 Daniel Peña-Oyarzún1,2,3†
Daniel Peña-Oyarzún1,2,3† Carla San Martin2†
Carla San Martin2† María Paz Hernández-Cáceres3†
María Paz Hernández-Cáceres3† Sergio Lavandero4,5,6†Eugenia Morselli7,8†
Sergio Lavandero4,5,6†Eugenia Morselli7,8† Mauricio Budini3,8†
Mauricio Budini3,8† Patricia V. Burgos8,9,10,11†
Patricia V. Burgos8,9,10,11† Alfredo Criollo3,4,8*†
Alfredo Criollo3,4,8*†- 1Physiology Department, Faculty of Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Interdisciplinary Center for Research in Territorial Health of the Aconcagua Valley (CIISTe Aconcagua), School of Medicine, Faculty of Medicine, San Felipe Campus, Universidad de Valparaíso, San Felipe, Chile
- 3Instituto de Investigación en Ciencias Odontológicas (ICOD), Facultad de Odontología, Universidad de Chile, Santiago, Chile
- 4Advanced Center for Chronic Diseases (ACCDiS), Facultad de Ciencias Químicas y Farmacéuticas & Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 5Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Santiago, Chile
- 6Cardiology Division, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 7Department of Basic Sciences, Faculty of Medicine and Sciences, Universidad San Sebastián, Santiago de Chile, Chile
- 8Autophagy Research Center, Universidad de Chile, Santiago de Chile, Chile
- 9Centro de Biología Celular y Biomedicina (CEBICEM), Facultad de Medicina y Ciencia, Universidad San Sebastián, Santiago, Chile
- 10Centro de Envejecimiento y Regeneración (CARE-UC), Facultad de Ciencias Biológicas, Pontificia Universidad Católica, Santiago, Chile
- 11Centro Ciencia & Vida, Fundación Ciencia & Vida, Santiago, Chile
Autophagy is an intracellular degradation mechanism that allows recycling of organelles and macromolecules. Autophagic function increases metabolite availability modulating metabolic pathways, differentiation and cell survival. The oral environment is composed of several structures, including mineralized and soft tissues, which are formed by complex interactions between epithelial and mesenchymal cells. With aging, increased prevalence of oral diseases such as periodontitis, oral cancer and periapical lesions are observed in humans. These aging-related oral diseases are chronic conditions that alter the epithelial-mesenchymal homeostasis, disrupting the oral tissue architecture affecting the quality of life of the patients. Given that autophagy levels are reduced with age, the purpose of this review is to discuss the link between autophagy and age-related oral diseases.
Introduction
Macroautophagy (hereafter “autophagy”) is an intracellular degradation mechanism, evolutionarily conserved from yeast to mammals and present in basal conditions in all cells of the human body. During autophagy, intracellular organelles and macromolecules are engulfed in double membrane vesicles known as “autophagosomes”, which then fuse with a lysosome, to allow the recycling of the engulfed material (1). Cells undergo basal autophagy that recycles dysfunctional organelles and proteins, thereby maintaining cell homeostasis (2, 3). However, under stress conditions such as starvation or microorganism infections, autophagy may be upregulated to produce energy by catabolic degradation, or to remove the exogenous organisms (4, 5). This is critical for oral tissues, given the continuous exposure to bacteria and viruses in the oral cavity, the high metabolic requirement that allows the turnover of the oral mucosa cells and the constant physical stress that teeth are subjected to (6–8). Besides these pro-survival effects, autophagy is also referred to as type II programed cell death, meaning that when the pro-autophagic stimulus is extremely harsh, autophagy targets the whole cell for death (9). Therefore, the role of autophagy in oral diseases is very complex, and in many cases, depends on its levels and on the progression of the disease. An overview of autophagy is shown in Figure 1.
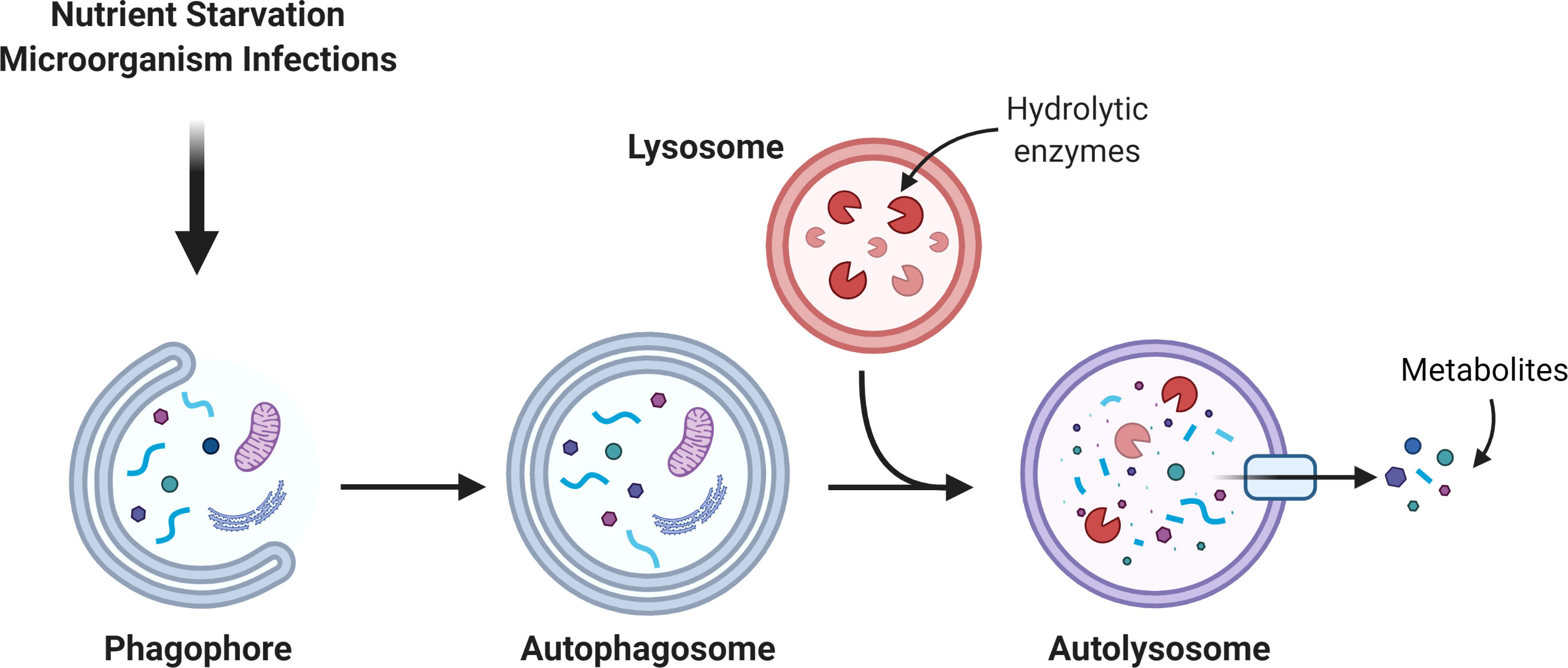
Figure 1 Autophagic pathway. The autophagosome is a double membrane organelle which sequesters intracellular material. Then, the autophagosome fuses with the lysosome to form the autolysosome where hydrolytic enzymes promote the degradation of the autolysosome cargo. The catabolism of the cargo generates simple new metabolites that turn back to the cytosol to be used in different metabolic pathways.
Aging is an irreversible biological phenomenon, affected by lifestyle, environment and genetics. The concept of aging goes beyond the concept of “lifespan”; it represents the different functional and anatomical changes in the tissues with time, which ultimately decreases the capacity to respond to internal and external stressors (10). As a homeostatic mechanism, autophagy is also affected by aging (11). A decreased autophagic tone is observed with age, while autophagy is enhanced during periodontitis, oral cancer, chronic oral infections and dental senescence (12). Studies on the physiological contribution of autophagy during aging of the oral tissues are scarce, however it has been reported that the dentin-secreting odontoblasts from old subjects (> 75 years old) show impaired fusion between autophagosomes and lysosomes, compared to those from young subjects (< 25 years old) (13). Additionally, autophagy is reduced in aged odontoblasts, which finally could affect cell homeostasis since odontoblasts are post-mitotic cells, highly dependent on cell homeostasis to promote tooth repair and healing (14). Similarly, autophagy is involved in other oral physiological responses, including tooth development and bacteria-host interaction (15). In this regard, the following question arises: Could the age-related decrease in autophagy explain the initiation and/or progression of age-dependent oral diseases, such as oral cancer, periodontitis and periapical lesions? Given that autophagy may promote either a pro- or an anti-survival effect on oral cells, and that autophagy has reciprocal control of over host immunity and energy expenditure, the answer to this question may not be as straightforward. Thus, the aim of this review is to discuss, the role of autophagy in human oral diseases associated with aging.
Aging of the oral cavity
Aging is a natural process, in which the functional ability to cope with external and internal stressors is progressively reduced (10). Biological aging hallmarks include genome alterations (i.e. cumulative DNA damage, decreased Histone H3 methylation at Lys-9 and Lys-27, and increased Histone H4 acetylation at Lys-16 and Lys-20); a stable cell cycle arrest, also known as cellular senescence, caused by telomere attrition and increased expression of the cyclin dependent kinase inhibitor 2A CDKN2A/P16INK4A; and deregulated nutrient sensing pathways, associated to higher expression of the mechanistic Target of Rapamycin, MTOR, and augmented reactive oxygen species, ROS, production (16). Indeed, pharmacological inhibition of MTOR with rapamycin, which is also a well-known autophagy activator, dramatically increases lifespan in mice (17).
Oral structures may be divided into two: the teeth and the oral mucosa. Resembling an iceberg, the roots of the teeth are inserted in the alveolar bone below the gingival lining, leaving the dental crown visible at the surface (18). The structure of the teeth is formed by layers; the outermost layer is the enamel, a very hard mineral-based cover of hydroxyapatite of 2.5 mm wide which covers the dental crown (19). Enamel is produced by cells called ameloblasts during tooth development, after which they undergo apoptosis (19). The outermost layer under the gingival lining is called cementum. Cementum is composed of hydroxyapatite, collagen and proteoglycans, resulting in a much thinner and less hard structure (20). The cementum links the dental root to the alveolar bone by attaching the periodontal ligaments (21). The central layer of teeth is the dentin, a bone-like structure formed by projections of differentiated odontoblasts, which senses external stimuli like caries (22). Odontoblast bodies are concentrated in the inner layer of the teeth, the dental pulp, where nerves and blood supply are found (23). Finally, the oral mucosa is a stratified epithelium covering all the structures in the oral cavity, including the tongue. The cells that compose the oral mucosa are the keratinocytes, which produce cytokeratin and form an epithelial barrier that separates the oral cavity from the environment (24).
During oral aging, increased ROS levels provokes a reduction of organic matrix in the enamel, yielding a crystal structure that is even harder than younger enamel (25). This decreases the susceptibility to develop caries, but weakens the tooth in case of physical insults, leading to increased incidence of fractures and cracks (26). Thus, dentin undergoes sclerosis, because senescent odontoblasts deposit secondary dentin, thereby reducing the sensing capacity and dental pulp space (27). Histological analysis of aged odontoblasts shows that they also switch from a columnar to cuboidal arrangement while exhibiting accumulation of lipofuscin, a brown-yellow pigment that indicates decreased lysosomal digestion of lipids, as well as diminished mitochondrial oxidative function (28). Gingival retraction occurs during aging, therefore the cementum that normally lies below the gingival lining is progressively exposed (29). This implies that despite reduced susceptibility to caries, observed in aged enamel, increased incidence of caries at the root occurs with age, as the cementum has no resistance against the acid in the oral cavity (30). Compared to young individuals, aged dental pulp has decreased stem cell density and increased cellular senescence, caused by secondary dentin deposition and dystrophic calcification that blocks pulpal arteries (31). Besides gingival retraction, aged oral mucosa undergoes epithelial atrophy and increased subepithelial deposit of collagen, while reducing elastin content (32). This is observed histologically by decreased epithelial ridges, a wave-like epithelial arrangement in contact with the connective tissue where proliferative keratinocytes are found, which explains the slower regeneration of the oral mucosa in older individuals (33, 34).
Aging-related oral diseases
Oral cancer
Oral cavity cancer is a highly lethal disease, with a mortality rate of 50% after 5 years and an average of diagnosis of 62 years, affecting more men than women (2:1) (35). The prevalence of oral cancer is over 300,000 cases per year worldwide; it is the sixth most common type of cancer (36). The combination of tobacco and alcohol is by far the main risk factor, while other risk factors are vitamin deficiencies, particularly those of the B complex, and the human papilloma virus, HPV (37, 38). The main type of oral cancer is oral squamous cell carcinoma, OSCC, and other types of oral cancers account for less than 5% (39). OSCC is commonly located at the mobile tongue, in 20% of the cases, and the floor of the mouth in 30% of the cases (40). The proportion of oral cancers that proceed from leukoplakias ranges between 17% and 35%, highlighting the fact that dental consultation is crucial for early cancer detection (41). Indeed, precancerous lesions, defined by the WHO as “morphologically altered tissue in which cancer occurs more often than in normal autologous tissue”, also known as oral dysplasia, precede initiation of OSCC (41, 42). The clinical presentation is highly variable; ulcerated lesions are the most frequent, but in some cases bleeding, pain, or numbness may also be present (43).
Periodontitis
Periodontitis is an extremely frequent disease, affecting nearly 70% of the global population (44). Periodontitis is a consequence of gingivitis, where a bacterial biofilm (dental plaque) forms on the gingival tissue, leading to inflammation and gingival retraction of the gum surrounding the tooth (45). In periodontitis, chronic inflammation and gingival retraction cause the migration of anaerobic gram negative bacteria such as Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans into the subgingival space (46). Since this continuous inflammation produces periodontal damage and alveolar bone resorption, diagnosis of periodontitis is commonly established when the probe depth of the gingival sulcus is over 3 mm (47, 48), which in severe cases can reach 6 mm (49). Prevalence of severe periodontitis is around 20% in adults between 35 and 44 years, while it is around 40% in adults over 60 years (50). Risk factors for periodontitis include smoking, diabetes mellitus, obesity, alcoholism, osteoporosis and stress (51). Infection with P. gingivalis disrupts oral epithelium arrangement and barrier function as proteolytic enzymes such as gingipains and collagenases are released (52). Also, during periodontal infections the antioxidant transcription factor NFE2 like bZIP transcription factor 2, NFE2L2/NRF-2, is severely downregulated, showing that oxidative stress is a key feature during periodontitis (53). Periodontal tissues show higher activation of nicotinamide adenine dinucleotide phosphate NADPH oxidase 4, NOX4, which catalyzes the production of superoxide anion after exposure with lipopolysaccharide, LPS, obtained from P. gingivalis (54). ROS like superoxide anion exhaust antioxidant catalase reserves, increasing progression of periodontal inflammation (55).
Periapical lesions
Susceptibility to enamel fractures and secondary dentination increases with age, leading to pulp exposure to external stressors (26). Periapical lesions are necrosis of the dental pulp tissue associated with an exacerbated inflammatory response due to infections, also known as periapical granuloma (56). Periapical granuloma is characterized by the persistence of microorganisms in the radicular system of the dental pulp, most of them bacteria like Acinetobacter johnsonii and Propionibacterium acnes, or fungi such as Candida albicans (57, 58). Infiltration of macrophages, lymphocytes and plasmatic cells is followed by the production of a neutrophil-rich exudate, resulting in an acute oral inflammation (59). Chronic inflammation is established after the organism attempts to repair the damaged tissue by production of new odontoblasts, mesenchymal cells and matrix proteins, but being compromised by the presence of the microorganisms, forming a granuloma tissue (60). As a consequence of periapical granuloma, radicular cysts appear, spaces filled with extracellular liquid derived from epithelial fragments of the periodontal ligament after necrosis of the dental pulp (61).
Mechanism of autophagy
Autophagy is a process in which cellular organelles called autophagosomes are formed to sequester and degrade intracellular material and macromolecules. Autophagy can be divided into five stages: initiation, nucleation, elongation, closure and fusion (62). The proteins that participate in the formation of autophagosomes are known as autophagy-related proteins or ATGs (63). In the initiation stage, signaling pathways like starvation, pathogen invasion, oxidative stress, among others, inhibit MTOR and/or activate AMP-dependent protein kinase, AMPK (64, 65). While AMPK-dependent phosphorylation activates unc-51 like autophagy activating kinase 1, ULK1/ATG1, MTOR-dependent phosphorylation of ULK1 inhibits it (66). Active ULK1 traffics to endomembrane domains, where it phosphorylates the protein Beclin 1 (BECN1) at Ser-14 (67). BECN1 allows the formation of the class III phosphatidylinositol 3-phosphate kinase, PtdIns3KC3, which phosphorylates the phosphatidylinositol lipids, creating “nucleation” signals for the recruitment of other ATGs (68, 69). Elongation of the autophagosome requires the incorporation of the Microtubule Associated Protein 1 Light Chain 3, MAP1LC3, (or just LC3) into the autophagosome membrane (70), which is previously cleaved by the ATG4 protease and conjugated with phosphatidylethanolamine by the ATG5-ATG12-ATG16 complex to forming LC3-II (71, 72). Measurement of the LC3-I to LC3-II conversion or quantification of LC3-II levels are common strategies to study autophagy (73). Other ATGs proteins such as ATG2 and ATG9 are involved in the trafficking of lipids allowing expansion of the autophagosome (74). Finally, the autophagosome membrane encloses and fuses with a lysosome that contains hydrolytic enzymes and low pH, to allow cargo degradation (75). Therefore, autophagic flux, known as the complete process beginning with autophagosome formation and degradation of the enclosed material, is usually evaluated by the use of autophagosome-lysosome fusion inhibitors (i.e. chloroquine or bafilomycin-A1) or by the tracking of the adaptor protein SQSTM1/p62, which targets poly-ubiquitinated proteins to LC3 on the autophagosome, allowing their degradation (76). Indeed, lower levels of SQSTM1/p62, reflect higher autophagic flux (77, 78). The mechanism of autophagy is depicted in Figure 2.
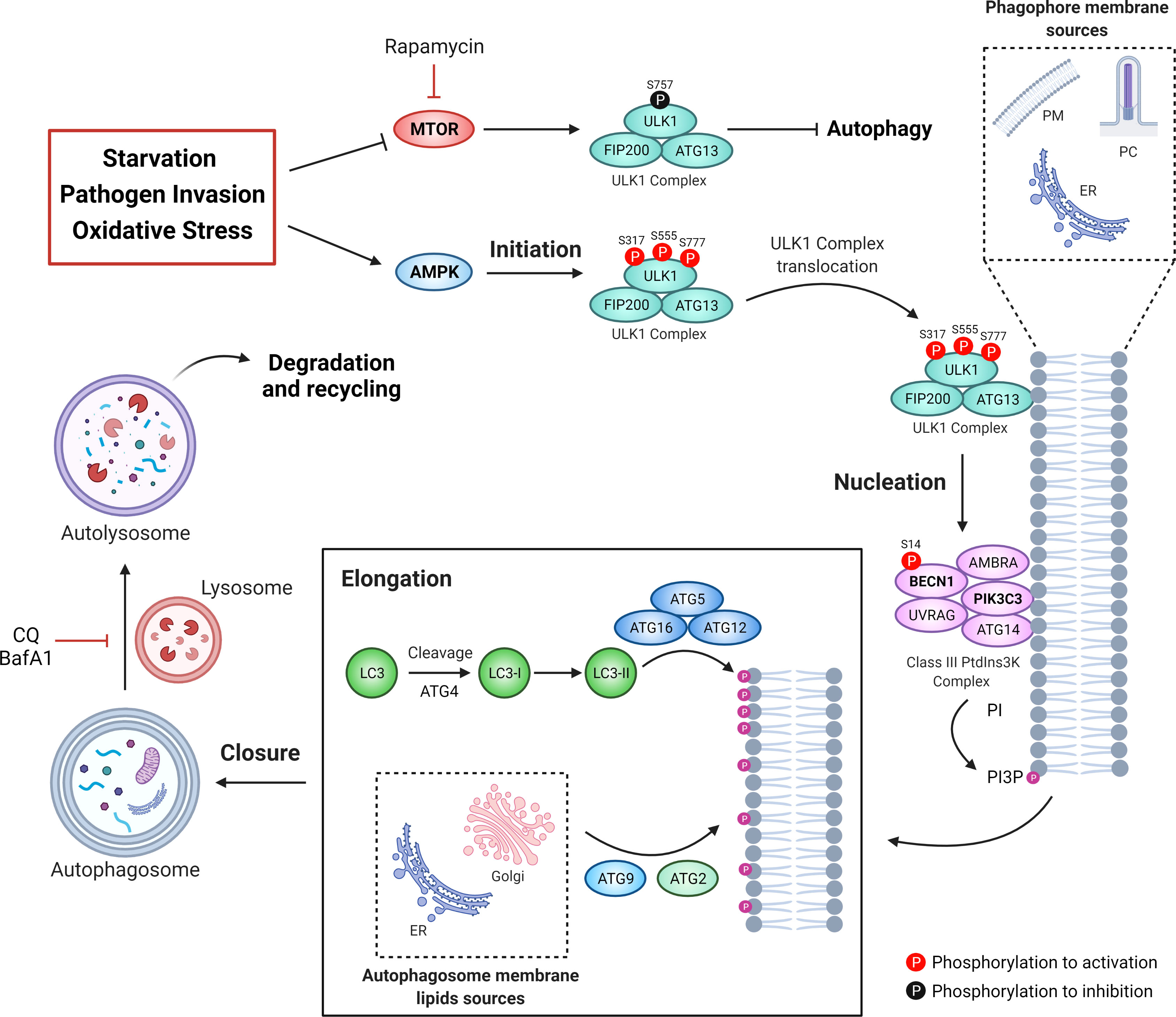
Figure 2 The autophagic machinery. Different types of stressors can be sensed by MTOR and AMPK. MTOR inhibits the ULK1 complex kinase activity phosphorylating its Ser757. Under stress conditions, AMPK activates ULK1 by phosphorylation on Ser317, Ser555, and Ser777, leading to the activation of the class III phosphatidylinositol 3-phosphate kinase complex (PtdIns3KC3). Then, active PtdIns3KC3 increases the levels of phosphatidylinositol 3-phosphate (PtdIns3P) in specific membrane micro domains, allowing the recruitment of proteins like the WD repeat domain, phosphoinositide interacting (WIPI). Thus, elongation of the phagophore membrane is mediated by two ubiquitination-like systems: the complex ATG12-ATG5-ATG16 and the conjugate LC3-phosphatidylethanolamine (PE), known as LC3-II. Additionally, both ATG2 and ATG9 participate in the elongation of the autophagosome through the trafficking of lipids from the endoplasmic reticulum or the Golgi apparatus. Once that the autophagosome membrane engulf intracellular components it encloses itself and then fuses with lysosomes to form the autolysosome. Chloroquine (CQ) or bafilomycin A1 (BafA1) can be used to block autophagosome-to-lysosome fusion, allowing the accumulation of autophagosomes.
Autophagy and age-related oral diseases
Reduced autophagy is observed in almost all tissues during aging, including oral cells. For instance, odontoblasts from 75 year-old individuals show accumulation of autophagic vesicles compared to odontoblasts from 25 year-old individuals (13). In contrast to younger odontoblasts, older odontoblasts display higher co-localization of mitochondria and lysosomes, accompanied by accumulation of lypofusin, suggesting lysosomal dysfunction (13). Thus, autophagic vesicles accumulation in older individuals is a result of decreased autophagy flux. In fact, reestablishing autophagy with rapamycin, attenuates aged-induced periodontal bone loss and gingival inflammation in mice, suggesting that autophagy-based pharmacological treatment could delay oral aging (79). In the next sections, we will describe the contribution of autophagy in age-related oral diseases.
Oral cancer
The role of autophagy in most cancers is complex and controversial. Indeed, autophagy has been extensively described as a “double-edged sword”, with different roles during carcinogenesis and cancer progression (80). Despite oral cancer has been proposed to follow this double-edged sword behavior (81), some considerations need to be addressed when oral and non-oral cancers are compared.
Previous work shows that both heterozygous deletion of ATG5 and specific homozygous deletion of hepatic ATG7 in mice, result in hepatomegaly and liver tumor formation, respectively, in 6-month-old mice (82). These tumors accumulate SQSTM1/p62 and ubiquitin aggregates, as well as 8-hydroxydeoxyguanosine, 8-OHdG, suggesting that reduced autophagic degradation is associated with oxidative stress (82). Similarly, spontaneous neoplastic formations in lung and liver have been found in mice with a heterologous deletion of BECN1 (83, 84), implying that impaired autophagy promotes carcinogenesis in non-oral tissues. In oral carcinogenesis, tumor xenografts in mice obtained by subcutaneous injection of TSCC (human tongue squamous cell carcinoma) cells downregulated for BECN1 display significant increase in volume and weight, compared to control TSCC xenografts (85). Additionally, in mice treated for 16 weeks with 4-nitroquinoline N-oxide, 4-NQO, a cigarette-smoke compound (86), progression of the oral mucosa from normal to dysplastic, and then from dysplastic to cancerous positively correlates with the increase in LC3 and SQSTM1/p62 levels, suggesting that the malignant transformation of normal oral cells is associated with decreased autophagy (87). Altogether these studies indicate that both in oral and non-oral tissues, inhibition of autophagy is involved in cancer initiation.
During progression of non-oral cancers, cancer cells increase their autophagic tone to overcome the stress of crowding, hypoxia and nutrient deprivation. For instance, it has been demonstrated that deletion of scribble/scrib (the ortholog of human scribble planar cell polarity protein, SCRIB) in Drosophila melanogaster (scrib KO flies), a well-known model of eye tumor that invades the central nervous system, reduces lipid droplet content in the adipose tissue and increases muscle atrophy measured by micro-computerized tomography, as well as increases LC3 processing in all the aforementioned tissues, suggesting that scrib-deficient tumor cells obtain nutrients by wasting host organs (88). Autophagy is required for this systemic organ wasting, as scrib KO flies that do not express ATG13, part of the ULK1 kinase complex, show reduced muscle atrophy and increased lipid droplet content, compared to scrib KO flies (88). Similarly, treatment of pancreatic adenocarcinoma xenografts in vitro with the inhibitor of the autophagic flux chloroquine, a chemical compound that blocks the fusion between lysosome and autophagosome, decreases oxygen consumption and impairs tumor growth (89). Consistently, treatment of MCF7 breast cancer xenografts with chloroquine reduces tumor viability (90). Given that these cells display higher levels of LC3-II and autophagosome vesicles under serum deprivation, authors conclude that autophagy is induced to support tumor growth in conditions of starvation (90).
On the other hand, progression of oral cancer should be interpreted with caution, or at least better dissected in a specific time frame. Indeed, the treatment of Cal 27, a human OSCC cell line, with up to 2 mM melatonin, increases LC3-I to LC3-II conversion and reduces SQSTM1/p62 levels, while increasing Caspase 3 cleavage, indicating that melatonin treatment induces both autophagy and apoptosis (91). Additionally, Cal 27 subcutaneous tumor xenografts in mice reduce their weight after treatment with 100 mg/kg of melatonin, suggesting that induction of autophagy promotes apoptosis in OSCC cells (91). In this model, autophagy is induced through the transcription factor binding to IGHM enhancer 3, TFE3, which upregulates the expression of autophagy-related genes, such as atg7 and lamp1 (92). Similarly, treatment of Cal 27 cells with sepantronium bromide, a chemical inhibitor of Survivin, not only leads to apoptosis, but also triggers autophagy, as it has been observed by the increase in LC3 lipidation and SQSTM1/p62 degradation (93, 94). Autophagy is upregulated in this model due to decreased MTOR activity, as reduced of MTOR auto-activating phosphorylation at Ser-2448, as well as diminished phosphorylation of ribosomal protein S6, RPS6, at Ser-235 and Ser-236, a downstream target of MTOR (93). Most importantly, tamoxifen-induced double knock-out mice for transforming growth factor β1 receptor, TGFBR1, and phosphatase and tensin homolog, PTEN, display a reduction in tumor growth, as well as a weak immunohistochemical staining for MTOR phosphorylation at Ser-2448 and SQSTM1/p62, after exposure with 5 mg/kg sepantronium bromide (93). All together, these findings indicate that induction of autophagy during oral cancer progression negatively affects tumor growth.
Other studies have shown that biopsies from patients with poorly differentiated OSCC show higher immunohistochemical staining against LC3, BECN1 and SQSTM1/p62, indicative of an impaired autophagy (95–97). However, given that increased levels of ATG5 and ATG9, and therefore higher autophagy, correlates to unfavorable overall survival of cancer patients (98), it is possible that oral cancer progression promoted by autophagy inhibition occurs only during advanced stages of OSCC, while in the early stages of oral cancer development autophagy is upregulated to support tumor progression. Wound healing and transwell assays in TSCC cells treated with rapamycin show reduced cell migration and invasion when compared to control cells, suggesting autophagy inhibition in the later stages of cancer progression promotes cell migration and invasion (99).
The change in the “autophagic behavior” of OSCC may be explained by the cancer microenvironment, particularly by the carcinoma-associated fibroblasts, CAFs (100). CAFs release interleukin-33, IL33, and chemokine (C-C motif) ligand 7, CCL7 inhibiting autophagy in OSCC cells and promoting proliferation and epithelial-mesenchymal transition, EMT (101, 102). CAFs also transfer their mitochondria to the OSCC cells and inhibit AMPK, explaining the metabolic switch of the OSCC cells to lactate production, their resistance to metformin treatment and the impairment of autophagy (103, 104). This is interesting because, given that oral autophagic status is decreased with age, OSCC cells may be more prone to acquire malignant traits on their own than from interacting with the oral mesenchymal cells. This also suggests that autophagy-based treatments for oral cancer should consider the age of the patients.
Periodontitis
During periodontitis, pathogens like P. gingivalis use the autophagic machinery of oral keratinocytes, dendritic cells and macrophages to survive and disseminate (105). After entering myeloid dendritic cells, P. gingivalis is transported within early endosomes, as indicated by co-localization between RAB5A positive vesicles and P. gingivalis (106). Inside the eukaryotic cell, several virulence factors of P. gingivalis increase LC3-II and BECN1 levels, while reducing caspase activation and annexin V staining, indicating that periodontal pathogens induce autophagosome formation and suppress apoptosis (107). For instance, the penta-acylated form of LPS from P. gingivalis not only increases LC3 vesicle formation, but also quadruples autophagosomes diameter, compared to the tetra-acylated form (108). Given that, contrary to the tetra-acylated LPS from P. gingivalis, the penta-acylated form is recognized by the toll-like receptor 4, TLR4, interaction of pathogen associated molecular patters, PAMP, with TLR molecules is crucial for P. gingivalis-dependent autophagosome formation (108). In addition, this penta-acylated LPS from P. gingivalis reduced melanoregulin, MREG, levels, a protein required for lysosomal hydrolase activity of Cathepsin D and β-N-Acetylglucosaminidase, suggesting that P. gingivalis PAMPs diminish lysosomal activity (108, 109). Similarly, the mannose content of the fimbria major subunit Mfa1, mfa1, of P. gingivalis is recognized by the C-type lectin receptor CD209/DC-SIGN inside the oral mucosa cells, ultimately downregulating the expression of lysosomal associated membrane protein 1, Lamp1 (106). P. gingivalis remains hidden inside the autophagosomes, from where it obtains the nutrients to its replication, but blocks the fusion with the lysosome, avoiding degradation (106, 110). This is relevant since, in the literature, increased autophagosome formation by P. gingivalis is commonly confused with reduced autophagy flux. In fact, recovering autophagy in P. gingivalis-infected macrophages with calcitriol, 1α, 25-dihydroxyvitamin D3, decreases the survival of bacteria while increasing lysosomal function (111). Similar results are observed after stimulation of P. gingivalis-infected myeloid dendritic cells with rapamycin (106). Nevertheless, butyrate, a short chain fatty acid produced by anaerobic bacteria of the dental plaque, induces autophagy, promoting caspase-independent cell death in oral keratinocytes (112). This suggests that some metabolic byproducts of the periodontal pathogens may increase autophagy in oral gingival tissues, while other structural components of periodontitis-related pathogens are important for increasing autophagosome formation but then impair the autophagic flux. Despite the contribution of the autophagosome to P. gingivalis survival within the intracellular eukaryotic microenvironment, not all P. gingivalis that enter through early endosomes are targeted by the autophagic system, as some bacteria will be routed to the recycling endosome pathway to allow exocytosis. Indeed, gingival epithelial cells knocked-down for recycling endosome marker RAB11, the small GTPase required for interaction with the exocyst components RAS like proto-oncogene A, RALA, or the exocyst complex components 2/3/84, EXOC2/3/84 and then infected with P. gingivalis, display decreased colony formation units, cfu, in agar when the extracellular culture medium of the cells is plated (113). Consistently, in the same experimental conditions, increased bacterial cfu is observed when plating the intracellular content of the infected gingival cells, suggesting impaired exocytosis (113). Given that RALB participates during starvation-induced formation of the autophagosome by assembling BECN1 components (114), it is possible that P. gingivalis proliferates within the autophagosome and then escapes to infect neighboring gingival cells using the exocyst pathway. However, this has not been proven yet.
On the other hand, the interaction between oral pathogens and oral host cells is modulated by aging. A study where the 16S rRNA of the oral microbiome from “clinically healthy” old (> 40 years) or young subjects (around 23 years), as well as from “clinically confirmed periodontitis” patients, was isolated and sequenced, showed that the oral microbiome from old (but healthy) individuals is an intermediate condition between healthy young subjects and periodontitis patients (115). Indeed, higher levels of P. gingivalis, Tannerella forsythia and Treponema denticola, together known as the “red complex pathogens,” involved in the development of chronic periodontitis, increased 48, 25 and 55 fold, respectively in old healthy subjects when compared to young healthy persons (115). It has been previously suggested that this increased prevalence in red complex pathogens with age is due to a low-grade pro-inflammatory condition or “inflammaging” (116). In fact, assessment of the gingival fluid from aged individuals (> 65 years) and young individuals (< 25 years), both groups without periodontitis, show augmented B cell infiltration and IgG3 levels, suggesting an immune shift towards chronic inflammation (117). The relation between autophagy and inflammaging has been comprehensively reviewed elsewhere (118). Briefly, autophagy recycles aged mitochondria, thus preventing the rise of ROS levels and blunting the activation of the danger sensor NLR family pyrin domain containing 3, NLRP3, a protein that mediates IL1B release through caspase 1 activation (118). Whether autophagy predisposes the development of chronic periodontitis in old subjects by reducing inflammaging is not known yet.
It is important to understand that increased ROS levels does not necessarily leads to periodontitis, since the physiologic response following bacterial infection of the oral mucosa and periodontal tissue is by oxidative stress-driven autophagy (119). Indeed, neutrophils contribute to ROS formation upon pathogen infection in a mechanism that is associated with higher expression of ATG12 and LC3 (120). Because treatment of periodontitis-related neutrophils with N-acetylcysteine avoids the gene expression of ATG12 and LC3, autophagy is actually induced by ROS in these cells, possibly aiding cell survival (120). This could be important to overcome the downregulation of NFE2L2 observed in periodontitis (53). As oral autophagy is decreased with age, increased periodontal toxicity by oxidative stress is observed, resulting in elevated periodontitis-induced bone resorption (121, 122).
Periapical lesions
Autophagy is crucial for odontogenesis, since it provides energy and removal of wasted intracellular components in enamel and oral epithelial cells (123). Compared to dental pulp cells from young rats, senescent dental pulp cells obtained from old rats display increased immunohistochemical staining of LC3 and BECN1, accompanied by decreased levels of peroxisome proliferator activated receptor gamma PPARγ (124), a transcriptional factor required to induce the expression of autophagy related proteins (125, 126). In this model, stimulation with LPS exhausts autophagy related proteins, suggesting that accumulation of LC3 and BECN1 is a consequence of an impaired autophagy flux (124). Indeed, adenoviral upregulation of PPARγ recovers autophagosome formation to cope with LPS stimulation (124). Additionally, dental pulp stem cells obtained from canine and human tooth root treated with C-X-C motif chemokine ligand 12, CXCL12, show increased levels of LC3-II and ATG5-ATG12 complex, as well as inhibition of the MTOR signaling, indicating that CXCL12 promotes autophagy in dental pulp stem cells (127). Authors demonstrated that CXCL12-dependent autophagy is required for stem cell migration as CXCL12 increased pore migration in transwell assay, but the phenomenon was prevented by treatment with the autophagic inhibitor chloroquine (127). Thus, in this model, high expression of ATG5-ATG12 complex and LC3, indicative of increased autophagy, is critical for development and regeneration of the dental pulp (128). Therefore, in old mice, autophagosomes are accumulated as a result of decreased autophagy flux, reducing periapical lesion repair. For instance, a rapid rise in LC3-II/LC3-I ratio, ATG12 levels and activating phosphorylations of ULK1 occur in murine odontoblasts after treatment with hydrogen peroxide, showing that increased autophagy is required for early dental repair (129). In the same model, treatment with simvastatin, an MTOR inhibitor of the statin-family drug for treatment of obesity, reduced dental destruction (129). Increased expression of ATG5-ATG12 and activating phosphorylation of AMPK, accompanied by augmented levels of hypoxia inducible factor 1 subunit alpha, HIF1α, has been observed in periapical granulomas (130). Given that the treatment of pre-odontoblast cell line mDPC6T with chloroquine reduces cell viability in presence of LPS, we can conclude that autophagy in periapical lesions promotes survival of odontoblasts under harmful stimulus (131). Therefore, as autophagy flux is reduced with age, dental repair capacity will be progressively limited, shifting the focus towards the search for novel polymers with better repair capacity and integration in older individuals (132).
Interestingly, eukaryotic pathogens like C. albicans associated with periapical lesions require autophagy for infection and virulence. Fungal expression of ATG9 has been shown to allow intracellular trafficking (133). Thus, VPS34, the fungal homologue of PIK3C3, forms a complex with the Vma7 subunit of H+-ATPase, contributing to vacuolar acidification and thereby increased autophagy-lysosome pathway activation (134). Additionally, C. albicans strains with deletion of VPS34 undergo cell death in stress conditions, such as during nitrogen starvation (134). Indeed, their survival is severely impaired in this condition, indicating that fungal autophagy is key for C. albicans virulence (134). This means that autophagy is increased in C. albicans during periapical lesions, but decreased in older individuals that are being infected. The role of autophagy in age-related oral diseases is summarized in Figure 3.
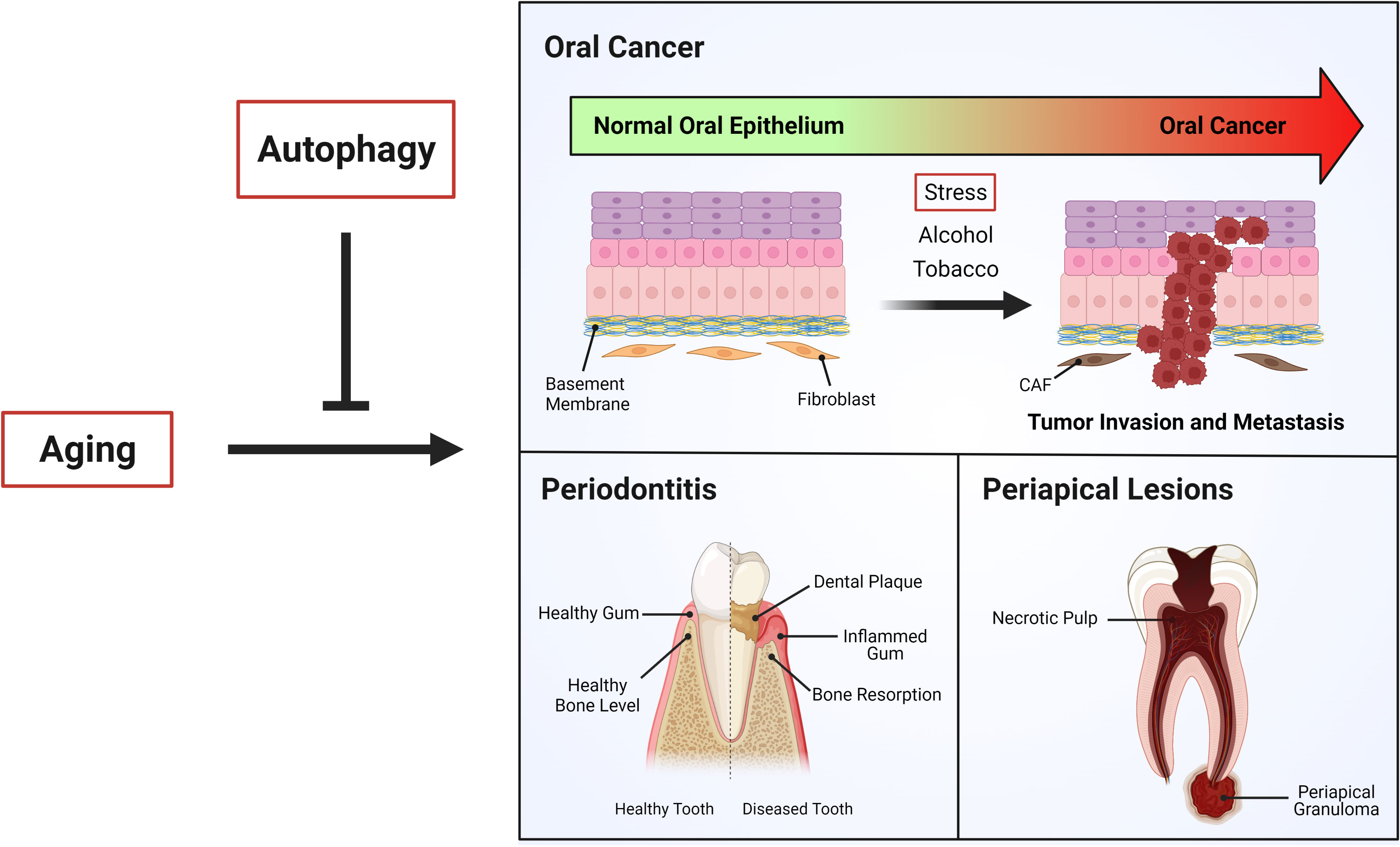
Figure 3 Autophagy and age-related oral diseases. Aging is a physiological process in which different cellular mechanism decline, leading in most of cases age-related diseases. “Autophagy”, which is impaired during aging, has been shown be critical to the control of age-related oral diseases such as oral cancer, periodontitis and periapical lesions.
Concluding remarks
Aging is a natural and irreversible process that all of us are experiencing. The complex interplay of the molecular mechanisms that are altered during aging finally leads to reduced sensitivity and adjustment capacity against external and internal changes in the cells. One of the main tools that cells have to control energy balance and recycling of wasted molecules is autophagy. Autophagy, as a manifestation of this reduced homeostatic capacity, begins to diminish with age. Susceptibility to certain pathologies also increases, especially those associated with the oral cavity, which is in constant stress by interaction with the environment, like oral cancer, periodontitis and periapical lesions. What is important to note is that age-dependent autophagic decrease is in most cases a result of impaired fusion between autophagosomes and lysosomes; this implies that autophagosome formation may actually occur, which for example favors the survival of P. gingivalis and C. albicans during periodontitis and periapical lesions. It seems that progression of aged-associated oral diseases is explained both by reduced lysosomal activity and by the accumulation of autophagosomes that protect foreign pathogens. The role of autophagy in oral cancer is difficult to establish, as it depends on whether we are observing the initiation, the progression, or the late development of the cancer. The orchestrated shift in autophagy in oral cancer cells may be defined by the tumor microenvironment in a specific temporal manner.
Author contributions
Writing: DP-O, CM, SL, EM, PB, AC Design of Figures: MH-C, AC. Writing, Corrections and Editing: MB. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Agencia Nacional de Investigación y Desarrollo de Chile (ANID, Chile): FONDECYT [1200499 to EM and 1211329 to AC]; PIA-ANID [ACT172066 to EM, MB, PB and AC]; FONDAP [15130011 to SL and AC]; FONDECYT Post-doctoral fellowship [3200313 to DP-O and 3210630 to MH-C]; ANID/BASAL/FB210008 and ANID/BASAL/ACE2100099 to PB. Programa de Movilidad Internacional Santander Universidades, Versión 2021 to AC
Acknowledgments
We sincerely thank everyone in the Criollo laboratories for discussion and constructive criticism. Figures made in BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol (2015) 16(8):461–72. doi: 10.1038/nrm4024
2. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ (2013) 20(1):31–42. doi: 10.1038/cdd.2012.81
3. Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol (2013) 8:105–37. doi: 10.1146/anharma
4. Sharma V, Verma S, Seranova E, Sarkar S, Kumar D. Selective autophagy and xenophagy in infection and disease. Front Cell Dev Biol (2018) 6:147. doi: 10.3389/fcell.2018.00147
5. He L, Zhang J, Zhao J, Ma N, Kim SW, Qiao S, et al. Autophagy: The last defense against cellular nutritional stress. Adv Nutr (2018) 9(4):493–504. doi: 10.1093/advances/nmy011
6. Deo PN, Deshmukh R. Oral microbiome: Unveiling the fundamentals. J Oral Maxillofac Pathol (2019) 23(1):122–8. doi: 10.4103/jomfp.JOMFP_304_18
7. Costea DE, Loro LL, Dimba EA, Vintermyr OK, Johannessen AC. Crucial effects of fibroblasts and keratinocyte growth factor on morphogenesis of reconstituted human oral epithelium. J Invest Dermatol (2003) 121(6):1479–86. doi: 10.1111/j.1523-1747.2003.12616.x
8. Koc D, Dogan A, Bek B. Bite force and influential factors on bite force measurements: a literature review. Eur J Dent (2010) 4(2):223–32. doi: 10.1055/s-0039-1697833
9. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol (2008) 9(12):1004–10. doi: 10.1038/nrm2529
10. Khan SS, Singer BD, Vaughan DE. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell (2017) 16(4):624–33. doi: 10.1111/acel.12601
11. Martinez-Lopez N, Athonvarangkul D, Singh R. Autophagy and aging. Adv Exp Med Biol (2015) 847:73–87. doi: 10.1007/978-1-4939-2404-2_3
12. Tan YQ, Zhang J, Zhou G. Autophagy and its implication in human oral diseases. Autophagy (2017) 13(2):225–36. doi: 10.1080/15548627.2016.1234563
13. Couve E, Osorio R, Schmachtenberg O. The amazing odontoblast: activity, autophagy, and aging. J Dent Res (2013) 92(9):765–72. doi: 10.1177/0022034513495874
14. Rajan S, Ljunggren A, Manton DJ, Björkner AE, McCullough M. Post-mitotic odontoblasts in health, disease, and regeneration. Arch Oral Biol (2020) 109:104591. doi: 10.1016/j.archoralbio.2019.104591
15. Zhuang H, Ali K, Ardu S, Tredwin C, Hu B. Autophagy in dental tissues: a double-edged sword. Cell Death Dis (2016) 7(4):e2192. doi: 10.1038/cddis.2016.103
16. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer: G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
17. Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell (2014) 13(3):468–77. doi: 10.1111/acel.12194
18. Matsuda K, Haga-Tsujimura M, Yoshie S, Shimomura-Kuroki J. Characteristics of alveolar bone associated with physiological movement of molar in mice: a histological and histochemical study. Odontology (2014) 102(1):98–104. doi: 10.1007/s10266-012-0093-y
19. Lacruz RS, Habelitz S, Wright JT, Paine ML. Dental enamel formation and implications for oral health and disease. Physiol Rev (2017) 97(3):939–93. doi: 10.1152/physrev.00030.2016
20. Yamamoto T, Hasegawa T, Yamamoto T, Hongo H, Amizuka N. Histology of human cementum: Its structure, function, and development. Jpn Dent Sci Rev (2016) 52(3):63–74. doi: 10.1016/j.jdsr.2016.04.002
21. Nottmeier C, Liao N, Simon A, Decker MG, Luther J, Schweizer M, et al. Wnt1 promotes cementum and alveolar bone growth in a time-dependent manner. J Dent Res (2021) 100(13):1501–9. doi: 10.1177/00220345211012386
22. Goldberg M, Kulkarni AB, Young M, Boskey A. Dentin: structure, composition and mineralization. Front Biosci (Elite Ed) (2011) 3(2):711–35. doi: 10.2741/e281
23. Franca CM, Riggers R, Muschler JL, Widbiller M, Lococo PM, Diogenes A, et al. 3D-imaging of whole neuronal and vascular networks of the human dental pulp via CLARITY and light sheet microscopy. Sci Rep (2019) 9(1):10860. doi: 10.1038/s41598-019-47221-5
25. Park S, Wang DH, Zhang D, Romberg E, Arola D. Mechanical properties of human enamel as a function of age and location in the tooth. J Mater Sci Mater Med (2008) 19(6):2317–24. doi: 10.1007/s10856-007-3340-y
26. Yahyazadehfar M, Zhang D, Arola D. On the importance of aging to the crack growth resistance of human enamel. Acta Biomater (2016) 32:264–74. doi: 10.1016/j.actbio.2015.12.038
27. Xu H, Zheng Q, Shao Y, Song F, Zhang L, Wang Q, et al. The effects of ageing on the biomechanical properties of root dentine and fracture. J Dent (2014) 42(3):305–11. doi: 10.1016/j.jdent.2013.11.025
28. Couve E, Schmachtenberg O. Autophagic activity and aging in human odontoblasts. J Dent Res (2011) 90(4):523–8. doi: 10.1177/0022034510393347
29. Hartmann R, Muller F. Clinical studies on the appearance of natural anterior teeth in young and old adults. Gerodontology (2004) 21(1):10–6. doi: 10.1111/j.1741-2358.2004.00009.x
30. Griffin SO, Griffin PM, Swann JL, Zlobin N. Estimating rates of new root caries in older adults. J Dent Res (2004) 83(8):634–8. doi: 10.1177/154405910408300810
31. Ersahan S, Sabuncuoglu FA. Effect of age on pulpal blood flow in human teeth during orthodontic movement. J Oral Sci (2018) 60(3):446–52. doi: 10.2334/josnusd.17-0316
32. Breustedt A. Age-induced changes in the oral mucosa and their therapeutic consequences. Int Dent J (1983) 33(3):272–80.
33. Abu Eid R, Sawair F, Landini G, Saku T. Age and the architecture of oral mucosa. Age (Dordr) (2012) 34(3):651–8. doi: 10.1007/s11357-011-9261-1
34. Zhang J, An Y, Gao LN, Zhang YJ, Jin Y, Chen FM. The effect of aging on the pluripotential capacity and regenerative potential of human periodontal ligament stem cells. Biomaterials (2012) 33(29):6974–86. doi: 10.1016/j.biomaterials.2012.06.032
35. Montero PH, Patel SG. Cancer of the oral cavity. Surg Oncol Clin N Am (2015) 24(3):491–508. doi: 10.1016/j.soc.2015.03.006
36. Warnakulasuriya S. Living with oral cancer: epidemiology with particular reference to prevalence and life-style changes that influence survival. Oral Oncol (2010) 46(6):407–10. doi: 10.1016/j.oraloncology.2010.02.015
37. Petersen PE. Oral cancer prevention and control–the approach of the world health organization. Oral Oncol (2009) 45(4-5):454–60. doi: 10.1016/j.oraloncology.2008.05.023
38. Spence T, Bruce J, Yip KW, Liu FF. HPV associated head and neck cancer. Cancers (Basel) (2016) 8(8):75–86. doi: 10.3390/cancers8080075
39. Rivera C, Venegas B. Histological and molecular aspects of oral squamous cell carcinoma (Review). Oncol Lett (2014) 8(1):7–11. doi: 10.3892/ol.2014.2103
40. Pires FR, Ramos AB, Oliveira JB, Tavares AS, Luz PS, Santos TC. Oral squamous cell carcinoma: clinicopathological features from 346 cases from a single oral pathology service during an 8-year period. J Appl Oral Sci (2013) 21(5):460–7. doi: 10.1590/1679-775720130317
41. Warnakulasuriya S, Johnson NW, van der Waal I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J Oral Pathol Med (2007) 36(10):575–80. doi: 10.1111/j.1600-0714.2007.00582.x
42. Ranganathan K, Kavitha L. Oral epithelial dysplasia: Classifications and clinical relevance in risk assessment of oral potentially malignant disorders. J Oral Maxillofac Pathol (2019) 23(1):19–27. doi: 10.4103/jomfp.JOMFP_13_19
43. Zini A, Czerninski R, Sgan-Cohen HD. Oral cancer over four decades: epidemiology, trends, histology, and survival by anatomical sites. J Oral Pathol Med (2010) 39(4):299–305. doi: 10.1111/j.1600-0714.2009.00845.x
44. Dye BA. Global periodontal disease epidemiology. Periodontol (2012) 58(1):10–25. doi: 10.1111/j.1600-0757.2011.00413.x
45. Kinane DF, Stathopoulou PG, Papapanou PN. Periodontal diseases. Nat Rev Dis Primers (2017) 3:17038. doi: 10.1038/nrdp.2017.38
46. Arora N, Mishra A, Chugh S. Microbial role in periodontitis: Have we reached the top? some unsung bacteria other than red complex. J Indian Soc Periodontol (2014) 18(1):9–13. doi: 10.4103/0972-124X.128192
47. Graves DT, Li J, Cochran DL. Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res (2011) 90(2):143–53. doi: 10.1177/0022034510385236
48. Meseli SE, Kuru B, Kuru L. Relationships between initial probing depth and changes in the clinical parameters following non-surgical periodontal treatment in chronic periodontitis. J Istanb Univ Fac Dent (2017) 51(3):11–7. doi: 10.17096/jiufd.40993
49. Preshaw PM. Detection and diagnosis of periodontal conditions amenable to prevention. BMC Oral Health (2015) 15 Suppl:1, S5. doi: 10.1186/1472-6831-15-S1-S5
50. Nazir MA. Prevalence of periodontal disease, its association with systemic diseases and prevention. Int J Health Sci (Qassim) (2017) 11(2):72–80.
51. Van Dyke TE, Sheilesh D. Risk factors for periodontitis. J Int Acad Periodontol (2005) 7(1):3–7.
52. Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon's knife to a meat chopper-like brutal degradation of proteins. Periodontol (2010) 54(1):15–44. doi: 10.1111/j.1600-0757.2010.00377.x
53. Sima C, Aboodi GM, Lakschevitz FS, Sun C, Goldberg MB, Glogauer M. Nuclear factor erythroid 2-related factor 2 down-regulation in oral neutrophils is associated with periodontal oxidative damage and severe chronic periodontitis. Am J Pathol (2016) 186(6):1417–26. doi: 10.1016/j.ajpath.2016.01.013
54. Golz L, Memmert S, Rath-Deschner B, Jager A, Appel T, Baumgarten G, et al. LPS from p. gingivalis and hypoxia increases oxidative stress in periodontal ligament fibroblasts and contributes to periodontitis. Mediators Inflamm (2014) 2014:986264. doi: 10.1155/2014/986264
55. Petelin M, Pavlica Z, Ivanusa T, Sentjurc M, Skaleric U. Local delivery of liposome-encapsulated superoxide dismutase and catalase suppress periodontal inflammation in beagles. J Clin Periodontol (2000) 27(12):918–25. doi: 10.1034/j.1600-051x.2000.027012918.x
56. Lopez-Marcos JF. Aetiology, classification and pathogenesis of pulp and periapical disease. Med Oral Patol Oral Cir Bucal (2004) 9 Suppl:58–62; 52-7.
57. Mussano F, Ferrocino I, Gavrilova N, Genova T, Dell'Acqua A, Cocolin L, et al. Apical periodontitis: preliminary assessment of microbiota by 16S rRNA high throughput amplicon target sequencing. BMC Oral Health (2018) 18(1):55. doi: 10.1186/s12903-018-0520-8
58. Ashraf H, Samiee M, Eslami G, Ghodse Hosseini MR. Presence of candida albicans in root canal system of teeth requiring endodontic retreatment with and without periapical lesions. Iran Endod J (2007) 2(1):24–8.
59. Alptekin NO, Ari H, Ataoglu T, Haliloglu S, Alptekin T, Serpek B. Neutrophil elastase levels in periapical exudates of symptomatic and asymptomatic teeth. J Endod (2005) 31(5):350–3. doi: 10.1097/01.don.0000140567.25382.cd
60. Holland R, Gomes JEF, Cintra LTA, Queiroz IOA, Estrela C. Factors affecting the periapical healing process of endodontically treated teeth. J Appl Oral Sci (2017) 25(5):465–76. doi: 10.1590/1678-7757-2016-0464
61. Tsesis I, Krepel G, Koren T, Rosen E, Kfir A. Accuracy for diagnosis of periapical cystic lesions. Sci Rep (2020) 10(1):14155. doi: 10.1038/s41598-020-71029-3
62. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
63. Klionsky DJ. Look people, "Atg" is an abbreviation for "autophagy-related." That's it. Autophagy (2012) 8(9):1281–2. doi: 10.4161/auto.21812
64. Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal (2013) 25(1):50–65. doi: 10.1016/j.cellsig.2012.09.020
65. Meijer AJ, Lorin S, Blommaart EF, Codogno P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids (2015) 47(10):2037–63. doi: 10.1007/s00726-014-1765-4
66. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–41. doi: 10.1038/ncb2152
67. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol (2013) 15(7):741–50. doi: 10.1038/ncb2757
68. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol (2008) 182(4):685–701. doi: 10.1083/jcb.200803137
69. Proikas-Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci (2015) 128(2):207–17. doi: 10.1242/jcs.146258
70. Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol (2008) 445:77–88. doi: 10.1007/978-1-59745-157-4_4
71. Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem (2004) 279(46):47704–10. doi: 10.1074/jbc.M407016200
72. Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy (2013) 9(3):424–5. doi: 10.4161/auto.22931
73. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy (2008) 4(2):151–75. doi: 10.4161/auto.5338
74. Gomez-Sanchez R, Rose J, Guimaraes R, Mari M, Papinski D, Rieter E, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol (2018) 217(8):2743–63. doi: 10.1083/jcb.201710116
75. Lorincz P, Juhasz G. Autophagosome-lysosome fusion. J Mol Biol (2020) 432(8):2462–82. doi: 10.1016/j.jmb.2019.10.028
76. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci (2017) 18(9):1865–77. doi: 10.3390/ijms18091865
77. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy (2011) 7(3):279–96. doi: 10.4161/auto.7.3.14487
78. Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol (2009) 452:181–97. doi: 10.1016/S0076-6879(08)03612-4
79. An JY, Kerns KA, Ouellette A, Robinson L, Morris HD, Kaczorowski C, et al. Rapamycin rejuvenates oral health in aging mice. Elife (2020) 9:e54318. doi: 10.7554/eLife.54318
80. Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene (2018) 37(9):1142–58. doi: 10.1038/s41388-017-0046-6
81. Alexandra T, Marina IM, Daniela M, Ioana SI, Maria B, Radu R, et al. Autophagy-a hidden but important actor on oral cancer scene. Int J Mol Sci (2020) 21(23):9325–36. doi: 10.3390/ijms21239325
82. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25(8):795–800. doi: 10.1101/gad.2016211
83. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest (2003) 112(12):1809–20. doi: 10.1172/JCI20039
84. Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene (2013) 32(18):2261–72, 2272e 1-11. doi: 10.1038/onc.2012.252
85. Weng J, Wang C, Wang Y, Tang H, Liang J, Liu X, et al. Beclin1 inhibits proliferation, migration and invasion in tongue squamous cell carcinoma cell lines. Oral Oncol (2014) 50(10):983–90. doi: 10.1016/j.oraloncology.2014.06.020
86. Hawkins BL, Heniford BW, Ackermann DM, Leonberger M, Martinez SA, Hendler FJ. 4NQO carcinogenesis: a mouse model of oral cavity squamous cell carcinoma. Head Neck (1994) 16(5):424–32. doi: 10.1002/hed.2880160506
87. Wu JS, Li L, Wang SS, Pang X, Wu JB, Sheng SR, et al. Autophagy is positively associated with the accumulation of myeloid−derived suppressor cells in 4−nitroquinoline−1−oxide−induced oral cancer. Oncol Rep (2018) 40(6):3381–91. doi: 10.3892/or.2018.6747
88. Khezri R, Holland P, Schoborg TA, Abramovich I, Takáts S, Dillard C, et al. Host autophagy mediates organ wasting and nutrient mobilization for tumor growth. EMBO J (2021) 40(18):e107336. doi: 10.15252/embj.2020107336
89. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev (2011) 25(7):717–29. doi: 10.1101/gad.2016111
90. Sanchez CG, Penfornis P, Oskowitz AZ, Boonjindasup AG, Cai DZ, Dhule SS, et al. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis (2011) 32(7):964–72. doi: 10.1093/carcin/bgr029
91. Fan T, Pi H, Li M, Ren Z, He Z, Zhu F, et al. Inhibiting MT2-TFE3-dependent autophagy enhances melatonin-induced apoptosis in tongue squamous cell carcinoma. J Pineal Res (2018) 64(2). doi: 10.1111/jpi.12457
92. Fan T, Wang X, Zhang S, Deng P, Jiang Y, Liang Y, et al. NUPR1 promotes the proliferation and metastasis of oral squamous cell carcinoma cells by activating TFE3-dependent autophagy. Signal Transduct Target Ther (2022) 7(1):130. doi: 10.1038/s41392-022-00939-7
93. Zhang L, Zhang W, Wang YF, Liu B, Zhang WF, Zhao YF, et al. Dual induction of apoptotic and autophagic cell death by targeting survivin in head neck squamous cell carcinoma. Cell Death Dis (2015) 6(5):e1771. doi: 10.1038/cddis.2015.139
94. Kelly RJ, Thomas A, Rajan A, Chun G, Lopez-Chavez A, Szabo E, et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann Oncol (2013) 24(10):2601–6. doi: 10.1093/annonc/mdt249
95. Tang JY, Fang YY, Hsi E, Huang YC, Hsu NC, Yang WC, et al. Immunopositivity of beclin-1 and ATG5 as indicators of survival and disease recurrence in oral squamous cell carcinoma. Anticancer Res (2013) 33(12):5611–6.
96. Tang JY, Hsi E, Huang YC, Hsu NC, Chu PY, Chai CY. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum Pathol (2013) 44(11):2558–62. doi: 10.1016/j.humpath.2013.06.017
97. Liu JL, Chen FF, Lung J, Lo CH, Lee FH, Lu YC, et al. Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br J Cancer (2014) 111(5):944–54. doi: 10.1038/bjc.2014.355
98. Tang JY, Hsi E, Huang YC, Hsu NC, Chen YK, Chu PY, et al. ATG9A overexpression is associated with disease recurrence and poor survival in patients with oral squamous cell carcinoma. Virchows Arch (2013) 463(6):737–42. doi: 10.1007/s00428-013-1482-5
99. Wang Y, Wang C, Tang H, Wang M, Weng J, Liu X, et al. Decrease of autophagy activity promotes malignant progression of tongue squamous cell carcinoma. J Oral Pathol Med (2013) 42(7):557–64. doi: 10.1111/jop.12049
100. Dourado MR, Guerra ENS, Salo T, Lambert DW, Coletta RD. Prognostic value of the immunohistochemical detection of cancer-associated fibroblasts in oral cancer: A systematic review and meta-analysis. J Oral Pathol Med (2018) 47(5):443–53. doi: 10.1111/jop.12623
101. Ding L, Ren J, Zhang D, Li Y, Huang X, Hu Q, et al. A novel stromal lncRNA signature reprograms fibroblasts to promote the growth of oral squamous cell carcinoma via LncRNA-CAF/interleukin-33. Carcinogenesis (2018) 39(3):397–406. doi: 10.1093/carcin/bgy006
102. Jung DW, Kim J, Che ZM, Oh ES, Kim G, Eom SH, et al. A triazine compound S06 inhibits proinvasive crosstalk between carcinoma cells and stromal fibroblasts via binding to heat shock protein 90. Chem Biol (2011) 18(12):1581–90. doi: 10.1016/j.chembiol.2011.10.001
103. Zhang Z, Gao Z, Rajthala S, Sapkota D, Dongre H, Parajuli H, et al. Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell Mol Life Sci (2020) 77(6):1115–33. doi: 10.1007/s00018-019-03209-y
104. Zhang Z, Liang X, Fan Y, Gao Z, Bindoff LA, Costea DE, et al. Fibroblasts rescue oral squamous cancer cell from metformin-induced apoptosis via alleviating metabolic disbalance and inhibiting AMPK pathway. Cell Cycle (2019) 18(9):949–62. doi: 10.1080/15384101.2019.1598727
105. Lee K, Roberts JS, Choi CH, Atanasova KR, Yilmaz O. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence (2018) 9(1):845–59. doi: 10.1080/21505594.2018.1454171
106. El-Awady AR, Miles B, Scisci E, Kurago ZB, Palani CD, Arce RM, et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PloS Pathog (2015) 10(2):e1004647. doi: 10.1371/journal.ppat.1004647
107. Hirasawa M, Kurita-Ochiai T. Porphyromonas gingivalis induces apoptosis and autophagy via ER stress in human umbilical vein endothelial cells. Mediators Inflammation (2018) 2018:1967506. doi: 10.1155/2018/1967506
108. Blasi I, Korostoff J, Dhingra A, Reyes-Reveles J, Shenker BJ, Shahabuddin N, et al. Variants of porphyromonas gingivalis lipopolysaccharide alter lipidation of autophagic protein, microtubule-associated protein 1 light chain 3, LC3. Mol Oral Microbiol (2016) 31(6):486–500. doi: 10.1111/omi.12141
109. Damek-Poprawa M, Diemer T, Lopes VS, Lillo C, Harper DC, Marks MS, et al. Melanoregulin (MREG) modulates lysosome function in pigment epithelial cells. J Biol Chem (2009) 284(16):10877–89. doi: 10.1074/jbc.M808857200
110. Yamatake K, Maeda M, Kadowaki T, Takii R, Tsukuba T, Ueno T, et al. Role for gingipains in porphyromonas gingivalis traffic to phagolysosomes and survival in human aortic endothelial cells. Infect Immun (2007) 75(5):2090–100. doi: 10.1128/IAI.01013-06
111. Niu L, Chen S, Yang X, Ma C, Pan C, Wang H, et al. Vitamin d decreases porphyromonas gingivalis internalized into macrophages by promoting autophagy. Oral Dis (2021) 27(7):1775–88. doi: 10.1111/odi.13696
112. Tsuda H, Ochiai K, Suzuki N, Otsuka K. Butyrate, a bacterial metabolite, induces apoptosis and autophagic cell death in gingival epithelial cells. J Periodontal Res (2010) 45(5):626–34. doi: 10.1111/j.1600-0765.2010.01277.x
113. Takeuchi H, Furuta N, Morisaki I, Amano A. Exit of intracellular porphyromonas gingivalis from gingival epithelial cells is mediated by endocytic recycling pathway. Cell Microbiol (2011) 13(5):677–91. doi: 10.1111/j.1462-5822.2010.01564.x
114. Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell (2011) 144(2):253–67. doi: 10.1016/j.cell.2010.12.018
115. Lenartova M, Tesinska B, Janatova T, Hrebicek O, Mysak J, Janata J, et al. The oral microbiome in periodontal health. Front Cell Infect Microbiol (2021) 11:629723. doi: 10.3389/fcimb.2021.629723
116. Feres M, Teles F, Teles R, Figueiredo LC, Faveri M. The subgingival periodontal microbiota of the aging mouth. Periodontol (2016) 72(1):30–53. doi: 10.1111/prd.12136
117. Fransson C, Mooney J, Kinane DF, Berglundh T. Differences in the inflammatory response in young and old human subjects during the course of experimental gingivitis. J Clin Periodontol (1999) 26(7):453–60. doi: 10.1034/j.1600-051x.1999.260707.x
118. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) (2012) 4(3):166–75. doi: 10.18632/aging.100444
119. Wang Y, Andrukhov O, Rausch-Fan X. Oxidative stress and antioxidant system in periodontitis. Front Physiol (2017) 8:910. doi: 10.3389/fphys.2017.00910
120. Bullon P, Cordero MD, Quiles JL, Ramirez-Tortosa Mdel C, Gonzalez-Alonso A, Alfonsi S, et al. Autophagy in periodontitis patients and gingival fibroblasts: unraveling the link between chronic diseases and inflammation. BMC Med (2012) 10:122. doi: 10.1186/1741-7015-10-122
121. Linden GJ, McClean KM, Woodside JV, Patterson CC, Evans A, Young IS, et al. Antioxidants and periodontitis in 60-70-year-old men. J Clin Periodontol (2009) 36(10):843–9. doi: 10.1111/j.1600-051X.2009.01468.x
122. Aung KT, Akiyama K, Kunitomo M, Mun AY, Tosa I, Nguyen HTT, et al. Aging-affected MSC functions and severity of periodontal tissue destruction in a ligature-induced mouse periodontitis model. Int J Mol Sci (2020) 21(21):8103–22. doi: 10.3390/ijms21218103
123. Lee YH, Lee HY, Kim TG, Lee NH, Yu MK, Yi HK. PPARgamma maintains homeostasis through autophagy regulation in dental pulp. J Dent Res (2015) 94(5):729–37. doi: 10.1177/0022034515573833
124. Lee YH, Lee HY, Kim TG, Lee NH, Yu MK, Yi HK. PPARγ maintains homeostasis through autophagy regulation in dental pulp. J Dent Res (2015) 94(5):729–37. doi: 10.1177/0022034515573833
125. Liu J, Yao Q, Xiao L, Ma W, Li F, Lai B, et al. PPARγ induces NEDD4 gene expression to promote autophagy and insulin action. FEBS J (2020) 287(3):529–45. doi: 10.1111/febs.15042
126. Yan S, Yang X, Chen T, Xi Z, Jiang X. The PPARγ agonist troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther (2014) 21(5):188–93. doi: 10.1038/cgt.2014.16
127. Yang JW, Zhang YF, Wan CY, Sun ZY, Nie S, Jian SJ, et al. Autophagy in SDF-1α-mediated DPSC migration and pulp regeneration. Biomaterials (2015) 44:11–23. doi: 10.1016/j.biomaterials.2014.12.006
128. Yang JW, Zhang YF, Wan CY, Sun ZY, Nie S, Jian SJ, et al. Autophagy in SDF-1alpha-mediated DPSC migration and pulp regeneration. Biomaterials (2015) 44:11–23. doi: 10.1016/j.biomaterials.2014.12.006
129. Lai EH, Hong CY, Kok SH, Hou KL, Chao LH, Lin LD, et al. Simvastatin alleviates the progression of periapical lesions by modulating autophagy and apoptosis in osteoblasts. J Endod (2012) 38(6):757–63. doi: 10.1016/j.joen.2012.02.023
130. Huang HY, Wang WC, Lin PY, Huang CP, Chen CY, Chen YK, et al. (2018) 51 Suppl:2, e125–e145. doi: 10.1111/iej.12782
131. Pei F, Lin H, Liu H, Li L, Zhang L, Chen Z. Dual role of autophagy in lipopolysaccharide-induced preodontoblastic cells. J Dent Res (2015) 94(1):175–82. doi: 10.1177/0022034514553815
132. Huang Y, Song B, Zhou X, Chen H, Wang H, Cheng L. Dental restorative materials for elderly populations. Polymers (Basel) (2021) 13(5):828–40. doi: 10.3390/polym13050828
133. Palmer GE, Kelly MN, Sturtevant JE. Autophagy in the pathogen candida albicans. Microbiol (Reading) 153(Pt (2007) 1):51–8. doi: 10.1099/mic.0.2006/001610-0
134. Eck R, Nguyen M, Gunther J, Kunkel W, Zipfel PF. The phosphatidylinositol 3-kinase Vps34p of the human pathogenic yeast candida albicans is a multifunctional protein that interacts with the putative vacuolar h+ -ATPase subunit Vma7p. Int J Med Microbiol (2005) 295(1):57–66. doi: 10.1016/j.ijmm.2004.12.007
Keywords: autophagy, oral diseases, aging, periodontitis, oral cancer, periapical lesions
Citation: Peña-Oyarzún D, San Martin C, Hernández-Cáceres MP, Lavandero S, Morselli E, Budini M, Burgos PV and Criollo A (2022) Autophagy in aging-related oral diseases. Front. Endocrinol. 13:903836. doi: 10.3389/fendo.2022.903836
Received: 24 March 2022; Accepted: 13 July 2022;
Published: 05 August 2022.
Edited by:
Oliana Carnevali, Marche Polytechnic University, ItalyReviewed by:
Luciana Dini, Sapienza University of Rome, ItalyAngelica Giuliani, Marche Polytechnic University, Italy
Kunihiro Sakuma, Tokyo Institute of Technology, Japan
Copyright © 2022 Peña-Oyarzún, San Martin, Hernández-Cáceres, Lavandero, Morselli, Budini, Burgos and Criollo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfredo Criollo, YWxjcmlvbGxvQHUudWNoaWxlLmNs
†ORCID: Daniel Peña-Oyarzún, https://orcid.org/0000-0001-8881-8074
Carla San Martin, https://orcid.org/0000-0001-5411-5207
María Paz Hernández-Cáceres, https://orcid.org/0000-0003-2365-9652
Sergio Lavandero, http://orcid.org/0000-0003-4258-1483
Eugenia Morselli, http://orcid.org/0000-0002-7840-8351
Mauricio Budini, http://orcid.org/0000-0003-1821-291X
Patricia Burgos, https://orcid.org/0000-0003-4521-7978
Alfredo Criollo, https://orcid.org/0000-0002-2737-7751