 Xiaoning Wang1,2†
Xiaoning Wang1,2† Xin Zhang2†
Xin Zhang2† Yidan Han2†
Yidan Han2† Xinwei Duan2Jianchang Wang1Hui Yan1
Xinwei Duan2Jianchang Wang1Hui Yan1 Shanshan Wang1
Shanshan Wang1 Yunteng Xu2Zaishi Zhu2Lili Wang2,3
Yunteng Xu2Zaishi Zhu2Lili Wang2,3 Yanfeng Huang2Qing Lin2
Yanfeng Huang2Qing Lin2 Xue Tan2
Xue Tan2 Junkuan Zhuo2
Junkuan Zhuo2 Haifeng Zhang2Min Mao2Weiying Gou2Zhouping Yi2
Haifeng Zhang2Min Mao2Weiying Gou2Zhouping Yi2 Xihai Li1,3*
Xihai Li1,3*- 1College of Integrative Medicine, Fujian University of Traditional Chinese Medicine, Fuzhou, China
- 2Academy of Integrative Medicine, Fujian University of Traditional Chinese Medicine, Fuzhou, China
- 3Fujian Key Laboratory of Integrative Medicine on Geriatrics, Fujian University of Traditional Chinese Medicine, Fuzhou, China
Bone immunity regulates osteoclast differentiation and bone resorption and is a potential target for the treatment of postmenopausal osteoporosis (PMOP). The molecular network between bone metabolism and the immune system is complex. However, the molecular mechanism underlying the involvement of the major histocompatibility complex class II (MHC-II) molecule protein presentation pathway in PMOP remains to be elucidated. The MHC-II molecule is a core molecule of the protein presentation pathway. It is combined with the processed short peptide and presented to T lymphocytes, thereby activating them to become effector T cells. T-cell-derived inflammatory factors promote bone remodeling in PMOP. Moreover, the MHC-II molecule is highly expressed in osteoclast precursors. MHC-II transactivator (CIITA) is the main regulator of MHC-II gene expression and the switch for protein presentation. CIITA is also a major regulator of osteoclast differentiation and bone homeostasis. Therefore, we hypothesized that the MHC-II promotes osteoclast differentiation, providing a novel pathogenic mechanism and a potential target for the treatment of PMOP.
Introduction
Postmenopausal osteoporosis (PMOP), a major public health concern, is attributed to an imbalance of bone metabolism in postmenopausal women. The condition is characterized by decreasing bone strength and bone mineral density (1–3). There are several pathological mechanisms involved in the development of PMOP, such as imbalances in osteogenic/adipogenic differentiation of bone marrow mesenchymal stem cells (BMSCs), osteogenic and/or osteoclast (OC) coupling, and bone immunity. Among them, bone immunity imbalance has recently attracted more attention.
The relationship between bone metabolism disorder and the immune system is complex (4). Major histocompatibility complex class II molecules (MHC-II) play essential roles in the adaptive immune response and participate in the immune regulation of bone health (5–7). However, the molecular mechanism underlying the regulatory effects of MHC-II on bone metabolism imbalance remains unclear. Therefore, this review summarizes the currently available knowledge regarding this molecular mechanism. The following aspects are discussed. Activation of T and B lymphocytes requires MHC-II molecules, and loss of estrogen (E2) leads to the conversion of T cells to effector T cells (TE) and the chronic production of related inflammatory cytokines. In turn, TE promote bone remodeling by releasing inflammatory factors. The main cytokines produced by TE affect bone resorption and bone formation. MHC-II gene expression is inseparable from the regulation of MHC-II transactivator (CIITA) and phagolysosomal membrane integrity. Moreover, bioinformatics revealed the potential pathologic association between PMOP and T activation, as well as the protein presentation pathway. Understanding the mechanisms through which MHC-II molecules regulate bone homeostasis may facilitate the development of targeted drugs for the treatment of PMOP.
There are two types of MHC molecules involved in adaptive immune response in mammals, namely, MHC-I and MHC-II. The former is distributed in almost all nucleated cells in the body; the latter is distributed in professional antigen-presenting cells (APCs), such as monocytes/macrophages, dendritic cells (DCs), and B cells. Both types are tightly regulated and involved in the activation process of effector T lymphocytes (TE) (8–10).
MHC-II molecules as key immune molecules participating in bone remodeling
OC precursors highly express MHC-II molecules
As OC precursors, monocytes/macrophages are a type of professional APCs. Hence, it is more meaningful to investigate the effect of the MHC-II molecule protein presentation pathway on bone metabolism in PMOP.
Activation of T and B lymphocytes requires MHC-II molecules
How can the initial T cells and memory T cells (TM) be activated and induce inflammation? A series of processes are required for T cells to induce adaptive immune responses and inflammation. The initial activation of T cells and TM is followed by further proliferation and differentiation into TE. The activation of the initial T cells and TM requires signals from the interaction between the T-cell receptor and the MHC-antigenic peptide complex. MHC molecules monitor different proteolytic machineries in APCs and transport the hydrolyzed peptide to the surface of APCs for the identification and combination of initial T cells and TM (11–13). DCs are the most powerful APCs in the body, particularly mature DCs whose main feature is the high expression of MHC-II molecules. Therefore, the MHC-II molecular presentation pathway plays an important role in activating the initial T cells and TM to become TE and may be closely related to the pathogenesis of PMOP.
It is well established that extracellular proteins enter the APCs and participate in the formation of phagosomes. Phagosomes combine with intracellular lysosomes to form phagolysosomes, where proteins are hydrolyzed into short peptides. MHC-II molecule–peptide complexes migrate to the surface of APCs for identification by CD4+ T cells, thereby activating them to become T helper 1 (Th1), Th2, Th17, etc. It is established that Th2 provides CD40L for the activation of B lymphocytes. Activated T and B lymphocytes can produce interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), interleukin-17A (IL-17A), and receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) (14), which are important immune molecules involved in bone remodeling in PMOP. This evidence further shows that MHC-II molecules are closely related to the occurrence and development of PMOP.
E2 deficiency leads to conversion of T cells into TE and chronic production of related inflammatory cytokines
Recently, a new pathway has been described, indicating that E2 loss results in chronic production of TNF-α and IL-17 by converting TM into TE. IL-7 and IL-15 are involved in the process; both are mainly secreted by bone marrow dendritic cells (BMDCs), which are important APCs in the bone marrow (BM) (15). Animal experiments have shown the absence of bone loss in specially treated mice, in which TM cannot convert into TE (16).
Physiologically, E2 can induce apoptosis of BMDCs and TM through the Fas ligand pathway (15). In the absence of E2, BMDCs exist for a prolonged period of time. This effect leads to antigen-independent activation of TM to produce TNF-α and IL-17A (16). In general, the classical activation and conversion of TM into TE require antigen stimulation (17). However, the activation induced by loss of E2 differs from that model (15), and this difference may be associated with chronic inflammation in PMOP. We hypothesize that, in postmenopausal women, the lifespan of DCs and TM is extended with the decline in E2. This further contributes to the conversion of initial T cells and TM activation into TE, resulting in the release of various inflammatory factors and causing PMOP.
TE promotes bone remodeling by releasing inflammatory factors
The adaptive immune system is fundamental to the progression of PMOP (15). Adaptive immune responses are composed of cellular immunity and humoral immunity. The former is mainly mediated by T lymphocytes, while the latter is mainly mediated by B lymphocytes.
The deficiency of E2 promotes persistent inflammation, which is conducive to the development of PMOP. Mechanistic studies on the relationship between menopausal E2 loss and activation of T cells have primarily been performed in rodents with ovariectomy (OVX); the key results obtained from these investigations have been verified in human studies (15). Bone loss was decreased in T-cell-deficient mature mice with OVX, demonstrating that T cells are required to promote bone resorption in PMOP (18–22). TE can secrete numerous cytokines, such as IFN-γ, TNF-α, and IL-17A.
Main cytokines produced by TE affect bone resorption
OCs are multinucleated giant cells formed by the fusion of multiple mononuclear macrophages differentiated from myeloid progenitor cells in the BM. OCs appear to be sensitive to cytokines produced by TE, such as IFN-γ, TNF-α, and IL-17A.
IFN-γ derived from TE can regulate the RANKL signal pathway during OC differentiation (20). Th1 cells are one of the main TE in T-cell immunity and the major producers of IFN-γ. Initially, bone loss due to inflammation was attributed to a Th1-mediated pathological process. However, it was later demonstrated that Th17 cells are the main drivers of bone loss (23).
It has been reported that mature monocytes/macrophages differentiate into OCs in an IFN-γ-rich microenvironment and promote cell fusion (24). Moreover, IFN-γ could readily induce monocyte aggregation, leading to the formation of multinuclear giant cells (25). In addition, it has been shown that TNF-α can directly act on OCs and their precursors, and it cooperates with the RANKL signal pathway in osteoclastogenesis (26–29). TNF-α can recruit TNF receptor-associated factors to sequentially activate NF-κB p50, and, c-Fos, and nuclear factor of activated T cells 1 (NFATC1) for the promotion of OC differentiation. This process is similar to and independent of the RANKL pathway (30). Blockade of the pathways by which lymphocytes migrate to the BM reduced the levels of TNF-α and Th17 cells in the BM after OVX in mice. These effects were accompanied by trabecular bone loss in this model (31).
E2 deficiency can increase the number of Th17 cells and TNF-α-producing T cells in the BM, and this process is dependent on the gut microbiome. Subsequently, BM IL-17A and TNF-α stimulate RANKL expression and activity, causing bone loss. Demonstrating the functional relevance of T-cell trafficking, blockade of Th17 cells and TNFα-producing T cells from the gut or their influx into the BM prevented OVX-induced bone loss. Therefore, it can be concluded that T cells in the gut are proximal targets of E2 deficiency-induced bone loss in PMOP (32).
Additionally, as an immune cytokine, IL-17A participates in the regulation of bone remodeling (15). IL-17A is mainly secreted by a special subtype of TE, namely, Th17 cells (33), and promotes bone destruction (34–36). Of note, there is a one-quarter amino acid sequence homology of IL-17A between humans and mice (37). It is thought that IL-17A participates in inflammation and may be mainly derived from the activated memory CD4+ T cells (TM), which subsequently differentiate into Th17 cells (37, 38). The local cytokine environment can promote or protect against bone loss. In addition, the mechanisms through which TNF-α and IL-17A affect bone metabolism via OCs have been studied extensively (39, 40).
IL-17A signaling plays a role through the IL-17A receptor (IL-17AR); however, the role of IL-17A signaling in OCs remains elusive (41). Different concentrations of IL-17A exert varied effects on OCs. A low concentration of IL-17A (0.5 ng/ml) can promote OC differentiation via RANKL-JUN N-terminal kinase (RANKL-JNK) signaling and reduce the apoptosis of OCs through the RANKL–beclin 1 (BECN1)–autophagy–TRAF3 pathway. IL-17A increases the number of OC precursors to influence subsequent RANKL-dependent OC differentiation. However, a high concentration of IL-17A (5–50 ng/ml) could inhibit OC differentiation and stimulate the apoptosis of OCs via the two aforementioned pathways (42, 43). Interestingly, a higher concentration of IL-17A (100 ng/ml) increases the number of OC precursors and induces OC formation (34). IL-17A also indirectly targets the OC-supporting cells, such as BMSCs, osteoblasts (OBs), and osteocytes, to produce various cytokines and molecules for the regulation of OC differentiation (4). Binding of IL-17A to its receptor IL-17AR on pre-OC triggers Act1 adaptor protein and may activate the downstream Janus kinase 2-signal transducer and activator of transcription 3 (JAK2-STAT3) signal, which can promote RANKL expression (44–47). The upregulation of RANKL and the increase in the RANKL/osteoprotegerin ratio could promote OC differentiation.
Theories on the activity of IL-17A in OC differentiation remain controversial; thus, it is imperative to explore the specific underlying mechanisms (48). Specific subtypes of TE express TNF-α, which increases OB apoptosis and indirectly stimulates osteoclastogenesis via B-cell-produced RANKL, thereby triggering bone loss during PMOP (49).
The main cytokines derived from TE show different effects on bone formation
As discussed above, inflammation affects bone resorption. However, studies on the role of inflammatory factors in restraining bone formation are currently limited. Physiologically, under coupled bone remodeling conditions, there is a dynamic balance between bone resorption and formation. For example, increasing resorption is accompanied by the recruitment of BMSCs and their conversion into OBs (15). However, this process appears to be impaired in the presence of inflammatory cytokines (i.e., TNF-α and IL-17). Therefore, bone formation is reduced versus bone resorption, which is consistent with the pathological mechanism of PMOP. OBs and BMSCs are sensitive to TNF-α and IL-17A.
TNF-α can inhibit bone formation by suppressing OB differentiation. It can inhibit the expression of osterix (OSX) and runt-related transcription factor 2 (RUNX2), which are vital to OB differentiation (50, 51). RUNX2 is a specific transcription factor that could commit BMSCs to the OB pathway. It has been demonstrated that TNF-α could restrict the differentiation of BMSCs into OBs by regulating RUNX2 expression (15). OSX is another key transcription factor for OB maturation, and TNF-α also can target OSX expression (52–54). Furthermore, OB differentiation can be regulated via the mechanistic target of rapamycin (mTOR) pathway (55–58). Studies have shown that TNF-α can preferentially regulate cellular metabolism in adipocytes and muscle cells (59–61). OBs, adipocytes, and muscle cells originate from BMSCs through different directions of differentiation. The mechanism by which TNF-α regulates OB cellular metabolism is currently unknown. In-vitro studies have shown that TNF-α can regulate autophagy and apoptosis via the NF-κB signal pathway in OBs (62–64), both of which are controlled by the mTOR.
In ankylosing spondylitis, a study on human pre-OB has indicated that IL-17A could promote bone-derived cells to differentiate into OBs through the JAK2/STAT3 signal pathway (65). IL-17A can promote the differentiation of BMSCs into OBs and the mineralization of OBs by upregulating the expression of bone formation-related gene alkaline phosphatase and RUNX2 (66). IL-17A and bone morphogenetic protein 2 (BMP2) could promote the osteogenic differentiation of BMSCs (67). OBs and adipocytes are both differentiated from a common pluripotent precursor, namely, BMSCs. The decision for the differentiation of BMSCs into OBs or adipocytes is delicately balanced and there is competition. IL-17A may steer BMSCs into OBs. Moreover, it can activate cyclooxygenase 2 (COX2)-induced prostaglandin E2 (PGE2) to inhibit lipid-related proteins, such as peroxisome proliferator-activated receptor gamma (PPARγ) and adiponectin. This process leads to a reduction in the differentiation of BMSCs into adipocytes (68). Therefore, IL-17A may exert different effects on OCs and OBs and can induce extensive bone turnover in PMOP.
It has been demonstrated that IL-17A could affect the differentiation of BMSCs into OBs and the functions of mature OBs (40). Th17 cells release IL-17A, which directs mesenchymal stem cell differentiation toward the osteogenic lineage but also indirectly increases OC differentiation (49).
In summary, bone metabolism is sensitive to chronic inflammation induced by the activation of T cells in PMOP. Specifically, deficiency of E2 promotes the conversion of TM into TE in the BM.
Key aspects of MHC-II molecule protein complexes presented to T cells
Proteins enter APCs and participate in the formation of phagosomes. Phagosomes combine with intracellular lysosomes to form phagolysosomes, where proteins are hydrolyzed into short peptides. Meanwhile, MHC-II molecules synthesized in the endoplasmic reticulum are transported to MHC class II-containing compartments, where peptides are loaded in the peptide-binding groove of MHC-II molecules (69). Phagolysosomes associate with MHC class II-containing compartments to form terminal lysosomes in APCs. MHC-II molecule–peptide complexes migrate to the cell surface for identification by CD4+ T cells and activation of adaptive immune responses. Therefore, the expression of MHC-II molecules and the integrity of the phagolysosomal membrane are critical for the presentation of the MHC-II molecule protein complexes to T cells.
Expression of MHC-II genes is dependent on the regulation of CIITA
It has been shown that CIITA is a master regulator of MHC-II genes in APCs, which are critical for the activation of T cells and the induction of adaptive immune response (8, 9).
Under physiological conditions, CIITA is the master regulator of MHC-II genes (8, 9). It is a non-DNA-binding co-activator, which can specifically regulate the expression of MHC-II molecules. CIITA deficiency could result in rare human immunodeficiency disease (70). Overexpression of CIITA induces severe spontaneous osteoporosis by an increase in the number of OCs and bone resorption (7).
The classical MHC-II molecules in humans (HLA-DR, HLA-DP, and HLA-DQ) are the major target genes of CIITA. The factors which regulate MHC-II expression play roles via the promoters that drive transcription of the MHC2TA gene encoding CIITA. As OC precursors, macrophages constitutively and highly express MHC-II molecules (8, 9). In the presence of low levels of CIITA, the synthesis of MHC-II molecules can be limited, and the presentation of MHC-II molecule proteins in DCs (a type of APC) is impaired (18). The expression of genes which encode accessory proteins required for MHC-II molecule protein presentation can also be regulated by CIITA. Thus, CIITA is a central regulator controlling the response to proteins that will be processed and the maintenance of tolerance in the immune system (8, 9). This coordinated regulation of MHC-II and other genes necessary for its function is unique; hence, CIITA has been termed the “master regulator” of MHC-II molecules and the protein presentation pathway (8, 9).
Therefore, it is hypothesized that overexpression of CIITA will lead to overexpression of MHC-II molecules, which in turn can cause overactivation of the protein presentation pathway. Subsequently, more initial T cells and TM are activated to become TE, resulting in excessive immune response and inflammation in PMOP. As monocytes/macrophages are a type of professional APCs, we hypothesized that the relationship between CIITA and MHC-II may be in the OC precursors.
Phagolysosomal membrane integrity determines whether MHC-II molecule–peptide complexes can be presented
Phagolysosome is a critical endocytic organelle in the MHC-II molecular protein presentation pathway. Its membrane integrity determines whether MHC-II molecule–peptide complexes can be presented. The rupture of the membrane before the presentation of the MHC-II molecule–peptide complexes will lead to APC death and lack of T-cell activation.
Following the completion of the MHC-II molecule–peptide complexes, the membrane of the phagolysosome fuses with the cell membrane. At the same time, the peptide-binding region of the MHC-II molecule can bind to processed peptides, and its immunoglobulin-like region can be specifically recognized by CD4 molecules expressed on T cells.
The effect of phagolysosomal membrane integrity on the MHC class II molecular protein presentation pathway is critical. However, the mechanism regulating the integrity of the phagolysosomal membrane is unknown. The maintenance of phagolysosomal membrane integrity is regulated by numerous factors, such as the osmotic control of membrane tension, lipid bilayer modifications and renitence vacuoles, and membrane-stabilizing proteins (71).
The membrane-stabilizing proteins are of particular interest. It has been reported that the Bin–amphiphysin–Rvs (BAR) domain-containing protein family plays important roles in scaffolding and stabilizing the curved membranes (72). These proteins will increase the surface of the endocytic organelle, thereby facilitating the rapid export of osmolytes that can diffuse into the tubules and access the membrane solute carriers (73, 74).
Mature endocytic organelles, including phagolysosomes, require transmembrane proteins to protect the membrane from the harsh luminal environment (75, 76). The lysosome-associated membrane proteins (LAMP1 and LAMP2) and the lysosome integral membrane protein 2 (LIMP2) have been well investigated (76, 77). Loss of LAMP1 and LAMP2 does not affect the lysosomal membrane integrity (78, 79). In contrast, loss of LIMP2 results in severe damage to the lysosomes (76). The homolog of the human LIMP2, SCAV-3, has been identified as an important regulator of lysosome integrity (76). Loss of SCAV-3 can lead to the rupture of lysosome membranes. Therefore, SCAV-3 is vital for preserving lysosomal membrane stability. Notably, modulation of lysosome integrity by the insulin/insulin-like growth factor 1 (insulin/IGF1) signaling pathway affects longevity (76).
In summary, we conclude that phagolysosomal membrane integrity determines whether MHC-II molecule–peptide complexes can be presented to T cells. This affects their activation and conversion into TE, thereby influencing the release of numerous cytokines and the development of PMOP.
MHC-II may promote OC differentiation
It has been demonstrated that CIITA is a key regulator of OC differentiation and bone remodeling (6). Previous studies have shown that CIITA exerts an indirect effect on bone homeostasis during E2 deficiency-induced bone loss, which may be associated with its effects on protein presentation. In OVX mice, an increase in the expression of CIITA increased the expression of MHC-II molecules and enhanced activation-induced T-cell proliferation (75). Hence, it is vital to activate T cells for bone loss caused by OVX, and CIITA could be regulated by the presence of E2. The regulation of CIITA is dependent on IFN-γ, because OVX leads to increased levels of IFN-γ derived from TE. Of note, it has been shown that CIITA was not upregulated in IFN-γR-/p-mice (80). Therefore, it would be meaningful to examine the expression of CIITA in OC precursors during PMOP.
CIITA is a key regulator of the activation of T cells and should be considered an important factor in the relationship between the immune response and bone health (7). However, the effect of the MHC-II molecule on OC differentiation is currently unknown. We hypothesize that MHC-II molecules could also promote OC differentiation.
Bioinformatics revealed a potential pathologic association between PMOP and T activation, as well as the protein presentation pathway
In the article, we used bioinformatics analysis methods (Supplementary 1) to integrate multiple databases for the screening of different genes involved in PMOP. Next, we performed enrichment analysis of the Kyoto Encyclopedia of Genes and Genomes pathway. Of the top 30 pathways, some pathways associated with immunity in PMOP were enriched (e.g., cytokine–cytokine receptor interaction, JAK-STAT signaling pathway, Th17 cell differentiation, OC differentiation, T-cell receptor signaling pathway, and TNF signaling pathway) (Figure 1A). The top 20 Gene Ontology enrichment candidate targets of the different genes associated with immunity in PMOP are shown in Figure 1B. There was a pathologic crosstalk of core cytokine networks involved in PMOP and immunity. Gene Ontology functional enrichment analysis of common differentially expressed genes in PMOP and immunity was performed, including the cellular component. Figure 1B shows the enrichment of the MHC-II protein complex.
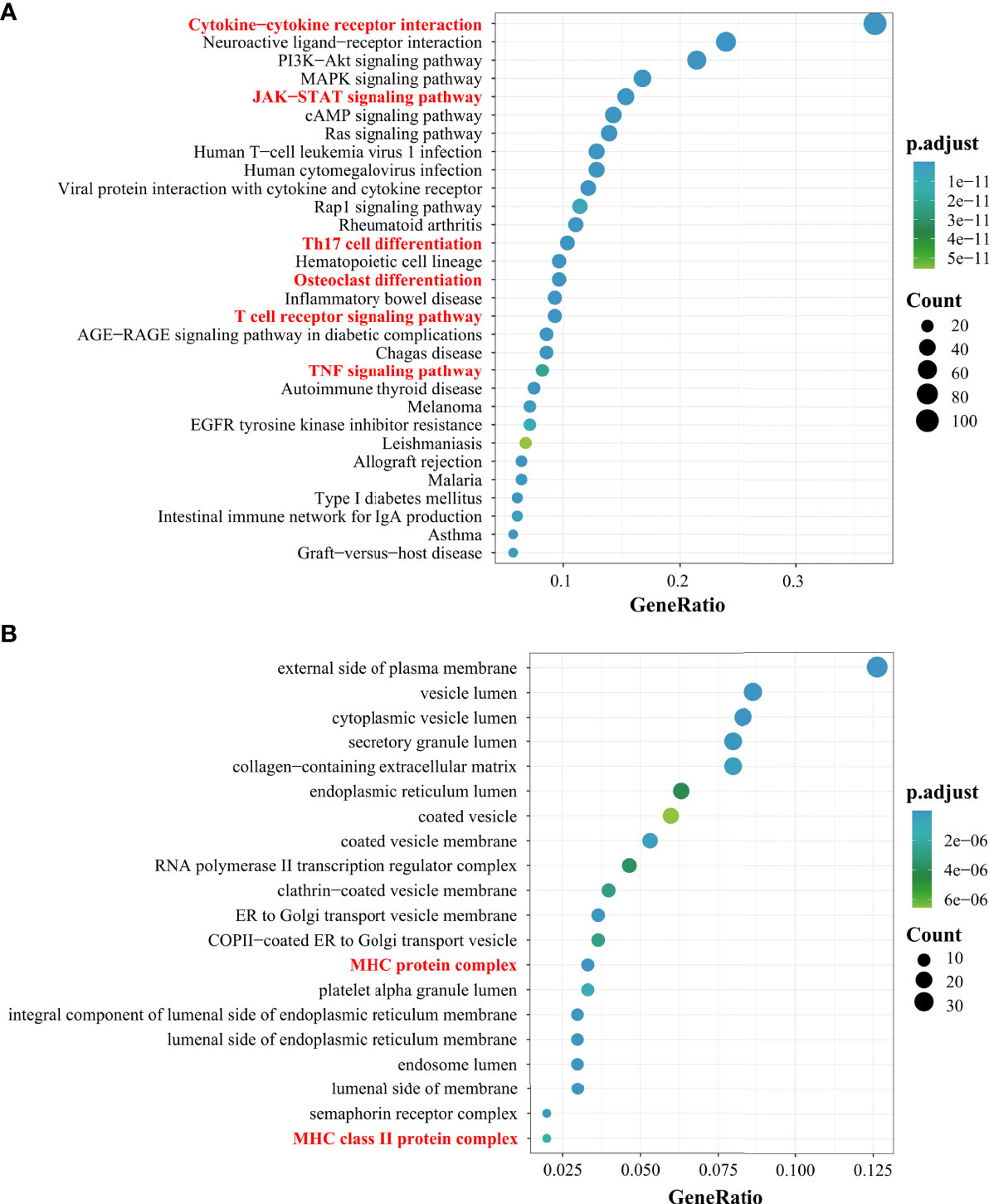
Figure 1 Results of the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. The top 30 KEGG pathway enrichment candidate targets of the target genes (A). Pathways with significant changes (FDR < 0.05) were identified. The vertical coordinates represent the KEGG pathway with significant enrichment, and the horizontal coordinates represent the gene ratio which refers to the ratio of enriched genes to all target genes. The top 20 GO enrichment candidate targets of the target genes (B). The color of the bubble graph indicates the categories of “cellular components” in the GO of the target genes (FDR < 0.05), and the horizontal coordinates represent the gene ratio which refers to the ratio of enriched genes to all target genes.
In addition, protein–protein interaction network topology analysis was conducted to identify common differentially expressed genes in PMOP and immunity genes (Figure 2). IL-17A, TNF, and IFN are derived from TE; IGF is associated with the membrane integrity of phagolysosomes; PPAR is associated with the differentiation of BMSCs into adipocytes; CD40L can be expressed only on activated T cells; and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) can provide inhibitory information for T-cell activation.
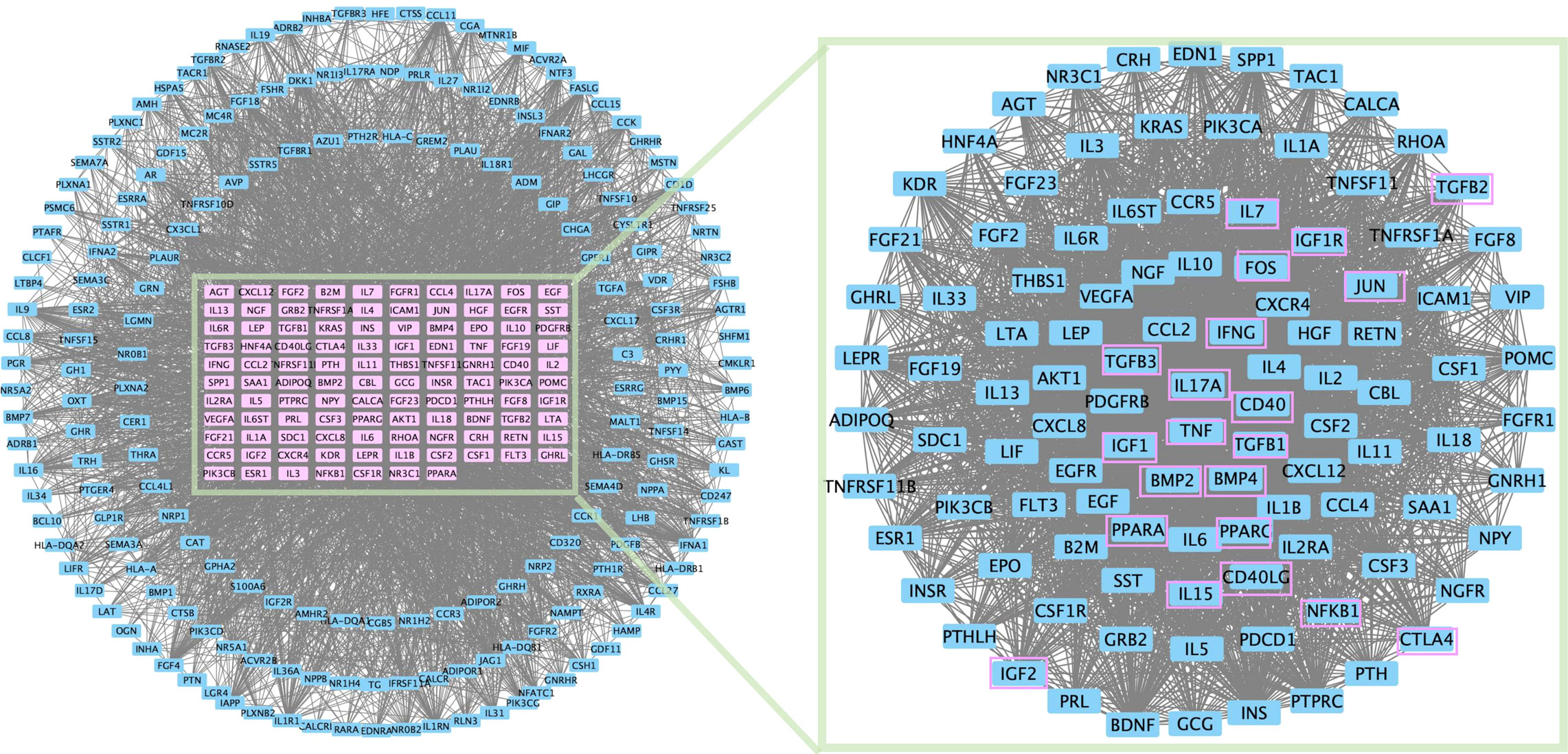
Figure 2 PPI network topology analysis was conducted for common differential genes in PMOP differential genes and immunity genes.
These results indicate that MHC-II molecules may promote OC differentiation and play crucial roles in the development of PMOP. They mainly act through various cytokines produced by TE cells, some may act on osteoblasts, and some may act on osteoclasts (Figure 3).
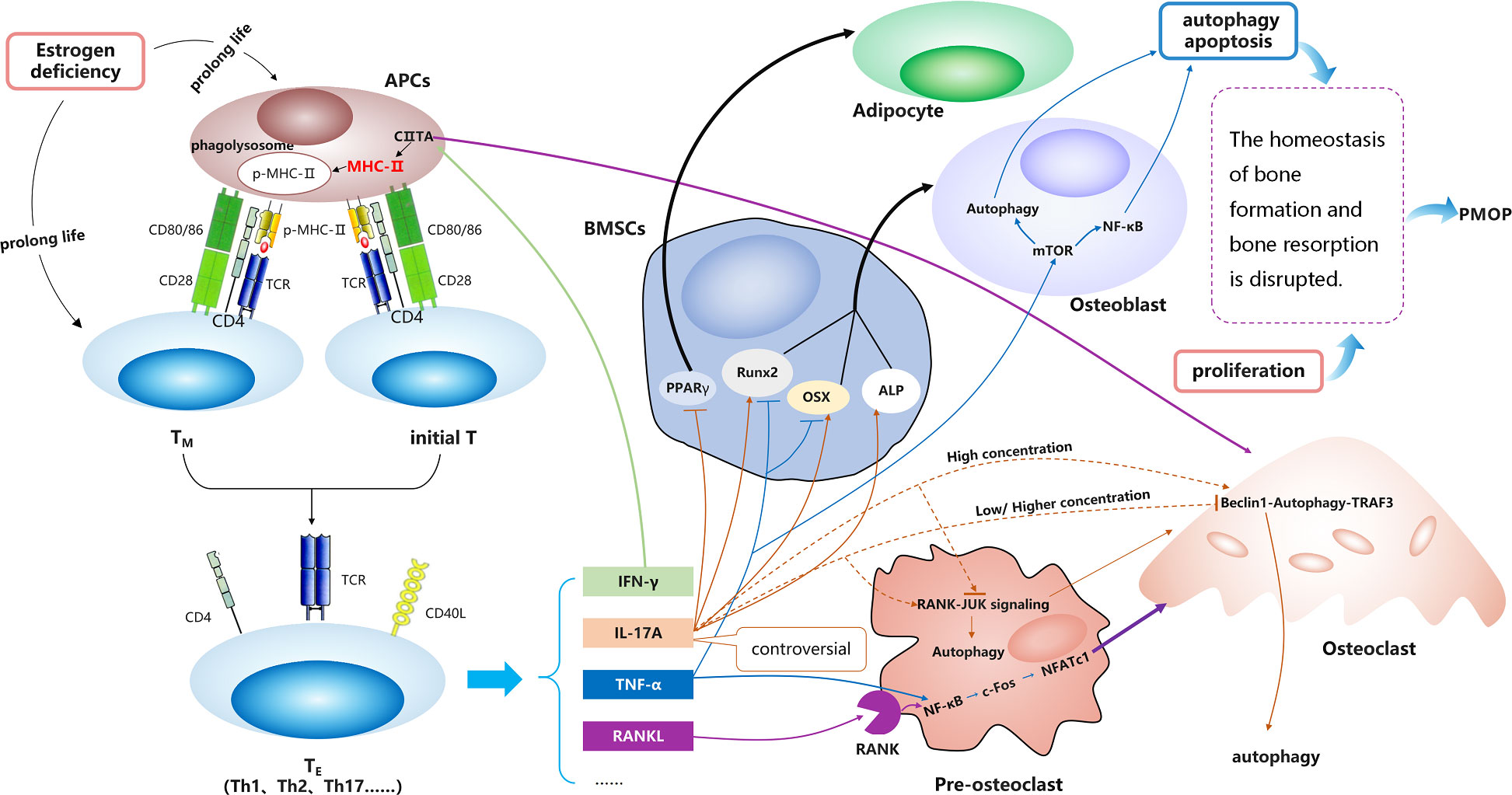
Figure 3 The relationship between MHC-II and PMOP.
Conclusion
This review focused on the MHC-II molecular protein presentation pathway. The phagolysosomal membrane integrity and CIITA are critical to the MHC-II molecular protein presentation pathway. The evidence suggests that MHC-II molecules play a key role in OC differentiation, providing a new direction for revealing the pathological mechanism underlying the development of PMOP. Such knowledge may provide potential therapeutic targets for the prevention and treatment of PMOP. According to the network pharmacology analysis, the common differential genes in PMOP differential genes and immunity genes were found. However, which genes are upregulated and which are downregulated and their specific roles in bone immunity need to be further explored.
Author contributions
The manuscript of this review was composed by XW, XZ, Y-DH, XD, JW, HY, SW, YX, ZZ, LW, Y-FH, QL, XT, JZ, HZ, MM, WG, and ZY under the guidance and advice of XL. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (No. 82074461), the Natural Science Foundation of Fujian Province (No. 2020J01719), and the Chen Keji Integrated Chinese and Western Medicine Development Fund-supported projects (No.CKJ2021012).
Acknowledgments
We would like to thank the two reviewers for their helpful suggestions which helped us to improve the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.876067/full#supplementary-material
References
1. Tella SH, Gallagher JC. Prevention and treatment of postmenopausal osteoporosis. J Steroid Biochem Mol Biol (2014) 142:155e70. doi: 10.1016/j.jsbmb.2013.09.008
2. Li L, Chen X, Lv S, Dong M, Zhang L, Tu J, et al. Influence of exercise on bone remodeling-related hormones and cytokines in ovariectomized rats: a model of postmenopausal osteoporosis. PloS One (2014) 9:e112845. doi: 10.1371/journal.pone.0112845
3. Kwun S, Laufgraben MJ, Gopalakrishnan G. Prevention and treatment of postmenopausal osteoporosis. Obstet Gynaecol (2012) 14:251e6. doi: 10.1111/j.1744-4667.2012.00135.x
4. Tang M, Lu L, Yu X. Interleukin-17A interweaves the skeletal and immune systems. Front Immunol (2021) 11:625034. doi: 10.3389/fimmu.2020.625034
5. Letendre C, Auger J-P, Lemire P, Galbas T, Gottschalk M, Thibodeau J, et al. Streptococcus suis serotype 2 infection impairs interleukin-12 production and the MHC-II-Restricted antigen presentation capacity of dendritic cells. Front Immunol (2018) 9:1199. doi: 10.3389/fimmu.2018.01199
6. Benasciutti E, Mariani E, Oliva L, Scolari M, Perilli E, Barras E, et al. MHC class II transactivator is an in vivo regulator of osteoclast differentiation and bone homeostasis co-opted from adaptive immunity. J Bone Miner Res (2014) 29(2):290–303. doi: 10.1002/jbmr.2090
7. Mary C, Nakamura MD. CIITA: a master regulator of adaptive immunity shows its innate side in the bone. J Bone Miner Res (2014) 29(2):287–89. doi: 10.1002/jbmr.2161
8. Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol (2005) 5:793–806. doi: 10.1038/nri1708
9. Krawczyk M, Reith W. Regulation of MHC class II expression, a unique regulatory system identified by the study of a primary immunodeficiency disease. Tissue Antigens (2006) 67:183–97. doi: 10.1111/j.1399-0039.2006.00557.x
10. Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, et al. Structure of the human MHC-I peptide-loading complex. Nature (2017) 551:525–28. doi: 10.1038/nature24627
11. Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol (2005) 23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538
12. Dersh D, Holly J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol (2020) 20:644. doi: 10.1038/s41577-020-00445-3
13. Moore TV, Nishimura MI. Improved MHC II epitope prediction - a step towards personalized medicine. Nat Rev Clin Oncol (2020) 17:71–2. doi: 10.1038/s41571-019-0315-0
14. Neale Weitzmann M. Bone and the immune system. Toxicol Pathol (2017) 45(7):911–24. doi: 10.1177/0192623317735316
15. Wu D, Cline-Smith A, Shashkova E, Perla A, Katyal A, Aurora R. T-Cell mediated inflammation in postmenopausal osteoporosis. Front Immunol (2021) 12:687551. doi: 10.3389/fimmu.2021.687551
16. Cline-Smith A, Axelbaum A, Shashkova E, Chakraborty M, Sanford J, Panesar P, et al. Ovariectomy activates chronic low-grade inflammation mediated by memory T-cells which promotes osteoporosis in mice. J Bone Miner Res (2020) 35(6):1174–87. doi: 10.1002/jbmr.3966
17. MacLeod MK, Kappler JW, Marrack P. Memory CD4 T cells: Generation, reactivation and re-assignment. Immunology (2010) 130(1):10–5. doi: 10.1111/j.1365-2567.2010.03260.x
18. Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y-H, Qian W-P, et al. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci USA (2003) 100(18):10405–10. doi: 10.1073/pnas.1533207100
19. Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest (2000) 106(10):1229–37. doi: 10.1172/JCI11066
20. Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, et al. UpRegulation of TNF-producing T cells in the bone marrow: A key mechanism by which estrogen deficiency induces bone loss In vivo. Proc Natl Acad Sci USA (2001) 98(24):13960–65. doi: 10.1073/pnas.251534698
21. Roggia C, Tamone C, Cenci S, Pacifici R, Isaia GC. Role of TNF-alpha producing T-cells in bone loss induced by estrogen deficiency. Minerva Med (2004) 95(2):125–32.
22. Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: An inflammatory tale. J Clin Invest (2006) 116(5):1186–94. doi: 10.1172/JCI28550
23. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med (2006) 203(12):2673–82. doi: 10.1084/jem.20061775
24. Kim JW, Lee MS, Lee CH, Kim HY, Chae SU, Kwak HB, et al. Effect of interferon-gamma on the fusion of mononuclear osteoclasts into bone-resorbing osteoclasts. BMB Rep (2012) 45(5):281–86. doi: 10.5483/BMBRep.2012.45.5.281
25. Biros E, Malabu UH, Vangaveti VN, Birosova E, Moran CS. The IFN-γ/miniTrpRS signaling axis: An insight into the pathophysiology of osteoporosis and therapeutic potential. Cytokine Growth Factor Rev (2022) 64:7–11. doi: 10.1016/j.cytogfr.2022.01.005
26. Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J Exp Med (2012) 209(2):319–34. doi: 10.1084/jem.20111566
27. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-a induces differentiation of and bone resorption by osteoclasts. J Biol Chem (2000) 275(7):4858–64. doi: 10.1074/jbc.275.7.4858
28. Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor a stimulates osteoclast differentiation by a mechanism independent of the Odf/Rankl–rank interaction. J Exp Med (2000) 191(2):275–86. doi: 10.1084/jem.191.2.275
29. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-a induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest (2000) 106(12):1481–88. doi: 10.1172/JCI11176
30. Yao Z, Getting SJ, Locke IC. Regulation of TNF-induced osteoclast differentiation. Cells (2021) 11(1):132. doi: 10.3390/cells11010132
31. Lorenzo J. From the gut to bone: connecting the gut microbiota with Th17 T lymphocytes and postmenopausal osteoporosis. J Clin Invest (2021) 131(5):e146619. doi: 10.1172/JCI146619
32. Yu M, Pal S, Paterson CW, Li JY, Tyagi AM, Adams J, et al. Ovariectomy induces bone loss via microbial-dependent trafficking of intestinal TNF+ T cells and Th17 cells. J Clin Invest (2021) 131(4):e143137. doi: 10.1172/JCI143137
33. Miossec P. IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect (2009) 11(5):625–30. doi: 10.1016/j.micinf.2009.04.003
34. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: A new candidate in the pathogenesis of osteoporosis. PloS One (2012) 7(9):e44552. doi: 10.1371/journal.pone.0044552
35. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
36. Zhao R, Wang X, Feng F. Upregulated cellular expression of IL-17 by CD4+ T-cells in osteoporotic postmenopausal women. Ann Nutr Metab (2016) 68(2):113–18. doi: 10.1159/000443531
37. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff B M, Spriggs MK, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol (1995) 155(12):5483–86.
38. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T Cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med (1996) 183(6):2593–603. doi: 10.1084/jem.183.6.2593
39. Zhao B. TNF and bone remodeling. Curr Osteoporos Rep (2017) 15(3):126–34. doi: 10.1007/s11914-017-0358-z
40. Tang M, Lu L, Yu X. Interleukin-17A interweaves the skeletal and immune systems. Front Immunol (2021) 11(3841):625034. doi: 10.3389/fimmu.2020.625034
41. Roberts JL, Mella-Velazquez G, Dar HY, Liu G, Drissi H. Deletion of IL-17ra in osteoclast precursors increases bone mass by decreasing osteoclast precursor abundance. Bone (2022) 157:116310. doi: 10.1016/j.bone.2021.116310
42. Ke D, Fu X, Xue Y, Wu H, Zhang Y, Chen X, et al. IL-17A regulates the autophagic activity of osteoclast precursors through RANKL-JNK1 signaling during osteoclastogenesis in vitro. Biochem Biophys Res Commun (2018) 497(3):890–96. doi: 10.1016/j.bbrc.2018.02.164
43. Xue Y, Liang Z, Fu X, Wang T, Xie Q, Ke D. IL-17A modulates osteoclast precursors’ apoptosis through autophagy-TRAF3 signaling during osteoclastogenesis. Biochem Biophys Res Commun (2019) 508(4):1088–92. doi: 10.1016/j.bbrc.2018.12.029
44. DeSelm CJ, Takahata Y, Warren J, Chappel JC, Khan T, Li X, et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem (2012) 113(9):2895–902. doi: 10.1002/jcb.24165
45. Zhang F, Wang CL, Koyama Y, Mitsui N, Shionome C, Sanuki R, et al. Compressive force stimulates the gene expression of IL-17s and their receptors in MC3T3-E1 cells. Connective Tissue Res (2010) 51(5):359–69. doi: 10.3109/03008200903456942
46. Funaki Y, Hasegawa Y, Okazaki R, Yamasaki A, Sueda Y, Yamamoto A, et al. Resolvin E1 inhibits osteoclastogenesis and bone resorption by suppressing IL-17-induced RANKL expression in osteoblasts and RANKL-induced osteoclast differentiation. Yonago Acta Med (2018) 61(1):8–18. doi: 10.33160/yam.2018.03.002
47. Wang Z, Tan J, Lei L, Sun W, Wu Y, Ding P, et al. The positive effects of secreting cytokines IL-17 and IFN-g on the early-stage differentiation and negative effects on the calcification of primary osteoblasts in vitro. Int Immunopharmacol (2018) 57:1–10. doi: 10.1016/j.intimp.2018.02.002
48. Song L, Tan J, Wang Z, Ding P, Tang Q, Xia M, et al. Interleukin-17A facilitates osteoclast differentiation and bone resorption via activation of autophagy in mouse bone marrow macrophages. Mol Med Rep (2019) 19(6):4743–52. doi: 10.3892/mmr.2019.10155
49. Fischer V, Haffner-Luntzer M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. Semin Cell Dev Biol (2022) 123:14–21. doi: 10.1016/j.semcdb.2021.05.014
50. Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-alpha on bone homeostasis. Front Immunol (2014) 5:48. doi: 10.3389/fimmu.2014.00048
51. Algate K, Haynes DR, Bartold PM, Crotti TN, Cantley MD. The effects of tumour necrosis factor-alpha on bone cells involved in periodontal alveolar bone loss; osteoclasts, osteoblasts and osteocytes. J Periodontal Res (2016) 51:549–66. doi: 10.1111/jre.12339
52. Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology (2000) 141(11):3956–64. doi: 10.1210/endo.141.11.7739
53. Gilbert L, He X, Farmer P, Rubin J, Drissi H, Wijnen AJ, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/Aml3/ Pebp2alpha a) is inhibited by tumor necrosis factor-alpha. J Biol Chem (2002) 277(4):2695–701. doi: 10.1074/jbc.M106339200
54. Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa b pathways. J Biol Chem (2006) 281(10):6297–306. doi: 10.1074/jbc.M507804200
55. Chen J, Long F. mTORC1 signaling promotes osteoblast differentiation from preosteoblasts. PloS One (2015) 10(6):e0130627. doi: 10.1371/journal.pone.0130627
56. Chen J, Holguin N, Shi Y, Rubin J, Nanes MS. mTORC2 signaling promotes skeletal growth and bone formation in mice. J Bone Miner Res (2015) 30(2):369–78. doi: 10.1002/jbmr.2348
57. Fitter S, Matthews MP, Martin SK, Xie J, Ooi SS, Walkley CR, et al. mTORC1 plays an important role in skeletal development by controlling preosteoblast differentiation. Mol Cell Biol (2017) 37(7):e00668–16. doi: 10.1128/MCB.00668-16
58. Schaub T, Gurgen D, Maus D, Lange C, Tarabykin V, Dragun D, et al. mTORC1 and mTORC2 differentially regulate cell fate programs to coordinate osteoblastic differentiation in mesenchymal stromal cells. Sci Rep (2019) 9(1):20071. doi: 10.1038/s41598-019-56237-w
59. Ciaraldi TP, Carter L, Mudaliar S, Kern PA, Henry RR. Effects of tumor necrosis factor-a on glucose metabolism in cultured human muscle cells from nondiabetic and type 2 diabetic subjects. Endocrinology (1998) 139(12):4793–800. doi: 10.1210/endo.139.12.6368
60. Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factor-a induces skeletal muscle insulin resistance in healthy human subjects Via inhibition of akt substrate 160 phosphorylation. Diabetes (2005) 54(10):2939–45. doi: 10.2337/diabetes.54.10.2939
61. Hauner H, Petruschke T, Russ M, Röhrig K, Eckel J. Effects of tumour necrosis factor alpha (Tnfa) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia (1995) 38(7):764–71. doi: 10.1007/s001250050350
62. Chen L, Bao J, Yang Y, Wang Z, Xia M, Tan J, et al. Autophagy was involved in tumor necrosis factor-Alpha-Inhibited osteogenic differentiation of murine calvarial osteoblasts through wnt/beta-catenin pathway. Tissue Cell (2020) 67:101401. doi: 10.1016/j.tice.2020.101401
63. Zheng LW, Wang WC, Mao XZ, Luo YH, Tong ZY, Li D, et al. TNF-alpha regulates the early development of avascular necrosis of the femoral head by mediating osteoblast autophagy and apoptosis via the P38 MAPK/NFkappaB signaling pathway. Cell Biol Int (2020) 44(9):1881–89. doi: 10.1002/cbin.11394
64. Zheng L, Wang W, Ni J, Mao X, Song D, Liu T, et al. Role of autophagy in tumor necrosis factor-Alpha-Induced apoptosis of osteoblast cells. J Investig Med (2017) 65(6):1014–20. doi: 10.1136/jim-2017-000426
65. Jo S, Wang SE, Lee YL, Kang S, Lee B, Han J, et al. IL-17A induces osteoblast differentiation by activating JAK2/STAT3 in ankylosing spondylitis. Arthritis Res Ther (2018) 20(1):115. doi: 10.1186/s13075-018-1582-3
66. Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ (2009) 16(10):1332–43. doi: 10.1038/cdd.2009.74
67. Croes M, Öner FC, van Neerven D, Sabir E, Kruyt MC, Blokhuis TJ, et al. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone (2016) 84:262–70. doi: 10.1016/j.bone.2016.01.010
68. Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates proinflammatory responses in adipocytes. Biochem Pharmacol (2009) 77(12):1835–44. doi: 10.1016/j.bcp.2009.03.008
69. Pos W, Sethi DK, Call MJ, et al. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell (2012) 151:1557–68. doi: 10.1016/j.cell.2012.11.025
70. Reith WMB. The bare lymphocyte syndrome and the regulation of mhc expression. Ann Rev Immunol (2001) 19:331–73. doi: 10.1146/annurev.immunol.19.1.331
71. Childs E, Henry CM, Canton J, Sousa CR. Maintenance and loss of endocytic organelle integrity: mechanisms and implications for antigen cross-presentation. Open Biol (2021) 11(11):210194. doi: 10.1098/rsob.210194
72. Simunovic M, Evergren E, Callan-Jones A, Bassereau P. Curving cells inside and out: roles of BAR domain proteins in membrane shaping and its cellular implications. Annu Rev Cell Dev Biol (2019) 35:111–29. doi: 10.1146/annurev-cellbio-100617-060558
73. Freeman SA, Grinstein S. Resolution of macropinosomes, phagosomes and autolysosomes: osmotically driven shrinkage enables tubulation and vesiculation. Traffic (2018) 19:965–74. doi: 10.1111/tra.12614
74. Linkner J, Witte G, Zhao H, Junemann A, Nordholz B, Runge-Wollmann P, et al. The inverse BAR domain protein IBARa drives membrane remodeling to control osmoregulation, phagocytosis and cytokinesis. J Cell Sci (2014) 127:1279–92. doi: 10.1242/jcs.140756
75. Fukuda M. Lysosomal membrane glycoproteins: structure, biosynthesis, and intracellular trafficking. J Biol Chem (1991) 266:21327–30. doi: 10.1016/S0021-9258(18)54636-6
76. Li Y, Chen B, Zou W, Wang X, Wu Y, Zhao D, et al. The lysosomal membrane protein SCAV-3 maintains lysosome integrity and adult longevity. J Cell Biol (2016) 215:167–85. doi: 10.1083/jcb.201602090
77. Heybrock S, Kanerva K, Meng Y, Ing C, Liang A, Xiong Z, et al. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat Commun (2019) 10:3521. doi: 10.1038/s41467-019-11425-0
78. Eskelinen E-L, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell (2004) 15:3132–45. doi: 10.1091/mbc.e04-02-0103
79. Fehrenbacher N, Bastholm L, Kirkegaard-Sørensen T, Rafn B, Bøttzauw T, Nielsen C, et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res (2008) 68:6623–33. doi: 10.1158/0008-5472.CAN-08-0463
Keywords: postmenopausal osteoporosis, MHC-II molecule, protein presentation pathway, effector T cells, bone immunity
Citation: Wang X, Zhang X, Han Y, Duan X, Wang J, Yan H, Wang S, Xu Y, Zhu Z, Wang L, Huang Y, Lin Q, Tan X, Zhuo J, Zhang H, Mao M, Gou W, Yi Z and Li X (2022) Role of the major histocompatibility complex class II protein presentation pathway in bone immunity imbalance in postmenopausal osteoporosis. Front. Endocrinol. 13:876067. doi: 10.3389/fendo.2022.876067
Received: 15 February 2022; Accepted: 13 July 2022;
Published: 11 August 2022.
Edited by:
Ling-Qing Yuan, Second Xiangya Hospital, Central South University, ChinaReviewed by:
Qiu-Shi Wei, Guangzhou University of Chinese Medicine, ChinaHongting Jin, Zhejiang Chinese Medical University, China
Qian-qian Liang, Shanghai University of Traditional Chinese Medicine, China
Copyright © 2022 Wang, Zhang, Han, Duan, Wang, Yan, Wang, Xu, Zhu, Wang, Huang, Lin, Tan, Zhuo, Zhang, Mao, Gou, Yi and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xihai Li, bGl4aWhhaWZ6QDE2My5jb20=
†These authors have contributed equally to this work