 Steven G. Waguespack
Steven G. Waguespack- Department of Endocrine Neoplasia and Hormonal Disorders and the Children’s Cancer Hospital, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Multiple endocrine neoplasia type 1 (MEN1), an autosomal-dominantly inherited tumor syndrome, is classically defined by tumors arising from the “3 Ps”: Parathyroids, Pituitary, and the endocrine Pancreas. From its earliest descriptions, MEN1 has been associated with other endocrine and non-endocrine neoplastic manifestations. High quality evidence supports a direct association between pathogenic MEN1 variants and neoplasms of the skin (angiofibromas and collagenomas), adipose tissue (lipomas and hibernomas), and smooth muscle (leiomyomas). Although CNS tumors, melanoma, and, most recently, breast cancer have been reported as MEN1 clinical manifestations, the published evidence to date is not yet sufficient to establish causality. Well-designed, multicenter prospective studies will help us to understand better the relationship of these tumors to MEN1, in addition to verifying the true prevalence and penetrance of the well-documented neoplastic associations. Nevertheless, patients affected by MEN1 should be aware of these non-endocrine manifestations, and providers should be encouraged always to think beyond the “3 Ps” when treating an MEN1 patient.
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is a rare, autosomal-dominantly inherited tumor syndrome defined clinically by glandular hyperplasia and benign or malignant neoplasms involving two or more endocrine glands in a single individual (1, 2). It is caused by inactivating pathogenic DNA variants in the MEN1 gene, located on chromosome 11q13, which functions as a tumor suppressor and encodes menin, a ubiquitous nuclear protein that plays a role in transcriptional regulation, genome stability, cell division and proliferation (3, 4). MEN1 represents an archetypal example of the "two hit" mechanism of disease, first described by Knudson in hereditary retinoblastoma (5). A heterozygous pathogenic germline MEN1 variant is insufficient to induce tumor formation and thus a somatic chromosomal loss or loss-of-function mutation (the “second hit” affecting the wild type MEN1 allele, thus causing biallelic loss) is required to cause disease (6–8). The “second hit” causes a loss of heterozygosity (LOH) at the MEN1 locus in at-risk tissues and, in turn, decreased menin expression and attenuation of the ordinary constraints by menin on cell growth. LOH affecting the MEN1 gene in a tumor is highly suggestive of causality but not always conclusive, given that genomic loss of 11q13 can also occur in sporadic neuroendocrine tumors (9–11) and also due to the possibility that deletions of other genes contiguous to MEN1 on chromosome 11q13 may actually be responsible for a particular clinical phenotype.
Classically defined by tumors in the “3 Ps”: Parathyroids, Pituitary, and the endocrine Pancreas, other endocrine neoplastic manifestations include adrenocortical tumors, extra-pancreatic foregut neuroendocrine tumors, and very rarely pheochromocytoma. Even though MEN1 is considered an endocrine tumor syndrome, other neoplastic manifestations affecting the skin, adipose tissue, smooth muscle, central nervous system, and breast have been reported. The purpose of the current paper is to look beyond the 3 Ps and the other endocrine manifestations of MEN1 to provide a critical appraisal and state-of-the-art review of the often-overlooked, non-endocrine components of this fascinating hereditary syndrome.
Methodology
Through July 2022, the author identified articles published in English through the U.S. National Library of Medicine (PubMed®; https://www.ncbi.nlm.nih.gov/pubmed) employing a systematic search using the following terms: “multiple endocrine neoplasia type 1”, “death”, “survival”, “series”, “breast cancer/carcinoma”, “dermatologic”, “skin”, “cutaneous”, “angiofibroma”, “collagenoma”, “lipoma”, “hibernoma”, “melanoma”, “leiomyoma”, “fibroid”, “ependymoma”, and “meningioma”. A secondary review of reference lists and subsequent manuscripts citing previously published papers led to the identification of additional relevant articles.
Cutaneous tumors (angiofibromas and collagenomas)
MEN1 is one of several hereditary endocrine tumor syndromes associated with dermatologic manifestations (12). Long recognized but often unappreciated as a component of the clinical phenotype, the skin manifestations of MEN1 include angiofibromas and collagenomas (Table 1; Figures 1-3). Usually multiple and tending to be more commonly identified with increasing age in MEN1 patients (13–15), these dermatological manifestations can also be diagnosed in children (17–19) (Figure 1). In fact, they can be the earliest or only manifestation of MEN1 (13, 17, 20–23). Like other MEN1 manifestations, there is no clear genotype-phenotype correlation (24, 25). By age 40, the penetrance of angiofibromas and collagenomas in MEN1 patients has previously been estimated to be 85% and 70%, respectively (26). However, the true prevalence is difficult to ascertain due to the lack of large multi-institutional and prospective studies of MEN1 patients undergoing a thorough dermatologic examination. The first study that systematically examined unselected patients with MEN1 for skin manifestations was published in 1997 from the National Institutes of Health (NIH) (13). In 32 patients, angiofibromas and/or collagenomas were highly prevalent (29/32; 91%), including one patient in whom the clinical diagnosis of MEN1 was made before the biochemical confirmation of primary hyperparathyroidism (PHPT); no patients in a control group were identified to have multiple angiofibromas or collagenomas. A second study from the NIH was published in 2004 and included a systematic dermatologic examination in 110 consecutive patients with sporadic and MEN1-associated gastrinomas (15). In this study, 39/48 (81%) MEN1 patients had an angiofibroma or collagenoma, which were much more commonly seen in MEN1 patients compared with sporadic gastrinoma patients.
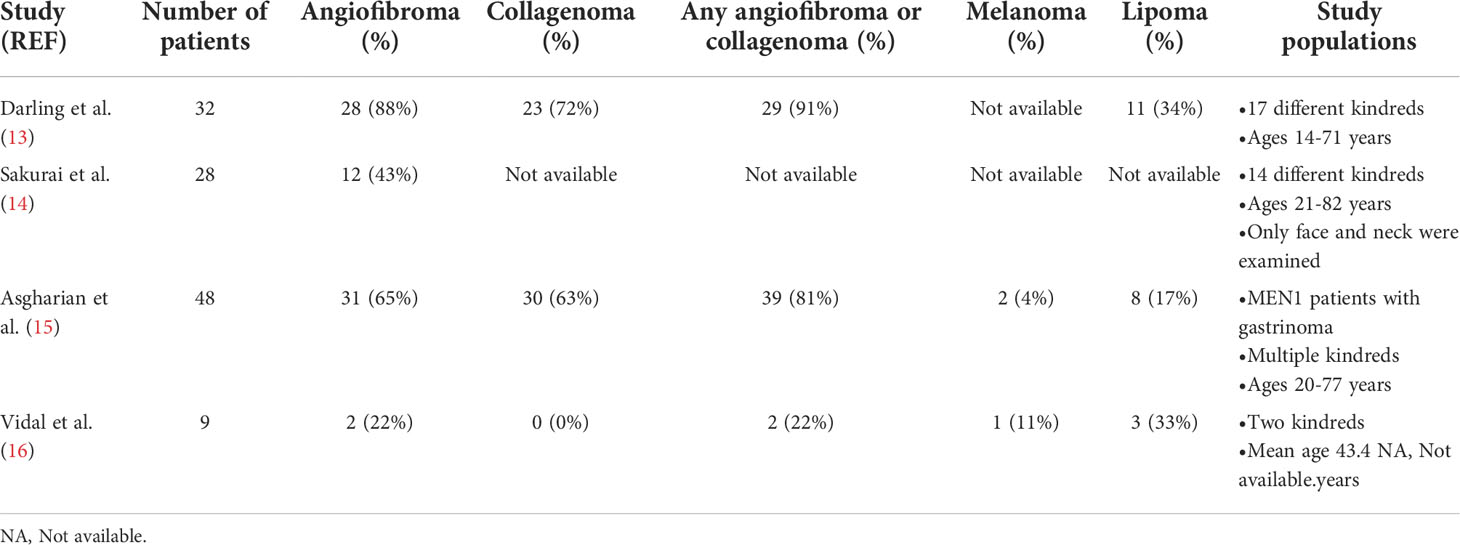
Table 1 Prevalence of cutaneous and adipose tissue neoplasms in patients with MEN1 having a comprehensive dermatologic evaluation.
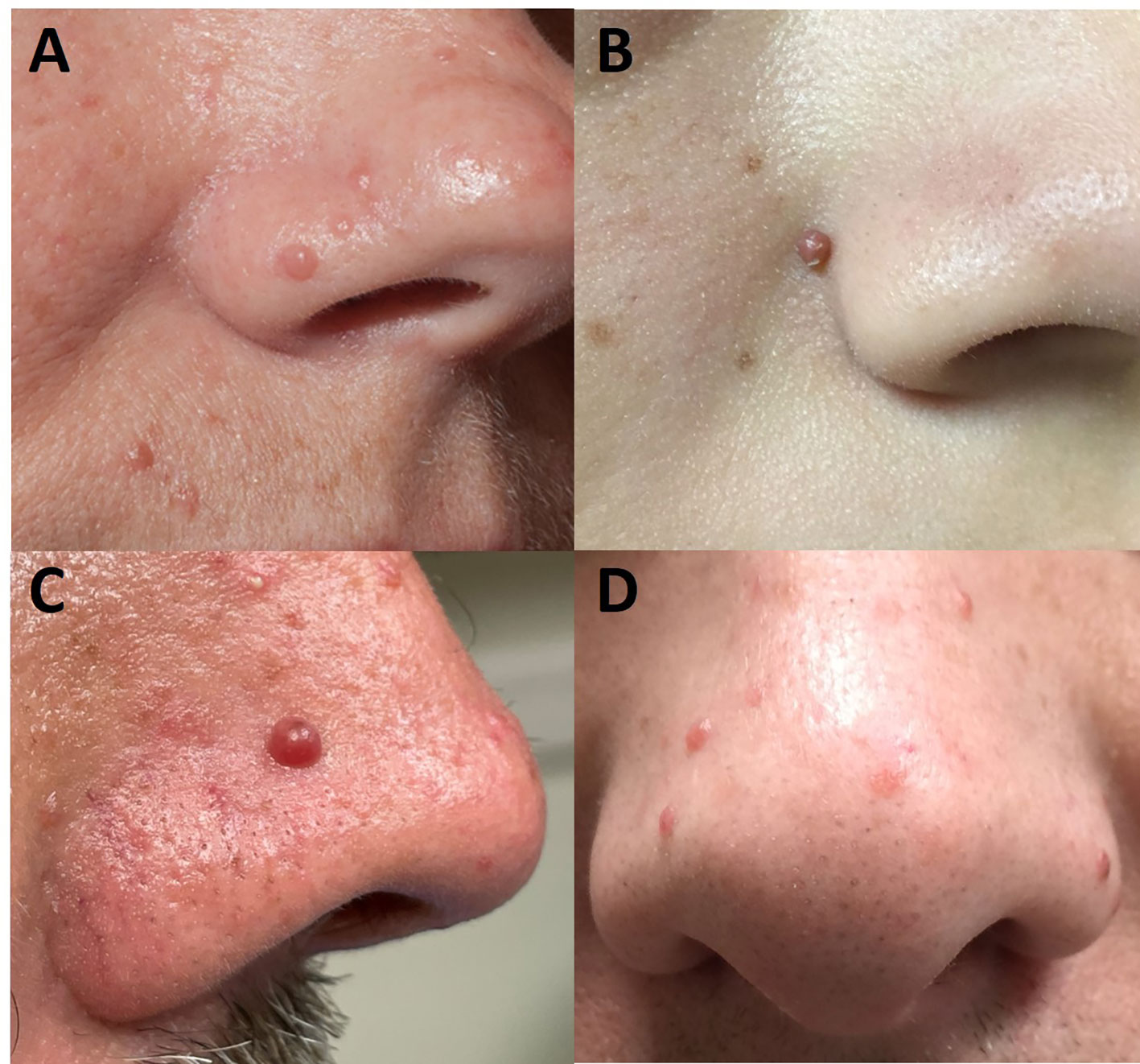
Figure 1 Facial angiofibromas as seen in a 39-year-old woman (A), 14-year-old girl (B), 48-year-old man (C), and 43-year-old man (D) with multiple endocrine neoplasia type 1. Angiofibromas are telangiectatic, skin colored to pink to red-brown, dome-shaped papules that are histologically characterized by fibrous tissue and vascular proliferation. They are distributed primarily on the central part of the face, especially the nose.
Similar to the defining MEN1 “3Ps”, these dermatologic neoplasms have also been shown to have LOH with allelic deletion of the MEN1 gene (27, 28), a finding not seen in a melanocytic nevus or acrochordon from MEN1 patients (27), in an angiofibroma from a patient with tuberous sclerosis (27), or in 19 sporadic angiofibromas (29), although two of the sporadic tumors were identified to have somatic missense MEN1 mutations.
The identification of these neoplasms in someone with an MEN1-associated endocrine tumor can help to detect a patient with MEN1 (15, 30). In the study of patients with gastrinoma (with and without MEN1), the presence of > three angiofibromas and any collagenoma had high sensitivity (75%) and specificity (95%) for identifying patients with MEN1 (15). Notably, MEN4, which occurs secondary to mutations in CDKN1B, has not been associated with cutaneous neoplasms (31) despite other phenotypic overlap with MEN1.
Angiofibroma
Angiofibromas (Figure 1) are benign fibroblastic tumors that histologically comprise bland spindle cells in a variably myxoid to collagenous stroma with a prominent vascular network. They present as telangiectatic, skin colored to pink to red-brown, dome-shaped papules with a glistening surface that primarily occur in the central part of the face, especially the nose. They may be mistaken for acne but do not spontaneously resolve. Facial angiofibromas may also resemble a wide variety of other skin disorders, including rosacea, multiple trichoepitheliomas, eruptive generalized keratoacanthomas (Grzybowski type), and Muir-Torre syndrome (32). MEN1-related angiofibromas are true neoplasms that arise from cells with a mesenchymal immunophenotype that are concentrated in a perivascular location (28).
The prevalence of angiofibromas in comprehensively screened MEN1 patients ranges from 22-88% depending on the population (13–16). At the NIH (13, 15), 65-88% of patients were affected, which may reflect the likely more severe MEN1 cases that were referred to their center, whereas the lower prevalence (43%) in Japan (14) may relate to ethnicity and the low rate (22%) in Spain (16) might be a reflection of only nine patients from two kindreds being studied. Notably, in all studies, facial angiofibromas were more prevalent in MEN1 patients compared with their control populations. In MEN1 patients with any angiofibroma, multiple lesions are identified in most patients: 33% have ≥ three (14), 77% have > three (15), and 57% have > four angiofibromas (13). Generally ranging in size from 1-4 mm, angiofibromas can less frequently number >10 and even up to 50 (13, 14).
Besides MEN1, the genodermatoses associated with facial angiofibromas include the tuberous sclerosis complex (TSC), in which facial angiofibromas (formerly called adenoma sebaceum) is a defining clinical feature, and the Birt-Hogg-Dubé Syndrome [See GeneReviews® (33)]. MEN1-related angiofibromas differ from those seen in TSC, in which the lesions are larger, more numerous, and earlier in onset; in TSC, the angiofibromas are also predominantly in a malar distribution and do not appear on the upper lip and its vermilion border as can be seen in MEN1 patients (13, 34). Angiofibromas are also not pathognomonic for MEN1 or TSC and can occur sporadically (35).
An angiofibroma diagnosis is usually made clinically in a patient with known MEN1 but a biopsy can facilitate the diagnosis in someone not known to be affected. Treatment may be desired for cosmetic reasons and therapeutic approaches would be similar to patients with TSC (32).
Collagenoma
Collagenomas (Figures 2, 3) are benign connective tissue nevi, which are hamartomas of the dermis, that have a dominant collagen component (36). Histologically, these lesions appear as unencapsulated areas of dense, thick collagen bundles arranged in a haphazard array within the reticular dermis below a region of normal-appearing papillary dermis (13, 37). They present as well-circumscribed, raised (and sometimes pedunculated), round to oval dome-shaped papules that are hypopigmented or skin-colored. They are usually subcentimeter in size (37) but can be larger (17) and are usually found on the neck, shoulders, and trunk (13); however, they have also been found on the face (38). In addition to MEN1, the genodermatoses associated with collagenomas include familial cutaneous collagenoma, Birt-Hogg-Dubé Syndrome, Buschke-Ollendorff syndrome, proteus syndrome, and TSC, in which the collagenoma is known as a shagreen patch (33, 36). The diagnosis of collagenoma is primarily a clinical one and, if treatment is required, surgery or intralesional glucocorticoids can be considered (39).
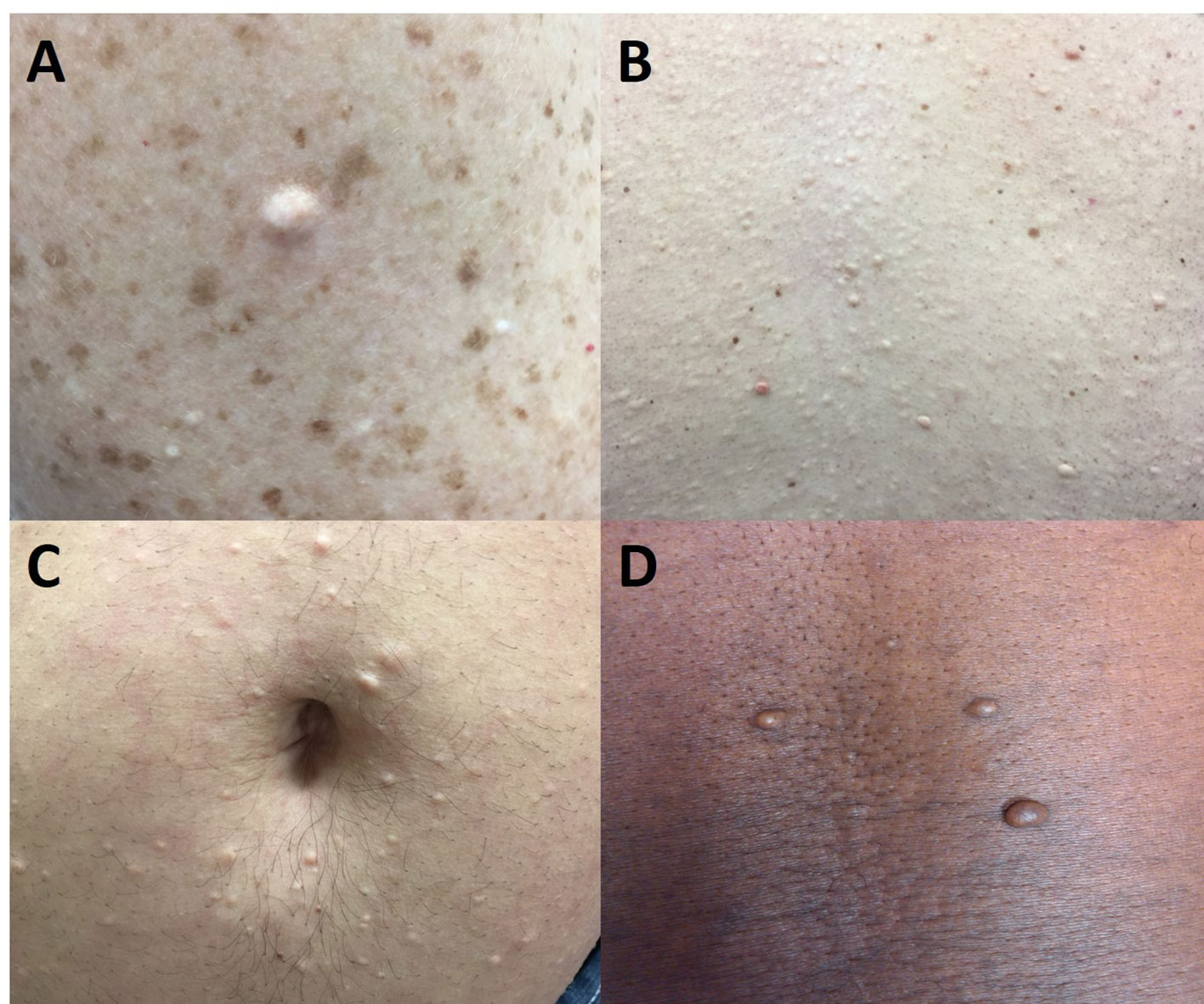
Figure 2 Multiple endocrine neoplasia type 1-associated collagenomas, which are benign connective tissue nevi with a dominant collagen component. They are hypopigmented or skin-colored and usually found on the neck, shoulder, and trunk. (A) 46-year-old woman with multiple collagenomas in addition to pigmented nevi on the arm. (B) 46-year-old man with innumerable collagenomas on the back. (C) 33-year-old man with multiple peri-umbilical collagenomas. (D) 46-year-old man with truncal collagenomas.
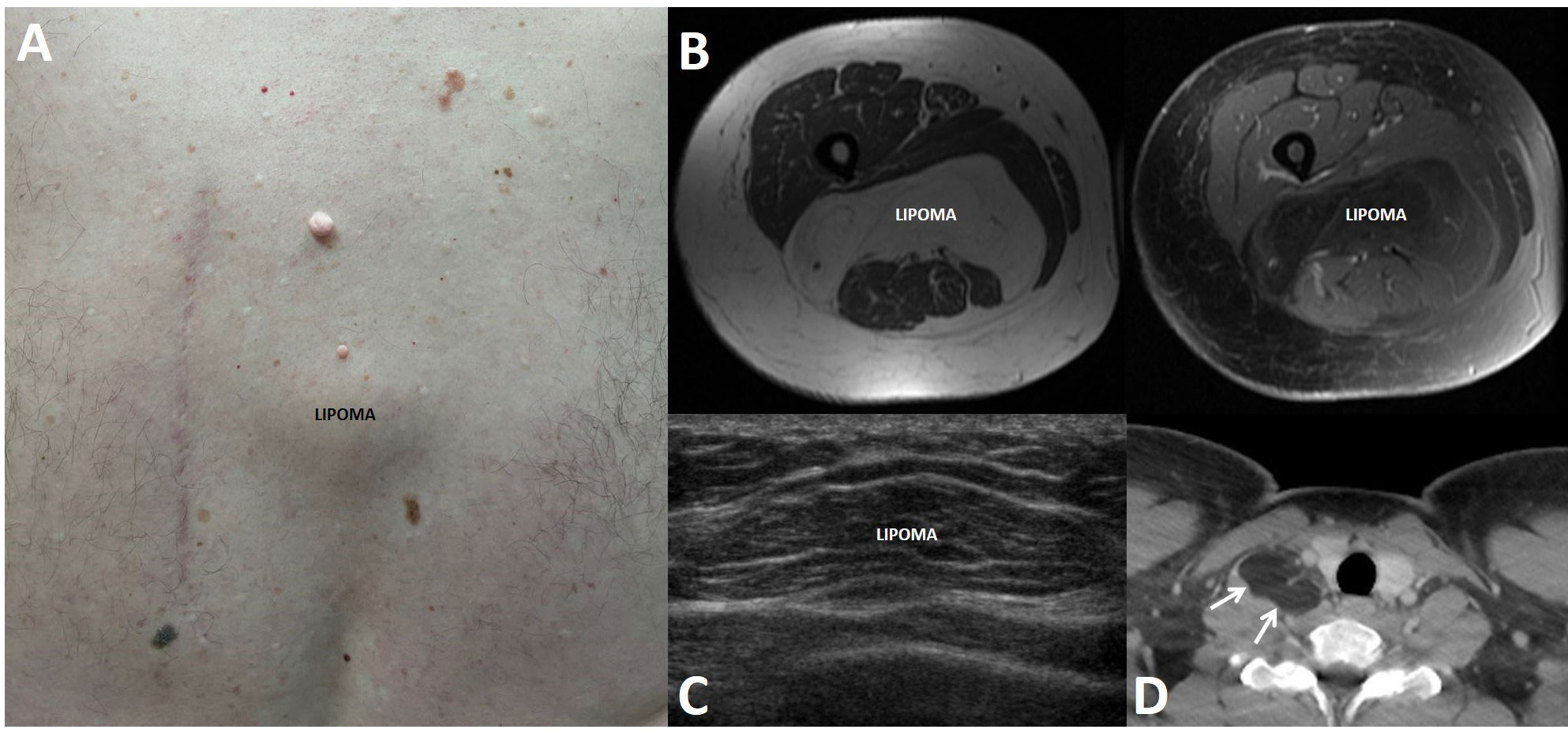
Figure 3 Lipomas in patients with multiple endocrine neoplasia type 1. Lipomas are benign tumors made of mature adipocytes that can arise from anywhere that fat is located. (A) 62-year-old man with a subcutaneous lipoma located over the spine in the mid back. Also seen is an adjacent surgical scar related to the prior resection of a large lipoma and scattered collagenomas. (B) 49-year-old woman with multifocal intramuscular lipomas, including a large posterior thigh lipoma as seen on axial T1-weighted pre- (left panel) and post-contrast (right) magnetic resonance imaging. (C) 16-year-old boy with a mobile soft tissue mass arising over the lower anterior chest. Ultrasound showed a 3.7 cm lesion consistent with lipoma. (D) 25-year-old man with a 3.6 cm lipoma (arrows) in the right scalene muscle, adjacent to the right thyroid lobe, as seen on contrast-enhanced computed tomography.
The prevalence of collagenomas in larger studies of comprehensively screened MEN1 patients ranges from 63-72% (13, 15) whereas one small study of nine patients from two Spanish kindreds did not identify any (16). In the NIH studies, these lesions were clearly more common in MEN1 patients compared with their control populations. In a more recent report from India comprised of 18 MEN1 patients from 14 unrelated families, the prevalence of collagenomas was 28% (40); however, these patients were not systematically examined by a dermatologist and the true prevalence may have been underestimated. Similar to MEN1-associated angiofibromas, collagenomas are usually multiple with 83% of MEN1 patients diagnosed with a collagenoma having at least three or four lesions (13, 15).
Melanoma
Several publications have reported the diagnosis of melanoma in MEN1 patients. The earliest reports were single cases included in larger MEN1 series (41–44). In 2000, Nord et al. described seven cases of melanoma in unrelated MEN1 patients, including two patients with an unknown primary site of melanoma who died of disseminated disease (45). Two of the four studies primarily focusing on the dermatologic manifestations of MEN1(Table 1) reported patients with melanoma: 2/48 (4%) in an NIH study (15) and 1/9 (11%) in the small Spanish study (16). Another study from the NIH in 2004 described the characteristics of 107 MEN1 patients with Zollinger-Ellison syndrome. Melanoma was found in three patients (3%), but in a concomitant literature review of 1009 cases of MEN1 and Zollinger-Ellison syndrome, there were no melanomas found (46). Subsequently, two additional case reports were published (47, 48). Melanoma has been reported as a cause of death in some studies (44, 45, 49) whereas other publications studying mortality in MEN1 are notable for no deaths from melanoma (50–52).
The MEN1 gene has not been definitively implicated in melanoma pathogenesis. Böni et al. analyzed 23 primary sporadic cutaneous melanomas and 17 metastases for MEN1 mutations and for LOH using polymorphic markers closely linked to the MEN1 gene (53). None of the tumors demonstrated LOH at the MEN1 locus or a pathogenic MEN1 variant. Nord et al. subsequently published a study including 39 sporadic melanomas, 13 melanoma cell lines, and melanomas from 20 unrelated familial melanoma kindreds that did not have germline mutations in the hereditary melanoma genes CDKN2A and CDK4 (45). Only one somatic MEN1 mutation was identified in a sporadic tumor whereas LOH including the MEN1 gene locus was found in 4/19 (21%) sporadic melanomas. No somatic mutations were identified in the cell lines or in the familial cases. Unfortunately, in this study, the authors were unable to test for LOH in the melanomas from MEN1 patients. In one later case report of melanoma in an MEN1 patient, LOH was not found using polymorphic DNA markers that map to the MEN1 gene locus (47). Therefore, there are insufficient data looking for somatic alterations in the MEN1 gene in melanomas removed from MEN1 patients. More recently, there has been some published evidence to suggest that MEN1 may suppress the malignant phenotype of melanoma cells (54) and act as a melanoma tumor suppressor (55). However, in whole exome sequencing of 331 cutaneous melanoma patients studied in The Cancer Genome Atlas program, MEN1 variants were not reported (56).
Cutaneous melanoma is a relatively common malignancy that is increasing in frequency and will be diagnosed in about 2% of people during their lifetimes in the United States (57). The major risk factors are being a non-Hispanic White and exposure to ultraviolet radiation. Given the very few reports in MEN1 (that don’t appear to exceed the prevalence of the general population) and the lack of supporting data that the MEN1 gene directly plays a role in melanoma pathogenesis, it appears that melanoma diagnosed in an MEN1 patient is more likely to be coincidence.
Summary of cutaneous tumors
There are high quality data to support that angiofibromas and collagenomas are causally related to alterations affecting the MEN1 gene, but cutaneous melanoma should not be considered part of the MEN1 clinical spectrum. Angiofibromas/collagenomas can rarely be an initial manifestation of MEN1 and identifying these lesions in a patient presenting with another MEN1-defining tumor or having a strong family history of MEN1 can help to secure the clinical diagnosis. Most likely to be multiple in MEN1 patients and with no clear genotype-phenotype correlations, angiofibromas and collagenomas are primarily identified in adults. In most cases, these lesions do not require intervention, although some patients may want to seek treatment for cosmetic reasons. The initial and ongoing evaluation of an MEN1 patient should include a comprehensive skin examination. Patients should be educated about any skin manifestations they may have and be encouraged to practice preventative skin care and to seek consultation with a dermatologist if there is any visually concerning or symptomatic lesion.
Lipomas and hibernomas
Lipomas, benign mesenchymal tumors made of mature adipocytes, can arise from anywhere that fat is located. They are either solitary or multiple and present as slow-growing, soft, mobile, and painless masses. In addition to occurring sporadically in the general population, lipomas can arise in various genetic disorders, including MEN1, encephalocraniocutaneous lipomatosis, KCNK9 Imprinting Syndrome, PTEN Hamartoma Tumor Syndrome, PIK3CA-Related Overgrowth Spectrum, Birt-Hogg-Dubé Syndrome, and Myoclonic Epilepsy Associated with Ragged Red Fibers [See GeneReviews® (33)].
In 1964, lipomas were first proposed by Ballard et al. (58) to be a component of MEN1, then called multiple endocrine adenomatosis, based on their finding of multicentric lipomas in 11 patients in addition to at least six previously published clinical MEN1 cases with lipomas dating back as far as 1927. Lipomas from MEN1 patients have been shown in multiple reports to have LOH at the MEN1 locus (8, 27, 59–62), consistent with a causal relationship with MEN1, and there are no clear genotype-phenotype correlations (24, 25). Lipomas in MEN1 patients (Figure 3) are not infrequently multiple and are typically subcutaneous, but they can also be found more deeply and reach large sizes that may require surgical extirpation (21, 63–66). Lipomas can also be the first clinical manifestation in MEN1 patients, diagnosed as young as 9 years (24, 67), but there is not a clear correlation with patient age or disease duration (15). A single case of liposarcoma has been reported (68).
The penetrance by age 40 of lipomas in MEN1 patients has previously been estimated to 30% (26). However, like angiofibromas and collagenomas, the exact prevalence is difficult to ascertain due to the lack of large studies of MEN1 patients undergoing a comprehensive evaluation. In the literature, lipomas are reported to be found in 0.9-34% of MEN1 patients (13, 15, 16, 24, 25, 42, 46, 69–72). In the studies incorporating a comprehensive dermatologic evaluation of MEN1 patients (13, 15, 16) (Table 1), the prevalence was 17-34%. Other series from referral centers or large MEN1 databases reported a lipoma prevalence of 12-30% (24, 67, 70–74), but not all studies have shown a significant lipoma prevalence (42, 46, 69).
Hibernomas are rare, benign adipocytic tumors that have brown fat differentiation and can demonstrate uptake of 18F-fluorodeoxyglucose on positron emission tomography, potentially leading to a concern for sarcoma (75–78). Histologically they can be confused with atypical lipomatous tumors, but they do not recur after complete resection, nor do they metastasize (79). To date there have been seven published cases of hibernomas occurring in MEN1 patients (60, 75–78, 80–82), and these have primarily been located in the pelvic region and thigh. The association with MEN1 may not purely be coincidental given that LOH was identified in two 11q13 markers in a resected tumor from an MEN1 patient (60) and several publications have documented deletions of the MEN1 gene in apparently sporadic hibernomas (83–85). However, these deletions are large and not limited to MEN1. Importantly, in the Nord et al. study (85), concomitant loss of the AIP gene, implicated in the syndrome of familial isolated pituitary adenomas, was also identified and a later study concluded that loss of AIP is responsible for the brown fat phenotype (86). Thus, it appears that a large deletion of 11q13 including both MEN1 and AIP is necessary for hibernoma development.
In summary, lipomas are clearly established as an MEN1 clinical manifestation, occurring in up to one third of patients. Hibernomas may also very rarely be diagnosed in MEN1 patients. Because sporadic lipomas are not uncommon in the general population, the finding of a lipoma in a patient suspected to have MEN1 is not sensitive enough to make the diagnosis (15). Lipomas can grow quite large and may need to be resected due to symptoms or for cosmetic reasons, but around 70% of MEN1 patients with lipomas can be managed conservatively (67).
Smooth muscle tumors
Smooth muscle tumors include benign leiomyomas (Figure 4) and their malignant counterpart, leiomyosarcoma. These mesenchymal neoplasms can arise throughout the body but most commonly affect the uterus where they are colloquially referred to as uterine fibroids. Several genetic syndromes have been associated with smooth muscle tumors, including FH Tumor Predisposition Syndrome (Hereditary Leiomyomatosis and Renal Cell Cancer [HLRCC]), Alport syndrome, PTEN Hamartoma Tumor Syndrome, MEN1, CDC73-Related Disorders/Hyperparathyroidism-jaw tumor syndrome, and the Birt-Hogg-Dubé Syndrome [See GeneReviews® (33)]. Leiomyomas appear to occur in 10% or less of MEN1 patients (25, 26, 46, 52, 87) and 12.6% of women with MEN1 (24), but there has been no systematic evaluation of the prevalence of these tumors in MEN1 patients and so the true prevalence remains unknown, especially noting that uterine leiomyomas are very common in the general population and thus may not necessarily be an MEN1-related neoplasm in female patients.
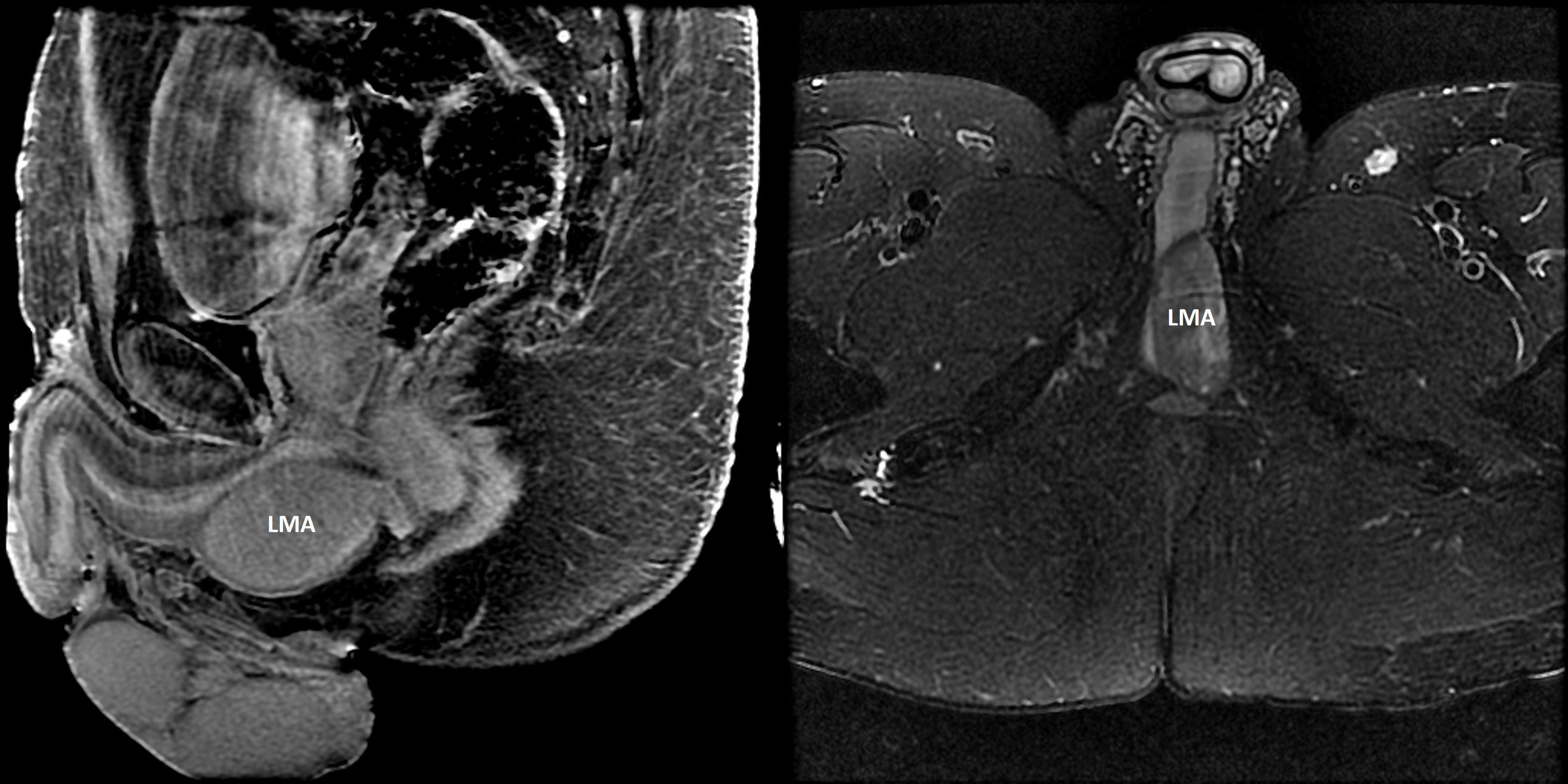
Figure 4 Leiomyoma (LMA). A 41-year-old man with MEN1 presented with an enlarging, painless perineal swelling. Magnetic resonance imaging (Left panel: contrast–enhanced sagittal T1-weighted fat suppressed image; right panel: axial T2-weighted fat suppressed image) identified a 5.8 x 2.6 x 3.8 cm mass in the perineum abutting the crus of the penis and the bulbous urethra. The mass was completely excised and histologic evaluation confirmed a leiomyoma. Somatic testing of the leiomyoma revealed the known germline pathogenic MEN1 variant and evidence for biallelic loss (loss of heterozygosity) at chromosome 11q (next-generation sequencing performed by Foundation Medicine; Cambridge, Massachusetts, USA).
In 1959 and 1962, necropsy reports from two men with clinical MEN1 was notable for the finding of genitourinary leiomyomata (scrotum and periprostatic) (88, 89). Subsequent early case reports of leiomyomata in patients with clinical MEN1 included a patient with an esophageal leiomyoma (58), a 39-year-old female patient with a uterine leiomyoma (90), a 43-year-old man with a pilar (cutaneous) leiomyoma derived from an arrector pili muscle (91), and a 45-year-old woman with a leiomyoma of the broad ligament (65). Ballard et al., in their 1964 publication, also reported other previously published cases of smooth muscle tumors in patients with multiple endocrine adenomatosis (58). The possible association of leiomyoma with MEN1 was first hypothesized in 1997 in two publications: a case report of a woman with clinical MEN1 and a history of uterine leiomyomatosis/leiomyosarcoma who was diagnosed with a lung lymphangioleiomyoma at age 60 years (92) and, as part of a larger study of 13 MEN1 patients, a 56-year-old female with MEN1 who had an esophageal leiomyoma (60). In the latter case, LOH at the putative MEN1 locus was not detected using 10 polymorphic markers spanning 11q13, but the MEN1 gene had not yet been cloned and so it was hypothesized that there could have been a small intragenic deletion or point mutation in the putative MEN1 gene that was not detectable by conventional LOH analysis. Shortly thereafter, a second case of two distinct esophageal leiomyomata was reported in a 36-year-old woman who was ultimately diagnosed with MEN1 and confirmed to have a germline pathogenic variant (93). In both leiomyomas, there was LOH at the MEN1 locus and the authors proposed that esophageal leiomyoma is associated with alterations in the MEN1 gene. Additional studies further solidified the role of MEN1 in the development of both esophageal and uterine leiomyomata (94), although LOH was not detected in four of seven leiomyomas from a single patient who had lung, esophageal and uterine tumors. Other cases of uni- and multifocal smooth muscle tumors in MEN1 patients have been reported in the English literature (24, 25, 52, 87, 95–104) and, in addition to the aforementioned sites, these tumors have arisen in the ureter (96), bladder (97), epididymis (87), ventricle (87), and small bowel (87, 103). Most cases have been in females and these neoplasms occur more rarely in men (87–89, 91, 96) (Figure 4).
In summary, patients with MEN1 are at risk for the development of benign and occasionally malignant (52, 92, 95) smooth muscle tumors. Females appear to be at increased risk compared with males and these tumors primarily arise in the upper gastrointestinal and genitourinary tracts. Given the primarily asymptomatic and benign nature of these tumors, routine prospective clinical screening is not warranted.
Central nervous system tumors
Meningioma
Meningiomas, typically slow-growing benign tumors attached to the dura mater, are believed to originate from arachnoid cells (105, 106). Exposure to ionizing radiation is a major risk factor, and patients with somatotroph pituitary adenomas also appear to have a higher risk (107). Meningiomas are located throughout the central nervous system (CNS) and symptoms relate to mass effect and are determined by tumor size and location (108). Although clinical behavior is mostly benign (WHO grade I), meningiomas can also be atypical (WHO grade II) and frankly malignant (WHO grade III). Thus, depending on their grade and location, these tumors can cause considerable patient morbidity and mortality.
The most common intracranial tumor, meningiomas comprise 39% of all tumors and 54.5% of non-malignant primary brain and other CNS tumors; median age of diagnosis is 66 years, and the incidence of meningioma increases with age (109–111). Meningiomas are more commonly diagnosed in females and Blacks compared with males and Whites, respectively (109, 110). Meningiomas are an incidental finding in 0.9% of MRI scans in patients over age 45 years with a prevalence of 0.5% in 45- to 59-year-olds to 1.6% in persons 75 years of age or older (110). Across all ages, meningiomas are incidentally identified in 0.6% of brain MRIs, but not before age 20 years; the peak incidence is 17/1000 scans (95% CI 4–37) in 80-year-olds (111).
Various genetic syndromes have been associated with the development of meningiomas, most commonly neurofibromatosis type 2, but also the PTEN hamartoma tumor syndrome, Werner syndrome, BAP1 tumor predisposition syndrome, and Rubinstein-Taybi syndrome, among others (33, 105, 106, 112). From an MEN1 perspective, initial reports of a meningioma in patients with clinical MEN1 were published in 1984 and 1996 (113, 114). In 2004, in a study of 74 eligible MEN1 patients prospectively evaluated at the National Institutes of Health (NIH), there were six cases (8.1%) of meningioma diagnosed at a mean age of 50.8 years (range 29-76) (115). All patients were asymptomatic and the meningiomas incidentally identified. One had received prior pituitary irradiation and 50% of the patients had a pituitary adenoma, but it is unknown if any of these were somatotroph adenomas. There was no meningioma growth in 60% of patients on serial imaging with a mean FU of 3.6 ± 1.8 years, but one patient ultimately died from a progressive meningioma (52). Meningioma was found late in the clinical course with a mean time of 17.6 years after the onset of MEN1. In one excised tumor, LOH was identified by all six polymorphic markers spanning the MEN1 locus. Since the original publication from the NIH suggesting causality, there has been an additional case report of the diagnosis of meningioma in a 35-year-old woman with MEN1 and PHPT, pancreatic neuroendocrine tumor (PNET), and a somatotroph pituitary adenoma (116). No other series of MEN1 patients and meningioma has yet been published to confirm the NIH findings, although patients with meningiomas have been reported in other publications (24, 25, 43, 87, 117), including one patient who died of a meningioma (117). In the Florentine database, the prevalence of meningiomas was 2.1% (24).
Thus far there has been no strong evidence supporting a role of MEN1 in the pathogenesis of meningiomas (118). Zhu and colleagues recently studied tumors from patients diagnosed with both pituitary adenoma and meningioma (PAM) (119, 120). In their first study of 57 PAM patients, tumors from PAM patients, compared with “sporadic” pituitary adenomas or meningiomas, had lower MEN1 expression and, in turn, hyperactivation of the mTOR signaling pathway (119). A follow up study reported that 5/23 patients with PAM harbored a germline missense MEN1 variant (c.1523G>A; p.G508D), but none of those patients had other MEN1 clinical manifestations and thus did not have MEN1 (120). This variant is also considered benign or likely benign by ClinVar (121).
In summary, meningiomas have been proposed as a component of MEN1 yet data are few to strongly support the role of loss of MEN1 function in meningioma pathogenesis. The high incidence of meningioma in the general population also makes it more likely that the finding of meningioma may be coincidental, with the understanding that a patient with MEN1 might have additional risk factors for meningioma development such as growth hormone excess and a history of ionizing radiation exposure. Screening for meningioma is not recommended in MEN1 patients, but if one is identified, it should be managed similarly to patients without MEN1.
Ependymoma
Ependymal tumors are a subtype of glioma that can occur throughout the CNS (supratentorial, infratentorial, and spinal), and they are defined by their anatomic locations and underlying molecular profiles (122–125). These tumors comprise 1.6% of all brain and other CNS tumors and are malignant in approximately 57% of all cases; proportionally they are more common (5.3%) in children < age 15 years, the group that also has the highest malignancy rate (89%) (109). Males and Whites have a higher incidence and the median age is 45 years (109); about 87% of pediatric ependymomas occur intracranially (mean age 5.0-7.8 years) whereas adult tumors more commonly (64%) occur in the spinal cord at a mean age of 45.5 years (126).
In 1991, in a study profiling the large Tasmanian MEN1 kindred, a case of spinal ependymoma was reported in a 46-year-old man, the brother of a woman with MEN1, who ultimately developed hypercalcemia and was thus diagnosed with MEN1 (65). Several years later, the first case proposing a link between MEN1 and ependymoma was published: a 51-year-old man who presented with gait disturbance and hypoesthesia was found to have a benign spinal ependymoma and was ultimately diagnosed with clinical MEN1 (PHPT, PNET, and pituitary microadenoma) (127). The authors hypothesized that there might be a common genetic etiology given the association of 11q13 translocations with ependymoma and the fact that the putative MEN1 gene had been mapped to the same chromosomal locus. Shortly thereafter, a third case of spinal ependymoma was reported in a 29-year-old female MEN1 patient who had genetic confirmation of a pathogenic MEN1 variant (128). Furthermore, LOH studies performed on the resected tumor showed LOH in the MEN1 region involving the loss of the wild type alleles. The authors concluded that ependymoma is a feature of MEN1, although uncommon compared with other MEN1-related tumors. The first case of an intracranial ependymoma in an MEN1 patient was in 2010 (129). In this case, a 44-year-old woman followed for de novo MEN1 presented with memory loss and disorientation and was found to have a large tumor causing obstructive hydrocephalus, a tumor that ultimately led to her death. A second, well-documented case of an intracranial ependymoma arising from the cervicomedullary junction was reported in 2017 in a 33-year-old man with genetically confirmed MEN1 (130). Further testing of the tumor, which had a DNA methylation profile clustering with that of spinal ependymomas, identified somatic loss of the remaining wildtype allele due to a chromosome 11 deletion. The authors concluded that ependymoma can arise as part of MEN1 and should be potentially screened for in patients with this syndrome. Other published cases include a 53-year-old man with clinical MEN1 (no MEN1 variant provided) and a tanycytic ependymoma arising from the filum terminale, but there was no evident abnormality at chromosome 11q13 (131). This report also referred to another case report previously published in Japanese of a 34-year-old man with a ventricular ependymoma and MEN1 (132).
The data that ependymoma may rarely arise as a component of MEN1 remain scarce and limited to seven case reports, two of which had documented LOH involving the MEN1 locus (128, 130), and generic reports of death from ependymoma in larger series of MEN1 patients (52, 133). The average age of the published cases is 41.4 years, not dissimilar from the average age of ependymoma diagnosis in the general population (109). Previous publications of familial ependymoma not associated with MEN1 (134) would suggest that there could be other not-yet-identified susceptibility genes. In addition, somatic alterations of chromosome 11 can be found in sporadic ependymomas (129, 135, 136), thus lessening the impact of finding LOH in any given tumor. Finally, given the current molecular understanding of ependymal tumors (122–125), it would be prudent to re-evaluate the published cases in this context before assigning causality to aberrations in the MEN1 gene. Given the above, routine screening for ependymoma is not recommended in MEN1 patients but if a patient presents with new neurological signs and symptoms, appropriate imaging should be ordered to look for this very rare possibility.
Breast carcinoma
Most recently, it has been proposed that the risk of breast carcinoma is heightened in females with MEN1. To date, there have been approximately 90 or fewer cases of breast carcinoma in women with MEN1 reported in the literature (24, 43, 44, 51, 72, 81, 87, 95, 99, 137–150); breast cancer has not yet been reported in a man with MEN1. The exact number of cases is difficult to determine due to the likely overlap of cases in the published literature and the lack of specific case numbers in one publication. Table 2 highlights 21 individual cases in women with MEN1 for which more-detailed clinical information has been published. In these cases, the median age of diagnosis is 45 years (33-69 years), and most cases are invasive ductal carcinomas, similar to the general population. Other histologies include ductal carcinoma in situ, invasive lobular carcinoma, invasive micropapillary carcinoma, and lobular carcinoma in situ, noting that the last histology is no longer considered to be a malignant lesion (151). The vast majority of breast cancers in MEN1 females are unilateral and unifocal, and there is no consistent pattern of hormone receptor or human epidermal growth factor receptor 2 (HER2) expression, although over 80% express the estrogen receptor. Furthermore, the tumors mostly appear to be smaller and without lymph node or distant metastases. The French and Belgian Groupe d’Etude des Tumeurs Endocrines reported five female patients with MEN1 who died of breast cancer (51). However, the number of breast cancer deaths in their MEN1 population was similar to the estimated breast cancer mortality in the general population and thus did not a priori suggest a heightened risk of aggressive breast cancer in MEN1 patients. Deaths due to breast carcinoma in patients with MEN1 have also been reported in other publications (44, 52, 87, 137, 141), but the numbers are small and, on the surface, don’t appear to exceed that which might be identified in the general population without MEN1. Furthermore, other papers looking at mortality in MEN1 are notable for the lack of breast cancer cases (49, 50, 52, 117, 152–154).
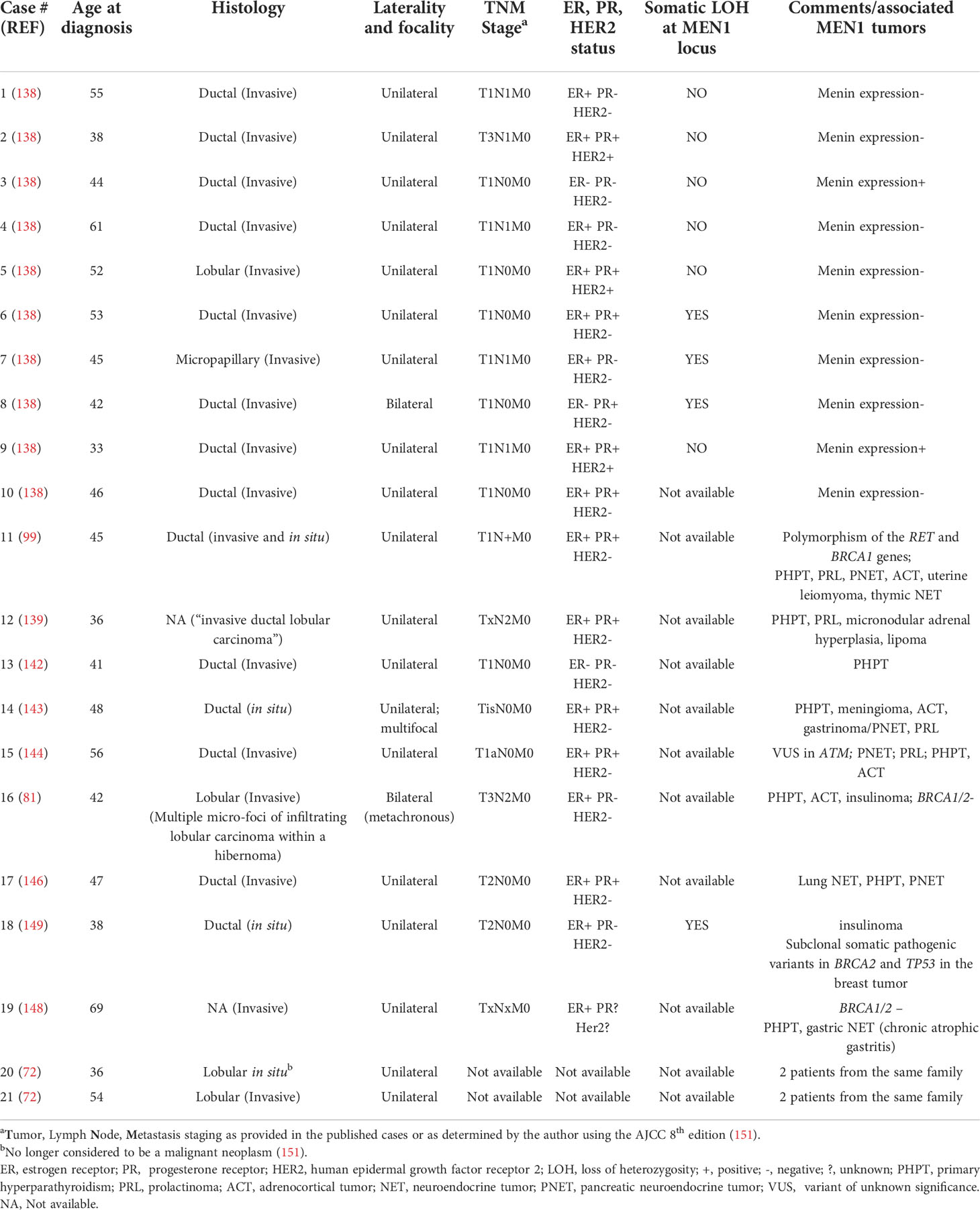
Table 2 Clinical details of 21 cases of breast cancer in women with genetically confirmed MEN1.
The first postulation that the MEN1 gene might play a role in breast cancer pathogenesis was made in 2004 by Honda et al. who reported a case of a 44-year-old woman with a parathyroid adenoma, aldosteronoma, and a scirrhous (ductal) breast carcinoma (155). The patient was found to have a common germline MEN1 single nucleotide polymorphism (thus she did not have MEN1), and the breast and parathyroid tumors showed LOH at the MEN1 locus.
In 2014, a study from the Netherlands reported the incidence of breast cancer from the Dutch longitudinal MEN1 database (138). In 190 female patients, 12 (6.3%) developed invasive, primarily ductal, breast carcinomas. The relative risk (RR) of breast cancer was 2.83 (p<0.001) with a standardized incidence ratio (SIR) of 2.14 (95% confidence interval [CI] 1.18-3.86). The mean age at diagnosis was 48.0 ± 8.8 years, compared with an age of 60-65 years in the general Dutch population. This observation was similar in three independent MEN1 cohorts from France (RR 2.33; p=0.03), the United States (RR 2.40; p=0.11), and Australia (Tasmania) (RR 2.31; p=0.22), although only one of these comparisons was significant, likely due to the small numbers of breast cancer patients in those MEN1 registries. Combining the three verification cohorts, the SIR was 1.96 (95% CI, 1.33 to 2.88). In total, amongst all the databases, there were 44 cases of breast cancer in 865 female MEN1 patients (5.1%) at an average age of diagnosis of 50 years. In evaluable tumors from the Dutch women with confirmed germline MEN1 mutations, LOH at the MEN1 locus was demonstrated in three of nine cancers and reduction of nuclear menin expression >50% was identified in eight of ten tumors.
Interestingly, three out of the 12 Dutch breast cancer patients had a history of hyperprolactinemia. There appears to be a positive association between elevated levels of prolactin and the development of invasive breast cancer (RR 1.42; CI 1.24-1.60) (156). Therefore, the risk of breast cancer could theoretically be heightened inwomen with MEN1 who concomitantly have hyperprolactinemia/prolactinoma.
In 2017, a follow-up cross-sectional case control study within the Dutch cohort showed that the increased risk of breast cancer was not associated with other known risk factors (age at menarche, age at first child birth, parity, oral contraception use, obesity, breast feeding, alcohol consumption, and smoking) or familial breast cancer occurrence (140). In this study, the median age at breast cancer diagnosis in 22 women (11 genetically confirmed; 11 obligate carriers) was 45 years (range 30 to 80 years). In contrast, relatives without MEN1 and breast cancer had a median age of diagnosis of 57.5 years (range 40 to 85 years; p=0.03), closer to the mean age at breast cancer diagnosis (61.2 years) in the Dutch population. No women in this study had been diagnosed with a prolactinoma. The authors also assessed exposure to radiation from computerized tomography (CT) scans done for MEN1 surveillance and the frequency of CT scans was similar for women with and without breast cancer. In mutation-negative, clinical MEN1 patients, the age of breast carcinoma diagnosis was not different from the general Dutch population.
From a basic science perspective, there are data to suggest a role for the MEN1 gene in hormone-dependent breast cancer (157–160). Menin is a coactivator for ERα-mediated transcription and the majority of disease-related MEN1 mutations prevent menin-ERα interaction (161, 162). In a mouse model, MEN1-disrupted mammary glands are significantly more likely to develop mammary intraepithelial neoplasia (MIN) that, in most cases, display complete menin inactivation (163). In this same study, reduced menin expression was also found in a large proportion of two independent cohorts of breast carcinoma patients. There are also data to suggest that menin expression might in fact promote tamoxifen resistance: in 65 ER-positive breast cancer samples from women treated with adjuvant tamoxifen for 2–5 years, menin-positive tumors were found to have a worse relapse-free survival compared with menin-negative ones (162). An older study of 24 breast cancers did not find mutations in MEN1 exon 2 (where most mutations had been described up to that date) (164). In The Cancer Genome Atlas study of 510 breast tumors published in 2012, mutations in MEN1 were not reported (165). In a later study looking at the molecular landscape in 560 breast cancers, somatic MEN1 mutations were extraordinarily rare (166). Most recently, in a large study sequencing 34 putative germline susceptibility genes in 60,466 women with breast cancer and 53,461 controls, protein-truncating (odds ratio 0.37 [95% CI 0.07–1.97; p=0.24]) and rare missense MEN1 variants (odds ratio 0.86 [95% CI 0.66–1.12; p=0.25]) were not associated with an increased breast cancer risk (167).
Intensified breast cancer screening for women with MEN1 was suggested by Dreijerink et al. in their initial publication (138) but the role of enhanced screening in this population was subsequently questioned (168). In 2017, the Dutch group proposed that women with MEN1 start biennial screening at age 40, which is 10 years before the Dutch screening program that starts at age 50 years (140). As stated by Dreijerink et al. in a letter to the editor (169), the decision to intensify breast cancer screening in MEN1 patients should not be taken lightly, and the dilemma is whether the benefits of detecting early stage cancers will outweigh the potential harms from the surveillance (i.e. earlier mammography and its attendant increased lifetime exposure to radiation) and the risk of false positives that can lead to unnecessary procedures, patient anxiety, and increased health care costs.
Although the data derived from the Dutch MEN1 cohort are compelling, causality has not yet been unequivocally demonstrated (168). First, LOH affecting chromosome 11q occurs in sporadic breast cancer (170, 171) and thus the finding of LOH in the published cases may be incidental. As suggested by Brennan (168), more extensive LOH studies are required to determine the extent of chromosome 11q loss in breast tumors from MEN1 patients compared with matched, sporadic breast cancer controls. Second, the rare possibility of patients coincidentally having pathogenic germline variants in other breast cancer susceptibility genes needs to be considered (172, 173). Third, in more recent population studies, germline MEN1 variants are not clearly associated with a higher risk of breast cancer (167), and other publications have not reported a breast cancer risk in MEN1 patients greater than that of the general population (51, 145). Fourth, for a tumor syndrome classically associated with multifocality, it is striking that there are not more multifocal or bilateral breast cancers in the published cases, not to mention the lack of male breast cancer, the risk of which should theoretically also be increased. Fifth, although the onset of breast cancer in Dutch MEN1 patients is approximately 15 years earlier than the general population (140), the overall point prevalence of breast cancer [5.1% in 865 women from four databases (138); 4.2% in the Florentine database (24)] may not be higher than the lifetime risk of breast cancer in any woman. In the United States, for example, a woman has a 12.9% probability (1 in 8) of developing invasive breast cancer from birth to death (57). Sixth, prior studies from Sweden identified a significant association between PHPT and subsequent incidence of breast cancer in women (174, 175) and another case series found an association of presumed familial isolated PHPT and breast cancer (176). There may also be an association between insulinoma diagnosis and breast cancer risk (150). Therefore, a link between breast cancer and MEN1 may not necessarily be directly related to a pathogenic MEN1 variant but rather to an MEN1 clinical manifestation such as prolactinoma, hyperparathyroidism, or insulinoma. Finally, the earlier age of diagnosis may be more related to a surveillance bias (via routine chest imaging for neuroendocrine tumor surveillance) rather than more aggressive biology. Further understanding of how these patients came to be diagnosed with breast cancer would be important.
In summary, it has recently been suggested that the risk of breast carcinoma is increased in women with MEN1, but further studies are required to prove causality with more certainty. If breast cancer is indeed part of the MEN1 tumor spectrum, it appears to be one with a relatively low clinical penetrance and without a clearly heightened risk of death from metastatic disease. Women with MEN1 should be counseled about this possible increased risk and be encouraged to be “breast aware” and perform routine breast self-examination. Whether or not formal screening mammography should commence earlier than the general population remains an area of uncertainty, and the age at which to begin screening should also consider a woman’s unique familial and personal risk factors for breast carcinoma.
Conclusion
MEN1 is a rare, autosomal-dominantly inherited tumor syndrome classically defined by tumors arising from the “3 Ps”: Parathyroids, Pituitary, and the endocrine Pancreas. From its earliest descriptions, MEN1 has been associated with other endocrine and non-endocrine neoplastic manifestations, and the data strongly support an association with neoplasms of the skin (angiofibromas and collagenomas), adipose tissue (lipomas and hibernomas), and smooth muscle (leiomyomas). Although CNS tumors, melanoma, and, most recently, breast cancer have been reported as MEN1 clinical manifestations, the published evidence to date is not yet of sufficient high quality to include these tumors in the MEN1 clinical spectrum. Well-designed, multicenter studies will help us to understand better the relationship of these tumors to MEN1, in addition to verifying the true prevalence and penetrance of the well-documented neoplastic associations. Nevertheless, patients affected by MEN1 should be aware of these non-endocrine manifestations and providers should be encouraged always to think beyond the “3 Ps” when treating an MEN1 patient.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Acknowledgments
The author sincerely thanks the patients and families with MEN1 who have taught him so much and who continue to do so at each encounter. The author is also indebted to Maria Luisa Brandi, Rajesh Thakker, and Gerlof Valk for their review of the manuscript.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97(9):2990–3011. doi: 10.1210/jc.2012-1230
2. Giusti F, Marini F, Brandi ML. Multiple endocrine neoplasia type 1. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al. editors. GeneReviews(R). (Seattle: University of Washington) (1993):1993-2022.
3. Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat (2008) 29(1):22–32. doi: 10.1002/humu.20605
4. Agarwal SK. The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer (2017) 24(10):T119–T34. doi: 10.1530/ERC-17-0199
5. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U.S.A (1971) 68(4):820–3. doi: 10.1073/pnas.68.4.820
6. Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature (1988) 332(6159):85–7. doi: 10.1038/332085a0
7. Thakker RV, Bouloux P, Wooding C, Chotai K, Broad PM, Spurr NK, et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. N Engl J Med (1989) 321(4):218–24. doi: 10.1056/NEJM198907273210403
8. Pannett AA, Thakker RV. Somatic mutations in MEN type 1 tumors, consistent with the Knudson "two-hit" hypothesis. J Clin Endocrinol Metab (2001) 86(9):4371–4. doi: 10.1210/jcem.86.9.7844
9. Tanaka C, Kimura T, Yang P, Moritani M, Yamaoka T, Yamada S, et al. Analysis of loss of heterozygosity on chromosome 11 and infrequent inactivation of the MEN1 gene in sporadic pituitary adenomas. J Clin Endocrinol Metab (1998) 83(8):2631–4. doi: 10.1210/jc.83.8.2631
10. Swarts DR, Ramaekers FC, Speel EJ. Molecular and cellular biology of neuroendocrine lung tumors: Evidence for separate biological entities. Biochim Biophys Acta (2012) 1826(2):255–71. doi: 10.1016/j.bbcan.2012.05.001
11. Oberg K. Genetics and molecular pathology of neuroendocrine gastrointestinal and pancreatic tumors (gastroenteropancreatic neuroendocrine tumors). Curr Opin endocrinol diabetes Obes (2009) 16(1):72–8. doi: 10.1097/MED.0b013e328320d845
12. Saggini A, Brandi ML. Skin lesions in hereditary endocrine tumor syndromes. Endocr Pract (2011) 17 Suppl 3:47–57. doi: 10.4158/EP11055.RA
13. Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol (1997) 133(7):853–7. doi: 10.1001/archderm.1997.03890430067009
14. Sakurai A, Matsumoto K, Ikeo Y, Nishio SI, Kakizawa T, Arakura F, et al. Frequency of facial angiofibromas in Japanese patients with multiple endocrine neoplasia type 1. Endocr J (2000) 47(5):569–73. doi: 10.1507/endocrj.47.569
15. Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: Prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab (2004) 89(11):5328–36. doi: 10.1210/jc.2004-0218
16. Vidal A, Iglesias MJ, Fernandez B, Fonseca E, Cordido F. Cutaneous lesions associated to multiple endocrine neoplasia syndrome type 1. J Eur Acad Dermatol Venereol. (2008) 22(7):835–8. doi: 10.1111/j.1468-3083.2008.02578.x
17. Witchel SF, Ranganathan S, Kilpatrick M, Carty SE. Reverse referral: From pathology to endocrinology. Endocr Pathol (2009) 20(1):78–83. doi: 10.1007/s12022-009-9059-1
18. Simonds WF, Varghese S, Marx SJ, Nieman LK. Cushing's syndrome in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf). (2012) 76(3):379–86. doi: 10.1111/j.1365-2265.2011.04220.x
19. Makri A, Bonella MB, Keil MF, Hernandez-Ramirez L, Paluch G, Tirosh A, et al. Children with MEN1 gene mutations may present first (and at a young age) with cushing disease. Clin Endocrinol (Oxf). (2018) 89(4):437–43. doi: 10.1111/cen.13796
20. Sakurai A, Hashizume K, Fukushima Y. Facial angiofibroma as an initial manifestation in multiple endocrine neoplasia type 1. Intern Med (2008) 47(11):1067–8. doi: 10.2169/internalmedicine.47.1127
21. Zeller S, Marx SJ, Lungu AO, Cowen EW, Turner ML. Multiple angiofibromas and collagenomas in a 45-year-old man with recurrent nephrolithiasis, fatigue, and vision loss. J Am Acad Dermatol (2009) 61(2):319–22. doi: 10.1016/j.jaad.2009.01.026
22. Simi S, Narayanan B, Nandakumar G. Not just skin deep: a case report of multiple endocrine neoplasia type 1. Indian J Dermatol (2012) 57(4):304–7. doi: 10.4103/0019-5154.97679
23. Roman JW, Logemann NF, Adams E. Incidental angiofibromas prompt a diagnosis of multiple endocrine neoplasia type-1 (MEN-1). Dermatol Online J (2014) 20(9). doi: 10.5070/D3209023911
24. Marini F, Giusti F, Brandi ML. Multiple endocrine neoplasia type 1: extensive analysis of a large database of Florentine patients. Orphanet J Rare Dis (2018) 13(1):1–18. doi: 10.1186/s13023-018-0938-8
25. Marini F, Giusti F, Fossi C, Cioppi F, Cianferotti L, Masi L, et al. Multiple endocrine neoplasia type 1: Analysis of germline MEN1 mutations in the Italian multicenter MEN1 patient database. Endocrine (2018) 62(1):215–33. doi: 10.1007/s12020-018-1566-8
26. Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, et al. Multiple endocrine neoplasia type 1 (MEN1): Not only inherited endocrine tumors. Genet Med (2009) 11(12):825–35. doi: 10.1097/GIM.0b013e3181be5c97
27. Pack S, Turner ML, Zhuang Z, Vortmeyer AO, Boni R, Skarulis M, et al. Cutaneous tumors in patients with multiple endocrine neoplasia type 1 show allelic deletion of the MEN1 gene. J Invest Dermatol (1998) 110(4):438–40. doi: 10.1046/j.1523-1747.1998.00140.x
28. Vortmeyer AO, Boni R, Pack SD, Darling TN, Zhuang Z. Perivascular cells harboring multiple endocrine neoplasia type 1 alterations are neoplastic cells in angiofibromas. Cancer Res (1999) 59(2):274–8.
29. Boni R, Vortmeyer AO, Pack S, Park WS, Burg G, Hofbauer G, et al. Somatic mutations of the MEN1 tumor suppressor gene detected in sporadic angiofibromas. J Invest Dermatol (1998) 111(3):539–40. doi: 10.1046/j.1523-1747.1998.00317.x
30. Shirali AS, Pieterman CRC, Lewis MA, Hyde SM, Makawita S, Dasari A, et al. It's not a mystery, it's in the history: Multidisciplinary management of multiple endocrine neoplasia type 1. CA: Cancer J Clin (2021) 71(5):369–80. doi: 10.3322/caac.21673
31. Alrezk R, Hannah-Shmouni F, Stratakis CA. MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer (2017) 24(10):T195–208. doi: 10.1530/ERC-17-0243
32. Schwartz RA, Fernandez G, Kotulska K, Jozwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol (2007) 57(2):189–202. doi: 10.1016/j.jaad.2007.05.004
33. Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, et al eds. GeneReviews®. Seattle: University of Washington (1993-2022). Available at: https://www.ncbi.nlm.nih.gov/books/NBK1116/.
34. Jozwiak S, Schwartz RA, Janniger CK, Michalowicz R, Chmielik J. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol (1998) 37(12):911–7. doi: 10.1046/j.1365-4362.1998.00495.x
35. Hunter AG, Nezarati MM, Velsher L. Absence of signs of systemic involvement in four patients with bilateral multiple facial angiofibromas Am J Med Genet Part A (2010) 152A(3):657–64. doi: 10.1002/ajmg.a.33320
36. Arora H, Falto-Aizpurua L, Cortes-Fernandez A, Choudhary S, Romanelli P. Connective tissue nevi: A review of the literature. Am J dermatopathol (2017) 39(5):325–41. doi: 10.1097/DAD.0000000000000638
37. Xia Y, Darling TN. Rapidly growing collagenomas in multiple endocrine neoplasia type I. J Am Acad Dermatol (2007) 56(5):877–80. doi: 10.1016/j.jaad.2006.11.005
38. Cottenden J, Green PJ, Stein JD, Walsh NM. Facial papules in a young woman with a history of pancreatic and parathyroid surgery. Am J dermatopathol (2017) 39(4):316–7. doi: 10.1097/DAD.0000000000000516
39. Saki N, Dorostkar A, Heiran A, Aslani FS. Satisfactory treatment of a large connective tissue nevus with intralesional steroid injection. Dermatol Pract conceptual. (2018) 8(1):12–4. doi: 10.5826/dpc.0801a03
40. Goroshi M, Bandgar T, Lila AR, Jadhav SS, Khare S, Shrikhande SV, et al. Multiple endocrine neoplasia type 1 syndrome: Single centre experience from western India. Fam Cancer (2016) 15(4):617–24. doi: 10.1007/s10689-016-9891-7
41. Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. Am J Med (1987) 82(4):731–7. doi: 10.1016/0002-9343(87)90008-8
42. Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM (1996) 89(9):653–69. doi: 10.1093/qjmed/89.9.653
43. Giraud S, Zhang CX, Serova-Sinilnikova O, Wautot V, Salandre J, Buisson N, et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet (1998) 63(2):455–67. doi: 10.1086/301953
44. Doherty GM, Olson JA, Frisella MM, Lairmore TC, Wells SA Jr., Norton JA. Lethality of multiple endocrine neoplasia type I. World J Surg (1998) 22(6):581–6. doi: 10.1007/s002689900438
45. Nord B, Platz A, Smoczynski K, Kytola S, Robertson G, Calender A, et al. Malignant melanoma in patients with multiple endocrine neoplasia type 1 and involvement of the MEN1 gene in sporadic melanoma. Int J Cancer. (2000) 87(4):463–7. doi: 10.1002/1097-0215(20000815)87:4<463::AID-IJC1>3.0.CO;2-8
46. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and zollinger-Ellison syndrome: A prospective study of 107 cases and comparison with 1009 cases from the literature. Med (Baltimore). (2004) 83(1):43–83. doi: 10.1097/01.md.0000112297.72510.32
47. Baldauf C, Vortmeyer AO, Koch CA, Sticherling M. Combination of multiple skin malignancies with multiple endocrine neoplasia type 1: Coincidental or pathogenetically related? Dermatology (2009) 219(4):365–7. doi: 10.1159/000193058
48. Brown GT, Cowen EW, Lee CC. Malignant melanoma masquerading as an angiofibroma in a patient with MEN-1. JAMA Dermatol (2015) 151(1):105–6. doi: 10.1001/jamadermatol.2014.2186
49. Geerdink EA, van der Luijt RB, Lips CJ. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol (2003) 149(6):577–82. doi: 10.1530/eje.0.1490577
50. Wilkinson S, Teh BT, Davey KR, McArdle JP, Young M, Shepherd JJ. Cause of death in multiple endocrine neoplasia type 1. Arch Surg (1993) 128(6):683–90. doi: 10.1001/archsurg.1993.01420180085016
51. Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des tumeurs endocrines) cohort study among 758 patients. World J Surg (2010) 34(2):249–55. doi: 10.1007/s00268-009-0290-1
52. Ito T, Igarashi H, Uehara H, Berna MJ, Jensen RT. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: A prospective study: Comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Med (Baltimore). (2013) 92(3):135–81. doi: 10.1097/MD.0b013e3182954af1
53. Boni R, Vortmeyer AO, Huang S, Burg G, Hofbauer G, Zhuang Z. Mutation analysis of the MEN1 tumour suppressor gene in malignant melanoma. Melanoma Res (1999) 9(3):249–52. doi: 10.1097/00008390-199906000-00006
54. Gao SB, Feng ZJ, Xu B, Chen Y, Zheng HH, Yin P, et al. Menin represses malignant phenotypes of melanoma through regulating multiple pathways. J Cell Mol Med (2011) 15(11):2353–63. doi: 10.1111/j.1582-4934.2010.01222.x
55. Fang M, Xia F, Mahalingam M, Virbasius CM, Wajapeyee N, Green MR. MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Mol Cell Biol (2013) 33(13):2635–47. doi: 10.1128/MCB.00167-13
56. Cancer Genome Atlas N. Genomic classification of cutaneous melanoma. Cell. (2015) 161(7):1681–96. doi: 10.1016/j.cell.2015.05.044
57. Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER cancer statistics review, 1975-2017. In: November 2019 SEER data submission. Bethesda, MD: National Cancer Institute (2020). Available at: https://seer.cancer.gov/csr/1975_2016/.
58. Ballard HS, Fame B, Hartsock RJ. Familial multiple endocrine adenoma-peptic ulcer complex. Med (Baltimore). (1964) 43:481–516. doi: 10.1097/00005792-196407000-00003
59. Morelli A, Falchetti A, Weinstein L, Fabiani S, Tomassetti P, Enzi G, et al. RFLP analysis of human chromosome 11 region q13 in multiple symmetric lipomatosis and multiple endocrine neoplasia type 1-associated lipomas. Biochem Biophys Res Commun (1995) 207(1):363–8. doi: 10.1006/bbrc.1995.1196
60. Dong Q, Debelenko LV, Chandrasekharappa SC, Emmert-Buck MR, Zhuang Z, Guru SC, et al. Loss of heterozygosity at 11q13: Analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (1997) 82(5):1416–20. doi: 10.1210/jcem.82.5.3944
61. Vortmeyer AO, Boni R, Pak E, Pack S, Zhuang Z. Multiple endocrine neoplasia 1 gene alterations in MEN1-associated and sporadic lipomas. J Natl Cancer Inst (1998) 90(5):398–9. doi: 10.1093/jnci/90.5.398
62. Rusconi D, Valtorta E, Rodeschini O, Giardino D, Lorenzo I, Predieri B, et al. Combined characterization of a pituitary adenoma and a subcutaneous lipoma in a MEN1 patient with a whole gene deletion. Cancer Genet (2011) 204(6):309–15. doi: 10.1016/j.cancergen.2011.03.006
63. Sturiale A, Giudici F, Alemanno G, Cavalli T, Addasi R, Santomaggio C, et al. Massive intrathoracic lipoma in men1 syndrome. Int J Surg Case Rep (2015) 6C:247–50. doi: 10.1016/j.ijscr.2014.10.071
64. Marshall AH, Sloper JC. Pluriglandular adenomatosis of the pituitary, parathyroid and pancreatic islet cells associated with lipomatosis. J Pathol bacteriol (1954) 68(1):225–9. doi: 10.1002/path.1700680127
65. Shepherd JJ. The natural history of multiple endocrine neoplasia type 1. highly uncommon or highly unrecognized? Arch Surg (1991) 126(8):935–52. doi: 10.1001/archsurg.1991.01410320017001
66. Fushimi Y, Kamei S, Tatsumi F, Sanada J, Shimoda M, Kimura T, et al. Multiple endocrine neoplasia type 1 with a frameshift mutation in its gene accompanied by a giant cervical lipoma and multiple fatty deposits in the pancreas: case report. BMC endocrine Disord (2021) 21(1):164. doi: 10.1186/s12902-021-00821-7
67. Giusti F, Cianferotti L, Boaretto F, Cetani F, Cioppi F, Colao A, et al. Multiple endocrine neoplasia syndrome type 1: Institution, management, and data analysis of a nationwide multicenter patient database. Endocrine. (2017) 58(2):349–59. doi: 10.1007/s12020-017-1234-4
68. Johnson GJ, Summerskill WH, Anderson VE, Keating FR Jr. Clinical and genetic investigation of a large kindred with multiple endocrine adenomatosis. N Engl J Med (1967) 277(26):1379–85. doi: 10.1056/NEJM196712282772601
69. Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet (1998) 62(2):232–44. doi: 10.1086/301729
70. Kouvaraki MA, Lee JE, Shapiro SE, Gagel RF, Sherman SI, Sellin RV, et al. Genotype-phenotype analysis in multiple endocrine neoplasia type 1. Arch Surg (2002) 137(6):641–7. doi: 10.1001/archsurg.137.6.641
71. Carty SE, Helm AK, Amico JA, Clarke MR, Foley TP, Watson CG, et al. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery (1998) 124(6):1106–13. doi: 10.1067/msy.1998.93107
72. Febrero B, Segura P, Ruiz-Manzanera JJ, Teruel E, Ros I, Rios A, et al. Uncommon tumors in multiple endocrine neoplasia (MEN) type 1: Do they have a relationship with the prognosis of these patients? J Endocrinol Invest (2021) 44(6):1327–30. doi: 10.1007/s40618-020-01414-2
73. Samaan NA, Ouais S, Ordonez NG, Choksi UA, Sellin RV, Hickey RC. Multiple endocrine syndrome type i. clinical, laboratory findings, and management in five families. Cancer (1989) 64(3):741–52. doi: 10.1002/1097-0142(19890801)64:3<741::aid-cncr2820640329>3.0.co;2-f
74. Snyder N 3rd, Scurry MT, Deiss WP. Five families with multiple endocrine adenomatosis. Ann Intern Med (1972) 76(1):53–8. doi: 10.7326/0003-4819-76-1-53
75. Ognong Boulemo A, Roch JA, Ricard F, Fontaine Hommell J, Cotton F. Hibernoma: Don't be caught out by a PET scan! Diagn interventional Imaging (2013) 94(6):649–51. doi: 10.1016/j.diii.2013.02.002
76. Karrouz W, Kamoun M, Odou M-F, Pigny P, Caiazzo R, Pattou F, et al. Hibernoma and type 1 multiple endocrine neoplasia (MEN1)? a metabolic link? Annales d'Endocrinol (2013) 74(2):157. doi: 10.1016/j.ando.2013.03.013
77. Hedayati V, Thway K, Thomas JM, Moskovic E. MEN1 syndrome and hibernoma: An uncommonly recognised association? Case Rep Med (2014) 2014:804580. doi: 10.1155/2014/804580
79. Al Hmada Y, Schaefer IM, Fletcher CDM. Hibernoma mimicking atypical lipomatous tumor: 64 cases of a morphologically distinct subset. Am J Surg Pathol (2018) 42(7):951–7. doi: 10.1097/PAS.0000000000001061
80. Marchand L, Decaussin-Petrucci M, Giraud S, Cotton F, Thivolet C, Simon C. Hibernoma and multiple endocrine neoplasia type 1 syndrome: A non-fortuitous association? A case report and literature review. Ann Endocrinol (Paris). (2017) 78(3):194–7. doi: 10.1016/j.ando.2017.03.001
81. Deguidi G, Mirandola S, Nottegar A, Tsvetkova V, Bianchi B, Pellini F. Axillary hibernoma in woman with lobular breast cancer and MEN1 syndrome: A case report. Int J Surg Case Rep (2020) 77:834–8. doi: 10.1016/j.ijscr.2020.11.073
82. Al-Salameh A, Cadiot G, Calender A, Goudet P, Chanson P. Clinical aspects of multiple endocrine neoplasia type 1. Nat Rev Endocrinol (2021) 17(4):207–24. doi: 10.1038/s41574-021-00468-3
83. Gisselsson D, Hoglund M, Mertens F, Dal Cin P, Mandahl N. Hibernomas are characterized by homozygous deletions in the multiple endocrine neoplasia type I region. Metaphase fluorescence in situ hybridization reveals complex rearrangements not detected by conventional cytogenetics. Am J Pathol (1999) 155(1):61–6. doi: 10.1016/S0002-9440(10)65099-7
84. Maire G, Forus A, Foa C, Bjerkehagen B, Mainguene C, Kresse SH, et al. 11q13 alterations in two cases of hibernoma: Large heterozygous deletions and rearrangement breakpoints near GARP in 11q13.5. Genes Chromosomes Cancer (2003) 37(4):389–95. doi: 10.1002/gcc.10223
85. Nord KH, Magnusson L, Isaksson M, Nilsson J, Lilljebjorn H, Domanski HA, et al. Concomitant deletions of tumor suppressor genes MEN1 and AIP are essential for the pathogenesis of the brown fat tumor hibernoma. Proc Natl Acad Sci U S A (2010) 107(49):21122–7. doi: 10.1073/pnas.1013512107
86. Magnusson L, Hansen N, Saba KH, Nilsson J, Fioretos T, Rissler P, et al. Loss of the tumour suppressor gene AIP mediates the browning of human brown fat tumours. J pathol (2017) 243(2):160–4. doi: 10.1002/path.4945
87. Vierimaa O, Ebeling TM, Kytola S, Bloigu R, Eloranta E, Salmi J, et al. Multiple endocrine neoplasia type 1 in northern finland; clinical features and genotype phenotype correlation. Eur J Endocrinol (2007) 157(3):285–94. doi: 10.1530/EJE-07-0195
88. Cohen BD, Lubash GD, Rubin AL. Parathyroid and pancreatic adenomas. Am J Med (1959) 26(5):801–7. doi: 10.1016/0002-9343(59)90238-4
89. Williams ED, Celestin LR. The association of bronchial carcinoid and pluriglandular adenomatosis. Thorax (1962) 17(2):120–7. doi: 10.1136/thx.17.2.120
90. Berg B, Biorklund A, Grimelius L, Ingemansson S, Larsson LI, Stenram U, et al. A new pattern of multiple endocrine adenomatosis: Chemodectoma, bronchial carcinoid, GH-producing pituitary adenoma, and hyperplasia of the parathyroid glands, and antral and duodenal gastrin cells. Acta Med Scand (1976) 200(4):321–26. doi: 10.1111/j.0954-6820.1976.tb08239.x
91. Burton JL, Hartog M. Multiple endocrine adenomatosis (Type 1) with cutaneous leiomyomata and cysts of moll. Br J Dermatol (1977) 97 Suppl 15:74–5. doi: 10.1111/j.1365-2133.1977.tb14343.x
92. Carnevale V, Romagnoli E, Remotti D, D'Erasmo E, Spagna G, Pisani D, et al. Pulmonary lymphangioleiomyoma in a patient with multiple endocrine neoplasia type I. J Endocrinol Invest. (1997) 20(5):282–5. doi: 10.1007/BF03350301
93. Vortmeyer AO, Lubensky IA, Skarulis M, Li G, Moon YW, Park WS, et al. Multiple endocrine neoplasia type 1: Atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol (1999) 12(9):919–24.
94. McKeeby JL, Li X, Zhuang Z, Vortmeyer AO, Huang S, Pirner M, et al. Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol (2001) 159(3):1121–7. doi: 10.1016/S0002-9440(10)61788-9
95. Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: Clinical and genetic topics. Ann Intern Med (1998) 129(6):484–94. doi: 10.7326/0003-4819-129-6-199809150-00011
96. Ikota H, Tanimoto A, Komatsu H, Ozawa Y, Matsushita H. Ureteral leiomyoma causing hydronephrosis in type 1 multiple endocrine neoplasia. Pathol Int (2004) 54(6):457–9. doi: 10.1111/j.1440-1827.2004.01642.x
97. Choi H, Kim S, Moon JH, Lee YH, Rhee Y, Kang ES, et al. Multiple endocrine neoplasia type 1 with multiple leiomyomas linked to a novel mutation in the MEN1 gene. Yonsei Med J (2008) 49(4):655–61. doi: 10.3349/ymj.2008.49.4.655
98. Takeda A, Sakurai A, Imoto S, Nakamura H. Parasitic myomas after laparoscopic-assisted myomectomy in multiple endocrine neoplasia type 1. J obstetrics gynaecol Res (2013) 39(5):1098–102. doi: 10.1111/jog.12009
99. Jeong YJ, Oh HK, Bong JG. Multiple endocrine neoplasia type 1 associated with breast cancer: A case report and review of the literature. Oncol letters. (2014) 8(1):230–4. doi: 10.3892/ol.2014.2144
100. Hall MJ, Innocent J, Rybak C, Veloski C, Scott WJ, Wu H, et al. Bilateral granulosa cell tumors: A novel malignant manifestation of multiple endocrine neoplasia 1 syndrome found in a patient with a rare menin in-frame deletion. Appl Clin Genet (2015) 8:69–73. doi: 10.2147/TACG.S72223
101. El-Maouche D, Welch J, Agarwal SK, Weinstein LS, Simonds WF, Marx SJ. A patient with MEN1 typical features and MEN2-like features. Int J endocrine Oncol (2016) 3(2):89–95. doi: 10.2217/ije-2015-0008
102. Misgar RA, Sahu D, Purra S, Wani AI, Bashir MI. Multiple uterine leiomyomas in multiple endocrine neoplasia type 1 with a novel MEN1 gene mutation. J Hum Reprod Sci (2020) 13(1):75–7. doi: 10.4103/jhrs.JHRS_42_19
103. Alpert L, Al-Sabti R, Graham RP, Pai RK, Gonzalez RS, Zhang X, et al. Smooth muscle tumors of the gastrointestinal tract: An analysis of prognostic features in 407 cases. Mod Pathol (2020) 33(7):1410–9. doi: 10.1038/s41379-020-0492-5
104. Chen F, Xu Q, Yue W, Yu X, Shao S. A MEN1 patient presenting with multiple parathyroid adenomas and transient hypercortisolism: A case report and literature review. Front Endocrinol (Lausanne). (2022) 13:802453. doi: 10.3389/fendo.2022.802453
105. Huntoon K, Toland AMS, Dahiya S. Meningioma: A review of clinicopathological and molecular aspects. Front Oncol (2020) 10:579599. doi: 10.3389/fonc.2020.579599
106. Ogasawara C, Philbrick BD, Adamson DC. Meningioma: A review of epidemiology, pathology, diagnosis, treatment, and future directions. Biomedicines (2021) 9(3):1–23. doi: 10.3390/biomedicines9030319
107. Engelhardt J, Nunes ML, Pouchieu C, Ferriere A, San-Galli F, Gimbert E, et al. Increased incidence of intracranial meningiomas in patients with acromegaly. Neurosurgery (2020) 87(4):639–46. doi: 10.1093/neuros/nyz438
108. Glenn CA, Tullos HJ, Sughrue ME. Natural history of intracranial meningiomas. In: Handbook of clinical neurology (Netherlands, United Kingdom, and United States:Elsevier) 169. (2020). p. 205–27. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32553291.
109. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro-oncology. (2021) 23 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noab200
110. Vernooij MW, Ikram MA, Tanghe HL, Vincent AJ, Hofman A, Krestin GP, et al. Incidental findings on brain MRI in the general population. N Engl J Med (2007) 357(18):1821–8. doi: 10.1056/NEJMoa070972
111. Sunny DE, Amoo M, Al Breiki M, Teng EDW, Henry J, Javadpour M. Prevalence of incidental intracranial findings on magnetic resonance imaging: A systematic review and meta-analysis. Acta neurochirurgica (2022) 164(10):2751–65. doi: 10.1007/s00701-022-05225-7
112. Kerr K, Qualmann K, Esquenazi Y, Hagan J, Kim DH. Familial syndromes involving meningiomas provide mechanistic insight into sporadic disease. Neurosurgery (2018) 83(6):1107–18. doi: 10.1093/neuros/nyy121
113. Banik S, Hasleton PS, Lyon RL. An unusual variant of multiple endocrine neoplasia syndrome: A case report. Histopathology (1984) 8(1):135–44. doi: 10.1111/j.1365-2559.1984.tb02328.x
114. Chigot JP, Bendib S, Turpin G, Benlian P. [Characteristic pathological associations in multiple endocrine neoplasia type 1]. Presse Med (1996) 25(27):1229–33.
115. Asgharian B, Chen YJ, Patronas NJ, Peghini PL, Reynolds JC, Vortmeyer A, et al. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res (2004) 10(3):869–80. doi: 10.1158/1078-0432.CCR-0938-3
116. Herrero-Ruiz A, Villanueva-Alvarado HS, Corrales-Hernandez JJ, Higueruela-Minguez C, Feito-Perez J, Recio-Cordova JM. Coexistence of GH-producing pituitary macroadenoma and meningioma in a patient with multiple endocrine neoplasia type 1 with hyperglycemia and ketosis as first clinical sign. Case Rep Endocrinol (2017) 2017:2390797. doi: 10.1155/2017/2390797
117. Horiuchi K, Okamoto T, Iihara M, Tsukada T. Analysis of genotype-phenotype correlations and survival outcomes in patients with primary hyperparathyroidism caused by multiple endocrine neoplasia type 1: the experience at a single institution. Surg Today (2013) 43(8):894–9. doi: 10.1007/s00595-012-0354-y
118. Birzu C, Peyre M, Sahm F. Molecular alterations in meningioma: prognostic and therapeutic perspectives. Curr Opin Oncol (2020) 32(6):613–22. doi: 10.1097/CCO.0000000000000687
119. Zhu H, Miao Y, Shen Y, Guo J, Xie W, Zhao S, et al. The clinical characteristics and molecular mechanism of pituitary adenoma associated with meningioma. J Trans Med (2019) 17(1):354. doi: 10.1186/s12967-019-2103-0
120. Zhu H, Miao Y, Shen Y, Guo J, Xie W, Zhao S, et al. Germline mutations in MEN1 are associated with the tumorigenesis of pituitary adenoma associated with meningioma. Oncol letters (2020) 20(1):561–8. doi: 10.3892/ol.2020.11601
121. National Center for Biotechnology Information. ClinVar; [VCV000136166.15]. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000136166.15 (Accessed June 11, 2022).
122. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell (2015) 27(5):728–43. doi: 10.1016/j.ccell.2015.04.002
123. Larrew T, Saway BF, Lowe SR, Olar A. Molecular classification and therapeutic targets in ependymoma. Cancers. (2021) 13(24):1–29. doi: 10.3390/cancers13246218
124. Kresbach C, Neyazi S, Schuller U. Updates in the classification of ependymal neoplasms: The 2021 WHO classification and beyond. Brain Pathol (Zurich Switzerland). (2022) 32(4):e13068. doi: 10.1111/bpa.13068
125. Ruda R, Bruno F, Pellerino A, Soffietti R. Ependymoma: Evaluation and management updates. Curr Oncol Rep (2022) 24(8):985–93. doi: 10.1007/s11912-022-01260-w
126. McGuire CS, Sainani KL, Fisher PG. Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study. J Neurosurg (2009) 110(4):725–9. doi: 10.3171/2008.9.JNS08117
127. Kato H, Uchimura I, Morohoshi M, Fujisawa K, Kobayashi Y, Numano F, et al. Multiple endocrine neoplasia type 1 associated with spinal ependymoma. Intern Med (1996) 35(4):285–9. doi: 10.2169/internalmedicine.35.285
128. Giraud S, Choplin H, Teh BT, Lespinasse J, Jouvet A, Labat-Moleur F, et al. A large multiple endocrine neoplasia type 1 family with clinical expression suggestive of anticipation. J Clin Endocrinol Metab (1997) 82(10):3487–92. doi: 10.1210/jc.82.10.3487
129. Al-Salameh A, Francois P, Giraud S, Calender A, Bergemer-Fouquet AM, de Calan L, et al. Intracranial ependymoma associated with multiple endocrine neoplasia type 1. J Endocrinol Invest (2010) 33(5):353–6. doi: 10.1007/BF03346599
130. Cuevas-Ocampo AK, Bollen AW, Goode B, Pajtler KW, Chavez L, Sharma T, et al. Genetic confirmation that ependymoma can arise as part of multiple endocrine neoplasia type 1 (MEN1) syndrome. Acta neuropathol (2017) 133(4):661–3. doi: 10.1007/s00401-017-1689-7
131. Funayama T, Sakane M, Yoshizawa T, Takeuchi Y, Ochiai N. Tanycytic ependymoma of the filum terminale associated with multiple endocrine neoplasia type 1: first reported case. Spine J (2013) 13(8):e49–54. doi: 10.1016/j.spinee.2013.02.066
132. Tobimatsu T, Kaji H, Naito J, Iu MF, Nomura R, Sowa H, et al. (Case report No.143: one case of multiple endosecretory-adenomatosis accompanied with ventricula ependymoma). Clin calcium (2004) 14(10):150–3.
133. Thevenon J, Bourredjem A, Faivre L, Cardot-Bauters C, Calender A, Murat A, et al. Higher risk of death among MEN1 patients with mutations in the JunD interacting domain: A groupe d'etude des tumeurs endocrines (GTE) cohort study. Hum Mol Genet (2013) 22(10):1940–8. doi: 10.1093/hmg/ddt039
134. Dimopoulos VG, Fountas KN, Robinson JS. Familial intracranial ependymomas. Rep three cases Family Rev literature. Neurosurg focus. (2006) 20(1):E8. doi: 10.3171/foc.2006.20.1.9
135. Lamszus K, Lachenmayer L, Heinemann U, Kluwe L, Finckh U, Hoppner W, et al. Molecular genetic alterations on chromosomes 11 and 22 in ependymomas. Int J Cancer (2001) 91(6):803–8. doi: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1134>3.0.CO;2-P
136. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta neuropathol (2019) 138(6):1075–89. doi: 10.1007/s00401-019-02056-2
137. Dean PG, van Heerden JA, Farley DR, Thompson GB, Grant CS, Harmsen WS, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg (2000) 24(11):1437–41. doi: 10.1007/s002680010237
138. Dreijerink KM, Goudet P, Burgess JR, Valk GD. International breast cancer in MENSG. breast-cancer predisposition in multiple endocrine neoplasia type 1. N Engl J Med (2014) 371(6):583–4. doi: 10.1056/NEJMc1406028
139. Elezovic V, Petakov M, Macut D, Ognjanovic S, Isailovic T, Milicevic I, et al. Breast cancer in multiple endocrine neoplasia type 1. Endocrine Abstracts (2014) 35:P299. doi: 10.1530/endoabs.35.P299
140. van Leeuwaarde RS, Dreijerink KM, Ausems MG, Beijers HJ, Dekkers OM, de Herder WW, et al. MEN1-dependent breast cancer: Indication for early screening? results from the Dutch MEN1 study group. J Clin Endocrinol Metab (2017) 102(6):2083–90. doi: 10.1210/jc.2016-3690
141. Donegan D, Singh Ospina N, Rodriguez-Gutierrez R, Al-Hilli Z, Thompson GB, Clarke BL, et al. Long-term outcomes in patients with multiple endocrine neoplasia type 1 and pancreaticoduodenal neuroendocrine tumours. Clin Endocrinol (Oxf). (2017) 86(2):199–206. doi: 10.1111/cen.13264
142. Herranz-Antolin S, Gil-Garcia S, Alvarez-de Frutos V. Multiple endocrine neoplasia type 1 and breast cancer. an association to consider. Endocrinol Diabetes Nutr (Engl Ed). (2018) 65(8):468–9. doi: 10.1016/j.endinu.2018.05.008
143. Kim SH, Park JH. Adrenal incidentaloma, breast cancer and unrecognized multiple endocrine neoplasia type 1. Acta endocrinol (Bucharest Romania 2005). (2019) 15(4):513–7. doi: 10.4183/aeb.2019.513
144. Larouche V, Akirov A, Thain E, Kim RH, Ezzat S. Co-Occurrence of breast cancer and neuroendocrine tumours: New genetic insights beyond multiple endocrine neoplasia syndromes. Endocrinol Diabetes Metab (2019) 2(4):e00092. doi: 10.1002/edm2.92
145. Manoharan J, Bollmann C, Kann PH, Di Fazio P, Bartsch DK, Albers MB. Gender differences in multiple endocrine neoplasia type 1: Implications for screening? Visceral Med (2020) 36(1):3–9. doi: 10.1159/000505498
146. Maria BC, Quadros AC, Alves N, Coutinho J. Incidental finding of MEN-1 syndrome during staging and follow-up of breast carcinoma. BMJ Case Rep (2020) 13(12):1–6. doi: 10.1136/bcr-2020-238784
147. Oleinikov K, Uri I, Jacob H, Epshtein J, Benson A, Ben-Haim S, et al. Long-term outcomes in MEN-1 patients with pancreatic neuroendocrine neoplasms: An Israeli specialist center experience. Endocrine. (2020) 68(1):222–9. doi: 10.1007/s12020-020-02217-4
148. Chaves C, Nunes da Silva T, Dias Pereira B, Anselmo J, Claro I, Cavaco BM, et al. A case report of multiple endocrine neoplasia type 1 and autoimmune disease: Coincidence or correlation? Med (Baltimore) (2021) 100(49):e28145. doi: 10.1097/MD.0000000000028145
149. Cheah SK, Bisambar CR, Pitfield D, Giger O, Hoopen RT, Martin JE, et al. Breast cancer in multiple endocrine neoplasia type 1 (MEN1). Endocrinol Diabetes Metab Case Rep (2021) 2021:1–7. doi: 10.1530/EDM-20-0196
150. Peltola E, Hannula P, Huhtala H, Metso S, Sand J, Laukkarinen J, et al. Long-term morbidity and mortality in patients diagnosed with an insulinoma. Eur J Endocrinol (2021) 185(4):577–86. doi: 10.1530/EJE-21-0230
151. AJCC cancer staging manual (2017). Springer International Publishing/American College of Surgeons (Accessed et al).
152. Cadiot G, Vuagnat A, Doukhan I, Murat A, Bonnaud G, Delemer B, et al. Prognostic factors in patients with zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. groupe d'Etude des neoplasies endocriniennes multiples (GENEM and groupe de recherche et d'Etude du syndrome de zollinger-Ellison (GRESZE). Gastroenterology. (1999) 116(2):286–93. doi: 10.1016/S0016-5085(99)70124-1
153. de Laat JM, van der Luijt RB, Pieterman CR, Oostveen MP, Hermus AR, Dekkers OM, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med (2016) 14(1):182. doi: 10.1186/s12916-016-0708-1
154. Vinault S, Mariet AS, Le Bras M, Mirallie E, Cardot-Bauters C, Pattou F, et al. Metastatic potential and survival of duodenal and pancreatic tumors in multiple endocrine neoplasia type 1: A GTE and AFCE cohort study (Groupe d'etude des tumeurs endocrines and association francophone de chirurgie endocrinienne). Ann Surg (2020) 272(6):1094–101. doi: 10.1097/SLA.0000000000003162
155. Honda M, Tsukada T, Horiuchi T, Tanaka R, Yamaguchi K, Obara T, et al. Primary hyperparathyroidism associatiated with aldosterone-producing adrenocortical adenoma and breast cancer: Relation to MEN1 gene. Intern Med (2004) 43(4):310–4. doi: 10.2169/internalmedicine.43.310
156. Aranha AF, Dos Anjos LG, Turri JAO, Simoes RS, Maciel GAR, Baracat EC, et al, et al. Impact of the prolactin levels in breast cancer: a systematic review and meta-analysis. Gynecol Endocrinol. (2022) 38(5):385–90. doi: 10.1080/09513590.2022.2047173
157. Dreijerink KMA, Groner AC, Vos ESM, Font-Tello A, Gu L, Chi D, et al. Enhancer-mediated oncogenic function of the menin tumor suppressor in breast cancer. Cell Rep (2017) 18(10):2359–72. doi: 10.1016/j.celrep.2017.02.025
158. Abou Ziki R, Luo Y, Vlaeminck-Guillem V, Le Romancer M, Zhang CX. Involvement of the MEN1 gene in hormone-related cancers: Clues from molecular studies, mouse models, and patient investigations. Endocrines (2020) 1(2):58–81. doi: 10.3390/endocrines1020007
159. Teinturier R, Abou Ziki R, Kassem L, Luo Y, Malbeteau L, Gherardi S, et al. Reduced menin expression leads to decreased ERalpha expression and is correlated with the occurrence of human luminal b-like and ER-negative breast cancer subtypes. Breast Cancer Res Treat (2021) 190(3):389–401. doi: 10.1007/s10549-021-06339-9
160. Abou Ziki R, Teinturier R, Luo Y, Cerutti C, Vanacker JM, Poulard C, et al. MEN1 silencing triggers the dysregulation of mTORC1 and MYC pathways in ER+ breast cancer cells. Endocr Relat Cancer (2022) 29(8):451–65. doi: 10.1530/ERC-21-0337
161. Dreijerink KM, Mulder KW, Winkler GS, Hoppener JW, Lips CJ, Timmers HT. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res (2006) 66(9):4929–35. doi: 10.1158/0008-5472.CAN-05-4461
162. Imachi H, Murao K, Dobashi H, Bhuyan MM, Cao X, Kontani K, et al. Menin, a product of the MENI gene, binds to estrogen receptor to enhance its activity in breast cancer cells: possibility of a novel predictive factor for tamoxifen resistance. Breast Cancer Res Treat (2010) 122(2):395–407. doi: 10.1007/s10549-009-0581-0
163. Seigne C, Auret M, Treilleux I, Bonnavion R, Assade F, Carreira C, et al. High incidence of mammary intraepithelial neoplasia development in Men1-disrupted murine mammary glands. J pathol (2013) 229(4):546–58. doi: 10.1002/path.4146
164. Costa SC, Nascimento LS, Ferreira FJ, Mattos PS, Camara-Lopes LH, Ward LS. Lack of mutations of exon 2 of the MEN1 gene in endocrine and nonendocrine sporadic tumors. Braz J Med Biol Res = Rev Bras pesquisas medicas e biologicas. (2001) 34(7):861–5. doi: 10.1590/S0100-879X2001000700004
165. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
166. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature (2016) 534(7605):47–54. doi: 10.1038/nature17676
167. Breast Cancer Association C, Dorling L, Carvalho S, Allen J, Gonzalez-Neira A, Luccarini C, et al. Breast cancer risk genes - association analysis in more than 113,000 women. N Engl J Med (2021) 384(5):428–39. doi: 10.1056/NEJMoa1913948
168. Brennan P. Breast cancer risk in MEN1 - a cancer genetics perspective. Clin Endocrinol (Oxf). (2015) 82(3):327–229. doi: 10.1111/cen.12614
169. Dreijerink KM, Valk GD. Reply to: Breast cancer risk in MEN1–a cancer genetics perspective. Clin Endocrinol (Oxf). (2015) 83(1):141. doi: 10.1111/cen.12679
170. Zhuang Z, Merino MJ, Chuaqui R, Liotta LA, Emmert-Buck MR. Identical allelic loss on chromosome 11q13 in microdissected in situ and invasive human breast cancer. Cancer Res (1995) 55(3):467–71.
171. Loo LW, Ton C, Wang YW, Grove DI, Bouzek H, Vartanian N, et al. Differential patterns of allelic loss in estrogen receptor-positive infiltrating lobular and ductal breast cancer. Genes Chromosomes Cancer (2008) 47(12):1049–66. doi: 10.1002/gcc.20610
172. Ghataorhe P, Kurian AW, Pickart A, Trapane P, Norton JA, Kingham K, et al. A carrier of both MEN1 and BRCA2 mutations: case report and review of the literature. Cancer Genet Cytogenet (2007) 179(2):89–92. doi: 10.1016/j.cancergencyto.2007.08.009
173. Papi L, Palli D, Masi L, Putignano AL, Congregati C, Zanna I, et al. Germline mutations in MEN1 and BRCA1 genes in a woman with familial multiple endocrine neoplasia type 1 and inherited breast-ovarian cancer syndromes: a case report. Cancer Genet Cytogenet (2009) 195(1):75–9. doi: 10.1016/j.cancergencyto.2009.06.019
174. Michels KB, Xue F, Brandt L, Ekbom A. Hyperparathyroidism and subsequent incidence of breast cancer. Int J Cancer (2004) 110(3):449–51. doi: 10.1002/ijc.20155
175. Nilsson IL, Zedenius J, Yin L, Ekbom A. The association between primary hyperparathyroidism and malignancy: Nationwide cohort analysis on cancer incidence after parathyroidectomy. Endocr Relat Cancer (2007) 14(1):135–40. doi: 10.1677/erc.1.01261
Keywords: MEN1, angiofibroma, collagenoma, meningioma, ependymoma, lipoma, leiomyoma, breast cancer
Citation: Waguespack SG (2022) Beyond the “3 Ps”: A critical appraisal of the non-endocrine manifestations of multiple endocrine neoplasia type 1. Front. Endocrinol. 13:1029041. doi: 10.3389/fendo.2022.1029041
Received: 26 August 2022; Accepted: 26 September 2022;
Published: 17 October 2022.
Edited by:
Natalia Simona Pellegata, Helmholtz Association of German Research Centres (HZ), GermanyReviewed by:
Thomas Cuny, Aix-Marseille Université, FranceDelmar Muniz Lourenco Jr., University of São Paulo, Brazil
Copyright © 2022 Waguespack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven G. Waguespack, c3dhZ3Vlc0BtZGFuZGVyc29uLm9yZw==