 William F. Simonds
William F. Simonds- Metabolic Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
Cushing’s syndrome (CS) resulting from endogenous hypercortisolism can be sporadic or can occur in the context of familial disease because of pituitary or extra-pituitary neuroendocrine tumors. Multiple endocrine neoplasia type 1 (MEN1) is unique among familial endocrine tumor syndromes because hypercortisolism in this context can result from pituitary, adrenal, or thymic neuroendocrine tumors and can therefore reflect either ACTH-dependent or ACTH-independent pathophysiologies. The prominent expressions of MEN1 include primary hyperparathyroidism, tumors of the anterior pituitary, gastroenteropancreatic neuroendocrine tumors, and bronchial carcinoid tumors along with several common non-endocrine manifestations such as cutaneous angiofibromas and leiomyomas. Pituitary tumors are present in about 40% of MEN1 patients, and up to 10% of such tumors secrete ACTH that can result in Cushing’s disease. Adrenocortical neoplasms occur frequently in MEN1. Although such adrenal tumors are mostly clinically silent, this category can include benign or malignant tumors causing hypercortisolism and CS. Ectopic tumoral ACTH secretion has also been observed in MEN1, almost exclusively originating from thymic neuroendocrine tumors. The range of clinical presentations, etiologies, and diagnostic challenges of CS in MEN1 are reviewed herein with an emphasis on the medical literature since 1997, when the MEN1 gene was identified.
Introduction
Endogenous Cushing’s syndrome (CS) can occur either sporadically or else within families as a result of pituitary or nonpituitary neuroendocrine tumors (1). Among familial endocrine tumor syndromes, multiple endocrine neoplasia type 1 (MEN1) is distinctive because CS in MEN1 can result from both ACTH-dependent and ACTH-independent causes (Figure 1). The former category includes Cushing’s disease (CD) and ectopic tumoral ACTH secretion.
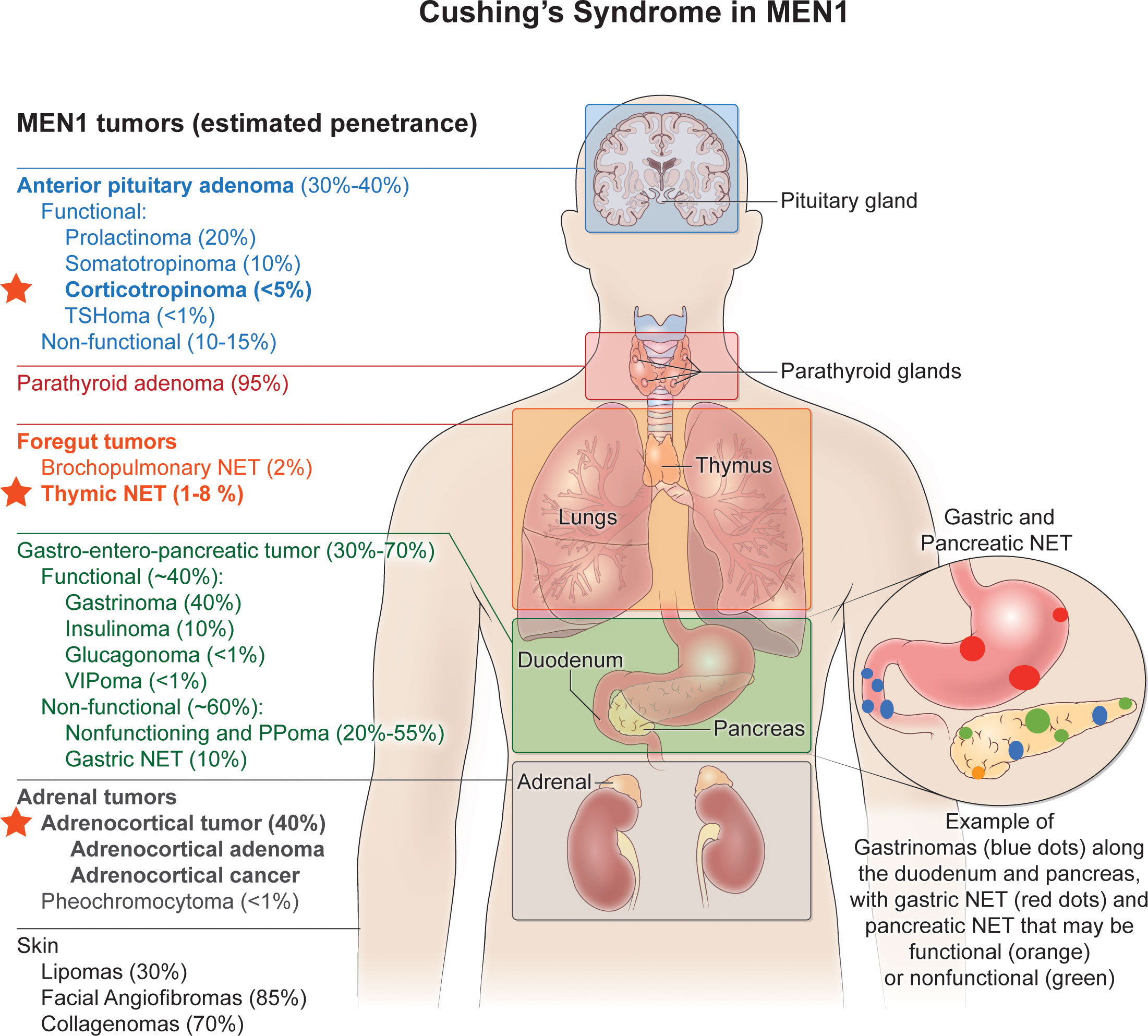
Figure 1 Cushing’s syndrome in MEN1. Multiple endocrine neoplasia type 1 (MEN1) is distinctive among familial endocrine tumor syndromes since Cushing’s syndrome in MEN1 can result from both ACTH-dependent and ACTH-independent causes. The former category includes Cushing’s disease (CD) and ectopic tumoral ACTH secretion, and the latter includes adrenocortical adenomas or carcinomas that secrete cortisol. The red stars indicate the MEN1-associated endocrine tumors that have been associated with either ACTH-dependent (pituitary corticotropinoma; thymic NETs) or ACTH-independent (adrenocortical adenoma or cancer) Cushing’s syndrome. See text for further details. Endocrine and non-endocrine tumors typical of MEN1 are listed on the left, with their estimated penetrance given in the parentheses. MEN1-associated endocrine tumors that can cause Cushing’s syndrome are in bold font. TSHoma, thyrotropinoma; NET, neuroendocrine tumor; PPoma, endocrine tumor that secretes pancreatic polypeptide.
MEN1 is an autosomal dominant familial cancer predisposition syndrome whose most prominent expressions include multiglandular primary hyperparathyroidism, anterior pituitary tumors, foregut carcinoid tumors, and tumors of gastroenteropancreatic neuroendocrine cells (2). Anterior pituitary tumors occur in about half of MEN1 patients; and a small fraction of these secrete ACTH (3). Both non-functional and functional adrenocortical tumors occur in MEN1, and among these are included benign and malignant functional tumors causing CS (4). Ectopic CS from ACTH-secreting thymic carcinoids has also been reported in MEN1 (5–8). The MEN1 syndrome also includes non-endocrine tumors in several tissues (9).
This monograph provides a review and update on the range of clinical presentations, etiologies, and diagnostic challenges of CS in MEN1 patients, emphasizing the medical literature over the last two and a half decades since the cloning of the MEN1 gene (10).
Cushing’s disease in MEN1
Tumors of the anterior pituitary tumor occur in about 30 to 40% of MEN1 patients. Approximately 4-10% of these pituitary tumors secrete ACTH (3, 11–13). Such tumors can secrete only ACTH or be pluri-hormonal. At the time of presentation, the pituitary tumors in MEN1 patients with CD may be either macro- or microadenomas. Which presentation may depend on how early in the disease course patients develop symptoms and seek medical attention. Another factor determining the size at initial detection of CD-associated pituitary adenomas is the extent and frequency of tumor surveillance in patients known to have, or to be at risk for, MEN1. Only a minority of MEN1 patients with CD develop visual symptoms or cavernous sinus invasion (12).
Although rare in sporadic CD, the presence of multiple synchronous or metachronous pituitary tumors may be discovered during the course of evaluation and/or surgery for CD in patients with MEN1. In a retrospective review of a large series of MEN1 patients that included 19 patients with CS, three of 11 MEN1 patients with CD (27%) had additional non-ACTH secreting pituitary microadenomas identified at surgery (14). This represented an incidence of multiple pituitary adenomas significantly higher than both that reported previously in a series of unselected sporadic pituitary adenomas (3 out of 116; 2.6%) (15) and that found in a large series of mainly sporadic CD cases (11 out of 658 total, excluding two MEN1 patients; approx. 2%) (16). Others have reported the presence of multiple synchronous or metachronous pituitary tumors in MEN1 patients both with and without CD (17–19).
Cushing’s disease can present in childhood or adolescence as the initial manifestation of MEN1 (Table 1) (20–22). The presenting signs and symptoms frequently include growth delay or retardation, weight gain and truncal obesity, headaches, and plethora. In females, hirsutism or menstrual irregularities may also be present at the time of diagnosis. Biochemical manifestations of hypercortisolism and pituitary imaging and localization studies resemble those of adults with CD (20–22).
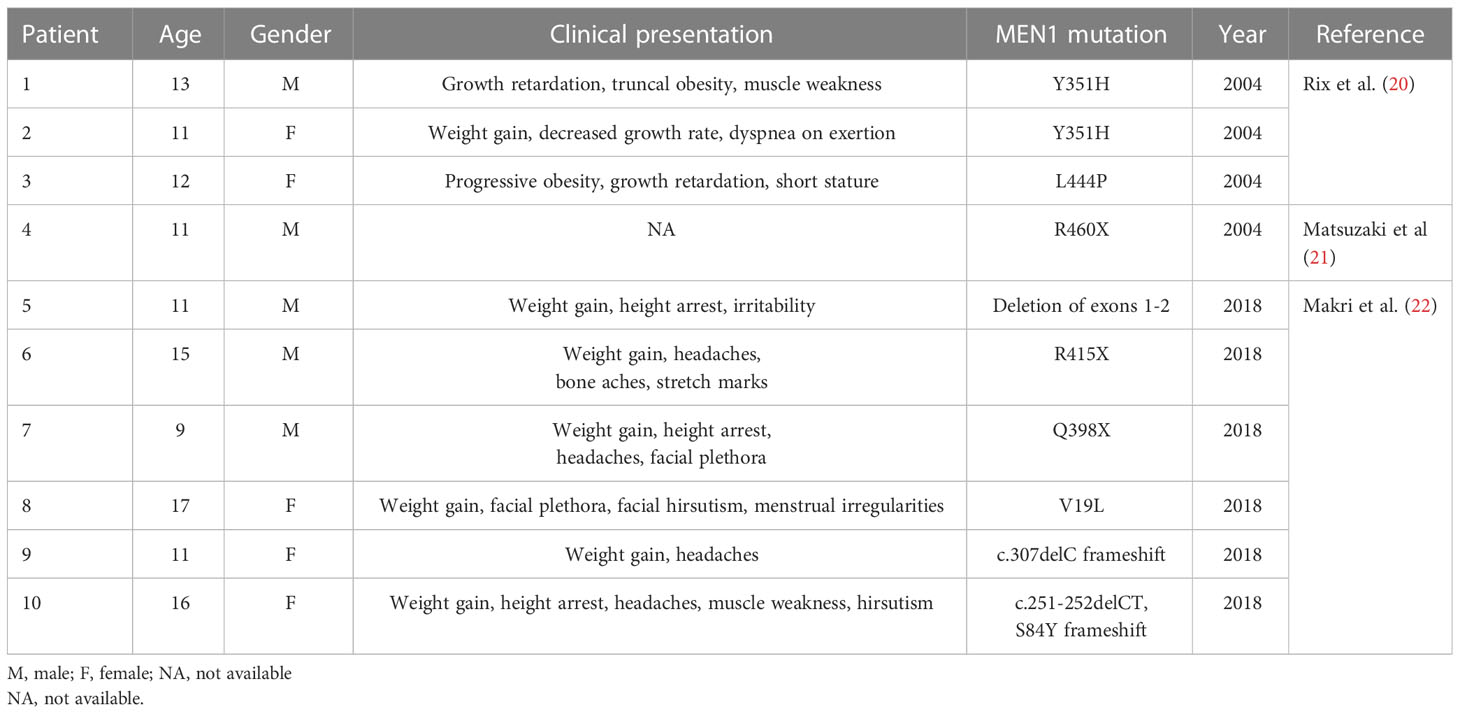
Table 1 Cushing’s disease as the presenting manifestation of MEN1.
In a large study of pediatric and adolescent patients with pituitary adenomas screened for predisposing germline mutations, two adolescents with recurrent or difficult-to-treat CD were found to harbor germline MEN1 mutations among 74 subjects with CD (23). In a large clinical and molecular analysis of 235 younger patients with apparently sporadic pituitary adenomas, two patients who presented with CD in their 20s were identified with the same germline variant of unknown significance in the MEN1 gene (24). It is controversial whether this particular variant, that encodes a R171Q change in the menin protein coding sequence, should be considered a polymorphism or mutation (25). A retrospective study of 80 children and adolescents with MEN1 included 18 patients with pituitary adenomas but none was found to have CD (26).
Somatic mutation of the MEN1 gene in sporadic pituitary tumors appears to be rare, including in those patients with sporadic CD (27). This contrasts with findings in the sporadic counterparts to other tumors typical of the MEN1 syndrome, such as parathyroid adenomas (28–32) and gastrinomas (33, 34) in which somatic MEN1 gene mutation is very common.
ACTH-independent Cushing’s syndrome in MEN1 due to adrenal disease
Besides functional tumors of pituitary corticotropes that may cause CS in MEN1, CS can also result from ACTH-independent causes originating in the adrenals. Adrenal gland enlargement and adrenal nodularity are very common in MEN1 (35–37). Although most adrenal lesions are non-functional, ACTH-independent CS due to benign or malignant adrenal tumors has been well-documented in MEN1 (14, 35, 38). Besides the more common benign adrenal tumors resulting in CS, adrenocortical carcinoma (ACC) has been previously reported in MEN1 patients with CS (4, 35, 38–40). For example, in a prospective series of 38 patients with proven germline MEN1 mutation, two had functional adrenal tumors resulting in CS and/or virilization: one had bilateral adenomas and one had ACC (35). In a retrospective analysis of MEN1 patients that included 19 patients with CS, three of 14 subjects with an identifiable cause for their CS (21%) had ACTH-independent CS due to adrenal tumors, two of these three from ACC (14). In general ACC is greater than 10-fold more common among MEN1 adrenal lesions than among sporadic adrenal incidentalomas (38).
ACTH-independent Cushing’s syndrome due to adrenal neoplasia can represent the presenting manifestation of MEN1. A previously healthy 16-year-old girl, lacking a known family history of MEN1, presented with progressive weight gain, hirsutism, acne, irregular menses, extensive skin striae, fatigue, and muscle weakness associated with a cortisol-producing adrenal adenoma, leading to identification of a frameshift mutation in MEN1 in the proband and multiple family members (41). A 43-year-old woman who presented with amenorrhea, hirsutism, and acne, associated with a large adrenal mass, was subsequently shown to have ACC, biochemical CS, and MEN1 (42).
Even though loss of heterozygosity at the MEN1 locus on chromosome 11q13 can be frequently documented in ACC, somatic mutation of the MEN1 gene in sporadic benign or malignant adrenocortical tumors appears to be rare (43–45). Widely metastatic ACC associated with somatic mutations in both TP53 and MEN1 and rapidly progressing CS has been reported however (46).
Less commonly, primary aldosteronism due to the presence of adrenocortical aldosteronoma has been documented in association with the MEN1 syndrome (38, 47, 48). In fact, cases of primary aldosteronism are more common among MEN1 adrenal lesions than among sporadic adrenal incidentalomas (38).
Cushing’s syndrome due to ectopic ACTH secretion in MEN1
Another cause of ACTH-dependent CS in MEN1 patients, apart from ACTH-secreting pituitary tumors causing CD, are extrapituitary neuroendocrine tumors that secrete ACTH ectopically. Ectopic production of ACTH causing CS is a recognized paraneoplastic phenomenon that has been documented in association with a wide variety of endocrine and non-endocrine neoplasms and that presents significant diagnostic and therapeutic challenges (49). That ectopic production of ACTH causing CS is rare in MEN1 is supported by the absence of any such patient among the 90 cases of ectopic CS reported in a large 20-year retrospective review (50). Similarly, no MEN1 patients were among 12 cases with ectopic CS and thymic neuroendocrine tumors (NETs) who underwent surgical resection in another large retrospective study from a single institution (51).
In the context of MEN1, CS resulting from ectopic ACTH production has nevertheless been associated almost exclusively with the presence of thymic NETs (“thymic carcinoids”) (5–8). Thymic NETs have been found in some 1 to 8% of MEN1 patients in several large series and are a major cause of morbidity and mortality (52–56). Risk factors for the development of thymic NETs include male gender and a history of smoking (52, 54, 56).
MEN1-related thymic NETs, including those associated with the ectopic production of ACTH causing CS, can be aggressive in nature with a high potential for metastasis and are associated with an increased mortality (53, 57). Analysis of tumor tissue from a patient with MEN1 operated on twice for a thymic NET associated with the ectopic production of ACTH causing CS showed an initial Ki-67 labeling index of 5% but on re-operation the tumor was found to be invasive with a Ki-67 index of 30% (7). Another patient with germline mutation-proven MEN1 associated with thymic NET and ectopic CS was found at surgery to have an unresectable, invasive anterior mediastinal mass with a Ki-67 index of >50% (58). Although not well studied in the context of MEN1, such high Ki-67 indices and the increase in Ki-67 index noted on sequential operations likely represent the dominance over time of the more aggressive MEN1-null thymic neuroendocrine tumor clones. Thymectomy in a male MEN1 patient with a smoking history, a thymic NET, and ectopic CS revealed tumor invasion of the right atrium and metastases to the mediastinal lymph nodes- the patient eventually died from metastatic disease (5).
Rarely, CS from the ectopic production of ACTH from thymic NET can be the presenting manifestation of MEN1. Amoli and co-workers described a 29-year-old man who presented with psychosis due to ectopic CS associated with a large anterior mediastinal cystic mass with diffuse lymphadenopathy (59). After excision of a large, invasive, thymic NET, the patient experienced an initial period of remission, but CS recurred after 2 years, this time associated with primary hyperparathyroidism and hyperprolactinemia, and a clinical and genetic diagnosis of MEN1 was confirmed (59).
Thymic NET, including cases associated with CS due to the ectopic production of ACTH, can cluster in MEN1 families (54, 56, 60). Two of four siblings with MEN1 in Chinese kindred developed ectopic CS due to the production of ACTH from thymic NETs, and a third sibling with MEN1 had a thymic NET that stained positive for ACTH upon immunohistochemical analysis (8).
Finding an etiology for Cushing’s syndrome in MEN1
Because of the potential clinical complexity in MEN1 and the fact that hypercortisolism can result from pituitary, adrenal, or other endocrine tumors, finding an etiology for MEN1-associated CS be challenging. In a retrospective series of 19 MEN1 patients with CS evaluated at one institution, an etiology could not be found in five patients, and among these the hypercortisolism appeared to resolve spontaneously in three (14). Al Brahim et al. describe an MEN1 patient with CS and two pituitary adenomas, one a corticotrope microadenoma, as well as ectopic production of corticotropin-releasing hormone from a thymic NET (17). Contributing to the diagnostic challenge is the recognition that periodic or cyclic CS may occur with all etiologies of CS (61).
Discussion
CS is an uncommon manifestation of the autosomal dominant familial cancer predisposition syndrome MEN-1 that can be caused by both ACTH-dependent and independent mechanisms of hypercortisolism. It is therefore important to consider all etiologies for patients with MEN and CS, since hypercortisolism due to CD, adrenal etiologies, and the ectopic secretion of ACTH are well-documented in MEN1 just as in non-familial cases.
In MEN1, the presence of multiple synchronous or metachronous pituitary tumors may be discovered during pre-operative evaluation or surgery for CD, consistent with other “multiplicities” that characterize this familial tumor syndrome. Such potential pituitary tumor multifocality must be kept in mind during preoperative tumor localization for CD in MEN1 patients, since visualized pituitary lesions may not necessarily correspond to the culprit functional corticotrope adenoma.
Since both CD and ACTH-independent Cushing’s syndrome due to adrenal neoplasia can present in childhood or adolescence as its initial manifestation, MEN1 must be considered for all younger patients diagnosed with CS, especially in those with a potentially relevant family medical history. Early recognition of MEN1 in such patients could be beneficial by leading to earlier surveillance for other functional or non-functional endocrine and other tumors that might impact growth, development, and/or sexual maturation.
Even though most adrenal lesions are non-functional, adrenal gland enlargement and adrenal nodularity are very common in MEN1. Careful functional evaluation and determination of tumor imaging characteristics of adrenal lesions > 1 cm is important in MEN1, especially keeping in mind that ACC is > 10-fold more common among MEN1 adrenal lesions than among sporadic cases. That being the case, benign adrenal adenomas are still much more common among MEN1 patients with functional adrenal masses resulting in ACTH-independent CS.
Thymic NETs are the manifestation of MEN1 most often associated with the ectopic secretion of ACTH resulting in CS. Unfortunately, MEN1-related thymic NETs are frequently aggressive in nature with a high potential for metastasis and are associated with an increased mortality. This has led to recommendations for the performance of a prophylactic transcervical, near-total thymectomy concurrent with parathyroid surgery in MEN1 (62, 63), however strong data supporting the preventive efficacy of this intervention may be lacking (64). There have been rare reports of CS, extrapituitary in etiology but often difficult to distinguish from CD, resulting from ectopic production of corticotropin-releasing hormone (CRH), from pheochromocytomas, medullary thyroid cancers, and other neuroendocrine tumors (65–68). To date however, no such cases have been reported in the context of MEN1.
Because of the inherent endocrine complexity in many cases, it may not always be possible to determine the etiology of endogenous CS in MEN1. Yet we now have available advanced diagnostic tools that include bilateral inferior petrosal sinus sampling (69), optimized MRI protocols for pituitary lesions, such as fat-suppressed volumetric interpolated breath-hold examination (VIBE) imaging (70), high resolution computed tomography, functional imaging with somatostatin analogs and 18F-DOPA, and dynamic testing with desmopressin (71) and corticotrophin-releasing hormone. With this armamentarium, the etiology for most cases of CS in MEN1 should be determinable, especially when combined with careful observation and the clinical acumen of experienced medical endocrinologists, interventional radiologists, and endocrine surgeons. An even higher degree of diagnostic success may emerge from the future utilization of advanced artificial intelligence and machine learning algorithms, already being successfully applied in the field of endocrinology and metabolism (72, 73).
Author contributions
WS conceived, organized, and wrote the manuscript.
Funding
The intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases funded this study (ZIA DK043012-22).
Acknowledgments
We thank past and present members of the Metabolic Diseases Branch, NIDDK, Mr. Craig Cochran and the nurses and fellows of the endocrinology wards at the NIH Clinical Center for their excellent patient care.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367(9522):1605–17. doi: 10.1016/S0140-6736(06)68699-6
2. Marx SJ. Multiple endocrine neoplasia type 1. In: Kinzler KW, editor. The genetic basis of human cancer, 2nd ed. New York: McGraw-Hill (2002). p. 475–500.
3. Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab (2002) 87(2):457–65. doi: 10.1210/jcem.87.2.8145
4. Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg (2002) 26(8):891–6. doi: 10.1007/s00268-002-6492-4
5. Takagi J, Otake K, Morishita M, Kato H, Nakao N, Yoshikawa K, et al. Multiple endocrine neoplasia type I and cushing’s syndrome due to an aggressive ACTH producing thymic carcinoid. Intern Med (2006) 45(2):81–6. doi: 10.2169/internalmedicine.45.1427
6. Yano M, Fukai I, Kobayashi Y, Mizuno K, Konishi A, Haneda H, et al. ACTH-secreting thymic carcinoid associated with multiple endocrine neoplasia type 1. Ann Thorac Surg (2006) 81(1):366–8. doi: 10.1016/j.athoracsur.2004.07.084
7. Ghazi AA, Dezfooli AA, Mohamadi F, Yousefi SV, Amirbaigloo A, Ghazi S, et al. Cushing syndrome secondary to a thymic carcinoid tumor due to multiple endocrine neoplasia type 1. Endocr Pract (2011) 17(4):e92–6. doi: 10.4158/EP11038.CR
8. Li X, Su J, Zhao L, Wu J, Ding X, Fang F, et al. Familial cushing syndrome due to thymic carcinoids in a multiple endocrine neoplasia type 1 kindred. Endocrine (2014) 47(1):183–90. doi: 10.1007/s12020-013-0141-6
9. Waguespack SG. Beyond the “3 ps”: a critical appraisal of the non-endocrine manifestations of multiple endocrine neoplasia type 1. Front Endocrinol (Lausanne) (2022) 13:1029041. doi: 10.3389/fendo.2022.1029041
10. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science (1997) 276(5311):404–7. doi: 10.1126/science.276.5311.404
11. Shyamasunder AH, Pai R, Ramamoorthy H, Sakhti D, Manipadam MT, Kapoor N, et al. Clinical profile and mutations associated with multiple endocrine neoplasia-Type1 (MEN1) and their first-degree relatives at risk of developing MEN1: a prospective study. Horm Metab Res (2021) 53(4):245–56. doi: 10.1055/a-1402-0183
12. Cohen-Cohen S, Brown DA, Himes BT, Wheeler LP, Ruff MW, Major BT, et al. Pituitary adenomas in the setting of multiple endocrine neoplasia type 1: a single-institution experience. J Neurosurg (2020) 134(3):1132–8. doi: 10.3171/2020.1.JNS193538
13. Wu Y, Gao L, Guo X, Wang Z, Lian W, Deng K, et al. Pituitary adenomas in patients with multiple endocrine neoplasia type 1: a single-center experience in China. Pituitary (2019) 22(2):113–23. doi: 10.1007/s11102-019-00939-x
14. Simonds WF, Varghese S, Marx SJ, Nieman LK. Cushing’s syndrome in multiple endocrine neoplasia type 1. Clin Endocrinol (2012) 76(3):379–86. doi: 10.1111/j.1365-2265.2011.04220.x
15. Magri F, Villa C, Locatelli D, Scagnelli P, Lagonigro MS, Morbini P, et al. Prevalence of double pituitary adenomas in a surgical series: clinical, histological and genetic features. J Endocrinol Invest (2010) 33(5):325–31. doi: 10.3275/671610.1007/BF03346594
16. Ratliff JK, Oldfield EH. Multiple pituitary adenomas in cushing’s disease. J Neurosurg (2000) 93(5):753–61. doi: 10.3171/jns.2000.93.5.0753
17. Al Brahim NY, Rambaldini G, Ezzat S, Asa SL. Complex endocrinopathies in MEN-1: diagnostic dilemmas in endocrine oncology. Endocr Pathol (2007) 18(1):37–41. doi: 10.1007/s12022-007-0008-6
18. Mete O, Alshaikh OM, Cintosun A, Ezzat S, Asa SL. Synchronous multiple pituitary neuroendocrine tumors of different cell lineages. Endocr Pathol (2018) 29(4):332–8. doi: 10.1007/s12022-018-9545-4
19. Sahdev A, Jager R. Bilateral pituitary adenomas occurring with multiple endocrine neoplasia type one. AJNR Am J Neuroradiol (2000) 21(6):1067–9.
20. Rix M, Hertel NT, Nielsen FC, Jacobsen BB, Hoejberg AS, Brixen K, et al. Cushing’s disease in childhood as the first manifestation of multiple endocrine neoplasia syndrome type 1. Eur J Endocrinol (2004) 151(6):709–15. doi: 10.1530/eje.0.1510709
21. Matsuzaki LN, Canto-Costa MH, Hauache OM. Cushing’s disease as the first clinical manifestation of multiple endocrine neoplasia type 1 (MEN1) associated with an R460X mutation of the MEN1 gene. Clin Endocrinol (2004) 60(1):142–3. doi: 10.1111/j.1365-2265.2004.01943.x
22. Makri A, Bonella MB, Keil MF, Hernandez-Ramirez L, Paluch G, Tirosh A, et al. Children with MEN1 gene mutations may present first (and at a young age) with cushing disease. Clin Endocrinol (2018) 89(4):437–43. doi: 10.1111/cen.13796
23. Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet (2010) 78(5):457–63. doi: 10.1111/j.1399-0004.2010.01406.x
24. Martinez de LaPiscina I, Portillo Najera N, Rica I, Gaztambide S, Webb SM, Santos A, et al. Clinical and genetic characteristics in patients under 30 years with sporadic pituitary adenomas. Eur J Endocrinol (2021) 185(4):485–96. doi: 10.1530/EJE-21-0075
25. De Carlo E, Pilon C, Zatelli MC, degli Uberti EC, Fallo F. Isolated R171Q amino acid change in MEN1 gene: polymorphism or mutation? Clin Endocrinol (2008) 69(3):511. doi: 10.1111/j.1365-2265.2008.03193.x
26. Shariq OA, Lines KE, English KA, Jafar-Mohammadi B, Prentice P, Casey R, et al. Multiple endocrine neoplasia type 1 in children and adolescents: clinical features and treatment outcomes. Surgery (2022) 171(1):77–87. doi: 10.1016/j.surg.2021.04.041
27. Poncin J, Stevenaert A, Beckers A. Somatic MEN1 gene mutation does not contribute significantly to sporadic pituitary tumorigenesis. Eur J Endocrinol (1999) 140(6):573–6. doi: 10.1530/eje.0.1400573
28. Miedlich S, Krohn K, Lamesch P, Muller A, Paschke R. Frequency of somatic MEN1 gene mutations in monoclonal parathyroid tumours of patients with primary hyperparathyroidism. Eur J Endocrinol (2000) 143(1):47–54. doi: 10.1530/eje.0.1430047
29. Uchino S, Noguchi S, Sato M, Yamashita H, Yamashita H, Watanabe S, et al. Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res (2000) 60(19):5553–7.
30. Scarpelli D, D’Aloiso L, Arturi F, Scillitani A, Presta I, Bisceglia M, et al. Novel somatic MEN1 gene alterations in sporadic primary hyperparathyroidism and correlation with clinical characteristics. J Endocrinol Invest (2004) 27(11):1015–21. doi: 10.1007/BF03345303
31. Vierimaa O, Villablanca A, Alimov A, Georgitsi M, Raitila A, Vahteristo P, et al. Mutation analysis of MEN1, HRPT2, CASR, CDKN1B, and AIP genes in primary hyperparathyroidism patients with features of genetic predisposition. J Endocrinol Invest (2009) 32(6):512–8. doi: 10.1007/BF03346498
32. Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet (1997) 16(4):375–8. doi: 10.1038/ng0897-375
33. Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham TA, Wang C, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res (1997) 57(21):4682–6.
34. Kawamura J, Shimada Y, Komoto I, Okamoto H, Itami A, Doi R, et al. Multiple endocrine neoplasia type 1 gene mutations in sporadic gastrinomas in Japan. Oncol Rep (2005) 14(1):47–52.
35. Waldmann J, Bartsch DK, Kann PH, Fendrich V, Rothmund M, Langer P. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg (2007) 392(4):437–43. doi: 10.1007/s00423-006-0124-7
36. Schaefer S, Shipotko M, Meyer S, Ivan D, Klose KJ, Waldmann J, et al. Natural course of small adrenal lesions in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Eur J Endocrinol (2008) 158(5):699–704. doi: 10.1530/EJE-07-0635
37. Whitley SA, Moyes VJ, Park KM, Brooke AM, Grossman AB, Chew SL, et al. The appearance of the adrenal glands on computed tomography in multiple endocrine neoplasia type 1. Eur J Endocrinol (2008) 159(6):819–24. doi: 10.1530/EJE-08-0516
38. Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, et al. Adrenal involvement in MEN1. analysis of 715 cases from the groupe d’etude des tumeurs endocrines database. Eur J Endocrinol (2012) 166(2):269–79. doi: 10.1530/EJE-11-0679
39. Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Theodorsson E, et al. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (1992) 75(1):76–81. doi: 10.1210/jcem.75.1.1352309
40. Skogseid B, Rastad J, Gobl A, Larsson C, Backlin K, Juhlin C, et al. Adrenal lesion in multiple endocrine neoplasia type 1. Surgery (1995) 118(6):1077–82. doi: 10.1016/s0039-6060(05)80117-5
41. Alzahrani AS, Al-Khaldi N, Shi Y, Al-Rijjal RA, Zou M, Baitei EY, et al. Diagnosis by serendipity: cushing syndrome attributable to cortisol-producing adrenal adenoma as the initial manifestation of multiple endocrine neoplasia type 1 due to a rare splicing site MEN1 gene mutation. Endocr Pract (2008) 14(5):595–602. doi: 10.4158/EP.14.5.595
42. Kharb S, Pandit A, Gundgurthi A, Garg MK, Brar KS, Kannan N, et al. Hidden diagnosis of multiple endocrine neoplasia-1 unraveled during workup of virilization caused by adrenocortical carcinoma. Indian J Endocrinol Metab (2013) 17(3):514–8. doi: 10.4103/2230-8210.111672
43. Heppner C, Reincke M, Agarwal SK, Mora P, Allolio B, Burns AL, et al. MEN1 gene analysis in sporadic adrenocortical neoplasms. J Clin Endocrinol Metab (1999) 84(1):216–9. doi: 10.1210/jcem.84.1.5388
44. Gortz B, Roth J, Speel EJ, Krahenmann A, De Krijger RR, Matias-Guiu X, et al. MEN1 gene mutation analysis of sporadic adrenocortical lesions. Int J Cancer (1999) 80(3):373–9. doi: 10.1002/(sici)1097-0215(19990129)80:3<373::aid-ijc7>3.0.co;2-b
45. Schulte KM, Heinze M, Mengel M, Simon D, Scheuring S, Kohrer K, et al. MEN I gene mutations in sporadic adrenal adenomas. Hum Genet (1999) 105(6):603–10. doi: 10.1007/s004399900193
46. Schreiber AR, Kar A, Goodspeed AE, Pozdeyev N, Somerset H, Raeburn CD, et al. Leptomeningeal metastasis from adrenocortical carcinoma: a case report. J Endocr Soc (2020) 4(3):bvaa017. doi: 10.1210/jendso/bvaa017
47. Zhu Y, Zhang HB, Lu L, Li NS, Fu Y, Wang O, et al. [Adrenal lesions in patients of multiple endocrine neoplasia type 1]. Zhonghua Yi Xue Za Zhi (2010) 90(38):2689–92.
48. Parisien-La Salle S, Corbeil G, El-Haffaf Z, Duranceau C, Latour M, Karakiewicz PI, et al. Genetic dissection of primary aldosteronism in a patient with MEN1 and ipsilateral adrenocortical carcinoma and adenoma. J Clin Endocrinol Metab (2022) 108(1):26–32. doi: 10.1210/clinem/dgac564
49. Alba EL, Japp EA, Fernandez-Ranvier G, Badani K, Wilck E, Ghesani M, et al. The mount Sinai clinical pathway for the diagnosis and management of hypercortisolism due to ectopic ACTH syndrome. J Endocr Soc (2022) 6(7):bvac073. doi: 10.1210/jendso/bvac073
50. Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the national institutes of health. J Clin Endocrinol Metab (2005) 90(8):4955–62. doi: 10.1210/jc.2004-2527
51. Neary NM, Lopez-Chavez A, Abel BS, Boyce AM, Schaub N, Kwong K, et al. Neuroendocrine ACTH-producing tumor of the thymus–experience with 12 patients over 25 years. J Clin Endocrinol Metab (2012) 97(7):2223–30. doi: 10.1210/jc.2011-3355
52. Teh BT, McArdle J, Chan SP, Menon J, Hartley L, Pullan P, et al. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Med (Baltimore) (1997) 76(1):21–9. doi: 10.1097/00005792-199701000-00002
53. Gibril F, Chen YJ, Schrump DS, Vortmeyer A, Zhuang Z, Lubensky IA, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2003) 88(3):1066–81. doi: 10.1210/jc.2002-021314
54. Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, Avenia N, et al. Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series. J Clin Endocrinol Metab (2005) 90(5):2603–9. doi: 10.1210/jc.2004-1155
55. Christakis I, Qiu W, Silva Figueroa AM, Hyde S, Cote GJ, Busaidy NL, et al. Clinical features, treatments, and outcomes of patients with thymic carcinoids and multiple endocrine neoplasia type 1 syndrome at MD Anderson cancer center. Hormones Cancer (2016) 7(4):279–87. doi: 10.1007/s12672-016-0269-y
56. Goudet P, Murat A, Cardot-Bauters C, Emy P, Baudin E, du Boullay Choplin H, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des tumeurs endocrines). World J Surg (2009) 33(6):1197–207. doi: 10.1007/s00268-009-9980-y
57. Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, et al. Risk factors and causes of death in MEN1 disease. a GTE (Groupe d’Etude des tumeurs endocrines) cohort study among 758 patients. World J Surg (2010) 34(2):249–55. doi: 10.1007/s00268-009-0290-1
58. Asemota IR, Ajiboye O, Nwaichi C, Mbachi C, Mba B. Cushing’s syndrome due to a functional thymic neuroendocrine tumor in multiple endocrine neoplasia type 1 syndrome. Cureus (2021) 13(10):e18590. doi: 10.7759/cureus.18590
59. Hasani-Ranjbar S, Rahmanian M, Ebrahim-Habibi A, Soltani A, Soltanzade A, Mahrampour E, et al. Ectopic cushing syndrome associated with thymic carcinoid tumor as the first presentation of MEN1 syndrome-report of a family with MEN1 gene mutation. Fam Cancer (2014) 13(2):267–72. doi: 10.1007/s10689-013-9692-1
60. Teh BT. Thymic carcinoids in multiple endocrine neoplasia type 1. J Intern Med (1998) 243(6):501–4. doi: 10.1046/j.1365-2796.1998.00329.x
61. Meinardi JR, Wolffenbuttel BH, Dullaart RP. Cyclic cushing’s syndrome: a clinical challenge. Eur J Endocrinol (2007) 157(3):245–54. doi: 10.1530/EJE-07-0262
62. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab (2001) 86(12):5658–71. doi: 10.1210/jcem.86.12.8070
63. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97(9):2990–3011. doi: 10.1210/jc.2012-1230
64. De Jong MC, Parameswaran R. Revisiting the evidence for routine transcervical thymectomy for the prevention of thymic carcinoid tumours in MEN-1 patients. Oncology (2022) 100(12):696–700. doi: 10.1159/000526775
65. Elliott PF, Berhane T, Ragnarsson O, Falhammar H. Ectopic ACTH- and/or CRH-producing pheochromocytomas. J Clin Endocrinol Metab (2021) 106(2):598–608. doi: 10.1210/clinem/dgaa488
66. Streuli R, Krull I, Brandle M, Kolb W, Stalla G, Theodoropoulou M, et al. A rare case of an ACTH/CRH co-secreting midgut neuroendocrine tumor mimicking cushing’s disease. Endocrinol Diabetes Metab Case Rep (2017) 2017:17–0058. doi: 10.1530/EDM-17-0058
67. Karageorgiadis AS, Papadakis GZ, Biro J, Keil MF, Lyssikatos C, Quezado MM, et al. Ectopic adrenocorticotropic hormone and corticotropin-releasing hormone co-secreting tumors in children and adolescents causing cushing syndrome: a diagnostic dilemma and how to solve it. J Clin Endocrinol Metab (2015) 100(1):141–8. doi: 10.1210/jc.2014-2945
68. Chrisoulidou A, Pazaitou-Panayiotou K, Georgiou E, Boudina M, Kontogeorgos G, Iakovou I, et al. Ectopic cushing’s syndrome due to CRH secreting liver metastasis in a patient with medullary thyroid carcinoma. Hormones (Athens) (2008) 7(3):259–62. doi: 10.1007/BF03401514
69. Oldfield EH, Doppman JL, Nieman LK, Chrousos GP, Miller DL, Katz DA, et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of cushing’s syndrome. N Engl J Med (1991) 325(13):897–905. doi: 10.1056/NEJM199109263251301
70. Micko A, Hosmann A, Marik W, Bartsch S, Weber M, Knosp E, et al. Optimizing MR imaging for intraoperative image guidance in sellar pathologies. Pituitary (2020) 23(3):266–72. doi: 10.1007/s11102-020-01035-1
71. Vassiliadi DA, Tsagarakis S. DIAGNOSIS OF ENDOCRINE DISEASE: the role of the desmopressin test in the diagnosis and follow-up of cushing’s syndrome. Eur J Endocrinol (2018) 178(5):R201–R14. doi: 10.1530/EJE-18-0007
72. Gubbi S, Hamet P, Tremblay J, Koch CA, Hannah-Shmouni F. Artificial intelligence and machine learning in endocrinology and metabolism: the dawn of a new era. Front Endocrinol (Lausanne) (2019) 10:185. doi: 10.3389/fendo.2019.00185
Keywords: familial neoplasia syndrome, ACTH - independent CS, Cushing's adenoma, pituitary tumor, MEN1 = multiple endocrine neoplasia Type 1, corticotropinoma
Citation: Simonds WF (2023) Expressions of Cushing’s syndrome in multiple endocrine neoplasia type 1. Front. Endocrinol. 14:1183297. doi: 10.3389/fendo.2023.1183297
Received: 09 March 2023; Accepted: 05 June 2023;
Published: 20 June 2023.
Edited by:
Ricardo Correa, University of Arizona, United StatesReviewed by:
Kinga Fulopne Nemeth, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Simonds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William F. Simonds, YmlsbHNAbmlkZGsubmloLmdvdg==