 Sooyeon Lee
Sooyeon Lee Benjamin Thilo Krüger
Benjamin Thilo Krüger Anita Ignatius
Anita Ignatius Jan Tuckermann
Jan Tuckermann- 1Institute for Comparative Molecular Endocrinology, University of Ulm, Ulm, Germany
- 2Institute of Orthopedic Research and Biomechanics, Trauma Research Center Ulm, Ulm University Medical Center, Ulm, Germany
Glucocorticoids (GCs) are steroid hormones that respond to stress and the circadian rhythm. Pharmacological GCs are widely used to treat autoimmune and chronic inflammatory diseases despite their adverse effects on bone after long-term therapy. GCs regulate bone homeostasis in a cell-type specific manner, affecting osteoblasts, osteoclasts, and osteocytes. Endogenous physiological and exogenous/excessive GCs act via nuclear receptors, mainly via the GC receptor (GR). Endogenous GCs have anabolic effects on bone mass regulation, while excessive or exogenous GCs can cause detrimental effects on bone. GC-induced osteoporosis (GIO) is a common adverse effect after GC therapy, which increases the risk of fractures. Exogenous GC treatment impairs osteoblastogenesis, survival of the osteoblasts/osteocytes and prolongs the longevity of osteoclasts. Under normal physiological conditions, endogenous GCs are regulated by the circadian rhythm and circadian genes display oscillatory rhythmicity in bone cells. However, exogenous GCs treatment disturbs the circadian rhythm. Recent evidence suggests that the disturbed circadian rhythm by continuous exogenous GCs treatment can in itself hamper bone integrity. GC signaling is also important for fracture healing and rheumatoid arthritis, where crosstalk among several cell types including macrophages and stromal cells is indispensable. This review summarizes the complexity of GC actions via GR in bone cells at cellular and molecular levels, including the effect on circadian rhythmicity, and outlines new therapeutic possibilities for the treatment of their adverse effects.
Introduction
Glucocorticoids (GCs) are steroid hormones that respond to stress and the circadian rhythm. Endogenous GCs are released by the adrenal glands upon activation of the hypothalamic-pituitary-adrenal (HPA) axis. Excessive or insufficient levels of endogenous GCs, Cushing's syndrome or Addison's disease, respectively, result in low bone mass and increased fracture risk (1–5). Due to their anti-inflammatory potential, exogenous GCs like dexamethasone, prednisolone, and many others are synthesized for pharmacological applications. Since the late 1940s they are widely used to treat autoimmune and chronic inflammatory diseases, recently they have been also utilized for Covid-19 treatment (6–8). However, long-term GC therapy can cause severe adverse effects in bone such as osteoporosis, and 30-50% of those patients experience fractures (9, 10).
Once GCs enter their target cell, they become activated by the 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) or deactivated by 11β-HSD2 (11, 12). After that initial step, the activated GCs bind to the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily. GR is ubiquitously expressed and acts as a monomer, homodimer or even a tetramer (7, 13). The ligand-bound GR translocates into the nucleus and induces transactivation or transrepression of target genes in several ways (7): 1) direct binding of GR homodimers or oligomers to DNA associated GC-response elements (GRE), 2) direct binding of GR monomers to GRE, 3) tethering as a GR monomer to other DNA-bound inflammatory transcription factors such as NF-κB, AP-1, IRF-3 or Stat3.
Despite this common mechanism of GCs via GR, endogenous and exogenous GCs act distinctively in bone and are dependent on pathophysiological environments. Thus, it is necessary to understand the role of GCs in bone cells and their mechanism of action in several bone diseases. This review summarizes the status of current studies on cellular and molecular, endogenous and exogenous GC actions via the GR in bone cells. Additionally, it describes the effect of circadian rhythmicity in GC actions, and outlines new therapeutic possibilities for the treatment of their adverse effects.
Endogenous GC Action in Bone Homeostasis
Endogenous GCs directly regulate bone homeostasis via the GR in a cell-type specific manner.
Several animal models have proved that GC signaling in osteoblast-lineage cells is critical to maintain bone mass. The effect of inactivated GC signaling in mature osteoblasts and osteocytes was investigated by overexpression of 11β-HSD2, the responsible enzyme for GC inactivation. A 2.3 kb or 3.6 kb fragment of Col1a1 promoter-driven overexpression of 11β-HSD2 (Col2.3-HSD2 or Col3.6-HSD2) reduced cortical and trabecular bone mass in mice, which suggests the importance of GC signaling in osteoblast-lineage cells to regulate bone mass (14–16). Interestingly, another mouse model blocking GC action in osteoblast-lineage cells by osteocalcin promoter-driven overexpression of 11β-HSD2 (OG2-11β-HSD2) did not show any alteration in the bone under normal physiological conditions (17). These discrepancies among different mouse models could be explained by determining the specific stages of osteoblast-lineage cells or investigating cell-type specific conditional knock-out mouse models. Notably, GR deficiency in mice using cre overexpression under the control of early committed osteoblast progenitor markers (Runx2 or Osx1) resulted in decreased bone mass (18, 19). Taken together, endogenous GC signaling in osteoblast-lineage cells is essential in bone mass regulation. However, osteocyte-specific endogenous GC action remains inexplicit.
Osteoclasts, another key cell type for bone mass regulation, are not affected by endogenous GC signaling. Osteoclastogenesis and bone formation were normal in mice with the GR deleted in osteoclast progenitor cells (GRLysMCre) (18). Osteoclast-specific overexpression of 11β-HSD2 using the tartrate-resistant acid phosphatase (TRAP) promoter (TRAP-HSD2) did not alter bone mass in mice (20). Collectively, endogenous GC signaling does not affect osteoclastogenesis under normal physiological conditions.
However, GCs have a profound effect on bone loss that is induced by a model of microgravity- the hindlimb unloading (HU), a model that was developed for simulating the environment of astronauts during space voyages. In this HU model, rodents showed an elevated endogenous corticosterone level (21), which led to a decreased bone mass due to decreased osteoblastogenesis, and increased apoptosis of osteoblasts and osteocytes (22). However, blocking of GC signaling in mature osteoblasts and osteocytes using Col2.3-HSD2 transgenic mice did not alter cortical bone mass in the HU model (22). Osteoclastogenesis and bone resorption were enhanced during HU due to enhanced receptor activator of nuclear factor-κB ligand (RANKL) production in osteocytes (22). This outlines the importance of endogenous GC signaling in mature osteoblasts and osteocytes, in response to mechanical loading.
Excessive Exogenous GC Action in Bone and GC-Induced Osteoporosis
Long-term GC therapy is the most common cause of secondary osteoporosis, which leads to an increased risk of fractures (23, 24). In patients, exogenous GCs with doses higher than 2.5 mg for more than 3 months are shown to weaken bone quality (25). There is also clear evidence that exogenous GCs inhibit osteogenesis (6, 10). Bone marrow stromal cells (BMSCs) isolated from patients with corticosteroid-induced osteonecrosis showed impaired osteogenesis (26). Similarly, BMSCs isolated from a rat GIO model displayed decreased proliferation and osteogenic differentiation (27). Application of exogenous GCs in vivo suppressed proliferation and differentiation of osteoblasts and induced apoptosis of osteoblasts and osteocytes, resulting in a low bone mass (17, 18). This side effect could partially be rescued by leukemia inhibitory factor (LIF) treatment that activated Stat3, Mapk/Erk, and Akt signaling in GC-treated cells (28). Despite long-term exposure to high dose GCs, osteoblast lineage-specific GR deficient mice (GRRunx2cre) displayed normal bone formation and unaltered osteoblast and osteocyte numbers (18). This is corroborated by studies with GC inactivation in mature osteoblasts and osteocytes, using mice overexpressed 11β-HSD2 under the osteocalcin gene 2 (OG2) promoter (OG2-11β-HSD2). In these mice, GC-mediated increased apoptosis of osteoblasts and osteocytes is abrogated as well (17). These studies show that exogenous GC treatment leading to GC excess impairs osteoblastogenesis, the survival of osteoblasts, and osteocytes.
GCs affect the cross-talk among bone cells. Exposure to high doses of GCs results in an increased amount of RANKL secreted by osteoblasts and osteocytes. In turn, this increases the RANKL to osteoprotegerin (OPG) ratio and enhances bone resorption by osteoclasts (29–31).
Excessive GCs can also directly affect osteoclastogenesis (20, 32). During the initial phase of the therapy, GCs increase bone resorption by promoting osteoclast proliferation, osteoclast differentiation, and prolonging their life span (20, 33–35). However, the effect of long-term GC exposure on osteoclasts is still not entirely resolved. A few studies reported that long-term GC excess rather reduces osteoclast activity due to disrupted cytoskeleton of the osteoclasts (35, 36). However, several other studies addressed osteoclast apoptosis after long-term GC exposure (32, 34, 35, 37). Some studies showed that GCs reduce osteoclast apoptosis (34, 35), while others reported that GCs do not affect osteoclast apoptosis at all (32, 37). Collectively, pharmacological GCs affect osteoclastogenesis and bone resorption either directly, or via increased RANKL secretion from osteoblasts/osteocytes.
GCs in Skeletal Stem Cells
Skeletal stem cells are essential for bone development, growth, and maintenance (38). During the last decade, skeletal stem cell markers have been identified in humans and rodents (38–41). To date, however, the role of GCs in these cells has not yet been extensively explored. Earlier, a study demonstrated that GR deletion in mesenchymal tissues using Dermo1-Cre induces postnatal lethality due to defects in the lung and intestines (42). GR silencing on human BMSCs showed an inhibited osteogenic differentiation in vitro (43). These studies imply that GC signaling via the GR plays a key role in mesenchymal stem cells (MSCs) differentiation towards osteoblasts.
It is also known that GIO is clinically described by decreased bone mass along with increased marrow adiposity (24), indicating that GR regulates the balance between osteoblastogenesis and adipogenesis of MSCs (44). High GC doses (1 µM Dexamethasone) increased adipogenesis of human BMSCs regulated by c-Jun signaling (43). Other studies suggested that GCs induce adipogenic regulators. Adipogenesis was promoted in cortisol (1 µM) treated mouse bone marrow-derived stromal cell line ST-2, by increasing expression of Peroxisome proliferator-activated receptor-gamma2 (PPAR-γ2) and CCAAT/enhancer-binding protein (C/EBP) transcription factors that are the adipocyte master regulator (45). Similarly, C/EBPalpha expression was increased in bone of dexamethasone-treated mice (50 mg/kg daily for 5 weeks) as well as in primary BMSCs isolated from those mice (46). Dexamethasone treatment in rat BMSCs also increased PPAR-γ expression in a dose-dependent manner, whereas a PPAR-γ knockdown promoted osteogenesis (47). This GC-induced PPAR-γ expression increases Secreted frizzled-related protein 5 (SFRP5) expression which inhibits the Wnt/β-catenin pathway and thus suppresses osteogenesis (47).
Taken together, endogenous GCs promote osteoblastogenesis of MSCs, whereas exogenous or excessive GCs regulate the balance between osteoblastogenesis and adipogenesis of MSCs. Further studies are necessary to investigate the role of GCs in the fate decision of skeletal stem cells in vivo.
Circadian Rhythmicity and GCs
Endogenous GCs are released under the control of circadian rhythms, that are modulated by the central circadian clock in the suprachiasmatic nucleus (SCN) of the hypothalamus (48). The daily rhythmicity of plasma GC levels modulates physiological processes in many peripheral tissues including bone (48, 49).
Indeed, diurnal rhythm appears in some bone metabolic markers such as the bone resorption marker C-terminal cross-linked telopeptide of type I collagen (CTX), osteocyte function marker fibroblast growth factor 23 (FGF23), and turnover marker serum osteocalcin (50–53). Other bone markers such as sclerostin, procollagen type 1 N-terminal propeptide (P1NP), OPG, or soluble RANKL serum levels did not display rhythmicity (50, 52). However, the 24-hour serum profiles of men displayed that bone formation marker P1NP was significantly reduced after a long-term (3 weeks) disruption of the circadian rhythm despite no alteration of CTX level (54). In mice, disrupted circadian rhythm by weekly alternating light-dark cycles (10 or 15 weeks) led to a reduced level of both P1NP and CTX, implicating a decreased bone turnover due to disrupted circadian rhythm (55). This is likely due to the altered expression level of circadian locomotor output cycles kaput (Clock) genes that regulate the circadian rhythm in bone cells (55). Unexpectedly, unlike with the P1NP level, osteoblast surface increased in these mice (55). Together with decreased osteoclast surface, trabecular bone mass was increased in these mice despite altered Clock gene expression in the bone due to disrupted circadian rhythm (55). Nevertheless, this study indicated the importance of circadian rhythm in bone health. Investigations considering different ages and duration of circadian rhythm disruption would provide further insights into the effects of circadian rhythm in bone.
Furthermore, genetic deletion of Clock genes in mice leads to altered bone phenotypes (56–61). Under normal physiological conditions, brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein 1 (Bmal1) and period 1 (Per1) genes are expressed with oscillatory rhythmicity in bone (58, 61). Bmal1 knock-out mice and mice with Bmal1 deletion in Osx+ osteoblast precursors and their progeny showed a decreased bone mass with increased bone resorption, suggesting that Bmal1 regulates bone homeostasis by controlling osteoblast-mediated bone resorption (58). An osteoclast-specific Bmal1 knock-out mouse showed an increased bone mass due to reduced osteoclast differentiation, indicating Bmal1 also regulates osteoclast-mediated bone resorption (57). The Clock gene that forms heterodimers with Bmal1 or Bmal2 regulates bone formation via protein disulfide isomerase family A member 3 (Pdia3), shown by reduced bone formation and increased apoptosis of osteoblasts in Clock knock-out mice (56). On the other hand, physical stress-induced GC signaling induces only the Per1 gene in mouse liver, heart, lung, and stomach by binding the GR to the GRE in the Per1 promoter (62). However, it is not yet known if the GR directly binds to Bmal1, Clock, and Per1 promoters to modulate their actions in bone cells.
Conversely, a single injection of synthetic corticosteroids can reset the circadian time in the periphery such as the liver, kidney, and heart by modulating circadian gene expression (63–65). Short-term dexamethasone treatment (2 hours) synchronizes circadian gene expression in osteoblast and osteoclasts in vitro (66, 67). Upon GC treatment, this circadian rhythm was also observed in cultured osteoblasts of Per1::luciferase transgenic mice (58). A single injection of dexamethasone could restore the circadian rhythm of osteoclast-related genes such as cathepsin K (Ctsk) in adrenalectomized mice (66). Per2 knock-out mice could not restore the GC-induced bone loss despite a bisphosphonate (Zoledronic acid) treatment, although Per2 knock-out osteoblasts showed an increased proliferation capacity (68).
However, constant GC exposure by inserting slow-release corticosterone pellets led to a shutdown of the endogenous HPA axis due to negative feedback, and thus to a flattening of GC-mediated circadian rhythm mediated gene expression (69). This resulted in bone loss not only by the excessive effects of GCs but also due to disrupted circadian gene expression, increased circulating bone resorption marker, and decreased bone formation (69).
Taken together, daily endogenous GC rhythm is important for bone homeostasis. A single treatment with exogenous GCs can regulate circadian gene expression, whereas disrupted circadian rhythm by continuous GC exposure contributes in addition to direct GC effects on osteoporosis.
Influence of GCs on Bone Fracture Healing
It is well known that patients undergoing long-term GC medication are at a significantly increased risk for bone fractures (23, 70). Even though steroid use has not been found to be a major risk factor for non-union fracture healing in clinical studies (71), preclinical studies indicate that GCs also influence the complex fracture healing process (6, 72). This applies not only to GC therapy but also to endogenous GCs which control many physiological processes and, as stress hormones, are released upon a bone fracture. It can be anticipated that endogenous as well and exogenous or excessive GCs influence all stages of bone fracture healing, which necessitates a finely tuned interaction between multiple cell types, including immune, bone, and stromal cells which are all crucially regulated by GCs (6, 72). A fracture leads to the disruption of bone, blood vessels, soft tissues, and the release of danger-associated molecular patterns (DAMPS). These quickly trigger an innate immune response to contain the damage, and clear the wound site from tissue debris and pathogens (73–75). The initial response involves the activation of the complement system, the release of inflammatory chemokines and cytokines from local immune, endothelial and mesenchymal cells, as well as the recruitment and activation of further immune cells, mainly neutrophils, monocytes, and macrophages. Later, lymphocytes are also recruited to the fracture site and initiate an adaptive immune response. The inflammatory phase is regarded to promote the recruitment, proliferation, and differentiation of mesenchymal and endothelial precursor cells, which are essential for subsequent healing processes. This process comprises of the formation of a soft callus with fibrous and cartilaginous tissue, which is then continuously transformed into the bone by endochondral ossification. Finally, the hard callus is remodeled until the original bone structure is restored (73–75).
So far, only a few studies have addressed the role of endogenous GCs during fracture healing by using mouse models with impaired GC signaling (76–79). Fracture healing was significantly impaired when the endogenous GC action was globally eliminated by using mice with an inducible GR knock-out (GRgtROSACreERT2) (78). In these mice, the early systemic and local immune responses upon fracture were significantly increased. During callus formation, cartilage-to-bone transformation was disturbed, confirmed by persisting cartilage and reduced bony bridging of the fragments in GRgtROSACreERT2 mice. This study suggests a crucial role of endogenous GCs in all stages of fracture healing. Several studies showed the role of GC signaling in distinct cell types during bone regeneration. When GC signaling was disrupted in osteoblasts using Col2.3-11ß-HSD2 mice (76), intramembranous bone formation was not affected, whereas GR deletion in chondroblasts using GRCol2CreERT2 mice resulted in impaired endochondral bone healing, by increasing the cartilaginous fraction of the fracture callus (77). To investigate whether GR dimerization (which is regarded to be essential for the anti-inflammatory effects of GCs) is important for fracture healing, Hachemi et al used mice with a defective GR dimerization ability (GRdim) (79). Impaired GR dimerization had no significant effect on the healing process in a model of isolated femur fracture (79). However, in a model of compromised fracture healing, induced by hyperinflammation in a combined model of fracture and thoracic trauma, impaired GR dimerization in GRdim mice reduced inflammation and abolished the deleterious effects of posttraumatic hyperinflammation on fracture healing (79). In summary, these studies demonstrate that endogenous GCs promote fracture healing by controlling the immune response and by stimulating cartilage-to-bone transition.
In contrast to endogenous GCs, exogenously applied GCs can provoke negative effects on the fracture healing process as demonstrated in pre-clinical investigations in different species, including rabbits (80, 81), rats (82), and mice (83, 84). Consistently, these studies report impaired cartilage-to-bone transformation, reduced quality and structure of the newly formed bone, and poor biomechanical properties of the fracture callus. However, these studies are mostly descriptive and the molecular and cellular reasons for the delayed bone healing under long-term GC therapy are still not fully understood.
GCs action in Rheumatoid Arthritis
Although GCs are used to ameliorate the symptoms of rheumatoid arthritis (RA) since the 1950s, there are still surprises concerning the mode of action of GCs, their activating enzymes 11β-HSD1 and the GR requirement in distinct cell types. In RA and osteoarthritis, GCs are still in frequent use, in combination with other treatment regimens (85). Preclinical animal models for the GC modulating enzyme 11β-HSD1/2 and the GR in distinct cell types revealed distinct requirements of GC function in different cells depending on the model. First of all, the attenuation of complete GR dimerization by a knock-in of a point mutation into the second zinc finger demonstrates that an intact function of the GR allows gene regulation beyond the suppression of cytokines in different RA models (86, 87). Accordingly, global inhibition of the GC activating 11β-HSD1 abrogated the therapeutic response towards corticosterone by reduction of inflammatory symptoms in mice (88).
However, the definition of critical cell types for mediating GC action present in RA varied in distinct animal models. In antigen-induced arthritis, GR in T cells (presumably in Th17 cells) was critical to confer anti-inflammatory effects, since mice lacking the GR in T cells were completely resistant to the dexamethasone-mediated reduction of joint swelling (86). In serum transfer-induced arthritis, however, there was the surprising discovery that global GR deletion in hematopoietic cells by hematopoietic stem cell transfer into irradiated wild-type mice did not abrogate the therapeutic effects of dexamethasone (87). Vice versa GR global knock-out mice and mice with attenuated GR dimerization failed to respond to dexamethasone, even when their hematopoietic system was reconstituted by GR wild-type cells (87). These mice could not induce anti-inflammatory macrophages in the joint which are critical to resolve inflammation in RA (87). Elimination of the GR in fibroblasts (Col1a2CreERT2) attenuated the therapeutic response, strongly suggesting that GCs affect the fibroblast like-synovial cell (FLS) – macrophage crosstalk via the GR (87). Intriguingly, Hardy and colleagues showed that GC production in myeloid cells might be necessary for re-activating GC function (88). Thus, cellular cross-talk targeted by systemic and locally produced GCs seems to underly the therapeutic actions of GCs, which need to be further elucidated. Given that FLS exists in pro-inflammatory and anti-inflammatory subsets (89, 90) and interstitial/lining macrophages are existing with different fates in arthritis (91), this raises the complexity and fine-tuning of GR cross-talk.
Conclusions and Perspectives
GCs are frequently used drugs in clinics despite their detrimental effects on bone after long-term use. They act in cell-type specific manner, and via cellular crosstalk mechanisms, which are still partially unknown. Currently, some drugs are applied to treat GIO by either inhibiting osteoclast activity (Bisphosphonates and Denosumab) or stimulating osteoblast activity (Teriparatide) (92). However, the utilization of drugs to treat unwanted effects caused by other drugs is not ideal for patients. Thus, it is of utmost importance to develop new therapies with a cell-type specific delivery of GCs, and/or targeting downstream molecules to avoid or minimize the detrimental effects. Further understanding of the controversial effects of endogenous and exogenous/excessive GCs on the bone that are both anabolic and catabolic will help to develop therapeutic concepts (Figures 1A, B). Daily GC rhythm should be considered during GC therapy. Chronotherapy when administering GCs could help to increase therapeutic efficacy, and reduce detrimental effects, although further investigations are required considering that the drug half-life and bioavailability can be inflexible (93). In addition, preclinical models considering factors such as physical stress, aging, and diseases can be introduced to investigate diverse clinical settings. Advanced technologies such as single-cell RNA sequencing and lineage-tracing animal models will allow us to map the alteration of specific cell types present in bone in response to GCs. It will be also helpful to determine dynamic spatial profile and crosstalk among bone cells in clinically relevant models such as fracture healing and RA. Further studies are needed to understand how GC rhythm affects such disease models. These actions in combination will ultimately broaden our scope to approach innovative therapies.
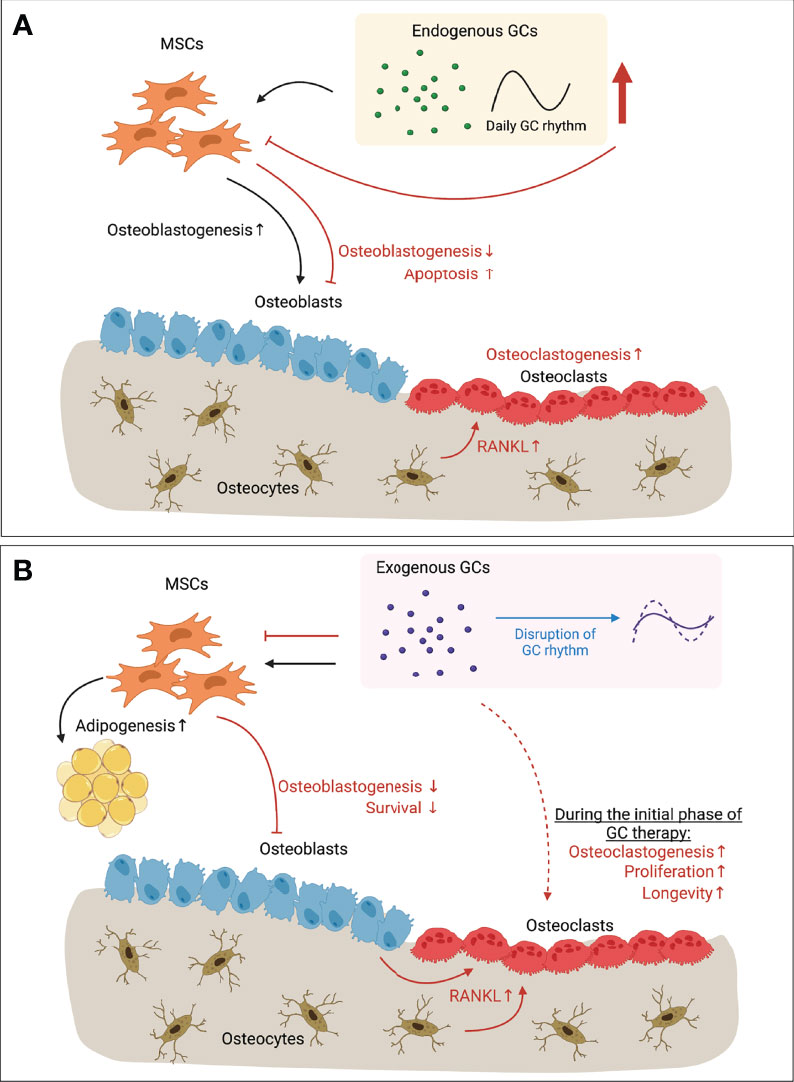
Figure 1 Paradoxical effects of GCs in bone. (A) Endogenous GCs regulated by circadian rhythm (and expressing daily GC rhythm accordingly) have anabolic effects on osteoblastogenesis (black arrows). When endogenous GC level is increased upon stress (e.g. mechanical unloading), however, bone mass is decreased due to inhibited osteoblastogenesis, increased apoptosis of osteoblasts and osteocytes, and enhanced osteoclastogenesis due to the increased RANKL secreted by apoptotic osteocytes (red arrows). (B) Long-term exogenous GC therapy inhibits osteoblastogenesis and survival of osteoblasts (red arrows). Increased RANKL secretion by osteoblasts and osteocytes let enhance bone resorption by osteoclasts (red arrows). Direct action of exogenous GCs on osteoclasts has showed with increased osteoclastogenesis, increased proliferation and longevity of osteoclasts during the initial phase of GC therapy (dotted red arrow). However, direct effects of long-term GC therapy on osteoclasts still remain elusive. Exogenous GCs also regulate the balance between osteoblastogenesis and adipogenesis of MSCs that is one of feature of GIO (black arrows). On the other hand, continuous exogenous GC therapy can flatten the endogenous GCs rhythm (blue arrow), resulting in disrupted circadian gene expression and levels of circulating bone turnover markers. Together, Long-term GC therapy leads to bone loss by its direct action on bone cells, and/or via disrupting GC rhythm. GC, Glucocorticoid; RANKL, Receptor activator of nuclear factor-κB ligand; MSC, Mesenchymal stem cell; GIO, GC-induced osteoporosis. This illustration was created with BioRender.com.
Author Contributions
SL, JT, BK, and AI wrote chapters of the article. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the following funding resources: German Research Foundation (DFG) Tu220/14-1, DFG (Ci 216/2-1), and DFG in the framework of Collaborative Research Center CRC1149 "Danger Response, Disturbance Factors and Regenerative Potential after Trauma" (251293561-CRC 1149, INST 40/492-1, and INST 40/492-2).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are very grateful to Peter Steele-Perkins for editing the manuscript.
References
1. Batista SL, De Araújo IM, Carvalho AL, Alencar MAVSD, Nahas AK, Elias J, et al. Beyond the Metabolic Syndrome: Visceral and Marrow Adipose Tissues Impair Bone Quantity and Quality in Cushing’s Disease. PLoS One (2019) 14:1–13. doi: 10.1371/journal.pone.0223432
2. Kaltsas G, Makras P. Skeletal Diseases in Cushing’s Syndrome: Osteoporosis Versus Arthropathy. Neuroendocrinology (2010) 92:60–4. doi: 10.1159/000314298
3. Løvs̊ K, Gjesdal CG, Christensen M, Wolff AB, Almås B, Svartberg J, et al. Glucocorticoid Replacement Therapy and Pharmacogenetics in Addison’s Disease: Effects on Bone. Eur J Endocrinol (2009) 160:993–1002. doi: 10.1530/EJE-08-0880
4. Björnsdottir S, Sääf M, Bensing S, Kämpe O, Michaëlsson K, Ludvigsson JF. Risk of Hip Fracture in Addison’s Disease: A Population-Based Cohort Study. J Intern Med (2011) 270:187–95. doi: 10.1111/j.1365-2796.2011.02352.x
5. Devogelaer JP, Crabbe J, De Deuxchaisnes CN. Bone Mineral Density in Addison’s Disease: Evidence for an Effect of Adrenal Androgens on Bone Mass. Br Med J (Clin Res Ed) (1987) 294:798–800. doi: 10.1136/bmj.294.6575.798
6. Ahmad M, Hachemi Y, Paxian K, Mengele F, Koenen M, Tuckermann J. A Jack of All Trades: Impact of Glucocorticoids on Cellular Cross-Talk in Osteoimmunology. Front Immunol (2019) 10:2460. doi: 10.3389/fimmu.2019.02460
7. Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, et al. Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol Rev (2016) 96:409–47. doi: 10.1152/physrev.00011.2015
8. Sterne JAC, Murthy S, Diaz JV, Slutsky AS, Villar J, Angus DC, et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-Analysis. JAMA - J Am Med Assoc (2020) 324:1330–41. doi: 10.1001/jama.2020.17023
9. Weinstein RS. Glucocorticoid-Induced Bone Disease. N Engl J Med (2011) 365:62–70. doi: 10.1056/NEJMcp1012926
10. Chotiyarnwong P, McCloskey EV. Pathogenesis of Glucocorticoid-Induced Osteoporosis and Options for Treatment. Nat Rev Endocrinol (2020) 16:437–47. doi: 10.1038/s41574-020-0341-0
11. Chapman K, Holmes M, Seckl J. 11B-Hydroxysteroid Dehydrogenases Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol Rev (2013) 93:1139–206. doi: 10.1152/physrev.00020.2012
12. Martin CS, Cooper MS, Hardy RS. Endogenous Glucocorticoid Metabolism in Bone: Friend or Foe. Front Endocrinol (Lausanne) (2021) 12:733611. doi: 10.3389/fendo.2021.733611
13. Presman DM, Ganguly S, Schiltz RL, Johnson TA, Karpova TS, Hager GL. DNA Binding Triggers Tetramerization of the Glucocorticoid Receptor in Live Cells. Proc Natl Acad Sci USA (2016) 113:8236–41. doi: 10.1073/pnas.1606774113
14. Sher LB, Woitge HW, Adams DJ, Gronowicz GA, Krozowski Z, Harrison JR, et al. Transgenic Expression of 11β-Hydroxysteroid Dehydrogenase Type 2 in Osteoblasts Reveals an Anabolic Role for Endogenous Glucocorticoids in Bone. Endocrinology (2004) 145:922–9. doi: 10.1210/en.2003-0655
15. Kalak R, Zhou H, Street J, Day RE, Modzelewski JR, Spies CM, et al. Endogenous Glucocorticoid Signalling in Osteoblasts Is Necessary to Maintain Normal Bone Structure in Mice. Bone (2009) 45:61–7. doi: 10.1016/j.bone.2009.03.673
16. Yang M, Trettel LB, Adams DJ, Harrison JR, Canalis E, Kream BE. Col3.6-HSD2 Transgenic Mice: A Glucocorticoid Loss-of-Function Model Spanning Early and Late Osteoblast Differentiation. Bone (2010) 47:573–82. doi: 10.1016/j.bone.2010.06.002
17. O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, et al. Glucocorticoids Act Directly on Osteoblasts and Osteocytes to Induce Their Apoptosis and Reduce Bone Formation and Strength. Endocrinology (2004) 145:1835–41. doi: 10.1210/en.2003-0990
18. Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids Suppress Bone Formation by Attenuating Osteoblast Differentiation via the Monomeric Glucocorticoid Receptor. Cell Metab (2010) 11:517–31. doi: 10.1016/j.cmet.2010.05.005
19. Pierce JL, Ding KH, Xu J, Sharma AK, Yu K, Arbona NM, et al. The Glucocorticoid Receptor in Osteoprogenitors Regulates Bone Mass and Marrow Fat. J Endocrinol (2019) 243:27–42. doi: 10.1530/JOE-19-0230
20. Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids Act Directly on Osteoclasts to Increase Their Life Span and Reduce Bone Density. Endocrinology (2006) 147:5592–9. doi: 10.1210/en.2006-0459
21. Wei LX, Zhou JN, Roberts AI, Shi YF. Lymphocyte Reduction Induced by Hindlimb Unloading: Distinct Mechanisms in the Spleen and Thymus. Cell Res (2003) 13:465–71. doi: 10.1038/sj.cr.7290189
22. Yang J, Li J, Cui X, Li W, Xue Y, Shang P, et al. Blocking Glucocorticoid Signaling in Osteoblasts and Osteocytes Prevents Mechanical Unloading-Induced Cortical Bone Loss. Bone (2020) 130:115108. doi: 10.1016/j.bone.2019.115108
23. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of Oral Corticosteroids and Risk of Fractures. J Bone Min Res (2000) 15:993–1000. doi: 10.1359/jbmr.2000.15.6.993
24. Weinstein RS. Glucocorticoid-Induced Osteoporosis and Osteonecrosis. Endocrinol Metab Clin North Am (2012) 41:595–611. doi: 10.1016/j.ecl.2012.04.004
25. Messina OD, Vidal LF, Vidal MV, Bultink IEM, Raterman HG, Lems W. Management of Glucocorticoid-Induced Osteoporosis. Aging Clin Exp Res (2021) 33:793–804. doi: 10.1007/s40520-021-01823-0
26. Houdek MT, Wyles CC, Packard BD, Terzic A, Behfar A, Sierra RJ. Decreased Osteogenic Activity of Mesenchymal Stem Cells in Patients With Corticosteroid-Induced Osteonecrosis of the Femoral Head. J Arthroplasty (2016) 31:893–8. doi: 10.1016/j.arth.2015.08.017
27. Zhou DA, Zheng HX, Wang CW, Shi D, Li JJ. Influence of Glucocorticoids on the Osteogenic Differentiation of Rat Bone Marrow-Derived Mesenchymal Stem Cells. BMC Musculoskelet Disord (2014) 15:1–7. doi: 10.1186/1471-2474-15-239
28. Lee S, Liu P, Ahmad M, Tuckermann JP. Leukemia Inhibitory Factor Treatment Attenuates the Detrimental Effects of Glucocorticoids on Bone in Mice. Bone (2021) 145:115843. doi: 10.1016/j.bone.2021.115843
29. Piemontese M, Xiong J, Fujiwara Y, Thostenson JD, O’Brien CA. Cortical Bone Loss Caused by Glucocorticoid Excess Requires RANKL Production by Osteocytes and Is Associated With Reduced OPG Expression in Mice. Am J Physiol - Endocrinol Metab (2016) 311:E587–93. doi: 10.1152/ajpendo.00219.2016
30. Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, et al. Stimulation of Osteoprotegerin Ligand and Inhibition of Osteoprotegerin Production by Glucocorticoids in Human Osteoblastic Lineage Cells: Potential Paracrine Mechanisms of Glucocorticoid-Induced Osteoporosis. Endocrinology (1999) 140:4382–9. doi: 10.1210/endo.140.10.7034
31. Swanson C, Lorentzon M, Conaway HH, Lerner UH. Glucocorticoid Regulation of Osteoclast Differentiation and Expression of Receptor Activator of Nuclear Factor-κb (NF-κb) Ligand, Osteoprotegerin, and Receptor Activator of NF-κb in Mouse Calvarial Bones. Endocrinology (2006) 147:3613–22. doi: 10.1210/en.2005-0717
32. Sivagurunathan S, Muir MM, Brennan TC, Seale JP, Mason RS. Influence of Glucocorticoids on Human Osteoclast Generation and Activity. J Bone Miner Res (2005) 20:390–8. doi: 10.1359/JBMR.041233
33. Hirayama T, Sabokbar A, Athanasou NA. Effect of Corticosteroids on Human Osteoclast Formation and Activity. J Endocrinol (2002) 175:155–63. doi: 10.1677/joe.0.1750155
34. Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, et al. Promotion of Osteoclast Survival and Antagonism of Bisphosphonate-Induced Osteoclast Apoptosis by Glucocorticoids. J Clin Invest (2002) 109:1041–8. doi: 10.1172/JCI0214538
35. Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, et al. Glucocorticoids Suppress Bone Formation via the Osteoclast. J Clin Invest (2006) 116:2152–60. doi: 10.1172/JCI28084
36. Hong JM, Teitelbaum SL, Kim TH, Ross FP, Kim SY, Kim HJ. Calpain-6, a Target Molecule of Glucocorticoids, Regulates Osteoclastic Bone Resorption via Cytoskeletal Organization and Microtubule Acetylation. J Bone Miner Res (2011) 26:657–65. doi: 10.1002/jbmr.241
37. Conaway HH, Henning P, Lie A, Tuckermann J, Lerner UH. Activation of Dimeric Glucocorticoid Receptors in Osteoclast Progenitors Potentiates RANKL Induced Mature Osteoclast Bone Resorbing Activity. Bone (2016) 93:43–54. doi: 10.1016/j.bone.2016.08.024
38. Ambrosi TH, Longaker MT, Chan CKF. A Revised Perspective of Skeletal Stem Cell Biology. Front Cell Dev Biol (2019) 7:189. doi: 10.3389/fcell.2019.00189
39. Chan CKF, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, et al. Identification and Specification of the Mouse Skeletal Stem Cell. Cell (2015) 160:285–98. doi: 10.1016/j.cell.2014.12.002
40. Chan CKF, Gulati GS, Sinha R, Tompkins JV, Lopez M, Carter AC, et al. Identification of the Human Skeletal Stem Cell. Cell (2018) 175:43–56.e21. doi: 10.1016/j.cell.2018.07.029
41. Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, et al. Resting Zone of the Growth Plate Houses a Unique Class of Skeletal Stem Cells. Nature (2018) 563:254–8. doi: 10.1038/s41586-018-0662-5
42. Li A, Hardy R, Stoner S, Tuckermann J, Seibel M, Zhou H. Deletion of Mesenchymal Glucocorticoid Receptor Attenuates Embryonic Lung Development and Abdominal Wall Closure. PloS One (2013) 8(5):e63578. doi: 10.1371/journal.pone.0063578
43. Cárcamo-Orive I, Gaztelumendi A, Delgado J, Tejados N, Dorronsoro A, Fernández-Rueda J, et al. Regulation of Human Bone Marrow Stromal Cell Proliferation and Differentiation Capacity by Glucocorticoid Receptor and AP-1 Crosstalk. J Bone Miner Res (2010) 25:2115–25. doi: 10.1002/jbmr.120
44. Han L, Wang B, Wang R, Gong S, Chen G, Xu W. The Shift in the Balance Between Osteoblastogenesis and Adipogenesis of Mesenchymal Stem Cells Mediated by Glucocorticoid Receptor. Stem Cell Res Ther (2019) 10:1–14. doi: 10.1186/s13287-019-1498-0
45. Pereira RC, Delany AM, Canalis E. Effects of Cortisol and Bone Morphogenetic Protein-2 on Stromal Cell Differentiation: Correlation With CCAAT-Enhancer Binding Protein Expression. Bone (2002) 30:685–91. doi: 10.1016/S8756-3282(02)00687-7
46. Li J, Zhang N, Huang X, Xu J, Fernandes JC, Dai K, et al. Dexamethasone Shifts Bone Marrow Stromal Cells From Osteoblasts to Adipocytes by C/Ebpαlpha Promoter Methylation. Cell Death Dis (2013) 4:1–11. doi: 10.1038/cddis.2013.348
47. He HP, Gu S. The PPAR-γ/SFRP5/Wnt/β-Catenin Signal Axis Regulates the Dexamethasone-Induced Osteoporosis. Cytokine (2021) 143:155488. doi: 10.1016/j.cyto.2021.155488
48. Oster H, Challet E, Ott V, Arvat E, de Kloet ER, Dijk DJ, et al. The Functional and Clinical Significance of the 24-Hour Rhythm of Circulating Glucocorticoids. Endocr Rev (2017) 38:3–45. doi: 10.1210/er.2015-1080
49. Chung S, Son GH, Kim K. Circadian Rhythm of Adrenal Glucocorticoid: Its Regulation and Clinical Implications. Biochim Biophys Acta - Mol Basis Dis (2011) 1812:581–91. doi: 10.1016/j.bbadis.2011.02.003
50. Swanson C, Shea SA, Wolfe P, Markwardt S, Cain SW, Munch M, et al. 24-Hour Profile of Serum Sclerostin and Its Association With Bone Biomarkers in Men. Osteoporos Int (2017) 28:3205–13. doi: 10.1007/s00198-017-4162-5
51. Qvist P, Christgau S, Pedersen BJ, Schlemmer A, Christiansen C. Circadian Variation in the Serum Concentration of C-Terminal Telopeptide of Type I Collagen (Serum CTx): Effects of Gender, Age, Menopausal Status, Posture, Daylight, Serum Cortisol, and Fasting. Bone (2002) 31:57–61. doi: 10.1016/S8756-3282(02)00791-3
52. Dovio A, Generali D, Tampellini M, Berruti A, Tedoldi S, Torta M, et al. Variations Along the 24-Hour Cycle of Circulating Osteoprotegerin and Soluble RANKL: A Rhythmometric Analysis. Osteoporos Int (2008) 19:113–7. doi: 10.1007/s00198-007-0423-z
53. Kawai M, Kinoshita S, Shimba S, Ozono K, Michigami T. Sympathetic Activation Induces Skeletal Fgf23 Expression in a Circadian Rhythm-Dependent Manner. J Biol Chem (2014) 289:1457–66. doi: 10.1074/jbc.M113.500850
54. Swanson CM, Shea SA, Wolfe P, Cain SW, Munch M, Vujović N, et al. Bone Turnover Markers After Sleep Restriction and Circadian Disruption: A Mechanism for Sleep-Related Bone Loss in Humans. J Clin Endocrinol Metab (2017) 102:3722–30. doi: 10.1210/jc.2017-01147
55. Schilperoort M, Bravenboer N, Lim J, Mletzko K, Busse B, van Ruijven L, et al. Circadian Disruption by Shifting the Light-Dark Cycle Negatively Affects Bone Health in Mice. FASEB J (2020) 34:1052–64. doi: 10.1096/fj.201901929R
56. Yuan G, Hua B, Yang Y, Xu L, Cai T, Sun N, et al. The Circadian Gene Clock Regulates Bone Formation Via Pdia3. J Bone Miner Res (2017) 32:861–71. doi: 10.1002/jbmr.3046
57. Xu C, Ochi H, Fukuda T, Sato S, Sunamura S, Takarada T, et al. Circadian Clock Regulates Bone Resorption in Mice. J Bone Miner Res (2016) 31:1344–55. doi: 10.1002/jbmr.2803
58. Takarada T, Xu C, Ochi H, Nakazato R, Yamada D, Nakamura S, et al. Bone Resorption Is Regulated by Circadian Clock in Osteoblasts. J Bone Miner Res (2017) 32:872–81. doi: 10.1002/jbmr.3053
59. Samsa WE, Vasanji A, Midura RJ, Kondratov RV. Deficiency of Circadian Clock Protein BMAL1 in Mice Results in a Low Bone Mass Phenotype. Bone (2016) 84:194–203. doi: 10.1016/j.bone.2016.01.006
60. Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G. The Molecular Clock Mediates Leptin-Regulated Bone Formation. Cell (2005) 122:803–15. doi: 10.1016/j.cell.2005.06.028
61. Hinoi E, Ueshima T, Hojo H, Iemata M, Takarada T, Yoneda Y. Up-Regulation of Per mRNA Expression by Parathyroid Hormone Through a Protein Kinase A-CREB-Dependent Mechanism in Chondrocytes. J Biol Chem (2006) 281:23632–42. doi: 10.1074/jbc.M512362200
62. Yamamoto T, Nakahata Y, Tanaka M, Yoshida M, Soma H, Shinohara K, et al. Acute Physical Stress Elevates Mouse Period1 mRNA Expression in Mouse Peripheral Tissues via a Glucocorticoid-Responsive Element. J Biol Chem (2005) 280:42036–43. doi: 10.1074/jbc.M509600200
63. Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, et al. Resetting of Circadian Time in Peripheral Tissues by Glucocorticoid Signaling. Science (80- ) (2000) 289:2344–7. doi: 10.1126/science.289.5488.2344
64. Pezük P, Mohawk JA, Wang LA, Menaker M. Glucocorticoids as Entraining Signals for Peripheral Circadian Oscillators. Endocrinology (2012) 153:4775–83. doi: 10.1210/en.2012-1486
65. Kamagata M, Ikeda Y, Sasaki H, Hattori Y, Yasuda S, Iwami S, et al. Potent Synchronization of Peripheral Circadian Clocks by Glucocorticoid Injections in PER2::LUC-Clock/Clock Mice. Chronobiol Int (2017) 34:1067–82. doi: 10.1080/07420528.2017.1338716
66. Fujihara Y, Kondo H, Noguchi T, Togari A. Glucocorticoids Mediate Circadian Timing in Peripheral Osteoclasts Resulting in the Circadian Expression Rhythm of Osteoclast-Related Genes. Bone (2014) 61:1–9. doi: 10.1016/j.bone.2013.12.026
67. Komoto S, Kondo H, Fukuta O, Togari A. Comparison of β-Adrenergic and Glucocorticoid Signaling on Clock Gene and Osteoblast-Related Gene Expressions in Human Osteoblast. Chronobiol Int (2012) 29:66–74. doi: 10.3109/07420528.2011.636496
68. Abe T, Sato T, Yoda T, Hoshi K. The Period Circadian Clock 2 Gene Responds to Glucocorticoids and Regulates Osteogenic Capacity. Regener Ther (2019) 11:199–206. doi: 10.1016/j.reth.2019.07.006
69. Schilperoort M, Kroon J, Kooijman S, Smit AE, Gentenaar M, Mletzko K, et al. Loss of Glucocorticoid Rhythm Induces an Osteoporotic Phenotype in Female Mice. Aging Cell (2021) 20:1–13. doi: 10.1111/acel.13474
70. Kobza AO, Herman D, Papaioannou A, Lau AN, Adachi JD. Understanding and Managing Corticosteroid-Induced Osteoporosis. Open Access Rheumatol Res Rev (2021) 13:177–90. doi: 10.2147/OARRR.S282606
71. Zura R, Mehta S, Della Rocca GJ, Steen RG. Biological Risk Factors for Nonunion of Bone Fracture. JBJS Rev (2016) 4:1–12. doi: 10.2106/JBJS.RVW.O.00008
72. Hachemi Y, Rapp AE, Picke AK, Weidinger G, Ignatius A, Tuckermann J. Molecular Mechanisms of Glucocorticoids on Skeleton and Bone Regeneration After Fracture. J Mol Endocrinol (2018) 61:R75–90. doi: 10.1530/JME-18-0024
73. Claes L, Recknagel S, Ignatius A. Fracture Healing Under Healthy and Inflammatory Conditions. Nat Rev Rheumatol (2012) 8:133–43. doi: 10.1038/nrrheum.2012.1
74. Einhorn TA, Gerstenfeld LC. Fracture Healing: Mechanisms and Interventions. Nat Rev Rheumatol (2015) 11:45–54. doi: 10.1038/nrrheum.2014.164
75. Wildemann B, Ignatius A, Leung F, Taitsman LA, Smith RM, Pesántez R, et al. Non-Union Bone Fractures. Nat Rev Dis Prim (2021) 7:57. doi: 10.1038/s41572-021-00289-8
76. Weber AJ, Li G, Kalak R, Street J, Buttgereit F, Dunstan CR, et al. Osteoblast-Targeted Disruption of Glucocorticoid Signalling Does Not Delay Intramembranous Bone Healing. Steroids (2010) 75:282–6. doi: 10.1016/j.steroids.2010.01.005
77. Tu J, Henneicke H, Zhang Y, Stoner S, Cheng TL, Schindeler A, et al. Disruption of Glucocorticoid Signaling in Chondrocytes Delays Metaphyseal Fracture Healing But Does Not Affect Normal Cartilage and Bone Development. Bone (2014) 69:12–22. doi: 10.1016/j.bone.2014.08.016
78. Rapp AE, Hachemi Y, Kemmler J, Koenen M, Tuckermann J, Ignatius A. Induced Global Deletion of Glucocorticoid Receptor Impairs Fracture Healing. FASEB J (2018) 32:2235–45. doi: 10.1096/fj.201700459RR
79. Hachemi Y, Rapp AE, Lee S, Dorn A, Krüger BT, Kaiser K, et al. Intact Glucocorticoid Receptor Dimerization Is Deleterious in Trauma-Induced Impaired Fracture Healing. Front Immunol (2021) 11:628287. doi: 10.3389/fimmu.2020.628287
80. Waters RV, Gamradt SC, Asnis P, Vickery BH, Avnur Z, Hill E, et al. Systemic Corticosteroids Inhibit Bone Healing in a Rabbit Ulnar Osteotomy Model. Acta Orthop Scand (2000) 71:316–21. doi: 10.1080/000164700317411951
81. Lyritis G, Papadopoulou Z, Nikiforidis P, Batrinos M, Varonos D. Effect of Cortisone and an Anabolic Steroid Upon Plasma Hydroxyproline During Fracture Healing in Rabbits. Acta Orthop Scand (1975) 46:25–30. doi: 10.3109/17453677508989188
82. Newman RJ, Francis MJ, Duthie RB. Nuclear Magnetic Resonance Studies of Experimentally Induced Delayed Fracture Union. Clin Orthop Relat Res (1987) (216):253–61. doi: 10.1097/00003086-198703000-00039
83. Liu Y-Z, Akhter MP, Gao X, Wang X-Y, Wang X-B, Zhao G, et al. Glucocorticoid-Induced Delayed Fracture Healing and Impaired Bone Biomechanical Properties in Mice. Clin Interv Aging (2018) 13:1465–74. doi: 10.2147/CIA.S167431
84. Sandberg OH, Aspenberg P. Glucocorticoids Inhibit Shaft Fracture Healing But Not Metaphyseal Bone Regeneration Under Stable Mechanical Conditions. Bone Joint Res (2015) 4:170–5. doi: 10.1302/2046-3758.410.2000414
85. Buttgereit F. Views on Glucocorticoid Therapy in Rheumatology: The Age of Convergence. Nat Rev Rheumatol (2020) 16:239–46. doi: 10.1038/s41584-020-0370-z
86. Baschant U, Frappart L, Rauchhaus U, Bruns L, Reichardt HM, Kamradt T, et al. Glucocorticoid Therapy of Antigen-Induced Arthritis Depends on the Dimerized Glucocorticoid Receptor in T Cells. Proc Natl Acad Sci USA (2011) 108:19317–22. doi: 10.1073/pnas.1105857108
87. Koenen M, Culemann S, Vettorazzi S, Caratti G, Frappart L, Baum W, et al. Glucocorticoid Receptor in Stromal Cells Is Essential for Glucocorticoid-Mediated Suppression of Inflammation in Arthritis. Ann Rheum Dis (2018) 77:1610–8. doi: 10.1136/annrheumdis-2017-212762
88. Fenton C, Martin C, Jones R, Croft A, Campos J, Naylor AJ, et al. Local Steroid Activation Is a Critical Mediator of the Anti-Inflammatory Actions of Therapeutic Glucocorticoids. Ann Rheum Dis (2021) 80:250–60. doi: 10.1136/annrheumdis-2020-218493
89. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch Signalling Drives Synovial Fibroblast Identity and Arthritis Pathology. Nature (2020) 582:259–64. doi: 10.1038/s41586-020-2222-z
90. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct Fibroblast Subsets Drive Inflammation and Damage in Arthritis. Nature (2019) 570:246–51. doi: 10.1038/s41586-019-1263-7
91. Culemann S, Grüneboom A, Nicolás-Ávila JÁ, Weidner D, Lämmle KF, Rothe T, et al. Locally Renewing Resident Synovial Macrophages Provide a Protective Barrier for the Joint. Nature (2019) 572:670–5. doi: 10.1038/s41586-019-1471-1
92. Agarwal A, Adachi JD. Therapies for Preventing Bone Loss With Glucocorticoid Treatment. Curr Osteoporos Rep (2021) 19:34–9. doi: 10.1007/s11914-020-00653-9
Keywords: glucocorticoid receptor, transgenic mice, osteoporosis, osteoblast, osteoclast
Citation: Lee S, Krüger BT, Ignatius A and Tuckermann J (2022) Distinct Glucocorticoid Receptor Actions in Bone Homeostasis and Bone Diseases. Front. Endocrinol. 12:815386. doi: 10.3389/fendo.2021.815386
Received: 15 November 2021; Accepted: 16 December 2021;
Published: 10 January 2022.
Edited by:
Ulrike Baschant, University Hospital Carl Gustav Carus, GermanyReviewed by:
Kaleen N. Hayes, Brown University, United StatesElizabeth Martha Winter, Leiden University Medical Center, Netherlands
Copyright © 2022 Lee, Krüger, Ignatius and Tuckermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Tuckermann, amFuLnR1Y2tlcm1hbm5AdW5pLXVsbS5kZQ==