 Isabella Santiago de Melo Miranda1*
Isabella Santiago de Melo Miranda1* Luciana Pinto Valadares2
Luciana Pinto Valadares2 Gustavo Barcelos Barra3
Gustavo Barcelos Barra3 Pedro Góes Mesquita3
Pedro Góes Mesquita3 Lidiana Bandeira de Santana1
Lidiana Bandeira de Santana1 Lucas Faria de Castro1
Lucas Faria de Castro1 Ticiane Henriques Santa Rita3
Ticiane Henriques Santa Rita3 Luciana Ansaneli Naves1
Luciana Ansaneli Naves1- 1Section of Endocrinology, University Hospital of Brasilia, Brasilia, Brazil
- 2SARAH Network Rehabilitation Hospitals, Brasilia, Brazil
- 3Genomics Section, Sabin Medicina Diagnóstica, Brasília, Brazil
Objective: Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant syndrome characterized by its clinical variability and complexity in diagnosis and treatment. We performed both clinical and molecular descriptions of four families with MEN1 in a follow-up at a tertiary center in Brasília.
Methods: From a preliminary review of approximately 500 medical records of patients with pituitary neuroendocrine tumor (PitNET) from the database of the Neuroendocrinology Outpatient Clinic of the University Hospital of Brasília, a total of 135 patients met the criteria of at least two affected family members. From this cohort, we have identified 34 families: only four with a phenotype of MEN1 and the other 30 families with the phenotype of familial isolated pituitary adenoma (FIPA). Eleven patients with a clinical diagnosis of MEN1 from these four families were selected.
Results: Variants in MEN1 gene were identified in all families. One individual from each family underwent genetic testing using targeted high-throughput sequencing (HTS). All patients had primary hyperparathyroidism (PHPT), and the second most common manifestation was PitNET. One individual had well-differentiated liposarcoma, which has been previously reported in a single case of MEN1. Three variants previously described in the database and a novel variant in exon 2 have been found.
Conclusions: The study allowed the genotypic and phenotypic characterization of families with MEN1 in a follow-up at a tertiary center in Brasília.
Introduction
Multiple endocrine neoplasia type 1 (MEN1; OMIM 131100) is an autosomal dominant syndrome characterized by its clinical variability and complexity in diagnosis and treatment. It is primarily defined by the occurrence of tumors in the parathyroid glands, gastroenteropancreatic (GEP) tract, and anterior pituitary (1, 2). Other endocrine and non-endocrine tumors can also occur, such as adrenocortical adenomas, carcinoids, angiofibromas, collagenomas, leiomyomas, and lipomas (3, 4). MEN1 gene contains 10 exons that encode a 610-amino-acid protein called MENIN, which acts as a tumor suppressor. MENIN seems to play a role in regulating DNA replication and transcription and in maintaining the integrity of the genome (5).
The delay in diagnosis occurs in most cases. These patients may already present with complications related to these tumors, increasing morbidity and mortality, thus leading to a worse prognosis (2). Therefore, it is important to recognize this great phenotypic variability.
Although there is no well-established genotype–phenotype correlation, genetic testing should be performed to confirm clinical diagnosis and identify asymptomatic first-degree relatives (2, 6). The absence of hot spots establishes the need for sequencing the entire MEN1 gene. Genetic evaluation using targeted high-throughput sequencing (HTS) analyses multiple coding regions and splicing sites simultaneously, in addition to having a lower cost when compared to the Sanger method (7, 8).
For this purpose, we performed both clinical and molecular descriptions of four families with MEN1 in a follow-up at a tertiary center in Brasília. The aim of this study is to emphasize the importance of recognizing the several clinical manifestations that may be present in individuals with MEN1 so that diagnosis and familial screening can be readily performed.
Materials and methods
Subjects and clinical diagnosis
From a preliminary review of approximately 500 medical records of patients with pituitary neuroendocrine tumor (PitNET) from the database of the Neuroendocrinology Outpatient Clinic of the University Hospital of Brasília, a total of 135 patients met the criteria of at least two affected family members. From this cohort, we have identified 34 families: only four with a phenotype of MEN1 and the other 30 families with the phenotype of familial isolated pituitary adenoma (FIPA). Eleven patients with a clinical diagnosis of MEN1 from these four families were selected.
Clinical diagnosis of MEN1 was defined by the presence of at least two of the three classical tumors (parathyroid, GEP tract, or PitNET). Laboratory tests (calcium, parathyroid hormone (PTH), prolactin, insulin-like growth factor 1 (IGF-1), luteinizing hormone (LH), follicle-stimulating hormone (FSH), estradiol, testosterone, and cortisol), imaging studies, pathological assessment, and relevant clinical events were collected and analyzed through a review of medical records.
The study was carried out in accordance with the Declaration of Helsinki and approved by the Ethics Committee for Research on Human Beings of the Faculty of Health Sciences, University of Brasília. The patients provided their written informed consent to participate in this study.
MEN1 gene mutation analysis
DNA was extracted from 200 μl of EDTA-whole blood from subjects in an EDTA tube using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and quantified on the Qubit 2.0 fluorometer system using Qubit™ 1X dsDNA HS (Life Technologies, Carlsbad, CA, USA). DNA samples were subjected to a paired-end sequencing process using the NextSeq 500 sequencer (Illumina, San Diego, CA, USA).
Each DNA library was prepared using the KAPA Hyperplus Library Preparation (Roche, Basel, Switzerland), and the coding region of approximately 4,000 genes associated with hereditary diseases and their respective splicing sites were enriched using SeqCap EZ inherited diseases panel (Roche, Basel, Switzerland) and Kapa HyperPrep kit (Roche, Basel, Switzerland). The size and quality of the pre-hybridization and post-hybridization DNA libraries were checked on D1000 ScreenTape using the 4200 TapeStation instrument (Agilent Technologies, Santa Clara, CA, USA) and quantified on the Qubit 2.0 Fluorometer System (Life Technologies, Carlsbad, CA, USA).
The enriched DNA library pool was subjected to paired-end sequencing using the 2 × 75 cycle NextSeq 500/550 V2 midi output kit (Illumina, San Diego, USA) on NextSeq 500 sequencer (Illumina, San Diego, USA).
After demultiplexing using bcl2fastq2 v2.20 conversion software (Base Space, Illumina, San Diego, USA), fastQ files were submitted to DRAGEN Germline 3.5.7 for mapping against the human genome (hg19) and variants calling. Dragen VCF files were uploaded to Varstation (www.varstation.com) for annotation and classification of variants. Twenty-two genes associated with endocrine tumors (AIP, APC, CDC73, CDKN1B, DICER1, FH, MAX, MEN1, MET, NF1, PRKAR1A, PTEN, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, TP53, VHL, and WRN) were filtered for analysis. Variants were classified according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) [PMID 25741868].
Results
Clinical features
We described four families with at least three patients presenting the clinical phenotype of MEN1 (Figure 1). The age of the first presentation ranged from 29 to 55 years with a median age of 42 years, and there was no difference in prevalence between genders. All patients that participated in the study (100%) had primary hyperparathyroidism (PHPT), and only three underwent parathyroidectomy (Table 1). Five of the six individuals with asymptomatic PHPT who underwent densitometry had low bone mass at diagnosis, and three (individuals III.1 from families 1 and 2 and individual III.2 from family 4) were younger than 30 years.
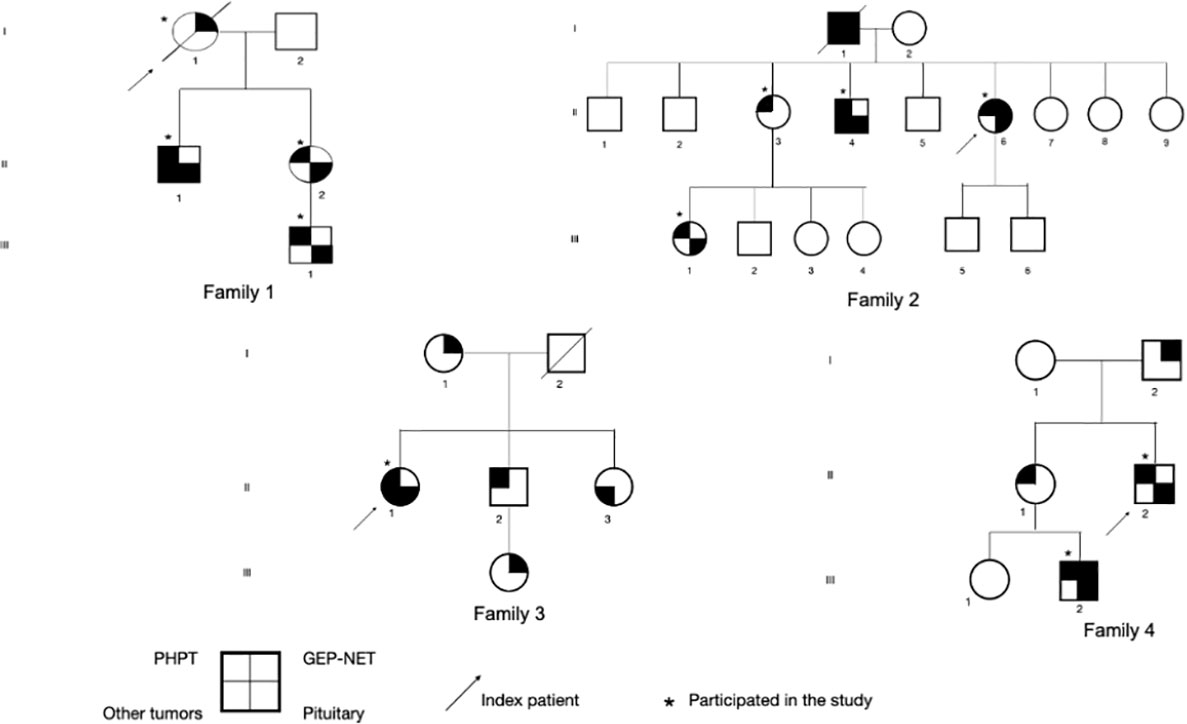
Figure 1 Representation of four pedigrees with clinical/familial MEN1. PHPT, primary hyperparathyroidism; GEP-NET, neuroendocrine tumors of gastroenteropancreatic tract; MEN1, multiple endocrine neoplasia type 1.
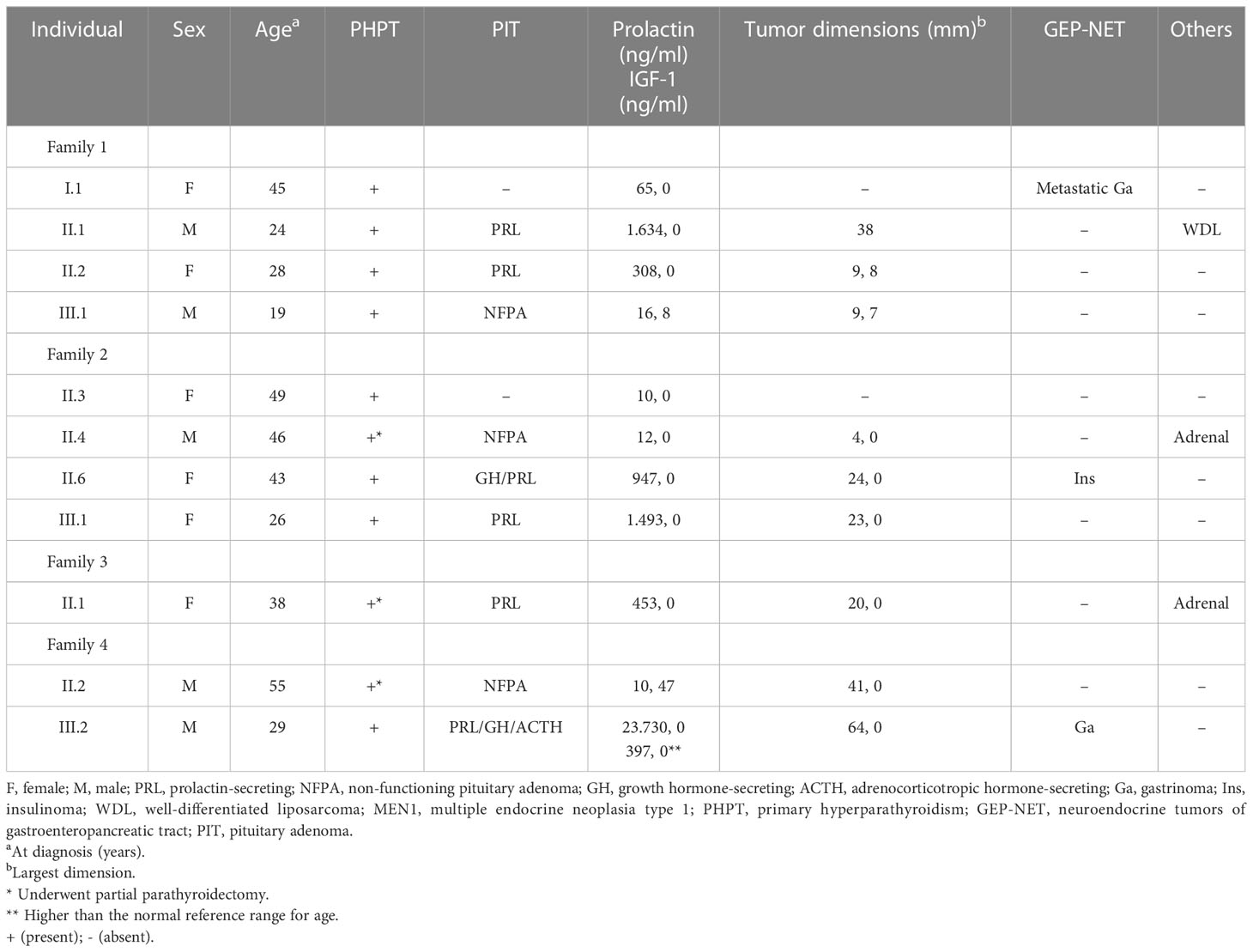
Table 1 Clinical characteristics of subjects from four families with clinical MEN1.
The second most frequent manifestation was a PitNET in nine of the 11 patients (81.8%), with the most common being macroadenomas, which were present in seven individuals (77.8%). Regarding subtypes, prolactinomas were the most common, present in four individuals (44.4%), followed by clinically non-functioning adenomas (33.3%). Three individuals had macroprolactinomas that grew during follow-up, despite a 3.5 mg weekly dose of cabergoline, thus requiring surgical treatment. Patient II.2 (family 1) presented with headache and impaired visual field, with a diagnosis of pituitary apoplexy, and underwent transsphenoidal surgery (Figure 2).

Figure 2 Coronal and sagittal MRI of individual II.1 (family 1). T1-weighted image with a 24 × 19 × 18 mm pituitary macroadenoma (A) that evolved after 6 months with hemorrhagic degeneration and compression of the optic chiasm (B). After surgery, a small remnant can be seen in the left half of the anterior pituitary gland (C).
Individual III.2 (family 4) had a giant pituitary adenoma resulting in intracranial hypertension and underwent craniotomy. He had hyperprolactinemia and IGF-1 higher than the normal reference range for age (Table 1). Cabergoline was initiated after surgery at a weekly dose of 3.5 mg. Both prolactin and IGF-1 levels dropped to the normal range after 6 months. Magnetic resonance imaging (MRI) showed a notable reduction in tumor size 6 months after surgery (Figure 3). Two individuals had co-secreting adenomas with one having immunohistochemistry showing positive staining for prolactin, growth hormone (GH), and adrenocorticotropic hormone (ACTH) (Figure 4).
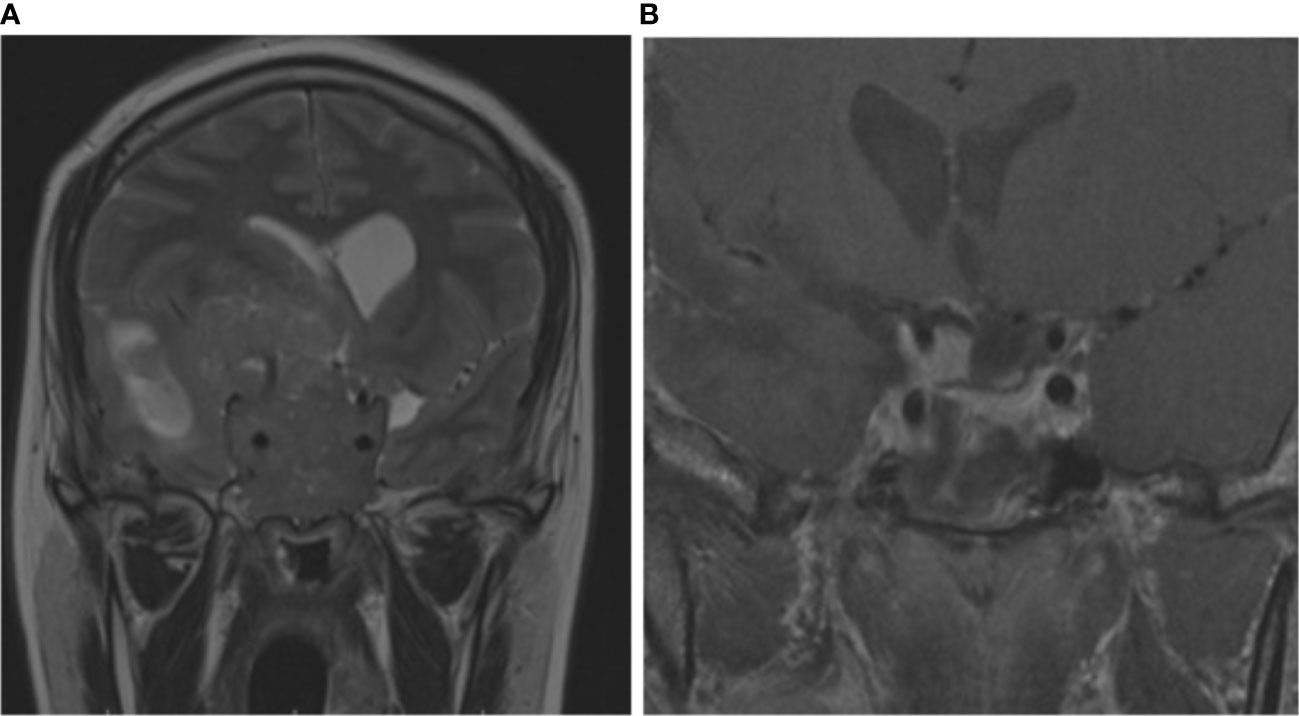
Figure 3 Coronal and sagittal MRI of individual III.2 (family 4). Giant pituitary adenoma with suprasellar extension reaching the right frontoparietal region, with invasion of cavernous sinus and third ventricle (A). Six months after surgery and cabergoline, the tumor exhibited significant shrinkage and necrosis (B).
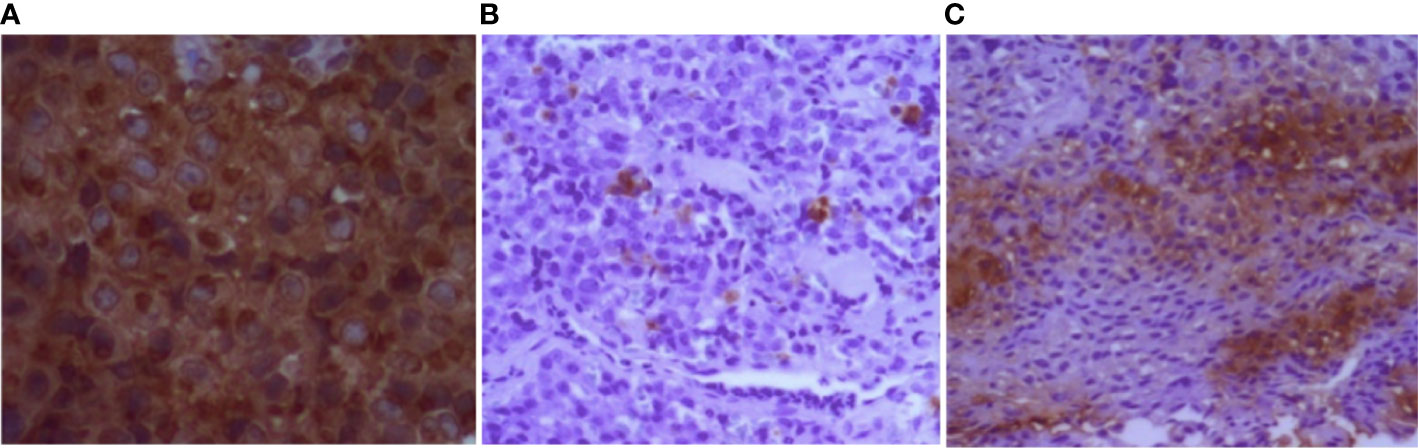
Figure 4 Immunohistochemistry of pituitary tumor (individual III.2, family 4). (A) Diffuse positive staining for prolactin. (B, C) Strong and focal positive staining for GH and ACTH, respectively. GH, growth hormone; ACTH, adrenocorticotropic hormone.
Only three patients had GEP tract tumors—two gastrinomas and one insulinoma—who underwent surgical resection. The only patient in our study who died was the index case (I.1) of family 1 who was diagnosed with metastatic gastrinoma. In this family, an uncommon association with well-differentiated liposarcoma was found in patient II.2. Two individuals had adrenal tumors with dimensions greater than 4 cm. Individual II.4 (family 2) had a bilateral lesion and subclinical hypercortisolism, and individual II.1 (family 3) had a non-functioning unilateral lesion (Figure 1), both treated surgically.
Genotyping analysis
Variants in MEN1 gene were identified in all families. In individual II.1 (family 1), a non-sense mutation (NM_130799.3:c.76G>T:p.Glu26Ter) has been found in exon 2. Missense mutations have been detected in individuals II.6 (family 2) and II.1 (family 3) (NM_130799.3:c.1021:p.Trp341Arg in exon 7 and NM_130799.3:c.124G>C:p.Gly42Arg in exon 2, respectively). Another variant has been identified in the splicing site (NM_130799.3:c.654+1G>T) in individual II.2 (family 4) (Figure 5). Allelic variants found in individuals II.1 (family 1), II.6 (family 2), and II.2 (family 4) were considered pathogenic after applying ACMG variant classification rules (12). A novel variant was found in individual II.1 (family 3) and classified as probably pathogenic (Table 2).

Figure 5 Distribution of germline mutations in MEN1 gene identified in individuals from each family of our study (NM_130799.3). F1, family 1; F2, family 2; F3, family 3; F4, family 4.
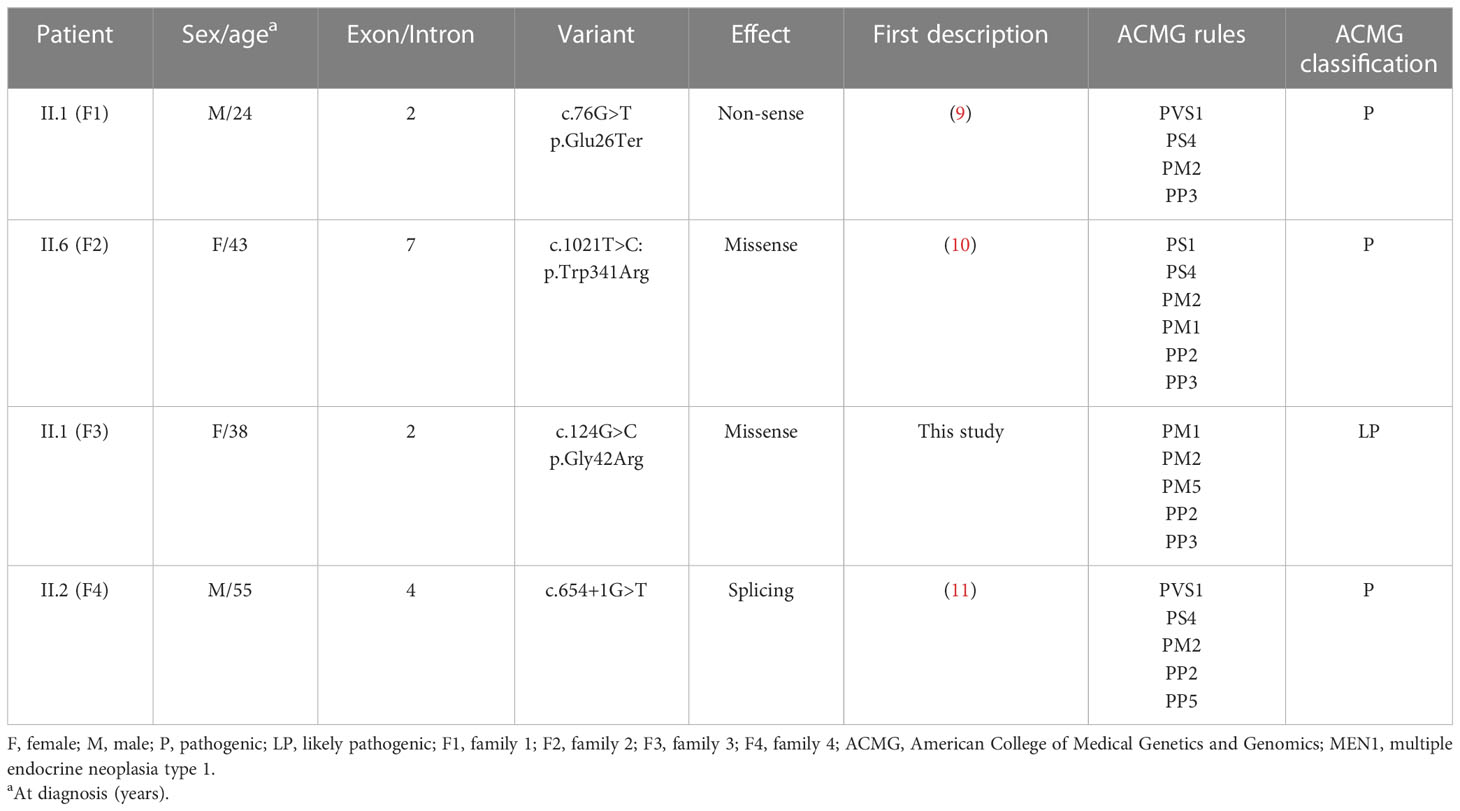
Table 2 Mutation analysis of each family with MEN1 (NM_130799.3).
Discussion
This study was the first to perform both clinical and molecular descriptions of four families with MEN1 in a follow-up at a tertiary center in Brasília. Patients with MEN1 have a reduced life expectancy when compared to individuals with sporadic tumors (1). The prognosis improves considerably when these tumors are identified at an early stage, especially in patients who are asymptomatic (2). In this regard, it is necessary for an interdisciplinary team that can clinically recognize the phenotypic variability found in these individuals, as well as perform family genetic screening.
The median age at diagnosis of the index cases was 45 years, which suggests a certain delay in diagnosis. It has also been observed in other countries and can occur because of both a delay in identifying the index case and the lag time to complete the family screening (13). A study using the database of patients with MEN1 in Japan showed that the mean age at diagnosis occurred in the fourth decade in 50% of cases (14). Another study using an Italian database also found that the mean age at diagnosis was also around the fourth decade (15).
Studies in patients with MEN1 have already shown that women aged between 20 and 35 years have a higher incidence of osteopenia and osteoporosis when compared to the general population of the same age group (16–18). PHPT was present in all patients of our study, and three asymptomatic individuals under 30 years old had low bone mass at diagnosis. A Brazilian study demonstrated impairment of bone mass in half of the individuals under the age of 30 years, despite being more frequent and severe in patients over 50 years old (19). Early development of PHPT, occurring about three decades before sporadic cases, leads to prolonged and persistent exposure to high levels of PTH. Therefore, peak bone mass is compromised, and, consequently, the risk of fractures increases (20).
Although PHPT is the first clinical manifestation in almost 90% of cases (15, 21), PitNETs were the first and most common manifestation in our series, occurring in five out of 11 individuals (45.5%). We believe that this was due to the fact that patients were referred to our neuroendocrinology outpatient clinic. Most of them had macroadenomas (77.8%), which are more common in individuals with MEN1 and generally present morphological invasion of adjacent structures compared to sporadic cases (22–24).
Prolactinomas were the most common, present in four individuals (44.4%), followed by non-functioning pituitary adenoma in three individuals (33.3%). The literature shows that prolactinomas are the most prevalent in the context of MEN1 (~65%), followed by somatotropinoma, ACTH-secreting adenoma, and non-functioning adenomas (21, 25). Some studies have shown that plurihormonal adenomas are more frequent in the context of MEN1 when compared to sporadic pituitary adenomas. Unusual associations may occur, such as the secretion of prolactin and ACTH (26, 27), which was present in one of our patients who had immunohistochemistry showing positive staining for prolactin, GH, and ACTH (individual III.2 of family 4).
A higher frequency of GEP tract tumors is expected in these individuals since it is the second most common manifestation in MEN1 (1, 3). However, only three individuals had GEP tract tumors: two gastrinomas and one insulinoma. It is possible that imaging studies missed some of these tumors, especially non-functioning ones. Furthermore, endoscopic ultrasound was not available in all radiology services.
Adrenal lesions have been described in approximately 36% to 73% of patients with MEN1 (28, 29). They are usually diagnosed as incidentalomas during radiological screening, and most are non-functioning (30). Previous studies have shown that most of these lesions are less than 4 cm in diameter (29, 31, 32). Two individuals had adrenocortical adenomas with dimensions greater than 4 cm (II.4 of family 2 and II.1 of family 3). Probably, the patients in our study had a later diagnosis, which may justify this finding.
A genotype–phenotype correlation in MEN1 has been difficult to demonstrate even among family relatives harboring the same variants (33). It appears that mutations play an uncertain role in the clinical features of the disease, such as the age of onset, recurrence, or aggressiveness markers (34, 35). By observing the clinical manifestations of the 11 participants in our study, we noticed the heterogeneity of phenotypes presented among members of the same family. Nevertheless, genetic evaluation reduces the morbidity and mortality of individuals such as MEN1, as it allows to identify cases in the clinical spectrum and to receive treatment in the early stages of the disease (4, 6).
There are still little data about the clinical and genetic features of MEN1 in Brazil. Therefore, most patients may be symptomatic at diagnosis. Some studies report clinical and genetic screenings of patients with MEN1 conducted at Hospital das Clínicas in São Paulo, which has become one of the reference services for this disease in Brazil (8, 36, 37). In our center, as a University Hospital, we offer genetic testing to all patients with clinical evidence of FIPA, MEN1, and MEN2.
Some studies used the Sanger method or multiplex binding-dependent probe amplification (MLPA) for MEN1 gene screening (38–40). We chose to perform targeted HTS to identify the variants because it analyses multiple coding regions and splicing sites simultaneously of MEN1 gene and other genes associated with MEN1-like phenotypes as CDKN1B and AIP, respectively, causing MEN4 and FIPA. Therefore, information acquisition occurs with greater speed and lower estimated cost when compared to the Sanger method (7, 8).
A non-sense variant has been identified in family 1 (c.76G>T:p.Glu26Ter), causing an early stop codon in transcription and thus reducing the reading frame by more than 90%. There is a loss of functional domains, such as nuclear localization sequences (NLSs) located in the C-terminal portion of MENIN, NLS-1 (codons 479–497), and NLS-2 (codons 588–608). In addition, there is a loss of interaction sites of MENIN and JunD protein, a member of the AP1 transcription factor family (protein 1 activator) (41). This variant was first described in a Danish family with PHPT and carcinoid tumor of the duodenum (9). There is also a description of the same mutation in a Hungarian family with two individuals who had PHPT and prolactinoma (42), a similar phenotype found in two individuals (II.1 and II.2) of family 1 in our study. The novel missense variant in exon 2 (c.124G>C:p.Gly42Arg) identified in family 3 seems to also affect the binding site of MENIN protein with JunD. There are descriptions of mutations that occurred at the same position but with different amino acids (43, 44).
Angiofibromas, collagenomas, and lipomas are common in MEN1 (1, 45). The incidence of lipomas can range from 0.9% to 34% in these patients (46, 47) and may present with an atypical location such as in the intrathoracic region (48). Well-differentiated liposarcomas are malignant mesenchymal neoplasms that usually affect proximal regions of the limbs and the retroperitoneum, occurring more frequently between the fifth and seventh decades of life (49). In addition to lipomas in the right thoracic and clavicular regions, patient II.1 of family 1 had well-differentiated liposarcoma in the right shoulder, an association that has been previously reported in a single case (50). Amplifications involving the region of chromosome 12 (12q13-15) have been found in these tumors by some authors, mainly involving the oncogenes MDM2 and CDK4 (51).
MENIN also interacts with Smad3, one of the receptor-regulated elements that mediate TGF-b (52). It seems that impairment of the MENIN protein blocks the transcriptional effects mediated by Smad3 and TGF-b, which can lead to inadequate cell growth and tumor onset (5, 52). The variant found in family 4 (c.654+1G>T) occurs at a splicing site leading to an early stop codon, and it has been described in several individuals with MEN1. The first report was in an Australian family (11) and later described in a Brazilian study in which the patient had PHPT, prolactinoma, and gastrinoma (53), a similar phenotype to that found in one of the individuals in family 4 (III.2). A case report recently published also describes a young individual with the same variant in MEN1 who had a giant pituitary adenoma and intracranial hypertension (54), similar to individual III.2 (family 4). Functional studies using lymphoblastoid cell lines from an individual with PHPT and gastrinoma were performed to assess the pathophysiological implications of this variant. It has been shown that the mutant MENIN was not able to bind to Smad3, resulting in a blockage in the TGF-b signaling pathway and, consequently, affecting its inhibitory actions on cell growth (55).
A missense variant in exon 7 has been identified in family 2 (c.1021:p.Trp341Arg), which was previously described in two individuals with clinical MEN1: one with PHPT, GEP tract tumor and a non-functioning adrenal tumor (10) and another with PHPT, GEP tract tumor, and pituitary tumors (56). There is also another report of the same mutation in one individual with familial isolated hyperparathyroidism (FIHP) (34). Although FIHP is described as a distinct genetic entity, it is also believed to be a variant of other familial neoplastic syndromes in which PHPT is the main clinical manifestation, as in MEN1 (57). This milder MEN1 phenotype characterized by FIHP has been described in some families and may be more related to mutations located between exons 3 and 7 of the MENIN protein gene (58, 59). Even though all individuals in family 2 who participated in the study had PHPT, they also had other endocrine tumors that are part of the clinical condition of MEN1, such as pituitary adenomas secreting prolactin and GH, insulinoma, and adrenal tumors.
Our study has limitations, as we evaluated a small number of families. Also, there was heterogeneity in complementary exams performed during screening, as many were carried out in different radiology and pathology services. Although uncommon, we did not investigate deletions or duplications since multiplex ligation-dependent probe amplification (MLPA) or copy number variation (CNV) analysis was not performed. However, the study allowed the genotypic and phenotypic characterization of families with MEN1 in a follow-up at a tertiary center in Brasília.
In conclusion, MEN1 is a rare condition expressed by variable combinations of endocrine and non-endocrine tumors. Due to its heterogeneous phenotype, the diagnosis is late in most cases. Hence, these patients may already present with complications resulting from these tumors, which increases morbidity and mortality. Multicenter and prospective studies are important for a better understanding of the clinical and molecular characteristics of patients from our region to improve care in terms of diagnosis, treatment, and follow-up.
Data availability statement
The data presented in the study are deposited in the BioSample database repository, using the accession number PRJNA923744; The links to the individual samples are as follows: https://www.ncbi.nlm.nih.gov/sra/SRX19034472; https://www.ncbi.nlm.nih.gov/sra/SRX19034471; https://www.ncbi.nlm.nih.gov/sra/SRX19034470; https://www.ncbi.nlm.nih.gov/sra/SRX19034469.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee for Research on Human Beings of the Faculty of Health Sciences, University of Brasília. The patients/participants provided their written informed consent to participate in this study.
Author contributions
IM and LN were major contributions in writing the manuscript. IM, LN, LV, LdS and LdC analyzed the patient data. GB, PM and TR performed and analyzed the molecular data. All authors contributed to manuscript revision, read, and approved the submitted version.
Acknowledgments
The authors would like to thank the National Council of Research (CNPq), the Nucleus of Research Support (NAP), and the Sabin Institute, which performed some of the hormonal and genomic tests. The authors would also like to thank Dr. Cristiane Jeyce Gomes Lima for her help in reviewing the article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97(9):2990–3011. doi: 10.1210/jc.2012-1230
2. Marx SJ. Recent topics around multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2018) 103(4):1296–301. doi: 10.1210/jc.2017-02340
3. Kamilaris CDC, Stratakis CA. Multiple endocrine neoplasia type 1 (MEN1): An update and the significance of early genetic and clinical diagnosis. Front Endocrinol (2019) 10:339. doi: 10.3389/fendo.2019.00339
4. Brandi ML, Agarwal SK, Perrier ND, Lines KE, Valk GD, Thakker RV. Multiple endocrine neoplasia type 1: Latest insights. Endocr Rev (2021) 42(2):133–70. doi: 10.1210/endrev/bnaa031
5. Balogh K, Rácz K, Patócs A, Hunyady L. Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab (2006) 17(9):357–64. doi: 10.1016/j.tem.2006.09.004
6. Concolino P, Costella A, Capoluongo E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet (2016) 209(1-2):36–41. doi: 10.1016/j.cancergen.2015.12.002
7. Marini F, Giusti F, Brandi ML. Genetic test in multiple endocrine neoplasia type 1 syndrome: An evolving story. World J Exp Med (2015) 5(2):124–9. doi: 10.5493/wjem.v5.i2.124
8. Carvalho RA, Urtremari B, Jorge AAL, Santana LS, Quedas EPS, Sekiya T, et al. Germline mutation landscape of multiple endocrine neoplasia type 1 using full gene next-generation sequencing. Eur J Endocrinol (2018) 179(6):391–407. doi: 10.1530/EJE-18-0430
9. Jäger AC, Friis-Hansen L, Hansen TV, Eskildsen PC, Sølling K, Knigge U, et al. (2006). Characteristics of the Danish families with multiple endocrine neoplasia type 1. Molecular Cellular Endocrinol 249(1-2);123–32. doi: 10.1016/j.mce.2006.02.008
10. Giraud S, Zhang CX, Serova-Sinilnikova O, Wautot V, Salandre J, Buisson N, et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet (1998) 63(2):455–67. doi: 10.1086/301953
11. Teh BT, Kytölä S, Farnebo F, Bergman L, Wong FK, Weber G, et al. Mutation analysis of the MEN1 gene in multiple endocrine neoplasia type 1, familial acromegaly and familial isolated hyperparathyroidism. J Clin Endocrinol Metab (1998) 83(8):2621–6. doi: 10.1210/jcem.83.8.5059
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG laboratory quality assurance committee. standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
13. de Laat JM, van Leeuwaarde RS, Valk GD. The importance of an early and accurate MEN1 diagnosis. Front Endocrinol (2018) 9:533. doi: 10.3389/fendo.2018.00533
14. Yamazaki M, Suzuki S, Kosugi S, Okamoto T, Uchino S, Miya A, et al. MEN consortium of japan. delay in the diagnosis of multiple endocrine neoplasia type 1: typical symptoms are frequently overlooked. Endocr J (2012) 59(9):797–807. doi: 10.1507/endocrj.ej12-0071
15. Marini F, Giusti F, Brandi ML. Multiple endocrine neoplasia type 1: extensive analysis of a large database of Florentine patients. Orphanet J Rare Dis (2018) 13(1):205. doi: 10.1186/s13023-018-0938-8
16. Eller-Vainicher C, Chiodini I, Battista C, Viti R, Mascia ML, Massironi S, et al. Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity. J Bone Miner Res (2009) 24(8):1404–10. doi: 10.1359/jbmr.090304
17. Burgess JR, David R, Greenaway TM, Parameswaran V, Shepherd JJ. Osteoporosis in multiple endocrine neoplasia type 1: severity, clinical significance, relationship to primary hyperparathyroidism, and response to parathyroidectomy. Arch Surg (1999) 134(10):1119–23. doi: 10.1001/archsurg.134.10.1119
18. Kann PH, Bartsch D, Langer P, Waldmann J, Hadji P, Pfützner A, et al. Peripheral bone mineral density in correlation to disease-related predisposing conditions in patients with multiple endocrine neoplasia type 1. J Endocrinol Invest. (2012) 35(6):573–9. doi: 10.3275/7880
19. Lourenço DM Jr, Coutinho FL, Toledo RA, Montenegro FL, Correia-Deur JE, Toledo SP. Early-onset, progressive, frequent, extensive, and severe bone mineral and renal complications in multiple endocrine neoplasia type 1-associated primary hyperparathyroidism. J Bone Miner Res (2010) 25(11):2382–91. doi: 10.1002/jbmr.125
20. Maraghelli D, Giusti F, Marini F, Brandi ML. Bone tissue and mineral metabolism in hereditary endocrine tumors: clinical manifestations and genetic bases. Orphanet J Rare Dis (2020) 15(1):102. doi: 10.1186/s13023-020-01380-1
21. Giusti F, Cianferotti L, Boaretto F, Cetani F, Cioppi F, Colao A, et al. Multiple endocrine neoplasia syndrome type 1: institution, management, and data analysis of a nationwide multicenter patient database. Endocrine (2017) 58(2):349–59. doi: 10.1007/s12020-017-1234-4
22. Daly AF, Tichomirowa MA, Beckers A. The epidemiology and genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab (2009) 23(5):543–54. doi: 10.1016/j.beem.2009.05.008
23. Cohen-Cohen S, Brown DA, Himes BT, Wheeler LP, Ruff MW, Major BT, et al. Pituitary adenomas in the setting of multiple endocrine neoplasia type 1: a single-institution experience. J Neurosurg (2020) 134(3):1132–8. doi: 10.3171/2020.1.JNS193538
24. Beckers A, Betea D, Valdes Socin H, Stevenaert A. The treatment of sporadic versus MEN1-related pituitary adenomas. J Intern Med (2003) 253(6):599–605. doi: 10.1046/j.1365-2796.2003.01164.x
25. Al-Salameh A, Baudry C, Cohen R. Update on multiple endocrine neoplasia type 1 and 2. Presse Med (2018) 47(9):722–31. doi: 10.1016/j.lpm.2018.03.005
26. Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, et al. Groupe d’études des tumeurs endocrines. pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol (2008) 32(4):534–43. doi: 10.1097/PAS.0b013e31815ade45
27. Wu Y, Gao L, Guo X, Wang Z, Lian W, Deng K, et al. Pituitary adenomas in patients with multiple endocrine neoplasia type 1: a single-center experience in China. Pituitary (2019) 22(2):113–23. doi: 10.1007/s11102-019-00939-x
28. Barzon L, Pasquali C, Grigoletto C, Pedrazzoli S, Boscaro M, Fallo F. Multiple endocrine neoplasia type 1 and adrenal lesions. J Urol. (2001) 166(1):24–7. doi: 10.1016/S0022-5347(05)66068-5
29. Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, et al. Adrenal involvement in MEN1. analysis of 715 cases from the groupe d’etude des tumeurs endocrines database. Eur J Endocrinol (2012) 166(2):269–79. doi: 10.1530/EJE-11-0679
30. Ventura M, Melo M, Carrilho F. Outcome and long-term follow-up of adrenal lesions in multiple endocrine neoplasia type 1. Arch Endocrinol Metab (2019) 63(5):516–23. doi: 10.20945/2359-3997000000170
31. Waldmann J, Bartsch DK, Kann PH, Fendrich V, Rothmund M, Langer P. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg (2007) 392(4):437–43. doi: 10.1007/s00423-006-0124-7
32. Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg (2002) 26(8):891–6. doi: 10.1007/s00268-002-6492-4
33. Agarwal SK. The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer (2017) 24(10):T119–34. doi: 10.1530/ERC-17-0199
34. Wautot V, Vercherat C, Lespinasse J, Chambe B, Lenoir GM, Zhang CX, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat (2002) 20(1):35–47. doi: 10.1002/humu.10092
35. Kouvaraki MA, Lee JE, Shapiro SE, Gagel RF, Sherman SI, Sellin RV, et al. Genotype-phenotype analysis in multiple endocrine neoplasia type 1. Arch Surg (2002) 137(6):641–7. doi: 10.1001/archsurg.137.6.641
36. Lourenço DM Jr, Toledo RA, Coutinho FL, Margarido LC, Siqueira SA, dos Santos MA, et al. The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1. Clinics (Sao Paulo). (2007) 62(4):465–76. doi: 10.1590/S1807-59322007000400014
37. Lourenço DM Jr, Toledo RA, Mackowiak II, Coutinho FL, Cavalcanti MG, Correia-Deur JE, et al. Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. Eur J Endocrinol (2008) 159(3):259–74. doi: 10.1530/EJE-08-0153
38. Falchetti A. Genetics of multiple endocrine neoplasia type 1 syndrome: what’s new and what’s old. F1000Res (2017) 6:F1000 Faculty Rev–73. doi: 10.12688/f1000research.7230.1
39. Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjöld M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab (2007) 92(9):3389–95. doi: 10.1210/jc.2007-0476
40. Kong J, Wang O, Nie M, Shi J, Hu Y, Jiang Y, et al. Clinical and genetic analysis of multiple endocrine neoplasia type 1-related primary hyperparathyroidism in Chinese. PloS One (2016) 11(11):e0166634. doi: 10.1371/journal.pone.0166634
41. Knapp JI, Heppner C, Hickman AB, Burns AL, Chandrasekharappa SC, Collins FS, et al. Identification and characterization of JunD missense mutants that lack menin binding. Oncogene (2000) 19(41):4706–12. doi: 10.1038/sj.onc.1203832
42. Balogh K, Hunyady L, Patocs A, Gergics P, Valkusz Z, Toth M, et al (2007). MEN1 gene mutations in Hungarian patients with multiple endocrine neoplasia type 1. Clin Endocrinol (2007) 67(5):727–34. doi: 10.1111/j.1365-2265.2007.02953.x
43. Itoh M, Saikawa Y. A novel MEN1 mutation in a Japanese adolescent with multiple endocrine neoplasia type 1. Clin Pediatr Endocrinol (2017) 26(1):25–8. doi: 10.1297/cpe.26.25
44. Koehler VF, Jungheim K, Groß U, Iacovazzo D, Mann A, Korbonits M. Novel germline p.Gly42Val MEN1 mutation in a family with multiple endocrine neoplasia type 1 - excellent response of prolactinoma to cabergoline. Ann Clin Lab Sci (2017) 47(5):606–10.
45. Vidal A, Iglesias MJ, Fernández B, Fonseca E, Cordido F. Cutaneous lesions associated to multiple endocrine neoplasia syndrome type 1. J Eur Acad Dermatol Venereol. (2008) 22(7):835–8. doi: 10.1111/j.1468-3083.2008.02578.x
46. Agarwal S, Monsaert RP. Clinical review article: Multiple endocrine neoplasia type 1 and lipomas. Hosp Physician. (2002) 38(5):51–4.
47. Waguespack SG. Beyond the “3 ps”: A critical appraisal of the non-endocrine manifestations of multiple endocrine neoplasia type 1. Front Endocrinol (2022) 13:1029041. doi: 10.3389/fendo.2022.1029041
48. Sturiale A, Giudici F, Alemanno G, Cavalli T, Addasi R, Santomaggio C, et al. Massive intrathoracic lipoma in men1 syndrome. Int J Surg Case Rep (2015) 6:247–50. doi: 10.1016/j.ijscr.2014.10.071
49. Thway K. Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin Diagn Pathol (2019) 36(2):112–21. doi: 10.1053/j.semdp.2019.02.006
50. Johnson GJ, Summerskill WH, Anderson VE, Keating FR Jr. Clinical and genetic investigation of a large kindred with multiple endocrine adenomatosis. N Engl J Med (1967) 277(26):1379–85. doi: 10.1056/NEJM196712282772601
51. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol (2018) 36(2):151–9. doi: 10.1200/JCO.2017.74.9598
52. Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A. (2001) 98(7):3837–42. doi: 10.1073/pnas.061358098
53. Toledo RA, Lourenço DM Jr, Coutinho FL, Quedas E, Mackowiack I, Machado MC, et al. Novel MEN1 germline mutations in Brazilian families with multiple endocrine neoplasia type 1. Clin Endocrinol (2007) 67(3):377–84. doi: 10.1111/j.1365-2265.2007.02895.x
54. Dantas NCB, Soares CEL, Martins MRA, Lourenço DM Jr, Quidute ARP. Giant prolactinoma causing hydrocephalus and intracranial hypertension as first manifestations of multiple endocrine neoplasia type 1. Front Endocrinol (2019) 10:582. doi: 10.3389/fendo.2019.00582
55. Canaff L, Vanbellinghen JF, Kaji H, Goltzman D, Hendy GN. Impaired transforming growth factor-β (TGF-β) transcriptional activity and cell proliferation control of a menin in-frame deletion mutant associated with multiple endocrine neoplasia type 1 (MEN1). J Biol Chem (2012) 287(11):8584–97. doi: 10.1074/jbc.M112.341958
56. Tso AW, Rong R, Lo CY, Tan KC, Tiu SC, Wat NM, et al. Multiple endocrine neoplasia type 1 (MEN1): genetic and clinical analysis in the southern Chinese. Clin Endocrinol (Oxf). (2003) 59(1):129–35. doi: 10.1046/j.1365-2265.2003.01812.x
57. Isakov O, Rinella ES, Olchovsky D, Shimon I, Ostrer H, Shomron N, et al. Missense mutation in the MEN1 gene discovered through whole exome sequencing co-segregates with familial hyperparathyroidism. Genet Res (2013) 95(4):114–20. doi: 10.1017/S0016672313000141
58. Kassem M, Kruse TA, Wong FK, Larsson C, Teh BT. Familial isolated hyperparathyroidism as a variant of multiple endocrine neoplasia type 1 in a large Danish pedigree. J Clin Endocrinol Metab (2000) 85(1):165–7. doi: 10.1210/jcem.85.1.6299
Keywords: multiple endocrine neoplasia type 1, primary hyperparathyroidism, pituitary adenoma, gastroenteropancreatic tumor, menin gene, variants, molecular test
Citation: Miranda ISdM, Valadares LP, Barra GB, Mesquita PG, de Santana LB, de Castro LF, Rita THS and Naves LA (2023) Clinical and molecular features of four Brazilian families with multiple endocrine neoplasia type 1. Front. Endocrinol. 14:1117873. doi: 10.3389/fendo.2023.1117873
Received: 07 December 2022; Accepted: 20 February 2023;
Published: 10 March 2023.
Edited by:
Marialuisa Appetecchia, Hospital Physiotherapy Institutes (IRCCS), ItalyReviewed by:
Delmar Muniz Lourenco Jr., Clinical Hospital, University of São Paulo, BrazilPauline Romanet, Aix-Marseille Université, France
Bernadette Van Nesselrooij, University Medical Center Utrecht, Netherlands
Copyright © 2023 Miranda, Valadares, Barra, Mesquita, de Santana, de Castro, Rita and Naves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabella Santiago de Melo Miranda, aXNhYmVsbGFzYW50aWFnb19AaG90bWFpbC5jb20=