JH, MacCagnan P, Abucham J. Changes in Serum Thyroid Hormones Levels and Their Mechanisms During Long-Term Growth Hormone (GH) Replacement Therapy in GH Deficient Children. Clin Endocrinol (2000) 53:183–9. doi: 10.1046/j.1365-2265.2000.01071.x
65. Pirazzoli P, Cacciari E, Mandini M, Sganga T, Capelli M, Cicognani A, et al. Growth and Thyroid Function in Children Treated With Growth Hormone. J Pediatr (1992) 121:210–3. doi: 10.1016/S0022-3476(05)81190-4
66. Melmed S, Yamashita S. Insulin-Like Growth Factor-I Action on Hypothyroid Rat Pituitary Cells: Suppression of Triiodothyronine-Induced Growth Hormone Secretion and Messenger Ribonucleic Acid Levels. Endocrinology (1986) 118:1483–90. doi: 10.1210/endo-118-4-1483
67. Aizenman Y, de Vellis J. Brain Neurons Develop in a Serum and Glial Free Environment: Effects of Transferrin, Insulin, Insulin-Like Growth Factor-I and Thyroid Hormone on Neuronal Survival, Growth and Differentiation. Brain Res (1987) 406:32–42. doi: 10.1016/0006-8993(87)90766-9
68. Audhya TK, Gibson KD. Enhancement of Somatomedin Titers of Normal and Hypopituitary Sera by Addition of L-triiodothyronone In Vitro At Physiological Concentrations. Proc Natl Acad Sci USA (1975) 72:604–8. doi: 10.1073/pnas.72.2.604
69. Wang L, Shao YY, Ballock RT. Thyroid Hormone-Mediated Growth and Differentiation of Growth Plate Chondrocytes Involves IGF-1 Modulation of Beta-Catenin Signaling. J Bone Miner Res (2010) 25:1138–46. doi: 10.1002/jbmr.5
70. Ortiz L, Zannini M, Di Lauro R, Santisteban P. Transcriptional Control of the Forkhead Thyroid Transcription Factor TTF-2 by Thyrotropin, Insulin, and Insulin-Like Growth Factor I. J Biol Chem (1997) 272:23334–9. doi: 10.1074/jbc.272.37.23334
71. Thelen MH, Muller A, Zuidwijk MJ, van der Linden GC, Simonides WS, van Hardeveld C. Differential Regulation of the Expression of Fast-Type Sarcoplasmic-Reticulum Ca(2+)-ATPase by Thyroid Hormone and Insulin-Like Growth Factor-I in the L6 Muscle Cell Line. Biochem J (1994) 303(Pt 2):467–74. doi: 10.1042/bj3030467
72. Susperreguy S, Miras MB, Montesinos MM, Mascanfroni ID, Muñoz L, Sobrero G, et al. Growth Hormone (GH) Treatment Reduces Peripheral Thyroid Hormone Action in Girls With Turner Syndrome. Clin Endocrinol (2007) 67:629–36. doi: 10.1111/j.1365-2265.2007.02936.x
73. Cacicedo L, de los Frailes MT, Lorenzo MJ, Sanchez Franco F. Pituitary and Peripheral Insulin-Like Growth Factor-I Regulation by Thyroid Hormone. Ann N Y Acad Sci (1993) 692:287–90. doi: 10.1111/j.1749-6632.1993.tb26236.x
74. Shapiro LE, Samuels HH, Yaffe BM. Thyroid and Glucocorticoid Hormones Synergistically Control Growth Hormone mRNA in Cultured GH1 Cells. Proc Natl Acad Sci USA (1978) 75:45–9. doi: 10.1073/pnas.75.1.45
75. Samuels MH, Wierman ME, Wang C, Ridgway EC. The Effect of Altered Thyroid Status on Pituitary Hormone Messenger Ribonucleic Acid Concentrations in the Rat. Endocrinology (1989) 124:2277–82. doi: 10.1210/endo-124-5-2277
76. Chernausek SD, Underwood LE, Utiger RD, Van Wyk JJ. Growth Hormone Secretion and Plasma Somatomedin-C in Primary Hypothyroidism. Clin Endocrinol (1983) 19:337–44. doi: 10.1111/j.1365-2265.1983.tb00007.x
77. Montecino-Rodriguez E, Clark R, Johnson A, Collins L, Dorshkind K. Defective B Cell Development in Snell Dwarf (Dw/Dw) Mice Can Be Corrected by Thyroxine Treatment. J Immunol (Baltimore Md: 1950) (1996) 157:3334–40.
78. Näntö-Salonen K, Muller HL, Hoffman AR, Vu TH, Rosenfeld RG. Mechanisms of Thyroid Hormone Action on the Insulin-Like Growth Factor System: All Thyroid Hormone Effects Are Not Growth Hormone Mediated. Endocrinology (1993) 132:781–8. doi: 10.1210/endo.132.2.7678799
79. Cavaliere H, Knobel M, Medeiros-Neto G. Effect of Thyroid Hormone Therapy on Plasma Insulin-Like Growth Factor I Levels in Normal Subjects, Hypothyroid Patients and Endemic Cretins. Horm Res (1987) 25:132–9. doi: 10.1159/000180644
80. Westermark K, Alm J, Skottner A, Karlsson A. Growth Factors and the Thyroid: Effects of Treatment for Hyper- and Hypothyroidism on Serum IGF-I and Urinary Epidermal Growth Factor Concentrations. Acta Endocrinol (Copenh) (1988) 118:415–21. doi: 10.1530/acta.0.1180415
81. Angervo M, Tiihonen M, Leinonen P, Välimäki M, Seppälä M. Thyroxine Treatment Increases Circulating Levels of Insulin-Like Growth Factor Binding protein-1: A Placebo-Controlled Study. Clin Endocrinol (1993) 38:547–51. doi: 10.1111/j.1365-2265.1993.tb00352.x
82. Näntö-Salonen K, Rosenfeld RG. Insulin-Like Growth Factor Binding Protein Expression in the Hypothyroid Rat Is Age Dependent. Endocrinology (1992) 131:1489–96. doi: 10.1210/endo.131.3.1380443
83. Angervo M, Toivonen J, Leinonen P, Välimäki M, Seppälä M. Thyroxine Withdrawal is Accompanied by Decreased Circulating Levels of Insulin-Like Growth Factor-Binding Protein-1 in Thyroidectomized Patients. J Clin Endocrinol Metab (1993) 76:1199–201. doi: 10.1210/jcem.76.5.7684392
84. Polychronakos C, Guyda HJ, Patel B, Posner BI. Increase in the Number of Type II Insulin-Like Growth Factor Receptors During Propylthiouracil-Induced Hyperplasia in the Rat Thyroid. Endocrinology (1986) 119:1204–9. doi: 10.1210/endo-119-3-1204
85. Ohlsson C, Nilsson A, Isaksson O, Bentham J, Lindahl A. Effects of Tri-Iodothyronine and Insulin-Like Growth Factor-I (IGF-I) on Alkaline Phosphatase Activity, [3H]Thymidine Incorporation and IGF-I Receptor mRNA in Cultured Rat Epiphyseal Chondrocytes. J Endocrinol (1992) 135:115–23. doi: 10.1677/joe.0.1350115
86. Matsuo K, Yamashita S, Niwa M, Kurihara M, Harakawa S, Izumi M, et al. Thyroid Hormone Regulates Rat Pituitary Insulin-Like Growth Factor-I Receptors. Endocrinology (1990) 126:550–4. doi: 10.1210/endo-126-1-550
87. Moreno B, Rodríguez-Manzaneque JC, Pérez-Castillo A, Santos A. Thyroid Hormone Controls the Expression of Insulin-Like Growth Factor I Receptor Gene At Different Levels in Lung and Heart of Developing and Adult Rats. Endocrinology (1997) 138:1194–203. doi: 10.1210/endo.138.3.5018
88. Eggo MC. Molecular Regulation of Thyroid Gland Function. Curr Opin Endocrinol Diabetes Obes (2010) 17:396–401. doi: 10.1097/MED.0b013e32833c8942
89. Büch TR, Biebermann H, Kalwa H, Pinkenburg O, Hager D, Barth H, et al. G13-Dependent Activation of MAPK by Thyrotropin. J Biol Chem (2008) 283:20330–41. doi: 10.1074/jbc.M800211200
90. Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Girnita A, Lefkowitz RJ, et al. {Beta}-Arrestin Is Crucial for Ubiquitination and Down-Regulation of the Insulin-Like Growth Factor-1 Receptor by Acting as Adaptor for the MDM2 E3 Ligase. J Biol Chem (2005) 280:24412–9. doi: 10.1074/jbc.M501129200
91. Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Vasilcanu D, Girnita A, et al. Beta-Arrestin and Mdm2 Mediate IGF-1 Receptor-Stimulated ERK Activation and Cell Cycle Progression. J Biol Chem (2007) 282:11329–38. doi: 10.1074/jbc.M611526200
92. Zheng H, Shen H, Oprea I, Worrall C, Stefanescu R, Girnita A, et al. β-Arrestin-biased Agonism as the Central Mechanism of Action for Insulin-Like Growth Factor 1 Receptor-Targeting Antibodies in Ewing’s Sarcoma. Proc Natl Acad Sci USA (2012) 109:20620–5. doi: 10.1073/pnas.1216348110
93. van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PM, Roovers RC. Crosstalk Between Epidermal Growth Factor Receptor- and Insulin-Like Growth Factor-1 Receptor Signaling: Implications for Cancer Therapy. Curr Cancer Drug Targets (2009) 9:748–60. doi: 10.2174/156800909789271495
94. Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-Like Growth Factor-I Stimulates the Growth of Rat Thyroid Cells in Culture and Synergizes the Stimulation of DNA Synthesis Induced by TSH and Graves’-Igg. Endocrinology (1986) 119:940–2. doi: 10.1210/endo-119-2-940
95. Takahashi S, Conti M, Prokop C, Van Wyk JJ, Earp HS 3rd. Thyrotropin and Insulin-Like Growth Factor I Regulation of Tyrosine Phosphorylation in FRTL-5 Cells. Interaction Between cAMP-Dependent and Growth Factor-Dependent Signal Transduction. J Biol Chem (1991) 266:7834–41. doi: 10.1016/S0021-9258(20)89525-8
96. Brenner-Gati L, Berg KA, Gershengorn MC. Thyroid-Stimulating Hormone and Insulin-Like Growth Factor-1 Synergize to Elevate 1,2-Diacylglycerol in Rat Thyroid Cells. Stimulation of DNA Synthesis Via Interaction Between Lipid and Adenylyl Cyclase Signal Transduction Systems. J Clin Invest (1988) 82:1144–8. doi: 10.1172/JCI113672
97. Tominaga T, Dela Cruz J, Burrow GN, Meinkoth JL. Divergent Patterns of Immediate Early Gene Expression in Response to Thyroid-Stimulating Hormone and Insulin-Like Growth Factor I in Wistar Rat Thyrocytes. Endocrinology (1994) 135:1212–9. doi: 10.1210/endo.135.3.8070365
98. Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, Sinha Hikim A, et al. Evidence for an Association Between Thyroid-Stimulating Hormone and Insulin-Like Growth Factor 1 Receptors: A Tale of Two Antigens Implicated in Graves’ Disease. J Immunol (Baltimore Md: 1950) (2008) 181:4397–405. doi: 10.4049/jimmunol.181.6.4397
99. Ock S, Ahn J, Lee SH, Kang H, Offermanns S, Ahn HY, et al. IGF-1 Receptor Deficiency in Thyrocytes Impairs Thyroid Hormone Secretion and Completely Inhibits TSH-Stimulated Goiter. FASEB J (2013) 27:4899–908. doi: 10.1096/fj.13-231381
100. Clement S, Refetoff S, Robaye B, Dumont JE, Schurmans S. Low TSH Requirement and Goiter in Transgenic Mice Overexpressing IGF-I and IGF-Ir Receptor in the Thyroid Gland. Endocrinology (2001) 142:5131–9. doi: 10.1210/endo.142.12.8534
101. Morshed SA, Latif R, Davies TF. Characterization of Thyrotropin Receptor Antibody-Induced Signaling Cascades. Endocrinology (2009) 150:519–29. doi: 10.1210/en.2008-0878
102. Dupont J, LeRoith D. Insulin and Insulin-Like Growth Factor I Receptors: Similarities and Differences in Signal Transduction. Horm Res (2001) 55 Suppl 2:22–6. doi: 10.1159/000063469
103. Morshed SA, Ando T, Latif R, Davies TF. Neutral Antibodies to the TSH Receptor Are Present in Graves’ Disease and Regulate Selective Signaling Cascades. Endocrinology (2010) 151:5537–49. doi: 10.1210/en.2010-0424
104. Latif R, Morshed SA, Zaidi M, Davies TF. The Thyroid-Stimulating Hormone Receptor: Impact of Thyroid-Stimulating Hormone and Thyroid-Stimulating Hormone Receptor Antibodies on Multimerization, Cleavage, and Signaling. Endocrinol Metab Clin North Am (2009) 38:319–41. doi: 10.1016/j.ecl.2009.01.006
105. Burikhanov R, Coulonval K, Pirson I, Lamy F, Dumont JE, Roger PP. Thyrotropin Via Cyclic AMP Induces Insulin Receptor Expression and Insulin Co-stimulation of Growth and Amplifies Insulin and Insulin-Like Growth Factor Signaling Pathways in Dog Thyroid Epithelial Cells. J Biol Chem (1996) 271:29400–6. doi: 10.1074/jbc.271.46.29400
106. Chen H, Mester T, Raychaudhuri N, Kauh CY, Gupta S, Smith TJ, et al. Teprotumumab, an IGF-1R Blocking Monoclonal Antibody Inhibits TSH and IGF-1 Action in Fibrocytes. J Clin Endocrinol Metab (2014) 99:E1635–40. doi: 10.1210/jc.2014-1580
107. Kurzrock R, Patnaik A, Aisner J, Warren T, Leong S, Benjamin R, et al. A Phase I Study of Weekly R1507, a Human Monoclonal Antibody Insulin-Like Growth Factor-I Receptor Antagonist, in Patients With Advanced Solid Tumors. Clin Cancer Res: An Off J Am Assoc Cancer Res (2010) 16:2458–65. doi: 10.1158/1078-0432.CCR-09-3220
108. Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, et al. R1507, a Monoclonal Antibody to the Insulin-Like Growth Factor 1 Receptor, in Patients With Recurrent or Refractory Ewing Sarcoma Family of Tumors: Results of a Phase II Sarcoma Alliance for Research Through Collaboration Study. J Clin Oncol: Off J Am Soc Clin Oncol (2011) 29:4541–7. doi: 10.1200/jco.2010.28.15_suppl.10000
109. Ramalingam SS, Spigel DR, Chen D, Steins MB, Engelman JA, Schneider CP, et al. Randomized Phase II Study of Erlotinib in Combination With Placebo or R1507, a Monoclonal Antibody to Insulin-Like Growth Factor-1 Receptor, for Advanced-Stage non-Small-Cell Lung Cancer. J Clin Oncol: Off J Am Soc Clin Oncol (2011) 29:4574–80. doi: 10.1200/JCO.2011.36.6799
110. Kawanami T, Takiguchi S, Ikeda N, Funakoshi A. A Humanized anti-IGF-1R Monoclonal Antibody (R1507) and/or Metformin Enhance Gemcitabine-Induced Apoptosis in Pancreatic Cancer Cells. Oncol Rep (2012) 27:867–72. doi: 10.3892/or.2011.1597
111. Mahadevan D, Sutton GR, Arteta-Bulos R, Bowden CJ, Miller PJ, Swart RE, et al. Phase 1b Study of Safety, Tolerability and Efficacy of R1507, a Monoclonal Antibody to IGF-1R in Combination With Multiple Standard Oncology Regimens in Patients With Advanced Solid Malignancies. Cancer Chemother Pharmacol (2014) 73:467–73. doi: 10.1007/s00280-013-2372-x
112. Paik JS, Kim SE, Kim JH, Lee JY, Yang SW, Lee SB. Insulin-Like Growth Factor-1 Enhances the Expression of Functional TSH Receptor in Orbital Fibroblasts From Thyroid-Associated Ophthalmopathy. Immunobiology (2020) 225:151902. doi: 10.1016/j.imbio.2019.151902
113. Smith TJ, Hegedus L. Graves’ Disease. New Engl J Med (2016) 375:1552–65. doi: 10.1056/NEJMra1510030
114. Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. Autoantibodies to IGF-1 Binding Sites in Thyroid Associated Ophthalmopathy. Autoimmunity (1993) 16:251–7. doi: 10.3109/08916939309014643
115. Pritchard J, Horst N, Cruikshank W, Smith TJ. Igs From Patients With Graves’ Disease Induce the Expression of T Cell Chemoattractants in Their Fibroblasts. J Immunol (Baltimore Md: 1950) (2002) 168:942–50. doi: 10.4049/jimmunol.168.2.942
116. Douglas RS, Gianoukakis AG, Kamat S, Smith TJ. Aberrant Expression of the Insulin-Like Growth Factor-1 Receptor by T Cells From Patients With Graves’ Disease may Carry Functional Consequences for Disease Pathogenesis. J Immunol (Baltimore Md: 1950) (2007) 178:3281–7. doi: 10.4049/jimmunol.178.5.3281
117. Douglas RS, Naik V, Hwang CJ, Afifiyan NF, Gianoukakis AG, Sand D, et al. B Cells From Patients With Graves’ Disease Aberrantly Express the IGF-1 Receptor: Implications for Disease Pathogenesis. J Immunol (Baltimore Md: 1950) (2008) 181:5768–74. doi: 10.4049/jimmunol.181.8.5768
118. Douglas RS, Brix TH, Hwang CJ, Hegedus L, Smith TJ. Divergent Frequencies of IGF-I Receptor-Expressing Blood Lymphocytes in Monozygotic Twin Pairs Discordant for Graves’ Disease: Evidence for a Phenotypic Signature Ascribable to Nongenetic Factors. J Clin Endocrinol Metab (2009) 94:1797–802. doi: 10.1210/jc.2008-2810
119. Smith TJ, Hoa N. Immunoglobulins From Patients With Graves’ Disease Induce Hyaluronan Synthesis in Their Orbital Fibroblasts Through the Self-Antigen, Insulin-Like Growth Factor-I Receptor. J Clin Endocrinol Metab (2004) 89:5076–80. doi: 10.1210/jc.2004-0716
120. Ma R, Li Q, Wang Z, Yuan Y, Gan L, Qian J. Modulation of Hyaluronan Polymer Size Regulates Proliferation of Perimysial Fibroblasts in Thyroid Eye Disease. Biochem Biophys Res Commun (2018) 496:1376–81. doi: 10.1016/j.bbrc.2018.02.037
121. Janssen J. New Insights From IGF-IR Stimulating Activity Analyses: Pathological Considerations. Cells (2020) 9. doi: 10.3390/cells9040862
122. Krieger CC, Boutin A, Jang D, Morgan SJ, Banga JP, Kahaly GJ, et al. Arrestin-β-1 Physically Scaffolds TSH and IGF1 Receptors to Enable Crosstalk. Endocrinology (2019) 160:1468–79. doi: 10.1210/en.2019-00055
123. Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, et al. Teprotumumab for Thyroid-Associated Ophthalmopathy. New Engl J Med (2017) 376:1748–61. doi: 10.1056/NEJMoa1614949
124. Douglas RS, Kahaly GJ, Patel A, Sile S, Thompson EHZ, Perdok R, et al. Teprotumumab for the Treatment of Active Thyroid Eye Disease. New Engl J Med (2020) 382:341–52. doi: 10.1056/NEJMoa1910434
125. Ma H, Zhang T, Shen H, Cao H, Du J. The Adverse Events Profile of anti-IGF-1R Monoclonal Antibodies in Cancer Therapy. Br J Clin Pharmacol (2014) 77:917–28. doi: 10.1111/bcp.12228
126. Qu X, Wu Z, Dong W, Zhang T, Wang L, Pang Z, et al. Update of IGF-1 Receptor Inhibitor (Ganitumab, Dalotuzumab, Cixutumumab, Teprotumumab and Figitumumab) Effects on Cancer Therapy. Oncotarget (2017) 8:29501–18. doi: 10.18632/oncotarget.15704
127. Terwee CB, Gerding MN, Dekker FW, Prummel MF, Wiersinga WM. Development of a Disease Specific Quality of Life Questionnaire for Patients With Graves’ Ophthalmopathy: The GO-QOL. Br J Ophthalmol (1998) 82:773–9. doi: 10.1136/bjo.82.7.773
128. Markham A. Teprotumumab: First Approval. Drugs (2020) 80:509–12. doi: 10.1007/s40265-020-01287-y
129. Ozzello DJ, Kikkawa DO, Korn BS. Early Experience With Teprotumumab for Chronic Thyroid Eye Disease. Am J Ophthalmol Case Rep (2020) 19:100744. doi: 10.1016/j.ajoc.2020.100744
130. Sears CM, Azad AD, Dosiou C, Kossler AL. Teprotumumab for Dysthyroid Optic Neuropathy: Early Response to Therapy. Ophthal Plast Reconstr Surg (2020). doi: 10.1097/IOP.0000000000001831
131. Slentz D, Smith TJ, Kim DS, Joseph SS. Teprotumumab For Optic Neuropathy in Thyroid Eye Disease. Arch Ophthal (In Press) (2020) 244–7 doi: 10.1001/jamaophthalmol.2020.5296
132. Aakaash Varma CR, Levitt J. Resolution of Pretibial Myxedema With Teprotumumab in a Patient With Graves’ Disease. JAAD Case Rep (2020) 1281–2 doi: 10.1016/j.jdcr.2020.09.003
133. Pritchard J, Tsui S, Horst N, Cruikshank WW, Smith TJ. Synovial Fibroblasts From Patients With Rheumatoid Arthritis, Like Fibroblasts From Graves’ Disease, Express High Levels of IL-16 When Treated With Igs Against Insulin-Like Growth Factor-1 Receptor. J Immunol (Baltimore Md: 1950) (2004) 173:3564–9. doi: 10.4049/jimmunol.173.5.3564
Keywords: growth factor, hormone, goiter, autoimmune, Graves’ disease, ophthalmopathy, thyroid
Citation: Smith TJ (2021) Insulin-Like Growth Factor Pathway and the Thyroid. Front. Endocrinol. 12:653627. doi: 10.3389/fendo.2021.653627
Received: 14 January 2021; Accepted: 20 April 2021;
Published: 04 June 2021.
Edited by:
Jeff M. P. Holly, University of Bristol, United KingdomReviewed by:
Leonard Girnita, Karolinska Institutet (KI), SwedenSyed A. Morshed, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2021 Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terry J. Smith, dGVycnlzbWlAbWVkLnVtaWNoLmVkdQ==