 Ryan J. Dashek1,2
Ryan J. Dashek1,2 R. Scott Rector
R. Scott Rector- 1Research Service, Harry S. Truman Memorial Veterans’ Hospital, Columbia, MO, United States
- 2Comparative Medicine Program, Department of Veterinary Pathobiology, University of Missouri, Columbia, MO, United States
- 3School of Medicine, University of Missouri, Columbia, MO, United States
- 4Division of Cardiology, Department of Medicine, University of Missouri, Columbia, MO, United States
- 5Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, United States
- 6Department of Medical Pharmacology and Physiology, University of Missouri, Columbia, MO, United States
- 7Department of Nutrition and Exercise Physiology, University of Missouri, Columbia, MO, United States
- 8Division of Gastroenterology and Hepatology, Department of Medicine, University of Missouri, Columbia, MO, United States
Non-alcoholic fatty liver disease (NAFLD) is a multimorbidity disorder ranging from excess accumulation of fat in the liver (steatosis) to steatohepatitis (NASH) and end-stage cirrhosis, and the development of hepatocellular carcinoma (HCC) in a subset of patients. The defining features of NASH are inflammation and progressive fibrosis. Currently, no pharmaceutical therapies are available for NAFLD, NASH and HCC; therefore, developing novel treatment strategies is desperately needed. Reversion Inducing Cysteine Rich Protein with Kazal motifs (RECK) is a well-known modifier of the extracellular matrix in hepatic remodeling and transition to HCC. More recently, its role in regulating inflammatory and fibrogenic processes has emerged. Here, we summarize the most relevant findings that extend our current understanding of RECK as a regulator of inflammation and fibrosis, and its induction as a potential strategy to blunt the development and progression of NASH and HCC.
Introduction
The extracellular matrix (ECM) is a complex and dynamic component of multicellular organisms, regulating crucial cellular processes such as proliferation, differentiation, migration, adhesion, and tissue remodeling (1). As such, dysregulation of the ECM has been linked to several pathological conditions, including cancer and fibrosis (1, 2). Therefore, regulators of the ECM play pivotal roles in these conditions and have been explored as potential therapeutic targets in a variety of diseases. One such regulator is Reversion Inducing Cysteine Rich Protein with Kazal Motifs (RECK), a membrane-anchored glycoprotein (3). At the NH3-terminal, there are five cysteine repeats followed by two epidermal growth factor (EGF)-like repeats that are hypothesized to be required for proper interaction between RECK and its targets (4, 5). Moreover, at the COOH terminus, there exists three serine protease inhibitor (SPI)-like domains, that play a role in inhibiting target peptides through ‘physical trapping’ or ‘reversible tight binding’ (4). RECK itself is anchored to the cell membrane via the GPI-anchor located at the COOH-terminal (4).
RECK primarily regulates the activity of several matrix metalloproteinases (MMPs) that play a role ECM remodeling (6). This regulation of ECM components, combined with the observation that RECK is downregulated in cancers that metastasize, prompted several studies aimed at understanding RECK’s potential as a metastasis-suppressor (5, 7–9). However, it is not known how RECK regulates inflammation and the fibrogenic pathways.
Both inflammation and fibrosis contribute to the progression of nonalcoholic steatohepatitis (NASH), a clinical condition characterized by the excess accumulation of fat in the liver. It affects a subset of patients with NAFLD (non-alcoholic fatty liver disease) and can ultimately lead to liver fibrosis (cirrhosis) and transition to HCC (hepatocellular carcinoma). NAFLD affects approximately a third of the adult population in developed countries, and NASH is expected to become the leading indicator of liver-related mortality within a few years (10). Unfortunately, to date, no specific pharmacological therapies exist for NASH or HCC. Reduced RECK is a characteristic feature of many cancers, including HCC (11), promoting progression and metastasis. Interestingly, reduced RECK may also be a component of NASH as reported by Peng, et al. (12), where the authors reported that RECK expression is downregulated in mice fed a methionine-choline-deficient diet (12). Furthermore, they found that RECK is a novel target of Farsenoid X Receptor (FXR), and that FXR activation induced RECK mRNA and protein expression (12). In fact, FXR agonists have been identified to inhibit NASH by reducing hepatic gluconeogenesis, lipogenesis, and steatosis (13, 14). The use of therapeutic bile acids as FXR agonists appear promising in human clinical trials, but additional studies related to their long-term safety are warranted (15–17). It is plausible that FXR agonists may improve NASH outcomes, in part, through upregulation of hepatic RECK expression, as hepatic RECK knock-down appears to worsen hepatic inflammation and fibrosis in animal models of Western diet-induced NASH (our unpublished data). Here we seek to describe reduced RECK in the context of hepatocellular inflammation, fibrosis, NASH, and HCC.
RECK and Cancer
The ECM plays a critical role in the development and progression of cancer. Dysregulated ECM remodeling by tumor cells alters cell signaling, angiogenesis, and tissue biomechanics (18). These changes in the tumor microenvironment allow for local tissue invasion as well as distal metastasis of cancer cells (18). Therefore, regulators of the ECM have drawn much interest in the oncological realm, and RECK, a key player in ECM remodeling, is no exception.
Reduced RECK is a characteristic feature of many cancers (19). In fact, interest in regulators of the gelatinases, MMP2 and MMP9, stems from their identification as prognostic indicators in a number of tumors, including liposarcoma (20), breast cancer (21), oral squamous cell carcinoma (22), and ovarian cancer (23). Tumors expressing higher concentrations of these gelatinases are in general linked to poorer prognosis and overall survival (1, 24, 25). Furthermore, RECK is known to complex with MT1-MMP, and inhibit its proteolytic activity at the cell membrane and internalization (26). Increased expression of MT1-MMP in tumors has also been linked to poorer prognosis and reduced overall survival, independent of other gelatinases (27). The underlying theory is that sustained activation of these gelatinases promotes excess ECM degradation allowing for local and distant tumor invasion, as well as allow for angiogenesis. For example, MMP9 is known to promote the release of vascular endothelial growth factor (VEGF), a pro-angiogenic mediator (4, 19, 28). Interestingly, RECK was first identified as a gelatinase inhibitor, reducing ECM breakdown and promoting angiogenesis in several tumor types (29–32), including colorectal, gastric, and HCC. Across all tumor categories, preserving RECK expression was shown to inhibit MMP2 and MMP9 activity, and improve prognosis by decreasing invasion and metastasis (5).
As mentioned above, RECK expression is either downregulated or undetected in various invasive cancers (4). By contrast, tumors that expressed normal or elevated RECK levels show reduced tissue invasion and metastasis (4). The mechanisms underlying RECK downregulation in cancer are hypothesized to be multifactorial and tumor specific; however, the general mechanism appears to involve increased Sp1 binding to the RECK promoter, resulting in its reduced transcription (5). It is, however, unclear whether RECK downregulation occurs prior to, concurrently, or following tumor metastasis. Regardless, reduced RECK expression appears to worsen prognosis (7, 9, 33). RECK has also been shown to interact and inhibit other cellular pathways involved in cancer progression and metastasis, such as Notch and EGFR/RAS. Both Notch (34) and EGFR/RAS (22, 23) are implicated in inflammation and fibrosis, which we will explore further below.
RECK and Liver-Related Tumorigenesis
Within the context of the liver, RECK’s role was assessed in the pathogenesis of HCC and cholangiocarcinoma (CCA). In line with other oncological studies, several groups have found that overall prognosis and survival were significantly improved when tumors – either HCC or CCA – expressed relatively greater amounts of RECK (11, 29, 33, 35). What remains unanswered, however, is how and when RECK expression is altered in these individuals – i.e., does RECK suppression precede tumorigenesis, or occurs during progression and metastasis?
Several mechanisms are implicated in RECK downregulation. For example, several single nucleotide polymorphisms (SNPs) are identified in the RECK gene within given populations (36–38). These SNPs appeared more frequently in patients diagnosed with HCC versus healthy controls (36, 39), with Chung et al. outlining specific SNPs in RECK that are relevant to liver cancer in humans. Their group identified two SNPs of interest in the development of HCC; individuals with the RECK promoter rs10814325 polymorphism saw increased risk of developing HCC compared to wild-type carriers, while HCC patients carrying the rs11788747 had higher risk of developing distant metastasis than wild-type carriers (36). This leads to the hypothesis that singular changes within the RECK protein structure itself or promoter polymorphisms could have a significant impact on its activity and ultimately on tumor progression. Hypermethylation of its promoter has also been shown to downregulate RECK expression (33). In addition, hypermethylation of RECK promoter led to poorer prognosis in individuals with HCC (33).
Several micro-RNAs (miRs) are also shown to target RECK in HCC. For example, miR-135b, upregulated in HCC tissues, not only targets RECK post-transcriptionally (40), but also promotes HCC cell motility and invasiveness in vitro (41). Huang, et al. reported increased miR-21 and reduced RECK expression in CCA patients with lymph node metastasis or perineural invasion (42). In that study, silencing miR-21 dramatically decreased CCA cell invasion and metastasis, which was rescued by the forced expression of RECK.
These studies suggest a direct link between reduced RECK expression and invasion and metastasis of liver cancers. However, several questions remain unanswered. For example, what other mechanisms play a role in RECK regulation, and when RECK expression is altered, i.e., is this a dynamic process that can change over time or is activity static and only serves as a predisposing factor in these cases? It is also unclear whether RECK expression is altered prior to the formation of HCC in situations of NAFLD or cirrhosis. Of note, Furumoto et al. found that approximately half of individuals with HCC recruited into the study had reduced RECK expression. However, they did not delineate cases based on predisposing factors leading up to HCC, such as which patient had NASH prior to recruitment into the study (11).
It is unknown whether RECK is already downregulated or silenced in NAFLD, causing exacerbation of symptoms, including development and progression towards NASH, cirrhosis, and HCC. Individual heterogeneity in RECK expression due to various genetic and environmental factors may govern the development of each, or even all these processes. Therefore, we further examined the literature to determine whether RECK was found to be involved in pathways and physiological processes leading up to and including progression of NASH and liver fibrosis.
RECK and Inflammation
RECK regulation of the ECM also modulates inflammation. For example, RECK/MMP-mediated ECM remodeling plays a role not only in tumor cell spread, but also leukocyte infiltration into tissues. RECK-mediated inhibition of MMP2 and MMP9 expression and activity (19, 43, 44) has been shown to regulate inflammation in a variety of tissues and models. In models of experimental autoimmune encephalomyelitis (EAE), CD4+T cell invasion requires local MMP2 and MMP9-mediated parenchymal basement membrane breakdown (45, 46). MMP2 and MMP9 knock-out (KO) mice have reduced inflammatory cell influx into bronchoalveolar lavage fluid in experimental asthma models (47, 48). In models of acute pyelonephritis, it is known that there is a direct correlation between levels of MMP2 and MMP9 in the kidney and the severity of inflammation (49). Nascimento et al. found that MMP9 was involved in the early phases of temporomandibular joint inflammation in a rodent model, while MMP2 was involved in later phases of inflammation of the joint capsule. Additionally, they found that using doxycycline, a non-specific MMP inhibitor, diminished the inflammatory response (50). Furthermore, MMP9 was established as a mediator of inflammation within the intestinal muscularis in rodent models of post-operative ileus; inhibition of MMP9 activity reduced immune cell infiltration into intestinal muscularis, and MMP9-KO mice were protected from the inflammation and dysmotility associated with post-operative ileus (51). Finally, Ries, et al. found that inflammatory cytokines upregulate MMP2 and MMP9 in cultured human mesenchymal stem cells, which in turn allowed for chemotactic migration through reconstituted basement membranes (52), suggesting a complex interplay between inflammatory cytokines, MMP activity, and immune cell infiltration through a basement membrane. Given such, RECK may be a central regulator in controlling leukocyte extravasation into other tissues as well, such as liver in NASH.
Chronic inflammation in obesity has been shown to closely associate with metabolic syndromes, such as NASH. In the context of obesity and inflammation, elevated MMP2 expression and MMP9 activity are found in a mouse model of obesity and positively correlated with inflammatory cytokine expression (53). Even more compelling is that MMP9 has already been shown to be involved in the active recruitment of CD11b+ leukocytes (54) and migration of neutrophils (55) in the post-ischemic liver. Lingwal, et al. examined swine islet cell transplantation into the liver of C57Bl/6 mice via the portal vein and found that the transplantation drove an increase in MMP9 activity, which corresponded with massive inflammation in the liver (56). Using MMP9-KO mice, they found hepatic inflammatory infiltrates were significantly lower. More specifically, a positive correlation was observed between hepatic MMP9 expression and activity and CD11b+ leukocyte infiltration. Further, using pharmacological gelatinase inhibitors in vitro and in vivo, they reported a significant decrease in Kupffer cell migration towards TNF-α or IL-1β expressing loci (56). These results suggest that the gelatinases, MMP2 and MMP9, are critical in the inflammatory processes of the liver, and, through inhibiting the activity of these matrixins, reduction in inflammatory infiltrates could be achieved. As the downregulation of RECK clearly disrupts ECM integrity in the liver through dysregulation of MMP activity – as evidenced by the spread and invasiveness of HCC and CCA when RECK concentrations are lowered, as well as in the Lingwal, et al. study (56) – we could ask two critical questions that need further investigation: (i) would RECK downregulation lends itself to increased invasion of inflammatory cells into the liver in cases of NAFLD and NASH? and (ii) could restoring RECK reduces the amount of inflammation in these patients?
Beyond MMPs, RECK is also a known inhibitor of ADAM17 (A Disintegrin and Metalloproteinase Domain-containing protein 17) (57). Known also as TNFα-Converting Enzyme (TACE), ADAM17 plays a pivotal role in inflammation (58, 59). Of note, TNF-α expression has been shown to be upregulated in NASH (60), and plays a role in the development and progression of NAFLD (61). Therefore, regulating TNF-α release by targeting ADAM17 may be an effective strategy to blunt hepatic inflammation. However, identifying a pharmaceutical inhibitor of this enzyme has remained a challenge. It is therefore plausible that sustaining or inducing RECK has the therapeutic potential to target ADAM17 and overt inflammation in the liver as a result of metabolic dysregulation.
In addition to ADAM17, RECK has also been shown to inhibit ADAM10, though both ADAMs are critical and play a role in the activation of the pro-inflammatory Notch signaling cascade (34, 57). In fact, RECK has been shown to inhibit Notch signaling in neural tissues (34) and during angiogenesis (62). An increases in the Notch signaling pathway has been implicated in several proinflammatory conditions, such as rheumatoid arthritis (63) and uveitis (64). Increased Notch activity, specifically Notch2, is known to regulate monocyte cell fate and inflammation in response to Toll Like Receptor (TLR) signaling (65). Both canonical and non-canonical Notch activity have been found to be increased in response to inflammatory mediators (66), thereby creating a positive feedback loop of Notch➔inflammation➔Notch signaling. In the realm of NAFLD, the number of hepatocytes expressing a major Notch outcome product – Hes Family BHLH Transcription Factor 1 (Hes1) – is significantly elevated in patients with severe NASH (67), suggesting overt activation of this pathway. Since RECK modulates the Notch pathway via direct regulation of ADAM17 and ADAM10, strategies that sustain or induce RECK expression have the therapeutic potential in NASH.
In addition to Notch signaling, both ADAM10 and ADAM17 are shown to be crucial in regulating the epidermal growth factor receptor (EGFR) signaling cascade. For example, RECK’s inhibition of the ADAMs could prevent the release of membrane-anchored EGFR ligands, such as amphiregulin, and suppress EGFR activation. Indeed, RECK’s ability to downregulate EGFR activity has already been reported (68, 69). This is of particular interest in the context of NASH, as EGFR has been implicated in hepatocyte and liver regeneration, and HCC development. EGFR signaling is also implicated in mitochondrial dysfunction, apoptosis of hepatocytes and hepatic stellate cells (HSCs), and liver necrosis (70–72). Pharmacological inhibition of EGFR has shown to reduce high-fat diet-induced liver injury in mouse models of NAFLD (73, 74), suggesting that targeting EGFR signaling may prove to have therapeutic potential in human NASH. Sustaining or inducing RECK may be a strategy to modulate EGFR activity and inhibit NASH.
RECK and Fibrogenesis
Fibrosis results from excess accumulation of ECM components. The downregulation of RECK has been linked to fibrosis in several tissues. In a mouse model of Western diet-induced obesity, RECK protein levels were found to be decreased in the kidney and correlated positively with renal fibrosis (75). We previously reported reduced RECK expression in the fibrotic heart. We also reported reduced RECK expression and increased angiotensin-II-induced fibroblast migration and proliferation, and their reversal by ectopic RECK overexpression (76–78).
As previously reported by us, RECK regulates fibrosis in part by inhibiting activation of MMP2 and MMP9 (76–78). These gelatinases perform a much wider range of functions than the cleavage of ECM components, and can have more of a ‘processing’ than ‘degradation’ role in maintaining the ECM (79). MMPs have been studied extensively in the context of hepatic fibrosis (80–83). During hepatic fibrogenesis, collagen deposition from HSCs is markedly increased, and paradoxically both MMP2 and MMP9 are highly upregulated in these cells (84). For example, MMP2 is an autocrine proliferator and activator of HSCs (85), promoting further ECM deposition. In an animal model of CCA where RECK was found to be decreased, increased MMP2 was associated with periductal fibrosis (29). Importantly, it was suggested that serum MMP2 levels could serve as a diagnostic marker to assess the level of liver fibrosis in patients with NASH (86). Furthermore, a positive correlation was reported between serum MMP2 concentrations and liver function as assessed via bilirubin and albumin production, and prothrombin time (87). While both gelatinases are upregulated in the context of fibrogenesis, paradoxically hepatic fibrosis was exacerbated in MMP2-KO mice (88). This suggests not only a complex relationship between gelatinase function and activity in the context of hepatic fibrosis, but also activation of compensatory mechanisms. Therefore, rather than ablating their expression, inhibiting MMP activity sequentially could blunt progression of fibrosis. As such, sustained RECK expression may have the therapeutic potential in NASH by targeting time-dependent or sequential activation of MMPs.
Notch signaling, and RECK’s modulation of this pathway, may further serve to alter fibrogenesis. Activation of HSCs, classically, is mediated through TGF-β signaling (89), promoting Notch activity and fibrosis. Importantly, pharmaceutical Notch inhibitors prevent TGF-β-mediated HSC activation in vitro (90) and limit HSC activation and hepatic fibrosis in an animal model of fibrosis (91). In fibroblasts, Hes1 was shown to promote Col1A1 and Col1A2 transcription, and type I collagen deposition (92); however, it is unclear whether this holds true in HSCs as well. As has already been outlined above, RECK inhibits the Notch pathway by targeting ADAM10 and ADAM17 activity; whether this is sufficient to alter fibrosis in NASH patients is unknown.
EGFR signaling is also involved in tissue fibrosis. Its increased activity positively correlated with several pulmonary pathologies; individuals affected by the SARS outbreak of 2003 saw extensive lung fibrosis, which was suggested to be induced by a hyperactive host response to EGFR-mediated lung injury (93). More specifically, in a review by Stolarczyke and Scholte examining chronic obstructive pulmonary disease and cystic fibrosis, extensive evidence was found linking hyperactivity of the EGFR/ADAM17 signaling axis to ADAM17-cleavage of amphiregulin, an EGFR ligand (94). In a rodent model of lung injury resulting from chronic allergies, Morimoto et al. found that amphiregulin/EGFR signaling activated eosinophils to an inflammatory state with enhanced production of osteopontin, an important profibrotic protein. Furthermore, they found that amphiregulin was produced by memory Th cells, further contributing to pulmonary fibrosis (95). Chronic kidney disease (CKD) is associated with fibrosis (96); EGFR is activated following renal injury, and studies have suggested its potential inhibition as a treatment for CKD (97).
In the context of NASH, it has been found that treatment of isolated Kupffer cells, the resident liver macrophages, with CXCL6 increases EGFR phosphorylation and TGF-β induction (98). These results were confirmed by the same authors in vivo using a carbon tetrachloride (CCl4) model of NASH (98). Increases in EGFR phosphorylation was observed in hepatocytes, activated HSCs, and macrophages in fibrotic livers in response to CCl4. Furthermore, Egfr gene ablation (EGFR-KO) markedly reduced hepatic fibrosis and α-SMA expression in livers in response to CCl4 (99). EGF and EGFR are also upregulated in humans with chronic liver injury. However, in rodent models, it was shown that EGF was downregulated in liver fibrosis, but amphiregulin and EGFR were significantly increased (100). Overall, these reports indicate that overactivation of the EGFR signaling pathway may be linked to overt ADAM17 activity and NASH progression. Due to RECK’s inhibition of ADAM17 and consequent downregulation in EGFR signaling, it is plausible that sustaining or inducing RECK has the potential to prevent or even reverse hepatic fibrosis seen in NASH. A more comprehensive analysis of potential signaling pathway is necessary to better understand the protective role of RECK in NAFLD, NASH and HCC.
Future Directions
RECK plays a central role in modulating ECM components involved in the progression of inflammation and fibrosis. Therefore, examining the activity of RECK in the context of inflammatory and fibrotic conditions, such as NASH, is paramount. Currently, RECK inducers are being explored in the context of cancer treatment (101, 102), thus expansion of this research into the area of liver disease could prove fruitful and should be considered. Furthermore, there is considerable variation in the context of RECK activity within the individual cell types of the liver. For example, would RECK activity in HSCs prevent activation and collagen deposition? Can RECK activity in Kupffer cells prevent TGF-β release and mobilization through MMP inhibition? Does RECK alter hepatic inflammation and fibrosis through other mechanisms? Further, is RECK expression altered in livers of patients with NASH? Does downregulation predispose individuals towards developing cirrhosis and/or HCC? Further investigations will elucidate RECK’s central role and therapeutic potential in NASH and HCC.
Conclusion
RECK is a unique membrane anchored regulator of various MMPs and ADAMs. Through modulation of MMPs and ADAMs, RECK could target several key inflammatory and fibrogenic pathways by modulating ECM, inflammatory cytokines, and several other cellular processes, which could influence the outcomes of diseases such as NAFLD and NASH (Figure 1; key cellular targets listed in Table 1). Further studies are necessary to better understand the regulation and protective role of RECK in the diseased liver. Examination of these pathways may help us develop novel RECK inducers as therapeutics in NAFLD, NASH and HCC.
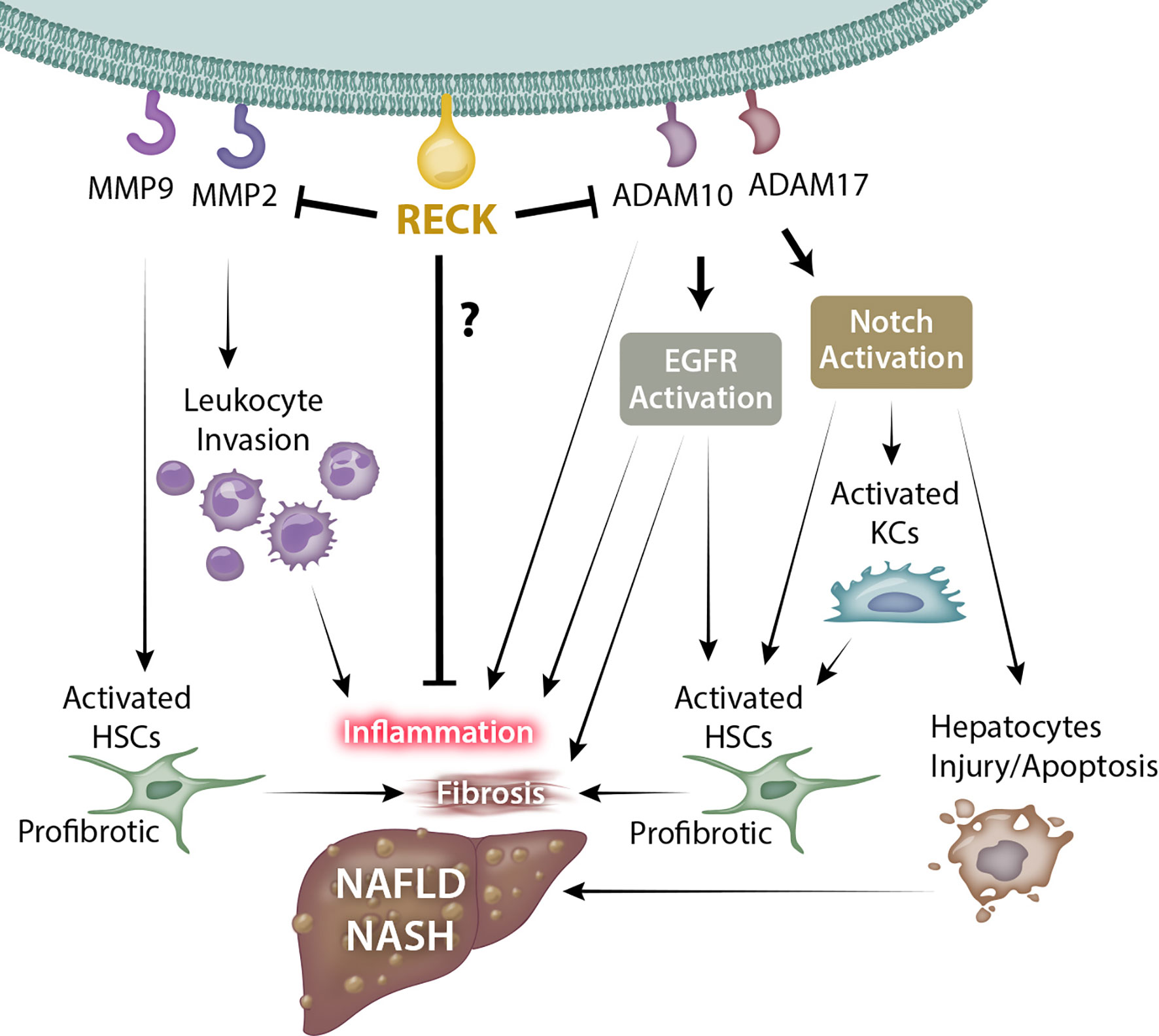
Figure 1 Possible mechanism through which RECK influences NAFLD/NASH development and pathogenesis. RECK’s inhibition of the gelatinases MMP2 and MMP9 may in turn reduce leukocyte invasion into the hepatic parenchyma and hepatic stellate cell activation. In addition, RECK inhibits the sheddases ADAM10 and ADAM17, which consequently may inhibit the release of proinflammatory cytokines from hepatic cells, as well as reduce activation of EGFR and Notch pathways, both of which contribute to inflammation and fibrosis of liver. © Copyright 2021 by The Curators of the University of Missouri, a public corporation.
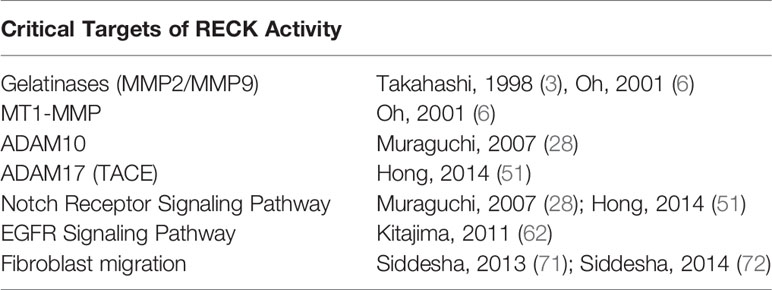
Table 1 Major targets of RECK activity.
Author Contributions
RD, BC, and RR conceived the manuscript idea. RD and CD wrote the manuscript. BC and RR provided critical editing and input. All authors have read and approved the final manuscript.
Funding
Support was provided by VA-Merit Grant I01BX003271 and NIH R01 DK113701-01 to RSR, and VA-Merit Grant (I01BX004220) and RCS (IK6004016) to BC. RD is supported by NIH T32 OD011126.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi: 10.1038/nrm3904
2. Paolillo M, Schinelli S. Extracellular Matrix Alterations in Metastatic Processes. Int J Mol Sci (2019) 20(19):4947. doi: 10.3390/ijms20194947
3. Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, et al. Regulation of Matrix Metalloproteinase-9 and Inhibition of Tumor Invasion by the Membrane-Anchored Glycoprotein RECK. Proc Natl Acad Sci USA (1998) 95(22):13221–6. doi: 10.1073/pnas.95.22.13221
4. Alexius-Lindgren M, Andersson E, Lindstedt I, Engström W. The RECK Gene and Biological Malignancy–Its Significance in Angiogenesis and Inhibition of Matrix Metalloproteinases. Anticancer Res (2014) 34(8):3867–73.
5. Clark JC, Thomas DM, Choong PF, Dass CR. RECK–a Newly Discovered Inhibitor of Metastasis With Prognostic Significance in Multiple Forms of Cancer. Cancer Metastasis Rev (2007) 26(3–4):675–83. doi: 10.1007/s10555-007-9093-8
6. Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, et al. The Membrane-Anchored MMP Inhibitor RECK Is a Key Regulator of Extracellular Matrix Integrity and Angiogenesis. Cell (2001) 107(6):789–800. doi: 10.1016/S0092-8674(01)00597-9
7. Chang CK, Hung WC, Chang HC. The Kazal Motifs of RECK Protein Inhibit MMP-9 Secretion and Activity and Reduce Metastasis of Lung Cancer Cells In Vitro and In Vivo. J Cell Mol Med (2008) 12(6B):2781–9. doi: 10.1111/j.1582-4934.2008.00215.x
8. Takemoto N, Tada M, Hida Y, Asano T, Cheng S, Kuramae T, et al. Low Expression of Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs (RECK) Indicates a Shorter Survival After Resection in Patients With Adenocarcinoma of the Lung. Lung Cancer (2007) 58(3):376–83. doi: 10.1016/j.lungcan.2007.07.004
9. Noda M, Takahashi C. Recklessness as a Hallmark of Aggressive Cancer. Cancer Sci (2007) 98(11):1659–65. doi: 10.1111/j.1349-7006.2007.00588.x
10. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD Disease Burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the Period 2016-2030. J Hepatol (2018) 69(4):896–904. doi: 10.1016/j.jhep.2018.05.036
11. Furumoto K, Arii S, Mori A, Furuyama H, Gorrin Rivas MJ, Nakao T, et al. RECK Gene Expression in Hepatocellular Carcinoma: Correlation With Invasion-Related Clinicopathological Factors and Its Clinical Significance. Reverse-Inducing–Cysteine-Rich Protein With Kazal Motifs. Hepatology (2001) 33(1):189–95. doi: 10.1053/jhep.2001.21048
12. Peng X, Wu W, Zhu B, Sun Z, Ji L, Ruan Y, et al. Activation of Farnesoid X Receptor Induces RECK Expression in Mouse Liver. Biochem Biophys Res Commun (2014) 443(1):211–6. doi: 10.1016/j.bbrc.2013.11.082
13. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile Acids Lower Triglyceride Levels via a Pathway Involving FXR, SHP, and SREBP-1c. J Clin Invest (2004) 113(10):1408–18. doi: 10.1172/JCI21025
14. Porez G, Prawitt J, Gross B, Staels B. Bile Acid Receptors as Targets for the Treatment of Dyslipidemia and Cardiovascular Disease. J Lipid Res (2012) 53(9):1723–37. doi: 10.1194/jlr.R024794
15. Oseini AM, Sanyal AJ. Therapies in Non-Alcoholic Steatohepatitis (NASH). Liver Int (2017) 37(Suppl 1):97–103. doi: 10.1111/liv.13302
16. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet (2015) 385(9972):956–65. doi: 10.1016/S0140-6736(14)61933-4
17. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic Acid for the Treatment of Non-Alcoholic Steatohepatitis: Interim Analysis From a Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet (2019) 394(10215):2184–96. doi: 10.1016/S0140-6736(19)33041-7
18. Mohan V, Das A, Sagi I. Emerging Roles of ECM Remodeling Processes in Cancer. Semin Cancer Biol (2020) 62:192–200. doi: 10.1016/j.semcancer.2019.09.004
19. Russell JJ, Grisanti LA, Brown SM, Bailey CA, Bender SB, Chandrasekar B. Reversion Inducing Cysteine Rich Protein With Kazal Motifs and Cardiovascular Diseases: The RECKlessness of Adverse Remodeling. Cell Signal (2021) 83:109993. doi: 10.1016/j.cellsig.2021.109993
20. Benassi MS, Gamberi G, Magagnoli G, Molendini L, Ragazzini P, Merli M, et al. Metalloproteinase Expression and Prognosis in Soft Tissue Sarcomas. Ann Oncol (2001) 12(1):75–80. doi: 10.1023/A:1008318614461
21. Jiang H, Li H. Prognostic Values of Tumoral MMP2 and MMP9 Overexpression in Breast Cancer: A Systematic Review and Meta-Analysis. BMC Cancer (2021) 21(1):149. doi: 10.1186/s12885-021-07860-2
22. Nishio K, Motozawa K, Omagari D, Gojoubori T, Ikeda T, Asano M, et al. Comparison of MMP2 and MMP9 Expression Levels Between Primary and Metastatic Regions of Oral Squamous Cell Carcinoma. J Oral Sci (2016) 58(1):59–65. doi: 10.2334/josnusd.58.59
23. Zeng L, Qian J, Zhu F, Wu F, Zhao H, Zhu H. The Prognostic Values of Matrix Metalloproteinases in Ovarian Cancer. J Int Med Res (2020) 48(1):300060519825983. doi: 10.1177/0300060519825983
24. Lotfi A, Mohammadi G, Tavassoli A, Mousaviagdas M, Chavoshi H, Saniee L. Serum Levels of MMP9 and MMP2 in Patients With Oral Squamous Cell Carcinoma. Asian Pac J Cancer Prev (2015) 16(4):1327–30. doi: 10.7314/APJCP.2015.16.4.1327
25. Rhee JS, Coussens LM. RECKing MMP Function: Implications for Cancer Development. Trends Cell Biol (2002) 12(5):209–11. doi: 10.1016/S0962-8924(02)02280-8
26. Miki T, Takegami Y, Okawa K, Muraguchi T, Noda M, Takahashi C. The Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs (RECK) Interacts With Membrane Type 1 Matrix Metalloproteinase and CD13/aminopeptidase N and Modulates Their Endocytic Pathways. J Biol Chem (2007) 282(16):12341–52. doi: 10.1074/jbc.M610948200
27. Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rossé C, Chavrier P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu Rev Cell Dev Biol (2016) 32:555–76. doi: 10.1146/annurev-cellbio-111315-125227
28. John A, Tuszynski G. The Role of Matrix Metalloproteinases in Tumor Angiogenesis and Tumor Metastasis. Pathol Oncol Res (2001) 7(1):14–23. doi: 10.1007/BF03032599
29. Namwat N, Puetkasichonpasutha J, Loilome W, Yongvanit P, Techasen A, Puapairoj A, et al. Downregulation of Reversion-Inducing-Cysteine-Rich Protein With Kazal Motifs (RECK) Is Associated With Enhanced Expression of Matrix Metalloproteinases and Cholangiocarcinoma Metastases. J Gastroenterol (2011) 46(5):664–75. doi: 10.1007/s00535-010-0345-y
30. Kang HG, Kim HS, Kim KJ, Oh JH, Lee MR, Seol SM, et al. RECK Expression in Osteosarcoma: Correlation With Matrix Metalloproteinases Activation and Tumor Invasiveness. J Orthop Res (2007) 25(5):696–702. doi: 10.1002/jor.20323
31. Liang QX, Liang YC, Xu ZY, Chen WL, Xie HL, Zhang B. RECK Overexpression Reduces Invasive Ability in Ameloblastoma Cells. J Oral Pathol Med (2014) 43(8):613–8. doi: 10.1111/jop.12179
32. van der Jagt MF, Sweep FC, Waas ET, Hendriks T, Ruers TJ, Merry AH, et al. Correlation of Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs (RECK) and Extracellular Matrix Metalloproteinase Inducer (EMMPRIN), With MMP-2, MMP-9, and Survival in Colorectal Cancer. Cancer Lett (2006) 237(2):289–97. doi: 10.1016/j.canlet.2005.06.009
33. Zhang C, Ling Y, Xu Y, Gao L, Li R, Zhu J, et al. The Silencing of RECK Gene Is Associated With Promoter Hypermethylation and Poor Survival in Hepatocellular Carcinoma. Int J Biol Sci (2012) 8(4):451–8. doi: 10.7150/ijbs.4038
34. Muraguchi T, Takegami Y, Ohtsuka T, Kitajima S, Chandana EP, Omura A, et al. RECK Modulates Notch Signaling During Cortical Neurogenesis by Regulating ADAM10 Activity. Nat Neurosci (2007) 10(7):838–45. doi: 10.1038/nn1922
35. Dong ZR, Chen ZQ, Yang XY, Ding ZN, Liu KX, Yan LJ, et al. RECK Expression Is Associated With Angiogenesis and Immunogenic Tumor Microenvironment in Hepatocellular Carcinoma, and Is a Prognostic Factor for Better Survival. J Cancer (2021) 12(13):3827–40. doi: 10.7150/jca.56167
36. Chung TT, Yeh CB, Li YC, Su SC, Chien MH, Yang SF, et al. Effect of RECK Gene Polymorphisms on Hepatocellular Carcinoma Susceptibility and Clinicopathologic Features. PLoS One (2012) 7(3):e33517. doi: 10.1371/journal.pone.0033517
37. Yu Y, Hu Y, Li K, Chen Z, Zhang H, Zhang L. RECK Gene Polymorphism Is Associated With Susceptibility and Prognosis of Wilms' Tumor in Chinese Children. Med Sci Monit (2015) 21:1928–33. doi: 10.12659/MSM.893606
38. Lei H, Hemminki K, Altieri A, Johansson R, Enquist K, Hallmans G, et al. Promoter Polymorphisms in Matrix Metalloproteinases and Their Inhibitors: Few Associations With Breast Cancer Susceptibility and Progression. Breast Cancer Res Treat (2007) 103(1):61–9. doi: 10.1007/s10549-006-9345-2
39. Bahgat DM, Shahin RM, Makar NN, Aziz AO, Hunter SS. Reversion-Inducing-Cysteine-Rich Protein With Kazal Motifs (RECK) Gene Single Nucleotide Polymorphism With Hepatocellular Carcinoma: A Case-Control Study. J Clin Lab Anal (2016) 30(1):36–40. doi: 10.1002/jcla.21806
40. Li Y, Xu D, Bao C, Zhang Y, Chen D, Zhao F, et al. MicroRNA-135b, a HSF1 Target, Promotes Tumor Invasion and Metastasis by Regulating RECK and EVI5 in Hepatocellular Carcinoma. Oncotarget (2015) 6(4):2421–33. doi: 10.18632/oncotarget.2965
41. Yang L, Jiang J. GAS5 Regulates RECK Expression and Inhibits Invasion Potential of HCC Cells by Sponging miR-135b. BioMed Res Int (2019) 2019:2973289. doi: 10.1155/2019/2973289
42. Huang Q, Liu L, Liu CH, You H, Shao F, Xie F, et al. MicroRNA-21 Regulates the Invasion and Metastasis in Cholangiocarcinoma and may be a Potential Biomarker for Cancer Prognosis. Asian Pac J Cancer Prev (2013) 14(2):829–34. doi: 10.7314/APJCP.2013.14.2.829
43. Takagi S, Simizu S, Osada H. RECK Negatively Regulates Matrix Metalloproteinase-9 Transcription. Cancer Res (2009) 69(4):1502–8. doi: 10.1158/0008-5472.CAN-08-2635
44. Mendes SR, Amo-Maestro LD, Marino-Puertas L, Diego I, Goulas T, Gomis-Rüth FX. Analysis of the Inhibiting Activity of Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs (RECK) on Matrix Metalloproteinases. Sci Rep (2020) 10(1):6317. doi: 10.1038/s41598-020-63338-4
45. Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan Is Selectively Cleaved at the Parenchymal Basement Membrane at Sites of Leukocyte Extravasation in Experimental Autoimmune Encephalomyelitis. J Exp Med (2006) 203(4):1007–19. doi: 10.1084/jem.20051342
46. Hannocks MJ, Zhang X, Gerwien H, Chashchina A, Burmeister M, Korpos E, et al. The Gelatinases, MMP-2 and MMP-9, as Fine Tuners of Neuroinflammatory Processes. Matrix Biol (2019) 75-76:102–13. doi: 10.1016/j.matbio.2017.11.007
47. Corry DB, Rishi K, Kanellis J, Kiss A, Song Lz LZ, Xu J, et al. Decreased Allergic Lung Inflammatory Cell Egression and Increased Susceptibility to Asphyxiation in MMP2-Deficiency. Nat Immunol (2002) 3(4):347–53. doi: 10.1038/ni773
48. Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, et al. Overlapping and Independent Contributions of MMP2 and MMP9 to Lung Allergic Inflammatory Cell Egression Through Decreased CC Chemokines. FASEB J (2004) 18(9):995–7. doi: 10.1096/fj.03-1412fje
49. Pevzner IB, Zorova LD, Galkin FA, Plotnikov EY, Zorov DB. Mitochondria-Associated Matrix Metalloproteinases 2 and 9 in Acute Renal Pathologies. Bull Exp Biol Med (2019) 166(3):334–8. doi: 10.1007/s10517-019-04345-y
50. Nascimento GC, Rizzi E, Gerlach RF, Leite-Panissi CR. Expression of MMP-2 and MMP-9 in the Rat Trigeminal Ganglion During the Development of Temporomandibular Joint Inflammation. Braz J Med Biol Res (2013) 46(11):956–67. doi: 10.1590/1414-431X20133138
51. Moore BA, Manthey CL, Johnson DL, Bauer AJ. Matrix Metalloproteinase-9 Inhibition Reduces Inflammation and Improves Motility in Murine Models of Postoperative Ileus. Gastroenterology (2011) 141(4):1283–92, 92.e1-4. doi: 10.1053/j.gastro.2011.06.035
52. Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. MMP-2, MT1-MMP, and TIMP-2 Are Essential for the Invasive Capacity of Human Mesenchymal Stem Cells: Differential Regulation by Inflammatory Cytokines. Blood (2007) 109(9):4055–63. doi: 10.1182/blood-2006-10-051060
53. Adapala VJ, Adedokun SA, Considine RV, Ajuwon KM. Acute Inflammation Plays a Limited Role in the Regulation of Adipose Tissue COL1A1 Protein Abundance. J Nutr Biochem (2012) 23(6):567–72. doi: 10.1016/j.jnutbio.2011.02.013
54. Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 Deficiency Protects Against Hepatic Ischemia/Reperfusion Injury. Hepatology (2008) 47(1):186–98. doi: 10.1002/hep.21922
55. D'Haese A, Wuyts A, Dillen C, Dubois B, Billiau A, Heremans H, et al. In Vivo Neutrophil Recruitment by Granulocyte Chemotactic Protein-2 Is Assisted by Gelatinase B/MMP-9 in the Mouse. J Interferon Cytokine Res (2000) 20(7):667–74. doi: 10.1089/107999000414853
56. Lingwal N, Padmasekar M, Samikannu B, Bretzel RG, Preissner KT, Linn T. Inhibition of Gelatinase B (Matrix Metalloprotease-9) Activity Reduces Cellular Inflammation and Restores Function of Transplanted Pancreatic Islets. Diabetes (2012) 61(8):2045–53. doi: 10.2337/db11-1143
57. Hong KJ, Wu DC, Cheng KH, Chen LT, Hung WC. RECK Inhibits Stemness Gene Expression and Tumorigenicity of Gastric Cancer Cells by Suppressing ADAM-Mediated Notch1 Activation. J Cell Physiol (2014) 229(2):191–201. doi: 10.1002/jcp.24434
58. Wong E, Cohen T, Romi E, Levin M, Peleg Y, Arad U, et al. Harnessing the Natural Inhibitory Domain to Control Tnfα Converting Enzyme (TACE) Activity In Vivo. Sci Rep (2016) 6:35598. doi: 10.1038/srep35598
59. Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: A Molecular Switch to Control Inflammation and Tissue Regeneration. Trends Immunol (2011) 32(8):380–7. doi: 10.1016/j.it.2011.05.005
60. Lopetuso LR, Mocci G, Marzo M, D'Aversa F, Rapaccini GL, Guidi L, et al. Harmful Effects and Potential Benefits of Anti-Tumor Necrosis Factor (TNF)-α on the Liver. Int J Mol Sci (2018) 19(8):2199. doi: 10.3390/ijms19082199
61. Cobbina E, Akhlaghi F. Non-Alcoholic Fatty Liver Disease (NAFLD) - Pathogenesis, Classification, and Effect on Drug Metabolizing Enzymes and Transporters. Drug Metab Rev (2017) 49(2):197–211. doi: 10.1080/03602532.2017.1293683
62. Gutiérrez J, Droppelmann CA, Salsoso R, Westermeier F, Toledo F, Salomon C, et al. A Hypothesis for the Role of RECK in Angiogenesis. Curr Vasc Pharmacol (2016) 14(1):106–15. doi: 10.2174/1570161113666151014130746
63. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch Signalling Drives Synovial Fibroblast Identity and Arthritis Pathology. Nature (2020) 582(7811):259–64. doi: 10.1038/s41586-020-2222-z
64. Wei H, Yin X, Tang H, Gao Y, Liu B, Wu Q, et al. Hypomethylation of Notch1 DNA Is Associated With the Occurrence of Uveitis. Clin Exp Immunol (2020) 201(3):317–27. doi: 10.1111/cei.13471
65. Gamrekelashvili J, Kapanadze T, Sablotny S, Ratiu C, Dastagir K, Lochner M, et al. Notch and TLR Signaling Coordinate Monocyte Cell Fate and Inflammation. Elife (2020) 9:e57007. doi: 10.7554/eLife.57007
66. Fazio C, Ricciardiello L. Inflammation and Notch Signaling: A Crosstalk With Opposite Effects on Tumorigenesis. Cell Death Dis (2016) 7(12):e2515. doi: 10.1038/cddis.2016.408
67. Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte Notch Activation Induces Liver Fibrosis in Nonalcoholic Steatohepatitis. Sci Transl Med (2018) 10(468):eaat0344. doi: 10.1126/scitranslmed.aat0344
68. Kitajima S, Miki T, Takegami Y, Kido Y, Noda M, Hara E, et al. Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs Interferes With Epidermal Growth Factor Receptor Signaling. Oncogene (2011) 30(6):737–50. doi: 10.1038/onc.2010.448
69. Lee YM, Lee SH, Lee KB, Nguyen MP, Lee MY, Park GH, et al. Silencing of Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs Stimulates Hyperplastic Phenotypes Through Activation of Epidermal Growth Factor Receptor and Hypoxia-Inducible Factor-2α. PLoS One (2013) 8(12):e84520. doi: 10.1371/journal.pone.0084520
70. Reinehr R, Häussinger D. Epidermal Growth Factor Receptor Signaling in Liver Cell Proliferation and Apoptosis. Biol Chem (2009) 390(10):1033–7. doi: 10.1515/BC.2009.106
71. Reinehr R, Häussinger D. CD95 Death Receptor and Epidermal Growth Factor Receptor (EGFR) in Liver Cell Apoptosis and Regeneration. Arch Biochem Biophys (2012) 518(1):2–7. doi: 10.1016/j.abb.2011.12.004
72. Bhushan B, Michalopoulos GK. Role of Epidermal Growth Factor Receptor in Liver Injury and Lipid Metabolism: Emerging New Roles for an Old Receptor. Chem Biol Interact (2020) 324:109090. doi: 10.1016/j.cbi.2020.109090
73. Bhushan B, Banerjee S, Paranjpe S, Koral K, Mars WM, Stoops JW, et al. Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Nonalcoholic Fatty Liver Disease in a Murine Fast-Food Diet Model. Hepatology (2019) 70(5):1546–63. doi: 10.1002/hep.30696
74. Liang D, Chen H, Zhao L, Zhang W, Hu J, Liu Z, et al. Inhibition of EGFR Attenuates Fibrosis and Stellate Cell Activation in Diet-Induced Model of Nonalcoholic Fatty Liver Disease. Biochim Biophys Acta Mol Basis Dis (2018) 1864(1):133–42. doi: 10.1016/j.bbadis.2017.10.016
75. Aroor AR, Habibi J, Nistala R, Ramirez-Perez FI, Martinez-Lemus LA, Jaffe IZ, et al. Diet-Induced Obesity Promotes Kidney Endothelial Stiffening and Fibrosis Dependent on the Endothelial Mineralocorticoid Receptor. Hypertension (2019) 73(4):849–58. doi: 10.1161/HYPERTENSIONAHA.118.12198
76. Siddesha JM, Valente AJ, Yoshida T, Sakamuri SS, Delafontaine P, Iba H, et al. Docosahexaenoic Acid Reverses Angiotensin II-Induced RECK Suppression and Cardiac Fibroblast Migration. Cell Signal (2014) 26(5):933–41. doi: 10.1016/j.cellsig.2014.01.005
77. Siddesha JM, Valente AJ, Sakamuri SS, Yoshida T, Gardner JD, Somanna N, et al. Angiotensin II Stimulates Cardiac Fibroblast Migration via the Differential Regulation of Matrixins and RECK. J Mol Cell Cardiol (2013) 65:9–18. doi: 10.1016/j.yjmcc.2013.09.015
78. Siddesha JM, Valente AJ, Sakamuri SS, Gardner JD, Delafontaine P, Noda M, et al. Acetylsalicylic Acid Inhibits IL-18-Induced Cardiac Fibroblast Migration Through the Induction of RECK. J Cell Physiol (2014) 229(7):845–55. doi: 10.1002/jcp.24511
79. Kurzepa J, Mądro A, Czechowska G, Celiński K, Kazmierak W, Slomka M. Role of MMP-2 and MMP-9 and Their Natural Inhibitors in Liver Fibrosis, Chronic Pancreatitis and Non-Specific Inflammatory Bowel Diseases. Hepatobiliary Pancreat Dis Int (2014) 13(6):570–9. doi: 10.1016/S1499-3872(14)60261-7
80. Feng M, Ding J, Wang M, Zhang J, Zhu X, Guan W. Kupffer-Derived Matrix Metalloproteinase-9 Contributes to Liver Fibrosis Resolution. Int J Biol Sci (2018) 14(9):1033–40. doi: 10.7150/ijbs.25589
81. Collazos J, Valle-Garay E, Suárez-Zarracina T, Montes AH, Cartón JA, Asensi V. Matrix Metalloproteases and Their Tissue Inhibitors in Non-Alcoholic Liver Fibrosis of Human Immunodeficiency Virus-Infected Patients. World J Virol (2017) 6(2):36–45. doi: 10.5501/wjv.v6.i2.36
82. Munsterman ID, Kendall TJ, Khelil N, Popa M, Lomme R, Drenth JPH, et al. Extracellular Matrix Components Indicate Remodelling Activity in Different Fibrosis Stages of Human Non-Alcoholic Fatty Liver Disease. Histopathology (2018) 73(4):612–21. doi: 10.1111/his.13665
83. Veidal SS, Nielsen MJ, Leeming DJ, Karsdal MA. Phosphodiesterase Inhibition Mediates Matrix Metalloproteinase Activity and the Level of Collagen Degradation Fragments in a Liver Fibrosis Ex Vivo Rat Model. BMC Res Notes (2012) 5(1):686. doi: 10.1186/1756-0500-5-686
84. Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in Liver Fibrosis - A Systematic Review With Special Emphasis on Anti-Fibrotic Strategies. J Hepatol (2007) 46(5):955–75. doi: 10.1016/j.jhep.2007.02.003
85. Benyon RC, Arthur MJ. Extracellular Matrix Degradation and the Role of Hepatic Stellate Cells. Semin Liver Dis (2001) 21(3):373–84. doi: 10.1055/s-2001-17552
86. Lieber CS, Weiss DG, Paronetto F. Value of Fibrosis Markers for Staging Liver Fibrosis in Patients With Precirrhotic Alcoholic Liver Disease. Alcohol Clin Exp Res (2008) 32(6):1031–9. doi: 10.1111/j.1530-0277.2008.00664.x
87. Kuyvenhoven JP, van Hoek B, Blom E, van Duijn W, Hanemaaijer R, Verheijen JH, et al. Assessment of the Clinical Significance of Serum Matrix Metalloproteinases MMP-2 and MMP-9 in Patients With Various Chronic Liver Diseases and Hepatocellular Carcinoma. Thromb Haemost (2003) 89(4):718–25. doi: 10.1055/s-0037-1613578
88. Onozuka I, Kakinuma S, Kamiya A, Miyoshi M, Sakamoto N, Kiyohashi K, et al. Cholestatic Liver Fibrosis and Toxin-Induced Fibrosis Are Exacerbated in Matrix Metalloproteinase-2 Deficient Mice. Biochem Biophys Res Commun (2011) 406(1):134–40. doi: 10.1016/j.bbrc.2011.02.012
89. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology (2020) 158(7):1913–28. doi: 10.1053/j.gastro.2019.11.311
90. Aimaiti Y, Yusufukadier M, Li W, Tuerhongjiang T, Shadike A, Meiheriayi A, et al. TGF-β1 Signaling Activates Hepatic Stellate Cells Through Notch Pathway. Cytotechnology (2019) 71(5):881–91. doi: 10.1007/s10616-019-00329-y
91. Bansal R, van Baarlen J, Storm G, Prakash J. The Interplay of the Notch Signaling in Hepatic Stellate Cells and Macrophages Determines the Fate of Liver Fibrogenesis. Sci Rep (2015) 5:18272. doi: 10.1038/srep18272
92. Hu M, Ou-Yang HF, Wu CG, Qu SY, Xu XT, Wang P. Notch Signaling Regulates Col1α1 and Col1α2 Expression in Airway Fibroblasts. Exp Biol Med (Maywood) (2014) 239(12):1589–96. doi: 10.1177/1535370214538919
93. Venkataraman T, Frieman MB. The Role of Epidermal Growth Factor Receptor (EGFR) Signaling in SARS Coronavirus-Induced Pulmonary Fibrosis. Antiviral Res (2017) 143:142–50. doi: 10.1016/j.antiviral.2017.03.022
94. Stolarczyk M, Scholte BJ. The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology. Mediators Inflammation (2018) 2018:1067134. doi: 10.1155/2018/1067134
95. Morimoto Y, Hirahara K, Kiuchi M, Wada T, Ichikawa T, Kanno T, et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity (2018) 49(1):134–50.e6. doi: 10.1016/j.immuni.2018.04.023
96. Panizo S, Martínez-Arias L, Alonso-Montes C, Cannata P, Martín-Carro B, Fernández-Martín JL, et al. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int J Mol Sci (2021) 22(1):408. doi: 10.3390/ijms22010408
97. Rayego-Mateos S, Rodrigues-Diez R, Morgado-Pascual JL, Valentijn F, Valdivielso JM, Goldschmeding R, et al. Role of Epidermal Growth Factor Receptor (EGFR) and Its Ligands in Kidney Inflammation and Damage. Mediators Inflamm (2018) 2018:8739473. doi: 10.1155/2018/8739473
98. Cai X, Li Z, Zhang Q, Qu Y, Xu M, Wan X, et al. CXCL6-EGFR-Induced Kupffer Cells Secrete TGF-β1 Promoting Hepatic Stellate Cell Activation via the SMAD2/BRD4/C-MYC/EZH2 Pathway in Liver Fibrosis. J Cell Mol Med (2018) 22(10):5050–61. doi: 10.1111/jcmm.13787
99. Scheving LA, Zhang X, Threadgill DW, Russell WE. Hepatocyte ERBB3 and EGFR Are Required for Maximal CCl4-Induced Liver Fibrosis. Am J Physiol Gastrointest Liver Physiol (2016) 311(5):G807–G16. doi: 10.1152/ajpgi.00423.2015
100. Fujii T, Fuchs BC, Yamada S, Lauwers GY, Kulu Y, Goodwin JM, et al. Mouse Model of Carbon Tetrachloride Induced Liver Fibrosis: Histopathological Changes and Expression of CD133 and Epidermal Growth Factor. BMC Gastroenterol (2010) 10:79. doi: 10.1186/1471-230X-10-79
101. Chen Y, Tseng SH. The Potential of RECK Inducers as Antitumor Agents for Glioma. Anticancer Res (2012) 32(7):2991–8.
Keywords: RECK, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, extracellular matrix, inflammation, fibrosis, hepatocellular carcinoma
Citation: Dashek RJ, Diaz C, Chandrasekar B and Rector RS (2021) The Role of RECK in Hepatobiliary Neoplasia Reveals Its Therapeutic Potential in NASH. Front. Endocrinol. 12:770740. doi: 10.3389/fendo.2021.770740
Received: 04 September 2021; Accepted: 04 October 2021;
Published: 20 October 2021.
Edited by:
Xavier Revelo, University of Minnesota Twin Cities, United StatesReviewed by:
Seonghwan Hwang, Pusan National University, South KoreaDavide Povero, Mayo Clinic, United States
Copyright © 2021 Dashek, Diaz, Chandrasekar and Rector. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Scott Rector, cmVjdG9yc0BoZWFsdGgubWlzc291cmkuZWR1