 Caio Leônidas Oliveira Andrade1
Caio Leônidas Oliveira Andrade1 Helton Estrela Ramos
Helton Estrela Ramos- 1Department of Life Sciences, University of the State of Bahia, Salvador, Brazil
- 2Medical School, Institute of Health Science-Federal University of Bahia, Salvador, Brazil
- 3Post-Graduate Program in Medicine and Health, Medical School of Medicine, Federal University of Bahia, Salvador, Brazil
- 4Postgraduate Program in Interactive Processes of Organs and Systems, Health & Science Institute, Federal University of Bahia, Salvador, Brazil
- 5Bioregulation Department, Health and Science Institute, Federal University of Bahia, Salvador, Brazil
Congenital hypothyroidism (CH) is an endocrine disease commonly found in newborns and is related to the absence or reduction of thyroid hormones (THs), which are essential for development since intrauterine life. Children with CH can develop hearing problems as THs are crucial for the auditory pathway’s development and maturation. Sensory deprivations, especially in hearing disorders at early ages of development, can impair language skills, literacy, and behavioral, cognitive, social, and psychosocial development. In this review we describe clinical and molecular aspects linking CH and hearing loss.
Introduction
The development of auditory pathways depends on the presence of adequate serum levels of thyroid hormones (TH) and their action on TH receptors (1–3). These hormones regulate proteins and enzymes responsible for the structural formation of the inner ear, being crucial for the proper performance of auditory function (4).
In fact, TH deficiency or TH defect action can cause severe changes in the development of the auditory system (2, 5). Clinical situations of reduction or absence of normal serum levels of TH, such as congenital hypothyroidism (CH), are frequently associated with hearing loss (6, 7). However, the incidence of hearing loss (HL) in individuals with CH is still uncertain, and it could affect ~20% of patients (8–10), occurring isolated or associated with vertigo and tinnitus (11).
It is well-known that when sensory deprivation events occur in the first months of life, a period considered critical for the maturation of biological functions, there is a high potential for subsequent significant delays in language, cognition, academic, emotional, and social development (12, 13). Therefore, the early detection and intervention of hearing problems, even in subclinical stages, allow individuals with auditory dysfunction to obtain sociolinguistic performances close to normal hearing (12, 14).
Given these facts and considering the scarcity of literature on the subject, the present study sought to achieve a narrative review on the probable dysfunctions of the auditory pathways connected to CH and TH deprivation in early neonatatal period, and its adverse impacts on social performance and language acquisition and development.
Clinical Aspects of Congenital
CH is an endocrine disease commonly found in newborns (15) with a worldwide occurrence of approximately 1:2,000 to 1:4,000 live births. It mainly affects females in the proportion of roughly 2:1 (16). CH is a public health issue, which can be detected with newborn screening (NS). The lack of an early diagnosis and adequate treatment can result in neurological and motor development changes and irreversible mental retardation (17). The main objective of neonatal screening is to promote the detection of congenital diseases before the symptomatic phase, enabling early treatment (18). CH does not usually present symptoms at birth, or there are only subtle manifestations of the disease, making clinical diagnosis difficult (19). The neonatal screening program recommends a TSH cut-off level of 10 mUI/L (7, 20). Newborns with high TSH in the test are referred for evaluation and confirmation of the diagnosis (21). Confirmation of the diagnosis of CH is made with laboratory exams, showing TSH greater than 15 mUI/L and total or free T4 with normal or low values (7, 17).
CH is normally classified as permanent or transient, whose etiology is classified into primary, secondary, and tertiary. The permanent condition requires lifelong treatment, as the hormonal deficiency is persistent. On the other hand, transient CH regains typical TH production in the initial months or years of life. In permanent primary cases, thyroid dysgenesis (TD) corresponds to 85% of cases, whereas dyshormonogenesis (DH) represents 10 to 15% of cases (22). In secondary CH, the lesion is in the pituitary; and in the tertiary form, the dysfunction is in the hypothalamus. The last two cases are extremely rare. Central CH (secondary or tertiary) is commonly associated with other pituitary hormonal deficiencies (15, 17). The absence of stimuli from pituitary TSH (Thyroid Stimulating Hormone) or hypothalamic TRH (Thyrotropin-Releasing Hormone) is the cause of deficient hormone production in the central CH (23).
The etiology and clinical phenotype of CH become essential in determining the severity, outcomes, and treatment of the disease, as patients may need therapy with higher doses and close monitoring, especially during early periods of life (24). THs are essential for adequate neurodevelopment since intrauterine life (25). Their absence leads to dysfunction of specific brain areas, affecting regions such as the posterior parietal, inferior temporal lobes, caudate nucleus, and hippocampus, which are responsible for, respectively, spatial location, object identification, attention, and memory (26, 27).
Action of Thyroid Hormones on Auditory Function
THs play an essential role in developing the inner ear during the embryonic period (28). Since the fetal period, the T3 is essential for auditory development, when the embryo in the first trimester depends exclusively on maternal THs, beginning its hormonal synthesis in the second half of the gestational period (2, 3). Triiodothyronine (T3) is mediated by the thyroid hormone receptor (THR), whose action on cochlear sensory cells is caused by the differential expression of thyroid hormone receptor alpha (THRα) and thyroid hormone receptor beta (THRβ) (21).
In the rodent cochlea, THRs are expressed in the sensory epithelium and other tissues from mid-gestation into the postnatal period and function as transcription factors playing important roles in control target genes relevant for auditory development and function, and the abnormal regulation of genes controlled by THRs has been assumed to be the origin of neurosensory deafness associated with CH (29).
The THRα gene is widely expressed throughout the spiral organ of corti, while the THRβ gene has its expression prominently in the greater epithelial ridges of sensitive hair cells (5, 30, 31). This gene expression pattern points out that the spiral organ is a direct-action site for TH and explains the scientific evidence of morphological and functional abnormalities of the structures that form the cochlea in cases of hypothyroidism (28, 32–37). Indeed, the THRs’ expression is timely coordinated in order to have a very precise signaling necessary for proper THR-dependent differentiation events, comprising complete inner sulcus, sensory epithelium, spiral ganglion, cochlea, and auditory nerve maturation (38).
Table 1 summarizes mouse models of TH action or production defects. Actually, THRα1 is considered non-essential for hearing, while defects on THRβ, in mice, present deafness linked to cochlear alterations. On animal models, THRβ-null mice show threshold elevations ranged from a few decibels to complete loss of auditory responsiveness. An isoform-specific importance ranking is observed, because only THRβ1 signaling defect is associated with retardation in the expression of the fast-activating potassium conductance in inner hair cells, whereas deletion of the THRβ2 isoform does not lead to anormal cochlear function (38).

Table 1 Mouse models for understanding the relevance of genes involved in thyroid development, hormone biosynthesis, and thyroid hormone action on hearing function.
Nonetheless, deletion of both THRβ1 and THRα1 produces exacerbated defects that simulate those provoked by hypothyroidism (38). In reality, human genetic alterations associated with loss of TRβ function, a condition named resistance to TH, also result in deafness (39).
The critical developmental time period of the middle and inner ears occurs in parallel to the natural elevation of TH serum plasma levels. Thyroxine (T4), liberated by the thyroid gland into the circulation, must be metabolically activated or inactivated by iodothyronine deiodinases, and 3,5,3’-triiodothyronine (T3) is the main ligand of the THRs. Therefore, TH adequate intracellular levels are accomplished after action of deiodinase type 2 (D2) and deiodinase type 1 (DI) encoded by Dio2 and Dio1, respectively (29).
Other evidence that suggests strong influences of TH in the cochlea is related to the expression of the SLC26a5 gene, which encodes the prestin protein. This protein is considered the outer hair cells (OHC) engine in the cochlear amplification process (40), which is reduced, immature, and with reduced distribution under hypothyroidism conditions (41–43). The gene expression encoding the K+ channels, KCNQ4, responsible for the endolymphatic potential formation, has also been discussed in the literature. Therefore, it has been shown that these ion channels are significantly reduced and poorly distributed under conditions of thyroid hypofunction (44). Figure 1 illustrates the molecular structures inherent in external hair cells, which are dependent on adequate serum levels for thyroid hormones in the body.
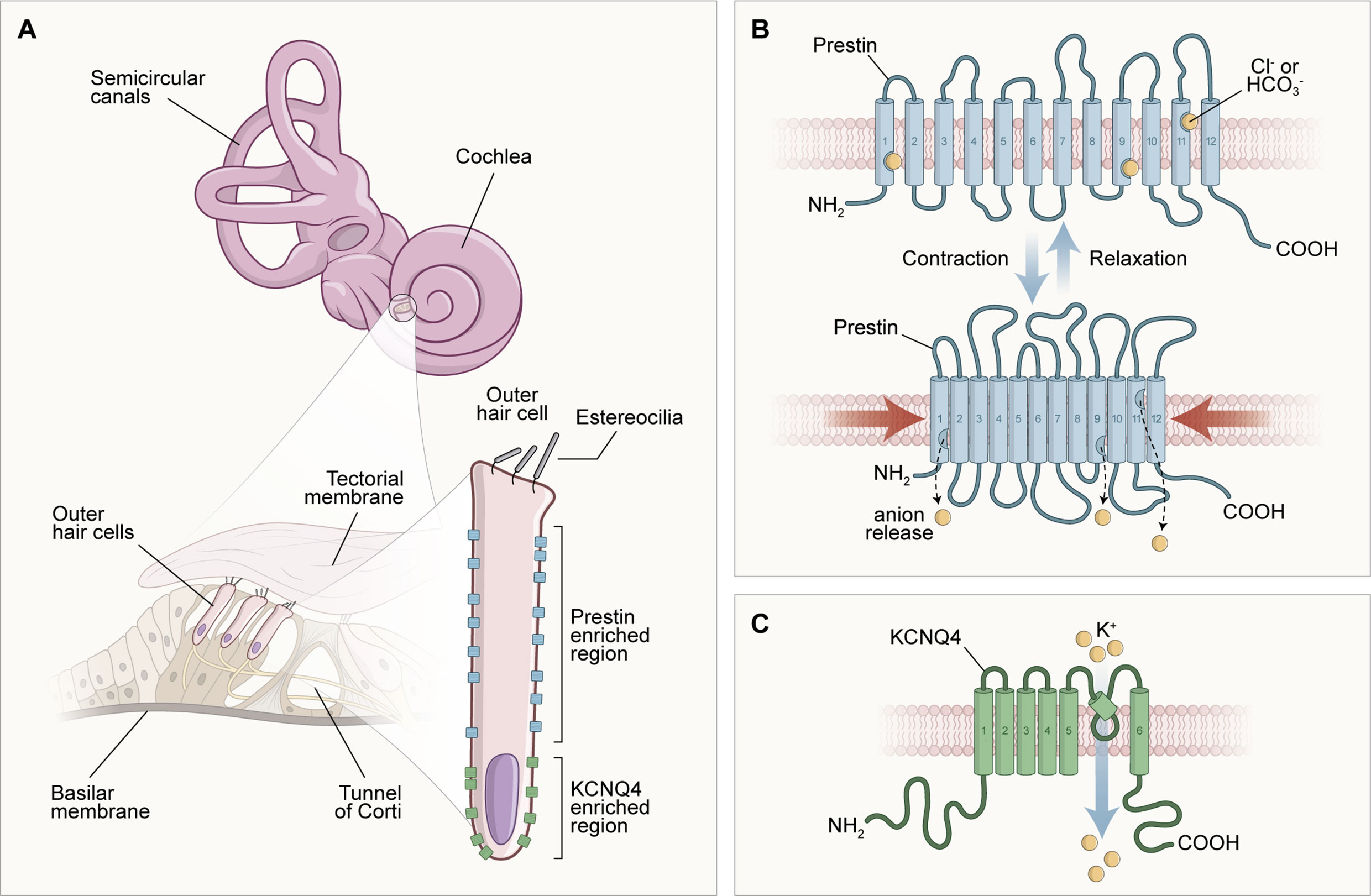
Figure 1 Scheme of the morphological configuration and special distribution of transmembrane proteins responsible for the endolymphatic potential (KCNQ4) generation and cochlear electromotility (prestin). In (A), the structures of the inner ear are illustrated in different parts: at the top of the image, there are the cochlea and semicircular canals, structures that compose the inner ear macroscopically; to the left of the image, in a microscopic analysis, there is the spiral organ (Tunnel of Corti) formed of the tectorial membrane, supporting and sensory cells; and to the right of the image, the morphology of the outer hair cell can be observed, delimiting the molecular location of the transmembrane proteins prestin (lateral) and KCNQ4 (basal). In (B), it is possible to observe the unique arrangement of the prestin protein and its physiology. Prestin is involved with the motor function of outer hair cells (OHC). This activity occurs when OHCs are depolarized through the influx of K+ positive electrical charges after a sound stimulation, creating a positive intracellular environment that favors the displacement of Cl- anions from the prestin's binding sites into the cytoplasm. This electrical charge movement causes a shortening of prestin and, consequently, a reduction in the size of the OHC, characterizing the active mechanism and the bio-electromotility of the OHC that occurs in the absence of calcium and ATP. In (C), after depolarization, an OHC enters the repolarization and hyperpolarization stage due to the exit of cations in the basal portion of the potassium channels formed by the transmembrane protein KCNQ4, which contributes to the generation of the endolymphatic potential.
Hormone deficiency can cause reductions in β-tectorin protein in the tectorial membrane, which explains structural abnormalities of the tectorial membrane and cochlear function. The OHCs are susceptible to serum thyroid hormone levels (36). Thus, serum levels of thyroid hormones circulating in the bloodstream can affect cell differentiation, which reduces the amount of organelles in the cytoplasm (37). These changes may be accompanied by abnormalities of the afferent dendrites and delayed growth of the efferent terminals that make direct connections with the OHC (4).
In the case of neural and central structures, studies in animal models have shown abnormalities in myelination and reduction of axons in the anterior commissure and corpus callosum (36). There is also a reduction in the number of microtubules in the neural cytoplasm and an altered distribution of apical dendrites of the pyramidal neurons (37). Added to this, there are records of a decline in the deoxyglucose levels of the metabolism marker in regions such as the cochlear nucleus, superior olivary complex, nuclei of the lateral lemniscus, inferior colliculus, medial geniculate body, and auditory cortex. Therefore, it is possible to state that TH deficiency will significantly affect the auditory pathway (42).
Consequences of CH on Auditory System
Hearing is one of the essential senses for human communication, and it is where the individual develops speech. In addition, it is through hearing that the process of acquisition and development of oral language occurs. The auditory system consists of a peripheral portion (outer, middle, and inner ears), which captures sounds and transforms them into electrical impulses, and a central portion (brain auditory pathways), responsible for the analysis and interpretation of what is heard (45). Any complication in one of these portions can result in hearing loss (46), compromising not only communication but also receptive and expressive language, literacy, school performance, and the child’s psychosocial development (45).
The functionality of the thyroid gland is crucial for the development of the auditory system (1). THs are vital for auditory pathway morphogenesis and maturation (3), and the deficiency of these hormones jeopardizes the development of hearing (47). Therefore, CH can result in hearing loss (27), and even with early treatment, small hearing changes can be observed in individuals with CH (48). This happens due to the cochlea’s susceptibility to metabolic disorders, resulting from its intense activity and low energy reserve (49). THs act in both systems (peripheral and central) in the auditory system, and they are responsible for forming key structures of the inner ear, such as the cochlear duct, organ of Corti, and tectorial membrane (50). Therefore, the shortage or lack of THs brings losses to these structures.
Audiological changes noted in CH patients are diverse. However, losses with sensorineural, bilateral, and symmetrical characteristics are often found, with degrees varying from mild to moderate (8, 9). Actually, the risk of hearing loss may be associated with the severity of CH (43). In the researched literature, hearing changes in CH are characterized as peripheral or central, of insidious occurrence, with impaired auditory abilities (cognitive functions related to hearing). These skills are essential to the development of oral and written language and social-emotional progress. Additionally, they affect the individual in periods considered critical to developing global skills and full stage of experimentation and interaction with the environment, compromising the quality of life. In this context, when a hearing disorder is detected early, even during the neonatal period, early intervention through speech therapy and indication of hearing aids, if necessary, may be required and performed, preventing future harm to the child.
Impact of Hearing Loss at Early Ages
In cases where TH deficiency occurs in early periods, as in CH, the risk of hearing loss in children is increased (8, 10). This data is significantly worrying when thinking about the harm that the reduction or absence of action of TH in the crucial periods of neurological development and maturation can bring. The central nervous system is one of the most affected (51), and it can alter the processing of the acoustic signal up to the cortex, causing difficulties in auditory skills (52) that will result in problems with behavioral, language, and social difficulties.
The crucial periods for the development of children’s hearing and oral language occur in early childhood. Nerve structures are already specialized in the brain of newborns with auditory cortical areas formed and ready to receive acoustic stimuli from the external environment. Consequently, the first contact with sounds is provided, instigating the mother tongue’s acquisition and increasing the linguistic repertoire (53, 54). Hence, when newborns have alterations in their auditory pathways that limit them to having an adequate sound sensation during the first 3 years of life, their linguistic and social potential will be low and reduced (53, 54). In the absence or deficit of sound stimuli at critical times, without adequate intervention, the child may present vital educational, social, and emotional delays (12).
The literature also shows that some language deficits, fine motor skills, visuospatial processing, attention and memory, and hearing disorders can persist in patients with CH even with early treatment (47, 48). Even moderate or mild hearing loss can alter the hearing perception of voiceless phonemes (55, 56), making the understanding of soft speech unintelligible, even in a quiet environment (57). As a result, phonological discrimination, phonological awareness, and phonological memory are compromised, consequently interfering in the learning processes of these children, directly affecting their quality of life and their families (58, 59).
Conclusion
THs are essential for brain and intellectual development, as well as for peripheral and central auditory functions that extend from the fetal period to 2 years of age, a period considered critical for typical development. Therefore, CH can be considered a potential risk factor for changes in acoustic signals’ processing mechanisms along the auditory pathway, which manifests itself as cognitive, language, and socioemotional delays.
Author Contributions
CLOA: conception, writing. CA: Editing, review. HR: Conception, writing. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CH, congenital hypothyroidism; T4, Hormone thyroxine; hypothalamic TRH, Thyrotropin-Releasing Hormone; NNSP, National Neonatal Screening Program; NS, Newborn screening; PAX8, Paired box gene 8; SECISBP2, Selenocysteine-insertion sequence binding protein 2; T3, Triiodothyronine; TH, thyroid hormones; TSH, Thyroid Stimulating Hormone; TR, Thyroid hormone receptor; THRα, thyroid hormone receptor alpha; THRβ, Thyroid hormone receptor beta; TSHR, Thyroid Stimulating Hormone receptor; DIO2, Type 2 deiodinase; OHC, Outer hair cells.
References
1. Sohmer H, Freeman S. The Importance of Thyroid Hormone for Auditory Development in the Fetus and Neonate. Audiol Neurootol (1996) 1(3):137–47. doi: 10.1159/000259194
2. Ng L, Kelley MW, Forrest D. Making Sense With Thyroid Hormone–the Role of T3 in Auditory Development. Nat Rev Endocrinol (2013) 9(5):296–307. doi: 10.1038/nrendo.2013.58
3. de Andrade CLO, Machado GC, Fernandes LC, Albuquerque JM, Casais-e-Silva LL, Ramos HE, et al. Mechanisms Involved in Hearing Disorders of Thyroid Ontogeny: A Literature Review. Arch Endocrinol Metab (2017) 61:p.501–05. doi: 10.1590/2359-3997000000292
4. Uziel A, Marot M, Rabie A. Corrective Effects of Thyroxine on Cochlear Abnormalities Induced by Congenital Hypothyroidism in the Rat. II. Electrophysiological Study. Brain Res (1985) 351(1):123–7. doi: 10.1016/0165-3806(85)90237-8
5. Knipper M, Zinn C, Maier H, Praetorius M, Rohbock K, Köpschall I, et al. Thyroid Hormone Deficiency Before the Onset of Hearing Causes Irreversible Damage to Peripheral and Central Auditory Systems. J Neurophysiol (2000) 83(5):3101–12. doi: 10.1152/jn.2000.83.5.3101
6. François M, Bonfils P, Leger J, Avan P, Czernichow P, Narcy P. Audiological Assessment of Eleven Congenital Hypothyroid Infants Before and After Treatment. Acta Otolaryngol (1993) 113:39–42. doi: 10.3109/00016489309135764
7. Trotsenburg PV, Stoupa A, Lége J, Rohrer TR, Peters C, Fugazzola L, et al. Congenital Hypothyroidism: A 2020-2021 Consensus Guidelines Update-An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid (2021) 31(3):387–419. doi: 10.1089/thy.2020.0333
8. Debruyne F, Vanderschueren-Lodeweyckx M, Bastijns P. Hearing in Congenital Hypothyroidism. Audiology (1983) 22(4):404–9. doi: 10.3109/00206098309072800
9. François MMD, Bonfils P, Leger J, Czernichow P, Narcy P. Role of Congenital Hypothyroidism in Hearing Loss in Children. J Pediatr (1994) 124(3):444–6. doi: 10.1016/S0022-3476(94)70373-6
10. Rovet J, Walker W, Bliss B, Buchanan L, Ehrlich R. Long-Term Sequelae of Hearing Impairment in Congenital Hypothyroidism. J Pediatr (1996) 128(6):776–83. doi: 10.1016/S0022-3476(96)70329-3
11. Lichtenberger-Geslin L, Santos S, Hassani Y, Ecosse E, Van Den Abbeele T, Léger J. Factors Associated With Hearing Impairment in Patients With Congenital Hypothyroidism Treated Since the Neonatal Period: A National Population-Based Study. J Clin Endocrinol Metab (2013) 98(9):3644–52. doi: 10.1210/jc.2013-1645
12. Stampa M. Aprendizagem E Desenvolvimento Das Habilidades Auditivas: Entendendo E Praticando Na Sala De Aula. Rio de Janeiro: WAK (2012). p. 128.
13. Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL. Language of Early and Later Identified Children With Hearing Loss. Pediatrics (1998) 102(5):1161–71. doi: 10.1542/peds.102.5.1161
14. Colunga JCM, Méndez JCA, Villarreal JMC, Zapico MJA, Estrada CM, Álvarez LMF, et al. Despistage De La Hipoacusia Neonatal: Resultados Después De 3 Años De Iniciar Nuestro Programa. Acta Otorrinolaringol Esp (2005) 55:55–8. doi: 10.1016/S0001-6519(05)78571-X
15. AmericanAcademyof Pediatrics (AAP), American Thyroid Association (ATA), Lawson WilkinsPediatric Endocrine Society (LWPES). Update of Newborn Screening and Therapy for Congenital Hypothyroidism. Pediatrics (2006) 117(6):2290–303. doi: 10.1542/peds.2006-0915
16. Eugène D, Djemli A, Van Vliet G. Sexual Dimorphism of Thyroid Function in Newborns With Congenital Hypothyroidism. J Clin Endocrinol Metab (2005) 90(5):2696–700. doi: 10.1210/jc.2004-2320
17. Maciel LMZ, Kimura ET, Nogueira CR, Mazeto GMFS, Magalhães PKR, Nascimento ML, et al. Hipotireoidismo Congênito: Recomendações do Departamento De Tireoide Da Sociedade Brasileira De Endocrinologia E Metabologia. Arq Bras Endocrinol Metab (2013) 57(3):184–92. doi: 10.1590/S0004-27302013000300004
18. Ford G, LaFranchi SH. Screening for Congenital Hypothyroidism: A Worldwide View of Strategies. Best Pract Res Clin Endocrinol Metab (2014) 28(2):175–87. doi: 10.1016/j.beem.2013.05.008
19. Alm J, Larsson A, Zetterström R. Congenital Hypothyroidism in Sweden Incidence and Age at Diagnosis. Acta Paediatr Scand (1978) 67(1):1–3. doi: 10.1111/j.1651-2227.1978.tb16268.x
20. Nascimento ML. Situação Atual Da Triagem Neonatal Para Hipotireoidismo Congênito: Críticas E Perspectivas. Arq Bras Endocrinol Metab (2011) 55:528–33. doi: 10.1590/S0004-27302011000800005
21. Sap J, Muñoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, et al. The C-Erb-A Protein is a High-Affinity Receptor for Thyroid Hormone. Nature (1986) 324(6098):635–40. doi: 10.1038/324635a0
22. Rastogi MV, Lafranchi SH. Congenital Hypothyroidism. Orphanet J Rare Dis (2010) 5(2):2–22. doi: 10.1186/1750-1172-5-17
23. Gupta V, Lee M. Central Hypothyroidism. Indian J Endocrinol Metab (2011) 15(Suppl2):S99–S106. doi: 10.4103/2230-8210.83337
24. Iranpour R, Hashemipour M, Amini M, Talaei SM, Kelishadi R, Hovsepian S, et al. [Tc]–99m Thyroid Scintigraphy in Congenital Hypothyroidism Screening Program. J Trop Pediatr (2006) 52(6):411–5. doi: 10.1093/tropej/fml038
25. Marti S, Alvarez M, Simoneau-Roy J, Leroux S, Van Vliet G, Robaey P. Effects of Early High–Dose Levothyroxine Treatment on. Auditory Brain. Event–Related Potentials at School Entry in. Children. with Congenital Hypothyroidism.. Horm Res (2006) 66(5):240–8. doi: 10.1159/000095069
26. Maniquéz MO, Lilianette BN, Ximena WV. Evolución Neurológica Em Pacientes Com Hipotireoidismo Congênito Disgnosticado Por Rastreo Neonatal. Rev Chil Pediatr (2008) 69:56–9. doi: 10.4067/S0370-41061998000200002
27. Rovet J. Long-Term Neuropsychological Sequelae of Early-Treated Congenital Hypothyroidism: Effects in Adolescence. Acta Paediatr Suppl (1999) 88(432):88–95. doi: 10.1111/j.1651-2227.1999.tb01168.x
28. Deol MS. The Role of Thyroxine in the Differentiation of the Organ of Corti. Acta Otolaryngol (1976) 81(5-6):429–35. doi: 10.3109/00016487609107497
29. Ng L, Goodyear RJ, Woods CA, Schneider MJ, Diamond E, Richardson GP, et al. Hearing Loss and Retarded Cochlear Development in Mice Lacking Type 2 Iodothyronine Deiodinase. Proc Natl Acad Sci USA (2004) 101(10):3474–9. 10.1073/pnas.0307402101
30. Bradley DJ, Towle HC, Young WS 3rd. Alpha and Beta Thyroid Hormone Receptor (TR) Gene Expression. During Auditory Neurogenesis: Evidence for TRisoform-Specific Transcriptional Regulation In Vivo. Proc Natl Acad Sci USA (1994) 91(2):439–43. doi: 10.1073/pnas.91.2.439
31. Lautermann J, ten Cate WJ. Postnatal Expression. Of the –Thyroid Hormone Receptor in. The Rat Cochlea. Hear Res (1997) 107(1-2):23–8. doi: 10.1016/S0378-5955(97)00014-2
32. O’Malley BW, Li D, Turner DS. Hearing Loss and Cochlear Abnormalities in the Congenital Hypothyroid (hyt/hyt) Mouse. Hear Res (1995) 88:181–9. doi: 10.1016/0378-5955(95)00111-G
33. Uziel A, Pujol R, Legranda C, Legranda J. Cochlear Synaptogenesis in the Hypothyroid Rat. Dev Brain Res (1983) 7:295–301. doi: 10.1016/0165-3806(83)90186-4
34. Deol MS. An. Experimental Approach to the Understanding and Treatment of Hereditary Syndromes With Congenital Deafness and Hypothyroidism. J Med Genet (1973) 10:235–42. doi: 10.1136/jmg.10.3.235
35. Uziel A, Gabrion J, Ohresser M, Legrand C. Effects of Hypothyroidism on the Structural Development of the Organ of Corti in the Rat. Acta Otolaryngologica (1981) 92:469–80. doi: 10.3109/00016488109133286
36. Berbel P, Guadaño-Ferraz A, Angulo A, Ramón Cerezo J. Role of Thyroid Hormones in the Maturation of Interhemispheric Connections in Rats. Behav Brain Res (1994) 64:9–14. doi: 10.1016/0166-4328(94)90114-7
37. Berbel P, Guadaño-Ferraz A, Martínez M, Quiles JA, Balboa R, Innocenti GM. Organization of Auditory Callosal Connections in Hypothyroid Adult Rats. Eur J Neurosci (1993) 5:1465–78. doi: 10.1111/j.1460-9568.1993.tb00214.x
38. Abel ED, Boers ME, Pazos-Moura C, Moura E, Kaulbach H, Zakaria M, et al. Divergent Roles for Thyroid Hormone Receptor Beta Isoforms in the Endocrine Axis and Auditory System. J Clin Invest (1999) 104(3):291–300. doi: 10.1172/JCI6397
39. Dumitrescu AM, Refetoff S. Impaired Sensitivity to Thyroid Hormone: Defects of Transport, Metabolism and Action. Feingold K. R., Anawalt B., Boyce A. editors. South Dartmouth (MA): MDText.com, Inc. (2000) Available from: https://www.ncbi.nlm.nih.gov/books/NBK279066/
40. Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin. Is the Motor Protein. Of Cochlear Outer Hair Cells. Nature (2000) 405:149–55. doi: 10.1038/35012009
41. Weber T, Zimmermann U, Winter H, Mack A, Köpschall T, Rohbock K, et al. Thyroid Hormone Is a Critical Determinant for the Regulation of the Cochlear Motor Protein Prestin. Proc Natl Acad Sci USA (2002) 99:2901–6. doi: 10.1073/pnas.052609899
42. Dow-Edwards D, Crane AM, Rosloff B, Kennedy C, Sokoloff L. Local Cerebral Glucose Utilization in the Adult Cretinous Rat. Brain Res (1986) 373:139–45. 1986. doi: 10.1016/0006-8993(86)90323-9
43. Bruno R, Aversa T, Catena M, Valenzise M, Lombardo F, De Luca F, et al. Even in the Era of Congenital Hypothyroidism Screening Mild and Subclinical Sensorineural Hearing Loss Remains a Relatively Common Complication of Severe Congenital Hypothyroidism. Hearing Res (2015) 327:43–7. doi: 10.1016/j.heares.2015.04.018
44. Winter H, Braig C, Zimmermann U, Geisler H, Fränzer J, Weber T, et al. Thyroid Hormone Receptors TRalpha1 and TRbeta Differentially Regulate Gene Expression. Of Kcnq4 and Prestin. During Final Differentiation. Of Outer Hair Cells. J Cell Sci (2006) 11914:2975–84. doi: 10.1242/jcs.03013
45. Bonaldi LV, Angelis MA, Smith RL. Anatomia Funcional do Sistema Vestibulococlear. In: Frota S, editor. Fundamentos De Fonoaudiologia: Audiologia, 2ª ed. Rio de Janeiro: Guanabara Koogan (2003). p. 1–17.
46. Correia RBF, Coelho JMS. Ações Em Saúde Auditiva Escolar No Município De Sobral-CE: Percepção De Fonoaudiólogos. Rev Bras em Promoção da Saúde (2012) 25(2):228–34. doi: 10.5020/18061230.2012.p228
47. Rovet JF. Children With Congenital Hypothyroidism and Their Siblings: Do They Really Differ? Pediatria (2005) 115(1):52–7. doi: 10.1542/peds.2004-1492
48. Bellman SC, Davies A, Fuggle PW, Grant DB, Smith I. Mild Impairmentof Neuro-Otological Function in Early Treated Congenital Hypothyroidism. Arch Dis Child (1996) 74:215–8. doi: 10.1136/adc.74.3.215
49. Azevedo MF. Emissões Otoacústicas. In: Figueiredo MS, editor. (Org.). Conhecimentos Essenciais Para Entender Bem Emissões Otoacústicas E BERA. São José dos Campos: Pulso (2003). p. 35–83.
50. Sedin G, Bulankina AV, Dietmar Riedel D, Moser T. Maturation of Ribbon Synapses in. Hair Cells is Driven. by Thyroid Hormone. J Neurosci (2007) 27:3163–73. doi: 10.1523/JNEUROSCI.3974-06.2007
51. Williams GR. Neurodevelopmental and Neurophysiological Actions of Thyroid Hormone. J Neuroendocrinol (2008) 20:784–94. doi: 10.1111/j.1365-2826.2008.01733.x
52. Figueiredo L, Lima MA, Vaisman M. Alterações Na Audiometria De Tronco Encefálico Em Mulheres Adultas Com Hipotireoidismo Subclínico. São Paulo: Sociedade Brasileira de Otorrinolaringologia (2003) p. 542–7. doi: 10.1590/S0034-72992003000400016
55. Tharpe AM, Bess F. Identification and Management of Children With Minimal Hearing Loss. Int J Pediatr Otorhinolatyngology (1991) 21:41–50. doi: 10.1016/0165-5876(91)90058-J
56. New ZelandGovernament. What is Mild Hearing Loss? (2014). Ministry of Health of New Zeland. Available at: https://www.nsu.govt.nz/system/files/resources/mild-hearing-loss.pdf (Accessed Acesso em 17 de dez de 2020).
57. Kaderavek JN, Pakulski LA. Minimal Hearing Loss Is Not Minimal. Teaching Exceptional Children. Council Exceptional Children (2002) 34:14–8. doi: 10.1177/004005990203400602
58. Davis A, Reeve K, Hind S, Bamford J. Children With Mild and Unilateral Hearing Impairment. Proceedings of the Second International Conference: A Sound Foundation Through Early Amplification.Section V. Chapter 14. (2002) 174–86.
Keywords: thyroid, congenital hypothyroidism, hearing loss, auditory system, hypothyroidism
Citation: Andrade CLO, Alves CAD and Ramos HE (2021) Congenital Hypothyroidism and the Deleterious Effects on Auditory Function and Language Skills: A Narrative Review. Front. Endocrinol. 12:671784. doi: 10.3389/fendo.2021.671784
Received: 24 February 2021; Accepted: 01 July 2021;
Published: 10 August 2021.
Edited by:
Cintia E. Citterio, CONICET Institute of Immunology, Genetics and Metabolism (INIGEM), ArgentinaReviewed by:
Anoop Arunagiri, University of Michigan, United StatesHannah Cooper, University College London, United Kingdom
Rudolf Hoermann, Klinikum Lüdenscheid, Germany
Copyright © 2021 Andrade, Alves and Ramos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helton Estrela Ramos, cmFtb3NoZWx0b25AZ21haWwuY29t; orcid.org/0000-0002-2900-2099