 Arnaud Molin1,2,3,4*
Arnaud Molin1,2,3,4* Sandrine Lemoine5,6
Sandrine Lemoine5,6 Martin Kaufmann7
Martin Kaufmann7 Pierre Breton1Marie Nowoczyn8,2
Pierre Breton1Marie Nowoczyn8,2 Céline Ballandonne3Nadia Coudray1Hervé Mittre1,2,4
Céline Ballandonne3Nadia Coudray1Hervé Mittre1,2,4 Nicolas Richard1,3Amélie Ryckwaert9
Nicolas Richard1,3Amélie Ryckwaert9 Alinoe Lavillaureix10
Alinoe Lavillaureix10 Glenville Jones7
Glenville Jones7 Justine Bacchetta6,11,12,13
Justine Bacchetta6,11,12,13 Marie-Laure Kottler1,2,3
Marie-Laure Kottler1,2,3- 1Caen University Hospital, Department of Genetics, Molecular Genetics Laboratory and Reference Center for Rare Diseases of Calcium and Phosphorus Metabolism (OSCAR), Caen, France
- 2Caen Normandy University, Medical School, Caen, France
- 3BioTARGEN, Caen Normandy University, Caen, France
- 4OeReCa, Caen Normandy University, Caen, France
- 5Department of Nephrology and Renal Functional Explorations, Edouard Herriot Hospital, Lyon, France
- 6University of Lyon, University of Lyon 1, Villeurbanne, France
- 7Queen’s University, Department of Biomedical and Molecular Sciences, Kingston, ON, Canada
- 8Caen University Hospital, Department of Biochemistry, Caen, France
- 9Department of Pediatrics, Rennes University Hospital, Rennes, France
- 10Department of Genetics, Rennes University Hospital, Rennes, France
- 11Reference Center for Rare Kidney Diseases (ORKID), Department of Pediatric Nephrology, Rhumatology and Dermatology, Woman Mother Children Hospital, Bron, France
- 12Reference Center for Rare Diseases of Calcium and Phosphorus Metabolism (OSCAR), Department of Pediatric Nephrology, Rhumatology and Dermatology, Woman Mother Children Hospital, Bron, France
- 13INSERM 1033, Bone Diseases Prevention, Lyon, France
Mutations in CYP24A1 (vitamin D 24-hydroxylase) and SLC34A1 (renal phosphate transporter NPT2a) cause autosomal recessive Infantile Hypercalcemia type 1 and 2, illustrating links between vitamin D and phosphate metabolism. Patients may present with hypercalciuria and alternate between chronic phases with normal serum calcium but inappropriately high 1,25-(OH)2D and appropriately low PTH, and acute phases with hypercalcemia with suppressed PTH. Mutations in SLC34A3 and SLC9A3R1 have been associated with phosphate wasting without hypercalcemia. The aims of this study were to evaluate the frequency of mutations in these genes in patients with a medical history suggestive of CYP24A1 mutation to search for a specific pattern. Using next generation sequencing, we screened for mutations in 185 patients with PTH levels < 20 pg/mL, hypercalcemia and/or hypercalciuria, and relatives. Twenty-eight (15%) patients harbored biallelic mutations in CYP24A1 (25) and SLC34A3 (3), mostly associated with renal disease (lithiasis, nephrocalcinosis) (86%). Hypophosphatemia was found in 7 patients with biallelic mutations in CYP24A1 and a normal phosphatemia was reported in 2 patients with biallelic mutations in SLC34A3. Rare variations in SLC34A1 and SLC34A3 were mostly of uncertain significance. Fifteen patients (8%) carried only one heterozygous mutation. Heterozygous relatives carrying SLC34A1 or SLC34A3 variation may present with biochemical changes in mineral metabolism. Two patients’ genotype may suggest digenism (heterozygous variations in different genes). No variation was found in SLC9A3R1. As no specific pattern can be found, patients with medical history suggestive of CYP24A1 mutation should benefit from SLC34A1 and SLC34A3 analysis.
Introduction
Hypersensitivity to vitamin D (HVD) may be defined as an inability to regulate vitamin D metabolism which leads to calcium homeostasis deregulation, especially in case of vitamin D supplementation (1). This phenotype was classically associated with Idiopathic Infantile Hypercalcemia (IIH), a condition firstly described in the early fifties (2). Children present with non-specific signs of hypercalcemia (failure to thrive, vomiting, polyuria and polydipsia) with appropriately low parathormone (PTH) and inappropriately high 1,25-(OH)2D, suggesting deregulation of vitamin D metabolism.
In 2011, mutations in CYP24A1 encoding the vitamin D 24-hydroxylase were identified as the cause of Infantile Hypercalcemia type 1 (IH1) (MIM 143880) with HVD phenotype (3, 4). This enzyme is responsible for inactivating 1,25-(OH)2D and 25-OH-D (5–7). A high 25-OH-D3:24,25-(OH)2D3 ratio (substrate over product ratio, R ratio), which directly reflects the enzymatic defect, has been specifically associated with this condition and proposed as an effective screening tool (8–10). The clinical spectrum has been broadened to older children, teenagers and adults with nephrocalcinosis, renal stones and chronic fluctuating high serum and urine calcium with decreased serum PTH (9, 11–15). Patients present with a lifelong susceptibility to acute hypercalcemia and hypercalciuria, especially in case of vitamin D supplementation (16) and sunshine exposure (17).
In 2016, mutations in SLC34A1, encoding the renal phosphate transporter NPT2a, were reported as the cause of Infantile Hypercalcemia type 2 (IH2) (MIM 616963) with renal phosphate wasting and hypophosphatemia (18). A few cases have been published thereafter with clinical phenotype similar to type 1 (19–21). These observations suggest that phosphate wasting disorders may lead to HVD.
Other genes have been associated with renal phosphate wasting. Mutations in SLC34A3 encoding the renal phosphate transporter NPT2c have been associated with autosomal recessive hypophosphatemic rickets (ARHR) with hypercalciuria (MIM 241530) (22, 23). Its phenotype also includes biochemical features of HVD (low PTH, high serum 1,25-(OH)2D, high urine calcium with renal stones and nephrocalcinosis) and normal to low FGF23 serum concentrations. Furthermore, two patients with idiopathic hypercalciuria without bone signs and biallelic variations in SLC34A3 were described suggesting a potential role of SLC34A3 in HVD phenotype (24). Lastly, SLC9A3R1, encoding NHERF1 (sodium-proton exchanger regulatory factor 1) protein, was previously associated with autosomal dominant phosphate wasting (25, 26). This protein is implicated in transport regulation of NPT2a to the apical membrane of the proximal tubular cells and control of its retrieval by PTH (27).
We have previously published a cohort of 72 patients (1-72, Supplementary Table S1) with HVD tested for CYP24A1 mutations (9). We highlighted a wider phenotype depending on vitamin D intakes, from a balanced state with normal or high normal serum calcium and hypercalciuria to an unbalanced state with hypercalcemia and hypercalciuria, but consistent low PTH. In the present study, we aimed to evaluate the respective contribution of above mentioned genes involved in phosphate wasting in this cohort enriched with 113 new patients. Our objective was to search for a diagnostic algorithm to guide candidate gene testing in patients with features of HVD.
Patients and Methods
Patients
Over a 5 years period, we enrolled for molecular analysis 185 patients (index cases) presenting with a serum PTH concentration <20 pg/mL (2nd generation PTH assay were considered, normal values: 15-65 pg/mL) and a medical history of acute or chronic hypercalcemia (>2.6 mmol/L) and/or chronic hypercalciuria. Patients with medical history of calcium homeostasis deregulation with PTH > 20 pg/mL were not considered.
Recruitment was national through the network of the National Center for rare diseases of calcium and phosphate metabolism.
After genetic counseling, relatives of patients carrying biallelic variations were studied for genetic screening. Clinical and biochemical data were available in 17 relatives.
Written informed consent was obtained from the patients and/or their parents for retrospective collection of clinical and laboratory data, and for DNA collection to conduct molecular studies.
Biochemical and Clinical Parameters
Data on clinical symptoms were collected retrospectively using records from hospitals or primary care physicians.
Data on biochemical parameters (calcium and phosphate in serum and urine, renal function: creatinine and estimation of glomerular filtration rate with CKD-EPI for adults (28), PTH and vitamin D concentrations) were collected retrospectively using the same procedure, and thus correspond to a collection of different methods from many clinical chemistry departments. Approximate normal values for 1,25-(OH)2D were used as follows considering the use of different assays inherent in such retrospective study: 65-134 pmol/L for children and 60-108 pmol/L for adults (29). As most of routine assays for vitamin D cannot discriminate between vitamin D3 and D2, we used the term “vitamin D” to describe these measurements. As serum phosphate decreases from birth to adulthood, values were expressed as Z-scores to avoid the effects of the age-related variations of reference values (24, 30, 31).
Routine biochemical assays were mostly performed at the time of the diagnosis of acute hypercalcemia, or during follow up after normalization of the calcium level.
For urinary calcium: creatinine ratio, the different cutpoints of normal level were as follow: adults <0.33; children under 6 months <2.4; children 7-12 months <1.7; children 1-5 years <1.1 and children above 5 years <0.7.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) analysis was performed as previously described using 100 µL of serum. Results were expressed as the molar ratio of 25-OH-D3: 24,25-(OH)2D3 (R ratio) (8). Values under 25 indicated no defect in 25-OH-D3-24-hydroxylase activity and were considered as normal.
Medullary nephrocalcinosis was assessed by renal ultrasonography (echogenic renal pyramids). Nephrolithiasis was defined as kidney stones on ultrasonography or a medical history of iterative renal colic.
Molecular Analysis
Genomic DNA was extracted from whole blood samples with QuickGene DNA Whole Blood kit (Kurabo Industries LTD, Osaka, Japan) and QuickGene-610L extraction system (Fujifilm LTD, Singapore, Republic of Singapore).
Coding exons and flanking regions of CYP24A1 (NM_000782.4), SLC34A1 (NM_003052.4), SLC34A3 (NM_001177317.1) and SLC9A3R1 (NM_004252.4) genes were studied by Massively Parallel Sequencing (MPS) using a custom Ampliseq library on the Ion Torrent Personal Genome Machine (PGM) (ThermoFisher Scientific, Waltham, Massachusetts, USA). Reads were aligned against the reference sequences and variants were annotated using IonSuit (ThermoFisher Scientific, Waltham, Massachusetts, USA) and Nextgene (SoftGenetics, State College, Pennsylvania, USA). A read depth (RD)-based approach was used to detect copy number variation (CNV) from targeted MPS data using the CovCop tool (32).
Sequences with coverage <30X and variations identified with MPS were studied with Sanger sequencing using Big Dye® Terminator v1.1 Cycle Sequencing kit (ThermoFisher Scientific, Waltham, Massachusetts, USA) on a ABI 3500 Sequencer (ThermoFisher Scientific, Waltham, Massachusetts, USA).
Variations of sequence were searched on Varsome database. Interpretation was based on standards and guidelines of the American College of Medical Genetics (33). Pathogenicity prediction programs PolyPhen-2, Align-GVGD, MutationTaster and SIFT were considered for in silico analyses. Allelic frequencies were evaluated through the GnomAD database. For research purposes, variations classified as of uncertain significance (III) were considered apart from mutations [i.e. likely pathogenic (IV) and pathogenic (V) variations] as some may represent pathogenic mutations and other benign variations.
Statistical Analysis
Data from patients with class IV and V variations were used to compare biochemical profiles.
Statistical analyses were made using Prism 7 (GraphPad Software Inc.). For each biochemical parameter, differences between groups were calculated using the Mann and Whitney nonparametric U test. A two-tailed p value <0.05 was considered statistically significant. Kruskal-Wallis test was used for multiple group comparison (one-way nonparametric ANOVA).
Sex was not considered a factor in the statistical analysis of the data.
Chi-squared test was used to compare the allelic frequency of recurrent variations in our cohort and in the general population (estimated from data of GnomAD database).
Results
Patients
We enrolled 185 patients [88 males (47%) and 97 females (53%)], including 150 children (81%) (range: 1 day – 16 years; mean 2.3 years) and 35 adults (19%) (range: 18-81 years; mean 42 years). Familial occurrence was recorded in 34 patients (18%).
Molecular Analysis: Variations
The frequency and the criteria in favor of pathogenicity of each variation are presented in Table 1.
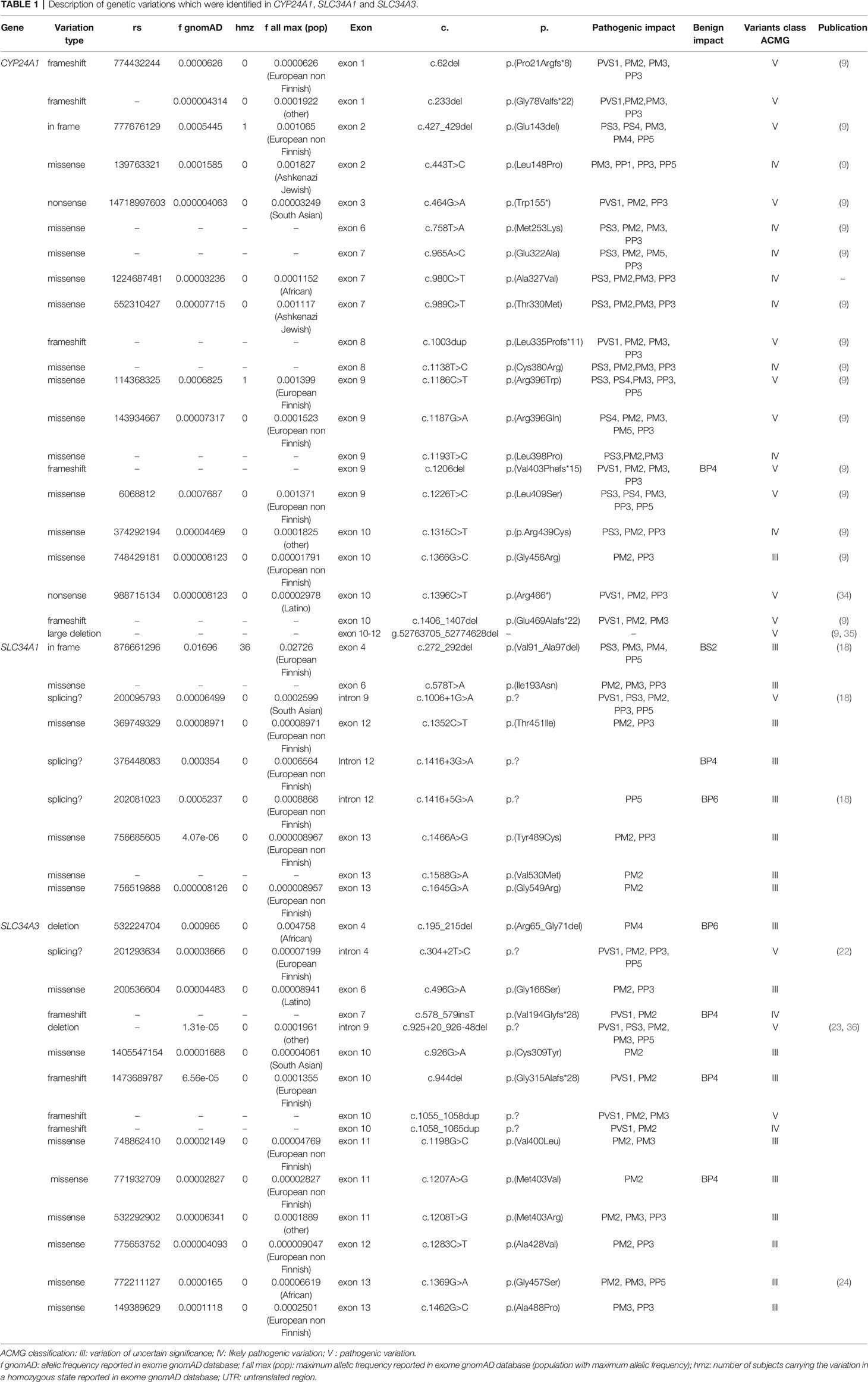
Table 1 Description of genetic variations which were identified in CYP24A1, SLC34A1 and SLC34A3.
We identified 20 mutations in CYP24A1, 1 mutation in SLC34A1 and 6 mutations in SLC34A3. Adding class III variations, the number of variations rose to 21 variations in CYP24A1, 9 variations in SLC34A1 and 15 variations in SLC34A3.
We described 2 new missenses pathogenic variations c.980C>T/p.(Ala327Val) and c.1193T>C/p.(Leu398Pro) in patients with elevated R ratio.
The recurrent variation in SLC34A1 c.272_292del/p.Val91_Ala97del (REC DEL in Supplementary Table S1) was identified in 10 patients (Table 1). In our cohort, its allelic frequency is 2.7%. A Chi-squared test showed that this frequency was not statistically different from the allelic frequency found in general population (1.696% in general population, 2.537% in European non-Finnish population according to GnomAD Exomes database).
Molecular Analysis: Genotype
Patients were classified into different groups according to their genotype (gene with variation, biallelic, i.e. homozygous or compound heterozygous, or monoallelic, i.e. heterozygous) (Table 2).
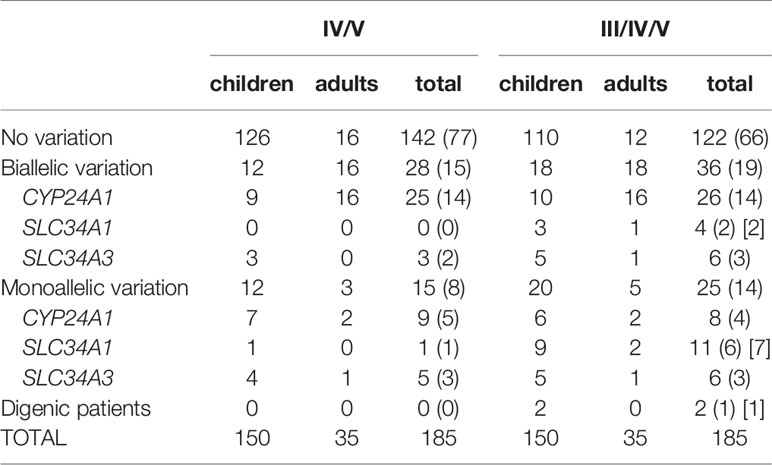
Table 2 Genotype in the cohort.
Depending on the inclusion of class III variations, we identified biallelic variations in 28-36 patients (15-19%) and monoallelic variations in 15-25 patients (8-14%) (heterozygous status) and no variation was found in 122-142 patients (66-77%) patients. Variations in CYP24A1 (biallelic as well as monoallelic) are more frequently found (18-19%) followed by those in SLC34A1 (1-8%) and SLC34A3 (5-6%). No variation was found in SLC9A3R1.
Digenic Patients
Two patients presented with one heterozygous class III SLC34A3 and a second variation in SLC34A1 (recurrent deletion) or CYP24A1 (the recurrent pathogenic variation p.Leu409Ser).
Especially, we reported 2 children of a non-consanguineous healthy couple who had experienced symptomatic neonatal hypercalcemia with similar biochemical profile (Figure 1). Both carried a SLC34A3 variation of uncertain significance inherited from their father and a CYP24A1 recurrent mutation inherited from their mother. The father presented with vertebral non-traumatic fracture; the mother was healthy.
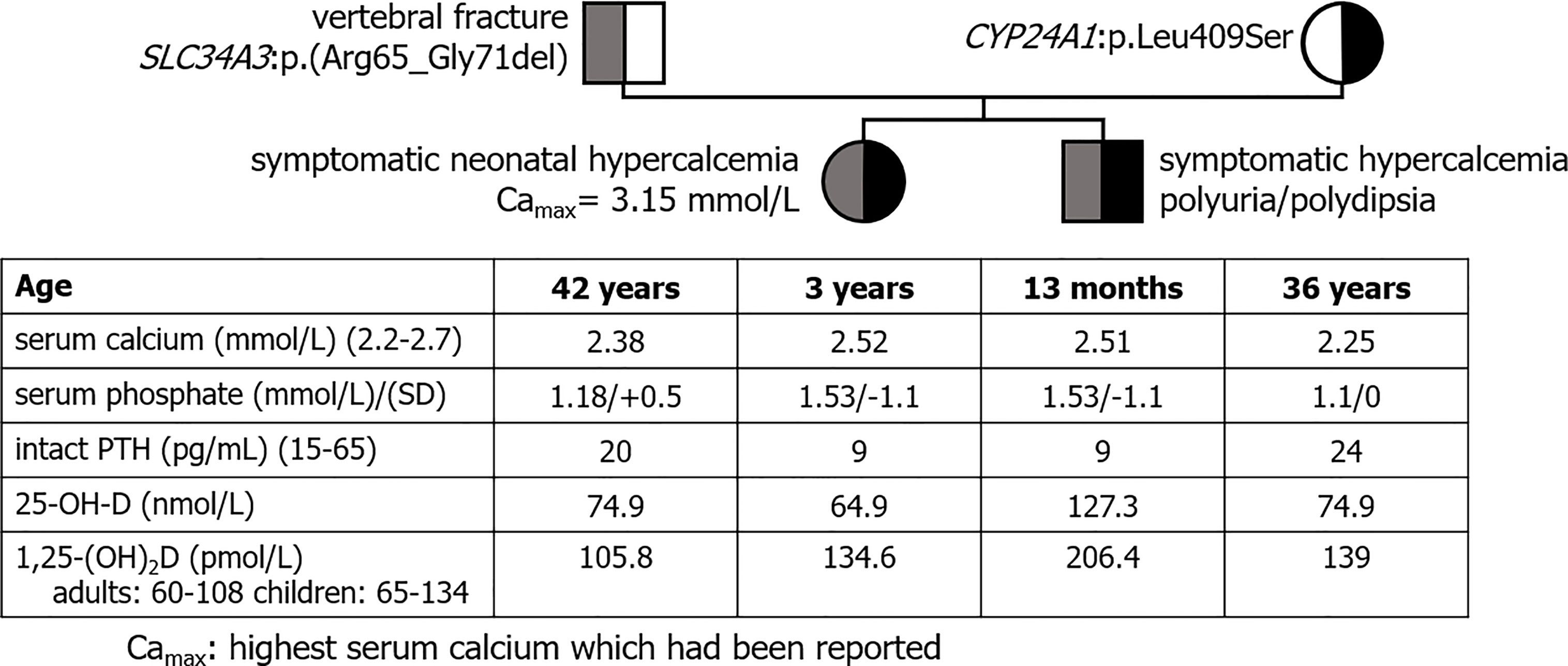
Figure 1 Pedigree tree of a family with suspected mechanism of digenism. Both children shared a biochemical profile evocative of HVD. Both parents shared a biochemical profile with normal serum calcium, high 1,25-(OH)2D, low PTH and a normal 25-OH-D evocative of mild HVD.
Phenotype of Patients With Biallelic Variation
Biallelic variations were found with a higher frequency in adult patients (45-51%; n= 16-18/35) than in children (8-12%; n= 12-18/150), most under 2 years (Table 2).
In patients with nephrocalcinosis (n=55; 44 children and 11 adults), we identified 18 (33%) patients with biallelic class IV or V variations, mostly in CYP24A1 (n=25/28) and also in SLC34A3 (n=3/28); 4 adults with biallelic CYP24A1 mutations presented chronic kidney disease (CKD-EPI<60mL/min). In patients with renal lithiasis (n=28; 18 children and 10 adults), we identified 6 (21%) patients with biallelic mutations in CYP24A1, mostly adults (n=5/6). Of note, CYP24A1 biallelic mutations were also found in 4 adult patients with medical history of hypercalcemia without nephrocalcinosis nor renal stones.
One patient was referred at the age of 5 for hypercalciuria and nephrocalcinosis, normal serum calcium and low PTH. At the age of 10, bowing of the lower limbs was evocative of rickets. He harbored a homozygous SLC34A3 mutation inherited from his consanguineous parents. His mother and his two brothers with the same mutation in a heterozygous state also presented nephrocalcinosis and no signs of bone disease.
Including class III variations, we identified 3 more patients with biallelic variations in SLC34A3 and nephrolithiasis (n=1) or nephrocalcinosis (n=2) and 4 patients with biallelic SLC34A1 variations, 2 of them (patients 103 and 170) having nephrocalcinosis detected as hyperechogenic kidney in utero (21).
Hypophosphatemia (serum phosphate Z-score < -2) was reported in 7 patients (27-28%) with CYP24A1 biallelic mutations and 1 patient with SLC34A3 biallelic mutations (Figure 2, Table 3 and Supplementary Table S1).
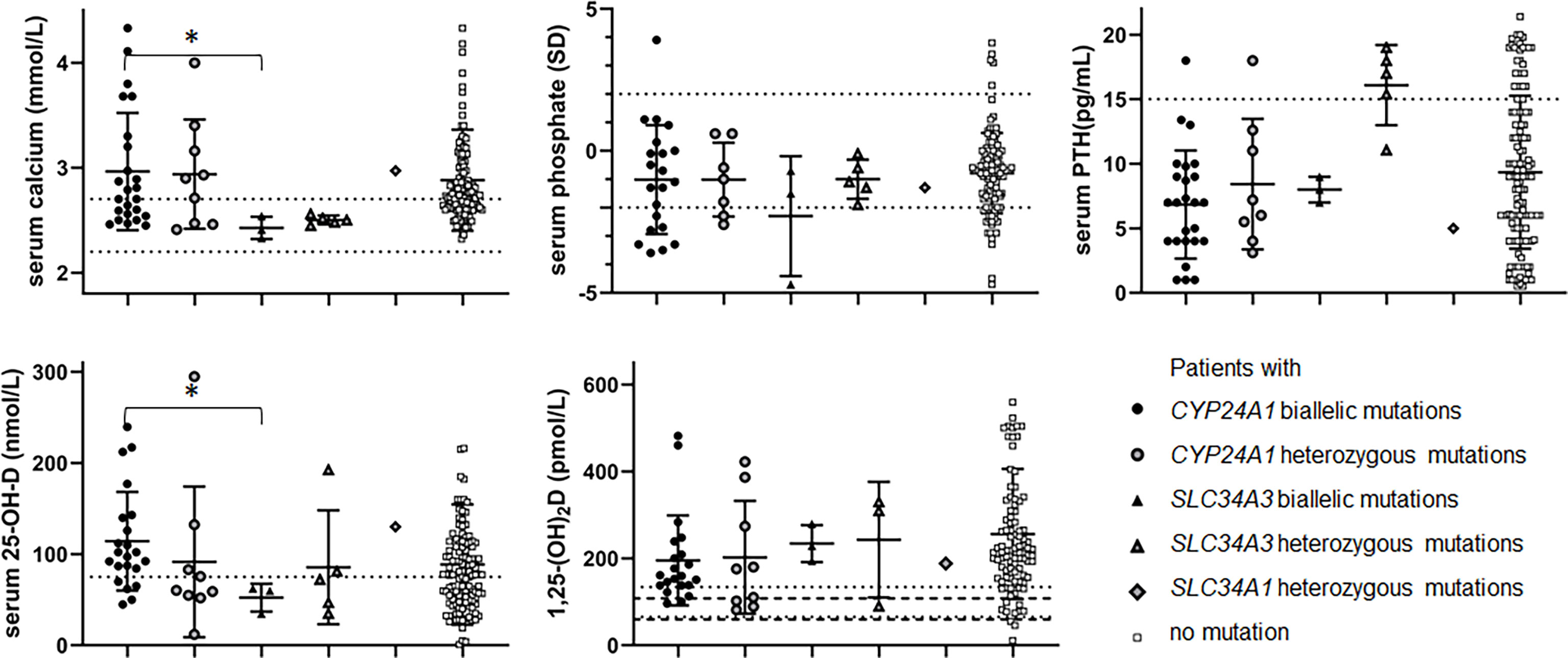
Figure 2 Biochemical data according to genotype: serum calcium (mmol/L), serum phosphate (SD), PTH (pg/mL) 25-OH-D (nmol/L) and 1,25-(OH)2D (pmol/L). Black dot: patients with biallelic CYP24A1 mutations. Grey dot: patients with heterozygous CYP24A1 mutations. Black triangle: patients with biallelic SLC34A3 mutations. Grey triangle: patients with heterozygous SLC34A3 mutations. Grey diamond: patients with heterozygous SLC34A1 mutations. White square: patients without variation. Serum calcium dot lines: normal values 2.2-2.7 mmol/L. Serum phosphate dot lines: -2-+2SD. Serum PTH dot line: 15 pg/mL. 25-OH-D dot line: 75 nmol/L. 1,25-(OH)2D dot and hyphen lines: approximative normal values for children and adults. *p ≤ 0.05.
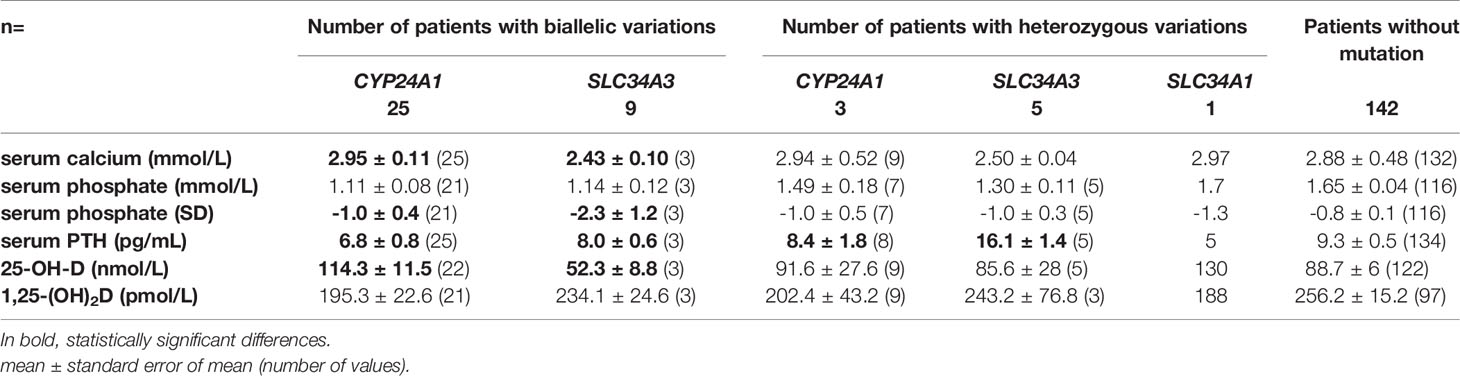
Table 3 Biochemical data according to genotype.
Serum calcium in patients with biallelic SLC34A3 mutation was lower than in patients with biallelic CYP24A1 mutation (p=0.0123*). Serum 25-OH-D in patients with biallelic CYP24A1 mutation was higher than in patients with biallelic SLC34A3 mutation (p=0.0139*). There was no other statistically significant difference between these two groups of patients. Serum phosphate (SD) tended to be lower in patients with biallelic SLC34A3 mutation than in patients with biallelic CYP24A1 mutation.
Patients With Mutation in a Heterozygous State
Mutations in a heterozygous state were found in patients with renal stones (n=6), nephrocalcinosis (n=4) and no renal complications (n=5), mostly in CYP24A1 (n=9) and SLC34A3 (n=5).
Including class III variations, we identified 11 patients with a SLC34A1 variation (7 of them carrying the recurrent c.272_292del/p.Val91_Ala97del) and 3 with a SLC34A3 variation. Among this 14 patients with a heterozygous class III variation, 6 had nephrocalcinosis, 1 nephrolithiasis and 7 no renal disease. Especially, 3 patients with a heterozygous c.272_292del/p.Val91_Ala97del had nephrocalcinosis.
Serum PTH of probands with a SLC34A3 mutation in a heterozygous state was higher than serum PTH in patients with biallelic SLC34A3 and CYP24A1 mutations (p=0.0357* and p=0.0003*** respectively). Biochemical profile of relatives and probands with a SLC34A3 mutation in a heterozygous state did not differ statistically, although PTH tended to be higher and 25-OH-D and 1,25-(OH)2D tended to be lower in relatives. We observed typical HVD biochemical features in SLC34A3 relatives’ group, namely hypercalciuria in 8/11 cases, low serum PTH in 6/11 cases, high 1,25-(OH)2D in 9/11 cases. Similarly, 2/4 relatives carrying a SLC34A1 class III variation presented biochemical changes, 1 having a high urinary calcium excretion and 1 having hypophosphatemia (<-2 Z-score).
Discussion
This report presents extensive clinical, biochemical and molecular data on a large cohort of patients with HVD phenotype and suspicion of IH1. We found: i) Mutations in genes associated with renal phosphate wasting can be found with a lower frequency than loss-of-function mutations in CYP24A1 and digenism may represent a rare mechanism of HVD. ii) Our data indicate that hypophosphatemia is not specific to phosphate wasting defects, also observed in CYP24A1 mutation, explaining a wide phenotypic overlap.
In our population, we found biallelic variants in CYP24A1, SLC34A1 and SLC34A3, in accordance with an autosomal recessive inheritance, in 28-36 patients (15-19%), but no mutation in SLC9A3R1, suggesting that mutations in this latter gene do not cause HVD phenotype. We observed a majority of biallelic mutations in CYP24A1, in accordance with our inclusion criteria (low serum PTH and medical history of hypercalcemia and/or hypercalciuria). We found a high number of variants of uncertain significance in SLC34A1 and SLC34A3 (17/24) with difficulties interpreting their pathogenicity in a context of routine molecular diagnosis. Even the presence of the recurrent c.272_292del/p.Val91_Ala97del variant previously described in IH2 may still be difficult to interpret because of a high allelic frequency in general population and in our cohort, suggesting a likely benign variant rather than a pathogenic loss of function one. Although its biological activity was preserved, it had been reported as a pathogenic variant responsible for cellular localization defect (18, 37). Its association with another variant in SLC34A1 in several patients (18) cannot exclude pathogenicity, maybe as an hypomorphic allele. Interpretation was more complex considering the high number of rare variants reported in a heterozygous state in these genes in online population databases (i.e. more than 700 SLC34A3 variations in GnomAD). In vitro functional studies are not feasible in the context of routine diagnosis, justifying the need for specific clinical screening or diagnostic criteria and the collection of molecular data.
Most patients with biallelic variants presented with renal complications including chronic hypercalciuria, renal stones (6/28) or nephrocalcinosis (18/28); and were adults or children over 2 years old (24/28). The lower diagnosis rate in children (8-12% versus 45-51%) suggested that most of the children in our cohort may present with another disease or a transient HVD which may be due to the initial prematurity in renal function during early infancy (3). Indeed, relative renal immaturity may lead to a delay in the adaptation of mineral metabolism from a prenatal to a postnatal context, especially regarding the vitamin D 24-hydroxylase expression. Patients with biallelic variants in CYP24A1 and SLC34A1 presented a significant phenotypic overlap. However, these two disorders can be easily differentiated on the basis of the serum 25-OH-D3:24,25-(OH)2D3 ratio, which is distinctly elevated in patients with biallelic CYP24A1 variants reflecting the vitamin D 24-hydroxylase defect; whereas the ratio remains normal in patients with biallelic SLC34A1 variants (8–10). More comprehensive profiling of serum vitamin D metabolite profiles by LC-MS/MS including 23- and 24-hydroxylated metabolites (23,25,26-(OH)3D3, 25-OH-D3-26,23-lactone and 1,24,25-(OH)3D3), strengthen the differences in vitamin D metabolism in these two diseases, as well as other causes of vitamin D-related hypercalcemia (38). A few clinical criteria may also help distinguish CYP24A1 mutation from other diseases. The presence of prenatal hyperechogenic kidneys in 2 patients of this cohort with SLC34A1 class III variants and 2 patients in the literature (21) suggested a more precocious disease of the urinary tract. To the best of our knowledge, no patient with prenatal hyperechogenic kidneys and CYP24A1 mutation were mentioned in the literature or observed in our patients. Mutations in SLC34A3 are responsible for ARHR, with characteristic high urine calcium and serum 1,25-(OH)2D, but hypercalcemia in these patients was not documented to date. Among patients with biallelic SLC34A3 variations, we reported 2 patients with fortuitously discovered hypercalcemia: A patient (n°152) with initial clinical features of HVD whose clinical presentation evolved into classic ARHR (with short stature and legs bowing); and a patient (n°88) with HVD with nephrocalcinosis without bone disease nor hypophosphatemia and normal height. These observations widen the phenotypic spectrum of SLC34A3 mutations to HVD. Whether the HVD presentation corresponds to a sustainable mild form of phosphate tubular reabsorption defect or a precocious presentation of ARHR requires further investigation.
In our cohort, only 2 patients with biallelic variations in SLC34A3 (class V) or SLC34A1 (class III) presented with hypophosphatemia. Thus, most of them did not present with a biochemical profile suggestive of a phosphate wasting disorder prima facie, including 2 patients with biallelic SLC34A3 pathogenic variations. Similarly, approximately one third of patients (5/16) did not present with hypophosphatemia in the IH2 cohort published by Schlingmann et al. (18) and patients with biallelic mutations in SLC34A3 and normal serum phosphate have also been published (24). Case reports have thus suggested that supplementation with oral phosphate could help correct calcium metabolism in patients with SLC34A1 mutation (18). More surprisingly, 27-28% of our patients with CYP24A1 biallelic mutations also presented with hypophosphatemia.
It is important to consider these findings in relation to the role of FGF23 in HVD, also described by Brancatella et al. (18, 39). FGF23 is a key regulator of phosphate and vitamin D homeostasis, up-regulated by serum phosphate and 1,25-(OH)2D (40, 41). Binding of FGF23 to FGF receptors induces the retrieval of renal phosphate transporters (responsible for phosphate tubular reabsorption) and decreases the expression of vitamin D 1α-hydroxylase (CYP27B1) (42). In IH1, we suggest that reduced clearance of 1,25-(OH)2D up-regulates FGF23 production and consequently leads to phosphate wasting and hypophosphatemia. In contrast, in NaPiIIa/NPT2a/NaPiIIc/NPT2c defects, renal phosphate wasting is responsible for a decrease in serum phosphate which in turn down-regulates FGF23 and up-regulates 1α-hydroxylase. As a consequence, the increase in 1,25-(OH)2D favors phosphate and calcium intestinal absorption. The negative feedback exerted by the resulting increase in serum calcium is supported by the low serum PTH. Such mechanism had previously been proposed and corroborated by murine Slc34a1-/- studies, illustrating the crucial role of FGF23 as a key regulator of vitamin D metabolism (18). Measurement of FGF23 in serum of HVD patients may be useful in future studies to specify this mechanism.
In 15-25 patients, we identified heterozygous variants, which represented diagnostic challenges in the context of a classic autosomal recessive disease. We could not exclude a second undetected variant (e.g. localized in non-coding sequences or a complex rearrangement) or contributory effect of the heterozygous variant as a risk factor. Although an autosomal dominant trait (7) had been proposed for heterozygous CYP24A1 loss-of-function mutation (43), our previous work showed that heterozygotes (proband as well as relatives) had normal 24-hydroxylase activity (9, 13). Thus, a patient with a CYP24A1 mutation in a heterozygous state and a high 25-OH-D3:24,25-(OH)2D3 ratio may benefit from extensive genetic exploration. Considering heterozygous variations in other genes, we observed a typical HVD phenotype in SLC34A3 heterozygous relatives which had previously not been diagnosed as HVD patients. Similarly, it was suggested that SLC34A1 heterozygous mutation may be a risk factor for kidney stone disease (18, 44) and heterozygous relatives may have abnormal biochemical data. It is likely that heterozygous phenotype may depend on additional factors, including genetics and environmental factors. Indeed, families in which the probands harbored a variation in two different genes suggested that heterozygous variations in CYP24A1, SLC34A1 and SLC34A3 may have an impact on vitamin D metabolism with a deleterious synergistic effect as previously proposed (45), as illustrated by the lower serum PTH in these patients compared to their heterozygous parents. Such a digenic mechanism, with both heterozygous SLC34A1 and SLC34A3 mutations, has been postulated as the cause of hypophosphatemic rickets with renal stone disease in an American family (46).
No mutation was identified in 66-77% of this cohort, especially in children (n= 110-126/150, 73-84%; adults: n=12-16/35, 34-46%). In our hands, these differences between children and adults may rely on obvious differences in the aims of genetic analysis in children and adults. In most children (78% or 117/150 children under 2 years), the first aim was to quickly distinguish CYP24A1 mutations from other causes with transient HVD. We may wonder if these children had consistent phenotype over the years but this work does not include follow up data. Repeated evaluation of mineral metabolism including PTH after initial discontinuation of vitamin D supplementation, may be useful to detect a normalization of serum PTH (not in favor of a genetic defect) or a consistent low serum PTH (in favor of a genetic defect). Evaluation of 24-hydroxylated vitamin D metabolites may also represent an interesting tool in this purpose (8, 10, 38). On the other hand, genetic analyses in adult were conducted in a different context of longer medical history to explain a phenotype. We cannot assume that patients without mutation in CYP24A1, SLC34A1, SLC34A3 or SLC9A3R1 did not carry variations in other genes or regulating sequences which were not analyzed.
No mutation was identified in SLC3A3R1 encoding NHERF1 in our cohort, which may be explained by our inclusion criterion as serum PTH in patients with SLC3A3R1 mutation were above 20 pg/mL in previous publications (25, 26).
Unfortunately, unlike genetic data obtained from the same procedure, the data collected in this retrospective cohort may include bias due to the aggregation of biochemical results from various assays and routine medical laboratories, which may impact the interpretation of 1,25-(OH)2D particularly. Appropriate exploration of phosphate metabolism including serum FGF23 evaluation would be of great interest to further characterize this pathophysiologic mechanism in HVD. Lastly, several patients without mutation (n=24) presented with hypophosphatemia suggesting the existence of other different mechanisms of calcium, vitamin D and phosphate metabolism deregulation.
Conclusion
With the exception of CYP24A1, specific biochemical profiles associated with individual candidate gene are lacking, making diagnosis of HVD patients challenging. Our results emphasize the importance of assessing SLC34A1 and SLC34A3 during genetic exploration of HVD in patients without vitamin 24-hydroxylase deficiency. While personalized treatment approaches for HVD such as hydrochlorothiazide, ketoconazole and fluconazole, phosphate oral supplementation have been described; our findings are anticipated to assist physicians with rapidly arriving at the correct diagnosis in the context of overlapping phenotypes in HVD patients so that appropriate treatment can be given.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by CPP Nord Ouest III. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
AM and M-LK designed the study. CB and NC realized molecular study. AM, NR, and HM interpreted molecular study. AM, SL, JB, PB, AL, and MN collected the clinical and biochemical data. MK and GJ realized LC-MS/MS studies. AM analyzed the data, drafted the paper and made all tables, and figures. M-LK, JB, SL, GJ, and MK revised the paper. All authors contributed to the article and approved the submitted version.
Funding
Délégation Recherche Clinique et Innovation (DRCI) - CHU de Caen APRIM - 16-048.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors acknowledge the patients and their families, all the physicians who collaborate with our center and Délégation Recherche Clinique et Innovation (DRCI) - CHU de Caen for its financial support (APRIM - 16-048).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.736240/full#supplementary-material
Supplementary Table 1 | Raw dataset.
References
1. Smith DW, Blizzard RM, Harrison HE. Idiopathic Hypercalcemia; A Case Report With Assays of Vitamin D in the Serum. Pediatrics (1959) 24:258–69.
2. Lightwood R, Stapleton T. Idiopathic Hypercalcaemia in Infants. Lancet (London England) (1953) 265:255–6. doi: 10.1016/S0140-6736(53)90187-1
3. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and Idiopathic Infantile Hypercalcemia. N Engl J Med (2011) 365:410–21. doi: 10.1056/NEJMoa1103864
4. Dauber A, Nguyen TT, Sochett E, Cole DEC, Horst R, Abrams SA, et al. Genetic Defect in CYP24A1, the Vitamin D 24-Hydroxylase Gene, in a Patient With Severe Infantile Hypercalcemia. J Clin Endocrinol Metab (2012) 97:1–7. doi: 10.1210/jc.2011-1972
5. Makin G, Lohnes D, Byford V, Ray R, Jones G. Target Cell Metabolism of 1,25-Dihydroxyvitamin D3 to Calcitroic Acid. Evidence for a Pathway in Kidney and Bone Involving 24-Oxidation. Biochem J (1989) 262:173–80. doi: 10.1042/bj2620173
6. Ohyama Y, Okuda K. Isolation and Characterization of a Cytochrome P-450 From Rat Kidney Mitochondria That Catalyzes the 24-Hydroxylation of 25-Hydroxyvitamin D3. J Biol Chem (1991) 266:8690–5. doi: 10.1016/S0021-9258(18)31501-1
7. Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-Hydroxylase (CYP24A1): Its Important Role in the Degradation of Vitamin D. Arch Biochem Biophys (2012) 523:9–18. doi: 10.1016/j.abb.2011.11.003
8. Kaufmann M, Gallagher JC, Peacock M, Schlingmann KP, Konrad M, DeLuca HF, et al. Clinical Utility of Simultaneous Quantitation of 25-Hydroxyvitamin D and 24,25-Dihydroxyvitamin D by LC-MS/MS Involving Derivatization With DMEQ-TAD. J Clin Endocrinol Metab (2014) 99:2567–74. doi: 10.1210/jc.2013-4388
9. Molin A, Baudoin R, Kaufmann M, Souberbielle JC, Ryckewaert A, Vantyghem MC, et al. CYP24A1 Mutations in a Cohort of Hypercalcemic Patients: Evidence for a Recessive Trait. J Clin Endocrinol Metab (2015) 100:E1343–52. doi: 10.1210/jc.2014-4387
10. Kaufmann M, Morse N, Molloy BJ, Cooper DP, Schlingmann KP, Molin A, et al. Improved Screening Test for Idiopathic Infantile Hypercalcemia Confirms Residual Levels of Serum 24,25-(OH)2 D3 in Affected Patients. J Bone Miner Res (2017) 32:1589–96. doi: 10.1002/jbmr.3135
11. Nesterova G, Malicdan MC, Yasuda K, Sakaki T, Vilboux T, Ciccone C, et al. 1,25-(OH)2D-24 Hydroxylase (CYP24A1) Deficiency as a Cause of Nephrolithiasis. Clin J Am Soc Nephrol (2013) 8:649–57. doi: 10.2215/CJN.05360512
12. Dinour D, Beckerman P, Ganon L, Tordjman K, Eisenstein Z, Holtzman E. Loss-Of-Function Mutations of CYP24A1, the Vitamin D 24-Hydroxylase Gene, Cause Long-Standing Hypercalciuric Nephrolithiasis and Nephrocalcinosis. J Urol (2013) 190:552–7. doi: 10.1016/j.juro.2013.02.3188
13. Meusburger E, Mündlein A, Zitt E, Obermayer-Pietsch B, Kotzot D, Lhotta K. Medullary Nephrocalcinosis in an Adult Patient With Idiopathic Infantile Hypercalcaemia and a Novel CYP24A1 Mutation. Clin Kidney J (2013) 6:211–5. doi: 10.1093/ckj/sft008
14. Jacobs TP, Kaufman M, Jones G, Kumar R, Schlingmann KP, Shapses S. Bilezikian JP. A Lifetime of Hypercalcemia and Hypercalciuria, Finally Explained. J Clin Endocrinol Metab (2014) 99:708–12. doi: 10.1210/jc.2013-3802
15. Wolf P, Müller-Sacherer T, Baumgartner-Parzer S, Winhofer Y, Kroo J, Gessl A, et al. A Case of “Late-Onset” Idiopathic Infantile Hypercalcemia Secondary to Mutations in the CYP24A1 Gene. Endocr Pract (2014) 20:e91–5. doi: 10.4158/EP13479.CR
16. Castanet M, Mallet E, Kottler ML. Lightwood Syndrome Revisited With a Novel Mutation in CYP24 and Vitamin D Supplement Recommendations. J Pediatr (2013) 163:1208–10. doi: 10.1016/j.jpeds.2013.04.056
17. Figueres M-L, Linglart A, Bienaime F, Allain-Launay E, Roussey-Kessler G, Ryckewaert A, et al. Kidney Function and Influence of Sunlight Exposure in Patients With Impaired 24-Hydroxylation of Vitamin D Due to CYP24A1 Mutations. Am J Kidney Dis (2015) 65:122–6. doi: 10.1053/j.ajkd.2014.06.037
18. Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, et al. Autosomal-Recessive Mutations in SLC34A1 Encoding Sodium-Phosphate Cotransporter 2a Cause Idiopathic Infantile Hypercalcemia. J Am Soc Nephrol (2016) 27:604–14. doi: 10.1681/ASN.2014101025
19. Pronicka E, Ciara E, Halat P, Janiec A, Wójcik M, Rowińska E, et al. Biallelic Mutations in CYP24A1 or SLC34A1 as a Cause of Infantile Idiopathic Hypercalcemia (IIH) With Vitamin D Hypersensitivity: Molecular Study of 11 Historical IIH Cases. J Appl Genet (2017) 58:349–53. doi: 10.1007/s13353-017-0397-2
20. Fearn A, Allison B, Rice SJ, Edwards N, Halbritter J, Bourgeois S, et al. Clinical, Biochemical, and Pathophysiological Analysis of SLC34A1 Mutations. Physiol Rep (2018) 6:e13715. doi: 10.14814/phy2.13715
21. Hureaux M, Molin A, Jay N, Saliou AH, Spaggiari E, Salomon R, et al. Prenatal Hyperechogenic Kidneys in Three Cases of Infantile Hypercalcemia Associated With SLC34A1 Mutations. Pediatr Nephrol (2018) 33(10):1723–29. doi: 10.1007/s00467-018-3998-z
22. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary Hypophosphatemic Rickets With Hypercalciuria Is Caused by Mutations in the Sodium-Phosphate Cotransporter Gene SLC34A3. Am J Hum Genet (2006) 78:193–201. doi: 10.1086/499410
23. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 Mutations in Patients With Hereditary Hypophosphatemic Rickets With Hypercalciuria Predict a Key Role for the Sodium-Phosphate Cotransporter NaPi-IIc in Maintaining Phosphate Homeostasis. Am J Hum Genet (2006) 78:179–92. doi: 10.1086/499409
24. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutations in SLC34A3/NPT2c Are Associated With Kidney Stones and Nephrocalcinosis. J Am Soc Nephrol (2014) 25:2366–75. doi: 10.1681/ASN.2013101085
25. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 Mutations and Responsiveness of Renal Parathyroid Hormone. N Engl J Med (2008) 359:1128–35. doi: 10.1056/NEJMoa0802836
26. Courbebaisse M, Leroy C, Bakouh N, Salaün C, Beck L, Grandchamp B, et al. A New Human NHERF1 Mutation Decreases Renal Phosphate Transporter NPT2a Expression by a PTH-Independent Mechanism. PloS One (2012) 7:1–8. doi: 10.1371/journal.pone.0034764
27. Prié D, Ureña Torres P, Friedlander G. Latest Findings in Phosphate Homeostasis. Kidney Int (2009) 75:882–9. doi: 10.1038/ki.2008.643
28. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, et al. A New Equation to Estimate Glomerular Filtration Rate. Ann Intern Med (2009) 150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006
29. Feldman D, Pike JW, Bouillon R, Giovannucci E, Goltzman D, Hewison M. Vitamin D. 4th ed. Feldman D, editor. London: Academic Press, Elsevier (2017). doi: 10.1016/C2015-0-05921-4
30. Colantonio DA, Kyriakopoulou L, Chan MK, Daly CH, Brinc D, Venner AA, et al. Closing the Gaps in Pediatric Laboratory Reference Intervals: A Caliper Database of 40 Biochemical Markers in a Healthy and Multiethnic Population of Children. Clin Chem (2012) 58:854–68. doi: 10.1373/clinchem.2011.177741
31. Bailey D, Colantonio D, Kyriakopoulou L, Cohen AH, Chan MK, Armbruster D, et al. Marked Biological Variance in Endocrine and Biochemical Markers in Childhood: Establishment of Pediatric Reference Intervals Using Healthy Community Children From the CALIPER Cohort. Clin Chem (2013) 59:1393–405. doi: 10.1373/clinchem.2013.204222
32. Derouault P, Parfait B, Moulinas R, Barrot C-C, Sturtz F, Merillou S, et al. “Cov’cop” Allows to Detect CNVs Responsible for Inherited Diseases Among Amplicons Sequencing Data. Bioinformatics (2017) 33:1586–8. doi: 10.1093/bioinformatics/btx017
33. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
34. Güven A, Konrad M, Schlingmann KP. Idiopathic Infantile Hypercalcemia: Mutations in SLC34A1 and CYP24A1 in Two Siblings and Fathers. J Pediatr Endocrinol Metab (2020) 33:1353–8. doi: 10.1515/jpem-2020-0169
35. Molin A, Nowoczyn M, Coudray N, Ballandone C, Abéguilé G, Mittre H, et al. Molecular Characterization of a Recurrent 10.9 Kb CYP24A1 Deletion in Idiopathic Infantile Hypercalcemia. Eur J Med Genet (2019) 62:103577. doi: 10.1016/j.ejmg.2018.11.011
36. Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ. Intronic Deletions in the SLC34A3 Gene Cause Hereditary Hypophosphatemic Rickets With Hypercalciuria. J Clin Endocrinol Metab (2006) 91:4022–7. doi: 10.1210/jc.2005-2840
37. Lapointe J-Y, Tessier J, Paquette Y, Wallendorff B, Coady MJ, Pichette V, et al. NPT2a Gene Variation in Calcium Nephrolithiasis With Renal Phosphate Leak. Kidney Int (2006) 69:2261–7. doi: 10.1038/sj.ki.5000437
38. Kaufmann M, Schlingmann K-P, Berezin L, Molin A, Sheftel J, Vig M, et al. Differential Diagnosis of Vitamin D-Related Hypercalcemia Using Serum Vitamin D Metabolite Profiling. J Bone Miner Res (2021) 36(7):1340–50. doi: 10.1002/jbmr.4306
39. Brancatella A, Cappellani D, Kaufmann M, Borsari S, Piaggi P, Baldinotti F, et al. Do the Heterozygous Carriers of a CYP24A1 Mutation Display a Different Biochemical Phenotype Than Wild Types? J Clin Endocrinol Metab (2021) 106:708–17. doi: 10.1210/clinem/dgaa876
40. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast Growth Factor 23 Is a Counter-Regulatory Phosphaturic Hormone for Vitamin D. J Am Soc Nephrol (2006) 17:1305–15. doi: 10.1681/ASN.2005111185
41. Saito H, Maeda A, Ohtomo S-I, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 Is Regulated by 1alpha,25-Dihydroxyvitamin D3 and Phosphorus In Vivo. J Biol Chem (2005) 280:2543–9. doi: 10.1074/jbc.M408903200
42. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 Is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J Bone Miner Res (2004) 19:429–35. doi: 10.1359/JBMR.0301264
43. Tebben PJ, Milliner DS, Horst RL, Harris PC, Singh RJ, Wu Y, et al. Hypercalcemia, Hypercalciuria, and Elevated Calcitriol Concentrations With Autosomal Dominant Transmission Due to CYP24A1 Mutations: Effects of Ketoconazole Therapy. J Clin Endocrinol Metab (2012) 97:E423–7. doi: 10.1210/jc.2011-1935
44. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis and Osteoporosis Associated With Hypophosphatemia Caused by Mutations in the Type 2a Sodium-Phosphate Cotransporter. N Engl J Med (2002) 347:983–91. doi: 10.1056/NEJMoa020028
45. Tabibzadeh N, Cheddani L, Daudon M, Haymann J-P, Toussaint A, Silve C, et al. The Case | Epistasis and Urolithiasis. Kidney Int (2017) 92:523–4. doi: 10.1016/j.kint.2017.01.026
Keywords: hypersensitivity to vitamin D, calcitriol induced hypercalcemia, phosphate wasting diseases, vitamin D, CYP24A1- hydroxylase, SLC34A1 gene, SLC34A3 gene
Citation: Molin A, Lemoine S, Kaufmann M, Breton P, Nowoczyn M, Ballandonne C, Coudray N, Mittre H, Richard N, Ryckwaert A, Lavillaureix A, Jones G, Bacchetta J and Kottler M-L (2021) Overlapping Phenotypes Associated With CYP24A1, SLC34A1, and SLC34A3 Mutations: A Cohort Study of Patients With Hypersensitivity to Vitamin D. Front. Endocrinol. 12:736240. doi: 10.3389/fendo.2021.736240
Received: 04 July 2021; Accepted: 07 September 2021;
Published: 13 October 2021.
Edited by:
Catherine Chaussain, Université de Paris, FranceReviewed by:
Eleanor DeLand Lederer, University of Texas Southwestern Medical Center, United StatesPaula H. Stern, Northwestern University, United States
Copyright © 2021 Molin, Lemoine, Kaufmann, Breton, Nowoczyn, Ballandonne, Coudray, Mittre, Richard, Ryckwaert, Lavillaureix, Jones, Bacchetta and Kottler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arnaud Molin, YXJuYXVkLm1vbGluQHVuaWNhZW4uZnI=