 Xiao Zhai1,2
Xiao Zhai1,2 Lian Duan1,2
Lian Duan1,2 Yong Yao3
Yong Yao3 Bing Xing3Kan Deng3
Bing Xing3Kan Deng3 Linjie Wang1,2Feng Feng4
Linjie Wang1,2Feng Feng4 Zhiyong Liang5Hui You4Hongbo Yang1,2
Zhiyong Liang5Hui You4Hongbo Yang1,2 Lin Lu1,2Shi Chen1,2
Lin Lu1,2Shi Chen1,2 Renzhi Wang3
Renzhi Wang3 Hui Pan1,2
Hui Pan1,2 Huijuan Zhu1,2*
Huijuan Zhu1,2*- 1Key Laboratory of Endocrinology of National Health Commission, Department of Endocrinology, State Key Laboratory of Complex Severe and Rare Diseases Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 2Department of Endocrinology, Peking Union Medical College Hospital, Chinese Academy of Medical Science (CAMS), Beijing, China
- 3Department of Neurosurgery, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 4Department of Radiology, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 5Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
Background: McCune-Albright syndrome is a rare disorder characterized by fibrous dysplasia, café au lait skin spots, and hyperfunctioning endocrinopathies. The coexistence of precocious puberty and growth hormone excess in McCune-Albright syndrome is rare. Both conditions can manifest as accelerated growth, and treatments can be more challenging for such patients. This study aimed to describe the clinical manifestations of combined GH excess and PP in the context of McCune-Albright syndrome and analyze the clinical features and treatments of these patients.
Method: Clinical data from 60 McCune-Albright syndrome patients from Peking Union Medical College Hospital were obtained. The demographic characteristics, growth hormone, insulin-like growth factor-1, prolactin, alkaline phosphatase, and sex hormone levels; growth velocity; and bone age data were obtained. The growth velocity Z-score, bone age over chronological age ratio, and predicted adult height Z-score were calculated before and after treatment. Published studies and case reports were systemically searched, and data on demographic, clinical, and biochemical characteristics and treatment outcomes were obtained.
Results: We reviewed seven patients among 60 McCune-Albright syndrome patients at Peking Union Medical College Hospital (5 female) and 39 patients (25 female) from the published literature. Six of the seven patients from Peking Union Medical College Hospital and half of the patients from the published studies were pediatric patients. These patients had increased growth velocity Z-scores and bone age over chronological age ratios. After good control of both conditions, the growth velocity Z-score and bone age over chronological age ratio decreased significantly, and the predicted adult height Z-score increased. The final heights and predicted adult height Z-scores were not impaired in patients with gigantism. All the patients had craniofacial fibrous dysplasia associated with optic and otologic complications.
Conclusion: McCune-Albright syndrome with growth hormone excess and precocious puberty is more common in girls. Patients have accelerated linear growth and advanced skeletal age, and early and good control of both conditions leads to a reduced growth velocity and stabilized bone age. The predicted adult and final heights are not negatively affected when growth hormone excess is diagnosed in pediatric patients.
Background
McCune-Albright syndrome (MAS) is a sporadic disease caused by somatic activating mutations of the GNAS1 gene encoding the α subunit of guanine nucleotide-binding protein (1), which lead to constitutive receptor activation and dysregulated production of cAMP (2). MAS is characterized by the triad of monostotic/polyostotic fibrous dysplasia (FD), café au lait skin pigmentation, and hyperfunctioning endocrinopathies, including gonadotropin-independent precocious puberty (PP), thyrotoxicosis, growth hormone (GH) excess, hyperprolactinemia, or neonatal hypercortisolism (2–6).
MAS is a rare disease, and the estimated prevalence is between 1/100,000 and 1/1,000,000 (7). Gonadotropin-independent sexual puberty is the most common endocrinopathy in MAS (4), affecting 50% of girls, and is far more common in girls than in boys with MAS (8). MAS-associated precocious puberty (PP) is a rare cause of PP, and the estimated prevalence of PP (including gonadotropin-dependent and gonadotropin-independent PP) in the general population from a Danish national registry was 0.2% in female patients and less than 0.05% in males (9). Progression to gonadotropin-dependent PP over time has also been documented in some patients (10, 11). MAS-associated PP in girls is caused by recurrent, unilateral autonomously functioning ovarian cysts, which leads to episodic estrogen production with suppressed gonadotropins (12). Girls typically present with breast development and painless vaginal bleeding. PP in boys with MAS is associated with a premature penile growth and bilateral testicular enlargement, secondary to Leydig cell hyperplasia and elevated testosterone production (5). Additionally, PP causes growth acceleration and skeletal advancement (13), leading to impaired adult height. GH excess, which is present in 20~30% of patients with MAS (8, 14–16), presents with increased growth velocity in children and adolescents, and facial and acral dysmorphia in adults. GH excess is usually accompanied by serious craniofacial FD complications—e.g., visual, hearing, and olfactory injuries (16–22).
The co-occurrence of PP and GH excess in MAS is rare. To date, only a few case reports regarding GH excess and PP in MAS have been reported. The diagnosis and treatment of GH excess may become challenging when PP is also present. GH excess and PP both present with accelerated linear growth, and facial dysmorphia is often difficult to assess because of craniofacial fibrous dysplasia (CFFD). Therefore, it is easy to miss the diagnosis of GH excess because of the coexistence of PP and CFFD. Additionally, choosing the appropriate treatment of GH excess and PP is essential for children and adolescents to achieve normal adult height.
This study aimed to retrospectively analyze the clinical manifestations, treatments, and outcomes of combined GH excess and PP in MAS patients from Peking Union Medical College Hospital (PUMCH), review all reported cases, and analyze the clinical features and treatments of these patients.
Subjects and Methods
Patients
This study was conducted in accordance with the rules of the hospital medical ethics committee, and informed consent was obtained. A retrospective study was performed on seven MAS patients who had a combined diagnosis of GH excess (including gigantism and acromegaly) and PP over 10 years (2010~2020) at PUMCH. The inclusion and exclusion process is shown in Figure 1.

Figure 1 Flowchart of the study population.
Literature Review
All the studies and case reports of GH excess and PP with MAS (GH excess, acromegaly, gigantism, precocious puberty, and McCune-Albright syndrome were the keywords) were systematically sought in the world literature up to September 2020. Studies and case reports were identified in the PubMed, Embase, Google Scholar, and Chinese Biomedical (CBM) databases, and those with an English abstract were included in the analysis.
Data Extraction
We extracted data from medical records in PUMCH and published studies and analyzed each patient in case reports and case series. Pretreatment data were routinely collected before the initiation of treatment, and posttreatment data were collected at the last consultation for patients under therapy. The following data were extracted (1): demographic characteristics (2); age at MAS, GH excess, and PP diagnosis (3); type of fibrous dysplasia: craniofacial fibrous dysplasia (CFFD) versus polyostotic FD (4); pituitary imaging findings (5); GH, insulin-like growth factor (IGF-1), and prolactin (PRL) levels, bone age (BA) before and after treatment (6); alkaline phosphatase (ALP) levels before and after treatment (7); transabdominal pelvic (females) or testicular ultrasounds (males) (ovarian and testicular volumes were calculated using the formula volume = length × width × thickness × 0.52) (8); treatment for GH excess (9); treatment for PP; and (10) final heights and his/her parents’ heights.
Diagnosis and Treatment of MAS, GH Excess, and PP
A diagnosis of MAS was made when at least two of the following major features existed: fibrous dysplasia of bone, café au lait skin pigmentation, and hyperfunctioning endocrinopathies.
The diagnosis of GH excess was based on the presence of typical clinical manifestations (accelerated linear growth, facial dysmorphia, shoe size modification, visual defects, and/or headaches), elevated IGF-1 levels (as assessed using the IMMULITE 2000 IGF-1 analyzer; Siemens Healthcare Diagnostic Inc.), and GH nadir ≥1 µg/L following documented hyperglycemia during an oral glucose load. The IGF-1 Z-scores were calculated according to the normal values of serum IGF-1 (5th and 95th percentiles) with adjustment for age and sex (15, 23, 24), and Z-scores greater than 2.0 were considered elevated. Gigantism is defined when GH excess leads to linear growth acceleration before the end of puberty and epiphyseal closure, while acromegaly is determined when GH excess is present in individuals after epiphyseal closure. Complete remission of GH excess is defined as a normal IGF-1 level for age and sex (Z score<2.0), and GH nadir<1 µg/L after the oral glucose tolerance test (OGTT).
Gonadotropin-independent PP is defined as the onset of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys (25), with excess secretion of sex hormones (estrogens or androgens) and suppressed production of gonadotropins. The gonadotropin-releasing hormone (GnRH) stimulation test was performed to exclude gonadotropin-dependent PP. Girls typically present with painless vaginal bleeding with breast development in early childhood, and boys present with a premature increase in penile size and mild bilateral testicular enlargement (13).
Height and Skeletal Measurements
Height measurements were taken at PUMCH using a stadiometer and reported as the average of three consecutive morning values. BAs were determined using the Greulich and Pyle atlas reading method (26). The predicted adult height (PAH) was calculated according to the Bayley-Pinneau method (27). The baseline BA and height measurements were obtained before the initiation of treatment. Height and growth velocity (GV) Z-scores were determined based on normative reference data for children and adolescents in China (28, 29). Height Z-scores from the literature review were based on normative reference data for children and adolescents of the World Health Organization (30). Posttreatment data were obtained at the time of treatment discontinuation or last evaluation for those remaining on treatment. Additionally, the predicted height was calculated as the mean parental height plus 6.5 cm for male patients and minus 6.5 cm for female patients. The ALP Z-scores were based on the reported distributions of ALP levels in the Chinese population (31).
Statistical Analysis
Statistical analyses were performed using SPSS 23.0, and figures were prepared using GraphPad Prism, version 6 (GraphPad Software Inc.). The data are presented as the means (minimal, max) or medians (minimal, max) as appropriate depending on the normality of the distribution. Paired samples t-tests were performed to compare patients before and after treatment. Independent sample t-tests were performed to make comparisons between GH excess patients diagnosed with MAS with or without PP. P values <0.05 were considered statistically significant.
Results
Clinical Characteristics and Treatment Outcomes in PUMCH
Baseline Demographic and Clinical Features
Seven MAS patients from PUMCH who had been diagnosed with GH excess and PP were included in this study; five were female (71.4%), and the mean age was 5.4 years (min,max 2.1, 8.9 years) of the diagnosis of MAS (Table 1). Café au lait spots were present in five individuals (71.4%). The mean age of the individuals at the initial development of signs of PP was 4.3 years (1.0, 7.0 years). In girls, the symptoms prompting evaluation were vaginal bleeding in three subjects (patients 1, 4, and 7) and breast development in two subjects (patients 3 and 5). Pelvic ultrasound showed unilateral (patients 1, 5, and 7) or bilateral (patient 3) ovarian cysts. In boys, the diagnosis was suggested by acne, scrotum enlargement, and erection. Testicular ultrasound showed bilateral testicular enlargement in patients 2 and 6 and focal hyperechoic lesions in patient 6. Subjects showed advanced skeletal maturation [median BA over chronological age (BA/CA) ratio: 1.44; 1.39, 1.98]. Six of the seven patients had gigantism, and the mean age at diagnosis was 6.5 years (4.7, 8.9 years). The presenting sign of the patients was accelerated linear growth. Patient 4 had experienced intermittent vaginal bleeding and focal bone dysplasia since the age of 7 years, and she was diagnosed with acromegaly at the age of 27 years in routine assessment during follow-up for MAS. CFFD was present in all the patients from PUMCH, among whom three patients (42.9%) had visual field deficits, two patients (28.6%) had conductive hearing loss, and one patient (14.3%) had olfactory dysfunction. The clinical characteristics of the patients from PUMCH are listed in Table 1.
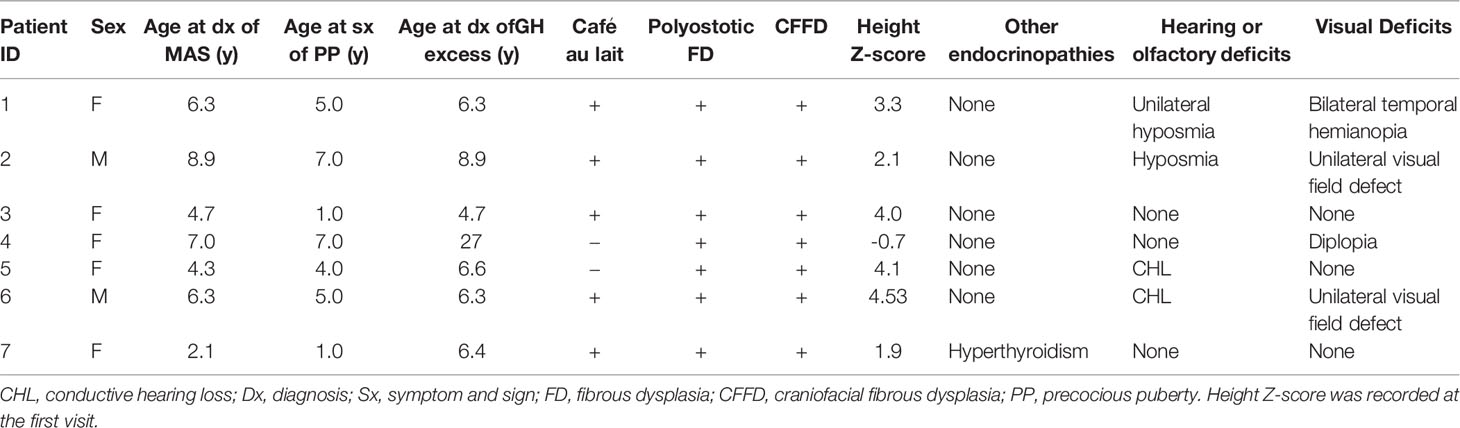
Table 1 Demographic and clinical characteristics of PUMCH patients.
Hormone Measurements and Imaging Findings
For hormone measurements, the mean random GH level, GH nadir after OGTT, and IGF-1 Z-score at the diagnosis of GH excess at PUMCH were 14.1 µg/L (4.9, 33.6), 13.2 µg/L (5.2, 35.3), and 6.4 (2.0, 10.2), respectively. Hyperprolactinemia was present in four patients (57.1%, mean 70.5 ng/ml; 47.7, 204.4), and a value greater than 200 ng/ml was only observed in patient 1. Magnetic resonance imaging (MRI) data were available for six individuals (Figure 2), revealing three macroadenomas and one microadenoma; patient 5 had multiple microadenomas, and patient 7 had normal MRI imaging findings. MRI imaging of patient 1 before and after surgery and pharmacological treatment is illustrated in Figure 3. Regarding other endocrinopathies, hyperthyroidism was present in one patient from PUMCH who had endocrinopathies accompanied by GH excess and PP in MAS (1/7; 14.3%).
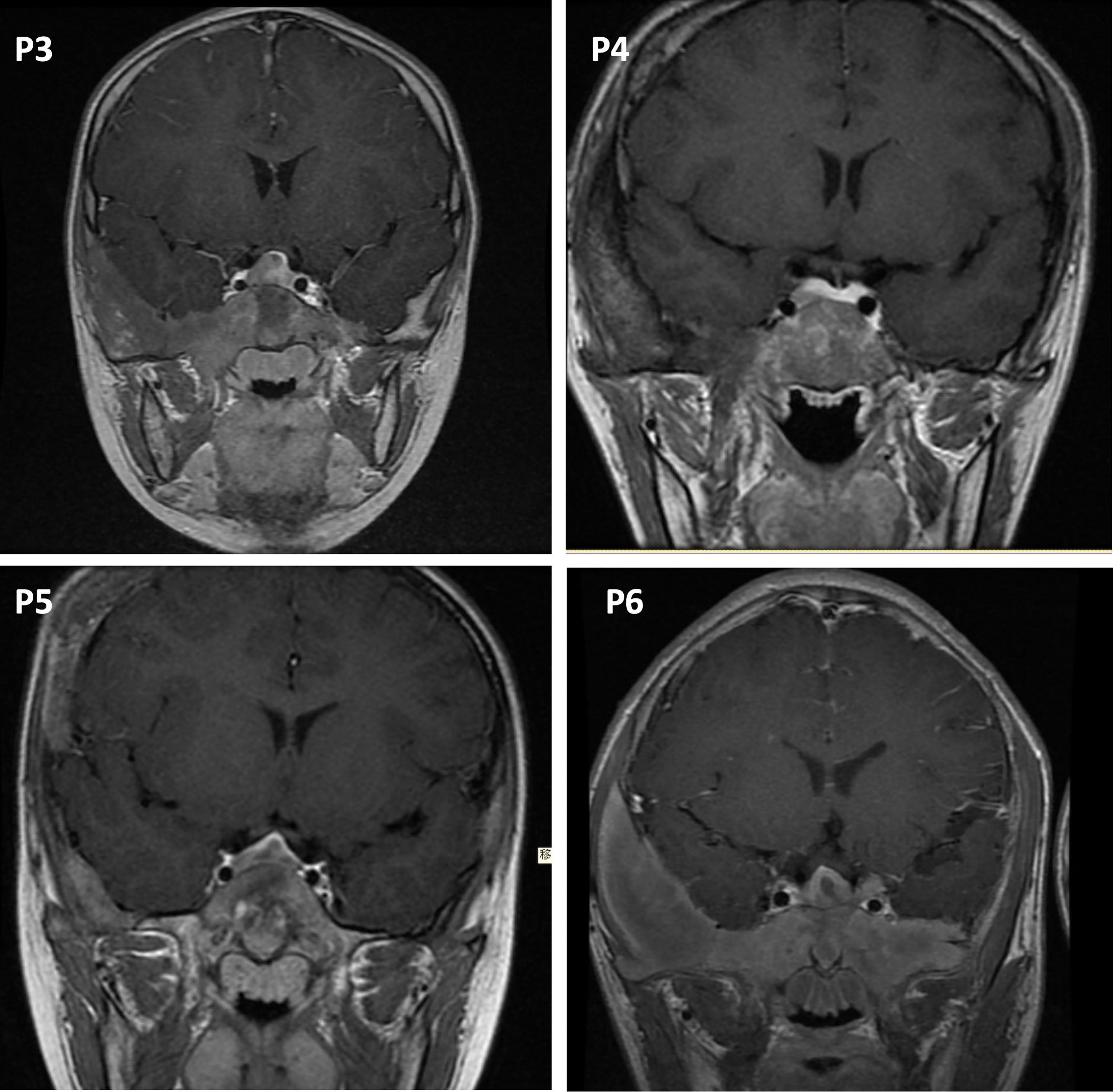
Figure 2 Pituitary MRI in the coronal view. P3, patient 3, macroadenoma. P4, patient 4, microadenoma. P5, patient 5, multiple microadenomas. P6, patient 6, macroadenoma with a cystic zone.
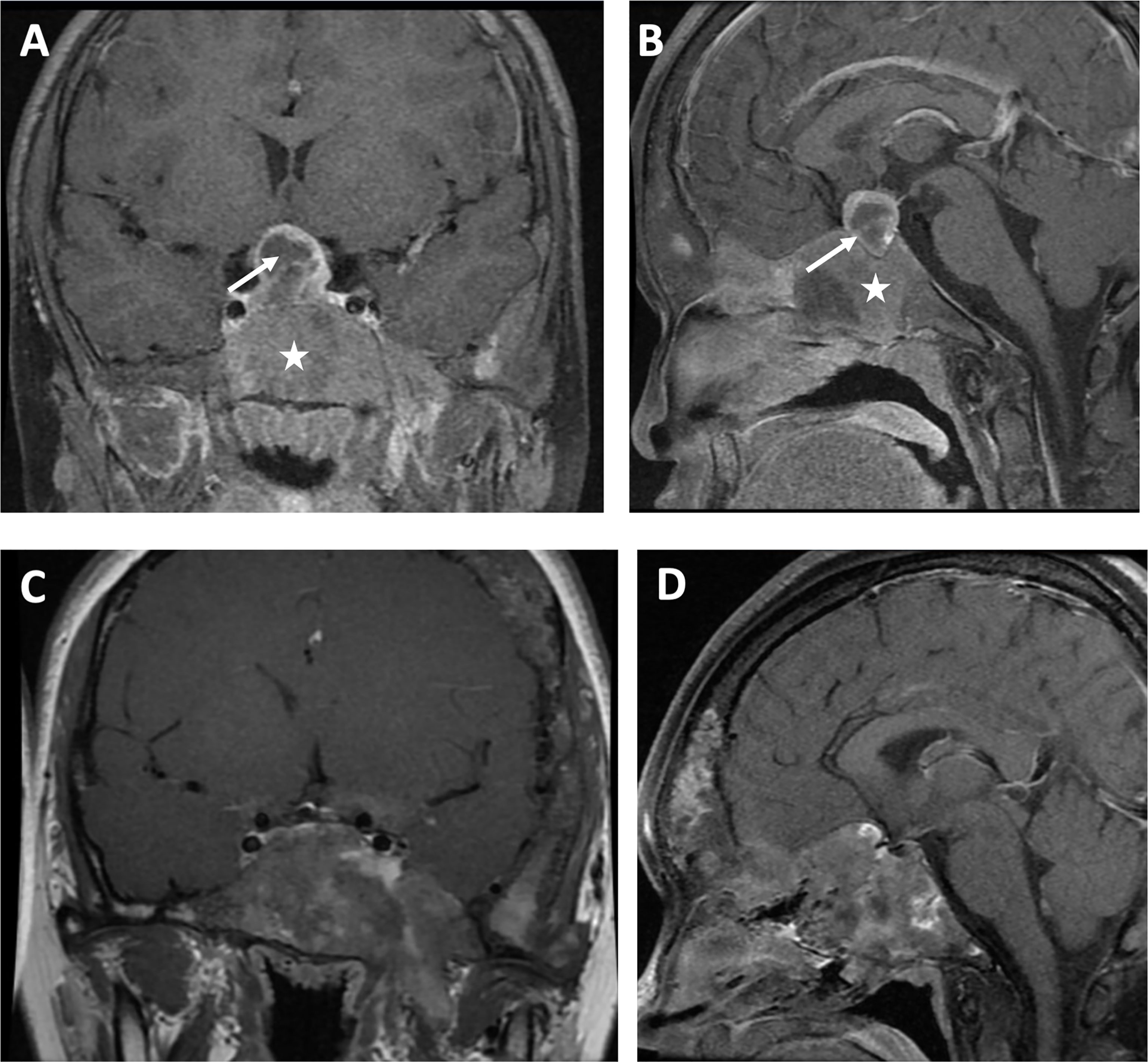
Figure 3 Visualization by MRI in the coronal view (A) and sagittal view (B) of a macroadenoma (arrow) surrounded by cranial fibrous dysplasia (star) in a patient with MAS and GH excess before surgery and pharmacologic treatment. Coronal view (C) and sagittal view (D) after surgery and pharmacologic treatment.
Treatment of PP and GH Excess
The treatment for PP varied at PUMCH (the clinical characteristics and treatment outcomes of PP at PUMCH are listed in Table 2). Letrozole was used in two of seven patients, either alone (patient 6) or in combination with medroxyprogesterone (patient 7). Patient 5 was treated with tamoxifen. Patient 2 was treated with a GnRH agonist (GnRHa) because of central PP secondary to peripheral PP. However, after 3 months of treatment, patient 2 discontinued GnRHa because of financial restriction. Patients 1 and 3 were closely monitored without medical treatment. Their symptoms were relatively stable, and their BA/CA ratios were close to the normal range.
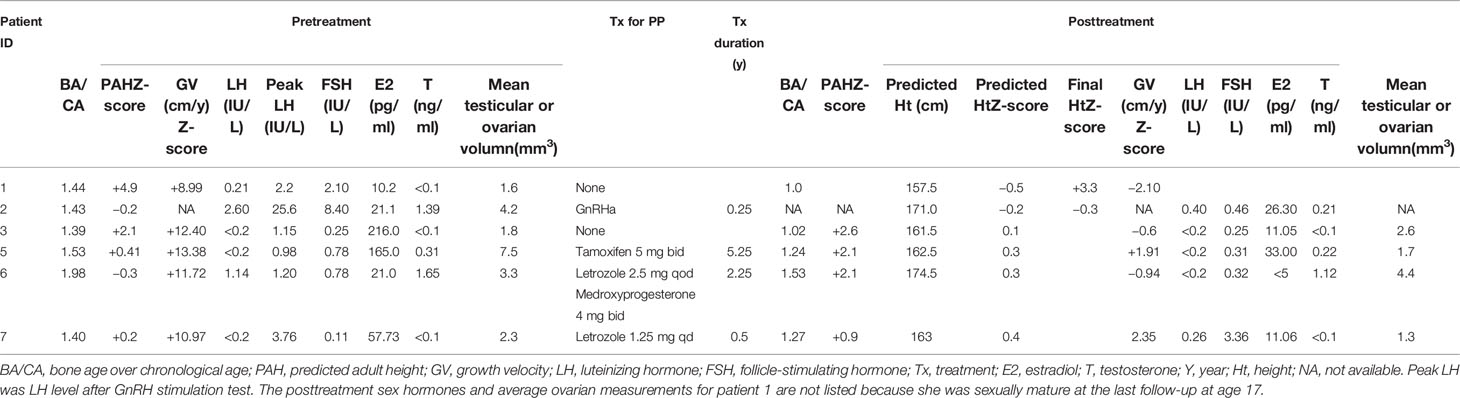
Table 2 Clinical and biochemical characteristics and treatment outcomes of patients with PP from PUMCH.
Five patients (71.4%) from PUMCH had undergone navigation-assisted transsphenoidal pituitary tumor resection, and two patients achieved complete remission (the clinical characteristics and treatment outcomes of patients with GH excess are listed in Table 3). Three patients (patients 1, 3, and 6) had partial remission after surgery and continued pharmacological treatment. Patient 1 then received treatment with dopamine agonist (DA) bromocriptine and long-acting somatostatin analog (LAR) octreotide (Sandostatin LAR, Novartis). The random GH level fell to 3.7 ng/ml, and the IGF-1 level was 601 ng/ml (IGF-1 Z-score 0.3). LAR was also effective in further lowering the IGF-1 levels after surgery in patient 3, but she had a partial response. Patient 6 was treated with bromocriptine alone after surgery, and the patient’s GH excess was partially controlled. Patient 7 was closely followed up without surgical or pharmacological treatment because her IGF-1 Z-score was slightly elevated and her GV was within the normal range. Visual and auditory complications remained stable after treatment. The ALP Z-score decreased significantly after successful treatment with GH excess and PP (P=0.04, Figure 4A).
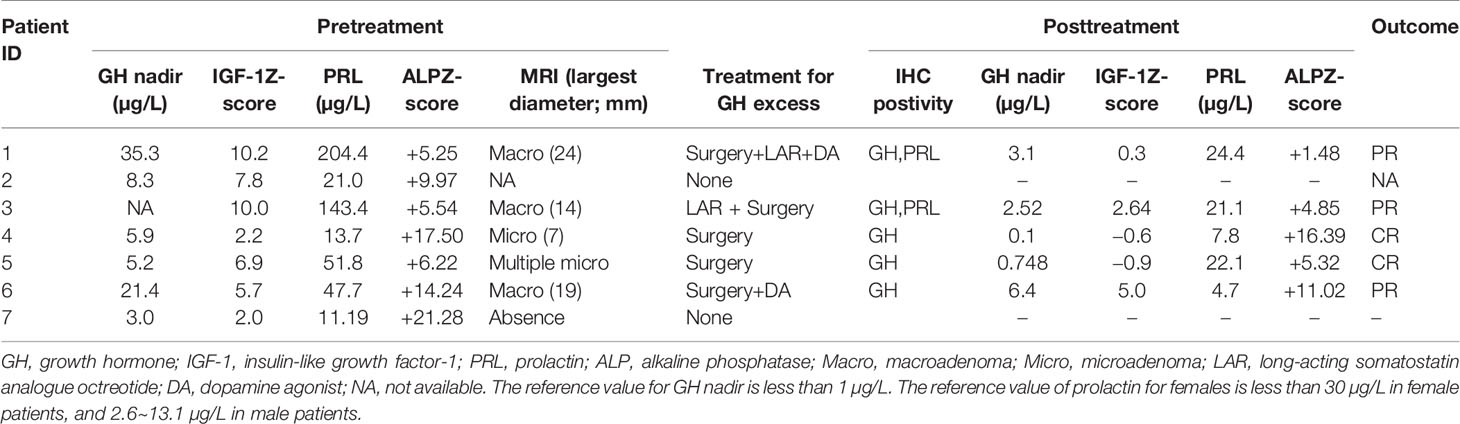
Table 3 Clinical and biochemical characteristics and treatment outcomes of patients with GH excess and PP from PUMCH.

Figure 4 ALP-Z score (A), GV Z-score (B), BA/CA ratio (C), PAH Z-score (D) before and after treatment. The post-Tx data were collected at the last consultation for patients under therapy. (E) Final height or PAH Z-scores of patients diagnosed with gigantism and PP of MAS versus their predicted adult height. (F) Final height Z-scores of patients diagnosed with gigantism and PP versus final height or PAH Z-scores of patients diagnosed with gigantism without PP. Tx, treatment; Pre-Tx, pretreatment; Post-Tx,posttreatment; ALP, alkaline phosphatase; GV, growth velocity; BA/CA, bone age over chronological age; PAH, predicted adult height. ***P < 0.001, **P < 0.01, *P < 0.05.
Growth Outcome
For patients diagnosed before the age of 16 years at PUMCH (patients 1, 3, 5, 6, and 7 were included; patient 2 was not included in the analysis because of a relatively short follow-up time), there was accelerated linear growth (mean GV Z-score: 11.5; 9.0, 13.4) and overall advanced skeletal maturation (mean BA/CA ratio: 1.53; 1.39, 1.98). After good control of GH excess and PP, the mean GV Z-score decreased from 11.49 to 0.12, which was statistically significant (P <0.001) (Figure 4B). The median BA/CA ratio also decreased significantly (1.44 to 1.24; P =0.005) (Figure 4C). The posttreatment PAH or final height Z-score (mean PAH or final height Z-score 1.8) was higher than the predicted height Z score (mean predicted height Z-score 0.1; P=0.03) (Figure 4E). The PAH Z-scores were normal (−2 to +2) in four of six patients (66.7%; patients 2, 5, 6, and 7), and patients 1 and 3 had increased PAH Z-scores. Most of the patients had PAH Z-scores that increased after treatment except for patient 1. The mean PAH Z-score increased from 1.46 to 2.2 after treatment, with no statistical significance (Figure 4D). The GV Z-score of patient 1 decreased from 8.99 to −2.1 after surgical and pharmacological treatment. Patient 4, diagnosed with acromegaly at the age of 27, had a final height of 156 cm (Z-score −0.7) and a predicted height of 159 cm (Z-score −0.3).
Six patients were diagnosed with GH excess without PP in our cohort (the clinical characteristics and final height of these patients are illustrated in Table 4), four of six had symptoms of gigantism before the age of 16 (without a diagnosis or treatment until adulthood), and two of six were diagnosed in adulthood. The final height Z-score of gigantic patients without PP (mean final height Z-score: 3.6; 2.2, 4.9) was higher than the final height Z-score or posttreatment PAH Z-score of patients codiagnosed with gigantism and PP (mean Z-score: 1.8; −0.3, 3.3; P=0.064) (Figure 4F).
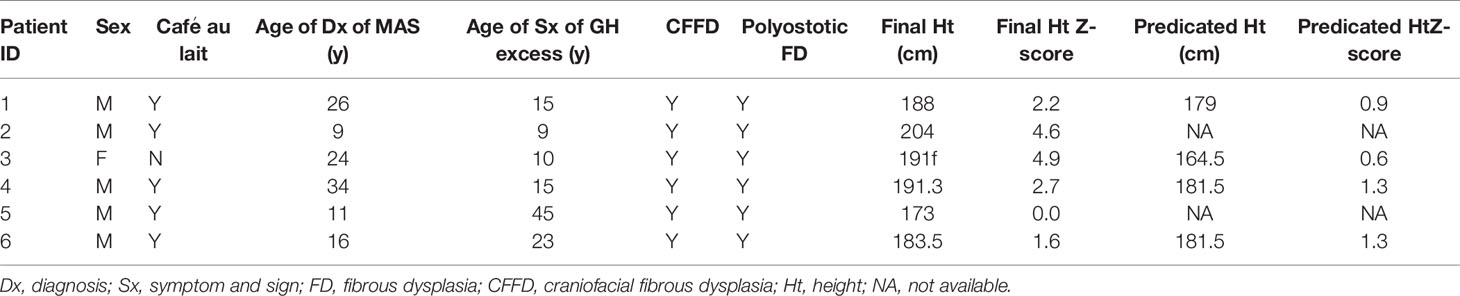
Table 4 Clinical characteristics and final height of patients diagnosed with GH excess without PP from PUMCH.
Clinical Characteristics and Treatment Outcomes From the Literature Review
The clinical characteristics, treatment, and outcomes of patients with MAS with GH excess and PP from previous case series and reports are summarized in Tables 5 and 6. Thirty-nine (25 females, 64.1%) patients were analyzed. Regarding GH excess, 13 patients were diagnosed before the age of 16 years old (mean age: 7.7 years; 4, 13), and 13 patients (50%) were diagnosed in adulthood, with an average age of 27.1 years (17.8, 38). The mean random GH level and IGF-1 Z-score were 39.0 µg/L (2.8, 290) and 5.9 (−0.2, 17.7), respectively. MRI data were available for 27 of 39 cases (69.2%), and CT and X-ray scans were performed for eight patients, revealing 14 macroadenomas (51.9%) and 7 (25.9%) microadenomas. For craniofacial FD complications, 11 patients had various degrees of visual impairment, and five patients had olfactory impairment. Five patients received treatment for PP among the 19 patients with available data. Aromatase inhibitors, tamoxifen, GnRHas, testolactone, anastrozole, and medroxyprogesterone acetate were administered (cases 3, 15, 16, 27, and 34). Syptoms of PP were well controlled in cases 15 and 16, and partially controlled in case 34. The response to treatment of PP was not mentioned in case 3 or 27. Eight (36.4%) out of 22 patients for whom data were available achieved complete remission, 10 (45.5%) individuals had partial remission, GH excess was not controlled in 4 (18.2%) individuals, and 1 (4.5%) patient died after surgery from postoperative hemorrhage. Among the patients with complete disease resolution, one patient was cured by surgery; one patient was treated with a DA alone; one patient was cured by an LAR and DA; and the remaining five patients received combination treatment with either pegvisomant, an LAR and DA or pegvisomant and fractioned radiotherapy.
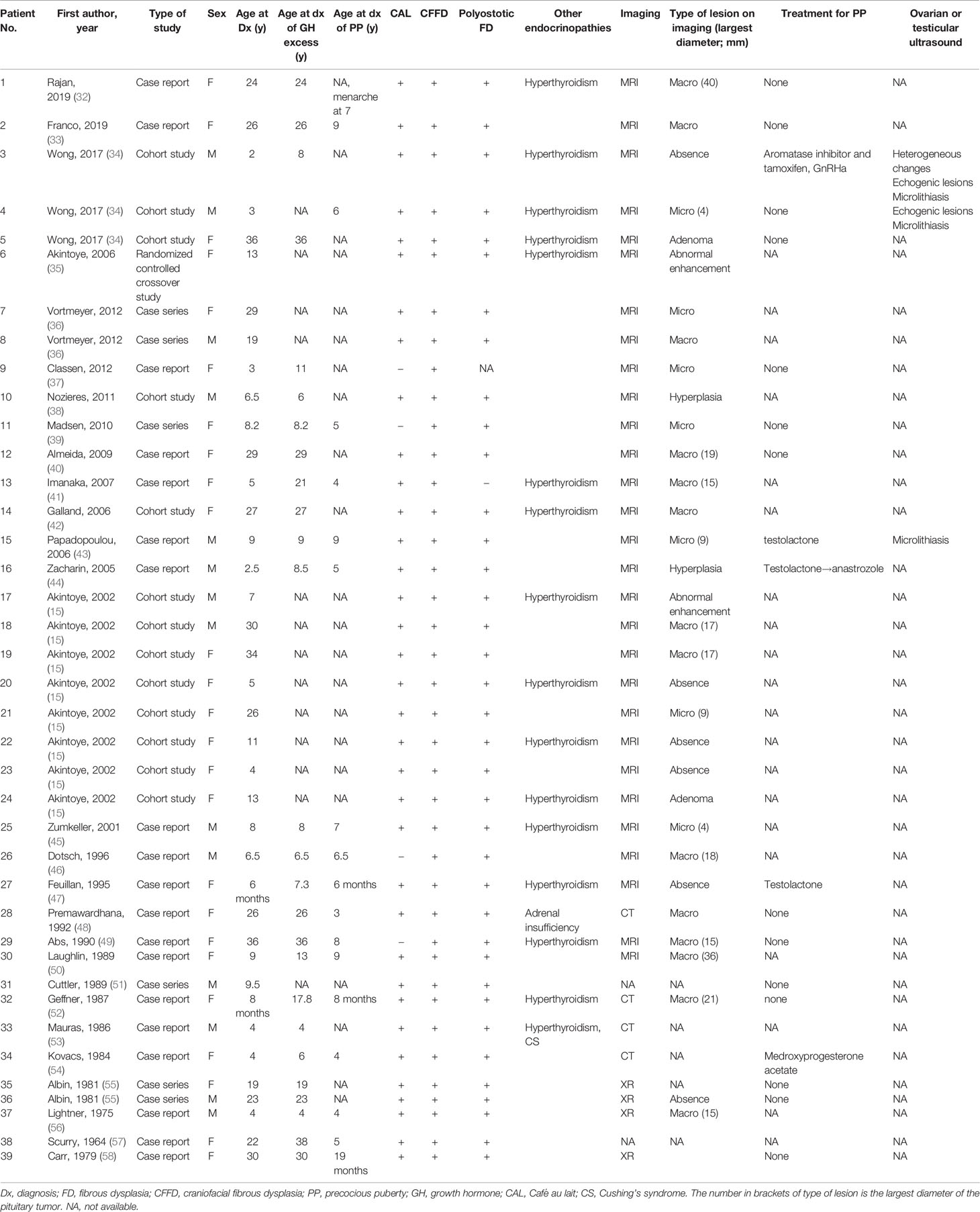
Table 5 Clinical characteristics of patients from the literature review.
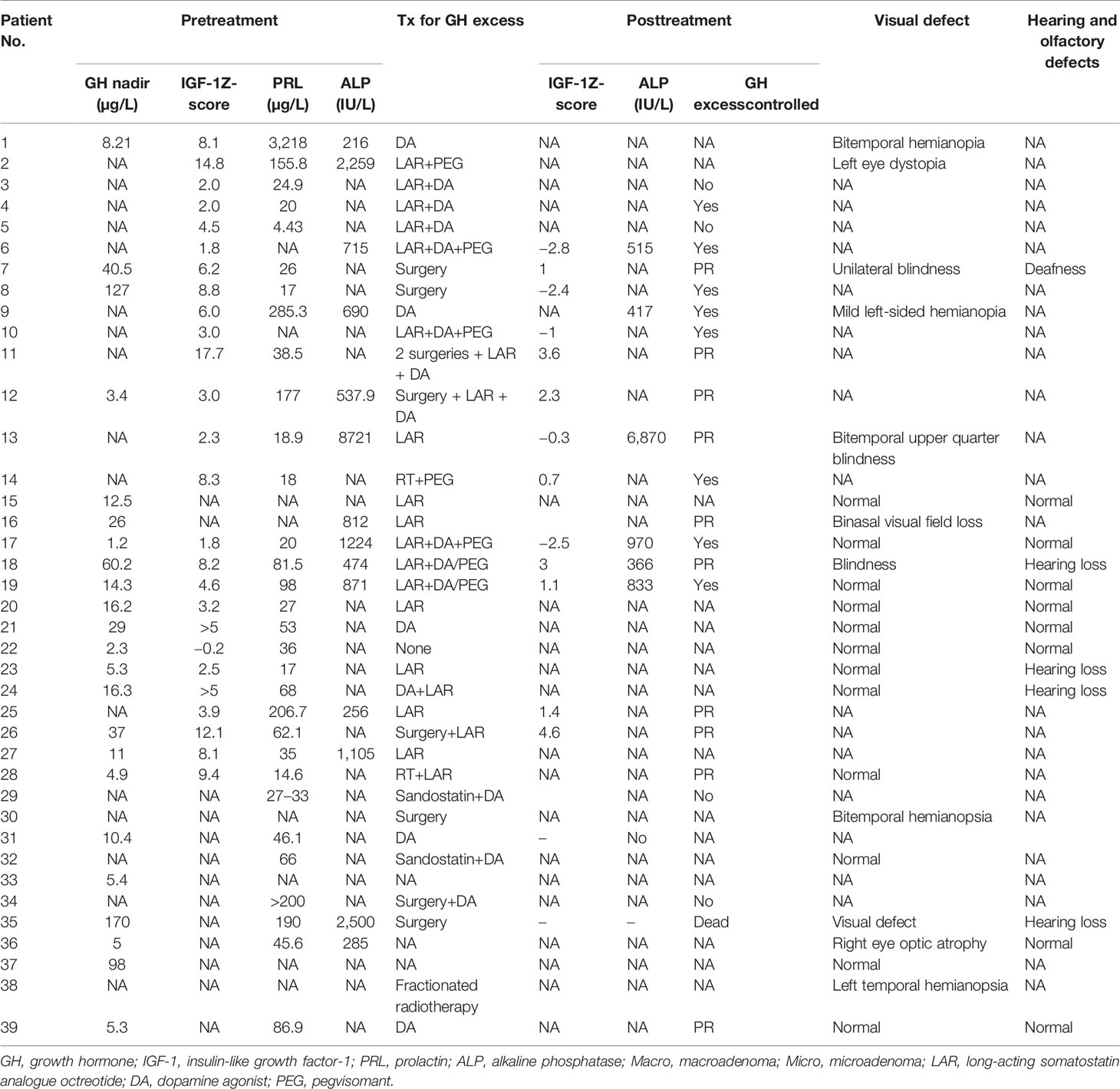
Table 6 Biochemical characteristics and treatment outcomes of patients with GH excess from the literature review.
In the literature review, for patients who had been diagnosed with gigantism, for whom the final height was thoroughly documented, the final heights were 180 cm, 183 cm, and 169 cm, and the height Z-scores were 2.2, 2.6, and −0.9 for cases 9, 11, and 15, respectively. Additionally, the predicted heights were 172.5 and 175 cm for cases 11 and 15, respectively (the parental height was not mentioned in case 9). For patients who had been diagnosed with acromegaly, three of six patients (cases 1, 12, 14, 32, 38, and 39) had short stature (139 cm, Z-score= −3.2 in case 14; 147.5 cm, Z-score= −2.1 in case 32; and 140 cm, Z-score= −3.1 in case 39).
Discussion
This study is the first case series to illustrate the clinical manifestations, treatment, and predicted or final height and the status of FD of combined GH excess and PP in the context of MAS patients. We found that the proportion of women with GH excess and PP of MAS was higher, whereas GH excess in MAS (14, 16) and “classic” acromegaly (59) affected almost equal proportions of women and men. This difference is likely due to PP affecting more female patients than male patients with MAS (8). In this case series, the number of MAS patients from PUMCH with gigantism was higher than the number with acromegaly, while approximately half of the literature review patients had gigantism. The average age at the acromegaly diagnosis in patients with MAS and PP in the literature review was 27.1 years, which is slightly lower than 30.1 years, the mean age of patients with acromegaly and MAS reported in a previous study (9); however, both of these ages were much younger than the median age of 40~50 years observed in “classic” acromegaly (60–62).
The data from PUMCH demonstrated that patients with a diagnosis of gigantism and PP have growth rate acceleration and skeletal maturation, and good control of both conditions leads to normalized GV and stabilized bone age. However, the data from PUMCH and a literature review showed that these patients had unaffected final heights. For those who were diagnosed with gigantism, the PAH Z-scores or final height Z-scores were higher than the predicted adult height Z-score. For patients diagnosed with acromegaly, one patient from PUMCH and approximately half of the patients reviewed in the literature had a short stature. Previous findings (15, 16) indicated that patients with a history of gigantism and PP achieve normal adult height despite early epiphyseal fusion, while those diagnosed with PP and acromegaly can end up with a significantly shorter adult height (63). The growth outcome in our cohort was that the height of MAS patients with PP and gigantism was higher than their predicted heights. This is probably because, in our cohort, PP was well controlled, while gigantism was partially relieved in some patients. This phenomenon indicates that sex hormones and GH have cumulative effects on the growth plate, and the time point at which GH excess appears is related to the final adult height.
The diagnosis and treatment of the coexistence of GH excess and PP is challenging. Under these circumstances, BA can better indicate the degree of sexual maturity. When height and BA are not parallel, we should consider comorbid gigantism. Laboratory screening for GH excess helps confirm the diagnosis. Good control of GH excess and PP is essential to reduce the growth rate and stabilize bone maturity.
Although patients with gigantism and PP in both our cohort and the literature review have unaffected final heights, early detection and treatment are still important because uncontrolled gigantism worsens CFFD. Bone turnover is increased in patients with GH excess, and they have increased levels of markers of bone formation and resorption (64). These biomarkers, particularly ALP, are usually related to the scope and intensity of bone involvement in MAS (65). Previous studies have already shown that uncontrolled gigantism is associated with the aggravation of CFFD and an increased risk of optic neuropathy (16–18) and hearing loss (19). Studies have also shown that early treatment during the pediatric period decreases the risk of these morbidities (15, 18) and results in lower serum ALP levels (14). In the present study, CFFD lesions were present in all patients from PUMCH, and polyostotic FD lesions were present in most patients, suggesting that they may be correlated with excess GH. Additionally, after effective treatment of GH excess, the visual field, hearing, and olfactory functions remained stable, and the ALP Z-score decreased after treatment. These results indicate that early successful treatment of GH excess might help ameliorate FD.
In addition to aggravating FD, GH excess is also related to impaired glucose tolerance, hypertension, cardiomyopathy, and an increased risk of tumors (22). Therefore, choosing the proper treatment is critical. Conventional treatments for GH excess include surgery, medications (somatostatin receptor ligands, Das, and the GH receptor antagonist pegvisomant), and radiotherapy. According to previous studies, medical therapy is considered the first-line treatment because pituitary surgery is technically difficult in patients with MAS because of the massive thickening of the skull base with FD (16, 35). However, technological progress has been made in this field in recent years, and we suggest that transphenoidal tumor resection under the guidance of neuronavigation might be feasible. An earlier cohort study from PUMCH revealed that transsphenoidal complete tumor excision with neuronavigational guidance is effective in patients with MAS with GH excess. In the present study, five of seven patients from PUMCH had undergone navigation-assisted transsphenoidal pituitary surgery, and two out of the five patients achieved full recovery. The remaining three patients had a partial response and then continued to receive long-acting somatostatin and bromocriptine. None of our patients had hypopituitarism after surgery. Therefore, from the experience of our center, navigation-assisted transsphenoidal pituitary surgery could be an effective and relatively safe choice for patients with MAS diagnosed with GH excess and PP. Radiotherapy is generally not advised because of the risk of fatal malignant transformation of CFFD in bones (66–68).
Uncontrolled PP can also cause psychological problems in children, and long-term exposure to high levels of estrogen elevates the risk of breast cancer and endometrial cancer. It has already been demonstrated that letrozole, a 3rd-generation aromatase inhibitor, and tamoxifen, an estrogen receptor modulator, are the most evidenced treatments (69). Medroxyprogesterone is also effective for controlling vaginal bleeding and breast development in the short term (13, 70). When secondary central PP develops, GnRHa is beneficial for blocking the hypothalamus-pituitary-gonadal (HPG) axis (71). In this study, the treatments for PP ranged from observation only to combinations of two medications. Letrozole and tamoxifen were the most frequently used. One patient developed secondary central PP and was treated with GnRHa for 3 months. Medroxyprogesterone was also used in combination with letrozole in one 6-year-old boy. During follow-up, we need to focus on changes in BA or the BA/CA ratio, not only changes in the growth rate, because the decline in the growth rate may only benefit from controlling excessive GH.
The co-occurrence of GH excess and PP in MAS is extremely rare. This study is the first to analyze the clinical features of this condition, and the diagnosis and treatment of both conditions pose considerable challenges. First, the diagnosis of gigantism may be overlooked because PP can also cause accelerated linear growth and atypical facial dysmorphia due to CFFD. Additionally, when evaluating the effectiveness of PP treatment in these patients, attention should be given to not only changes in GV but also changes in BA. A comprehensive consideration of GV, BA, and IGF-1 levels may lead to an accurate diagnosis and smooth follow-up.
However, this study still had several limitations. First, the sample size from PUMCH was small because the prevalence of this condition was extremely low, and patient 2 was lost of follow-up because of financial problem. We added 39 cases from the literature as supplementation. Additionally, when analyzing the association of GH excess with FD, we did not include a control group, and the severity of bone lesions and incidence of optic, hearing, and olfactory complications could not be compared with those in patients without GH excess.
Conclusion
MAS with GH excess and PP is rare, and these patients present with increased growth velocity and advanced bone age. Early diagnosis and proper treatments are essential. The predicated or final height of patients with gigantism is not impaired, while the final height of patients with acromegaly is shorter.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
This study is in compliance with the Helsinki Declaration, and the parents signed the informed consent form, with the approval of the Ethics Committee of Peking Union Medical College Hospital (Reference number: JS-1663). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
XZ, LD, and HZ participated in the design of the study. XZ carried out the study and collected important background information. LW and HYa carried out literature research. LL, SC, and HP provided assistance for statistical analysis. YY, BX, KD, and RW provided assistance for surgical clinical data collection. FF and HYo helped with radiologic data collection. ZL participated in pathology data collection. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Key Research and Development Program of China (No. 2016YFC0901501) and CAMS Innovation Fund for Medical Science (CAMS-2016-I2M-1-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boyce AM, Collins MT. Fibrous Dysplasia/McCune-Albright Syndrome: A Rare, Mosaic Disease of Gα s Activation. Endocr Rev (2020) 41(2):345–70. doi: 10.1210/endrev/bnz011
2. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating Mutations of the Stimulatory G Protein in the McCune–Albright Syndrome. New Engl J Med (1991) 325(24):1688–95. doi: 10.1056/NEJM199112123252403
3. Albright F, Butler AM, Hampton AO, Smith P. Syndrome Characterized by Osteitis Fibrosa Disseminata, Areas of Pigmentation and Endocrine Dysfunction, With Precocious Puberty in Females: Report of Five Cases. New Engl J Med (1937) 216(17):727–46. doi: 10.1056/NEJM193704292161701
4. Boyce AM, Florenzano P, de Castro LF, Collins MT. Fibrous Dysplasia/Mccune-Albright Syndrome. University of Washington, Seattle: GeneReviews (2019).
5. Tufano M, Ciofi D, Amendolea A, Stagi S. Auxological and Endocrinological Features in Children With McCune Albright Syndrome: A Review. Front Endocrinol (Lausanne) (2020) 11:522. doi: 10.3389/fendo.2020.00522
6. Holbrook L, Brady R. McCune Albright Syndrome. StatPearls. Treasure Island (FL: StatPearls Publishing LLC (2021). StatPearls Publishing Copyright © 2021.
7. Dumitrescu CE, Collins MT. McCune-Albright Syndrome. Orphanet J Rare Dis (2008) 3(1):12. doi: 10.1186/1750-1172-3-12
8. Collins MT, Singer FR, Eugster E. McCune-Albright Syndrome and the Extraskeletal Manifestations of Fibrous Dysplasia. Orphanet J rare Dis (2012) 7(Suppl 1):S4. BioMed Central. doi: 10.1186/1750-1172-7-S1-S4
9. Teilmann G, Pedersen CB, Jensen TK, Skakkebaek NE, Juul A. Prevalence and Incidence of Precocious Pubertal Development in Denmark: An Epidemiologic Study Based on National Registries. Pediatrics (2005) 116(6):1323–8. doi: 10.1542/peds.2005-0012
10. Schmidt H, Kiess W. Secondary Central Precocious Puberty in a Girl With McCune-Albright Syndrome Responds to Treatment With GnRH Analogue. J Pediatr Endocrinol Metab (1998) 11(1):77–81. doi: 10.1515/jpem.1998.11.1.77
11. Pasquino AM, Tebaldi L, Cives C, Maciocci M, Boscherini B. Precocious Puberty in the McCune-Albright Syndrome. Progression From Gonadotrophin-Independent to Gonadotrophin-Dependent Puberty in a Girl. Acta Paediatr Scand (1987) 76(5):841–3. doi: 10.1111/j.1651-2227.1987.tb10576.x
12. Corica D, Aversa T, Pepe G, De Luca F, Wasniewska M. Peculiarities of Precocious Puberty in Boys and Girls With McCune-Albright Syndrome. Front Endocrinol (Lausanne) (2018) 9:337. doi: 10.3389/fendo.2018.00337
13. Schoelwer M, Eugster EA. Treatment of Peripheral Precocious Puberty. Puberty From Bench to Clinic Vol. 29. Basel: Karger Publishers (2016) p. 230–9.
14. Yao Y, Liu Y, Wang L, Deng K, Yang H, Lu L, et al. Clinical Characteristics and Management of Growth Hormone Excess in Patients With McCune-Albright Syndrome. Eur J Endocrinol (2017) 176(3):295–303. doi: 10.1530/EJE-16-0715
15. Akintoye SO, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, et al. Characterization of Gsp-Mediated Growth Hormone Excess in the Context of McCune-Albright Syndrome. J Clin Endocrinol Metab (2002) 87(11):5104–12. doi: 10.1210/jc.2001-012022
16. Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright Syndrome. J Clin Endocrinol Metab (2014) 99(6):1955–69. doi: 10.1210/jc.2013-3826
17. Boyce AM, Glover M, Kelly MH, Brillante BA, Butman JA, Fitzgibbon EJ, et al. Optic Neuropathy in McCune-Albright Syndrome: Effects of Early Diagnosis and Treatment of Growth Hormone Excess. J Clin Endocrinol Metab (2013) 98(1):E126–34. doi: 10.1210/jc.2012-2111
18. Tessaris D, Boyce AM, Zacharin M, Matarazzo P, Lala R, De Sanctis L, et al. Growth Hormone—Insulin-Like Growth Factor 1 Axis Hyperactivity on Bone Fibrous Dysplasia in McCune-Albright Syndrome. Clin Endocrinol (2018) 89(1):56–64. doi: 10.1111/cen.13722
19. Boyce AM, Brewer C, DeKlotz TR, Zalewski CK, King KA, Collins MT, et al. Association of Hearing Loss and Otologic Outcomes With Fibrous Dysplasia. JAMA Otolaryngol–Head Neck Surg (2018) 144(2):102–7. doi: 10.1001/jamaoto.2017.2407
20. Burke AB, Collins MT, Boyce AM. Fibrous Dysplasia of Bone: Craniofacial and Dental Implications. Oral Dis (2017) 23(6):697–708. doi: 10.1111/odi.12563
21. Özcan İ, Ünsal G, Koca RB, Orhan K. Craniofacial Fibrous Dysplasia Involvements of Mccune-Albright Syndrome: A Review With an Additional Case. Curr Med Imaging (2020) 17(7):864–70. doi: 10.2174/1573405616666201209102418
22. Raborn LN, Pan KS, FitzGibbon EJ, Collins MT, Boyce AM. Optic Disc Edema in Fibrous Dysplasia/McCune-Albright Syndrome: Prevalence, Etiologies, and Clinical Implications. Bone (2021) 143:115661. doi: 10.1016/j.bone.2020.115661
23. Zhu H, Xu Y, Gong F, Shan G, Yang H, Xu K, et al. Reference Ranges for Serum Insulin-Like Growth Factor I (IGF-I) in Healthy Chinese Adults. PloS One (2017) 12(10):e0185561. doi: 10.1371/journal.pone.0185561
24. Xu S, Gu X, Pan H, Zhu H, Gong F, Li Y, et al. Reference Ranges for Serum IGF-1 and IGFBP-3 Levels in Chinese Children During Childhood and Adolescence. Endocr J (2010) 57(3):221–8. doi: 10.1507/endocrj.K09E-200
25. Adashi EY, Rock JA, Rosenwaks Z. Reproductive Endocrinology, Surgery, and Technology. Philadelphia: Lippincott-Raven (1996).
26. Greulich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and Wrist. Redwood City: Stanford University Press (1959).
27. Bayley N, Pinneau SR. Tables for Predicting Adult Height From Skeletal Age. Revised for Use With the Greulich-Pyle Hand Standards. J Pediatr (1952) 40(4):423–41. doi: 10.1016/S0022-3476(52)80205-7
28. Li H, Ji C, Zong X, Zhang Y. Height and Weight Standardized Growth Charts for Chinese Children and Adolescents Aged 0 to 18 Years. Zhonghua er ke za zhi= Chin J Pediatr (2009) 47(7):487–92. doi: 10.3760/cma.j.issn.0578-1310.2009.07.003
29. Zong X, Li H. General Growth Patterns and Simple Mathematic Models of Height and Weight of Chinese Children. Zhonghua er ke za zhi= Chin J Pediatr (2009) 47(5):371–5. doi: 10.3760/cma.j.issn.0578-1310.2009.05.017
30. de Onis M, Onyango AW, Borghi E, Siyam A, Nishida C, Siekmann J. Development of a WHO Growth Reference for School-Aged Children and Adolescents. Bull World Health Organ (2007) 85(9):660–7. doi: 10.2471/BLT.07.043497
31. Lu Q, Jia Z. Reference Values of Serum Alkaline Phosphatase for Chinese Children and Adolescents Aged 0 to 18 Years. Clinics Lab Med (2009) 6:1069–70. doi: 10.3969/j.issn.1672-9455.2009.13.026
32. Rajan R, Cherian KE, Asha HS, Paul TV. McCune Albright Syndrome: An Endocrine Medley. BMJ Case Rep CP (2019) 12(7):e229141. doi: 10.1136/bcr-2018-229141
33. Franco JMPL, Rêgo RD, Gomes GMF, da Silveira Santos DF, Santos ES. Surgical Treatment of Polyostotic Craniomaxillofacial Fibrous Dysplasia Associated With Acromegalia: The McCune–Albright Syndrome. J Craniofacial Surg (2019) 30(6):1806–8. doi: 10.1097/SCS.0000000000005498
34. Wong SC, Zacharin M. Long-Term Health Outcomes of Adults With McCune-Albright Syndrome. Clin Endocrinol (2017) 87(5):627–34. doi: 10.1111/cen.13419
35. Akintoye SO, Kelly MH, Brillante B, Cherman N, Turner S, Butman JA, et al. Pegvisomant for the Treatment of Gsp-Mediated Growth Hormone Excess in Patients With McCune-Albright Syndrome. J Clin Endocrinol Metab (2006) 91(8):2960–6. doi: 10.1210/jc.2005-2661
36. Vortmeyer AO, Gläsker S, Mehta GU, Abu-Asab MS, Smith JH, Zhuang Z, et al. Somatic GNAS Mutation Causes Widespread and Diffuse Pituitary Disease in Acromegalic Patients With McCune-Albright Syndrome. J Clin Endocrinol Metab (2012) 97(7):2404–13. doi: 10.1210/jc.2012-1274
37. Classen CF, Mix M, Kyank U, Hauenstein C, Haffner D. Pamidronic Acid and Cabergoline as Effective Long-Term Therapy in a 12-Year-Old Girl With Extended Facial Polyostotic Fibrous Dysplasia, Prolactinoma and Acromegaly in McCune-Albright Syndrome: A Case Report. J Med Case Rep (2012) 6(1):32. doi: 10.1186/1752-1947-6-32
38. Nozières C, Berlier P, Dupuis C, Raynaud-Ravni C, Morel Y, Chazot FB, et al. Sporadic and Genetic Forms of Paediatric Somatotropinoma: A Retrospective Analysis of Seven Cases and a Review of the Literature. Orphanet J Rare Dis (2011) 6(1):67. doi: 10.1186/1750-1172-6-67
39. Madsen H, Borges MT, Kerr JM, Lillehei KO, Kleinschmidt-Demasters B. McCune–Albright Syndrome: Surgical and Therapeutic Challenges in GH-Secreting Pituitary Adenomas. J Neuro-Oncol (2011) 104(1):215–24. doi: 10.1007/s11060-010-0461-9
40. Almeida JPC, Albuquerque LAF, Ferraz CL, Mota Í, Gondim J, Ferraz TM. McCune-Albright Syndrome and Acromegaly: Hormonal Control With Use of Cabergoline and Long-Acting Somatostatin-Case Report. Arquivos Brasileiros Endocrinologia Metabologia (2009) 53(1):102–6. doi: 10.1590/S0004-27302009000100015
41. Imanaka M, Iida K, Nishizawa H, Fukuoka H, Takeno R, Takahashi K, et al. McCune-Albright Syndrome With Acromegaly and Fibrous Dysplasia Associated With the GNAS Gene Mutation Identified by Sensitive PNA-Clamping Method. Internal Med (2007) 46(18):1577–83. doi: 10.2169/internalmedicine.46.0048
42. Galland F, Kamenicky P, Affres H, Reznik Y, Pontvert D, Le Bouc Y, et al. McCune-Albright Syndrome and Acromegaly: Effects of Hypothalamopituitary Radiotherapy and/or Pegvisomant in Somatostatin Analog-Resistant Patients. J Clin Endocrinol Metab (2006) 91(12):4957–61. doi: 10.1210/jc.2006-0561
43. Papadopoulou M, Doula S, Kitsios K, Kaltsas T, Kosta K. A Boy With McCune-Albright Syndrome Associated With GH Secreting Pituitary Microadenoma. Clinical Findings and Response to Treatment. Hormones-Athens- (2006) 5(3):205. doi: 10.14310/horm.2002.11186
44. Zacharin M. Paediatric Management of Endocrine Complications in McCune-Albright Syndrome. J Pediatr Endocrinol Metab (2005) 18(1):33–42. doi: 10.1515/JPEM.2005.18.1.33
45. Zumkeller W, Jassoy A, Lebek S, Nagel M. Clinical, Endocrinological and Radiological Features in a Child With McCune-Albright Syndrome and Pituitary Adenoma. J Pediatr Endocrinol Metab (2001) 14(5):553–60. doi: 10.1515/JPEM.2001.14.5.553
46. Dötsch J, Kiess W, Hänze J, Repp R, Lüdecke D, Blum W, et al. Gs Alpha Mutation at Codon 201 in Pituitary Adenoma Causing Gigantism in a 6-Year-Old Boy With McCune-Albright Syndrome. J Clin Endocrinol Metab (1996) 81(11):3839–42. doi: 10.1210/jcem.81.11.8923825
47. Feuillan PP, Jones J, Ross JL. Growth Hormone Hypersecretion in a Girl With McCune-Albright Syndrome: Comparison With Controls and Response to a Dose of Long-Acting Somatostatin Analog. J Clin Endocrinol Metab (1995) 80(4):1357–60. doi: 10.1210/jcem.80.4.7714111
48. Premawardhana L, Vora J, Mills R, Scanion M. Acromegaly and Its Treatment in the McCune-Albright Syndrome. Clin Endocrinol (1992) 36(6):605–8. doi: 10.1111/j.1365-2265.1992.tb02272.x
49. Abs R, Beckers A, Van de Vyver F, De Schepper A, Stevenaert A, Hennen G. Acromegaly, Multinodular Goiter and Silent Polyostotic Fibrous Dysplasia, A Variant of the McCune—Albright Syndrome. J Endocrinological Invest (1990) 13(8):671–5. doi: 10.1007/BF03349592
50. O'Laughlin RL, Selinger SE, Moriarty PE. Pituitary Adenoma in McCune-Albright Syndrome: MR Demonstration. J Comput Assisted Tomography (1989) 13(4):685–8. doi: 10.1097/00004728-198907000-00026
51. Cuttler L, Jackson JA, Uz-Zafar MS, Levitsky LL, Mellinger RC, Frohman LA. Hypersecretion of Growth Hormone and Prolactin in McCune-Albright Syndrome. J Clin Endocrinol Metab (1989) 68(6):1148–54. doi: 10.1210/jcem-68-6-1148
52. Geffner ME, Nagel RA, Dietrich RB, Kaplan SA. Treatment of Acromegaly With a Somatostatin Analog in a Patient With McCune-Albright Syndrome. J Pediatr (1987) 111(5):740–3. doi: 10.1016/S0022-3476(87)80258-5
53. Mauras N, Blizzard RM. The McCune-Albright Syndrome. Eur J Endocrinol (1986) 113(4_Suppl):S207–17. doi: 10.1530/acta.0.112S207
54. Kovacs K, Horvath E, Thorner MO, Rogol AD. Mammosomatotroph Hyperplasia Associated With Acromegaly and Hyperprolactinemia in a Patient With the McCune-Albright Syndrome. Virchows Archiv A (1984) 403(1):77–86. doi: 10.1007/BF00689340
55. Albin J, Wu R. Abnormal Hypothalamic-Pituitary Function in Polyostotic Fibrous Dysplasia. Clin Endocrinol (1981) 14(5):435–43. doi: 10.1111/j.1365-2265.1981.tb00632.x
56. Lightner ES, Penny R, Frasier SD. Growth Hormone Excess and Sexual Precocity in Polyostotic Fibrous Dysplasia (McCune-Albright Syndrome): Evidence for Abnormal Hypothalamic Function. J Pediatr (1975) 87(6):922–7. doi: 10.1016/S0022-3476(75)80906-1
57. Scurry MT, Bicknell JM, Fajans SS. Polyostotic Fibrous Dysplasia and Acromegaly. Arch Internal Med (1964) 114(1):40–5. doi: 10.1001/archinte.1964.03860070086008
58. Carr D, Mathie I, Manners A, Colman C. Hyperprolactinaemia In A Patient With The Mccune-Albright Syndrome. BJOG: Int J Obstetrics Gynaecol (1979) 86(4):330–1. doi: 10.1111/j.1471-0528.1979.tb11264.x
59. Colao A, Grasso LF, Giustina A, Melmed S, Chanson P, Pereira AM, et al. Acromegaly. Nat Rev Dis Primers (2019) 5(1):1–17. doi: 10.1038/s41572-019-0071-6
60. Gruppetta M, Mercieca C, Vassallo J. Prevalence and Incidence of Pituitary Adenomas: A Population Based Study in Malta. Pituitary (2013) 16(4):545–53. doi: 10.1007/s11102-012-0454-0
61. Dal J, Feldt-Rasmussen U, Andersen M, Kristensen LO, Laurberg P, Pedersen L, et al. Acromegaly Incidence, Prevalence, Complications and Long-Term Prognosis: A Nationwide Cohort Study. Eur J Endocrinol (2016) 175(3):181–90. doi: 10.1530/EJE-16-0117
62. Fernandez A, Karavitaki N, Wass JA. Prevalence of Pituitary Adenomas: A Community-Based, Cross-Sectional Study in Banbury (Oxfordshire, Uk). Clin Endocrinol (2010) 72(3):377–82. doi: 10.1111/j.1365-2265.2009.03667.x
63. Haddad N, Eugster E. An Update on the Treatment of Precocious Puberty in McCune-Albright Syndrome and Testotoxicosis. J Pediatr Endocrinol Metab (2007) 20(6):653–62. doi: 10.1515/JPEM.2007.20.6.653
64. Scillitani A, Chiodini I, Carnevale V, Giannatempo GM, Frusciante V, Villella M, et al. Skeletal Involvement in Female Acromegalic Subjects: The Effects of Growth Hormone Excess in Amenorrheal and Menstruating Patients. J Bone Miner Res (1997) 12(10):1729–36. doi: 10.1359/jbmr.1997.12.10.1729
65. Collins MT, Kushner H, Reynolds JC, Chebli C, Kelly MH, Gupta A, et al. An Instrument to Measure Skeletal Burden and Predict Functional Outcome in Fibrous Dysplasia of Bone. J Bone Miner Res (2005) 20(2):219–26. doi: 10.1359/JBMR.041111
66. Ruggieri P, Sim FH, Bond JR, Krishnan Unni K. Malignancies in Fibrous Dysplasia. Cancer (1994) 73(5):1411–24. doi: 10.1002/1097-0142(19940301)73:5<1411::AID-CNCR2820730516>3.0.CO;2-T
67. Hansen MR, Moffat JC. Osteosarcoma of the Skull Base After Radiation Therapy in a Patient With McCune-Albright Syndrome: Case Report. Skull Base (2003) 13(02):079–84. doi: 10.1055/s-2003-40597
68. Fuyi L, Wenting L, Yong Y, Guilin L, Yi Y, Wanchen D, et al. A Case of McCune-Albright Syndrome Associated With Pituitary GH Adenoma: Therapeutic Process and Autopsy. J Pediatr Endocrinol Metab (2011) 24(5-6):283–7. doi: 10.1515/jpem.2011.178
69. Neyman A, Eugster EA. Treatment of Girls and Boys With McCune-Albright Syndrome With Precocious Puberty–Update 2017. Pediatr Endocrinol reviews: PER (2017) 15(2):136. doi: 10.17458/per.vol15.2017.nau.treatmentgirlsboys
70. Holland FJ. Gonadotropin-Lndependent Precocious Puberty. Endocrinol Metab Clinics North Am (1991) 20(1):191–210. doi: 10.1016/S0889-8529(18)30288-3
Keywords: McCune-Albright syndrome, GH excess, precocious puberty (PP), gigantism, acromegaly
Citation: Zhai X, Duan L, Yao Y, Xing B, Deng K, Wang L, Feng F, Liang Z, You H, Yang H, Lu L, Chen S, Wang R, Pan H and Zhu H (2021) Clinical Characteristics and Management of Patients With McCune-Albright Syndrome With GH Excess and Precocious Puberty: A Case Series and Literature Review. Front. Endocrinol. 12:672394. doi: 10.3389/fendo.2021.672394
Received: 25 February 2021; Accepted: 11 October 2021;
Published: 29 October 2021.
Edited by:
Run Yu, UCLA David Geffen School of Medicine, United StatesReviewed by:
Domenico Corica, University of Messina, ItalyFu Qiong Chen, Huazhong University of Science and Technology, China
Copyright © 2021 Zhai, Duan, Yao, Xing, Deng, Wang, Feng, Liang, You, Yang, Lu, Chen, Wang, Pan and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijuan Zhu, c2hlbmd4aW4yMDA0QDE2My5jb20=