 Amaara Marzook1
Amaara Marzook1 Alejandra Tomas
Alejandra Tomas Ben Jones
Ben Jones- 1Section of Endocrinology and Investigative Medicine, Imperial College London, London, United Kingdom
- 2Section of Cell Biology and Functional Genomics, Imperial College London, London, United Kingdom
The glucagon-like peptide 1 receptor (GLP-1R) is a class B G protein-coupled receptor (GPCR) which mediates the effects of GLP-1, an incretin hormone secreted primarily from L-cells in the intestine and within the central nervous system. The GLP-1R, upon activation, exerts several metabolic effects including the release of insulin and suppression of appetite, and has, accordingly, become an important target for the treatment for type 2 diabetes (T2D). Recently, there has been heightened interest in how the activated GLP-1R is trafficked between different endomembrane compartments, controlling the spatial origin and duration of intracellular signals. The discovery of “biased” GLP-1R agonists that show altered trafficking profiles and selective engagement with different intracellular effectors has added to the tools available to study the mechanisms and physiological importance of these processes. In this review we survey early and recent work that has shed light on the interplay between GLP-1R signalling and trafficking, and how it might be therapeutically tractable for T2D and related diseases.
Introduction
Recent estimates suggest a global prevalence of approximately 422 million people living with diabetes, a number expected to rise to 629 million by 2045 (1). Type 2 diabetes (T2D), the commonest form, arises due to a combination of genetic and lifestyle factors that contribute to defective production of insulin from pancreatic beta cells and resistance to its action in peripheral and central tissues (2). Prolonged elevation of blood glucose, as well as related adverse metabolic features such as dyslipidaemia and chronic inflammation, lead ultimately to a range of serious health consequences, including cardiovascular disease, limb amputation, blindness, and kidney failure. Existing anti-diabetic drugs contribute to improve glycaemic control by either increasing the body’s sensitivity to insulin, enhancing insulin secretion or reducing renal glucose reabsorption. However, these drugs are reported to cause a myriad of adverse effects such as weight gain, oedema, intestinal discomfort and hypoglycaemia (3).
Glucagon-like peptide 1 receptor agonists (GLP-1RAs) have emerged as safe and effective treatments for diabetes (4). The GLP-1R, once activated, initiates effects that lead to an overall decrease in blood glucose levels, including potentiation of insulin secretion, reduced glucagon secretion, and weight loss via satiety induction that results in increased insulin sensitivity (5). GLP-1RAs work to stimulate insulin secretion in a glucose-dependent manner, therefore carrying a low risk of hypoglycaemia. To date, they represent the only G protein-coupled receptor (GPCR) ligands approved as glucose-lowering agents.
Once activated, GLP-1Rs are transported by the cellular endocytic machinery to different subcellular compartments, allowing qualitative and quantitative fine-tuning of intracellular signalling responses linked to insulin secretion and other downstream effects (6). Understanding these complex trafficking mechanisms could potentially provide an avenue for further development of compounds with optimised therapeutic characteristics. In particular, there has been substantial recent interest in agonists that have a high selectivity for particular signalling effectors or pathways, a concept also known as “biased agonism” (7). This phenomenon is strongly associated with alterations to GLP-1R trafficking, although the causal relationships are not fully understood. In this review we focus on the role of GLP-1R trafficking as a critical component of GLP-1R agonism, and how it may be possible to harness some of these processes to improve therapeutic targeting of the GLP-1R in T2D.
GLP-1R Signalling in Pancreatic Beta Cells
The GLP-1R is a class B (secretin family) GPCR. Activation of the GLP-1R in pancreatic beta cells results in insulin synthesis, potentiation of glucose-stimulated insulin secretion (GSIS), and longer-term effects on beta cell survival, proliferation and neogenesis. An early study used RNA nuclease protection combined with Southern blotting of reverse-transcribed PCR products to map Glp1r in rat tissues, identifying several sources of expression including pancreas, lung, kidney, stomach, intestine and brain (8). GLP1R mRNA transcripts were also reported in the human retina and retinas of db/db mice (9), and in immune cells such as invariant natural killer T (iKNT) cells (10). Historically, immunohistochemical confirmation has been hampered by the poor specificity of anti-GLP-1R antibodies (11), but better performing antibodies have enabled the distribution of GLP-1R to be more confidently ascertained in rodents, monkeys and humans (12–14). Certain species differences are apparent from these studies, with much higher levels of GLP-1R in rodent compared to human lung tissue, and absent expression in human thyroid. Comparisons of mRNA versus protein distribution typically deliver congruent results, with exceptions including GLP-1R protein detected on neuronal projections distant from the their respective Glp1r transcript-containing cell bodies (14), and instances where low abundance Glp1r transcripts, e.g. in pancreatic delta cells, do not yield detectable protein expression (15). Reporter mice, in which active Glp1r promoter-dependent Cre recombinase leads to fluorescent protein expression, have also been used to map GLP-1R-containing cell types (16–19). The most recent study by Andersen et al. (19) revealed additional GLP-1R-expressing cell types such as tracheal cartilage chondrocytes and skin fibroblasts that had not been identified with the original Glp1r reporter mouse (16); it is not known if these observations are applicable to humans. A further approach involves the use of radiolabelled (20) or, more recently, fluorescent peptides (21) to define GLP-1R ligand binding sites, revealing for example sub-populations of GLP-1R-expressing pancreatic alpha cells.
Human GLP-1R comprises 463 amino acids, including an N-terminal signal peptide that is cleaved on delivery to the plasma membrane (22). GLP-1R and related class B GPCRs are characterised by a stereotyped extracellular N-terminal domain crucial for ligand recognition, an intracellular C-terminus, and seven transmembrane (7TM) α-helices spanning the plasma membrane that are connected by three intracellular loops (ICL1-ICL3) and three extracellular loops (ECL1-ECL3) on either side of the membrane. Class B GPCRs typically bind their peptide ligands through a mechanism known as the two-domain model, in which the extracellular domain (ECD) binds to the C-terminal end of the ligand first, enabling a second interaction between the N-terminus of the ligand and the 7TM domains of the receptor (22). Recent advances in GPCR structural biology have provided insights into GLP-1R-ligand interactions that underpin binding and activation processes (23–26).
Gαs/cAMP/PKA Signalling in Beta Cells
Canonical GLP-1R signalling occurs via heterotrimeric G proteins, comprising an independent Gα subunit and a Gβ/γ dimer bound reversibly to Gα. The Gα subunits can be classified into 4 subtypes: Gαi, Gα0, Gαs, and Gαq/11, based on the nature of their interaction with specific downstream effectors. Agonist-activated GPCRs act as guanyl nucleotide exchange factors (GEFs) for G proteins. The nucleotide binding pocket opens, allowing exchange of inactive guanine diphosphate (GDP) for guanine triphosphate (GTP). GTP binding induces conformational changes within the Gα subunit that lead the Gβγ to dissociate, so that both subunits are free to activate downstream effector proteins, thereby initiating a signalling cascade (27).
The GLP-1R is well known to couple with the Gαs subunit, resulting in adenylate cyclase (AC) activation and cyclic adenosine monophosphate (cAMP) production (Figure 1). GLP-1R-stimulated pathways in pancreatic beta cells act within seconds to increase cAMP levels and minutes to potentiate GSIS. A rapid increase in cAMP is accompanied by activation of exchange protein activated by cAMP-2 (Epac2) and protein kinase A (PKA) (28). Activation of Epac2 reduces the concentration of ATP required to achieve closure of KATP channels promoting membrane depolarisation, Ca2+ influx and subsequent Ca2+-induced Ca2+ release from intracellular stores, insulin priming and finally insulin granule exocytosis (29–31). Activated PKA also promotes membrane depolarisation by directly phosphorylating the sulfonylurea receptor 1 (SUR1) and a regulatory subunit of KATP channels (32).
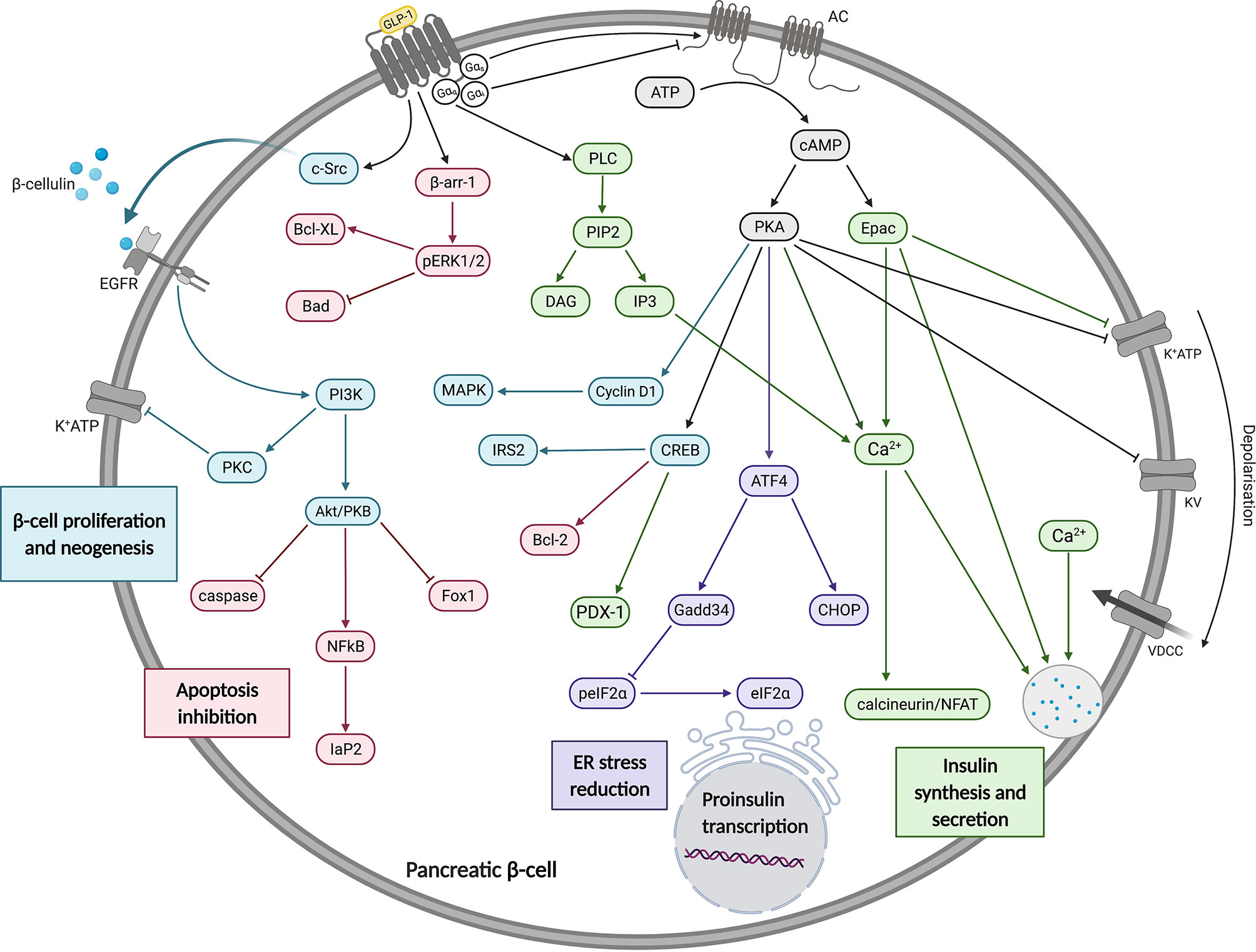
Figure 1 A summary of the effects of GLP-1 on insulin synthesis, secretion, beta cell proliferation, neogenesis and apoptosis inhibition. In pancreatic beta cells, GLP-1R signalling is predominantly via Gαs which mediates increases in cAMP to activate Epac and PKA. These have a range of effects, as described in the main text and depicted in this figure. These and other pathways promote increases in insulin gene transcription, synthesis and secretion. Figure created using BioRender.com.
Additionally, PKA-mediated activation of cyclin D1 and MAPK was found to be crucial in the G1/S phase transition during the cell cycle, promoting beta cell neogenesis (33). GLP-1R signalling also attenuates the development of ER stress in beta cells through cAMP-dependent potentiation of Activating Transcription Factor 4 (ATF4) translation (34). These findings further support the claim that sustained GLP-1R agonism may result in disease modifying activity in people with T2D through enhancement or preservation of functional beta cell mass (35). GLP-1R also activates cAMP response binding element (CREB) which stimulates the expression of insulin transcription factor pancreatic and duodenal hombox gene-1 (PDX-1) in a cAMP/PKA-dependent manner (36). CREB was also shown to stimulate Insulin Receptor Substrate 2 (IRS2) gene expression which is essential for beta cell growth and survival in isolated beta cells and rodent models (37).
Non-Gαs Signalling Events in Beta Cells
GLP-1R is able to couple not only to Gαs, but also to other Gα subtypes including Gαi and Gαq. The former acts in opposition to Gαs, leading to adenylate cyclase inhibition and reduced cAMP production. Whilst Gαi coupling has been detected in response to GLP-1R activation (25, 38), it is not clear whether this is a significant factor in beta cells or an artefact of overexpression systems. There is more evidence that GLP-1R can couple to Gαq in beta cells, as demonstrated by inositol triphosphate (IP3) turnover studies (39), diacylglycerol (DAG) production (40) and most convincingly, a Gαq FRET biosensor (41). The latter study is interesting as it reported that the effects of GLP-1 on GSIS were predominantly Gαs-dependent under normoglycaemic conditions, but chronic beta cell depolarisation due to sulphonylurea treatment or hyperglycaemia led to an apparent switch in coupling preference to Gαq. Activation of Gαq leads to phospholipase C (PLC) activation, allowing phosphatidylinositol 4,5-bisphosphate (PIP2) cleavage to generate DAG and IP3, with resultant effects on protein kinase C (PKC) activation and Ca2+ release from intracellular stores via IP3 receptor activation. Specifically, the putative Gαq-mediated mechanism is thought to involve PKC-mediated activation of an inward depolarising current carried by TRPM4/5 channels (42). The studies are not totally conclusive as cAMP can also activate PLC leading to intracellular Ca2+ increases (43).
Numerous reports show that GPCRs also induce distinct cellular responses via separate, non-G protein-dependent signalling cascades facilitated by the recruitment of β-arrestins, which act as scaffolds to promote interaction with and activation of mitogen-activated protein kinases (MAPKs) such as extracellular regulated kinase 1/2 (ERK1/2). Interestingly, a study using HEK293 cells with total elimination of all Gα subunits by a combination of CRISPR/Cas9 deletion and pharmacological inhibition suggested that, in fact, G proteins are essential for ERK1/2 phosphorylation and β-arrestins are not (44). However, it was also suggested that these results may have been artefacts of clonal selection rather than a true representation of the relative importance of G proteins and β-arrestins in this process (45). For the GLP-1R, β-arrestin-1 knockdown reduces ERK1/2 signalling in INS-1 cells, identifying a potential G protein-independent signalling axis required for GLP-1R-induced insulin secretion and coupling to protection against apoptosis (46, 47). In a study using INS-1 cells, GLP-1 facilitated β-cellulin release from the plasma membrane via β-arrestin-1-mediated recruitment of c-Src, leading to transactivation of the epidermal growth factor receptor (EGFR), a β-cellulin cognate receptor. This, in turn, sequentially activated phosphoinositide 3-kinase (PI3K) and downstream effectors protein kinase B (PKB/Akt), p38 MAPK and PKC contributing to DNA synthesis, gene expression, insulin synthesis and reduced apoptosis (48). Furthermore, mediated via PKB and nuclear factor-κB (NF-κB), GLP-1 also exhibits protective effects on beta cell glucotoxicity, lipotoxicity, glucolipotoxicity and stimulates transcription of the antiapoptotic genes Iap-2 and Bcl-2 (49). On the other hand, mice with beta cell-specific knockout of β-arrestin-1 or -2 did not show any defect in acute insulin secretory response to GLP-1RAs or alteration to beta cell mass (50, 51), although this was not investigated in detail for β-arrestin-2.
Biased GLP-1R Agonism
The pleiotropic interactions between GPCRs and multiple signalling proteins has led to an important concept, particularly relevant to drug discovery, that efficacy has a quality as well as a magnitude. This concept, referred to as “biased agonism” or “signal bias”, is defined by ligands of the same receptor that show favoured and distinct coupling preferences to particular cellular effectors or pathways (7). Although detailed molecular mechanisms underpinning biased signalling are not yet completely understood, it is generally believed that biased ligands preferentially stabilise particular GPCR conformations for selective engagement with specific signalling pathways (52). One of the first reports in which the apparent biased behaviour of ligands was explicitly recognised showed that [Sar1,Ile4,Ile8]-AngII was unable to activate G protein signalling but still triggered β-arrestin recruitment and ERK1/2 phosphorylation at the angiotensin 2 type 1 receptor (53). In fact, further examples of apparently biased ligands can be found in the historical literature, pre-dating the emergence of signal bias as a fashionable pharmacological concept. One notable example relevant to class B GPCR pharmacology is desHis1,glu9-glucagon, a glucagon receptor “antagonist” which was found to stimulate IP3 production without cAMP generation (54).
Whilst the majority of currently approved therapeutic GLP-1RAs show balanced agonism between most pathways measured (55), an increasing number of biased GLP-1RAs have now been described in the preclinical literature. In line with major advances in the understanding of the structural basis of GLP-1R activation (24), the peptide N-terminus has emerged as an important determinant of biased agonism, with several studies reporting that β-arrestin recruitment can be selectively diminished by amino acid sequence substitutions and novel chemical entities in this region (56–60). Despite the putative role of β-arrestins as GLP-1R effectors coupled to both insulin release and protection against beta cell apoptosis (46, 47), most biased GLP-1RA studies have shown that reduced β-arrestin recruitment is associated with enhanced insulin secretion, at least under conditions of sustained stimulation. This is presumed to be a consequence of β-arrestin-mediated desensitisation and/or downregulation of the GLP-1R (61). Studies in rodent models of T2D show improved glucose tolerance with G protein-biased compared to balanced GLP-1RA treatments, with these benefits progressively apparent later in the dosing period, consistent with a desensitisation-related mechanism. Recently, tirzepatide, a clinical candidate dual incretin receptor agonist that targets both the GLP-1R and the closely related glucose-dependent insulinotropic polypeptide receptor (GIPR), was found to show profound biased agonism characterised by minimal β-arrestin recruitment at the GLP-1R (62, 63). Tirzepatide appears to be highly effective for T2D in human trials (64), although it is not yet possible to know how much the biased GLP-1R agonism contributes to this.
Endocytic GLP-1R Trafficking
Most ligand-activated receptors undergo endocytosis, a process recognised to serve a number of purposes, including regulation of cell surface receptor levels to modulate cellular responsiveness to continued ligand exposure, and alternative sorting of receptors towards one of the cell surface recycling pathways for sustained signalling, or towards lysosomal targeting for receptor degradation to achieve signal termination and long term desensitization (or down-regulation) (65). The endocytic pathway is composed of a continuum of intracellular vesicular compartments showing varying levels of maturation, including early or sorting endosomes, multivesicular or late endosomes, endocytic recycling compartments and lysosomes (66), which are in turn interconnected with a number of other intracellular organelles including the Golgi apparatus, the ER and mitochondria (67–70). Activated GLP-1R undergoes rapid endocytosis, with almost complete disappearance of surface receptors within 15 minutes of exposure to a maximal agonist concentration (55). Proposed molecular determinants of GLP-1R trafficking are summarised in Figure 2 and discussed in more detail below.
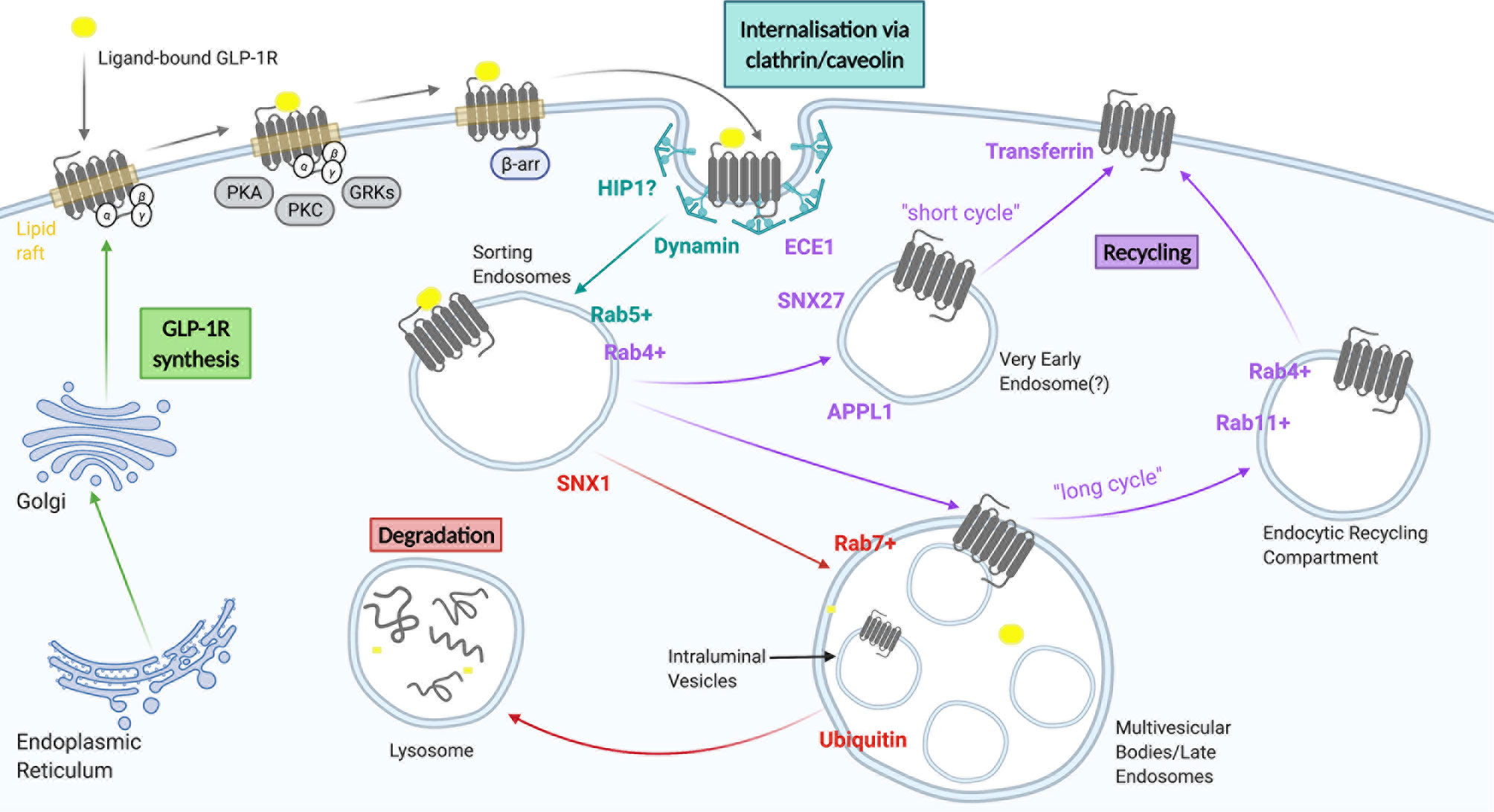
Figure 2 A summary of GLP-1R trafficking. GLP-1 receptors are internalised through clathrin-dependent and/or clathrin-independent mechanisms and subsequently transported to sorting endosomes/early endosomes. From here they can either be recycled back to the plasma membrane via a fast recycling pathway (short cycle), a slow recycling (long cycle), or be targeted for degradation to lysosomes via multi vesicular bodies/late endosomes. Figure created using BioRender.com.
GLP-1R Endocytosis – Clathrin-Dependent or -Independent?
The GLP-1R has been previously reported to follow a dynamin-dependent, but either clathrin-mediated (CME) or clathrin-independent (CIE) endocytosis pathway (71–73), which represent the two main mechanisms underlying receptor endocytosis. Studies performed in CHO and CHL cells recombinantly expressing GLP-1R revealed internalisation via clathrin-coated pits (74). GLP-1R also appeared to internalise via clathrin-coated pits in pancreatic beta cells (71). This process, which followed clustering of immobilized receptors at membrane hotspots, involved cargo selection, clathrin-coat assembly, membrane bending, vesicle scission and uncoating (75). This canonical GPCR internalisation mechanism has been typically associated with β-arrestin recruitment, which can target GPCRs for internalisation via interaction with the β2 subunit of the AP2 clathrin adaptor, promoting movement of the GPCR-β-arrestin complex to vesicular pits where β-arrestin, AP2 and clathrin form a tripartite interaction (76, 77). However, it has also been observed that depletion of β-arrestins has a minimal impact on agonist-induced GLP-1R internalisation (55, 61, 71). The GLP-1R was found to possess an AP2-binding domain in its C-terminal tail allowing it to directly interact with AP2, minimising the need for additional intermediaries (78). The effects of β-arrestin-1 knockdown in INS-1 cells did not support a role for β-arrestin-1 in GLP-1R trafficking (46). Additionally, the endocytic accessory protein huntingtin-interactin protein 1 (HIP1) has been implicated in GLP-1R endocytosis (79); HIP-1 is a CME accessory protein that forms a bridging interaction between clathrin and AP2, thereby stimulating clathrin coated pit assembly (80).
In contrast to the above evidence supporting primarily a CME-dependent endocytosis pathway, experiments with small interfering RNA targeting clathrin did not appear to affect GLP-1R endocytosis in HEK293 cells, suggesting the existence of an alternative CIE-mediated mechanism (81). It is plausible that the GLP-1R can utilize more than one type of endocytosis mechanism, enabling it to deviate towards one of these alternative entry pathways in the event of blockade of its main pathway for internalization. While the nature of these alternative CIE pathways has not been elucidated, there have been some suggestions of a possible role for caveolin-1 in this process (72). GLP-1R contains a classic caveolin-1-binding motif within ICL2 enabling it to interact and colocalise with the protein intracellularly. Occurrence of this interaction was supported by using inhibitors of caveolin-1, with this mechanism of GLP-1R internalisation proposed to occur through activation of Gαq proteins, followed by activation of PKC (69). It has also been suggested that β-arrestin-2 may play a role in GLP-1R trafficking mediated through caveolin-1 (46). Interestingly however, it has been reported that beta cell lines which display normal levels of GLP-1R endocytosis do not express caveolin-1, raising questions about the role of this protein in the control of GLP-1R internalisation (79). More generally, accumulating evidence within the general field of endocytosis has called into question whether a caveolin-mediated endocytic pathway exists at all (82), and the interaction between caveolin-1 and the GLP-1R might reflect alternative roles for this protein such as in the biosynthetic transport and targeting of mature receptors at the plasma membrane (60).
The Role of Membrane Nanodomains in GLP-1R Endocytosis
The local organisation of GPCRs within plasma membrane cholesterol- and sphingolipid-enriched nanodomains (historically known as liquid-ordered domains or “lipid rafts”) can be dynamically regulated by ligand-induced activation, and is a key regulator of GPCR behaviours through compartmentalisation of both receptor and signalling effectors into membrane “hotspots” (83). Indeed, Gαs, the main G protein signalling subunit for the GLP-1R, was predominantly associated with cholesterol-rich detergent-resistant membrane fractions in beta cells (71). Moreover, adequate plasma membrane cholesterol levels were essential for efficient GLP-1R endocytosis via either clathrin-dependent and clathrin-independent pathways, as demonstrated by the virtual abolition of agonist-induced GLP-1R internalisation following cholesterol extraction with MβCD (methyl-β-cyclodextrin) in beta cells (71). Moreover, the GLP-1R undergoes agonist-dependent palmitoylation at its C-terminus, providing a mechanism for altered interaction and targeting of active receptors to these cholesterol-rich membrane regions (71). As well as a role in concentrating active GPCRs in particular membrane nanodomains enriched in signalling and trafficking proteins, direct interactions with cholesterol may alter the binding affinity of GPCRs (84, 85), suggesting that this lipid might allosterically modulate receptors by limiting their conformational flexibility, a possibility that remains to be investigated for the GLP-1R.
Post-Endocytic GLP-1R Sorting/Trafficking
Post-endocytic GPCR trafficking occurs through a series of dynamically interconnected organelles crucial for the sorting of receptors to different intracellular compartments (86). The passage of GLP-1R through different beta cell endosomal compartments has been examined using electron microscopy (55, 87). Sorting endosomes, also known as early endosomes (EE), are the first organelles receiving the endocytosed receptor-containing vesicles, and are typically marked by Rab5 (88). The expression of a mutant Rab5 in beta cells inhibited internalisation of the GLP-1R, suggesting a crucial role for the sorting of receptors to Rab5-positive endosomes (89). From the limiting membrane of EEs, GPCRs may be recycled back to the plasma membrane via a “short/fast cycle” of trafficking from a Rab5-positive to a Rab4-positive endocytic compartment, via a “long/slow cycle” by entering Rab11-positive perinuclear recycling endosomes, or towards lysosomal degradation via retention in intraluminal vesicles (ILVs) of multivesicular bodies (MVBs), generated by maturation of EEs, a process that also causes termination of signalling, and subsequent trafficking to Rab7-positive late endosomes and lysosomes (90). High efficacy GLP-1RAs such as exendin-4 and GLP-1, when applied at high concentrations, target GLP-1Rs primarily towards retention in ILVs and lysosomal degradation (55, 79, 87). A retromer-associated factor, sorting nexin 1 (SNX1), was found to play a crucial role in targeting GLP-1R towards lysosomal degradation by restricting receptor recycling (79), but other molecular events governing this process are not well understood. For example, preliminary experiments have shown that receptor ubiquitination, a process typically involved in the canonical Endosomal Sorting Complex Required for Transport (ESCRT)-dependent targeting of endosomal proteins to ILVs within MVBs as a committing step towards lysosomal fusion, does not appear to apply to the GLP-1R, although alternative ESCRT-dependent but ubiquitin-independent, as well as ESCRT-independent mechanisms of sorting into ILVs have also been described, including for some GPCRs such as the protease-activated receptor PAR1 (91). Agonist-internalised GLP-1R colocalises with transferrin, a marker for recycling (92), and about 10 to 30% of internalised GLP-1R returns to the cell surface per hour after exendin-4 or GLP-1 treatment, respectively (55, 87). It has been shown in beta cells that another retromer-associated factor, sorting nexin 27 (SNX27), plays a role in cargo selection and GLP-1R sorting from early to recycling endosomes, with SNX27-knockdown significantly reducing GLP-1R recycling (79). This is due to its association with actin/sorting/nexin/retromer tubule (ASRT) and the Wiskott-Aldrich syndrome protein and SCAR homologue complex (WASH) (93). A “two-barcode” mechanism for GPCR recycling was hypothesised, acting through the endosomal ASRT pathway, involving both a PDZ-binding domain and phosphorylation of specific serine residues in the C-terminal domain (94). However, the GLP-1R does not possess a canonical PDZ-binding domain (79). Another aspect that requires further investigation is the possibility of GLP-1R undergoing retromer-dependent endosome-to-Golgi retrograde transport, as well as possible interactions with alternative retromer-like endosomal retrieval complexes involved in the control of receptor recycling such as the recently described retriever and commander complexes (95).
Once internalized, the acidic endosomal pH disrupts ligand-receptor interactions and influences post-endocytic GPCR trafficking, presumably as ligand-bound versus apo-state GPCR conformations show altered engagement, directly or indirectly, with local trafficking effectors. Accordingly, ligand binding affinity appears to be an important determinant of GLP-1R trafficking. High affinity agonists that remain bound to GLP-1R within the endosomal pathway tend to result in greater lysosomal targeting of the receptor, whereas lower affinity agonists preferentially lead to receptor recycling (87). As a further factor likely to influence ligand-receptor complex stability, the metalloprotease endothelin-converting enzyme-1 (ECE1) was also shown to colocalise with internalised GLP-1Rs, and presumed to facilitate GLP-1R recycling via proteolytic processing of endosomal GLP-1 (96), as previously shown for other GPCRs (97). Indeed, slow-recycling exendin-4 is highly resistant to ECE1, whereas GLP-1, which leads to faster GLP-1R recycling, is rapidly degraded by ECE1 at low pH (87). GLP-1RAs that carry a fatty acid chain to promote reversible binding to albumin, e.g. liraglutide and semaglutide, participate indirectly in recycling of albumin via the neonatal Fc receptor (FcRn) (98); and whilst this is an important factor determining the long half-life of these GLP1-RAs in the circulation, it is not known whether this phenomenon is relevant to GLP-1R recycling in beta cells.
Caveats to GLP-1R Trafficking Studies Reported in the Literature
It is important to note that many studies investigating GLP-1R trafficking have used immortalised cell lines overexpressing the GLP-1R, typically stimulated with high agonist concentrations that are well above the physiological range and may even exceed the likely peak concentrations of pharmacological GLP-1RAs in vivo. These experimental conditions are to a certain extent mandated by the limitations of available methods to monitor the redistribution of the endogenous, agonist-stimulated GLP-1R. For example, “feeding” of GLP-1R extracellular-domain-recognising antibodies to monitor endocytosis is precluded as they compete for the orthosteric ligand binding site (99), and fluorescent agonist ligands dissociate from the receptor after entering the endocytic pathway so are informative only for the initial internalisation phase. Appending biorthogonal tags such as HALO- or SNAP-tags to label the endogenous GLP-1R genomic sequence could allow a better understanding of trafficking events without the use of exogenous expression, but has not been reported to date. The use of high ligand concentrations leads to large, easy to measure changes in GLP-1R redistribution, but it is possible that mode of endocytosis could shift as receptor occupancy increases (100). More sophisticated imaging-based approaches to monitor individual endocytosis events e.g. using total internal reflection fluorescence microscopy (TIRFM) combined with single particle tracking (SPT) would allow these phenomena to be observed at physiological agonist concentrations. Additionally, adapting assays for longer stimulation times but with lower ligand concentrations would aid in understanding how GLP-1R agonist drugs influence GLP-1R trafficking over the many hours or days that they persist in the circulation.
GPCR Trafficking in the Spatiotemporal Control of Intracellular Signalling
Once thought to be solely constrained to the plasma membrane, GPCR signalling is now known to originate from various intracellular locations (101–103). Endocytic trafficking alters the location and duration of certain receptor-mediated signalling pathways and is thus a crucial regulator of specific cellular responses (104, 105). The possibility of endosomal cAMP generation has attracted considerable interest as a potential means to achieve sustained intracellular responses (106). Whilst the broad categories of “plasma membrane signalling” versus “endosomal signalling” are now well established, it should be acknowledged that subclassification of signals at point source is possible, e.g. from liquid-ordered versus liquid-disordered plasma membrane nanodomains (71).
Endosomal signalling by the GLP-1R has now begun to be explored. Internalised GLP-1Rs were shown to participate in compartmentalised cAMP generation, contributing to insulin granule exocytosis in cultured pancreatic beta cells before being directed to lysosomes for degradation (107). A follow up study showed that Gαs is recruited to activated GLP-1R-containing Rab5-positive EEs (89). Inhibition of GLP-1R internalisation using dominant negative dynamin constructs reduced GLP-1R cAMP responses (73, 108). Additionally, A-kinase anchoring proteins (AKAPs) target PKA to discrete subcellular locations of cAMP production, compartmentalising Ca2+ and cAMP signalling, a crucial mechanism for the regulation of insulin secretion (109, 110).
Notwithstanding the importance of signal compartmentalization and endosomal signalling, long term reduction in GPCR abundance at the cell surface as a result of sustained agonist-mediated endocytosis without sufficient compensatory recycling (i.e. downregulation) is a clear potential limiting factor in the capacity of a cell to generate prolonged signalling responses in the face of chronic ligand exposure. Compared to other class B GPCRs, including the glucagon and glucose-dependent insulinotropic polypeptide receptors (GCGR, GIPR), endocytosis of the GLP-1R is rapid and extensive (61, 73), which could quickly lead to depletion of plasma membrane receptors, particularly when combined with preferential targeting to lysosomal degradation, a situation in which the biosynthetic pathway might not be able to replenish the pool of membrane receptors with sufficient efficiency. GLP-1R endocytosis and subsequent endocytic trafficking therefore may both enhance and attenuate signalling, with numerous factors such as ligand efficacy, concentration and exposure period as well as receptor clustering and nanodomain segregation all potential determinants of which process dominates.
The Importance of Receptor Trafficking in Biased GLP-1R Agonism
The endocytic trafficking effects of a number of biased GLP-1RAs have been reported (55, 57–59, 87, 111). Typically, G protein-biased peptides with reduced β-arrestin recruitment efficacy also show attenuations in GLP-1R internalisation propensity, along with faster recycling back to the plasma membrane and reduced lysosomal degradation. Despite the canonical role of β-arrestins in clathrin-mediated endocytosis, it does not appear that the deficient β-arrestin response of biased agonists is directly responsible for the their distinct trafficking profiles, as agonist-induced GLP-1R endocytosis is minimally affected by the total absence of both β-arrestin isoforms (55, 61, 71). Instead, the reduced β-arrestin recruitment of slow-internalising, fast-recycling agonists is rather considered as a bystander marker for this type of trafficking behaviour, whose main role is in preventing rapid receptor desensitisation. An important characteristic that appears to determine the propensity to induce GLP-1R internalisation is the binding affinity displayed by the different agonists, a process that is intimately linked with their capacity to trigger receptor clustering and nanodomain segregation prior to internalisation (71). Indeed, allosteric modulation of binding affinity can render a weakly internalising agonist fast internalising properties (71). Additionally, different intra-endosomal GLP-1R-ligand dissociation rates observed with different biased GLP-1RAs is one factor that influences the receptor post-endocytic targeting, with faster dissociating biased GLP-1RAs tending to lead to more receptor recycling (55, 87). However, the precise biophysical mechanisms and receptor-effector interactions that differ between different biased GLP-1RAs and explain their differential endocytic rates have not been fully elucidated.
The fact that GLP-1R endocytosis promotes endosomal cAMP signalling but is also ultimately a route to receptor downregulation means that, irrespective of the mechanisms by which biased GLP-1RAs achieve their altered trafficking responses, they are likely to contribute to their overall signalling profiles. Endosomal versus plasma membrane signalling with a panel of GLP-1RAs was compared using targeted FRET cAMP biosensors (pmEpac2 and cytoEpac2), revealing that exendin-4 is biased in favour of cytoplasmic cAMP production compared to liraglutide (108). Aside from this study, the endosomal signalling characteristics of more recently invented biased GLP-1RAs with profoundly altered trafficking profiles have not been reported. This is a key gap in the literature, as it is not yet established whether the steady state preservation of surface GLP-1Rs seen with some biased GLP-1RAs is the primary result of a failure to enter the endocytic pathway (which would preclude endosomal signalling), or the accelerated rapid recycling from early endosomal compartments (which may permit endosomal signalling), or a combination of both processes. Targeted biosensor studies to investigate this phenomenon are required to address this issue. It is known however that lipid nanodomain-specific cAMP generation, measured using the raft-anchored cAMP sensor TEpacVV-Lyn (112), is selectively enhanced by the β-arrestin-biased GLP-1RA exendin-asp3 over the G protein-biased exendin-phe1, matching the relative propensity of these agonists to stimulate segregation of GLP-1R into lipid rafts (71).
Whilst their endosomal signalling characteristics are not yet known, it is well established that some of the biased GLP-1RAs with reduced tendency to promote GLP-1R internalisation do indeed cause less beta cell GLP-1R degradation over prolonged periods of stimulation (55, 87). As long periods of receptor-drug exposure are maintained with therapeutic GLP-1RAs that have extended pharmacokinetic stability, these studies provide useful information on the likely differential impact of biased GLP-1RAs on target downregulation. Whilst this phenomenon has not yet been studied in vivo, various reports demonstrate that the net result is that poorly internalising GLP-1RAs preserve the responsiveness of pancreatic beta cells, as measured by cumulative insulin secretion in vitro over many hours and by rechallenge experiments to reveal different rates of homologous desensitisation (55, 87), and hence have a beneficial net effect despite potential losses of intracellular signalling. The implication of these findings is that, even if endosomal signalling is an important component of GLP-1R agonism, the “benefits” of increased steady state plasma membrane receptors with certain biased GLP-1RAs might exceed the potential reductions in endosomal signalling that might accompany this phenomenon.
Conclusions
The GLP-1R plays a pivotal role in glucose homeostasis, insulin synthesis and potentiation of GSIS and has accordingly become a major therapeutic avenue for the treatment of T2D. It is apparent that the GLP-1R signalling-trafficking system is highly complex, comprising of many steps and can provide a platform for exquisite control of signalling events. The development of biased GLP-1RAs has provided a suite of tools for the reprogramming of GLP-1R endocytosis and post-endocytic sorting and a means to achieve distinct signalling patterns at different subcellular locations. The physiological and therapeutic implications of these processes are only now beginning to be explored.
Substantial progress has been made in our understanding of GLP-1R signalling and trafficking, such as the perpetuation and termination of signalling within endosomal compartments. However, significant gaps still remain to be resolved. The molecular mechanisms governing GLP-1R endocytosis are yet to be clearly defined, with a major role for typical candidates such as β-arrestins being, surprisingly, not supported by most of the evidence. Whilst the differences in post-endocytic GLP-1R sorting and trafficking with different GLP-1R ligands has been observed and described using an expanding number of imaging techniques, a comprehensive mechanistic explanation of these processes is still lacking. Moreover, the majority of studies have been performed in heterologous or insulinoma cell lines, often at high ligand concentrations, and confirmation in primary beta cells and in vivo will ultimately be required to determine the physiological importance of these processes. Finally, translating the study of the mechanisms governing GLP-1R trafficking to other cellular systems beyond the pancreas represents a further frontier in our understanding of the importance of these processes in GLP-1R biology. For example, it is postulated that GLP-1R-mediated endocytic carriage across tanycytes or endothelial cells is a specific mechanism by which GLP-1RAs access the brain (55, 113–115), although non-receptor-mediated mechanisms such as adsorptive transcytosis are also suggested to be important (116).
Overall, the promising results observed using biased GLP-1RAs with distinct trafficking profiles in preclinical studies have raised the possibility that this is a therapeutically viable strategy to confer greater efficacy and/or tolerability for the treatment of T2D. Dedicated studies in humans are now required to test this hypothesis.
Author Contributions
AM, AT, and BJ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The Section of Endocrinology and Investigative Medicine is funded by grants from the MRC, BBSRC, NIHR, and is supported by the NIHR Biomedical Research Centre Funding Scheme. The views expressed are those of the authors and not necessarily those of the funders. AT acknowledges funding from Diabetes UK. BJ acknowledges support from the Academy of Medical Sciences, Society for Endocrinology, The British Society for Neuroendocrinology, the European Federation for the Study of Diabetes, and an EPSRC capital award. BJ and AT also received funding from the MRC (MR/R010676/1) and the European Federation for the Study of Diabetes. The authors declare having received separate funding from Sun Pharmaceuticals. This funder had no involvement in the present study.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. Idf Diabetes Atlas: Global Estimates for the Prevalence of Diabetes for 2015 and 2040. Diabetes Res Clin Pract (2017) 128:40–50. doi: 10.1016/j.diabres.2017.03.024
2. Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of Type 2 Diabetes Mellitus. Int J Mol Sci (2020) 21:6275. doi: 10.3390/ijms21176275
3. Lau DCW, Teoh H. Impact of Current and Emerging Glucose-Lowering Drugs on Body Weight in Type 2 Diabetes. Can J Diabetes (2015) 39 Suppl 5:S148–54. doi: 10.1016/j.jcjd.2015.09.090
4. Andersen A, Lund A, Knop FK, Vilsbøll T. Glucagon-Like Peptide 1 in Health and Disease. Nat Rev Endocrinol (2018) 14:390–403. doi: 10.1038/s41574-018-0016-2
5. Holst JJ. The Physiology of Glucagon-Like Peptide 1. Physiol Rev (2007) 87:1409–39. doi: 10.1152/physrev.00034.2006
6. Manchanda Y, Bitsi S, Kang Y, Jones B, Tomas A. Spatiotemporal Control of GLP-1 Receptor Activity. Curr Opin Endocr Metab Res (2021) 16:19–27. doi: 10.1016/j.coemr.2020.07.003
7. Kenakin T. Biased Receptor Signaling in Drug Discovery. Pharmacol Rev (2019) 71:267–315. doi: 10.1124/pr.118.016790
8. Dunphy JL, Taylor RG, Fuller PJ. Tissue Distribution of Rat Glucagon Receptor and GLP-1 Receptor Gene Expression. Mol Cell Endocrinol (1998) 141:179–86. doi: 10.1016/S0303-7207(98)00096-3
9. Hernández C, Bogdanov P, Corraliza L, García-Ramírez M, Solà-Adell C, Arranz JA, et al. Topical Administration of GLP-1 Receptor Agonists Prevents Retinal Neurodegeneration in Experimental Diabetes. Diabetes (2016) 65:172–87. doi: 10.2337/db15-0443
10. Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, et al. Inkt Cells Induce FGF21 for Thermogenesis and Are Required for Maximal Weight Loss in GLP1 Therapy. Cell Metab (2016) 24:510–9. doi: 10.1016/j.cmet.2016.08.003
11. Pyke C, Knudsen LB. The Glucagon-Like Peptide-1 Receptor–or Not? Endocrinology (2013) 154:4–8. doi: 10.1210/en.2012-2124
12. Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 Receptor Localization in Monkey and Human Tissue: Novel Distribution Revealed With Extensively Validated Monoclonal Antibody. Endocrinology (2014) 155:1280–90. doi: 10.1210/en.2013-1934
13. Heppner KM, Kirigiti M, Secher A, Paulsen SJ, Buckingham R, Pyke C, et al. Expression and Distribution of Glucagon-Like Peptide-1 Receptor mRNA, Protein and Binding in the Male Nonhuman Primate (Macaca Mulatta) Brain. Endocrinology (2015) 156:255–67. doi: 10.1210/en.2014-1675
14. Jensen CB, Pyke C, Rasch MG, Dahl AB, Knudsen LB, Secher A. Characterization of the Glucagonlike Peptide-1 Receptor in Male Mouse Brain Using a Novel Antibody and In Situ Hybridization. Endocrinology (2018) 159:665–75. doi: 10.1210/en.2017-00812
15. Gray SM, Xin Y, Ross EC, Chazotte BM, Capozzi ME, El K, et al. Discordance Between GLP-1R Gene and Protein Expression in Mouse Pancreatic Islet Cells. J Biol Chem (2020) 295:11529–41. doi: 10.1074/jbc.RA120.014368
16. Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, et al. Identification and Characterization of GLP-1 Receptor-Expressing Cells Using a New Transgenic Mouse Model. Diabetes (2014) 63:1224–33. doi: 10.2337/db13-1440
17. Cork SC, Richards JE, Holt MK, Gribble FM, Reimann F, Trapp S. Distribution and Characterisation of Glucagon-like Peptide-1 Receptor Expressing Cells in the Mouse Brain. Mol Metab (2015) 4:718–31. doi: 10.1016/j.molmet.2015.07.008
18. Williams EK, Chang RB, Strochlic DE, Umans BD, Lowell BB, Liberles SD. Sensory Neurons That Detect Stretch and Nutrients in the Digestive System. Cell (2016) 166:209–21. doi: 10.1016/j.cell.2016.05.011
19. Andersen DB, Grunddal KV, Pedersen J, Kuhre RE, Lund ML, Holst JJ, et al. Using a Reporter Mouse to Map Known and Novel Sites of GLP-1 Receptor Expression in Peripheral Tissues of Male Mice. Endocrinology (2021) 162. doi: 10.1210/endocr/bqaa246
20. Kanse SM, Kreymann B, Ghatei MA, Bloom SR. Identification and Characterization of Glucagon-Like Peptide-1 7-36 Amide-Binding Sites in the Rat Brain and Lung. FEBS Lett (1988) 241:209–12. doi: 10.1016/0014-5793(88)81063-9
21. Ast J, Arvaniti A, Fine NHF, Nasteska D, Ashford FB, Stamataki Z, et al. Super-Resolution Microscopy Compatible Fluorescent Probes Reveal Endogenous Glucagon-Like Peptide-1 Receptor Distribution and Dynamics. Nat Commun (2020) 11:467. doi: 10.1038/s41467-020-14309-w
22. de Graaf C, Donnelly D, Wootten D, Lau J, Sexton PM, Miller LJ, et al. Glucagon-Like Peptide-1 and Its Class B G Protein-Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol Rev (2016) 68:954–1013. doi: 10.1124/pr.115.011395
23. Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, et al. Cryo-EM Structure of the Activated GLP-1 Receptor in Complex With a G Protein. Nature (2017) 546:248–53. doi: 10.1038/nature22394
24. Liang Y-L, Khoshouei M, Glukhova A, Furness SGB, Zhao P, Clydesdale L, et al. Phase-Plate cryo-EM Structure of a Biased Agonist-Bound Human GLP-1 receptor-Gs Complex. Nature (2018) 555:121–5. doi: 10.1038/nature25773
25. Zhao P, Liang Y-L, Belousoff MJ, Deganutti G, Fletcher MM, Willard FS, et al. Activation of the GLP-1 Receptor by a non-Peptidic Agonist. Nature (2020) 577:432–6. doi: 10.1038/s41586-019-1902-z
26. Hoare SRJ, Tewson PH, Quinn AM, Hughes TE. A Kinetic Method for Measuring Agonist Efficacy and Ligand Bias Using High Resolution Biosensors and a Kinetic Data Analysis Framework. Sci Rep (2020) 10:1–19. doi: 10.1038/s41598-020-58421-9
27. Oldham WM, Hamm HE. Heterotrimeric G Protein Activation by G-protein-coupled Receptors. Nat Rev Mol Cell Biol (2008) 9:60–71. doi: 10.1038/nrm2299
28. Tomas A, Jones B, Leech C. New Insights Into Beta-Cell Glp-1 Receptor and cAMP Signaling. J Mol Biol (2019) 28: 432(5):1347–66. doi: 10.1016/j.jmb.2019.08.009
29. Kang G, Chepurny OG, Holz GG. cAMP-regulated Guanine Nucleotide Exchange Factor II (Epac2) Mediates Ca2+-induced Ca2+ Release in INS-1 Pancreatic Beta-Cells. J Physiol (Lond) (2001) 536:375–85. doi: 10.1111/j.1469-7793.2001.0375c.xd
30. Kang G, Leech CA, Chepurny OG, Coetzee WA, Holz GG. Role of the cAMP Sensor Epac as a Determinant of KATP Channel ATP Sensitivity in Human Pancreatic Beta-Cells and Rat INS-1 Cells. J Physiol (Lond) (2008) 586:1307–19. doi: 10.1113/jphysiol.2007.143818
31. Yosida M, Dezaki K, Uchida K, Kodera S, Lam NV, Ito K, et al. Involvement of cAMP/EPAC/TRPM2 Activation in Glucose- and Incretin-Induced Insulin Secretion. Diabetes (2014) 63:3394–403. doi: 10.2337/db13-1868
32. Gromada J, Ding WG, Barg S, Renström E, Rorsman P. Multisite Regulation of Insulin Secretion by cAMP-increasing Agonists: Evidence That Glucagon-Like Peptide 1 and Glucagon Act Via Distinct Receptors. Pflugers Arch (1997) 434:515–24. doi: 10.1007/s004240050431
33. Friedrichsen BN, Neubauer N, Lee YC, Gram VK, Blume N, Petersen JS, et al. Stimulation of Pancreatic β-Cell Replication by Incretins Involves Transcriptional Induction of Cyclin D1 Via Multiple Signalling Pathways. J Endocrinol (2006) 188:481–92. doi: 10.1677/joe.1.06160
34. Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, et al. GLP-1 Receptor Activation Improves Beta Cell Function and Survival Following Induction of Endoplasmic Reticulum Stress. Cell Metab (2006) 4:391–406. doi: 10.1016/j.cmet.2006.10.001
35. Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, et al. Glucagon-Like Peptide 1 Inhibits Cell Apoptosis and Improves Glucose Responsiveness of Freshly Isolated Human Islets. Endocrinology (2003) 144:5149–58. doi: 10.1210/en.2003-0323
36. Wang X, Zhou J, Doyle ME, Egan JM. Glucagon-Like Peptide-1 Causes Pancreatic Duodenal Homeobox-1 Protein Translocation From the Cytoplasm to the Nucleus of Pancreatic Beta-Cells by a Cyclic Adenosine Monophosphate/Protein Kinase A-dependent Mechanism. Endocrinology (2001) 142:1820–7. doi: 10.1210/endo.142.5.8128
37. Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, et al. cAMP Promotes Pancreatic Beta-Cell Survival Via CREB-mediated Induction of IRS2. Genes Dev (2003) 17:1575–80. doi: 10.1101/gad.1097103
38. Montrose-Rafizadeh C, Avdonin P, Garant MJ, Rodgers BD, Kole S, Yang H, et al. Pancreatic Glucagon-Like Peptide-1 Receptor Couples to Multiple G Proteins and Activates Mitogen-Activated Protein Kinase Pathways in Chinese Hamster Ovary Cells. Endocrinology (1999) 140:1132–40. doi: 10.1210/endo.140.3.6550
39. Wheeler MB, Lu M, Dillon JS, Leng XH, Chen C, Boyd AE. Functional Expression of the Rat Glucagon-Like Peptide-I Receptor, Evidence for Coupling to Both Adenylyl Cyclase and Phospholipase-C. Endocrinology (1993) 133:57–62. doi: 10.1210/endo.133.1.8391428
40. Shigeto M, Ramracheya R, Tarasov AI, Cha CY, Chibalina MV, Hastoy B, et al. GLP-1 Stimulates Insulin Secretion by PKC-dependent TRPM4 and TRPM5 Activation. J Clin Invest (2015) 125:4714–28. doi: 10.1172/JCI81975
41. Oduori OS, Murao N, Shimomura K, Takahashi H, Zhang Q, Dou H, et al. Gs/Gq Signaling Switch in β Cells Defines Incretin Effectiveness in Diabetes. J Clin Invest (2020) 130:6639–55. doi: 10.1172/JCI140046
42. Shigeto M, Cha CY, Rorsman P. Kaku K. A Role of PLC/PKC-dependent Pathway in GLP-1-stimulated Insulin Secretion. J Mol Med (Berl) (2017) 95:361–8. doi: 10.1007/s00109-017-1508-6
43. Liu M, Simon MI. Regulation by cAMP-dependent Protein Kinease of a G-protein-mediated Phospholipase C. Nature (1996) 382:83–7. doi: 10.1038/382083a0
44. Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, et al. Lack of Beta-Arrestin Signaling in the Absence of Active G Proteins. Nat Commun (2018) 9:341. doi: 10.1038/s41467-017-02661-3
45. Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, et al. Manifold Roles of β-Arrestins in GPCR Signaling Elucidated With siRNA and CRISPR/Cas9. Sci Signal (2018) 11:eaat7650. doi: 10.1126/scisignal.aat7650
46. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu J-C, Olefsky JM. Beta-Arrestin-1 Mediates Glucagon-Like Peptide-1 Signaling to Insulin Secretion in Cultured Pancreatic Beta Cells. Proc Natl Acad Sci USA (2008) 105:6614–9. doi: 10.1073/pnas.0710402105
47. Quoyer J, Longuet C, Broca C, Linck N, Costes S, Varin E, et al. GLP-1 Mediates Antiapoptotic Effect by Phosphorylating Bad Through a Beta-Arrestin 1-Mediated ERK1/2 Activation in Pancreatic Beta-Cells. J Biol Chem (2010) 285:1989–2002. doi: 10.1074/jbc.M109.067207
48. Buteau J, Foisy S, Joly E, Prentki M. Glucagon-Like Peptide 1 Induces Pancreatic Beta-Cell Proliferation Via Transactivation of the Epidermal Growth Factor Receptor. Diabetes (2003) 52:124–32. doi: 10.2337/diabetes.52.1.124
49. Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M. Glucagon-Like Peptide-1 Prevents Beta Cell Glucolipotoxicity. Diabetologia (2004) 47:806–15. doi: 10.1007/s00125-004-1379-6
50. Zhu L, Almaça J, Dadi PK, Hong H, Sakamoto W, Rossi M, et al. β-Arrestin-2 is an Essential Regulator of Pancreatic β-Cell Function Under Physiological and Pathophysiological Conditions. Nat Commun (2017) 8:14295. doi: 10.1038/ncomms14295
51. Barella LF, Rossi M, Zhu L, Cui Y, Mei FC, Cheng X, et al. β-Cell-intrinsic β-Arrestin 1 Signaling Enhances Sulfonylurea-Induced Insulin Secretion. J Clin Invest (2019) 129:3732–7. doi: 10.1172/JCI126309
52. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular Mechanism of β-Arrestin-Biased Agonism At Seven-Transmembrane Receptors. Annu Rev Pharmacol Toxicol (2012) 52:179–97. doi: 10.1146/annurev.pharmtox.010909.105800
53. Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, et al. β-Arrestin2-mediated Inotropic Effects of the Angiotensin II Type 1A Receptor in Isolated Cardiac Myocytes. PNAS (2006) 103:16284–9. doi: 10.1073/pnas.0607583103
54. Unson CG, Gurzenda EM, Merrifield RB. Biological Activities of des-His1[Glu9]glucagon Amide, a Glucagon Antagonist. Peptides (1989) 10:1171–7. doi: 10.1016/0196-9781(89)90010-7
55. Jones B, Buenaventura T, Kanda N, Chabosseau P, Owen BM, Scott R, et al. Targeting GLP-1 Receptor Trafficking to Improve Agonist Efficacy. Nat Commun (2018) 9:1602. doi: 10.1038/s41467-018-03941-2
56. Zhang H, Sturchler E, Zhu J, Nieto A, Cistrone PA, Xie J, et al. Autocrine Selection of a GLP-1R G-Protein Biased Agonist With Potent Antidiabetic Effects. Nat Commun (2015) 6:8918–13. doi: 10.1038/ncomms9918
57. Lucey M, Pickford P, Bitsi S, Minnion J, Ungewiss J, Schoeneberg K, et al. Disconnect Between Signalling Potency and In Vivo Efficacy of Pharmacokinetically Optimised Biased Glucagon-Like Peptide-1 Receptor Agonists. Mol Metab (2020) 37:100991. doi: 10.1016/j.molmet.2020.100991
58. Pickford P, Lucey M, Fang Z, Bitsi S, la Serna de JB, Broichhagen J, et al. Signalling, Trafficking and Glucoregulatory Properties of Glucagon-Like Peptide-1 Receptor Agonists Exendin-4 and Lixisenatide. Br J Pharmacol (2020) 177(17):3905–23. doi: 10.1111/bph.15134
59. Fremaux J, Venin C, Mauran L, Zimmer R, Koensgen F, Rognan D, et al. Ureidopeptide GLP-1 Analogues With Prolonged Activity In Vivo Via Signal Bias and Altered Receptor Trafficking. Chem Sci (2019) 10:9872–9. doi: 10.1039/c9sc02079a
60. Wang M, Yao P, Gao M, Jin J, Yu Y. Novel Fatty Chain-Modified GLP-1R G-Protein Biased Agonist Exerts Prolonged Anti-Diabetic Effects Through Targeting Receptor Binding Sites. RSC Adv (2020) 10:8044–53. doi: 10.1039/C9RA10593J
61. Jones B, McGlone ER, Fang Z, Pickford P, Corrêa IR, Oishi A, et al. Genetic and Biased Agonist-Mediated Reductions in β-Arrestin Recruitment Prolong cAMP Signalling At Glucagon Family Receptors. J Biol Chem (2020) 296:100133. doi: 10.1074/jbc.RA120.016334
62. Yuliantie E, Darbalaei S, Dai A, Zhao P, Yang D, Sexton PM, et al. Pharmacological Characterization of Mono-, Dual- and Tri- Peptidic Agonists At GIP and GLP-1 Receptors. Biochem Pharmacol (2020) 177:114001. doi: 10.1016/j.bcp.2020.114001
63. Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an Imbalanced and Biased Dual GIP and GLP-1 Receptor Agonist. JCI Insight (2020) 5:1202. doi: 10.1172/jci.insight.140532
64. Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and Safety of LY3298176, a Novel Dual GIP and GLP-1 Receptor Agonist, in Patients With Type 2 Diabetes: A Randomised, Placebo-Controlled and Active Comparator-Controlled Phase 2 Trial. Lancet (2018) 392:2180–93. doi: 10.1016/S0140-6736(18)32260-8
65. Ferguson SS. Evolving Concepts in G Protein-Coupled Receptor Endocytosis: The Role in Receptor Desensitization and Signaling. Pharmacol Rev (2001) 53:1–24.
66. Maxfield FR, McGraw TE. Endocytic Recycling. Nat Rev Mol Cell Biol (2004) 5:121–32. doi: 10.1038/nrm1315
67. Scorrano L, De Matteis MA, Emr S, Giordano F, Hajnóczky G, Kornmann B, et al. Coming Together to Define Membrane Contact Sites. Nat Commun (2019) 10:1287–11. doi: 10.1038/s41467-019-09253-3
68. Todkar K, Chikhi L, Germain M. Mitochondrial Interaction With the Endosomal Compartment in Endocytosis and Mitochondrial Transfer. Mitochondrion (2019) 49:284–8. doi: 10.1016/j.mito.2019.05.003
69. English AR, Voeltz GK. Endoplasmic Reticulum Structure and Interconnections With Other Organelles. Cold Spring Harb Perspect Biol (2013) 5:a013227–a013227. doi: 10.1101/cshperspect.a013227
70. Tu Y, Zhao L, Billadeau DD, Jia D. Endosome-to-TGN Trafficking: Organelle-Vesicle and Organelle-Organelle Interactions. Front Cell Dev Biol (2020) 8:163. doi: 10.3389/fcell.2020.00163
71. Buenaventura T, Bitsi S, Laughlin WE, Burgoyne T, Lyu Z, Oqua AI, et al. Agonist-Induced Membrane Nanodomain Clustering Drives GLP-1 Receptor Responses in Pancreatic Beta Cells. PloS Biol (2019) 17:e3000097. doi: 10.1371/journal.pbio.3000097
72. Syme CA, Zhang L, Bisello A. Caveolin-1 Regulates Cellular Trafficking and Function of the Glucagon-Like Peptide 1 Receptor. Mol Endocrinol (2006) 20:3400–11. doi: 10.1210/me.2006-0178
73. Roed SN, Nøhr AC, Wismann P, Iversen H, Bräuner-Osborne H, Knudsen SM, et al. Functional Consequences of Glucagon-Like Peptide-1 Receptor Cross-Talk and Trafficking. J Biol Chem (2015) 290:1233–43. doi: 10.1074/jbc.M114.592436
74. Widmann C, Dolci W, Thorens B. Agonist-Induced Internalization and Recycling of the Glucagon-Like Peptide-1 Receptor in Transfected Fibroblasts and in Insulinomas. Biochem J (1995) 310( Pt 1):203–14. doi: 10.1042/bj3100203
75. Kaksonen M, Roux A. Mechanisms of Clathrin-Mediated Endocytosis. Nat Rev Mol Cell Biol (2018) 19:313–26. doi: 10.1038/nrm.2017.132
76. Scott MGH, Benmerah A, Muntaner O, Marullo S. Recruitment of Activated G Protein-Coupled Receptors to Pre-Existing Clathrin-Coated Pits in Living Cells. J Biol Chem (2002) 277:3552–9. doi: 10.1074/jbc.M106586200
77. Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, et al. The beta2-adrenergic Receptor/Betaarrestin Complex Recruits the Clathrin Adaptor AP-2 During Endocytosis. Proc Natl Acad Sci USA (1999) 96:3712–7. doi: 10.1073/pnas.96.7.3712
78. Huang G, Li J, Fu H, Yan Z, Bu G, He X, et al. Characterization of Glucagon-Like Peptide 1 Receptor (GLP1R) Gene in Chickens: Functional Analysis, Tissue Distribution, and Identification of its Transcript Variants. Domest Anim Endocrinol (2012) 43:1–15. doi: 10.1016/j.domaniend.2012.01.005
79. Buenaventura T, Kanda N, Douzenis PC, Jones B, Bloom SR, Chabosseau P, et al. A Targeted Rnai Screen Identifies Endocytic Trafficking Factors That Control Glp-1 Receptor Signaling in Pancreatic β-Cells. Diabetes (2018) 67:385–99. doi: 10.2337/db17-0639
80. Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS, et al. HIP1 Functions in Clathrin-Mediated Endocytosis Through Binding to Clathrin and Adaptor Protein 2. J Biol Chem (2001) 276:39271–6. doi: 10.1074/jbc.C100401200
81. Thompson A, Kanamarlapudi V. Agonist-Induced Internalisation of the Glucagon-Like Peptide-1 Receptor is Mediated by the Gαq Pathway. Biochem Pharmacol (2015) 93:72–84. doi: 10.1016/j.bcp.2014.10.015
82. Parton RG, del Pozo MA, Vassilopoulos S, Nabi IR, Le Lay S, Lundmark R, et al. Caveolae: The Faqs. Traffic (2020) 21:181–5. doi: 10.1111/tra.12689
83. Sungkaworn T, Jobin M-L, Burnecki K, Weron A, Lohse MJ, Calebiro D. Single-Molecule Imaging Reveals Receptor-G Protein Interactions At Cell Surface Hot Spots. Nature (2017) 550:543–7. doi: 10.1038/nature24264
84. Manna M, Niemelä M, Tynkkynen J, Javanainen M, Kulig W, Müller DJ, et al. Mechanism of Allosteric Regulation of β2-Adrenergic Receptor by Cholesterol. Elife (2016) 5:128. doi: 10.7554/eLife.18432
85. Ansell TB, Song W, Sansom MSP. The Glycosphingolipid Gm3 Modulates Conformational Dynamics of the Glucagon Receptor. Biophys J (2020) 119:300–13. doi: 10.1016/j.bpj.2020.06.009
86. Cullen PJ, Steinberg F. To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat Rev Mol Cell Biol (2018) 19:679–96. doi: 10.1038/s41580-018-0053-7
87. Fang Z, Chen S, Manchanda Y, Bitsi S, Pickford P, David A, et al. Ligand-Specific Factors Influencing GLP-1 Receptor Post-Endocytic Trafficking and Degradation in Pancreatic Beta Cells. Int J Mol Sci (2020) 21:8404. doi: 10.3390/ijms21218404
88. Woodman PG. Biogenesis of the Sorting Endosome: The Role of Rab5. Traffic (2000) 1:695–701. doi: 10.1034/j.1600-0854.2000.010902.x
89. Girada SB, Kuna RS, Bele S, Zhu Z, Chakravarthi NR, Dimarchi RD, et al. Gαs Regulates Glucagon-Like Peptide 1 Receptor-mediated Cyclic AMP Generation At Rab5 Endosomal Compartment. Mol Metab (2017) 6:1173–85. doi: 10.1016/j.molmet.2017.08.002
90. Zerial M, McBride H. Rab Proteins as Membrane Organizers. Nat Rev Mol Cell Biol (2001) 2:107–17. doi: 10.1038/35052055
91. Dores MR, Paing MM, Lin H, Montagne WA, Marchese A, Trejo J. AP-3 Regulates PAR1 Ubiquitin-Independent MVB/lysosomal Sorting Via an ALIX-mediated Pathway. Mol Biol Cell (2012) 23:3612–23. doi: 10.1091/mbc.E12-03-0251
92. Roed SN, Wismann P, Underwood CR, Kulahin N, Iversen H, Cappelen KA, et al. Real-Time Trafficking and Signaling of the Glucagon-Like Peptide-1 Receptor. Mol Cell Endocrinol (2014) 382:938–49. doi: 10.1016/j.mce.2013.11.010
93. McGarvey JC, Xiao K, Bowman SL, Mamonova T, Zhang Q, Bisello A, et al. Actin-Sorting Nexin 27 (Snx27)-Retromer Complex Mediates Rapid Parathyroid Hormone Receptor Recycling. J Biol Chem (2016) 291:10986–1002. doi: 10.1074/jbc.M115.697045
94. Bahouth SW, Nooh MM. Barcoding of GPCR Trafficking and Signaling Through the Various Trafficking Roadmaps by Compartmentalized Signaling Networks. Cell Signal (2017) 36:42–55. doi: 10.1016/j.cellsig.2017.04.015
95. Chen K-E, Healy MD, Collins BM. Towards a Molecular Understanding of Endosomal Trafficking by Retromer and Retriever. Traffic (2019) 20:465–78. doi: 10.1111/tra.12649
96. Lu J, Willars GB. Endothelin-Converting Enzyme-1 Regulates Glucagon-Like Peptide-1 Receptor Signalling and Resensitisation. Biochem J (2019) 476:513–33. doi: 10.1042/BCJ20180853
97. Zhao P, Canals M, Murphy JE, Klingler D, Eriksson EM, Pelayo J-C, et al. Agonist-Biased Trafficking of Somatostatin Receptor 2A in Enteric Neurons. J Biol Chem (2013) 288:25689–700. doi: 10.1074/jbc.M113.496414
98. Bukrinski JT, Sønderby P, Antunes F, Andersen B, Schmidt EGW, Peters GHJ, et al. Glucagon-Like Peptide 1 Conjugated to Recombinant Human Serum Albumin Variants With Modified Neonatal Fc Receptor Binding Properties. Impact on Molecular Structure and Half-Life. Biochemistry (2017) 56:4860–70. doi: 10.1021/acs.biochem.7b00492
99. Hennen S, Kodra JT, Soroka V, Krogh BO, Wu X, Kaastrup P, et al. Structural Insight Into Antibody-Mediated Antagonism of the Glucagon-like Peptide-1 Receptor. Sci Rep (2016) 6:26236. doi: 10.1038/srep26236
100. Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, et al. Clathrin-Independent Endocytosis of Ubiquitinated Cargos. PNAS (2005) 102:2760–5. doi: 10.1073/pnas.0409817102
101. Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, et al. Persistent cAMP-signals Triggered by Internalized G-protein-coupled Receptors. PloS Biol (2009) 7:e1000172. doi: 10.1371/journal.pbio.1000172
102. Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, et al. Sustained Cyclic AMP Production by Parathyroid Hormone Receptor Endocytosis. Nat Chem Biol (2009) 5:734–42. doi: 10.1038/nchembio.206
103. Vilardaga J-P, Jean-Alphonse FG, Gardella TJ. Endosomal Generation of cAMP in GPCR Signaling. Nat Chem Biol (2014) 10:700–6. doi: 10.1038/nchembio.1611
104. Tsvetanova NG, Zastrow von M. Spatial Encoding of Cyclic AMP Signaling Specificity by GPCR Endocytosis. Nat Chem Biol (2014) 10:1061–5. doi: 10.1038/nchembio.1665
105. Costa-Neto CM, Parreiras-E-Silva LT. Bouvier M. A Pluridimensional View of Biased Agonism. Mol Pharmacol (2016) 90:587–95. doi: 10.1124/mol.116.105940
106. Gidon A, Feinstein TN, Xiao K, Vilardaga J-P. Studying the Regulation Of Endosomal cAMP Production in GPCR Signaling. Methods Cell Biol (2016) 132:109–26. doi: 10.1016/bs.mcb.2015.10.007
107. Kuna RS, Girada SB, Asalla S, Vallentyne J, Maddika S, Patterson JT, et al. Glucagon-Like Peptide-1 Receptor-Mediated Endosomal cAMP Generation Promotes Glucose-Stimulated Insulin Secretion in Pancreatic β-Cells. Am J Physiol Endocrinol Metab (2013) 305:E161–70. doi: 10.1152/ajpendo.00551.2012
108. Fletcher MM, Halls ML, Zhao P, Clydesdale L, Christopoulos A, Sexton PM, et al. Glucagon-Like Peptide-1 Receptor Internalisation Controls Spatiotemporal Signalling Mediated by Biased Agonists. Biochem Pharmacol (2018) 156:406–19. doi: 10.1016/j.bcp.2018.09.003
109. Lester LB, Langeberg LK, Scott JD. Anchoring of Protein Kinase A Facilitates Hormone-Mediated Insulin Secretion. Proc Natl Acad Sci USA (1997) 94:14942–7. doi: 10.1073/pnas.94.26.14942
110. Hinke SA, Navedo MF, Ulman A, Whiting JL, Nygren PJ, Tian G, et al. Anchored Phosphatases Modulate Glucose Homeostasis. EMBO J (2012) 31:3991–4004. doi: 10.1038/emboj.2012.244
111. Fang Z, Chen S, Pickford P, Broichhagen J, Hodson DJ, Corrêa IR, et al. The Influence of Peptide Context on Signaling and Trafficking of Glucagon-like Peptide-1 Receptor Biased Agonists. ACS Pharmacol Transl Sci (2020) 3:345–60. doi: 10.1021/acsptsci.0c00022
112. Klarenbeek JB, Goedhart J, Hink MA, Gadella TWJ. Jalink K. A mTurquoise-based cAMP Sensor for Both FLIM and Ratiometric Read-Out has Improved Dynamic Range. PloS One (2011) 6:e19170. doi: 10.1371/journal.pone.0019170
113. Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalboge LS, et al. The Arcuate Nucleus Mediates GLP-1 Receptor Agonist Liraglutide-Dependent Weight Loss. J Clin Invest (2014) 124:4473–88. doi: 10.1172/JCI75276
114. Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide Lowers Body Weight in Rodents Via Distributed Neural Pathways. JCI Insight (2020) 5:891. doi: 10.1172/jci.insight.133429
115. Fu Z, Gong L, Liu J, Wu J, Barrett EJ, Aylor KW, et al. Brain Endothelial Cells Regulate Glucagon-Like Peptide 1 Entry Into the Brain Via a Receptor-Mediated Process. Front Physiol (2020) 11:555. doi: 10.3389/fphys.2020.00555
Keywords: GLP-1, GLP-1R, biased agonism, receptor trafficking, pancreatic beta cells
Citation: Marzook A, Tomas A and Jones B (2021) The Interplay of Glucagon-Like Peptide-1 Receptor Trafficking and Signalling in Pancreatic Beta Cells. Front. Endocrinol. 12:678055. doi: 10.3389/fendo.2021.678055
Received: 08 March 2021; Accepted: 15 April 2021;
Published: 10 May 2021.
Edited by:
Peter Flatt, Ulster University, United KingdomReviewed by:
Jonathan Campbell, Duke University, United StatesCatherine Arden, Newcastle University, United Kingdom
Copyright © 2021 Marzook, Tomas and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Jones, YmVuLmpvbmVzQGltcGVyaWFsLmFjLnVr; Alejandra Tomas, YS50b21hcy1jYXRhbGFAaW1wZXJpYWwuYWMudWs=