 Manuel Gado
Manuel Gado Ulrike Baschant
Ulrike Baschant Lorenz C. Hofbauer1,2,3
Lorenz C. Hofbauer1,2,3 Holger Henneicke
Holger Henneicke- 1Center for Regenerative Therapies TU Dresden, Technische Universität Dresden, Dresden, Germany
- 2Department of Medicine III, University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 3Center for Healthy Aging, University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
Despite the continued development of specialized immunosuppressive therapies in the form of monoclonal antibodies, glucocorticoids remain a mainstay in the treatment of rheumatological and auto-inflammatory disorders. Therapeutic glucocorticoids are unmatched in the breadth of their immunosuppressive properties and deliver their anti-inflammatory effects at unparalleled speed. However, long-term exposure to therapeutic doses of glucocorticoids decreases bone mass and increases the risk of fractures – particularly in the spine – thus limiting their clinical use. Due to the abundant expression of glucocorticoid receptors across all skeletal cell populations and their respective progenitors, therapeutic glucocorticoids affect skeletal quality through a plethora of cellular targets and molecular mechanisms. However, recent evidence from rodent studies, supported by clinical data, highlights the considerable role of cells of the osteoblast lineage in the pathogenesis of glucocorticoid-induced osteoporosis: it is now appreciated that cells of the osteoblast lineage are key targets of therapeutic glucocorticoids and have an outsized role in mediating their undesirable skeletal effects. As part of this article, we review the molecular mechanisms underpinning the detrimental effects of supraphysiological levels of glucocorticoids on cells of the osteoblast lineage including osteocytes and highlight the clinical implications of recent discoveries in the field.
Introduction
Harvey Cushing first described the development of ‘osteoporosis of the skeleton’ in the spine of patients suffering from endogenous hypercortisolism 90 years ago (1). Two decades later, clinicians observed the same phenomenon in patients receiving synthetic glucocorticoids (GCs) (2). GC-induced osteoporosis (GIO) is considered the third most common condition of pathological bone loss following post-menopause and aging, and is the most frequent cause of secondary osteoporosis. For instance, in the Global Longitudinal Study of Osteoporosis in Women (GLOW), about 2.7-4.6% of women from 10 different countries received treatment with GCs (3). Although a considerable proportion of GC-induced fractures remain asymptomatic and thus difficult to detect, exposure to exogenous GCs has been linked to a high incidence of fractures, particularly in the spine. A rapid reduction in bone mineral density (BMD) is generally observed as early as 3-6 months after initiation of GC treatment and persists during continued GC exposure (4–9). Aside from the spine, typically locations of GC-induced fractures include the ribs and pelvis (8, 10–12), indicating that sites rich in trabecular bone are more affected than the cortical structures (10). Interestingly, some studies observed a rapid development of fractures in patients receiving GCs, even before any detectable decreases in the bone mineral density (9, 13, 14), suggesting that not just bone mass but also bone quality is compromised in the presence of supra-physiological levels of GCs (Box 1).
Several molecular mechanisms underlying GIO have been identified through in vivo and in vitro studies. Overall, the effects of excess GCs in the skeleton are complex owing to the multifaceted nature of interactions between local and systemic factors. Generally, GCs act via the glucocorticoid receptor (GR), which is ubiquitously expressed in all skeletal cell types. The molecular nature of GC-GR interactions and their interplay with target cells are manifold and complex. Briefly, upon ligand binding the GR translocates to the nucleus where it either acts as a dimer by binding directly to the DNA in the promotor region of target genes or it may act as a monomer by interfering with other transcription factors such as activator protein 1 (AP-1) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). A detailed review of the molecular action of the GC-GR complex is provided by Hartmann et al. (18) or Vandewalle et al. (19).
The skeletal effects of therapeutic GC use have to be separated from the role of physiological GCs in the skeleton. Physiological concentrations of GCs are critically required for differentiation of stromal progenitors towards the osteoblast lineage – and away from adipocytes – (20, 21) and thus support bone formation (22) and the accrual of bone mass (23–25). Overall, physiological concentrations of GCs exert anabolic effects throughout the skeleton particularly during growth, whereas supraphysiological (or therapeutic) levels of GCs result in loss of bone mass and quality (26, 27). Early studies on GIO have described several extra-skeletal effects, which may mechanistically underpin GC-induced bone loss, such as i) a dysregulation of calcium homeostasis through decreased intestinal calcium absorption and increased renal calcium clearance; ii) a reduction in the growth hormone/insulin-like growth factor axis; iii) alteration in gonadal steroid hormones; or iv) the potential development of secondary hyperparathyroidism. Also, the catabolic effects of GCs on skeletal muscle have been marked as a contributor to increased fracture risk via increased incidence of falls secondary to muscle weakness (28–30). Interestingly, over the last two decades, advances in mouse genetics have enabled the detailed characterization of the mechanisms of GC-induced bone loss. This led to the discovery that the direct effects of supra-physiological levels of GCs on bone cells represent a significant part of the pathogenesis of GIO. Generally, the pathogenesis of GIO is characterized by two phases: an initial phase of accelerated bone loss owing mainly to increased osteoclast-mediated bone resorption; followed by a slow but continuous phase of qualitative and quantitative bone loss as a result of the compromised function of both osteoblasts and osteocytes. While all skeletal cell types – namely osteoblasts, osteocytes and osteoclasts – are targeted by GCs, it is now understood that cells of the osteoblast lineage are the main effectors of GC-induced bone loss and the GC-induced rise in fracture risk.
Here we review the molecular and cellular targets of therapeutic doses of GCs with a particular focus on osteoblasts and osteocytes as well as the implications for clinical therapy of GIO.
The Osteoblast Lineage as a Key Target for Excess GCs
Skeletal cells continually interact with one another through the process of bone remodeling. Bone remodeling includes the coordinated processes of bone formation and bone resorption. Formation of new bone is performed by osteoblasts, whereas bone resorption is carried out by osteoclasts. Osteocytes act as mechanosensors and orchestrate the skeletal remodeling process by initiating and governing the remodeling cycle (31, 32). While exogenous GCs affect all cells of the remodeling process – either directly or indirectly (Figure 1) –, cells of the osteoblast lineage, and therefore bone formation, are key targets of GCs in the skeleton.
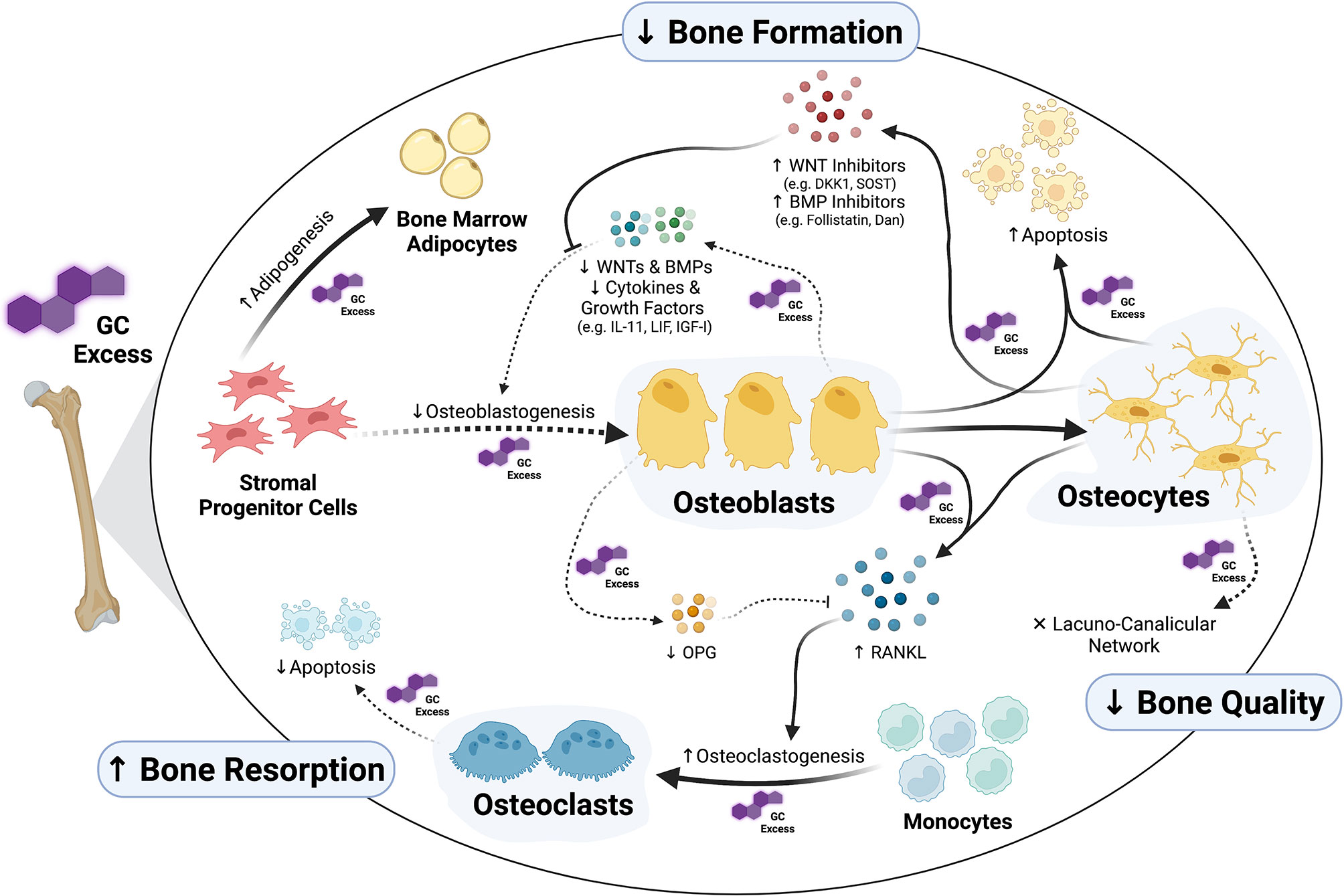
Figure 1 Osteoblasts and osteocytes as main targets of glucocorticoid (GC) excess in the skeleton. Exposure to supra-physiological levels of GCs affects many aspects of osteoblast formation and function. Whereas GCs inhibit osteogenic commitment of stromal progenitor cells by diversion into adipogenesis, they inhibit proliferation and differentiation of pre-osteoblasts through direct as well as autocrine/paracrine effects. Together with suppression of osteoblast function, all these GC-induced alterations in osteoblasts suppress bone formation. Additionally, GCs induce apoptosis of both osteoblasts and osteocytes and cause disruptions in osteocytic lacuna-canalicular network affecting bone quality. Osteoclast-mediated bone resorption is affected by GCs as well, especially through the regulation of the RANKL/OPG system via osteoblasts and osteocytes. The figure was created with BioRender.com.
Generally, exposure to supra-physiological levels of GCs results in a strong suppression of bone formation and the anabolic function of osteoblasts in both humans and rodents. Treatment of patients with therapeutic doses of GCs rapidly suppresses serum markers of bone formation such as osteocalcin, bone-specific alkaline phosphatase (ALP) and procollagen type I N-terminal propeptide (P1NP) (33–40). Similarly, prolonged exposure of rodents to excess GCs decreases the systemic markers of bone formation and the osteoblasts’ anabolic function, such as osteocalcin and P1NP (17, 41–46). Histomorphological analysis of bones from GC-treated rodents confirms these findings and reveals compromised bone formation and mineralization as well as a reduction in the number and surface of osteoblasts (17, 23, 43, 45, 47–49). Similar effects were observed in bone biopsies from GC-treated patients (50–53). Overall, GIO occurs in both rodents and humans with similar cellular and molecular features. Thus, rodents may act as a suitable model organism to investigate the molecular and cellular mechanism underlying GIO (54).
The significance of osteoblasts in the pathogenesis of GIO has been made clear through the utilization of genetically modified mouse models, in which GC-GR signaling has been disrupted in a cell-specific fashion. Protection of osteoblasts from excessive GC signaling by osteoblast-specific overexpression of the GC-inactivating enzyme, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), not only prevented GC-induced osteoblast apoptosis but also preserved osteoblast function and bone formation (43, 55). Similarly, specific deletion of GR in osteoblasts prevented both GC-driven bone loss as well as compromised bone formation (23). Some – though not all – studies investigating the disruption of GC signaling in osteoblasts/osteocytes during GC excess showed that not only osteoblast function and bone formation were preserved in this setting but also the GC-induced increase in osteoclast number and activity was prevented (43). Collectively, these results suggest that the adverse skeletal effects of exogenous GCs result to a large degree from their detrimental action on cells of the osteoblast lineage. Quantifying the overall contribution of osteoclasts to the development of GC-induced osteoporosis remains challenging. The selective abrogation of GC-GR signaling in osteoclasts (by GR knock-out) resulted in preserved bone resorption and preserved bone formation, indicating a prominent role for osteoclasts in GC-induced bone loss (56). However, – in the hands of different researchers – the osteoclast-specific disruption of GCs (either by 11β-HSD2 overexpression or conditional GR knockout) had no discernible protective effects against GC-induced bone loss since osteoblasts were readily affected by excess GCs (23, 57). Collectively, the weight of the evidence strongly points to the osteoblast lineage as a more impactful target of GCs in the skeleton compared to the cells of the osteoclast lineage.
The Effects of GC Excess on the Formation and Function of Osteoblasts
GCs cause alterations in the formation and apoptosis of osteoblasts as well as their function, all of which contribute to the pathogenesis of GIO. In vivo and in vitro studies have determined that supra-physiological levels of GCs exert their deleterious effects on cells of the osteoblast lineage at all stages of differentiation, leading to reduced osteoblast formation. Moreover, GCs limit both function and lifespan of osteoblasts, ultimately resulting in compromised bone formation. Furthermore, through the intrinsic link between bone formation and bone resorption, GCs may alter osteoblast activity and function through their action in osteoblasts and osteocytes. The effects of exogenous GCs on molecular pathways within osteoblasts are manifold and the relative contribution of each identified pathway is not always quantifiable. Nevertheless, the main effects of GCs on osteoblasts can be outlined as follows:
a) Decreased Osteogenic Cell Fate of Stromal Progenitor Cells
Given the multipotent nature of stromal progenitor cells in the bone marrow, supra-physiological levels of GCs induce diversion of these stem cells away from the osteoblast lineage towards the adipocyte lineage. Ultimately, this diversion of stem cell commitment leads to a decrease in the pool of osteoblast progenitors and limits bone formation. Accordingly, it has been shown that exposure to exogenous GCs in humans and rodents is associated with increased bone marrow adiposity (58–60). In line with these results, gene expression profiling of bone tissue from GC-treated mice displayed an induction of adipogenesis-related genes whereas osteogenic genes were downregulated (49). Moreover, bone marrow stromal progenitor cells from GC-treated rodents displayed reduced osteoblastogenesis ex vivo (23, 45, 48), with enhanced direction towards adipogenesis even in osteogenic media (59, 60). Similarly, exposure of bone marrow stromal progenitor cells to pharmacological levels of GCs results in decreased expression of essential osteogenic transcription factors such as runt-related transcription factor 2 (RUNX2), accompanied by concurrent increased expression of adipogenic transcription factors such as peroxisome proliferator- activated receptor gamma (PPARγ) and CCAAT-enhancer-binding protein alpha (C/EBPα) (61–66).
b) Suppressed Proliferation of Osteoprogenitors
Acting also on committed osteoblast precursors, GCs have been shown to inhibit and suppress their proliferation prior to full differentiation. In pre-osteoblast cultures, exposure to pharmacological ‘micromolar’ concentrations of GCs was associated with cell cycle arrest at the G1 phase due to downregulation of cell cycle activators such as Cyclin A, Cyclin D, Cyclin-dependent kinase 2 (CDK2), CDK4 and CDK6 (67–70) as well as upregulation of cell cycle inhibitors such as p53, p21 and p27 (67, 69, 71). In addition, GCs were shown to suppress the proliferation of osteoblast precursors through suppression of intracellular mitogenic signaling pathways, such as mitogen-activated protein kinase (MAPK) signaling via a rapid increase in the expression of a tyrosine phosphatase, MAPK phosphatase 1/dual specificity protein phosphatase 1 (MKP1/DUSP1), leading to dephosphorylation of extracellular-signal-regulated kinases (ERK), p38 and c-Jun N-terminal kinase (JNK) (72–74). Interestingly, while non-specific tyrosine phosphatase inhibition reversed GC-induced suppression of pre-osteoblasts in vitro and partly prevented deleterious bone effects (of GCs) in a rat model of GIO, Mkp1 knockout mice were not protected against the adverse effects of methylprednisolone treatment (72–76). In a different study Mkp1 deletion was shown to exacerbate inflammatory bone loss (77). These results suggest that targeting MKP1 may not represent a viable strategy for the prevention of GC-driven bone loss.
c) Inhibited Differentiation of Osteoblast Precursors Into Mature Osteoblasts
GC-induced inhibition of osteoblastogenesis is mediated mainly via suppression of signaling pathways involved in promoting osteoblast differentiation, importantly WNT and bone morphogenetic protein (BMP) pathways. First, GCs have been shown to inhibit the production of autocrine/paracrine WNT proteins, such as WNT7b, WNT10 and WNT16 (22, 78), as well as BMP proteins, such as BMP2, from mature osteoblasts (79–82). Conversely, the GC-driven suppression of osteoblast differentiation in vitro was corrected by supplementation of culture media with WNT and BMP proteins. Second, GCs increase the expression of inhibitory factors of the WNT and BMP signaling pathways from osteoblasts as well as osteocytes including WNT antagonists such as dickkopf1 (DKK1), sclerostin (SOST), secreted frizzled-related protein 1 (sRFP1) and axin-2 (22, 41, 49, 79, 83–89), as well as BMP antagonists, such as Follistatin and Dan (63, 79, 90). Third, exposure of pre-osteoblasts to supra-physiological levels of GCs suppresses the canonical WNT pathway through inducing degradation and inactivation of β-catenin, therefore inhibiting osteoblastogenesis (68, 91, 92). Moreover, suppression of growth factor pathways, such as insulin-like growth factor I (IGF-I), may contribute to the suppressive effects of GCs on osteoblastogenesis (93–96). GCs also suppress anabolic cytokines such as interleukin-11 (IL-11) and leukemia inhibitory factor (LIF) thereby reducing Janus kinase 2 (JAK2) – signal transducer and activator of transcription 3 (STAT3) signaling via inducing interaction of the monomeric glucocorticoid receptor with the transcription factor AP-1 (23, 97). Not only did supplementation of GC-treated osteoblasts with IL-11 (23, 97) and LIF (98) reverse the suppression in STAT3 signaling and osteoblast differentiation in vitro, treatment with LIF protected mice against GC-driven bone loss (98). Interestingly, reduced IL-11 expression was observed in other models of bone loss such as age-related suppression of bone formation, suggesting that IL-11 may be generally implicated in bone diseases (99, 100). Nevertheless, IL-11 is known to affect osteoclasts as well (101). Beside the direct targeting of key bone-anabolic pathways such as WNT and BMP signaling, GCs modulate the expression of miRNAs, including miR-29a, miR-34a-5p and miR-199a-5p, which regulate proliferation and differentiation of osteoblasts (102). A study by Wang and colleagues showed an association of GC-induced osteoporosis with miR-29a in rats, as GCs reduced the levels of miR-29a leading to a subsequent increase in deacetylation and ubiquitinylation of β-catenin, thus attenuating the pro-osteogenic impact of WNT signaling on differentiation of osteoblasts (103, 104). However, osteoblast-selective deletion of Dicer, an important enzyme in miRNA biogenesis, did not affect GC-induced suppression of osteogenesis both in vitro and in vivo (105).
d) Decreased Function of Osteoblasts
In addition to suppressed osteoblast formation, GCs decrease the anabolic function of osteoblasts, i.e., secretion of osteoid matrix proteins (e.g., collagen and osteocalcin) and subsequent mineralization of the matrix itself. For instance, GCs downregulate OCN (the gene encoding osteocalcin) gene expression in human and rat osteoblasts through direct binding of the GC-GR complex to a negative GC-response element (-GRE) in the enhancer region of the osteocalcin gene leading to trans-repression (106–108). Also, the expression of collagen from osteoblasts was shown to be suppressed by excess GCs via transcriptional and post-transcriptional mechanisms (109, 110). Apart from the synthesis of bone matrix proteins, supra-physiological levels of GCs were shown to provoke matrix degradation through upregulating expression of metalloproteinases such as matrix metalloproteinase 13 (MMP13) from osteoblasts (49, 111).
The Effects of Excess GCs on the Lifespan of Osteoblasts and Osteocytes
Aside from suppression of osteoblast differentiation and activity, exposure to pharmacological levels of GCs triggers apoptosis in osteoblasts as well as their descendants, osteocytes, limiting their lifespan. Apoptotic osteoblasts and osteocytes were clearly detectable in the bones not only from GC-treated rodents (17, 45, 48, 55, 112) but also from patients undergoing therapy with GCs (45, 52, 113). It may be inferred that the GC-induced osteoblast apoptosis, similarly to suppressed osteoblast differentiation, likely contributes to the compromised bone formation, ultimately leading to GC-induced loss of bone mass and increase in fracture risk. More importantly, prevention of GC-driven apoptosis in osteoblasts and osteocytes has been associated with preservation of bone mass as well as strength in mouse models of GIO. For instance, co-treatment of mice with bisphosphonates (48, 114), intermittent parathyroid hormone (PTH) (115) or osteoprotegerin (OPG) (116) alleviated the adverse effects of pharmacological GCs on osteoblast and osteocyte apoptosis as well as bone formation and mineralization resulting in protection from bone loss.
Despite the evidence outlined above, some studies failed to detect a GC-induced increase in apoptosis of osteoblasts and osteocytes despite the detrimental effects of GCs on bone formation (23). This might be related to differences in the mouse strain and/or the dose of GCs utilized in the study. Importantly, the induction of apoptosis in osteocytes and osteoblasts has been shown to be dose- and time-dependent. In response to low ‘nanomolar’ concentrations of GCs, osteocytes and osteoblasts rely on autophagy to repair cellular damage and maintain viability (112, 117–120). In mice treated with low dose GCs, an upregulation of the expression of anti-oxidant and autophagy genes as well as an appearance of autophagic osteocytes and osteoblasts was observed in the skeleton (112, 119). However, prolonged exposure and/or high ‘micromolar’ doses of GCs result in suppression of autophagy as well as excessive intracellular damage due to accumulation of autophagosomes inside osteocytes and osteoblasts, which ultimately lead to the activation of pro-apoptotic pathways and programmed cell death (112, 119, 121). Induction of autophagy in osteocytes and osteoblasts has been hypothesized to underpin a protective mechanism to preserve cellular viability (120, 122, 123); however, prolonged exposure to GCs is associated with suppressed autophagy leading to apoptosis (117, 123, 124). Indeed, enhancing autophagy in vivo by administration of the phytoecdysteroid, β-ecdysone, to GC-treated mice prevents GC-induced bone loss by reversing the suppression of bone formation and the induction of apoptosis in osteoblasts and osteocytes (121, 124). Likewise, pharmacological inhibition of autophagy was associated with an increase in GC-induced osteoblast apoptosis in vitro (117, 120). Nevertheless, the significance of autophagy in the detrimental effect of GCs on cells of the osteoblast lineage remains overwhelming (122, 125). Targeting apoptosis and autophagy of osteoblasts and osteocytes has been highlighted as a therapy for not only GC-driven bone loss (125), but also in age-related osteoporosis (126, 127).
Several studies using in vitro osteoblast and osteocyte cultures revealed some of the molecular mechanisms underpinning GC-induced apoptosis. Not only mechanisms related to regulation of transcription, but also rapid non-genomic mechanisms have been attributed to the apoptotic impact of GCs on the osteoblast lineage. The most evident subcellular apoptotic pathways in osteoblasts and/or osteocytes influenced by genomic GR actions have been upregulation of pro-apoptotic proteins such as BIM, BAK, p53 and p21 (67, 71, 128–130), as well as the suppression of survival, anti-apoptotic factors such as BCL-2, BCL-Xl and MCL-1 (67, 112, 131, 132). In addition, suppression of MAPK – ERK pathway through upregulation of MKP1/DUSP1 may act as another mechanism for GC-driven apoptosis in osteocytes and osteoblasts, as a non-selective protein tyrosine inhibitor was able to prevent GC-driven osteoblast apoptosis in vitro and in vivo (133). An increase in oxidative stress in the endoplasmic reticulum (ER) is one of the non-genomic pathways implicated in accumulation of reactive oxygen species (ROS), which may activate JNK signaling and programmed cell death in osteoblasts (84, 131, 134–136). Generally, prevention of oxidative stress exerts protective effects on osteoblasts and osteocytes thus preserving bone formation in addition to mediating anti-resorptive effects on osteoclasts (137). Prevention of ER stress and ROS accumulation via knocking down Eif2a (Eukaryotic Translation Initiation Factor 2A) not only prevented GC-induced apoptosis in vitro and in vivo, but also was associated with protection against bone loss (138). Inducing the protein tyrosine kinase 2 beta (PYK2) pathway and blocking focal adhesion kinase (FAK) signaling may contribute to GC-induced apoptosis in cells of the osteoblast lineage (136). In a recent report, genetic and pharmacological inactivation of Pyk2 signaling was proven effective in preventing not only apoptosis in osteoblasts and osteocytes, but also GC-induced bone loss, although reversing compromised osteoclast function was shown to likely contribute to such protective effects (139). Moreover, induction of Fas receptor/CD95 may advance apoptotic pathways in osteoblasts and osteocytes (140). Two recent studies hypothesized that long-non coding (lnc) RNAs are involved in GC-induced osteoblast apoptosis. Long-non coding RNAs are a large family of RNA molecules that are able to regulate protein expression and/or function. Lnc-MALAT1 and lnc-EPIC1 expression were shown to be altered in human osteoblasts treated with dexamethasone and to interact with AMP-activated protein kinase signaling and MYC [a regulator of osteoblast survival] (141, 142). However, the role of lncRNA in GIO remains to be validated in vivo.
The Effects of Excess GCs on the Function of Osteocytes
Osteocytes play a crucial role in bone homeostasis through modulating the formation and activity of osteoblasts and bone formation via the release of WNT signaling inhibitors, sclerostin and dickkopf1 (DKK1) (143). In a number of studies, an upregulation of sclerostin gene and protein expression has been observed in the cortical-rich bones from GC-treated mice, where osteocytes are generally more abundant than osteoblasts (39, 49, 87, 144). Strong evidence for the significant contribution of the GC-driven upregulation of sclerostin in osteocytes to GIO has come from studies of abrogated sclerostin action in rodent models of excess GCs. Administration of anti-sclerostin antibodies to rats and mice prevented the development of GC-induced bone loss largely via preserving the function and number of osteoblasts and maintaining bone formation and mineralization (46, 145). In addition, knocking out Sost (the gene encoding sclerostin) in mice provided protection from GC-driven bone loss (144). In humans, the contribution of sclerostin to GC-induced bone loss is less clear. One study described a trend increase in serum levels of sclerostin in patients receiving pharmacological GCs (36). However, the serum levels of sclerostin were decreased in the patients treated with GCs in comparison to matched controls (39), and similar results were observed after acute treatment with therapeutic GCs in another study (146). DKK1, another WNT inhibitor expressed in osteocytes, is upregulated in GC-treated animals, and anti-sense silencing of Dkk1 in mice was effective in preserving bone mass as well as bone formation during GC excess (49, 89). In a recent study, conditional knockout of Dkk1 in osteoblasts and/or osteocytes prevented the development of GC-induced bone loss via reversing the adverse effects of GCs on osteoblasts and bone formation (41). Notably, both sclerostin and DKK1 have emerged as promising therapeutic targets in a number of bone diseases (147), and may be utilized clinically for the management of GIO in the future.
Aside from affecting the regulatory role of osteocytes through sclerostin and DKK1, several alterations in the bone environment around the osteocyte-lacunar environment have been reported in response to pharmacological levels of GCs. In bones from GC-treated mice, changes in the bone matrix surrounding osteocyte lacunae were observed, specifically an increased lacunae size as well as perilacunar hypomineralization (17). Additionally, these effects were associated with compromised bone strength (17). Moreover, osteocyte perilacunar remodeling was shown to be adversely affected by exogenous GCs: a GC-induced suppression of the expression of matrix metalloproteinases (MMPs) leads to collagen disorganization and degeneration of the lacuno-canalicular network (148). In the in vitro setting, Gao et al. were able to show that the gap-junction connectivity of osteocytes was adversely affected by dexamethasone treatment of an osteocyte cell line (MLYO-cells). These dexamethasone-induced changes resulted in a suppressed amount of Connexin 43 due to degradation by autophagy, thus leading to shortening of osteocyte dendrites, which likely contributes to the compromised connectivity between osteocytes (149). Furthermore, GCs were shown to impair the skeletal vasculature leading to a reduction in solute transport from the circulation to the osteocyte-lacunar-canalicular network and a decrease in the interstitial fluid, thereby compromising bone strength (150). Interestingly, PTH treatment was able to rescue skeletal vascularity during GC exposure (151). More recently, two studies highlighted the role of the skeletal vasculature in the context of GCs during growth. GC-exposure in young mice (typically around 3 weeks of age) impaired angiogenesis and osteogenesis simultaneously (152, 153). Liu et al. were able to show that osteoclast-derived angiogenin was decreased in response to elevated levels of GCs, leading to an increase in blood vessel senescence (153).
In summary, GCs exert a detrimental impact on the function and lifespan of osteocytes leading not only to compromised bone formation but also to disruptions in the lacunar-canalicular network (Figure 1). The GC-induced dysfunction of the osteocyte-canalicular network may represent a potential mechanism underlying the predisposition to developing fractures shortly after initiation of GC treatment prior to any significant decreases in BMD – a frequent clinical observation (8). The role of the skeletal vasculature in GIO has been highlighted through recent studies and its role needs further exploration – particularly its connection to bone cells (i.e. osteoblast, osteocytes and osteoclasts) as well as its link to fracture risk.
The Effects of GC Excess on Osteoclasts
While the adverse effects of GCs on osteoblasts and osteocytes contribute to the long-term phase of bone loss and compromised bone strength in GIO, the initial rapid phase of bone loss typically observed in humans and rodents originates from a rapid induction of osteoclast-mediated bone resorption. In a number of in vivo studies, treatment of rodents with GCs results in a rapid elevation of systemic parameters of bone resorption including serum and/or urinary bone resorption markers, such as carboxy-terminal collagen crosslinks (CTX) and tartrate-resistant acid phosphatase-5b (TRAP-5b), upon exposure to supra-physiological levels of GCs (17, 41, 43, 46, 49). In addition, in the bones from GC-treated rodents, an increase in the number of osteoclasts, as well as an increase in gene expression of osteoclast-mediated bone resorption have been reported shortly after exposure to exogenous GCs (17, 45, 46, 48, 49). While some studies also showed upregulation of osteoclast activity and bone resorption markers at later time-points (41, 47, 154, 155), other studies failed to detect increases in bone resorption especially after prolonged GC exposure (45, 156). In addition, one study by Henneicke et al. showed that treatment with corticosterone affected osteoclasts in a site-specific manner in rodents: an increase in osteoclasts was detected in the endocortex, while they were reduced in the pericortex of tibia from GC-treated mice (43).
Several in vivo and in vitro studies have determined that the mechanisms of elevated osteoclast-mediated bone resorption in GIO originate not only from direct effects of GCs in osteoclasts, but also from indirect effects via the osteoblast lineage. It has been shown that the early increase in osteoclastic bone resorption may be accounted for by an increase in the survival of mature osteoclasts and reduced predisposition to apoptosis (48, 56, 57, 157). However, the direct impact of excess GCs on osteoclastogenesis and osteoclast activity has been controversially discussed due to conflicting results from in vitro studies. While some authors observed that pharmacological GCs augmented osteoclast formation and resorptive activity (158–160), others reported a reduction in proliferation of osteoclast precursors (56, 157). Additionally, bone marrow macrophages (osteoclast precursors) from GC-treated animals gave rise to a lower number of osteoclast precursors ex vivo than their placebo controls (45, 48). Furthermore, exposure of in vitro-formed osteoclasts to GCs increased their longevity, yet, in the same study, it decreased their resorptive function due to defects in cytoskeleton reorganization (56, 157). Interestingly, a recent study found that dexamethasone delayed the formation of multinucleated osteoclasts on plastic surfaces yet increased the formation of resorption pits on dentin slides (161). Ultimately, the contribution of direct effects of GCs on osteoclasts to the overall phenotype of GIO remains unclear due to the large amount of conflicting data.
In contrast, the indirect effects of GC excess on osteoclastogenesis and bone resorption have been well characterized across both in vivo and vitro studies. The receptor activator of NF-κB ligand (RANKL) – osteoprotegerin (OPG) system, which plays a crucial role in the differentiation of osteoclasts, is affected to a large degree by pharmacological levels of GCs. Several studies demonstrated that supraphysiological levels of GCs induce the expression and production of RANKL from osteoblasts in culture (162–165), a finding also confirmed in vivo (144, 166). Administration of a human anti-RANKL antibody to mice expressing human RANKL conferred protection from GC-induced bone loss (166). Some studies suggest that osteocytes – rather than osteoblasts – are the principle source of RANKL in vivo (167, 168); however, a more recent study failed to show an increase in RANKL in the osteocyte-enriched bones from GC-treated rodents (47). Interestingly, in the same study a genetic knockdown of Rankl specifically in osteocytes provided partial protection from GC-induced bone loss via reversal of the osteoclast induction (47).
Aside from RANKL, GCs have been shown to reduce the production of OPG, the decoy receptor of RANKL, from osteoblasts and/or osteocytes, which may aide GC-driven osteoclastogenesis (47, 144, 162–165, 169, 170). Additionally, administration of OPG was able to reduce GC-induced bone resorption in calvarial organ culture (165) as well as prevent GC-induced bone loss in rodents (116). Indeed, some studies suggest that the increase in the ratio between RANKL and OPG in bone may be largely due to suppressed OPG rather than due to increased RANKL (47, 144, 169). Other indirect contributors to GC-induced bone resorption include macrophage colony-stimulating factor (M-CSF): exposure of osteoblasts to pharmacological levels of GCs was shown to induce the expression of M-CSF, which acts as an essential factor for osteoclast differentiation (171).
In summary, GCs certainly exert direct effects on osteoclasts; however, whether these direct effects contribute to the phenotype of GC-induced bone loss remains controversial. In contrast, in vivo and in vitro studies clearly demonstrate that GCs readily induce osteoclast formation indirectly through upregulation of pro-osteoclastogenic factors derived from cells of the osteoblast lineage (Figure 1).
Targeting Osteoblasts as a Therapeutic Approach for the Management of GIO
As the mainstay of osteoporosis therapy anti-resorptive bisphosphonates have been widely used in the therapy of GIO. Generally, the use of bisphosphonate in GIO leads to an increase in bone mineral density compared to placebo or calcium and vitamin D supplements (15). Thus, three different bisphosphonates are currently approved for the treatment of GIO, namely risedronate (172, 173), alendronate (174) and zoledronic acid (175). Zoledronic acid has been shown to be superior to risedronate in GIO and postmenopausal osteoporosis (175) and is generally considered the most potent bisphosphonate. Although not an osteoanabolic therapy, denosumab, a RANKL inhibitor, counteracts a key mechanism of GCs in bone – the induction of RANKL release from osteoblasts and osteocytes. Clinical studies showed a larger increase in bone mineral density (BMD) during denosumab therapy compared to risedronate confirming its superiority to one of the bisphosphonates in GIO (176, 177). Unfortunately, denosumab has not yet been evaluated against the most potent bisphosphonate zoledronic acid in the context of GC use, but its value in the treatment of GIO is undeniable.
While bisphosphonates and denosumab have been successfully utilized to combat GIO, they only offset the GC-induced activation of osteoclasts – which is of particular importance during the initial stage of GC-therapy. However, as outlined above, bisphosphonates fail to address the suppression of osteoblast and osteocyte function, which are a crucial part of the pathogenesis of GIO. The development of targeted osteoporosis therapies opens up the possibility of targeting the mechanism underlying GIO more specifically.
Currently only one osteoanabolic agent, targeting bone formation directly, is approved for the treatment of GIO. As a parathyroid hormone (PTH) analog (1-34 PTH), teriparatide primarily stimulates bone formation – even though bone resorption is activated in response to teriparatide as well. However, bone resorption is initiated much later than bone formation resulting in an ‘anabolic window’, during which new bone is formed (178). Mechanistically, as an anabolic therapy it mitigates the GC-induced suppression of osteoblast (and osteocyte) activity, which forms a key part of the mechanism underpinning GIO. In the clinical setting, teriparatide has been shown to increase BMD to a larger extent than risedronate (179) and alendronate (180, 181) during GC exposure, thus highlighting the key role of osteoanabolic therapy for GIO. At this stage, no adequate comparison between teriparatide and denosumab exists during GIO (182), hence, no conclusions may be drawn regarding their relative potency in the context of GC therapy.
Novel osteoanabolic therapies such as the PTH-related protein analogue abaloparatide (183) and the anti-sclerostin antibody romosozumab (184, 185), which have been approved for the use in postmenopausal osteoporosis, have not yet been evaluated in GIO. Given their osteoanabolic properties, they may prove similarly effective as teriparatide.
In summary, all available pharmacological therapies are effective in GIO, this includes bisphosphonates, denosumab as well as teriparatide (Table 1). Therapies, which target the molecular and cellular mechanisms of GCs in the skeleton such as denosumab and teriparatide, have been shown to be superior to bisphosphonates in GIO. Some (186) but not all (187) guidelines reflect this by recommending the use of teriparatide in severe cases of GIO or following the occurrence of fractures under treatment with bisphosphonates.

Table 1 Current and future pharmacological GIO therapy.
Summary
Glucocorticoids affect the three main cell types within the skeleton – osteoblasts, osteocytes and osteoclasts – ultimately leading to a loss of bone mass and bone quality as well as causing a substantial increase in fracture risk. Preclinical studies have highlighted the key role of osteoblasts and osteocytes in the pathogenesis of glucocorticoid-induced osteoporosis and emerging clinical evidence supports the superiority of osteoblast-targeted therapies. Future studies should develop and evaluate therapeutic strategies that not only alleviate GC-induced bone resorption but also prevent the GC-induced damage to osteoblasts and osteocytes and activate bone formation. Furthermore, novel aspects of GIO such as the role of the skeletal vasculature ought to be explored in greater detail.
BOX 1. Bone mineral density as a surrogate parameter in GIO.
GCs have been shown to substantially increase fracture risk in humans. Interestingly, the increase in fracture risk manifest itself immediately after the commencement of GC therapy (8), leading to the hypothesis that GCs may damage bone beyond the loss of bone mass. And indeed, studies were able to establish that in patients suffering from GIO fractures occurred more frequently compared to patients with postmenopausal osteoporosis even when BMD scores were taken into account (13). Similarly, it has been established that the commonly used FRAX algorithm underestimates the occurrence of fractures in subjects treated with GCs (15). More recently the use of trabecular bone score (TBS) has been shown to potentially remedy some of these concerns (16); however, its use has not been widely adopted and/or established as a diagnostic tool in GIO. Overall, the predictive value of BMD is reduced in GIO compared to postmenopausal osteoporosis. This is of particular concern as virtually all studies assessing the use of anti-osteoporotic medication in GIO utilize BMD as a surrogate parameter for fractures. Studies were not adequately powered to allow for an analysis of fracture risk. This should be taken into account when evaluating the results of clinical trials comparing therapeutic agents in the context of GIO.
Preclinical studies have attempted to assess the underlying reason for the particularly high fracture risk in GIO compared to postmenopausal osteoporosis. Studies in rodents were able to link the high fracture risk in GIO as well as the rapid onset of fractures following commencement of GC-therapy to their detrimental effects on osteocytes. Lane et al. highlighted the role of the lacunar-canalicular network in this context, which is largely maintained by osteocytes (17). Others have built on this idea and highlighted the role of the skeletal vasculature in GIO, see section ‘The Effects of Excess GCs on the Function of Osteocytes’ for further details. However, the rapid increase in fracture risk with commencement of GC-therapy may also be the result of systemic effects of supraphysiological levels of GCs; i.e. GCs may decrease muscle strength and adversely affect coordination and/or lead to an increase in falls (and thus fractures) due to their effects in the central nervous system. Hence, whether the rapid and strong increase in fractures following commencement of therapeutic GCs is a result of bone-intrinsic effects of GCs or GC-action elsewhere in the body remains to be determined.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
Deutsche Forschungsgemeinschaft (Grant numbers: BA-6428/1-1 and HE-8391/1-1) and Else Kröner-Fresenius-Stiftung.
Conflict of Interest
HH has received travel support, speaking fees and honoraria for scientific consultation from Amgen, Takeda and Novo Nordisk. LH reports honoraria from Amgen, Alexion, Kyowa Kirin International, and UCB for scientific consultation to himself, and trial compensation from Ascendis, Novartis, and Takeda to his institution.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with one of the authors LH.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
11β-HSD2, 11β-hydroxysteroid dehydrogenase type 2; AP-1, activator protein 1; ALP, alkaline phosphatase; BMP, bone morphogenic protein; BMD, bone mineral density; CTX, carboxy-terminal collagen crosslinks; C/EBPα, CCAAT-enhancer-binding protein alpha; CDK, Cyclin-dependent Kinase; DUSP1, Dual-specificity phosphatase 1; DKK1, dickkopf1; ER, endoplasmic reticulum; ERK, extracellular-signal-regulated kinases; Eif2a, Eukaryotic Translation Initiation Factor 2A; FAK, focal adhesion kinase; GR, glucocorticoid receptor; GCs, glucocorticoids; -GRE, negative GC-response element; GIO, GC-induced osteoporosis; GLOW, Global Longitudinal Study of Osteoporosis in Women; IL-11, interleukin-11; IGF-1, insulin-like growth factor I; JNK, c-Jun N-terminal kinase; JAK2, Janus kinase 2; LncRNAs, long non-coding RNAs; LIF, leukemia inhibitory factor; MKP1, MAPK phosphatase 1; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; M-CSF, Macrophage colony-stimulating factor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; OPG, osteoprotegerin; OCN, osteocalcin; P1NP, procollagen type I N-terminal propeptide; PPARγ, Peroxisome proliferator-activated receptor gamma; PTH, parathyroid hormone; PYK2, Protein-tyrosine kinase 2 beta; RUNX2, runt-related transcription factor 2; RANKL, receptor activator of nuclear factor kappa-B ligand; ROS, reactive oxygen species; SOST, sclerostin; sRFP1, Secreted frizzled-related protein; STAT3, Signal Transducer And Activator Of Transcription 3; TRAP-5b, tartrate-resistant acid phosphatase-5b; TBS, trabecular bone score.
References
1. Cushing H. The Basophil Adenomas of the Pituitary Body and Their Clinical Manifestations (Pituitary Basophilism). Bull John Hopkins Hosp (1932), 137–95.
2. Freyberg RH, Traeger CH, Patterson M, Squires W, Adams CH, Stevenson C. Problems of Prolonged Cortisone Treatment for Rheumatoid Arthritis: Further Investigations. J Am Med Assoc (1951) 147:1538–43. doi: 10.1001/jama.1951.03670330030008
3. Díez-Pérez A, Hooven FH, Adachi JD, Adami S, Anderson FA, Boonen S, et al. Regional Differences in Treatment for Osteoporosis. The Global Longitudinal Study of Osteoporosis in Women (GLOW). Bone (2011) 49:493–8. doi: 10.1016/j.bone.2011.05.007
4. Amiche MA, Albaum JM, Tadrous M, Pechlivanoglou P, Lévesque LE, Adachi JD, et al. Fracture Risk in Oral Glucocorticoid Users: A Bayesian Meta-Regression Leveraging Control Arms of Osteoporosis Clinical Trials. Osteoporosis Int (2016) 27:1709–18. doi: 10.1007/s00198-015-3455-9
5. Donnan PT, Libby G, Boyter AC, Thompson P. The Population Risk of Fractures Attributable to Oral Corticosteroids. Pharmacoepidemiol Drug Saf (2005) 14:177–86. doi: 10.1002/pds.1075
6. Hoes JN, Jacobs JWG, Verstappen SMM, Bijlsma JWJ, van der Heijden GJMG. Adverse Events of Low- to Medium-Dose Oral Glucocorticoids in Inflammatory Diseases: A Meta-Analysis. Ann Rheum Dis (2009) 68:1833–8. doi: 10.1136/ard.2008.100008
7. Kalpakcioglu BB, Engelke K, Genant HK. Advanced Imaging Assessment of Bone Fragility in Glucocorticoid-Induced Osteoporosis. Bone (2011) 48:1221–31. doi: 10.1016/j.bone.2011.02.005
8. van Staa TP, Leufkens HGM, Abenhaim L, Zhang B, Cooper C. Use of Oral Corticosteroids and Risk of Fractures. J Bone Mineral Res (2000) 15:993–1000. doi: 10.1359/jbmr.2000.15.6.993
9. van Staa TP, Leufkens HGM, Cooper C. The Epidemiology of Corticosteroid-Induced Osteoporosis: A Meta-Analysis. Osteoporos Int (2002) 13:777–87. doi: 10.1007/s001980200108
10. Carbonare LD, Arlot ME, Chavassieux PM, Roux JP, Portero NR, Meunier PJ. Comparison of Trabecular Bone Microarchitecture and Remodeling in Glucocorticoid-Induced and Postmenopausal Osteoporosis. J Bone Mineral Res (2001) 16:97–103. doi: 10.1359/jbmr.2001.16.1.97
11. van Staa TP, Leufkens HGM, Abenhaim L, Zhang B, Cooper C. Oral Corticosteroids and Fracture Risk: Relationship to Daily and Cumulative Doses. Rheumatology (2000) 39:1383–9. doi: 10.1093/rheumatology/39.12.1383
12. Steinbuch M, Youket TE, Cohen S. Oral Glucocorticoid Use Is Associated With an Increased Risk of Fracture. Osteoporosis Int (2004) 15:323–8. doi: 10.1007/s00198-003-1548-3
13. van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone Density Threshold and Other Predictors of Vertebral Fracture in Patients Receiving Oral Glucocorticoid Therapy. Arthritis Rheum (2003) 48:3224–9. doi: 10.1002/art.11283
14. van Staa TP, Cooper C, Leufkens HGM, Bishop N. Children and the Risk of Fractures Caused by Oral Corticosteroids. J Bone Mineral Res (2003) 18:913–8. doi: 10.1359/jbmr.2003.18.5.913
15. Henneicke H, Gasparini SJ, Brennan-Speranza TC, Zhou H, Seibel MJ. Glucocorticoids and Bone: Local Effects and Systemic Implications. Trends Endocrinol Metab (2014) 25:197–211. doi: 10.1016/j.tem.2013.12.006
16. Florez H, Hernández-Rodríguez J, Muxi A, Carrasco JL, Prieto-González S, Cid MC, et al. Trabecular Bone Score Improves Fracture Risk Assessment in Glucocorticoid-Induced Osteoporosis. Rheumatol (Oxf) (2020) 59:1574–80. doi: 10.1093/rheumatology/kez464
17. Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, et al. Glucocorticoid-Treated Mice Have Localized Changes in Trabecular Bone Material Properties and Osteocyte Lacunar Size That Are Not Observed in Placebo-Treated or Estrogen-Deficient Mice. J Bone Mineral Res (2006) 21:466–76. doi: 10.1359/jbmr.051103
18. Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, et al. Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol Rev (2016) 96:409–47. doi: 10.1152/physrev.00011.2015
19. Vandewalle J, Luypaert A, Bosscher KD, Libert C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol Metab (2018) 29:42–54. doi: 10.1016/j.tem.2017.10.010
20. Shalhoub V, Conlon D, Tassinari M, Quinn C, Partridge N, Stein GS, et al. Glucocorticoids Promote Development of the Osteoblast Phenotype by Selectively Modulating Expression of Cell Growth and Differentiation Associated Genes. J Cell Biochem (1992) 50:425–40. doi: 10.1002/jcb.240500411
21. Zhou H, Mak W, Zheng Y, Dunstan CR, Seibel MJ. Osteoblasts Directly Control Lineage Commitment of Mesenchymal Progenitor Cells Through Wnt Signaling. J Biol Chem (2008) 283:1936–45. doi: 10.1074/jbc.m702687200
22. Mak W, Shao X, Dunstan CR, Seibel MJ, Zhou H. Biphasic Glucocorticoid-Dependent Regulation of Wnt Expression and its Inhibitors in Mature Osteoblastic Cells. Calcified Tissue Int (2009) 85:538–45. doi: 10.1007/s00223-009-9303-1
23. Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids Suppress Bone Formation by Attenuating Osteoblast Differentiation via the Monomeric Glucocorticoid Receptor. Cell Metab (2010) 11:517–31. doi: 10.1016/j.cmet.2010.05.005
24. Sher LB, Woitge HW, Adams DJ, Gronowicz GA, Krozowski Z, Harrison JR, et al. Transgenic Expression of 11β-Hydroxysteroid Dehydrogenase Type 2 in Osteoblasts Reveals an Anabolic Role for Endogenous Glucocorticoids in Bone. Endocrinology (2004) 145:922–9. doi: 10.1210/en.2003-0655
25. Kalak R, Zhou H, Street J, Day RE, Modzelewski JRK, Spies CM, et al. Endogenous Glucocorticoid Signalling in Osteoblasts Is Necessary to Maintain Normal Bone Structure in Mice. Bone (2009) 45:61–7. doi: 10.1016/j.bone.2009.03.673
26. Zhou H, Cooper MS, Seibel MJ. Endogenous Glucocorticoids and Bone. Bone Res (2013) 1:107–19. doi: 10.4248/BR201302001
27. Hardy RS, Zhou H, Seibel MJ, Cooper MS. Glucocorticoids and Bone: Consequences of Endogenous and Exogenous Excess and Replacement Therapy. Endocrine Rev (2018) 39:519–48. doi: 10.1210/er.2018-00097
28. Canalis E. Mechanisms of Glucocorticoid-Induced Osteoporosis. Curr Opin Rheumatol (2003) 15:454–7. doi: 10.1097/00002281-200307000-00013
29. Canalis E, Bilezikian JP, Angeli A, Giustina A. Perspectives on Glucocorticoid-Induced Osteoporosis. Bone (2004) 34:593–8. doi: 10.1016/j.bone.2003.11.026
30. Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-Induced Osteoporosis: Pathophysiology and Therapy. Osteoporosis Int (2007) 18:1319–28. doi: 10.1007/s00198-007-0394-0
31. Bellido T, Plotkin LI, Bruzzaniti A. Chapter 3 - Bone Cells. In: Burr DB, Allen MR, editors. Basic and Applied Bone Biology, 2nd ed. Academic Press (2019). p. 37–55. doi: 10.1016/B978-0-12-813259-3.00003-8
32. Rauner M, Stein N, Hofbauer LC. Basics of Bone Biology. In: Pietschmann P, editor. Principles of Osteoimmunology: Molecular Mechanisms and Clinical Applications. Vienna: Springer (2012). p. 1–26. doi: 10.1007/978-3-7091-0520-7_1
33. Cosman F, Nieves J, Herbert J, Shen V, Lindsay R. High-Dose Glucocorticoids in Multiple Sclerosis Patients Exert Direct Effects on the Kidney and Skeleton. J Bone Mineral Res (1994) 9:1097–105. doi: 10.1002/jbmr.5650090718
34. Devogelaer J-P, Durnez A, Gruson D, Manicourt DH. Bone Turnover Markers and Glucocorticoid Treatments. In: Patel V., Preedy V. (eds) Biomarkers in Bone Disease. Biomarkers in Disease: Methods, Discoveries and Applications. Dordrecht: Springer.
35. Gennari C. Differential Effect of Glucocorticoids on Calcium Absorption and Bone Mass. Br J Rheumatol (1993) 32(Suppl 2):11–4. doi: 10.1093/rheumatology/32.suppl_2.11
36. Gifre L, Ruiz-Gaspà S, Monegal A, Nomdedeu B, Filella X, Guañabens N, et al. Effect of Glucocorticoid Treatment on Wnt Signalling Antagonists (Sclerostin and Dkk-1) and Their Relationship With Bone Turnover. Bone (2013) 57:272–6. doi: 10.1016/j.bone.2013.08.016
37. Pearce G, Tabensky DA, Delmas PD, Baker HW, Seeman E. Corticosteroid-Induced Bone Loss in Men. J Clin Endocrinol Metab (1998) 83:801–6. doi: 10.1210/jcem.83.3.4621
38. Prummel MF, Wiersinga WM, Lips P, Sanders GT, Sauerwein HP. The Course of Biochemical Parameters of Bone Turnover During Treatment With Corticosteroids. J Clin Endocrinol Metab (1991) 72:382–6. doi: 10.1210/jcem-72-2-382
39. Thiele S, Hannemann A, Winzer M, Baschant U, Weidner H, Nauck M, et al. Regulation of Sclerostin in Glucocorticoid-Induced Osteoporosis (GIO) in Mice and Humans. Endocrine Connect (2019) 8:923–34. doi: 10.1530/EC-19-0104
40. Ton FN, Gunawardene SC, Lee H, Neer RM. Effects of Low-Dose Prednisone on Bone Metabolism. J Bone Mineral Res (2005) 20:464–70. doi: 10.1359/jbmr.041125
41. Colditz J, Thiele S, Baschant U, Garbe AI, Niehrs C, Hofbauer LC, et al. Osteogenic Dkk1 Mediates Glucocorticoid-Induced But Not Arthritis-Induced Bone Loss. J Bone Mineral Res (2019) 34:1314–23. doi: 10.1002/jbmr.3702
42. Gasparini SJ, Weber MC, Henneicke H, Kim S, Zhou H, Seibel MJ. Continuous Corticosterone Delivery via the Drinking Water or Pellet Implantation: A Comparative Study in Mice. Steroids (2016) 116:76–82. doi: 10.1016/j.steroids.2016.10.008
43. Henneicke H, Herrmann M, Kalak R, Brennan-Speranza TC, Heinevetter U, Bertollo N, et al. Corticosterone Selectively Targets Endo-Cortical Surfaces by an Osteoblast-Dependent Mechanism. Bone (2011) 49:733–42. doi: 10.1016/j.bone.2011.06.013
44. Lane NE. New Observations on Bone Fragility With Glucocorticoid Treatment. Results From an In Vivo Animal Model. J Musculoskelet Neuronal Interact (2005) 5:331–2.
45. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of Osteoblastogenesis and Promotion of Apoptosis of Osteoblasts End Osteocytes by Glucocorticoids Potential Mechanisms of Their Deleterious Effects on Bone. J Clin Invest (1998) 102:274–82. doi: 10.1172/JCI2799
46. Yao W, Dai W, Jiang L, Lay EYA, Zhong Z, Ritchie RO, et al. Sclerostin-Antibody Treatment of Glucocorticoid-Induced Osteoporosis Maintained Bone Mass and Strength. Osteoporosis Int (2016) 27:283–94. doi: 10.1007/s00198-015-3308-6
47. Piemontese M, Xiong J, Fujiwara Y, Thostenson JD, O’Brien CA. Cortical Bone Loss Caused by Glucocorticoid Excess Requires RANKL Production by Osteocytes and is Associated With Reduced OPG Expression in Mice. Am J Physiol - Endocrinol Metab (2016) 311:E587–93. doi: 10.1152/ajpendo.00219.2016
48. Weinstein RS, Chen J-R, Powers CC, Stewart SA, Landes RD, Bellido T, et al. Promotion of Osteoclast Survival and Antagonism of Bisphosphonate-Induced Osteoclast Apoptosis by Glucocorticoids. J Clin Invest (2002) 109:1041–8. doi: 10.1172/jci14538
49. Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid Excess in Mice Results in Early Activation of Osteoclastogenesis and Adipogenesis and Prolonged Suppression of Osteogenesis: A Longitudinal Study of Gene Expression in Bone Tissue From Glucocorticoid- Treated Mice. Arthritis Rheum (2008) 58:1674–86. doi: 10.1002/art.23454
50. Chappard D, Legrand E, Basle MF, Fromont P, Racineux Jl, Rebel A, et al. Altered Trabecular Architecture Induced by Corticosteroids: A Bone Histomorphometric Study. J Bone Mineral Res (1996) 11:676–85. doi: 10.1002/jbmr.5650110516
51. Dempster DW. Perspectives Bone Histomorphometry in Glucocorticoid-Induced Osteoporosis. J Bone Mineral Res (1989) 4:137–41. doi: 10.1002/jbmr.5650040202
52. Sambrook P, Hughes D, Nelson A, Robinson B, Mason R. Osteocyte Viability With Glucocorticoid Treatment: Relation to Histomorphometry. Ann Rheum Dis (2003) 62:1215–7. doi: 10.1136/ard.2003.008839
53. Vedi S, Elkin SL, Compston JE. A Histomorphometric Study of Cortical Bone of the Iliac Crest in Patients Treated With Glucocorticoids. Calcif Tissue Int (2005) 77:79–83. doi: 10.1007/s00223-004-0205-y
54. Wood CL, Soucek O, Wong SC, Zaman F, Farquharson C, Savendahl L, et al. Animal Models to Explore the Effects of Glucocorticoids on Skeletal Growth and Structure. J Endocrinol (2018) 236:R69–91. doi: 10.1530/JOE-17-0361
55. O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, et al. Glucocorticoids Act Directly on Osteoblasts and Osteocytes to Induce Their Apoptosis and Reduce Bone Formation and Strength. Endocrinology (2004) 145:1835–41. doi: 10.1210/en.2003-0990
56. Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, et al. Glucocorticoids Suppress Bone Formation via the Osteoclast. J Clin Invest (2006) 116:2152–60. doi: 10.1172/JCI28084
57. Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids Act Directly on Osteoclasts to Increase Their Life Span and Reduce Bone Density. Endocrinology (2006) 147:5592–9. doi: 10.1210/en.2006-0459
58. Berg BCV, Malghem J, Lecouvet FE, Devogelaer JP, Maldague B, Houssiau FA. Fat Conversion of Femoral Marrow in Glucocorticoid-Treated Patients: A Cross-Sectional and Longitudinal Study With Magnetic Resonance Imaging. Arthritis Rheum (1999) 42:1405–11. doi: 10.1002/1529-0131(199907)42:7<1405::AID-ANR14>3.0.CO;2-W
59. Li J, Zhang N, Huang X, Xu J, Fernandes JC, Dai K, et al. Dexamethasone Shifts Bone Marrow Stromal Cells From Osteoblasts to Adipocytes by C/Ebpαlpha Promoter Methylation. Cell Death Dis (2013) 4:e832–2. doi: 10.1038/cddis.2013.348
60. Sui B, Hu C, Liao L, Chen Y, Zhang X, Fu X, et al. Mesenchymal Progenitors in Osteopenias of Diverse Pathologies: Differential Characteristics in the Common Shift From Osteoblastogenesis to Adipogenesis. Sci Rep (2016) 6:30186. doi: 10.1038/srep30186
61. Cui Q, Wang GJ, Balian G. Steroid-Induced Adipogenesis in a Pluripotential Cell Line From Bone Marrow. J Bone Joint Surg Am (1997) 79:1054–63. doi: 10.2106/00004623-199707000-00012
62. Koromila T, Baniwal SK, Song YS, Martin A, Xiong J, Frenkel B. Glucocorticoids Antagonize RUNX2 During Osteoblast Differentiation in Cultures of ST2 Pluripotent Mesenchymal Cells. J Cell Biochem (2014) 115:27–33. doi: 10.1002/jcb.24646
63. Leclerc N, Luppen CA, Ho VV, Nagpal S, Hacia JG, Smith E, et al. Gene Expression Profiling of Glucocorticoid-Inhibited Osteoblasts. J Mol Endocrinol (2004) 33:175–93. doi: 10.1677/jme.0.0330175
64. Li X, Jin L, Cui Q, Wang GJ, Balian G. Steroid Effects on Osteogenesis Through Mesenchymal Cell Gene Expression. Osteoporosis Int (2005) 16:101–8. doi: 10.1007/s00198-004-1649-7
65. Pereira RC, Delany AM, Canalis E. Effects of Cortisol and Bone Morphogenetic Protein-2 on Stromal Cell Differentiation: Correlation With CCAAT-Enhancer Binding Protein Expression. Bone (2002) 30:685–91. doi: 10.1016/s8756-3282(02)00687-7
66. Shi XM, Blair HC, Yang X, McDonald JM, Cao X. Tandem Repeat of C/EBP Binding Sites Mediates Pparγ2 Gene Transcription in Glucocorticoid-Induced Adipocyte Differentiation. J Cell Biochem (2000) 76:518–27. doi: 10.1002/(sici)1097-4644(20000301)76:3<518::aid-jcb18>3.0.co;2-m
67. Chang JK, Li CJ, Liao HJ, Wang CK, Wang GJ, Ho ML. Anti-Inflammatory Drugs Suppress Proliferation and Induce Apoptosis Through Altering Expressions of Cell Cycle Regulators and Pro-Apoptotic Factors in Cultured Human Osteoblasts. Toxicology (2009) 258:148–56. doi: 10.1016/j.tox.2009.01.016
68. Gabet Y, Noh T, Lee C, Frenkel B. Developmentally Regulated Inhibition of Cell Cycle Progression by Glucocorticoids Through Repression of Cyclin a Transcription in Primary Osteoblast Cultures. J Cell Physiol (2011) 226:991–8. doi: 10.1002/jcp.22412
69. Rogatsky I, Trowbridge JM, Garabedian MJ. Glucocorticoid Receptor-Mediated Cell Cycle Arrest is Achieved Through Distinct Cell-Specific Transcriptional Regulatory Mechanisms. Mol Cell Biol (1997) 17:3181–93. doi: 10.1128/mcb.17.6.3181
70. Smith E, Redman RA, Logg CR, Coetzee GA, Kasahara N, Frenkel B. Glucocorticoids Inhibit Developmental Stage-Specific Osteoblast Cell Cycle. Dissociation of Cyclin A-Cyclin-Dependent Kinase 2 From E2F4-P130 Complexes. J Biol Chem (2000) 275:19992–20001. doi: 10.1074/jbc.M001758200
71. Li H, Qian W, Weng X, Wu Z, Li H, Zhuang Q, et al. Glucocorticoid Receptor and Sequential P53 Activation by Dexamethasone Mediates Apoptosis and Cell Cycle Arrest of Osteoblastic MC3T3-E1 Cells. PloS One (2012) 7:e37030. doi: 10.1371/journal.pone.0037030
72. Engelbrecht Y, Wet HD, Horsch K, Langeveldt CR, Hough FS, Hulley PA. Glucocorticoids Induce Rapid Up-Regulation of Mitogen-Activated Protein Kinase Phosphatase-1 and Dephosphorylation of Extracellular Signal-Regulated Kinase and Impair Proliferation in Human and Mouse Osteoblast Cell Lines. Endocrinology (2003) 144:412–22. doi: 10.1210/en.2002-220769
73. Horsch K, Wet H, Schuurmans MM, Allie-Reid F, Cato ACB, Cunningham J, et al. Mitogen-Activated Protein Kinase Phosphatase 1/Dual Specificity Phosphatase 1 Mediates Glucocorticoid Inhibition of Osteoblast Proliferation. Mol Endocrinol (2007) 21:2929–40. doi: 10.1210/me.2007-0153
74. Hulley PA, Gordon F, Hough FS. Inhibition of Mitogen-Activated Protein Kinase Activity and Proliferation of an Early Osteoblast Cell Line (MBA 15.4) by Dexamethasone: Role of Protein Phosphatases. Endocrinology (1998) 139:2423–31. doi: 10.1210/endo.139.5.6020
75. Hulley PA, Conradie MM, Langeveldt CR, Hough FS. Glucocorticoid-Induced Osteoporosis in the Rat is Prevented by the Tyrosine Phosphatase Inhibitor, Sodium Orthovanadate. Bone (2002) 31:220–9. doi: 10.1016/s8756-3282(02)00807-4
76. Conradie MM, Cato ACB, Ferris WF, de Wet H, Horsch K, Hough S. MKP-1 Knockout Does Not Prevent Glucocorticoid-Induced Bone Disease in Mice. Calcif Tissue Int (2011) 89:221–7. doi: 10.1007/s00223-011-9509-x
77. Sartori R, Li F, Kirkwood KL. MAP Kinase Phosphatase-1 Protects Against Inflammatory Bone Loss. J Dent Res (2009) 88:1125–30. doi: 10.1177/0022034509349306
78. Hildebrandt S, Baschant U, Thiele S, Tuckermann J, Hofbauer LC, Rauner M. Glucocorticoids Suppress Wnt16 Expression in Osteoblasts In Vitro and In Vivo. Sci Rep (2018) 8:8711. doi: 10.1038/s41598-018-26300-z
79. Hayashi K, Yamaguchi T, Yano S, Kanazawa I, Yamauchi M, Yamamoto M, et al. BMP/Wnt Antagonists are Upregulated by Dexamethasone in Osteoblasts and Reversed by Alendronate and PTH: Potential Therapeutic Targets for Glucocorticoid-Induced Osteoporosis. Biochem Biophys Res Commun (2009) 379:261–6. doi: 10.1016/j.bbrc.2008.12.035
80. Luppen CA, Smith E, Spevak L, Boskey AL, Frenkel B. Bone Morphogenetic Protein-2 Restores Mineralization in Glucocorticoid-Inhibited MC3T3-E1 Osteoblast Cultures. J Bone Mineral Res (2003) 18:1186–97. doi: 10.1359/jbmr.2003.18.7.1186
81. Luppen CA, Leclerc N, Noh T, Barski A, Khokhar A, Boskey AL, et al. Brief Bone Morphogenetic Protein 2 Treatment of Glucocorticoid-Inhibited MC3T3-E1 Osteoblasts Rescues Commitment-Associated Cell Cycle and Mineralization Without Alteration of Runx2. J Biol Chem (2003) 278:44995–5003. doi: 10.1074/jbc.M306730200
82. Luppen CA, Chandler RL, Noh T, Mortlock DP, Frenkel B. BMP-2 vs. BMP-4 Expression and Activity in Glucocorticoid-Arrested MC3T3-E1 Osteoblasts: Smad Signaling, Not Alkaline Phosphatase Activity, Predicts Rescue of Mineralization. Growth Factors (2008) 26:226–37. doi: 10.1080/08977190802277880
83. Butler JS, Queally JM, Devitt BM, Murray DW, Doran PP, O’Byrne JM. Silencing Dkk1 Expression Rescues Dexamethasone-Induced Suppression of Primary Human Osteoblast Differentiation. BMC Musculoskelet Disord (2010) 11:210. doi: 10.1186/1471-2474-11-210
84. Hurson CJ, Butler JS, Keating DT, Murray DW, Sadlier DM, O’Byrne JM, et al. Gene Expression Analysis in Human Osteoblasts Exposed to Dexamethasone Identifies Altered Developmental Pathways as Putative Drivers of Osteoporosis. BMC Musculoskelet Disord (2007) 8:12. doi: 10.1186/1471-2474-8-12
85. Ohnaka K, Taniguchi H, Kawate H, Nawata H, Takayanagi R. Glucocorticoid Enhances the Expression of Dickkopf-1 in Human Osteoblasts: Novel Mechanism of Glucocorticoid-Induced Osteoporosis. Biochem Biophys Res Commun (2004) 318:259–64. doi: 10.1016/j.bbrc.2004.04.025
86. Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid Suppresses the Canonical Wnt Signal in Cultured Human Osteoblasts. Biochem Biophys Res Commun (2005) 329:177–81. doi: 10.1016/j.bbrc.2005.01.117
87. Thiele S, Ziegler N, Tsourdi E, Bosscher KD, Tuckermann JP, Hofbauer LC, et al. Selective Glucocorticoid Receptor Modulation Maintains Bone Mineral Density in Mice. J Bone Mineral Res (2012) 27:2242–50. doi: 10.1002/jbmr.1688
88. Wang FS, Lin CL, Chen YJ, Wang CJ, Yang KD, Huang YT, et al. Secreted Frizzled-Related Protein 1 Modulates Glucocorticoid Attenuation of Osteogenic Activities and Bone Mass. Endocrinology (2005) 146:2415–23. doi: 10.1210/en.2004-1050
89. Wang FS, Ko JY, Yeh DW, Ke HC, Wu HL. Modulation of Dickkopf-1 Attenuates Glucocorticoid Induction of Osteoblast Apoptosis, Adipocytic Differentiation, and Bone Mass Loss. Endocrinology (2008) 149:1793–801. doi: 10.1210/en.2007-0910
90. Leclerc N, Noh T, Khokhar A, Smith E, Frenkel B. Glucocorticoids Inhibit Osteocalcin Transcription in Osteoblasts by Suppressing Egr2/Krox20-Binding Enhancer. Arthritis Rheum (2005) 52:929–39. doi: 10.1002/art.20872
91. Smith E, Frenkel B. Glucocorticoids Inhibit the Transcriptional Activity of LEF/TCF in Differentiating Osteoblasts in a Glycogen Synthase Kinase-3β-Dependent and -Independent Manner. J Biol Chem (2005) 280:2388–94. doi: 10.1074/jbc.m406294200
92. Smith E, Coetzee GA, Frenkel B. Glucocorticoids Inhibit Cell Cycle Progression in Differentiating Osteoblasts via Glycogen Synthase Kinase-3β. J Biol Chem (2002) 277:18191–7. doi: 10.1074/jbc.m109708200
93. Chevalley T, Strong DD, Mohan S, Baylink DJ, Linkhart TA. Evidence for a Role for Insulin-Like Growth Factor Binding Proteins in Glucocorticoid Inhibition of Normal Human Osteoblast-Like Cell Proliferation. Eur J Endocrinol (1996) 134:591–601. doi: 10.1530/eje.0.1340591
94. Delany AM, Canalis E. Transcriptional Repression of Insulin-Like Growth Factor I by Glucocorticoids in Rat Bone Cells. Endocrinology (1995) 136:4776–81. doi: 10.1210/endo.136.11.7588206
95. Delany AM, Durant D, Canalis E. Glucocorticoid Suppression of IGF I Transcription in Osteoblasts. Mol Endocrinol (2001) 15:1781–9. doi: 10.1210/mend.15.10.0704
96. Pereira RMR, Delany AM, Canalis E. Cortisol Inhibits the Differentiation and Apoptosis of Osteoblasts in Culture. Bone (2001) 28:484–90. doi: 10.1016/s8756-3282(01)00422-7
97. Rauch A, Gossye V, Bracke D, Gevaert E, Jacques P, Van Beneden K, et al. An Anti-Inflammatory Selective Glucocorticoid Receptor Modulator Preserves Osteoblast Differentiation. FASEB J (2011) 25:1323–32. doi: 10.1096/fj.10-173393
98. Lee S, Liu P, Ahmad M, Tuckermann JP. Leukemia Inhibitory Factor Treatment Attenuates the Detrimental Effects of Glucocorticoids on Bone in Mice. Bone (2021) 145:115843. doi: 10.1016/j.bone.2021.115843
99. Matsumoto T, Kuriwaka-Kido R, Kondo T, Endo I, Kido S. Regulation of Osteoblast Differentiation by Interleukin-11 via AP-1 and Smad Signaling. Endocr J (2012) 59:91–101. doi: 10.1507/endocrj.ej11-0219
100. Takeuchi Y, Watanabe S, Ishii G, Takeda S, Nakayama K, Fukumoto S, et al. Interleukin-11 as a Stimulatory Factor for Bone Formation Prevents Bone Loss With Advancing Age in Mice. J Biol Chem (2002) 277:49011–8. doi: 10.1074/jbc.M207804200
101. Kespohl B, Schumertl T, Bertrand J, Lokau J, Garbers C. The Cytokine Interleukin-11 Crucially Links Bone Formation, Remodeling and Resorption. Cytokine Growth Factor Rev (2021) 60:18–27. doi: 10.1016/j.cytogfr.2021.04.002
102. Komori T. Glucocorticoid Signaling and Bone Biology. Horm Metab Res (2016) 48:755–63. doi: 10.1055/s-0042-110571
103. Ko J-Y, Chuang P-C, Chen M-W, Ke H-C, Wu S-L, Chang Y-H, et al. MicroRNA-29a Ameliorates Glucocorticoid-Induced Suppression of Osteoblast Differentiation by Regulating β-Catenin Acetylation. Bone (2013) 57:468–75. doi: 10.1016/j.bone.2013.09.019
104. Wang F-S, Chuang P-C, Chung P-C, Lin C-L, Chen M-W, Ke H-J, et al. MicroRNA-29a Protects Against Glucocorticoid-Induced Bone Loss and Fragility in Rats by Orchestrating Bone Acquisition and Resorption. Arthritis Rheum (2013) 65:1530–40. doi: 10.1002/art.37948
105. Liu P, Baumgart M, Groth M, Wittmann J, Jäck H-M, Platzer M, et al. Dicer Ablation in Osteoblasts by Runx2 Driven cre-loxP Recombination Affects Bone Integrity, But Not Glucocorticoid-Induced Suppression of Bone Formation. Sci Rep (2016) 6:32112. doi: 10.1038/srep32112
106. Heinrichs AA, Banerjee C, Bortell R, Owen TA, Stein JL, Stein GS, et al. Identification and Characterization of Two Proximal Elements in the Rat Osteocalcin Gene Promoter That may Confer Species-Specific Regulation. J Cell Biochem (1993) 53:240–50. doi: 10.1002/jcb.240530309
107. Morrison N, Eisman J. Role of the Negative Glucocorticoid Regulatory Element in Glucocorticoid Repression of the Human Osteocalcin Promoter. J Bone Miner Res (1993) 8:969–75. doi: 10.1002/jbmr.5650080810
108. Morrison NA, Shine J, Fragonas JC, Verkest V, McMenemy ML, Eisman JA. 1,25-Dihydroxyvitamin D-Responsive Element and Glucocorticoid Repression in the Osteocalcin Gene. Science (1989) 246:1158–61. doi: 10.1126/science.2588000
109. Canalis E. Effect of Glucocorticoids on Type I Collagen Synthesis, Alkaline Phosphatase Activity, and Deoxyribonucleic Acid Content in Cultured Rat Calvariae. Endocrinology (1983) 112:931–9. doi: 10.1210/endo-112-3-931
110. Delany AM, Gabbitas BY, Canalis E. Cortisol Downregulates Osteoblast Alpha 1 (I) Procollagen mRNA by Transcriptional and Posttranscriptional Mechanisms. J Cell Biochem (1995) 57:488–94. doi: 10.1002/jcb.240570314
111. Delany AM, Jeffrey JJ, Rydziel S, Canalis E. Cortisol Increases Interstitial Collagenase Expression in Osteoblasts by Post-Transcriptional Mechanisms. J Biol Chem (1995) 270:26607–12. doi: 10.1074/jbc.270.44.26607
112. Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R, et al. Glucocorticoid Dose Determines Osteocyte Cell Fate. FASEB J (2011) 25:3366–76. doi: 10.1096/fj.11-182519
113. Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of Osteocytes in Glucocorticoid-Induced Osteonecrosis of the Hip. J Clin Endocrinol Metab (2000) 85:2907–12. doi: 10.1210/jc.85.8.2907
114. Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of Osteocyte and Osteoblast Apoptosis by Bisphosphonates and Calcitonin. J Clin Invest (1999) 104:1363–74. doi: 10.1172/JCI6800
115. Weinstein RS, Jilka RL, Almeida M, Roberson PK, Manolagas SC. Intermittent Parathyroid Hormone Administration Counteracts the Adverse Effects of Glucocorticoids on Osteoblast and Osteocyte Viability, Bone Formation, and Strength in Mice. Endocrinology (2010) 151:2641–8. doi: 10.1210/en.2009-1488
116. Weinstein RS, O’Brien CA, Almeida M, Zhao H, Roberson PK, Jilka RL, et al. Osteoprotegerin Prevents Glucocorticoid-Induced Osteocyte Apoptosis in Mice. Endocrinology (2011) 152:3323–31. doi: 10.1210/en.2011-0170
117. Han Y, Zhang L, Xing Y, Zhang L, Chen X, Tang P, et al. Autophagy Relieves the Function Inhibition and Apoptosis-Promoting Effects on Osteoblast Induced by Glucocorticoid. Int J Mol Med (2018) 41:800–8. doi: 10.3892/ijmm.2017.3270
118. Liu W, Zhao Z, Na Y, Meng C, Wang J, Bai R. Dexamethasone-Induced Production of Reactive Oxygen Species Promotes Apoptosis via Endoplasmic Reticulum Stress and Autophagy in MC3T3-E1 Cells. Int J Mol Med (2018) 41:2028–36. doi: 10.3892/ijmm.2018.3412
119. Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, et al. Glucocorticoid-Induced Autophagy in Osteocytes. J Bone Mineral Res (2010) 25:2479–88. doi: 10.1002/jbmr.160
120. Zhang S, Liu Y, Liang Q. Low-Dose Dexamethasone Affects Osteoblast Viability by Inducing Autophagy via Intracellular Ros. Mol Med Rep (2018) 17:4307–16. doi: 10.3892/mmr.2018.8461
121. Dai WW, Jiang L, Lay YAE, Chen H, Jin G, Zhang H, et al. Prevention of Glucocorticoid Induced Bone Changes With Beta-Ecdysone. Bone (2015) 74:48–57. doi: 10.1016/j.bone.2015.01.001
122. Wang L, Heckmann BL, Yang X, Long H. Osteoblast Autophagy in Glucocorticoid-Induced Osteoporosis. J Cell Physiol (2019) 234:3207–15. doi: 10.1002/jcp.27335
123. Yao W, Dai W, Jiang JX, Lane NE. Glucocorticoids and Osteocyte Autophagy. Bone (2013) 54:279–84. doi: 10.1016/j.bone.2013.01.034
124. Tang YH, Yue ZS, Li GS, Zeng LR, Xin DW, Hu ZQ, et al. Effect of P-Ecdysterone on Glucocorticoid-Induced Apoptosis and Autophagy in Osteoblasts. Mol Med Rep (2018) 17:158–64. doi: 10.3892/mmr.2017.7840
125. Wang T, Liu X, He C. Glucocorticoid-Induced Autophagy and Apoptosis in Bone. Apoptosis (2020) 25:157–68. doi: 10.1007/s10495-020-01599-0
126. Wang T, He H, Liu S, Jia C, Fan Z, Zhong C, et al. Autophagy: A Promising Target for Age-Related Osteoporosis. Curr Drug Targets (2019) 20:354–65. doi: 10.2174/1389450119666180626120852
127. Li X, Xu J, Dai B, Wang X, Guo Q, Qin L. Targeting Autophagy in Osteoporosis: From Pathophysiology to Potential Therapy. Ageing Res Rev (2020) 62:101098. doi: 10.1016/j.arr.2020.101098
128. Chen TL. Inhibition of Growth and Differentiation of Osteoprogenitors in Mouse Bone Marrow Stromal Cell Cultures by Increased Donor Age and Glucocorticoid Treatment. Bone (2004) 35:83–95. doi: 10.1016/j.bone.2004.03.019
129. Espina B, Liang M, Russell RGG, Hulley PA. Regulation of Bim in Glucocorticoid-Mediated Osteoblast Apoptosis. J Cell Physiol (2008) 215:488–96. doi: 10.1002/jcp.21335
130. Zhen Y-F, Wang G-D, Zhu L-Q, Tan S-P, Zhang F-Y, Zhou X-Z, et al. P53 Dependent Mitochondrial Permeability Transition Pore Opening Is Required for Dexamethasone-Induced Death of Osteoblasts. J Cell Physiol (2014) 229:1475–83. doi: 10.1002/jcp.24589
131. Deng S, Dai G, Chen S, Nie Z, Zhou J, Fang H, et al. Dexamethasone Induces Osteoblast Apoptosis Through ROS-PI3K/AKT/Gsk3β Signaling Pathway. BioMed Pharmacother (2019) 110:602–8. doi: 10.1016/j.biopha.2018.11.103
132. Lu NZ, Collins JB, Grissom SF, Cidlowski JA. Selective Regulation of Bone Cell Apoptosis by Translational Isoforms of the Glucocorticoid Receptor. Mol Cell Biol (2007) 27:7143–60. doi: 10.1128/mcb.00253-07
133. Conradie MM, Wet H, Kotze DDR, Burrin JM, Hough FS, Hulley PA. Vanadate Prevents Glucocorticoid-Induced Apoptosis of Osteoblasts In Vitro and Osteocytes In Vivo. J Endocrinol (2007) 195:229–40. doi: 10.1677/joe-07-0217
134. Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and Tumor Necrosis Factor α Increase Oxidative Stress and Suppress Wnt Protein Signaling in Osteoblasts. J Biol Chem (2011) 286:44326–35. doi: 10.1074/jbc.M111.283481
135. Feng Z, Zheng W, Tang Q, Cheng L, Li H, Ni W, et al. Fludarabine Inhibits STAT1-Mediated Up-Regulation of Caspase-3 Expression in Dexamethasone-Induced Osteoblasts Apoptosis and Slows the Progression of Steroid-Induced Avascular Necrosis of the Femoral Head in Rats. Apoptosis (2017) 22:1001–12. doi: 10.1007/s10495-017-1383-1
136. Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids Induce Osteocyte Apoptosis by Blocking Focal Adhesion Kinase-Mediated Survival: Evidence for Inside-Out Signaling Leading to Anoikis. J Biol Chem (2007) 282:24120–30. doi: 10.1074/jbc.M611435200
137. Domazetovic V, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Oxidative Stress in Bone Remodeling: Role of Antioxidants. Clin cases Miner Bone Metab (2017) 14:209–16. doi: 10.11138/ccmbm/2017.14.1.209
138. Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of Glucocorticoid Induced-Apoptosis of Osteoblasts and Osteocytes by Protecting Against Endoplasmic Reticulum (ER) Stress In Vitro and In Vivo in Female Mice. Bone (2015) 73:60–8. doi: 10.1016/j.bone.2014.12.012
139. Sato AY, Cregor M, McAndrews K, Li T, Condon KW, Plotkin LI, et al. Glucocorticoid-Induced Bone Fragility Is Prevented in Female Mice by Blocking Pyk2/Anoikis Signaling. Endocrinology (2019) 160:1659–73. doi: 10.1210/en.2019-00237
140. Kogianni G, Mann V, Ebetino F, Nuttall M, Nijweide P, Simpson H, et al. Fas/CD95 Is Associated With Glucocorticoid-Induced Osteocyte Apoptosis. Life Sci (2004) 75:2879–95. doi: 10.1016/j.lfs.2004.04.048
141. Fan J, Zhang Y, Liu W, Zhu X, Xu D, Zhao J, et al. Long Non-Coding RNA MALAT1 Protects Human Osteoblasts From Dexamethasone-Induced Injury via Activation of PPM1E-AMPK Signaling. CPB (2018) 51:31–45. doi: 10.1159/000495159
142. Zhang X-Y, Shan H-J, Zhang P, She C, Zhou X-Z. LncRNA EPIC1 Protects Human Osteoblasts From Dexamethasone-Induced Cell Death. Biochem Biophys Res Commun (2018) 503:2255–62. doi: 10.1016/j.bbrc.2018.06.146
143. Plotkin LI, Bellido T. Osteocytic Signalling Pathways as Therapeutic Targets for Bone Fragility. Nat Rev Endocrinol (2016) 12:593–605. doi: 10.1038/nrendo.2016.71
144. Sato AY, Cregor M, Delgado-Calle J, Condon KW, Allen MR, Peacock M, et al. Protection From Glucocorticoid-Induced Osteoporosis by Anti-Catabolic Signaling in the Absence of Sost/Sclerostin. J Bone Mineral Res (2016) 31:1791–802. doi: 10.1002/jbmr.2869
145. Achiou Z, Toumi H, Touvier J, Boudenot A, Uzbekov R, Ominsky MS, et al. Sclerostin Antibody and Interval Treadmill Training Effects in a Rodent Model of Glucocorticoid-Induced Osteopenia. Bone (2015) 81:691–701. doi: 10.1016/j.bone.2015.09.010
146. Maresova KB, Pavelka K, Stepan JJ. Acute Effects of Glucocorticoids on Serum Markers of Osteoclasts, Osteoblasts, and Osteocytes. Calcified Tissue Int (2013) 92:354–61. doi: 10.1007/s00223-012-9684-4
147. Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as Therapeutic Targets in Bone Diseases. Endocr Rev (2012) 33:747–83. doi: 10.1210/er.2011-1060
148. Fowler TW, Acevedo C, Mazur CM, Hall-Glenn F, Fields AJ, Bale HA, et al. Glucocorticoid Suppression of Osteocyte Perilacunar Remodeling is Associated With Subchondral Bone Degeneration in Osteonecrosis. Sci Rep (2017) 7:44618. doi: 10.1038/srep44618
149. Gao J, Cheng TS, Qin A, Pavlos NJ, Wang T, Song K, et al. Glucocorticoid Impairs Cell-Cell Communication by Autophagymediated Degradation of Connexin 43 in Osteocytes. Oncotarget (2016) 7:26966–78. doi: 10.18632/oncotarget.9034
150. Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien CA, et al. Endogenous Glucocorticoids Decrease Skeletal Angiogenesis, Vascularity, Hydration, and Strength in Aged Mice. Aging Cell (2010) 9:147–61. doi: 10.1111/j.1474-9726.2009.00545.x
151. Mohan G, Lay EY-A, Berka H, Ringwood L, Kot A, Chen H, et al. Lane NE. A Novel Hybrid Compound LLP2A-Ale Both Prevented and Rescued the Osteoporotic Phenotype in a Mouse Model of Glucocorticoid-Induced Osteoporosis. Calcif Tissue Int (2017) 100:67–79. doi: 10.1007/s00223-016-0195-6
152. Yang P, Lv S, Wang Y, Peng Y, Ye Z, Xia Z, et al. Preservation of Type H Vessels and Osteoblasts by Enhanced Preosteoclast Platelet-Derived Growth Factor Type BB Attenuates Glucocorticoid-Induced Osteoporosis in Growing Mice. Bone (2018) 114:1–13. doi: 10.1016/j.bone.2018.05.025
153. Liu X, Chai Y, Liu G, Su W, Guo Q, Lv X, et al. Osteoclasts Protect Bone Blood Vessels Against Senescence Through the Angiogenin/Plexin-B2 Axis. Nat Commun (2021) 12:1832. doi: 10.1038/s41467-021-22131-1
154. Lin NY, Chen CW, Kagwiria R, Liang R, Beyer C, Distler A, et al. Inactivation of Autophagy Ameliorates Glucocorticoid-Induced and Ovariectomy-Induced Bone Loss. Ann Rheumat Dis (2016) 75:1203–10. doi: 10.1136/annrheumdis-2015-207240
155. Shi J, Wang L, Zhang H, Jie Q, Li X, Shi Q, et al. Glucocorticoids: Dose-Related Effects on Osteoclast Formation and Function via Reactive Oxygen Species and Autophagy. Bone (2015) 79:222–32. doi: 10.1016/j.bone.2015.06.014
156. Weinstein RS, Jia D, Powers CC, Stewart SA, Jilka RL, Parfitt AM, et al. The Skeletal Effects of Glucocorticoid Excess Override Those of Orchidectomy in Mice. Endocrinology (2004) 145:1980–7. doi: 10.1210/en.2003-1133
157. Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, et al. Glucocorticoids and the Osteoclast. Ann N Y Acad Sci (2007) 1116:335–9. doi: 10.1196/annals.1402.057
158. Hirayama T, Sabokbar A, Athanasou NA. Effect of Corticosteroids on Human Osteoclast Formation and Activity. J Endocrinol (2002) 175:155–63. doi: 10.1677/joe.0.1750155
159. Sivagurunathan S, Muir MM, Brennan TC, Seale JP, Mason RS. Influence of Glucocorticoids on Human Osteoclast Generation and Activity. J Bone Mineral Res (2005) 20:390–8. doi: 10.1359/JBMR.041233
160. Takuma A, Kaneda T, Sato T, Ninomiya S, Kumegawa M, Hakeda Y. Dexamethasone Enhances Osteoclast Formation Synergistically With Transforming Growth Factor-β by Stimulating the Priming of Osteoclast Progenitors for Differentiation Into Osteoclasts. J Biol Chem (2003) 278:44667–74. doi: 10.1074/jbc.M300213200
161. Conaway HH, Henning P, Lie A, Tuckermann J, Lerner UH. Activation of Dimeric Glucocorticoid Receptors in Osteoclast Progenitors Potentiates RANKL Induced Mature Osteoclast Bone Resorbing Activity. Bone (2016) 93:43–54. doi: 10.1016/j.bone.2016.08.024
162. Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, et al. Stimulation of Osteoprotegerin Ligand and Inhibition of Osteoprotegerin Production by Glucocorticoids in Human Osteoblastic Lineage Cells: Potential Paracrine Mechanisms of Glucocorticoid-Induced Osteoporosis. Endocrinology (1999) 140:4382–9. doi: 10.1210/endo.140.10.7034
163. Humphrey EL, Williams JHH, Davie MWJ, Marshall MJ. Effects of Dissociated Glucocorticoids on OPG and RANKL in Osteoblastic Cells. Bone (2006) 38:652–61. doi: 10.1016/j.bone.2005.10.004
164. Kondo T, Kitazawa R, Yamaguchi A, Kitazawa S. Dexamethasone Promotes Osteoclastogenesis by Inhibiting Osteoprotegerin Through Multiple Levels. J Cell Biochem (2008) 103:335–45. doi: 10.1002/jcb.21414
165. Swanson C, Lorentzon M, Conaway HH, Lerner UH. Glucocorticoid Regulation of Osteoclast Differentiation and Expression of Receptor Activator of Nuclear Factor-κb (NF-κb) Ligand, Osteoprotegerin, and Receptor Activator of NF-κb in Mouse Calvarial Bones. Endocrinology (2006) 147:3613–22. doi: 10.1210/en.2005-0717
166. Hofbauer LC, Zeitz U, Schoppet M, Skalicky M, Schuler C, Stolina M, et al. Prevention of Glucocorticoid-Induced Bone Loss in Mice by Inhibition of RANKL. Arthritis Rheum (2009) 60:1427–37. doi: 10.1002/art.24445
167. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for Osteocyte Regulation of Bone Homeostasis Through RANKL Expression. Nat Med (2011) 17:1231–4. doi: 10.1038/nm.2452
168. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-Embedded Cells Control Osteoclast Formation. Nat Med (2011) 17:1235–41. doi: 10.1038/nm.2448
169. Piemontese M, Onal M, Xiong J, Wang Y, Almeida M, Thostenson JD, et al. Suppression of Autophagy in Osteocytes Does Not Modify the Adverse Effects of Glucocorticoids on Cortical Bone. Bone (2015) 75:18–26. doi: 10.1016/j.bone.2015.02.005
170. Rauner M, Goettsch C, Stein N, Thiele S, Bornhaeuser M, Bosscher KD, et al. Dissociation of Osteogenic and Immunological Effects by the Selective Glucocorticoid Receptor Agonist, Compound a, in Human Bone Marrow Stromal Cells. Endocrinology (2011) 152:103–12. doi: 10.1210/en.2010-0456
171. Rubin J, Biskobing DM, Jadhav L, Fan D, Nanes MS, Perkins S, et al. Dexamethasone Promotes Expression of Membrane-Bound Macrophage Colony-Stimulating Factor in Murine Osteoblast-Like Cells. Endocrinology (1998) 139:1006–12. doi: 10.1210/endo.139.3.5778
172. Cohen S, Levy RM, Keller M, Boling E, Emkey RD, Greenwald M, et al. Risedronate Therapy Prevents Corticosteroid-Induced Bone Loss: A Twelve-Month, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study. Arthritis Rheum (1999) 42:2309–18. doi: 10.1002/1529-0131(199911)42:11<2309::AID-ANR8>3.0.CO;2-K
173. Reid DM, Hughes RA, Laan RF, Sacco-Gibson NA, Wenderoth DH, Adami S, et al. Efficacy and Safety of Daily Risedronate in the Treatment of Corticosteroid-Induced Osteoporosis in Men and Women: A Randomized Trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res (2000) 15:1006–13. doi: 10.1359/jbmr.2000.15.6.1006
174. de Nijs RNJ, Jacobs JWG, Lems WF, Laan RFJ, Algra A, Huisman A-M, et al. Alendronate or Alfacalcidol in Glucocorticoid-Induced Osteoporosis. N Engl J Med (2006) 355:675–84. doi: 10.1056/NEJMoa053569
175. Reid DM, Devogelaer J-P, Saag K, Roux C, Lau C-S, Reginster J-Y, et al. Zoledronic Acid and Risedronate in the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis (HORIZON): A Multicentre, Double-Blind, Double-Dummy, Randomised Controlled Trial. Lancet (2009) 373:1253–63. doi: 10.1016/S0140-6736(09)60250-6
176. Saag KG, Wagman RB, Geusens P, Adachi JD, Messina OD, Emkey R, et al. Denosumab Versus Risedronate in Glucocorticoid-Induced Osteoporosis: A Multicentre, Randomised, Double-Blind, Active-Controlled, Double-Dummy, Non-Inferiority Study. Lancet Diabetes Endocrinol (2018) 6:445–54. doi: 10.1016/S2213-8587(18)30075-5
177. Saag KG, Pannacciulli N, Geusens P, Adachi JD, Messina OD, Morales-Torres J, et al. Denosumab Versus Risedronate in Glucocorticoid-Induced Osteoporosis: Final Results of a Twenty-Four-Month Randomized, Double-Blind, Double-Dummy Trial. Arthritis Rheumatol (2019) 71:1174–84. doi: 10.1002/art.40874
178. Canalis E, Giustina A, Bilezikian JP. Mechanisms of Anabolic Therapies for Osteoporosis. N Engl J Med (2007) 357:905–16. doi: 10.1056/NEJMra067395
179. Glüer C-C, Marin F, Ringe JD, Hawkins F, Möricke R, Papaioannu N, et al. Comparative Effects of Teriparatide and Risedronate in Glucocorticoid-Induced Osteoporosis in Men: 18-Month Results of the EuroGIOPs Trial. J Bone Miner Res (2013) 28:1355–68. doi: 10.1002/jbmr.1870
180. Saag KG, Shane E, Boonen S, Marín F, Donley DW, Taylor KA, et al. Teriparatide or Alendronate in Glucocorticoid-Induced Osteoporosis. N Engl J Med (2007) 357:2028–39. doi: 10.1056/NEJMoa071408
181. Saag KG, Zanchetta JR, Devogelaer J-P, Adler RA, Eastell R, See K, et al. Effects of Teriparatide Versus Alendronate for Treating Glucocorticoid-Induced Osteoporosis: Thirty-Six-Month Results of a Randomized, Double-Blind, Controlled Trial. Arthritis Rheum (2009) 60:3346–55. doi: 10.1002/art.24879
182. Hirooka Y, Nozaki Y, Inoue A, Li J, Shiga T, Kishimoto K, et al. Effects of Denosumab Versus Teriparatide in Glucocorticoid-Induced Osteoporosis Patients With Prior Bisphosphonate Treatment. Bone Rep (2020) 13:100293. doi: 10.1016/j.bonr.2020.100293
183. Miller PD, Hattersley G, Riis BJ, Williams GC, Lau E, Russo LA, et al. Effect of Abaloparatide vs Placebo on New Vertebral Fractures in Postmenopausal Women With Osteoporosis: A Randomized Clinical Trial. JAMA (2016) 316:722–33. doi: 10.1001/jama.2016.11136
184. Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, et al. Romosozumab Treatment in Postmenopausal Women With Osteoporosis. N Engl J Med (2016) 375:1532–43. doi: 10.1056/NEJMoa1607948
185. Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, et al. Romosozumab or Alendronate for Fracture Prevention in Women With Osteoporosis. N Engl J Med (2017) 377:1417–27. doi: 10.1056/NEJMoa1708322
186. Thomasius F, Baum E, Bernecker P, Böcker W, Brabant T, Clarenz P, et al. DVO Leitlinie 2017 Zur Prophylaxe, Diagnostik Und Therapie Der Osteoporose Bei Postmenopausalen Frauen Und Männern. Osteologie (2018) 27:154–60. doi: 10.1055/s-0038-1673537
Keywords: glucocorticoids, osteoblasts, osteocytes, glucocorticoid-induced osteoporosis (GIO), anti-resorptive treatment, osteo-anabolic treatment
Citation: Gado M, Baschant U, Hofbauer LC and Henneicke H (2022) Bad to the Bone: The Effects of Therapeutic Glucocorticoids on Osteoblasts and Osteocytes. Front. Endocrinol. 13:835720. doi: 10.3389/fendo.2022.835720
Received: 14 December 2021; Accepted: 10 February 2022;
Published: 31 March 2022.
Edited by:
Nicole Horwood, University of East Anglia, United KingdomReviewed by:
Alexander Rauch, University of Southern Denmark, DenmarkJan Tuckermann, University of Ulm, Germany
Copyright © 2022 Gado, Baschant, Hofbauer and Henneicke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holger Henneicke, aG9sZ2VyLmhlbm5laWNrZUB1a2RkLmRl