 Veronica Costiniti
Veronica Costiniti Guilherme H. Bomfim
Guilherme H. Bomfim Erna Mitaishvili
Erna Mitaishvili Ga-Yeon Son
Ga-Yeon Son Yi Li
Yi Li Rodrigo S. Lacruz
Rodrigo S. Lacruz- Department Molecular Pathobiology, College of Dentistry, New York University, New York, NY, United States
Most cells use calcium (Ca2+) as a second messenger to convey signals that affect a multitude of biological processes. The ability of Ca2+ to bind to proteins to alter their charge and conformation is essential to achieve its signaling role. Cytosolic Ca2+ (cCa2+) concentration is maintained low at ~100 nM so that the impact of elevations in cCa2+ is readily sensed and transduced by cells. However, such elevations in cCa2+ must be transient to prevent detrimental effects. Cells have developed a variety of systems to rapidly clear the excess of cCa2+ including Ca2+ pumps, exchangers and sequestering Ca2+ within intracellular organelles. This Ca2+ signaling toolkit is evolutionarily adapted so that each cell, tissue, and organ can fulfill its biological function optimally. One of the most specialized cells in mammals are the enamel forming cells, the ameloblasts, which also handle large quantities of Ca2+. The end goal of ameloblasts is to synthesize, secrete and mineralize a unique proteinaceous matrix without the benefit of remodeling or repair mechanisms. Ca2+ uptake into ameloblasts is mainly regulated by the store operated Ca2+ entry (SOCE) before it is transported across the polarized ameloblasts to reach the insulated enamel space. Here we review the ameloblasts Ca2+ signaling toolkit and address how the common electronegative non-metal fluoride can alter its function, potentially addressing the biology of dental fluorosis.
Introduction
Calcium (Ca2+) is the third most abundant metal in nature. The chemical properties of Ca2+, its radius (0.99 Å), hydration energy and charge, are such that they facilitate its role as a signaling messenger in cells favored over other cations, such as magnesium (Mg2+), that have a smaller radius and higher hydration energy (1). Its chemistry also allows Ca2+ to optimally accept binding sites of irregular geometry, in contrast with Mg2+, which requires octahedral-binding sites that are not commonly found in proteins. Instead, proteins offer a wide range of binding sites for Ca2+ (2) and therefore, Ca2+ has become a global phenomenon in cell signaling to the point that it is virtually impossible to consider a biological function where Ca2+ does not play a role (3). The impact of Ca2+ signaling on cell function is closely linked to changes in its concentration within cells. Cytosolic Ca2+ (cCa2+) concentration is maintained low at ~100 nM compared to ~2 mM outside the cell and therefore, small changes in cCa2+ have important effects (3). An excessively high elevation in cCa2+ could result in mitochondrial Ca2+ overloading, activation of proteases, activation of DNA-fragmenting enzymes and cell death, consequently, elevations in cCa2+ must be transient (4). Cells have adopted complex systems to clear excess of Ca2+ including pumps, exchangers, Ca2+-binding proteins as well as using intracellular organelles such as the endoplasmic reticulum (ER) and mitochondria as Ca2+ sinks (5).
Across cells and organs, the involvement of Ca2+ in signaling can be limited in scope, such as the elementary events involving Ca2+ exchange between the ER and mitochondria or can be more global such as the Ca2+ waves associated with muscle contraction (6). Although the signaling toolkit in mammalian cells is highly conserved, the physiological output of these signaling events is logically distinct across cells and organs. In some ways, it would be reasonable to consider that the role of Ca2+ in mineralizing cells, i.e. dental enamel and bone cells, would be well understood given their reliance on Ca2+ to form these tissues, however, this isn’t the case. The most highly calcified tissue of vertebrates, dental enamel, offers an attractive model for potentially decoding relevant Ca2+ signatures in mineralizing systems, yet knowledge of the identity and function of the Ca2+ toolkit of the enamel forming cells remains in its infancy. In this review, we will explore the most relevant aspects of what is known about Ca2+ transport in enamel cells, the ameloblasts, and will focus on the effects of a worldwide phenomenon caused by excessive amounts of another metal, fluoride, and how these two metals converge in a disease known as dental fluorosis.
Ca2+ in Enamel
Ca2+ is a key element in the composition of dental enamel (7), being required during the two stages of enamel development, namely the secretory and maturation stages. In the secretory stage, thin enamel crystals elongate within an organic matrix template formed by key enamel structural proteins (e.g. amelogenin, ameloblastin and enamelin) in a highly organized fashion (7). During the maturation or mineralization stage, crystals expand in width and thickness as the organic matrix is removed. Ca2+ requirements increase at this stage (7). Ameloblasts are epithelial cells of ectodermal origin that form and mineralize enamel. A mention of their morphological characteristics is relevant because secretory ameloblasts are ~65 µm in height and maturation are ~35 µm and both maintain a narrow diameter of ~5 µm. Therefore, ameloblasts are highly polarized cells and form a tight cell barrier, limiting intercellular passage of ions and minerals (7). The possibility of a passive Ca2+ transport system across the interstitial space is an unlikely scenario given the presence of tight junctions adjoining the distal pole of maturation stage ameloblasts (8). Passive transport also appears counterintuitive because the highly organized nature of the mineralized enamel crystals suggests a non-haphazard stoichiometric accumulation of ions in the enamel fluid (9).
Ameloblasts not only use Ca2+ as a signaling messenger, of which we have limited understanding, they must also transport Ca2+ safely across the cell. Hubbard indicated that the transcellular Ca2+ transport in ameloblasts is complex and requires an understanding of several steps (10): a) entry, b) transit and c) extrusion. For a very long time, the molecular pathways regulating these steps in ameloblasts had remained undefined. Recently, several studies of human mutations and rodent models indicate that the store operated Ca2+ entry (SOCE) pathway is essential for enamel mineralization, being the primary Ca2+ uptake systems in ameloblasts (11–13).
Store Operated Ca2+ Entry in Ameloblasts
SOCE, also known as Ca2+ release activated Ca2+ (CRAC) channels, is an essential and widely expressed Ca2+ influx channel to the extent that SOCE dominates the ability of non-excitable cells to uptake Ca2+ in physiological and pathological conditions (14). CRAC channels are formed by the Ca2+ sensors STIM1 and 2 (stromal interaction molecules) found in the membrane of the ER, and the channel pore formed by the ORAI proteins in the plasma membrane (ORAI1-3) (14–16). CRAC channels are activated following the stimulation of a cell surface receptor resulting in the production of PLC (phospholipase-C) and InsP3 (inositol 1,4,5 triphosphate), which in turns binds to its ER membrane receptors, the IP3R (17, 18). InsP3-IP3R interactions elicit the release of ER Ca2+ pools via the receptor channel (18, 19). A decline in luminal Ca2+ concentration in the ER triggers substantial conformational changes in STIM proteins leading to the binding and activation of the ORAI channel to allow a sustained Ca2+ influx (14, 20). Mutations in STIM1 or ORAI1 genes cause channelopathy including immune dysfunction and ectodermal dysplasia, amelogenesis imperfecta (AI), muscle weakness and anhidrosis (21, 22).
The links between SOCE and AI became evident in a series of papers published by the Feske laboratory reporting that patients with mutations in ORAI1 or STIM1 showed clear enamel defects described as type 2 hypomineralized AI (23, 24). Similar findings have been reported by other groups (25). Our studies using CRAC channels inhibitors such as synta-66, BTP-2 and GSK7975A, in rat ameloblasts, showed that these blockers markedly reduced or nearly abolished Ca2+ influx via SOCE (13, 26). We also showed that mice lacking STIM1/2 or ORAI1, which exhibited deficient SOCE in ameloblasts, had enamel defects raging from severe hypomineralization to disruptions in enamel crystal formation (11, 12). Combined, the human and mouse data highlight the critical role of SOCE as a key Ca2+ influx channel in enamel cells.
Potential Soce Modulators in Ameloblasts
Although STIM1/2 and ORAI1-3 proteins are the core components of SOCE, a number of molecular modulators have been described having various effects on SOCE, reviewed in (27). These include proteins that stabilize STIM–ORAI interactions, stimulate STIM1 conformational changes or induce slow Ca2+-dependent inactivation (27). A recent study reported that mutations in the solute carrier SLC10A7 gene results in hypomineralized enamel and AI (28). A more recent study suggested that SLC10A7 is a negative modulator of SOCE, because the knockdown of SLC10A7 resulted in increased SOCE (29). The mechanism of the interaction between SLC10A7 and SOCE is unknown, although some possibilities have been proposed: 1) disrupts sarcoendoplasmic reticulum calcium transport ATPase (SERCA) function, 2) destabilizes STIM1 oligomers or 3) interferes with ORAI. Based on human and mouse studies, a possible cause of the enamel defects caused by SLC10A7 mutations was associated with a deficiency in glycosaminoglycan (GAG) synthesis (28). GAGs are important components of extracellular matrix and when GAG degradation pathways are disrupted due to enzyme deficiency, GAGs accumulate causing skeletal dysplasia, and could also be the cause of enamel defects (30). However, this novel connection between SOCE and SLC10A7 suggests that Ca2+ could also play a role.
Another negative modulator of SOCE, known as SARAF (SOCE-associated regulatory factor), may be an important factor in SOCE modulation in ameloblasts. SARAF (also known as TMEM66) is an ER membrane protein that associates with STIM to promote Ca2+ dependent inactivation of SOCE (31). We found that Saraf was significantly upregulated in maturation stage ameloblasts relative to secretory ameloblasts (32), suggesting a possible function in enamel but no data is currently available.
Of significance are recent reports on Trpm7-inactive knock-in mutant mice which showed hypomineralized enamel in the heterozygous mice (33). The dental phenotype, as well as, other skeletal anomalies were ascribed to Mg2+ deficiency, which was required for alkaline phosphatase activity and mineralization (33). The transient receptor potential melastatin 7 (TRPM7) had been characterized as an ion channel permeable to divalent cations (Mg2+, Ca2+) linked to an intrinsic kinase domain, enabling it to modulate cellular functions (34). Faouzi et al. reported that although they did not consider TRPM7 a SOCE component, its kinase domain had a modulatory effect on SOCE (35). Our investigations into the role of TRPM7 in primary ameloblasts showed that TRPM7 potentiates Ca2+ influx via SOCE, and reported that its function is fully dependent on the prior activation of the ORAI1 channels (36).
Ca2+ Transit and Removal in Ameloblasts
Hubbard’s original groundbreaking studies on the identification of several Ca2+ proteins in ameloblasts and their upregulation in maturation (37–40), led him to suggest that Ca2+ may be transiting the ameloblasts via a type of safe tunneling mechanism which might implicate the ER tubules, a model he termed the “transcytosis” model (10, 41). The lack of enamel phenotype in mice with disrupted function of the Ca2+ binding proteins known as calbindins, reinforced this model and appeared to rule out a possible scenario where calbindins could ferry Ca2+ across the ameloblasts (42). Besides calbindins, ameloblasts express several other Ca2+ buffering proteins (8). However, how Ca2+ may be reaching the distal pole before it is extruded out of the cell, remains a significant gap in knowledge in enamel biology.
As highlighted above, it is important that elevations in cCa2+ concentrations are transient. To control this, cells employ two efficient systems to remove the Ca2+ from the cytosol: Ca2+ pumps and Ca2+ exchangers (3, 4, 43, 44).
Plasma membrane Ca2+-ATPases (PMCAs), or Ca2+ pumps, found in the cell membranes, that translocate Ca2+ from the inside to the outside of the cell, consuming ATP in the process (2, 45). PMCAs have high affinity for Ca2+ (Kd ~0.1 mM) but have low transport capacity, pumping 1 Ca2+ per ATP consumed (2, 45, 46). PMCAs are coded by the ATP2B genes and three of these genes (ATP2B1,3 and 4) are expressed in enamel cells, appearing to be upregulated during the secretory stage (47). However, whether PMCAs are functional in ameloblasts or whether there are differences across stages, has not been reported to date.
Ca2+ exchangers are also important in removing cCa2+ out of the cell and unlike PMCA, the exchangers do not require ATP hydrolysis. Instead, the Na+/Ca2+ exchangers use the chemical energy of the Na+ gradient (the Na+ concentration is much higher outside of the cell than inside the cell) to remove Ca2+ from the cytosol (3, 4). The exchangers have low Ca2+ affinity but high transport capacity with estimated values of ~5000 Hz for NCX (48). Because their function depends on electrochemical driving forces across the plasma membrane, the exchangers can operate in a forward mode (Ca2+ extrusion mode), which is their physiological function, as well as, in a reverse mode (Ca2+ uptake) (49).
There are two main families of Ca2+ exchangers, the NCX and NCKX. The NCX are Na+/Ca2+ exchangers in the plasma membrane, coded by the SLC8A genes, that remove 1 Ca2+ in exchange for 3 Na+ (49). By contrast, the NCKX family of proteins, coded by SLC24A genes, remove 1 Ca2+and 1 K+ in exchange for 4 Na+ (50). Unlike the NCX family, NCKXs require K+ to accomplish the Ca2+ exchange (50, 51). NCX were reported originally in ameloblasts in an outstanding paper by Okumura and colleagues (52). However, to date, there are no known mutations in the SLC8A genes causing AI. It was probably our identification of NCKX4 in ameloblasts (32, 53, 54) that improved our knowledge on the proteins having an essential role in Ca2+ extrusion in ameloblasts. We and others have suggested that NCKX4 is important in enamel mineralization (25, 54) because the expression of NCKX4 in maturation is high, the highest of the six NCKX family members in ameloblasts (54), and because mutations in the coding gene (SLC24A4) in humans and mouse models results in enamel defects (25, 55). Its localization in the distal pole of maturation ameloblasts would be consistent with a role in Ca2+ extrusion likely being more prominent in maturation (54). However, there are no currently published data on the functionality of NCKX4, and no data in maturation stage ameloblasts for NCX.
Disruptions on Ca2+ Homeostasis: The Case of Fluorosis
Having briefly described the broader picture of the Ca2+ handling system in ameloblasts, an interesting case study linking an enamel disease and disruptions in Ca2+ homeostasis is represented by dental fluorosis, a disease that arises when excessive amounts of fluoride are ingested during childhood, the time during which enamel development takes place (56, 57). The effects of fluoride on enamel are dose-dependent (58, 59) and result in pitted or discolored enamel prone to fracture, and increased wear and caries (56, 58). In chronic situations, fluorosis can lead to skeletal dysfunction causing bone breakage (58). The prevalence of dental fluorosis varies across countries. In the US, ~41% of adolescent population showed varying degrees of fluorosis (60). In India, dental fluorosis has a major impact with estimates of ~62 million people being affected (61). Clearly, dental fluorosis remains a world health issue, and despite decades of research, the proximate causes remain unclear.
Effects of Fluoride on Enamel
The benefits of fluoride in caries prevention have been known for decades. Therefore, the controlled supplementation of fluoride in drinking water or table salts has become a common practice in many countries. In the US, the most recent recommended concentration of fluoride in drinking water is ~0.7 parts per million (ppm), providing the best balance of protection from dental caries while limiting the risk of dental and/or skeletal fluorosis. At this concentration, fluoride has a positive effect on enamel strengthening chemical bonds in the formed enamel crystals, decreasing the risk of caries once the enamel crown has erupted (62–64). Excessive consumption of fluoride however leads to retention of enamel matrix proteins, e.g. amelogenin, and hypomineralization (58, 64). The effects on ameloblasts are stage-dependent with differences observed in the secretory and maturation stages (62–64). In the secretory stage, excess of fluoride inhibits protein secretion whereas in maturation it disrupts the cyclic modulation of ruffled-to-smooth ended ameloblasts which is important for ion transport (58, 59, 65). It has been reported that the ameloblast cell line LS8 treated with ≥1 mM concentrations of fluoride (NaF), results in ER stress and unfolded protein response (UPR) (66–69). This evidence suggests that excess of fluoride affects ameloblast biology, which could result in disruptions in crystal formation. Bronckers and colleagues suggested that fluoride causes hypermineralization of the enamel resulting in an increase in the proton load, a byproduct of crystal formation (65). Interestingly, they propose that this hypermineralization effect results in a barrier that prevents proper ion transport and protein recycling causing the hypomineralization effect associated with dental fluorosis (65).
One consideration that remains unclear is how fluoride access the ameloblasts. Fluoride could plausibly cross the ameloblast membranes via diffusion as hydrogen fluoride (HF), as reported in other cells (70). However, we have previously raised the possibility that it crosses the ameloblasts’ membrane via chloride channels, given their expression in ameloblasts (53, 71). Currently, the cellular uptake pathway of fluoride in enamel cells is not clear.
Interactions of Fluoride and Ca2+ Homeostasis
Associations between fluoride and Ca2+ have been reported (70). Rats drinking fluoridated water also receiving a dietary Ca2+ supplement showed ameliorated negative effects in their bones and kidney (analyzed histologically) than rats drinking the same fluoridated water but without receiving a Ca2+ supplement, strongly suggesting that Ca2+ has a protective role in fluorosis toxicity (72). It is also noteworthy that acute NaF exposure in rats resulted in hypocalcemia in plasma (73). Experiments conducted in osteoblasts and in rat proximal tubule showed that, in the presence of external Ca2+, fluoride stimulation at 10 µM concentration results in a rapid elevation of cCa2+ (74, 75). These elevations in cCa2+ were likely mediated by activation of G-protein-coupled receptors (GPCRs) (74, 75). These reports support the notion that fluoride modulates Ca2+ homeostasis in cells, as summarized in Figure 1.
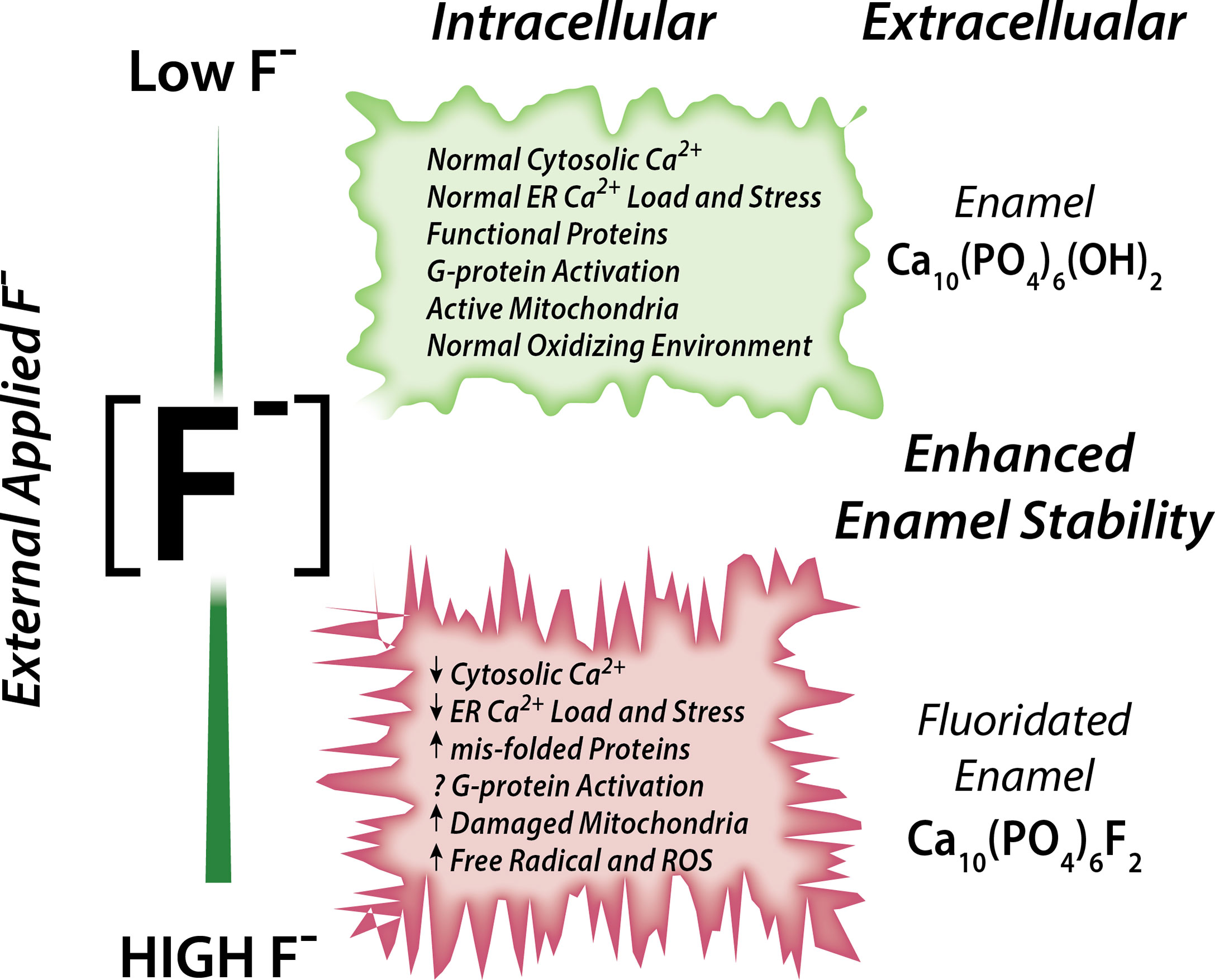
Figure 1 Schematic of the effects of fluoride in enamel cells. Fluoride modulates Ca2+ homeostasis with a dual effect. Low concentration leads to normal cytosolic and ER Ca2+ load, lack of ER stress leading to normal ER and mitochondrial function. High fluoride induces abnormal cytosolic and ER Ca2+, possibly via impairment of G-protein activation, and dysregulates cell metabolism.
The Disruptive Effects of Fluoride on Ameloblast Ca2+ Signaling
A recent study using rat primary ameloblasts and LS8 cells exposed to various concentrations of fluoride in vitro showed decreased internal Ca2+ in the ER and SOCE (76). These effects were observed when using high NaF concentrations (0.5 mM, 1 mM), equivalent to ~9 ppm and 18 ppm, respectively. In primary ameloblasts, these fluoride treatments decreased the ER Ca2+ within 30 minutes of incubation, and resulted in ER stress (76). LS8 cells exposed to bromide (NaBr) did not change these functions. An unexpected finding of the study was that treating LS8 and HEK-293 cells with the same concentration (1 mM) of NaF in similar conditions did not affect ER Ca2+ or SOCE in the latter. Moreover, NaF affected mitochondrial function in LS8 cells. Because this was not investigated in HEK-293 cells, we performed the seahorse mitochondrial assay like that was reported by Aulestia et al. in LS8 cells. Results show that while mitochondrial respiration in LS8 cells was negatively affected by NaF (1 mM) (Figure 2A), HEK-293 cells were not (Figure 2B) supporting the notion that mineralizing cells might be more sensitive to fluoride than other cells.
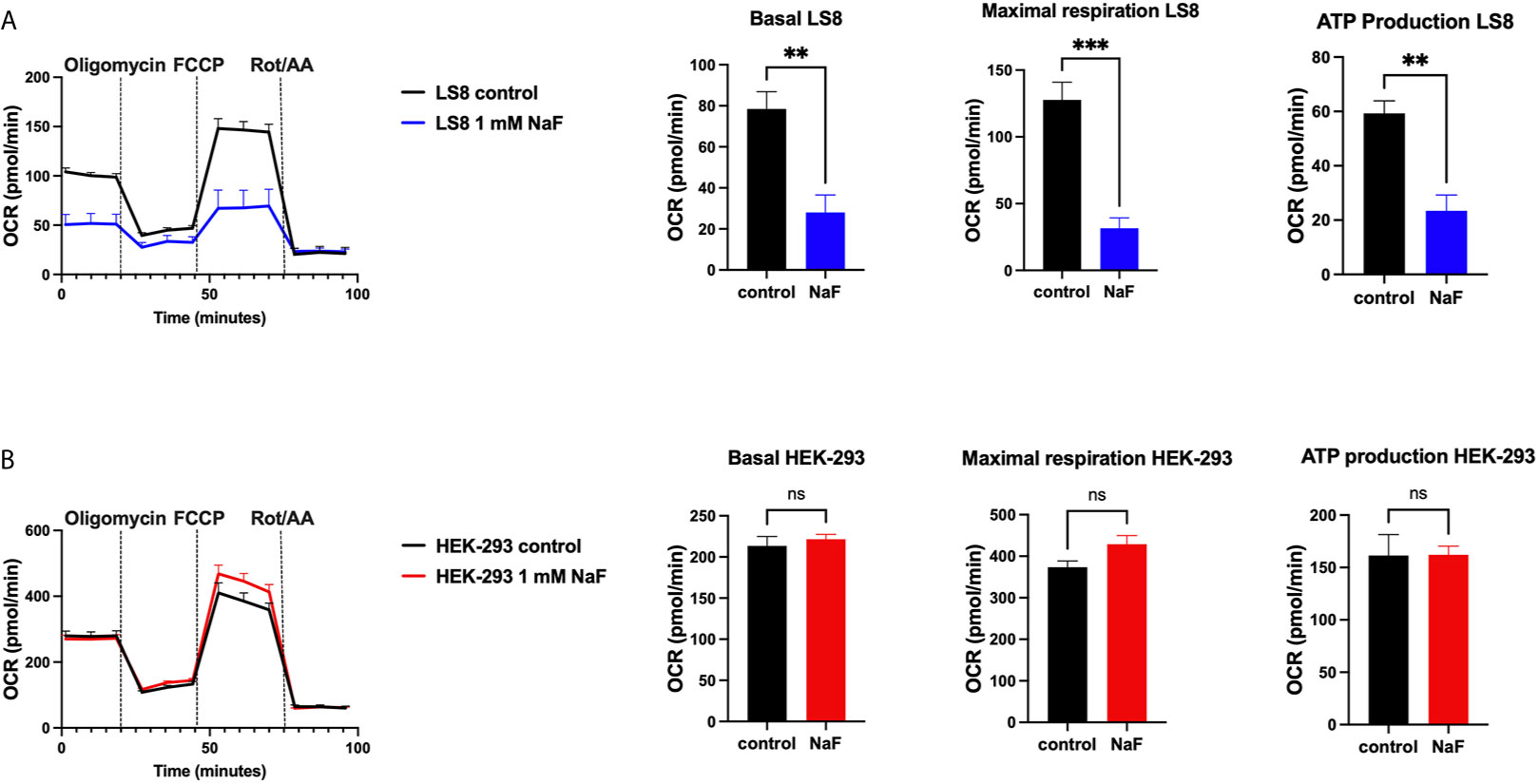
Figure 2 Mitochondrial OCR in fluoride treated cells. Oxygen consumption rate (OCR), basal respiration, ATP production and maximal respiration in LS8 cells (A) and in HEK-293 cells (B) after 4 hours of NaF (1 mM) pre-treatment. Oligomycin (1 µM), FCCP (1.5 µM) and rotenone/antimycin A (Rot/AA - 0.5 µM) were serially added in a Seahorse XFe24 Analyzer to assess differences in oxidative phosphorylation. Fluoride treatment affects OCR in the enamel LS8 cells but not HEK-293 cells (ns, not significant). Data represent the mean ± SEM of 3 independent experiments using unpaired Student’s t test. (**p < 0.01, ***p < 0.001).
The fluoride concentrations used above are relatively high and mimic the effects of fluorosis. However, we found that fluoride, at lower dosage, also had a negative effect on Ca2+ signaling in LS8 cells. When LS8 cells were treated for 24 hours with 10 µM of NaF, equivalent to ~0.2 ppm, it disrupted the function of the ER-localized Ca2+ channel IP3R and the activity of the SERCA pump during Ca2+ refilling of the ER (76). These data, we believe, provide a mechanism that can potentially address the biology of dental fluorosis or, at the very least, provide important information on the effects of fluoride in ameloblast Ca2+ physiology.
Summary
Enamel is a prototypical example of biologically controlled mineralization. Ameloblasts form a boundary that encloses the space of mineral formation and have the ability to control the introduction of ions into that space. The introduction of Ca2+ is essential for the mineralization of the long and thin hydroxyapatite enamel crystals. SOCE is the dominant system controlling the uptake of Ca2+ in ameloblasts. As Ca2+ crosses the ameloblast’s membrane, SERCA helps weather the effects of the Ca2+ storm but surely other mechanisms must be in place to ensure that ameloblasts protect cell function. However, little is known about these other processes including the capacity of mitochondria to uptake Ca2+ or the practicability of the clearance mechanisms by pumps and exchangers. Such information would be important to address the ins and outs of how enamel is mineralized by the ameloblasts. At any rate, it would appear that several components of the Ca2+ signaling toolkit of the ameloblasts are hindered by fluoride altering their physiology and function, likely affecting enamel mineralization.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was funded by NIH/National Institute of Dental and Craniofacial Research (NIDCR) awards (DE025639 and DE027679) to RL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Carafoli E, Krebs J. Why Calcium? How Calcium Became the Best Communicator. J Biol Chem (2016) 291(40):20849–57. doi: 10.1074/jbc.R116.735894
2. Cali T, Brini M, Carafoli E. Regulation of Cell Calcium and Role of Plasma Membrane Calcium ATPases. Int Rev Cell Mol Biol (2017) 332:259–96. doi: 10.1016/bs.ircmb.2017.01.002
3. Strehler EE. Plasma Membrane Ca2+ Pumps and Na+/Ca2+ Exchangers. Semin Cell Biol (1990) 1(4):283–95.
4. Guerini D. The Ca2+ Pumps and the Na+/Ca2+ Exchangers. Biometals (1998) 11(4):319–30. doi: 10.1023/a:1009210001608
5. Stephanopoulos G, Garefalaki ME, Lyroudia K. Genes and Related Proteins Involved in Amelogenesis Imperfecta. J Dent Res (2005) 84(12):1117–26. doi: 10.1177/154405910508401206
6. Berridge MJ, Bootman MD, Lipp P. Calcium–a Life and Death Signal. Nature (1998) 395(6703):645–8. doi: 10.1038/27094
7. Smith CE. Cellular and Chemical Events During Enamel Maturation. Crit Rev Oral Biol Med (1998) 9(2):128–61. doi: 10.1177/10454411980090020101
8. Nurbaeva MK, Eckstein M, Feske S, Lacruz RS. Ca2+ Transport and Signalling in Enamel Cells. J Physiol (2016) 595(10):3015–39 doi: 10.1113/JP272775
9. Lacruz RS. Enamel: Molecular Identity of its Transepithelial Ion Transport System. Cell Calcium (2017) 65:1–7. doi: 10.1016/j.ceca.2017.03.006
10. Hubbard MJ. Calcium Transport Across the Dental Enamel Epithelium. Crit Rev Oral Biol Med (2000) 11(4):437–66. doi: 10.1177/10454411000110040401
11. Eckstein M, Vaeth M, Aulestia FJ, Costiniti V, Kassam SN, Bromage TG, et al. Differential Regulation of Ca(2+) Influx by ORAI Channels Mediates Enamel Mineralization. Sci Signal (2019) 12(578). doi: 10.1126/scisignal.aav4663
12. Eckstein M, Vaeth M, Fornai C, Vinu M, Bromage TG, Nurbaeva MK, et al. Store-Operated Ca(2+) Entry Controls Ameloblast Cell Function and Enamel Development. JCI Insight (2017) 2(6):e91166. doi: 10.1172/jci.insight.91166
13. Nurbaeva MK, Eckstein M, Concepcion AR, Smith CE, Srikanth S, Paine ML, et al. Dental Enamel Cells Express Functional SOCE Channels. Sci Rep (2015) 5:15803. doi: 10.1038/srep15803
14. Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev (2015) 95(4):1383–436. doi: 10.1152/physrev.00020.2014
15. Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 Is an Essential Pore Subunit of the CRAC Channel. Nature (2006) 443(7108):230–3. doi: 10.1038/nature05122
16. Hoth M, Niemeyer BA. The Neglected CRAC Proteins: Orai2, Orai3, and STIM2. Curr Top Membr (2013) 71:237–71. doi: 10.1016/B978-0-12-407870-3.00010-X
17. Park HS, Betzenhauser MJ, Zhang Y, Yule DI. Regulation of Ca(2)(+) Release Through Inositol 1,4,5-Trisphosphate Receptors by Adenine Nucleotides in Parotid Acinar Cells. Am J Physiol Gastrointest Liver Physiol (2012) 302(1):G97–g104. doi: 10.1152/ajpgi.00328.2011
18. Wagner LE 2nd, Yule DI. Differential Regulation of the InsP(3) Receptor Type-1 and -2 Single Channel Properties by InsP(3), Ca(2)(+) and ATP. J Physiol (2012) 590(14):3245–59. doi: 10.1113/jphysiol.2012.228320
19. Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, et al. Isoform- and Species-Specific Control of Inositol 1,4,5-Trisphosphate (IP3) Receptors by Reactive Oxygen Species. J Biol Chem (2014) 289(12):8170–81. doi: 10.1074/jbc.M113.504159
20. Putney JW Jr. New Molecular Players in Capacitative Ca2+ Entry. J Cell Sci (2007) 120(Pt 12):1959–65. doi: 10.1242/jcs.03462
21. Lacruz RS, Feske S. Diseases Caused by Mutations in ORAI1 and STIM1. Ann N Y Acad Sci (2015) 1356:45–79. doi: 10.1111/nyas.12938
22. Robinson LJ, Mancarella S, Songsawad D, Tourkova IL, Barnett JB, Gill DL, et al. Gene Disruption of the Calcium Channel Orai1 Results in Inhibition of Osteoclast and Osteoblast Differentiation and Impairs Skeletal Development. Lab Invest (2012) 92(7):1071–83. doi: 10.1038/labinvest.2012.72
23. McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, et al. ORAI1 Deficiency and Lack of Store-Operated Ca2+ Entry Cause Immunodeficiency, Myopathy, and Ectodermal Dysplasia. J Allergy Clin Immunol (2009) 1241311-1318(6):e1317. doi: 10.1016/j.jaci.2009.10.007
24. Lian J, Cuk M, Kahlfuss S, Kozhaya L, Vaeth M, Rieux-Laucat F, et al. ORAI1 Mutations Abolishing Store-Operated Ca(2+) Entry Cause Anhidrotic Ectodermal Dysplasia With Immunodeficiency. J Allergy Clin Immunol (2017) 142(4):1297–310.e1211. doi: 10.1016/j.jaci.2017.10.031
25. Wang S, Choi M, Richardson AS, Reid BM, Seymen F, Yildirim M, et al. STIM1 and SLC24A4 Are Critical for Enamel Maturation. J Dent Res (2014) 93(7 suppl):94S–100S. doi: 10.1177/0022034514527971
26. Nurbaeva MK, Eckstein M, Devotta A, Saint-Jeannet JP, Yule DI, Hubbard MJ, et al. Evidence That Calcium Entry Into Calcium-Transporting Dental Enamel Cells Is Regulated by Cholecystokinin, Acetylcholine and ATP. Front Physiol (2018) 9:801. doi: 10.3389/fphys.2018.00801
27. Lopez JJ, Albarran L, Gomez LJ, Smani T, Salido GM, Rosado JA. Molecular Modulators of Store-Operated Calcium Entry. Biochim Biophys Acta (2016) 1863(8):2037–43. doi: 10.1016/j.bbamcr.2016.04.024
28. Dubail J, Huber C, Chantepie S, Sonntag S, Tuysuz B, Mihci E, et al. SLC10A7 Mutations Cause a Skeletal Dysplasia With Amelogenesis Imperfecta Mediated by GAG Biosynthesis Defects. Nat Commun (2018) 9(1):3087. doi: 10.1038/s41467-018-05191-8
29. Karakus E, Wannowius M, Muller SF, Leiting S, Leidolf R, Noppes S, et al. The Orphan Solute Carrier SLC10A7 Is a Novel Negative Regulator of Intracellular Calcium Signaling. Sci Rep (2020) 10(1):7248. doi: 10.1038/s41598-020-64006-3
30. Kubaski F, Osago H, Mason RW, Yamaguchi S, Kobayashi H, Tsuchiya M, et al. Glycosaminoglycans Detection Methods: Applications of Mass Spectrometry. Mol Genet Metab (2017) 120(1-2):67–77. doi: 10.1016/j.ymgme.2016.09.005
31. Palty R, Raveh A, Kaminsky I, Meller R. SARAF Inactivates the Store Operated Calcium Entry Machinery to Prevent Excess Calcium Refilling. Cell (2012) 149(2):425–38. doi: 10.1016/j.cell.2012.01.055
32. Lacruz RS, Smith CE, Bringas P Jr., Chen YB, Smith SM, Snead ML, et al. Identification of Novel Candidate Genes Involved in Mineralization of Dental Enamel by Genome-Wide Transcript Profiling. J Cell Physiol (2012) 227(5):2264–75. doi: 10.1002/jcp.22965
33. Nakano Y, Le MH, Abduweli D, Ho SP, Ryazanova LV, Hu Z, et al. A Critical Role of TRPM7 As an Ion Channel Protein in Mediating the Mineralization of the Craniofacial Hard Tissues. Front Physiol (2016) 7:258. doi: 10.3389/fphys.2016.00258
34. Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, et al. TRPM7 is Essential for Mg(2+) Homeostasis in Mammals. Nat Commun (2010) 1:109. doi: 10.1038/ncomms1108
35. Faouzi M, Kilch T, Horgen FD, Fleig A. The TRPM7 Channel Kinase Regulates Store-Operated Calcium Entry. J Physiol (2017) 595(10):3165–80. doi: 10.1113/JP274006
36. Bomfim GHS, Costiniti V, Li Y, Idaghdour Y, Lacruz RS. TRPM7 Activation Potentiates SOCE in Enamel Cells But Requires ORAI. Cell Calcium (2020) 87:102187. doi: 10.1016/j.ceca.2020.102187
37. Franklin IK, Winz RA, Hubbard MJ. Endoplasmic Reticulum Ca2+-ATPase Pump Is Up-Regulated in Calcium-Transporting Dental Enamel Cells: A non-Housekeeping Role for SERCA2b. Biochem J (2001) 358(Pt 1):217–24. doi: 10.1042/bj3580217
38. Hubbard MJ, McHugh NJ, Carne DL. Isolation of ERp29, a Novel Endoplasmic Reticulum Protein, From Rat Enamel Cells. Evidence for a Unique Role in Secretory-Protein Synthesis. Eur J Biochem (2000) 267(7):1945–57. doi: 10.1046/j.1432-1327.2000.01193.x
39. Hubbard MJ. Proteomic Analysis of Enamel Cells From Developing Rat Teeth: Big Returns From a Small Tissue. Electrophoresis (1998) 19(11):1891–900. doi: 10.1002/elps.1150191107
40. Hubbard MJ, McHugh NJ. Mitochondrial ATP Synthase F1-Beta-Subunit is a Calcium-Binding Protein. FEBS Lett (1996) 391(3):323–9. doi: 10.1016/0014-5793(96)00767-3
41. Hubbard MJ. Enamel Cell Biology. Towards a Comprehensive Biochemical Understanding. Connect Tissue Res (1998) 38(1-4):17–32. doi: 10.3109/03008209809017013
42. Hubbard MJ, McHugh NJ, Mangum JE. Exclusion of All Three Calbindins From a Calcium-Ferry Role in Rat Enamel Cells. Eur J Oral Sci (2011) 119:112–9. doi: 10.1111/j.1600-0722.2011.00890.x
43. Brini M, Cali T, Ottolini D, Carafoli E. Intracellular Calcium Homeostasis and Signaling. Met Ions Life Sci (2013) 12:119–68. doi: 10.1007/978-94-007-5561-1_5
44. Carafoli E. Intracellular Calcium Homeostasis. Annu Rev Biochem (1987) 56:395–433. doi: 10.1146/annurev.bi.56.070187.002143
45. Brini M, Cali T, Ottolini D, Carafoli E. Calcium Pumps: Why So Many? Compr Physiol (2012) 2(2):1045–60. doi: 10.1002/cphy.c110034
46. Brini M, Carafoli E. The Plasma Membrane Ca(2)+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb Perspect Biol (2011) 3(2). doi: 10.1101/cshperspect.a004168
47. Robertson SYT, Wen X, Yin K, Chen J, Smith CE, Paine ML. Multiple Calcium Export Exchangers and Pumps Are a Prominent Feature of Enamel Organ Cells. Front Physiol (2017) 8:336. doi: 10.3389/fphys.2017.00336
48. Niggli E, Lederer WJ. Activation of Na-Ca Exchange Current by Photolysis of "Caged Calcium". Biophys J (1993) 65(2):882–91. doi: 10.1016/S0006-3495(93)81105-6
49. Lytton J. Na+/Ca2+ Exchangers: Three Mammalian Gene Families Control Ca2+ Transport. Biochem J (2007) 406(3):365–82. doi: 10.1042/BJ20070619
50. Visser F, Lytton J. K+ -Dependent Na+/Ca2+ Exchangers: Key Contributors to Ca2+ Signaling. Physiol (Bethesda) (2007) 22:185–92. doi: 10.1152/physiol.00001.2007
51. Visser F, Valsecchi V, Annunziato L, Lytton J. Exchangers NCKX2, NCKX3, and NCKX4: Identification of Thr-551 as a Key Residue in Defining the Apparent K(+) Affinity of NCKX2. J Biol Chem (2007) 282(7):4453–62. doi: 10.1074/jbc.M610582200
52. Okumura R, Shibukawa Y, Muramatsu T, Hashimoto S, Nakagawa K, Tazaki M, et al. Sodium-Calcium Exchangers in Rat Ameloblasts. J Pharmacol Sci (2010) 112(2):223–30. doi: 10.1254/jphs.09267FP
53. Lacruz RS, Smith CE, Kurtz I, Hubbard MJ, Paine ML. New Paradigms on the Transport Functions of Maturation-Stage Ameloblasts. J Dent Res (2013) 92(2):122–9. doi: 10.1177/0022034512470954
54. Hu P, Lacruz RS, Smith CE, Smith SM, Kurtz I, Paine ML. Expression of the Sodium/Calcium/Potassium Exchanger, NCKX4, in Ameloblasts. Cells Tissues Organs (2012) 196(6):501–9. doi: 10.1159/000337493
55. Parry DA, Poulter JA, Logan CV, Brookes SJ, Jafri H, Ferguson CH, et al. Identification of Mutations in SLC24A4, Encoding a Potassium-Dependent Sodium/Calcium Exchanger, as a Cause of Amelogenesis Imperfecta. Am J Hum Genet (2013) 92(2):307–12. doi: 10.1016/j.ajhg.2013.01.003
56. Bartlett JD, Dwyer SE, Beniash E, Skobe Z, Payne-Ferreira TL. Fluorosis: A New Model and New Insights. J Dent Res (2005) 84(9):832–6. doi: 10.1177/154405910508400910
57. Denbesten P, Li W. Chronic Fluoride Toxicity: Dental Fluorosis. Monogr Oral Sci (2011) 22:81–96. doi: 10.1159/000327028
58. DenBesten PK, Thariani H. Biological Mechanisms of Fluorosis and Level and Timing of Systemic Exposure to Fluoride With Respect to Fluorosis. J Dent Res (1992) 71(5):1238–43. doi: 10.1177/00220345920710051701
59. Bronckers AL, Lyaruu DM, DenBesten PK. The Impact of Fluoride on Ameloblasts and the Mechanisms of Enamel Fluorosis. J Dent Res (2009) 88(10):877–93. doi: 10.1177/0022034509343280
60. Beltran-Aguilar ED, Barker L, Dye BA. Prevalence and Severity of Dental Fluorosis in the United States, 1999-2004. NCHS Data Brief (2010) 53):1–8.
61. Rao SM, Sherlin HJ, Anuja N, Pratibha R, Priya P, Chandrasekar T. Morphometry of Buccal Mucosal Cells in Fluorosis–a New Paradigm. Hum Exp Toxicol (2011) 30(11):1761–8. doi: 10.1177/0960327111400109
62. Robinson C, Connell S, Kirkham J, Brookes SJ, Shore RC, Smith AM. The Effect of Fluoride on the Developing Tooth. Caries Res (2004) 38(3):268–76. doi: 10.1159/000077766
63. Chen H, Czajka-Jakubowska A, Spencer NJ, Mansfield JF, Robinson C, Clarkson BH. Effects of Systemic Fluoride and In Vitro Fluoride Treatment on Enamel Crystals. J Dent Res (2006) 85(11):1042–5. doi: 10.1177/154405910608501113
64. Aoba T, Fejerskov O. Dental Fluorosis: Chemistry and Biology. Crit Rev Oral Biol Med (2002) 13(2):155–70. doi: 10.1177/154411130201300206
65. Lyaruu DM, Medina JF, Sarvide S, Bervoets TJ, Everts V, Denbesten P, et al. Barrier Formation: Potential Molecular Mechanism of Enamel Fluorosis. J Dent Res (2014) 93(1):96–102. doi: 10.1177/0022034513510944
66. Sharma R, Tsuchiya M, Bartlett JD. Fluoride Induces Endoplasmic Reticulum Stress and Inhibits Protein Synthesis and Secretion. Environ Health Perspect (2008) 116(9):1142–6. doi: 10.1289/ehp.11375
67. Sharma R, Tsuchiya M, Tannous BA, Bartlett JD. Measurement of Fluoride-Induced Endoplasmic Reticulum Stress Using Gaussia Luciferase. Methods Enzymol (2011) 491:111–25. doi: 10.1016/B978-0-12-385928-0.00007-9
68. Sierant ML, Bartlett JD. Stress Response Pathways in Ameloblasts: Implications for Amelogenesis and Dental Fluorosis. Cells (2012) 1(3):631–45. doi: 10.3390/cells1030631
69. Suzuki M, Bandoski C, Bartlett JD. Fluoride Induces Oxidative Damage and SIRT1/autophagy Through ROS-Mediated JNK Signaling. Free Radic Biol Med (2015) 89:369–78. doi: 10.1016/j.freeradbiomed.2015.08.015
70. Barbier O, Arreola-Mendoza L, Del Razo LM. Molecular Mechanisms of Fluoride Toxicity. Chem Biol Interact (2010) 188(2):319–33. doi: 10.1016/j.cbi.2010.07.011
71. Lacruz RS, Habelitz S, Wright JT, Paine ML. Dental Enamel Formation and Implications for Oral Health and Disease. Physiol Rev (2017) 97(3):939–93. doi: 10.1152/physrev.00030.2016
72. Hu Y, Smith CE, Cai Z, Donnelly LA, Yang J, Hu JC, et al. Enamel Ribbons, Surface Nodules, and Octacalcium Phosphate in C57BL/6 Amelx(-/-) Mice and Amelx(+/-) Lyonization. Mol Genet Genomic Med (2016) 4(6):641–61. doi: 10.1002/mgg3.252
73. Smith CEL, Kirkham J, Day PF, Soldani F, McDerra EJ, Poulter JA, et al. A Fourth KLK4 Mutation Is Associated With Enamel Hypomineralisation and Structural Abnormalities. Front Physiol (2017) 8:333. doi: 10.3389/fphys.2017.00333
74. Zerwekh JE, Morris AC, Padalino PK, Gottschalk F, Pak CY. Fluoride Rapidly and Transiently Raises Intracellular Calcium in Human Osteoblasts. J Bone Miner Res (1999) 5(Suppl 1):S131–136. doi: 10.1002/jbmr.5650051320
75. Mertz LM, Horn VJ, Baum BJ, Ambudkar IS. Calcium Entry in Rat Parotid Acini: Activation by Carbachol and Aluminum Fluoride. Am J Physiol (1990) 258(4 Pt 1):C654–661. doi: 10.1152/ajpcell.1990.258.4.C654
Keywords: Ca2+, fluoride, enamel, ameloblasts, store operated Ca2+ entry, amelogenesis imperfecta, fluorosis
Citation: Costiniti V, Bomfim GH, Mitaishvili E, Son G-Y, Li Y and Lacruz RS (2021) Calcium Transport in Specialized Dental Epithelia and Its Modulation by Fluoride. Front. Endocrinol. 12:730913. doi: 10.3389/fendo.2021.730913
Received: 25 June 2021; Accepted: 26 July 2021;
Published: 11 August 2021.
Edited by:
Gudrun Stenbeck, Brunel University London, United KingdomReviewed by:
Sadiq Umar, University of Illinois at Chicago, United StatesMichael Lansdell Paine, University of Southern California, United States
Copyright © 2021 Costiniti, Bomfim, Mitaishvili, Son, Li and Lacruz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo S. Lacruz, cnNsMTBAbnl1LmVkdQ==